
Abstract
The complexity of the intrinsic oxygen evolution reaction (OER) mechanism, particularly the precise relationships between the local coordination geometry of active metal centers and the resulting OER kinetics, remains to be fully understood. Herein, we construct a series of 3 d transition metal-incorporated cobalt hydroxide-based nanobox architectures for the OER which contain tetrahedrally coordinated Co(II) centers. Combination of bulk- and surface-sensitive operando spectroelectrochemical approaches reveals that tetrahedral Co(II) centers undergo a dynamic transformation into highly active Co(IV) intermediates acting as the true OER active species which activate lattice oxygen during the OER. Such a dynamic change in the local coordination geometry of Co centers can be further facilitated by partial Fe incorporation. In comparison, the formation of such active Co(IV) species is found to be hindered in CoOOH and Co-FeOOH, which are predominantly containing [CoIIIO6] and [CoII/FeIIIO6] octahedra, respectively, but no mono-μ-oxo-bridged [CoIIO4] moieties. This study offers a comprehensive view of the dynamic role of local coordination geometry of active metal centers in the OER kinetics.
Similar content being viewed by others


Introduction
Facilitating the kinetically demanding four-electron transfer process of the OER is the key to improving the efficiency of hydrogen production from water electrolysis1,2,3,4,5. To date, catalysts that deliver the most promising catalytic OER activities are restricted to the expensive platinum group metals, and should ideally be replaced with low-cost 3 d transition metals. Among the currently developed catalysts, transition metal-based oxides and (oxy)hydroxides display state-of-the-art catalytic activity for the OER, which benefit from the similarity of their active structural moieties to key OER intermediates6,7,8,9,10. However, a comprehensive understanding of the structure-activity relationships of oxide-based catalysts on the atomic level still remains to be established.
OER operating under alkaline conditions proceeds via diverse reaction pathways: (i) the adsorbate evolution mechanism (AEM) involves a proton-coupled electron transfer (PCET) process to generate the key MOOH intermediate (M = active site) for activating O2, however, the scaling relation of ΔGO - ΔGOH limits the theoretical OER overpotential of about 0.37 V3,11,12,13,14; (ii) the lattice oxygen mechanism (LOM) features a non-concerted proton-electron transfer (PT/ET) process and enables breaking of the ΔGO - ΔGOH scaling relation, moreover, the lattice oxygen participation during the OER process requires continuous structural stability of the catalysts. In some cases the direct coupling of two metal-oxo species into superoxo-like (O2−) species was reported to lead to a dual-site mechanism (DSM) pathway for the OER3,15,16,17,18,19,20,21. However, the dynamics of the local structural evolution of active metal centers during the catalytic reaction process render the establishment of clear OER pathways a great challenge3,16,17,22,23.
For example, Co-based catalysts have been widely investigated for the OER, and their activities exhibit a strong correlation with the local structural geometry of Co centers2,3,24,25. Octahedrally coordinated Co(III) edge sites commonly emerge as the dominant catalytically active species to trigger the OER2,3, but recent studies of secondary metal-substituted Co3O4 revealed that tetrahedrally coordinated Co(II) centers are more active than octahedrally coordinated Co(III) centers for the formation of active CoOx(OH)y species preceding the OER26. Additionally, combined experiments and theoretical studies demonstrate that the construction of µ2-OH-bridged Co(II)-Co(III) motifs is crucial for the catalytically active Co sites to drive the OER24. Such a fundamental understanding of the true catalytically active species and sites remains a complex analytical task that requires operando time-resolved monitoring of the catalysts under operational conditions. To this end, advanced in situ/operando spectroscopic characterization techniques, such as X-ray absorption spectroscopy (XAS), Raman spectroscopy, and UV–vis spectroscopy, enable researchers to precisely capture more and more key reactive intermediates during the catalytic reaction process2,3,24,27,28. Among them, in situ/operando XAS characterization is commonly implemented for the monitoring of OER catalysts due to its unique sensitivity to the local electronic structures and coordination environments of the active metal centers29,30. Unfortunately, the time resolution of most conventional XAS characterizations generally extends up to the level of minutes or hours depending on the as-prepared sample quality, and therefore fails to establish a full picture of the underlying OER kinetics25,31,32,33. Engineering complementary in situ/operando time-resolved characterization techniques is thus essential for a better understanding of the OER catalysts.
Herein, complementary in situ/operando time-resolved characterization techniques, such as quick-XAS (time resolution down to ca. 50 ms), Raman, UV–vis, electrochemical impedance spectroscopy (EIS), and pulse chronoamperometry, were implemented for systematically investigating inexpensive transition metal-based OER catalysts during the reaction process. These combined techniques allowed us to in situ unravel and identify the true chemical nature of the catalytically active species and sites. We started with nanostructured Co-based hydroxide-like materials (Co-NBs) containing a significant fraction of tetrahedral Co-based building blocks as model OER catalysts, followed by incorporating a second 3d-transition metal (M: Mn, Fe, Ni, Cu, and Zn) to adjust the local electronic structure and coordination environments of Co centers, resulting in a series of CoM-NB catalysts with tuned OER kinetics. Among all these elaborate catalysts, the as-prepared CoFe-NBs exhibit superior OER performance along with robust electrochemical stability for at least 1000 h.
Our operando time-resolved monitoring results reveal that the prepared Co-NBs (CoFe-NBs) undergo a dynamic local structural evolution from the pristine mono-μ-oxo-bridged Cotet(II)-Cooct(III) (Cotet(II)-Feoct(III)) moieties into high-valent CooctIV-O-CooctIII (CooctIV-O-FeoctIII) configurations, which account for the O2 release. In comparison, such a kinetic evolution in the local coordination environment of Co centers is found to be hindered in CoOOH and Co-FeOOH that are rich in di-μ-O(H) bridged [CoIIIO6] octahedra and a mixture of mono/di-μ-oxo- and di-μ-O(H)-bridged [CoIII/FeIIIO6] octahedra, respectively. Our combination of spectroscopic and pulse chronoamperometric investigations quantitatively unravels that partial Fe incorporation in Co-NBs not only promotes the generation of more OER active di-μ-oxo-bridged CooctIV- FeoctIII motifs but also stabilizes the crucial Co(IV) species during the OER cycling process. Results of pH-dependent and kinetic isotropic effect (KIE) investigations firmly suggest that the activation of lattice oxygen into peroxo-like (O22-) species accounts for the O2 generation. This key finding is corroborated by operando Raman characterizations with a clear observation of the according O22- vibrations. Most importantly, our systematic exploration and comprehensive understanding of the dynamics of active species and sites pave the way to the rational design of new heterogeneous catalysts for the OER.
Results
Materials synthesis and characterization
Mn-, Fe-, Ni-, Cu-, and Zn-incorporated cobalt hydroxide-based nanoboxes (CoM-NBs) were prepared through a self-templating strategy (see Experimental Methods) and were thoroughly characterized using a wide range of structural and morphological analysis tools (Supplementary Figs. 1–20). As demonstrated by the powder X-ray diffraction patterns (PXRD) (Supplementary Fig. 1), the observed significant diffraction peaks of Co-NBs matched well with Co1.176(OH)2Cl0.348(H2O)0.456 (COD-4343874)34,35. In sharp contrast, other CoM-NBs except for CoCu-NBs exhibit low-crystalline features with the presence of two main broadened diffraction peaks at 2θ ≈ 15.4° and 26.9° (Supplementary Figs. 5, 10, and 15), corresponding to the (012) and (110) crystal planes of Co1.176(OH)2Cl0.348(H2O)0.456. Such a difference in PXRD patterns between Co-NBs and CoM-NBs indicates that the partial incorporation of secondary metals into Co-NBs is likely to induce local structure distortions (detailed structural analysis: see section on ex situ XAS characterizations). Field-emission scanning electron (FESEM) and transmission electron microscopy (TEM) illustrate a representative nanobox architecture of the obtained CoM-NBs (Fig. 1 and Supplementary Figs. 2, 6, 11, and 16−18). Note that the synthetic parameters, i.e., reaction temperature, reaction solvents, and concentration of the introduced secondary metal ions, play a pivotal role in the construction of the Co-based nanobox architectures (Supplementary Figs. 1–6, 10, and 11 and see details in Discussion I in the SI). High-resolution TEM (HR-TEM) characterizations were further carried out for the morphological and structural characterizations of the prepared CoFe-NBs. The results (Fig. 1b and Supplementary Fig. 7) exhibit a mixture of ordered and disordered domains in CoFe-NBs, further uncovering its prevailing low-crystalline character. Additionally, an interplanar spacing of 2.51 Å was unraveled in CoFe-NBs, which can be assigned to the main exposed (012) crystal plane. This matches well with the corresponding selected area electron diffraction (SAED) pattern (Supplementary Fig. 8). Energy dispersive X-ray spectroscopy (EDX) elemental maps and line scan profiles (Fig. 1c and Supplementary Figs. 8 and 9) unravel a homogeneous elemental distribution of Fe, Co, O, and Cl signals throughout the entire nanobox particle. Similar results were also evident from the HR-TEM and scanning TEM(STEM)-EDX characterizations of CoNi-NBs (Supplementary Figs. 12 and 13). All these results support the successful formation of nanobox architectures of low-crystalline CoM-NBs.

Representative TEM a and HR-TEM b images of CoFe-NBs. c High-angle annular dark field-scanning transmission electron microscopy (HAADF-STEM) image and STEM-EDX elemental maps of CoFe-NBs. d, e Co K-edge XANES spectra and fitting of FT-EXAFS spectra of as-prepared samples vs. references. f WT contour profiles of Co K-edge EXAFS spectra of as-prepared samples vs. references (R = radial distance). g The G(r) functions of Co-NBs and CoFe-NBs derived from neutron PDF analysis (r = real interatomic distance).
To further highlight the OER performance metrics of engineered nanoarchitecture and to gain in-depth insights into the OER kinetics (detailed discussions: see section on operando characterizations), Co3O4, Fe-Co3O4, CoOOH, and Co-FeOOH references were prepared and systematically characterized through morphological and structural analysis (Supplementary Figs. 21−25). The results indicate that Co3O4 and Fe-Co3O4 retain their nanobox architectures. CoOOH displays representative nanoplatelet morphologies, in agreement with a previous study36. The as-prepared Co-FeOOH matches well with the Akaganeite-type phase (FeOOH, PDF no. 34-1266), showing a nanoneedle morphology.
Electronic structure and coordination environment analysis
The local electronic structure and coordination environment of Co-/CoFe-NBs and reference samples were analyzed through ex situ characterization techniques, namely XAS, X-ray photoelectron spectroscopy (XPS), Raman spectroscopy, and neutron pair distribution function (PDF) (Fig. 1d−g and Supplementary Figs. 26−32). As evident from the Co K-edge X-ray absorption near-structure (XANES) spectra, the spectral line shape of Co-NBs overlaps well with that of CoFe-NBs, suggesting a similar local structural geometry of Co centers. To obtain detailed insight into the Co valence states, a linear exploration of the rising absorption edge position derived from the investigated samples and reference cobalt standards is provided (inset plot in Fig. 1d) [Note: the edge positions were extracted from half-height of F/I0 = 0.5]. From the results, the average Co valence states in Co-NBs and CoFe-NBs were determined as +2.32 and +2.25, respectively. This is consistent with the XPS results (detailed discussion in Supplementary Fig. 30), supporting the presence of a mixture of Co(II) and Co(III) ions in the as-prepared Co-/CoFe-NBs. Analysis of Fe K-edge XANES spectra and XPS results offer strong evidence for the presence of Fe(III) ions in the as-prepared CoFe-NBs (Supplementary Figs. 28 and 31). Therefore, a slight decrease in the average Co valence states of CoFe-NBs compared to that of Co-NBs can be attributed to the partial replacement of Co(III) by Fe(III) centers.
Co and Fe K-edge extended X-ray absorption fine structure (EXAFS) spectra were further explored to gain in-depth insights into the local coordination environments of metal centers of the investigated materials (Supplementary Figs. 27 and 28c). As depicted by Co K-edge EXAFS spectra (Supplementary Fig. 27), Co-/CoFe-NBs exhibit similar oscillation features in the k range of 0 to 6 Å-1 compared to that of Co-FeOOH. However, their high k oscillations feature obvious profile differences either in the signal intensities or in the signal wavenumbers compared to all Co-based standards, i.e., Co(OH)2, Co3O4, Co-FeOOH, and CoOOH, reflecting disparate coordination environments of Co centers in Co-/CoFe-NBs compared to reference samples. Fitting of Co K-edge FT-EXAFS spectra (Fig. 1e and Supplementary Table 2) clearly indicated a 6-fold coordination environment of the first Co shells in reference Co(OH)2, Co-FeOOH, and CoOOH, in agreement with the previous PXRD results (Supplementary Fig. 23). The coordination number of the first coordination shell Co-O (CNCo-O) in Co3O4 was calculated as 5.33 due to the presence of mixed tetrahedral and octahedral Co geometries. For Co-NBs and CoFe-NBs, the CNCo-O1/O2 (Supplementary Table 2) was calculated as 5.33 and 4.93, respectively. Results obtained from the electron paramagnetic resonance (EPR) characterizations firmly excluded the appearance of oxygen vacancies in the prepared materials (Supplementary Fig. 30b). Thus, the observed low CNCo-O1/O2 (<6) values of Co-/CoFe-NBs can be attributed to the co-existence of tetrahedrally and octahedrally coordinated Co centers in the crystal structure. The results are also evident from the wavelet transform (WT) of Co K-edge EXAFS spectra. As expected, Co-/CoFe-NBs feature two similar intensity maxima at the k values of 4.1 and 6.2 Å−1 compared to those of reference Co3O4, which correspond to the Cotet/oct-O and Co-Co/Fe scatterings, respectively. Raman spectroscopy characterizations were further conducted to shed more light on the local structural geometries of the investigated samples. The results (detailed discussions in Supplementary Fig. 32) again unravel the existence of both tetrahedral and octahedral Co coordination environments in the investigated CoM-NBs.
Investigations of the Fe K-edge EXAFS spectra suggest a 6-fold coordination environment of the first coordination shells of Fe centers in CoFe-NBs, similar to that of Co-FeOOH (Supplementary Fig. 28 and Supplementary Table 3). This is a piece of evidence to corroborate that only Co ions can occupy the tetrahedral sites in the as-prepared CoFe-NBs, while the octahedral sites are occupied by both Co and Fe ions. A close inspection of the Fe K-edge FT-EXAFS spectra and the corresponding WT contour profiles (Supplementary Fig. 28d, e) notably evidenced a significant backscattering feature at R + ΔR of ca. 3.3 Å in Co-FeOOH, associated with the backscattering of the mono-μ-oxo-bridged Fe-Fe pairs. Such a Fe-Fe backscattering can not be detected in CoFe-NBs, manifesting that all the [FeO6] octahedra in CoFe-NBs are constructed through di-μ-O(H) bridges. From these results, one can also conclude that the mono-μ-oxo-bridged Cotet(II)-Feoct(III) scattering in CoFe-NBs features a similar interatomic distance as the di-μ-O(H)-bridged Fe-Fe scattering (Supplementary Figs. 33 and 34 and see details in Discussion III in the SI).
Taking into account all the structural analyses, structural diagrams of Co-/CoFe-NBs and Co-FeOOH are depicted in Supplementary Fig. 33. Evidently, the observed backscattering features within the interval R + ΔR = 2 to 3 Å in the Co(Fe) K-edge FT-EXAFS spectra of CoFe-NBs arise from the second shell Co-Co/Fe scatterings that exhibit 5 different types of configurations (the second shell Fe-Co/Fe scatterings with 3 different types of configurations). We further performed neutron pair distribution analysis (PDF) to unveil the nature of the local structural geometry of the investigated catalysts. As one can see from Fig. 1g, both Co-NBs and CoFe-NBs exhibit two scattering features at 2.79 and 3.11 Å. Accordingly, these two scattering features can be identified as arising from the di-μ-oxo-bridged Co(III)-Co(III) scattering and the sum of diverse Co(II)-Co(II)/Fe(III) scattering paths (Supplementary Fig. 33a). At the same time, it is also noteworthy that both Co-NBs and CoFe-NBs only feature prominent scattering signals within r = 0–8 Å, further pointing out the presence of short-range orderings within a quite disordered structure, which is largely in agreement with the previous PXRD and HR-TEM discussions (Fig. 1b and Supplementary Figs. 1, 5, and 7).
Electrochemical OER characterization
The as-prepared CoM-NBs were assembled as anodic electrodes and characterized in 1.0 M KOH for the OER (Supplementary Figs. 35−46). We first explored the impact of the synthetic parameters, typically the variations of atomic ratio of Co/Fe(Ni), on the OER activity of CoFe(Ni)-NBs (Supplementary Figs. 35−38). From the results, a distinct change in the OER performance of CoFe-NBs is observed when varying the atomic ratio of Fe/Co. In sharp contrast, the OER performance of CoNi-NBs is not sensitive to the changes in the atomic ratio of Ni/Co. A close inspection of the Tafel plots unravels facilitated OER kinetics along with an increase in the Fe content in CoFe-NBs (Supplementary Fig. 35b), highlighting the crucial role of the construction of mono-μ-oxo-bridged Cotet(II)-Feoct(III) moieties in the OER. A comprehensive understanding of the exact role of Fe incorporation in the OER kinetics of Co-NBs will be discussed in the following section on operando quick-XAS characterizations.
We further assessed the OER performance of Mn-/Fe-/Ni-/Cu-/Zn-incorporated Co-NBs, with similar atomic M/Co atomic ratios (Supplementary Fig. 20) vs. Co3O4, Fe-Co3O4, CoOOH, and Co-FeOOH references (Supplementary Figs. 39–46). Notably, to assess the intrinsic catalytic activities, the currents were normalized by electrochemically active surface area (ECSA) to exclude geometric effects (Supplementary Figs. 36, 38, 39, and 42)3,37,38,39. The overpotentials for achieving a specific current density of 0.02 mA/cmECSA2 are considered for investigation of the intrinsic activity. Figure 2a presents the CV curves of Co-/CoFe-NBs and Co-based references for the OER. To achieve a current density of 0.02 mA/cmECSA2, a minimum overpotential of 213 mV is essential for CoFe-NBs, lower than that of Co-FeOOH (296 mV), Fe-Co3O4 (300 mV), Co3O4 (312 mV), and CoOOH (333 mV). The OER activity of CoFe-NBs also outperforms the most recently studied CoFe-based OER electrocatalysts (Supplementary Table 8), which highlights the benefits of engineering nanoarchitecture catalysts for enhancing the OER activities40,41,42,43. Tafel plots and electrochemical impedance spectroscopy (EIS) measurements were also conducted to shed light on the intrinsic OER kinetics of the investigated catalysts. From the results, a Tafel slope value of 29.16 mV/dec with a charge transfer resistance of 6.95 Ω is observed in CoFe-NBs, which outperformed those of Co-NBs and other Co-based references (Fig. 2c and Supplementary Fig. 43), indicating facilitated OER kinetics with partial Fe incorporation into Co-NBs. To examine whether the observed currents primarily originate from the OER, the Faradaic efficiency was determined based on rotating-ring disk electrode (RRDE) experiments10,13,40. The RRDE results (Fig. 2d) reveal nearly 100% Faradaic efficiency at different disk currents for the investigated CoFe-NBs, thus manifesting that the OER is the primary reaction on the electrode surface.

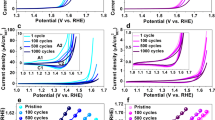
a Normalized CV curves with a 90% iR drop correction (currents were normalized by ECSA, scan rate = 5 mV/s, mass loading = ~ 0.25 mg/cm2, and rotation speed = 1600 rpm). b Comparison of overpotentials at 0.02 mA/cmECSA2 (error bars represent the standard deviation). c Tafel plots. d Faradaic efficiency of CoFe-NBs tested in Ar-saturated 1.0 M KOH through RRDE techniques. e Chronopotentiometry measurements of CoFe-NBs at different current densities. f Long-time stability measurements of CoFe-NBs loaded on carbon paper (Note: the electrolyte was refreshed after every 100 h).
Concerning the electrochemical OER stability tests, the investigated catalysts were first characterized with a standard chronopotentiometry test protocol using a glassy-carbon (GC) electrode as the working electrode (see experimental details and Supplementary Fig. 44). The results corroborate that the elaborate nanostructured CoM-NBs show negligible changes in their OER performance after 40 h stability measurements. In addition, we further performed the multi-potential step chronoamperometry tests to examine the electrochemical stability of the most promising CoFe-NB catalysts and the results again feature a robust OER stability (Fig. 2e). Most importantly, the elaborate nanostructured CoFe-NBs loaded on the carbon paper exhibit superior OER stability at a geometric current density of 20 mA/cm2, retaining their initial OER activity for 1000 h without any significant changes (Fig. 2f). Such effective and robust catalytic activity renders CoFe-NBs attractive OER candidates for practical water electrolysis (Supplementary Table 8).
Post OER characterizations
To gain in-depth insights into the origins of the observed highly active and robust OER activity of CoFe-NBs, ex situ characterizations were carried out for the catalysts after the OER tests (Fig. 3 and Supplementary Figs. 47–55) [Note: In the following section, discussions of the post-OER characterizations refer to the catalysts loaded on carbon paper after durability measurements for 1000 h, as shown in Fig. 2f. Additionally, both CV cycling and chronoamperometry methods were employed for exploring the catalysts after the durability test (Supplementary Fig. 45)]. The morphological properties of the post-catalytic CoFe-NBs were examined via both FESEM and TEM characterizations. The results demonstrated the integrity of the initial nanoscale cubic architectures in CoFe-NBs after the OER durability test (Supplementary Fig. 47). However, analysis of HR-TEM results (Supplementary Figs. 48–50) reveals an underlying phase transformation into (oxy)hydroxides in the post-catalytic CoFe-NBs.

a Co and Fe L-edge EELS and the corresponding STEM-EELS element maps of CoFe-NBs. (Note: EELS data were recorded through integration of the central part of the box particle, and the F signal arises from Nafion.) b, c Co K-edge XANES and FT-EXAFS spectra of CoFe-NBs. d WT contour profiles of Co K-edge EXAFS spectra of CoFe-NBs.
As the OER occurs at the interface between the catalysts and electrolytes, the nature of the local structural geometry of active metal centers plays a crucial role in their related catalytic reaction stability44,45,46,47,48,49,50,51. Most importantly, recent studies have reported that atomistic crystal phase or metal center segregation emerges as a considerable factor in influencing both the catalytic activity and stability of the OER catalysts, especially for Fe-containing catalyst systems3,48,49,51. This motivated us to explore the nature of Fe ions in CoFe-NBs after the long-time OER stability measurements. STEM-electron energy loss spectroscopy (EELS) elemental maps preliminarily allow for resolving the underlying Fe separation from the structure52. Our results (Fig. 3a) indicate homogeneous Co and Fe distributions throughout the entire nanocubic particle before and after the OER. Moreover, EELS analyses unravel similar Co and Fe L-edge features of CoFe-NBs before and after the OER, evidencing that no Fe separation occurred after the OER. The results (Supplementary Table 1) are also evident from ICP-MS characterization, and no significant amounts of Fe species can be detected in the electrolyte. Combination of FESEM/STEM-EDX and surface XPS characterizations shows that the atomic ratio of Co/Fe in CoFe-NBs remains almost unchanged before and after the OER stability tests (Supplementary Fig. 52). Examination of the second deconvoluted peak of the Co 2p3/2 XPS (Supplementary Figs. 29 and 51a) unravels that the post-catalytic CoFe-NBs display an energy shift towards lower binding energy compared to the pristine one, inferring the presence of a large fraction of Co(III) ions after the OER. Note that there is no significant change in the valence state of Fe ions in CoFe-NBs after the OER (Supplementary Figs. 31b and 51b), reflecting that the local structural changes around the Co ions account for the OER.
To further unravel the local electronic structure and coordination environments of CoFe-NBs after the OER stability measurements, Co and Fe K-edge XAS characterizations were carried out (Fig. 3b–d and Supplementary Figs. 53–55). Analysis of Co K-edge XANES spectra (Fig. 3b) reveals an obvious positive energy shift of the white line feature at ca. 7727 eV in all the post-catalytic CoFe-NBs compared to the pristine one, correspondingly their average Co valence state increased to close to +3 (+2.25 in the pristine sample) (Supplementary Fig. 53a). This again points out an underlying local structural evolution that occurred on the Co centers before the OER. Interpretations of Co K-edge EXAFS and FT-EXAFS spectra (Fig. 3c, d and Supplementary Fig. 53b) corroborated that an irreversible structural transformation into CoOOH-like species mostly contributes to the observed spectral feature changes of CoFe-NBs before and after the OER. Other than for the Co K-edge XAS data, we have also analyzed the Fe K-edge XAS data of CoFe-NBs before and after the OER. From the results (Supplementary Fig. 54a, b), a slightly positive energy shift can be still found in the white line features of Fe K-edge XANES spectra for the post-catalytic CoFe-NBs, but the average Fe oxidation state remains almost identical. This manifests that the coordination environment of the Fe centers only undergoes changes beyond the first coordination shell. Such a unique change can be confirmed by investigations of the Fe K-edge EXAFS and FT-EXAFS spectra (Supplementary Figs. 54c, d and 55), in which a newly formed second backscattering feature appears at R + ΔR of ca. 2.3 Å. Apparently, this newly emerging spectral feature cannot be reasonably assigned to the Fe-Fe backscattering, as demonstrated by the XAS analysis of reference Co-FeOOH. To precisely unveil the nature of the active species and sites of the investigated catalysts, operando time-resolved spectroscopic monitoring is required. One can notice that the observed changes in the local electronic structure and coordination environments of Co and Fe centers in the post-catalytic CoFe-NBs are not relevant to the OER stability test time or the way for the stability test (i.e., CV cycling or chronoamperometry). On the one hand, such a phenomenon suggests that the investigated CoFe-NBs mostly undergo a fast restructuring process during the OER, which is not easily captured by ex situ characterizations. On the other hand, our results also strongly validate that the in situ reconstructed species derived from pristine CoFe-NBs enable a quite stable local electronic structure and coordination environments during the long-time OER stability characterizations.
Dynamics of active species and sites derived from operando quick-XAS
The kinetics of local electronic structures and coordination environments of Co-/CoFe-NBs for the OER were explored through operando time-resolved quick-XAS characterizations, as well as reference CoOOH and Co-FeOOH (Fig. 4 and Supplementary Figs. 56–75). Compared to conventional XAS characterizations, the time resolution of quick-XAS characterizations can be down to ms and thus enables unraveling the dynamic role of the local coordination geometries of the active metal centers in the OER mechanisms of the investigated catalysts (Note: In this study, the time resolution of the operando quick-XANES spectra was extended to 1.5 s per spectrum for achieving a reasonable signal-to-noise ratio, as shown in Supplementary Fig. 56. For the operando quick-EXAFS spectra, its time resolution was further extended to 7.5 s per spectrum.)25,31,32. Moreover, in our study, the operando time-resolved Co and Fe K-edge quick-XAS signals were recorded simultaneously, which enables precise identification of the actual role of Co and Fe in the OER. We first performed operando time-resolved quick-XAS characterizations for Co-NBs to understand the exact role of tetrahedrally coordinated Co centers in the OER (Supplementary Figs. 57–60). Our results (see details in Discussion II in the SI) demonstrate that Co-NBs undergo a local structural transformation into the di-μ-oxo-bridged Cooct(IV)-Cooct(III/IV) motifs that act as the real catalytically active sites to trigger the OER. Importantly, the formation of such active sites primarily originates from the mono-μ-oxo-bridged Cotet(II)-Co(II/III) motifs, rather than the di-μ-OH-bridged octahedral Co(II) or di-μ-O(H)-bridged octahedral Co(III) configurations. To explore the crucial role of Fe incorporation in the OER kinetics of CoFe-NBs, operando time-resolved quick-XAS characterizations on the Co and Fe K-edges were further performed (Fig. 4 and Supplementary Figs. 61–66). For the operando time-resolved Co K-edge XANES monitoring (Fig. 4a), our results show a remarkable profile change of the white line peak features at ca. 7727 eV, associated with the dipolar transitions from Co 1 s to 4p hybridized orbitals29,40,41. In detail (Fig. 4b), from immersing into the electrolyte to the applied potentials of 1.3 V vs. RHE, there are no obvious variations on the white line features; however, an increase in the applied potentials from 1.3 to 1.45 V vs. RHE leads to the white line profile changes towards lower peak intensities and higher energy positions. After removing the applied potentials, a slight increase in the white line peak intensity along with a negative energy position shift can be observed. This suggests that CoFe-NBs undergo a partially reversible local structural evolution of Co centers to generate more active species for triggering the OER. Importantly, these in situ-formed active species exhibit partial redox reversibility and can partially convert into the OER resting species again after the reaction. Analysis of the average Co valence state (Fig. 4c) indicates that the aforementioned active species arise from the in situ generated high-valent Cooct(IV) species, and the Cooct(III) species emerge as the OER resting species. Note that the average Co valence state of CoFe-NBs reaches its maximum value of +3.28 at 1.45 V vs. RHE, which is obviously higher than that of Co-NBs (+3.07 at 1.55 V vs. RHE) (Supplementary Fig. 57c). This suggests that partial Fe incorporation facilitates access to the Cooct(IV) species in CoFe-NBs for the OER when compared to Co-NBs.

a Co K-edge XANES spectra; b 2D contour plots of Co K-edge XANES spectra in the energy ranges of 7720 to 7736 eV; c Calculated Co valence states against the OER reaction time and applied potentials; d 2D contour plots of Co K-edge EXAFS spectra; e 3D contour plots of Co K-edge FT-EXAFS spectra and schematic diagram of the structural kinetics; f Schematic illustration of the redox ability of diverse Co coordination geometries.
The local coordination environments of Co centers in CoFe-NBs were further investigated with operando time-resolved EXAFS and FT-EXAFS spectra (Fig. 4d, e and Supplementary Figs. 61a and 63). The results show that the investigated CoFe-NBs undergo similar kinetic changes in the local coordination environments of Co center compared to Co-NBs (see details in Discussion II in the SI). On this basis, at the OER-related potential regions, the newly formed second shell scattering at R + ΔR of ca. 2.4 Å in the Co K-edge FT-EXAFS spectra of CoFe-NBs can be assigned to the backscattering of high-valent Co-Co/Fe pairs (Fig. 4e and Supplementary Fig. 61b). The results are also evident from the fitting of the operando Co K-edge FT-EXAFS spectra (Supplementary Figs. 61b and 62 and Supplementary Table 5). Specifically, with the anodic polarizations, we observe that both CNCo-O1 and CNCo-Co1 experience an obvious increase, and RCo-O1 and RCo-Co1 exhibit trends toward shorter interatomic distance correspondingly (Supplementary Fig. 62). In sharp contrast, either CNCo-O2 or CNCo-Co2 displays a continuous decreasing tendency from 1.3 to 1.5 V vs. RHE but they do not completely vanish after the removal of the applied potentials, suggesting that the di-μ-oxo-bridged Co(II) octahedra are not active toward the OER in both Co-/CoFe-NBs (see details in Discussion II in the SI). Combining the operando monitoring results of Co-NBs and CoFe-NBs points out that the in situ generated CoIV-CoIII/IV/FeIII moieties perform the role of the OER in CoFe-NBs. Mostly, tetrahedral Co(II) sites account for the formation of these high-valent moieties. To precisely distinguish the scattering difference between Co-Co and Co-Fe pairs and to comprehensively understand the OER kinetics in CoFe-NBs, operando time-resolved quick-XAS characterizations on the Fe K-edge were carried out (Supplementary Figs. 64–66). Results from operando time-resolved Fe K-edge XANES spectra (Supplementary Fig. 64a–c) demonstrate that the oxidation states of Fe centers remain almost identical, suggesting that underlying deprotonation of Fe(III)-OH into Fe(IV)-O species is hindered. However, analysis of the Fe K-edge white line signature features slight profile changes on the transitions from Fe 1 s to 4p hybridized orbitals during the OER (Supplementary Fig. 64a, b). This validates that the aforementioned Fe K-edge white line profile changes can be thus ascribed to the underlying coordination environment variations of the second coordination shell of Fe centers, namely the changes arising from the mono-μ-oxo-bridged Fe-Co configurations (Supplementary Fig. 34). As expected, results from operando time-resolved Fe K-edge FT-EXAFS spectra (Supplementary Figs. 64e and 65b) clearly evidence the appearance of a prominent second scattering shell at ca. 2.3 Å, associated with the CooctIV-O-FeoctIII scattering in CoFe-NBs. Therefore, overall structural dynamics that occurred on the Co and Fe centers are depicted in Fig. 4e and Supplementary Fig. 64e together with the operando time-resolved Co and Fe K-edge FT-EXAFS spectra, in which the [CoIIO6] and [FeIIIO6] octahedra are supposed to serve as OER modulators in CoFe-NBs during the reaction process.
The plausible formation of the catalytically active CooctIV-O-CooctIII/FeoctIII moieties derived from the mono-μ-oxo-bridged Cotet(II) configurations in Co-/CoFe-NBs can be also evaluated from operando XAS characterizations of CoOOH and Co-FeOOH, which only contain [CoIIIO6] and [CoII/FeIIIO6] octahedra, respectively (Supplementary Figs. 67–75). Our operando investigations (see details in Discussion III in the SI) distinctly show that the generation of di-μ-oxo-bridged CooctIV-CooctIII (CooctIV-FeoctIII) configurations is found to be more difficult in CoOOH (Co-FeOOH) when they are compared with that of Co-/CoFe-NBs. With these results at hand, we propose that the redox accessibility of diverse Co coordination geometries that enables the formation of the catalytically active CoIV species follows the order as depicted in Fig. 4f, in which constructions of rich mono-μ-oxo-bridged Cotet(II)-Feoct(III) configurations are beneficial for facilitating the OER kinetics. This conclusion is also accordant with the above electrocatalytic characterizations of CoM-NBs for the OER (Fig. 2), where the as-prepared Mn-/Ni-/Cu-/Zn-incorporated Co-NBs display worse OER performance compared to Co-/CoFe-NBs due to their lack of Cotet(II)-Moct(III) moieties (detailed discussion in Supplementary Figs. 30 and 32).
The crucial role of high-valent CoIV species in the OER
To understand the pivotal role of the in situ formed high-valent Co(IV) species in the OER kinetics of Co-/CoFe-NBs, operando Raman characterizations were further performed. As shown in Fig. 5a and Supplementary Fig. 76, pristine Co-/CoFe-NBs feature the two most intense signatures at 522 and 689 cm−1, corresponding to the vibrations of Cotet(II)-O and Cooct(III)-O35,53,54,55,56,57. With the anodic polarizations, the vibration feature of Cotet(II)-O exhibits drastic profile changes and completely vanishes in the OER region. In comparison, the Cooct(III)-O variation retains its pristine spectral features almost during the entire operando monitoring. It is noteworthy that both in Co-NBs and CoFe-NBs their operando Raman spectra feature newly formed vibration features at ca. 464 and 566 cm−1 at the OER-related potentials (Fig. 5a and Supplementary Fig. 76). After removing the applied potentials, these newly formed spectral features change their profiles toward a lower peak intensity (Supplementary Fig. 76b, c). Based on previous studies, it is safe to assign the aforementioned spectral features to the vibration fingerprints of high-valent CoO2 species28,35,58,59,60,61,62,63,64,65. This key observation also supports that the dynamics of local geometric evolution of the tetrahedral Co(II) ions gives rise to the in situ generated catalytically active Co(IV) species.

a 2D contour plots of operando Raman spectra of CoFe-NBs in 1.0 M KOH for the OER. b Representative normalized differential operando UV–vis spectra of CoFe-NBs in 1.0 M KOH for the OER. The differential absorption spectra were obtained from the difference between the spectra at different potentials and in the electrolyte without any applied potentials. c Absorbances of CoFe-NBs at 500 and 540 nm upon CV scan. d Calculated surface accumulated charges of Co-NBs and CoFe-NBs against the OER applied potentials.
To quantitively probe the accumulations of the high-valent Co(IV) species in Co-/CoFe-NBs for the OER, we conducted operando UV–vis monitoring coupled with pulse chronoamperometry characterizations1,6,66,67. Fig. 5b represents the normalized differential absorption (ΔO.D.) spectra of CoFe-NBs for the OER (Co-NBs in Supplementary Fig. 77a). With the anodic polarizations close to the OER potentials, both Co-NBs and CoFe-NBs feature prominent peaks at ca. 500 nm, which undergo profile changes with a further increase in the applied potentials. According to previous studies, the appearance of newly formed absorption features within 500 to 600 nm is evidence for the oxidation of metal centers6,27,66,67. Combination of the above operando quick-XAS, Raman, and UV–vis spectroscopic monitoring (Figs. 4 and 5) clearly points out that the observed changes in the absorption feature at ca. 500 nm are due to the oxidation of Cotet(II) into the catalytically active Co(IV) species. We further recorded the operando UV–vis spectra of the catalysts at fixed wavelengths (500 and 540 nm) along CV scans to gain in-depth insights into the OER kinetics67,68. The results (Fig. 5c and Supplementary Fig. 77b) suggest that three distinct processes contribute to the absorption variations both at 500 and 540 nm. In detail, the absorbance in the first process (I) from 0.8 to 1.0 V vs. RHE remains almost identical, indicating no redox reactions occur in the Co-/CoFe-NBs. As the applied potentials increase higher than 1.0 V vs. RHE (referred to as process II), an increase in the absorbance at 500 and 540 nm is a sign of the oxidation of Cotet(II) into Co(III) species. In the OER-related potentials, the formation of active Co(IV) species contributes to the observed absorbance changes in the process (III), which is mainly due to the difference in the extinction coefficients between Co(III) and Co(IV) species6,27,68. In sharp contrast, with the cathodic CV scan, only one process was involved in both Co-NBs and CoFe-NBs, as confirmed by a similar decreasing absorbance trend recorded at 500 and 540 nm. By combining our previous operando XAS and Raman discussions (Figs. 4 and 5a and Supplementary Figs. 57–77), this decrease can be referred to as the reduction of Co(IV) to Co(III). These results altogether suggest that the in situ formed high-valent Cooct(IV) species are essential for the OER of the investigated catalysts.
With the above operando spectroscopic results at hand, the accumulated oxidized Co(IV) species can be quantitively determined by integrating the reduction currents at specific applied potentials based on the pulse chronoamperometry measurements (Supplementary Fig. 78)1,6,27,67. As shown in Fig. 5d, as the applied potentials increase, the total charges stored in the Co-/CoFe-NBs increase accordingly. Note that the variations of these accumulated total charges against the applied potentials can be divided into two disparate processes, correspondingly associated with the oxidation of Co(II) into Co(III) at lower potentials and Co(II/III) into Co(IV) at higher potentials, respectively, as evident from our above complementary operando monitoring (Figs. 4 and 5). Caution needs to be taken that the accumulated total charges in CoFe-NBs are much higher than those of Co-NBs, manifesting that the incorporation of Fe into Co-NBs facilitates the formation of a high fraction of high-valent Co(IV) species. This is consistent with our previous discussions on the dynamics of active species and sites in Co-/CoFe-NBs during the OER (Fig. 4 and also see details in Discussions II and III in the SI). We further conducted operando EIS tests to shed light on the critical role of Fe incorporation in the OER kinetics of Co-NBs. As shown in Supplementary Fig. 79, the Bode plots of Co-/CoFe-NBs are dominated by two distinct electrochemical processes, which correspond to the OER process at the low-frequency region (<10 Hz) and the surface oxidation process at the middle-frequency region (102–103 Hz)16,26,28,57,58,59. In our study, the surface oxidation processes are assigned to the redox reaction process of Co(III) to Co(IV). From the results (Supplementary Fig. 79), partial Fe incorporation not only shifts the OER process to a lower potential but also moves the surface oxidation process to a higher frequency region, highlighting the benefits of Fe incorporation in facilitating the OER kinetics.
Investigation of the intrinsic OER mechanism
pH dependent studies of the OER activity can offer in-depth insights into the reaction kinetics of the investigated catalysts13,14,15,62,69,70,71. Typically, the conventional AEM pathway involves four PCET processes with the formation of a key hydroperoxide intermediate, which is independent of the variations of pH conditions. In comparison, the generation of superoxo-like (O2−) or peroxo-like (O22-) intermediates contributes to the O-O bond formation in the LOM and the DSM pathways and thereafter results in a PT/ET process, which can be strongly affected by the pH variations. It should be also mentioned that the interaction of two metal-oxo intermediates derived from the redox reaction of individual metal centers contributes to the O-O coupling process in the DSM3,16,17,18,19,20. Our operando monitoring results (Figs. 4 and 5 and Supplementary Figs. 57–79) offer strong evidence for the dynamics of the local structural evolution of Co centers from the initial CotetII-O-CooctIII/FeoctIII into the in situ formed active CoIV-O-CoIII/FeIII moieties in the investigated catalysts before the OER, which excludes the DSM pathway therefore the OER primarily proceeds via the LOM pathway. Previous studies demonstrated that the O22- species is thermodynamically more stable compared to O2−13,18. Most importantly, negatively charged oxygenated O22- species can be identified through a chemical probing approach, such as with tetramethylammonium cations (TMA+)13,14,19,70,72.
As shown in Fig. 6a and Supplementary Fig. 80, it is apparent that both Co-NBs and CoFe-NBs display enhanced OER activity from pH 12.6 to 13.8. In addition, the proton reaction order was calculated as 1.17 for CoFe-NBs and 1.08 for Co-NBs (Fig. 6a and Supplementary Fig. 80a), respectively, suggesting that both catalysts undergo lattice oxygen activation before the generation of O214,62,72. When the OER was characterized in 1 M TMAOH, two catalysts featured a significant decline in their OER activity along with increased Tafel slope values (Fig. 6b and Supplementary Fig. 80b). This is because of the strong electrostatic interaction between the TMA+ ions and the O22- species, thus leading to an inhibition in the OER. KIE characterizations of the catalysts for the OER were performed in the 1 M KOD dissolved in D2O19,73,74,75. From the results (Fig. 6b and Supplementary Fig. 80b), a negligible KIE was observed both in Co-NBs and CoFe-NBs, pointing out that the cleavage of the O-H bond does not emerge as the rate-determining step (RDS). We further performed operando Raman characterizations of CoFe-NBs for the OER in 1 M KOH and 1 M KOD (D2O) solutions. As demonstrated in Fig. 6c, the use of KOD and D2O features similar Raman fingerprints within 450–700 cm−1 in CoFe-NBs during the OER compared to the one characterized in 1 M KOH solution, further supporting that the in situ generated CoO2 species accounts for the OER (Fig. 5a and Supplementary Fig. 76). A close inspection of the operando Raman spectra reveals a newly formed signature at ca. 1095 cm−1 in CoFe-NBs under the OER conditions, which are associated with the vibrations of key O22- intermediates generated in the LOM pathway19,62,76. Taking into account all information from our complementary operando monitoring and electrochemical characterizations, an overall electrochemically driven restructuring process coupled with the OER cycling process is depicted in Fig. 6d and Supplementary Fig. 81, where the RDS is attributed to the O-O bond formation atop the Co sites of the in situ generated CoIV-O-CoIII/FeIII moieties derived from the mono-μ-oxo-bridged CotetII- CooctIII/FeoctIII configurations (Fig. 6e). For a better comparison, a schematic illustration of the AEM pathway and the corresponding calculated free energy diagrams of OER intermediates are provided in Supplementary Figs. 82 and 83. The density functional theory (DFT) results (Fig. 6e and Supplementary Figs. 82 and 83) apparently indicate that the as-investigated catalysts are thermodynamically more favorable via a LOM pathway. We further calculated the Gibbs free energies of OER intermediates adsorbed on the Co sites derived from the octahedrally coordinated Co(II/III) centers in Co-NBs through both AEM and LOM pathways. The results (Fig. 6e and Supplementary Figs. 84 and 85) again manifest that the Cotet sites in Co-NBs are thermodynamically more favorable catalytic centers active compared with the Cooct sites. Notably, the above DFT calculations also suggest that partial Fe incorporation enables faster OER kinetics for facilitating the O-O bond formation (Supplementary Fig. 86), consistent with the key observations on the crucial role of Fe in the OER derived from our complementary operando time-resolved monitoring (Figs. 4 and 5).

a pH dependence of OER activities of CoFe-NBs. The inset plot shows the proton reaction order determined from the equation of ρRHE = (∂log(i)/∂pH). b CV curves of CoFe-NBs for the OER in 1 M KOH in H2O, 1 M KOD in D2O, and 1 M TMAOH in H2O. c Operando Raman spectra of CoFe-NBs recorded in different OER conditions vs. black GC electrode. d Schematic illustration of the pre-equilibrium and the OER cycling processes via the LOM pathways of CoFe-NBs (Note: the pre-equilibrium process represents the transition of Cotet into Cooct). e Calculated free energy diagrams of OER intermediates adsorbed on Co-/CoFe-NBs via the LOM pathways.
Discussion
In summary, we present a convenient secondary metal incorporation strategy to successfully construct a series of nanostructured hydroxide-like CoM-NBs (M: Mn, Fe, Ni, Cu, and Zn) with a substantial fraction of tetrahedrally coordinated Co(II) centers. The as-prepared CoFe-NBs were identified as highly active and robust electrocatalysts for O2 generation, which retain a current density of 20 mA/cm2 with a low overpotential of ca. 225 mV over 1000 h without any obvious changes.
To unravel the origins of the intrinsic high OER activities and to elucidate the nature of electrochemically-driven restructuring of the active species and sites, a combination of diverse operando time-resolved spectroscopic monitoring techniques coupled with electrochemical characterizations was applied. Using operando time-resolved quick-XAS characterization (time scale down to 50 ms) coupled with operando Raman spectroscopy, we observed a kinetic structural evolution in Co-/CoFe-NBs from the mono-μ-oxo-bridged CotetII-CooctIII/FeoctIII motif into the catalytically active CooctIV-CooctIII/FeoctIII configurations connected through di-μ-oxo bridges during the OER. These catalytically active moieties can be reversibly converted into the OER resting state of the di-μ-oxo-bridged CoIII-CoIII/FeIII motifs after the OER. Correspondingly, the OER kinetics of CoOOH and Co-FeOOH, which consist of [CoIIO6] and [CoII/FeIIIO6] octahedra, were explored as references with operando spectroelectrochemical monitoring. The results indicate that these catalysts feature a high energy barrier to trigger the generation of high-valent CooctIV species derived from the di-μ-O(H)-bridged Co octII/III-CooctII/III/FeoctIII configurations compared to that of Co-/CoFe-NBs with dominant mono-μ-oxo-bridged CotetII-CooctIII/FeoctIII configurations. Based on these results, the overall redox accessibility of the diverse local Co coordination geometries enabling the formation of the catalytically active CoIV species for O-O bond formation follows the order: CotetII-FeoctIII moieties (mono-μ-oxo bridge) > CotetII-CooctIII moieties (mono-μ-oxo bridge) > CooctIII-CooctIII moieties (di-μ-O(H) bridge) > CooctII-CooctII moieties (di-μ-OH bridge) > Co octII-FeoctIII moieties (di-μ-OH bridge). This highlights the importance of optimizing the local coordination geometries around the metal centers to improve OER kinetics.
Operando UV–vis spectrochronoamperometry studies quantitatively unraveled a strong correlation between the accumulated high-valent CoIV species and the observed OER activity. At the same time, partial Fe incorporation not only enables the formation of a high fraction of CoIV species but can also stabilize the CoIV species during the OER cycling process, thus offering high and robust OER activity. Furthermore, operando Raman spectra coupled with pH-dependent and KIE investigations demonstrate the formation of peroxo-like (O22-) species in CoFe-NBs during the OER, suggesting that the generation of O2 proceeds via a LOM pathway. The results furthermore agree with our DFT simulations. Most importantly, we provide a systematic investigation into the crucial role of the oxidation states and metal coordination geometries over extended OER periods (~1000 h stability tests). This will inspire future studies on OER catalysts via a kinetic optimization of the reactive mono-μ-oxo coordinations, i.e., through incorporation of different metal centers or tuning their oxidation states to facilitate the formation of high-valent species with fast OER kinetics. We expect that our comprehensive and mechanistic studies into the structure-activity relationships of OER catalysts will expedite the rational design of advanced water electrolysis electrocatalysts to sustainably generate clean hydrogen as an energy carrier.
Methods
Chemicals
Ammonium fluoride (≥98%), cetrimonium bromide (≥99%), cobalt(II) acetate tetrahydrate (≥98%), cobalt (II) nitrate hexahydrate (≥98%), cobalt(II) chloride (≥98%), copper(II) chloride (≥99%), deuterium oxide (≥99.9%), iron(II) chloride, iron(III) nitrate nonahydrate (≥98%), 2-methylimidazole (≥99%), manganese(II) chloride (≥99%), nickel(II) chloride (≥98%), potassium deuteroxide solution (40 wt.% in D2O), potassium hydroxide (≥99.99%), zinc(II) chloride (≥98%), hydrogen peroxide (≥33%), sodium hydroxide (≥97%), tetramethylammonium hydroxide solution (25 wt.% in H2O), urea (≥99.5%), and RuO2 (99.9%) were purchased from Sigma-Aldrich. All chemicals were used as received without any further purification. Carbon paper (TGP-H-60) was purchased from Alfa Aesar.
Synthesis of Co hydroxide-based nanoboxes (Co-NBs)
Co-NBs were prepared through a convenient self-templated approach using ZIF-67 precursors as template40. First, cubic ZIF-67 precursors were obtained based on a room-temperature coprecipitation method. In detail, a total of 580 mg of cobalt(II) nitrate hexahydrate and 10 mg of cetrimonium bromide were first dissolved into 20 mL of deionized (DI) water (denoted as solution A). Solution B was prepared by dissolving 9.08 g of 2-methylimidazole into 140 mL of DI water. Then, solution A was injected into solution B under magnetic stirring for 30 min at room temperature. ZIF-67 precursors were obtained through centrifugation and washing with ethanol several times. Afterward, the as-obtained ZIF-67 products were dried at 70 °C overnight for further use. To prepare Co-NBs, 40 mg of ZIF-67 precursors were homogeneously dispersed into 30 mL of ethanol via ultrasonication. Meanwhile, a total of 15 mg of CoCl2 was dissolved into 5 mL of DI water, and the as-prepared CoCl2 solution was then rapidly injected into ZIF-67 suspension under magnetic stirring for 20 min. The obtained Co-NB precursors were centrifuged, washed with DI water and ethanol at least three times, and dispersed into 30 mL of ethanol again. Finally, 5 ml of DI water was injected into the as-prepared suspension of Co-NBs, and the mixed suspension was heated to 75 °C and refluxed for 30 min under magnetic stirring. The obtained Co-NB precipitate was collected by centrifugation, washed several times with DI water and ethanol, and dried at 70 °C overnight. To explore the role of synthetic parameters on the structural and morphological properties of the targeted products, Co-NBs products were synthesized by varying the synthetic parameters, i.e., pure ethanol with refluxing at 75 °C, pure DI water with refluxing at 75 °C, and ethanol/DI water of 30:5 (mL) with room temperature stirring.
Synthesis of Co-NBs-OAc
The preparation of Co-NBs-OAc was similar to the synthetic strategy for Co-NBs, except for replacing cobalt(II) chloride precursor with cobalt(II) acetate tetrahydrate.
Synthesis of M-substituted cobalt hydroxide-based nanoboxes (CoM-NBs; M= Mn, Fe, Ni, Cu, and Zn)
The preparation of CoM-NBs was similar to the synthetic strategy for Co-NBs except for replacing CoCl2 with MCl2. The products obtained with different amounts of MCl2 (5 mg, 10 mg, 15 mg, and 20 mg) were denoted as CoM-NBs-1, CoM-NBs-2, CoM-NBs-3, and CoM-NBs-4. (Unless otherwise mentioned, the abbreviation CoM-NBs refers to the products prepared with the synthetic parameters of ethanol: water of 6: 1 (volume ratio), 15 mg of MCl2, and refluxing at 75 °C.)
Synthesis of Co3O4 and Fe-Co3O4
To prepare Co3O4 and Fe-Co3O4, the as-obtained Co-NBs and CoFe-NBs-3 precursors were placed into a muffle furnace and calcined at 350 °C in air for 2 h with a ramping rate of 2 °C min−1.
Synthesis of cobalt oxyhydroxide (CoOOH)
CoOOH was prepared as follows: a total of 580 mg of cobalt(II) nitrate hexahydrate was dissolved into 40 mL of DI water. Then, 25 mL of 0.1 M NaOH solution was added dropwise into the above Co-based solution under magnetic stirring for 15 min and held at 45 °C for 15 min. The obtained pink precipitate was collected by centrifugation, washed several times with DI water, and dispersed into 20 mL of DI water. Afterward, 5 mL of 8 M NaOH solution was added dropwise together with 10 mL of 30 wt.% H2O2 solution into the as-prepared Co-based suspension. The mixed suspension was stirred at 45 °C for 24 h. In the end, the as-obtained black precipitate was collected by centrifugation, washed several times with DI water, and dried at 70 °C overnight.
Synthesis of Co-substituted iron oxyhydroxide (Co-FeOOH)
Co-FeOOH was synthesized based on our previous study with a slight modification40. A total of 0.3 mmol of cobalt(II) nitrate hexahydrate and 0.25 mmol of iron(III) nitrate nonahydrate were dissolved into 12 mL of DI water along with 2 mmol of urea and 1 mmol of ammonium fluoride. Then, the obtained solution was sealed into a Teflon-lined stainless steel autoclave (20 mL) and was reacted at 120 °C for 15 h. Finally, Co-FeOOH products were obtained by centrifugation and dried at 70 °C overnight.
Materials characterizations
A STOE STADI P diffractometer (transmission mode, Ge monochromator, Mo Kα radiation (0.7093 Å), 50 kV and 40 mA) was used for recording the PXRD patterns. FESEM (Zeiss Supra 50 VP) and TEM (FEI Tecnai G2 Spirit) were used to characterize the morphological properties of the as-prepared products. The elemental compositions of the investigated samples were analyzed using FESEM/TEM-energy-dispersive X-ray spectroscopy (EDX) (Note: a 5 nm thick Pt/Au overlayer was coated on the samples to reduce the charging interference effect). HR-TEM, HAADF-STEM, EELS, and STEM-EDX element mappings were carried out on a FEI Titan Themis (300 kV) instrument. The instrument is equipped with a hexapole-type aberration corrector for STEM (CEOS DCOR) and a Super EDX system with a CEFID energy filter. Neutron diffraction patterns were performed at the D4c instrument of the Institute Laue-Langevin (Grenoble, France) using a Neutron wavelength (λ) of 0.4957 Å in a sealed vanadium cylindrical container at ambient temperature and pressure. The measurements were performed in the Q range of 0.38–23.53 Å–1. The PDF was analyzed based on Fourier transformed S(Q) using standard software at the ILL. A PerkinElmer PHI 1600 ESCA system with Mg Kα radiation (1253.6 eV) was used for the XPS studies. Room temperature EPR spectra were characterized based on a Bruker MiniScope MS 5000 spectrometer.
Ex situ and operando Raman measurements
A Renishaw Raman scope or InVia Qontor (Ar+ laser, 532) was employed for ex situ and operando Raman spectra studies40. Ex situ characterizations were performed with a solid power sample pressed on a quartz glass substrate. A screen-printed electrode system (SPCE, Dropsens, DRP-110) was used for conducting the operando Raman characterizations. To capture the OER intermediates, a thin layer of Au@SiO2 was covered on the surface of the investigated catalysts to acquire the surface-enhanced Raman signals. During the tests, a stepwise chronoamperometry method was applied to acquire the operando Raman signals.
Operando UV–vis measurements
Operando UV–vis spectra were recorded on a Lambda 650 S Perkin Elmer UV-visible spectrometer in the range of 350–800 nm using a HELLMA Quartz precision cell (10 mm). The operando measurements were performed in an in-house designed electrochemical quartz precision cell equipped with fluorine-doped tin oxide (FTO) as the working electrode (loading amounts of catalysts is ca. 0.05 mg/cm2). During the measurements, CV cycling and chronoamperometry with a stepwise increase of the applied potentials were performed to accumulate operando UV–vis spectroscopy signals.
Ex situ and operando XAS measurements
Ex situ and operando Co and Fe K-edge XAS experiments were performed at the SuperXAS-X10DA beamline at the Swiss Light Source (PSI, Switzerland) (ex situ sample of CoOOH, and ex situ and operando samples of Co-NBs and CoFe-NBs) and at the KMC-2 beamline at the Helmholtz Zentrum Berlin (HZB) BESSY II (Berlin, Germany) (ex situ and operando samples of CoOOH and Co-FeOOH). All the XAS data were recorded via transmission (TR, SuperXAS-X10DA) or fluorescence (FL, KMC-2) modes with a Si(111) double crystal monochromator (DCM) cooled with liquid nitrogen. The experiments at the SuperXAS-X10DA beamline were run in the quick-scanning extended X-ray absorption fine-structure (QEXAFS) mode with the monochromator oscillation frequency of 60 Hz, which allows for the collection of 120 spectra within the energy ranges from 7000 to 8450 eV per minute (up and down modes with different energy scan directions). Therefore, the time resolution of Co (7000 to 7650 eV) or Fe K-edge (7650 to 8450 eV) QXANES and QEXAFS spectra was determined as ca. 50 ms and ca. 250 ms, respectively. To achieve a reasonable signal-to-noise ratio of the collected QEXAFS spectra, several subsequently collected spectra need to be averaged together, as shown in Supplementary Fig. 56. Therefore, the resulting time resolution of QXANES and QEXAFS for Co-/CoFe-NBs was thus 1.5 s and 7.5 s, respectively. Operando experiments at KMC-2 were run in the conventional stepwise scan mode with an acquisition time of 10 min per spectrum, and each sample was measured three times.
For ex situ XAS characterizations, the solid powder samples were mixed with cellulose and pressed into pellets to achieve an absorption step of ca. 1. Energy was calibrated based on reference Co (7709 eV) and Fe (7112 eV) foils. Operando XAS experiments were performed with an in-house developed electrochemical cell, as reported in our previous study40. The operando XAS experiments were characterized with a standard three-electrode system with Hg/HgO (1.0 M KOH) reference electrode and graphite rod counter electrode. Carbon paper loaded with the catalysts (ca. 2.0 mg/cm2) was used as the working electrode. During the experiments, a chronoamperometry method with a stepwise increase of the applied potential within 1.0 to 1.525 V was used. Importantly, the collected XAS signals at the constant potential need to be repeated 3–5 times. The collected raw data from the SuperXAS-X10DA beamline were analyzed using the ProQEXAFS software, and the collected raw data from the KMC-2 were directly analyzed using the ATHENA software packages. The k2-weighted and Fourier transform (FT) EXAFS data were analyzed in the k-range from 0 to 12 Å−1 and R-range from 0 to 6 Å, respectively. No phase correction was applied in the EXAFS spectra. Fitting of FT-EXAFS spectra was conducted with the R-range within 1 to 3.5 Å. S02 was obtained based on the fitting of Co/Fe foil, which was determined as 0.9 (SuperXAS-X10DA) and 0.75 (KMC-2), respectively.
Preparation of working electrodes using a rotating disk electrode (RDE)
As a typical protocol for the preparation of the working electrode16, 3.0 mg of catalysts were dispersed in 600 μL of ethanol with 1.2 mg of carbon black and 30 μL of 5 wt% Nafion solution under ultrasonication for 1 h. Then, the 10 μL of the catalyst inks were loaded on a glassy carbon rotating disk electrode (GC-RDE, Metrohm, diameter = 5 mm, and loading mass ~ 0.25 mg/cm2). (Note: For the preparation of working electrodes of operando Raman, UV–vis, and XAS characterizations, the catalysts inks were prepared without carbon black additive).
Electrocatalytic performance measurements
Electrochemical OER characterizations were carried out in O2-saturated 1.0 M KOH (pH = 13.7 ± 0.02) at room temperature. The tests were performed with a standard three-electrode system (Metrohm Autolab PGSTAT302N potentiostat) with a reference Hg/HgO electrode (1.0 M KOH), counter graphite rod electrode, and working GC-RDE electrode, respectively (Note: The calibration of the reference Hg/HgO electrode was performed in an H2-saturated 1 M KOH electrolyte, where Pt rods served as both the working and counter electrodes. CV scans were then performed at a scan rate of 1 mV/s, and the potentials were determined at zero current. In this study, the reference Hg/HgO electrode potential was determined as 0.095 V vs. SHE (standard hydrogen electrode)). During the measurements, 20 cycles of CV with a fast scan rate of 50 mV/s were first applied without any rotations. Then, the CV curve with a low scan rate of 5 mV/s was collected under a rotation speed of 1600 rpm. Tafel analysis was performed based on the back scan of the CV curve with a low scan rate of 5 mV/s. All potentials were normalized to RHE based on E(RHE) = E + 0.095 + 0.059 × pH. In this study, a 90% iR-compensation was implemented for the collected electrochemical data, where R is the uncompensated ohmic contact resistance measured by EIS at the open-circuit voltage. As a typical example, the R value of commercial RuO2-loaded GC-RHE was determined as 6.62 ± 0.15 Ω. To ensure data reproducibility, all the tests were repeated at least 3 times. EIS measurements were conducted within a frequency range from 0.01 Hz to 10 kHz with a 5 mV amplitude. The ECSA was determined by CV methods in a non-Faradaic region. Then, the ECSA was calculated as follows: ECSA=geometric surface area × Cdl/C0 (Cdl is the double-layer capacitance (Cdl) measured from CV cycling and C0 is a constant of 0.04 mF/cm2). Faradaic efficiency was determined through rotating ring-disk electrode (RRDE) characterizations as follows: Faradaic efficiency=Iring/(Idisk*N). Iring and Idisk represent the ring and disk currents, respectively. N is the current collection efficiency of the RRDE with a constant value of 0.2. During the measurements, the applied potential of the rising electrode was set to a constant value of 0.3 V vs. RHE to ensure the oxygen reduction reaction. Durability tests were conducted with both GC-RDE (diameter of 5 mm, loading mass ~0.25 mg/cm2) and carbon paper electrodes (1 × 1 cm2, loading mass ~ 1 mg/cm2) through chronopotentiometry methods at 20 mA/cm2 and CV cycling for 1000 cycles.
DFT calculations
All DFT simulations were performed using the CP2K package. The Kohn-Sham DFT within the hybrid Gaussian and plane waves framework was applied for the electronic structure calculations. Goedecker-Teter-Hutter (GTH) pseudopotentials are used to describe the interactions within the atomic cores and the molecular orbitals of the valence electrons are expanded in Gaussian-type orbitals77. DZVP-MOLOPT-GTH basis sets were used for all the elements. The lane-wave cut-off energy was set up as 400 Ry. The main exchange and correlation scheme is PBE with D3 correction for the long-range dispersion interactions78. The theoretical model was constructed from the Co1.176(OH)2Cl0.348(H2O)0.456 (COD-4343874) (012) -terminated surface. Notably, according to our operando monitoring, all Cl atoms were replaced with O atoms, and the interatomic distances of Co-Co/Fe pairs were also adjusted correspondingly. Fe atoms were inserted afterward by substituting 1/2 octahedral Co atoms which are connected by the tetrahedral Co sites. A built 5 × 5 × 1 supercell with a total of 126 atoms was used for all simulations (Supplementary Data 1). Electronic structure optimizations are converged to an accuracy in energy of 5 × 10−7 Hartree. All proposed structures have been optimized with the Broyden-Fletcher-Goldfarb-Shanno (BFGS) scheme with a force threshold of 1 × 10−3 Hartree/Bohr. A 15 Å thickness of vacuum layer was added to the built supercell (both above and below) to remove the interaction effects arising from the surfaces.
Under alkaline conditions, the following equations (Eqs. 1–4) are used for describing the AEM pathway:
where * is active adsorption site, and OH*, O*, and OOH* represent the OER intermediates.
Gibbs free energy changes (ΔG) of the individual elementary step in the AEM pathway can be calculated as follows:
Herein, the theoretical overpotential ηtheory is calculated as:
In the above equations, EM* is the binding energy for the OER intermediates adsorption, ΔZPE is the difference in zero-point energy, and ΔS represents the changes of entropy corrections. U is the additional bias for computing the reaction-free energy in each step. In this study, the temperature effects are not considered, which is because all the reactions occurred at room temperature.
In comparison, the LOM pathway can be described by the following equations.
In the above equations, Ov is referred to as oxygen vacancy. The ΔG of the individual elementary step was calculated as follows:
Data availability
The authors declare that the data supporting the findings of this study are available within the Supplementary Information files. Source data are provided within the paper. The atomic coordinates of the optimized computational models generated in this study are provided in Supplementary Data 1. Source data are provided with this paper.
References
Nong, H. N. et al. Key role of chemistry versus bias in electrocatalytic oxygen evolution. Nature 587, 408–413 (2020).
Mefford, J. T. et al. Correlative operando microscopy of oxygen evolution electrocatalysts. Nature 593, 67–73 (2021).
Zhao, Y. et al. Oxygen evolution/reduction reaction catalysts: from in situ monitoring and reaction mechanisms to rational design. Chem. Rev. 123, 6257–6358 (2023).
Hausmann, J. N. et al. Understanding the formation of bulk- and surface-active layered (oxy)hydroxides for water oxidation starting from a cobalt selenite precursor. Energy Environ. Sci. 13, 3607–3619 (2020).
Chen, Z., Yang, H., Kang, Z., Driess, M. & Menezes, P. W. The pivotal role of s-, p-, and f-Block metals in water electrolysis: status quo and perspectives. Adv. Mater. 34, 2108432 (2022).
Rao, R. R. et al. Spectroelectrochemical Analysis of the water oxidation mechanism on doped nickel oxides. J. Am. Chem. Soc. 144, 7622–7633 (2022).
Tian, T., Zheng, M., Lin, J., Meng, X. & Ding, Y. Amorphous Ni–Fe double hydroxide hollow nanocubes enriched with oxygen vacancies as efficient electrocatalytic water oxidation catalysts. Chem. Commun. 55, 1044–1047 (2019).
Tian, T. et al. Study of the active sites in porous nickel oxide nanosheets by manganese modulation for enhanced oxygen evolution catalysis. ACS Energy Lett. 3, 2150–2158 (2018).
Reith, L. et al. In situ detection of iron in oxidation states ≥ IV in cobalt-iron oxyhydroxide reconstructed during oxygen evolution reaction. Adv. Energy Mater. 13, 2203886 (2023).
Wang, J. et al. Redirecting dynamic surface restructuring of a layered transition metal oxide catalyst for superior water oxidation. Nat. Catal. 4, 212–222 (2021).
Sun, Y. et al. Covalency competition dominates the water oxidation structure-activity relationship on spinel oxides. Nat. Catal. 3, 554–563 (2020).
Seh, Z. W. et al. Combining theory and experiment in electrocatalysis: Insights into materials design. Science 355, eaad4998 (2017).
Huang, Z.-F. et al. Chemical and structural origin of lattice oxygen oxidation in Co–Zn oxyhydroxide oxygen evolution electrocatalysts. Nat. Energy 4, 329–338 (2019).
Huang, Z.-F. et al. Tuning of lattice oxygen reactivity and scaling relation to construct better oxygen evolution electrocatalyst. Nat. Commun. 12, 3992 (2021).
Wei, Y. et al. Triggered lattice-oxygen oxidation with active-site generation and self-termination of surface reconstruction during water oxidation. Proc. Natl Acad. Sci. USA 120, e2312224120 (2023).
Zhao, Y. et al. Optimized NiFe-based coordination polymer catalysts: sulfur-tuning and operando monitoring of water oxidation. ACS Nano 16, 15318–15327 (2022).
Wan, W. et al. Mechanistic insight into the active centers of single/dual-atom Ni/Fe-based oxygen electrocatalysts. Nat. Commun. 12, 5589 (2021).
Zhang, N. & Chai, Y. Lattice oxygen redox chemistry in solid-state electrocatalysts for water oxidation. Energy Environ. Sci. 14, 4647–4671 (2021).
Yao, N. et al. Atomically dispersed Ru oxide catalyst with lattice oxygen participation for efficient acidic water oxidation. Chem 9, 1882–1896 (2023).
Zhang, T. et al. Spatial configuration of Fe–Co dual-sites boosting catalytic intermediates coupling toward oxygen evolution reaction. Proc. Natl Acad. Sci. USA 121, e2317247121 (2024).
Zhang, N. et al. Lattice oxygen activation enabled by high-valence metal sites for enhanced water oxidation. Nat. Commun. 11, 4066 (2020).
Chung, D. Y. et al. Dynamic stability of active sites in hydr(oxy)oxides for the oxygen evolution reaction. Nat. Energy 5, 222–230 (2020).
Zheng, W. & Lee, L. Y. S. Metal-organic frameworks for electrocatalysis: catalyst or precatalyst? ACS Energy Lett. 6, 2838–2843 (2021).
Bergmann, A. et al. Unified structural motifs of the catalytically active state of Co(oxyhydr)oxides during the electrochemical oxygen evolution reaction. Nat. Catal. 1, 711–719 (2018).
Timoshenko, J. et al. Deciphering the structural and chemical transformations of oxide catalysts during oxygen evolution reaction using quick X-ray absorption spectroscopy and machine learning. J. Am. Chem. Soc. 145, 4065–4080 (2023).
Wang, H.-Y. et al. In operando identification of geometrical-site-dependent water oxidation activity of spinel Co3O4. J. Am. Chem. Soc. 138, 36–39 (2016).
Francàs, L. et al. Spectroelectrochemical study of water oxidation on nickel and iron oxyhydroxide electrocatalysts. Nat. Commun. 10, 5208 (2019).
Moysiadou, A., Lee, S., Hsu, C.-S., Chen, H. M. & Hu, X. Mechanism of oxygen evolution catalyzed by cobalt oxyhydroxide: cobalt superoxide species as a key intermediate and dioxygen release as a rate-determining step. J. Am. Chem. Soc. 142, 11901–11914 (2020).
Timoshenko, J. & Roldan Cuenya, B. In Situ/Operando electrocatalyst characterization by X-ray absorption spectroscopy. Chem. Rev. 121, 882–961 (2021).
Risch, M., Morales, D. M., Villalobos, J. & Antipin, D. What X-Ray absorption spectroscopy can tell us about the active state of earth-abundant electrocatalysts for the oxygen evolution reaction. Angew. Chem. Int. Ed. 61, e202211949 (2022).
Müller, O., Nachtegaal, M., Just, J., Lützenkirchen-Hecht, D. & Frahm, R. Quick-EXAFS setup at the SuperXAS beamline for in situ X-ray absorption spectroscopy with 10 ms time resolution. J. Synchrotron Rad. 23, 260–266 (2016).
Kim, B.-J. et al. Functional role of Fe-Doping in Co-Based Perovskite oxide catalysts for oxygen evolution reaction. J. Am. Chem. Soc. 141, 5231–5240 (2019).
Lin, S.-C. et al. Operando time-resolved X-ray absorption spectroscopy reveals the chemical nature enabling highly selective CO2 reduction. Nat. Commun. 11, 3525 (2020).
Ma, R. et al. Tetrahedral Co(II) Coordination in α-Type cobalt hydroxide: rietveld refinement and X-ray absorption spectroscopy. Inorg. Chem. 45, 3964–3969 (2006).
Zhao, X. et al. Spatiotemporally and chemically resolved imaging of electrocatalytic oxygen evolution on single nanoplates of cobalt-layered hydroxide. J. Am. Chem. Soc. 145, 20897–20906 (2023).
Yang, J., Liu, H., Martens, W. N. & Frost, R. L. Synthesis and characterization of cobalt hydroxide, cobalt oxyhydroxide, and cobalt oxide nanodiscs. J. Phys. Chem. C. 114, 111–119 (2010).
Sun, S., Li, H. & Xu, Z. J. Impact of surface area in evaluation of catalyst activity. Joule 2, 1024–1027 (2018).
Huang, Z.-F. et al. Design of efficient bifunctional oxygen reduction/evolution electrocatalyst: recent advances and perspectives. Adv. Energy Mater. 7, 1700544 (2017).
Wei, C. et al. Recommended practices and benchmark activity for hydrogen and oxygen electrocatalysis in water splitting and fuel cells. Adv. Mater. 31, 1806296 (2019).
Zhao, Y. et al. Dynamics and control of active sites in hierarchically nanostructured cobalt phosphide/chalcogenide-based electrocatalysts for water splitting. Energy Environ. Sci. 15, 727–739 (2022).
Zhao, Y., Wan, W., Erni, R., Pan, L. & Patzke, G. R. Operando spectroscopic monitoring of metal chalcogenides for overall water splitting: new views of active species and sites. Angew. Chem. Int. Ed. 63, e202400048 (2024).
Wang, H., Zhuo, S., Liang, Y., Han, X. & Zhang, B. General self-template synthesis of transition-metal oxide and chalcogenide mesoporous nanotubes with enhanced electrochemical performances. Angew. Chem. Int. Ed. 55, 9055–9059 (2016).
Zhuo, S. et al. Dual-template engineering of triple-layered nanoarray electrode of metal chalcogenides sandwiched with hydrogen-substituted graphdiyne. Nat. Commun. 9, 3132 (2018).
Huang, Y. et al. In Situ/Operando Soft X-ray spectroscopic identification of a Co4+ intermediate in the oxygen evolution reaction of defective Co3O4 nanosheets. J. Phys. Chem. Lett. 13, 8386–8396 (2022).
Li, A. et al. Atomically dispersed hexavalent iridium oxide from MnO2 reduction for oxygen evolution catalysis. Science 384, 666–670 (2024).
Zhong, X. et al. Stabilization of layered lithium-rich manganese oxide for anion exchange membrane fuel cells and water electrolysers. Nat. Catal. 7, 546–559 (2024).
Hausmann, J. N. & Menezes, P. W. Effect of surface-adsorbed and intercalated (Oxy)anions on the oxygen evolution reaction. Angew. Chem. Int. Ed. 61, e202207279 (2022).
Kuai, C. et al. Phase segregation reversibility in mixed-metal hydroxide water oxidation catalysts. Nat. Catal. 3, 743–753 (2020).
Kuai, C. et al. Revealing the dynamics and roles of iron incorporation in nickel hydroxide water oxidation catalysts. J. Am. Chem. Soc. 143, 18519–18526 (2021).
Yang, F. et al. Spatially and chemically resolved visualization of Fe incorporation into NiO octahedra during the oxygen evolution reaction. J. Am. Chem. Soc. 145, 21465–21474 (2023).
Haase, F. T. et al. Role of Fe decoration on the oxygen evolving state of Co3O4 nanocatalysts. Energy Environ. Sci. 17, 2046–2058 (2024).
Ruiz Caridad, A., Erni, R., Vogel, A. & Rossell, M. D. Applications of a novel electron energy filter combined with a hybrid-pixel direct electron detector for the analysis of functional oxides by STEM/EELS and energy-filtered imaging. Micron 160, 103331 (2022).
Han, J. et al. Lattice oxygen activation through deep oxidation of Co4N by Jahn−Teller-active dopants for improved electrocatalytic oxygen. Angew. Chem. Int. Ed. 63, e202405839 (2024).
Zhang, R. et al. Tracking the role of defect types in Co3O4 structural evolution and active motifs during oxygen evolution reaction. J. Am. Chem. Soc. 145, 2271–2281 (2023).
Jing, C. et al. Electrocatalyst with dynamic formation of the dual-active site from the dual pathway observed by in situ Raman spectroscopy. ACS Catal. 12, 10276–10284 (2022).
Zheng, W., Liu, M. & Lee, L. Y. S. Electrochemical instability of metal–organic frameworks: in situ spectroelectrochemical investigation of the real active sites. ACS Catal. 10, 81–92 (2020).
Xiao, Z. et al. Operando identification of the dynamic behavior of oxygen vacancy-rich Co3O4 for oxygen evolution reaction. J. Am. Chem. Soc. 142, 12087–12095 (2020).
Hu, Y. et al. Interfacial evolution on Co-based oxygen evolution reaction electrocatalysts probed by using in situ surface-enhanced Raman spectroscopy. Anal. Chem. 95, 1703–1709 (2023).
Deng, X. et al. Understanding the roles of electrogenerated Co3+ and Co4+ in Selectivity-Tuned 5-Hydroxymethylfurfural Oxidation. Angew. Chem. Int. Ed. 60, 20535–20542 (2021).
Yao, N., Zhu, J., Jia, H., Cong, H. & Luo, W. Identification of in situ generated iron-vacancy induced oxygen evolution reaction kinetics on cobalt iron Oxyhydroxide†. Chin. J. Chem. 42, 343–350 (2024).
Huang, J. et al. Modifying redox properties and local bonding of Co3O4 by CeO2 enhances oxygen evolution catalysis in acid. Nat. Commun. 12, 3036 (2021).
Ye, P. et al. Lattice oxygen activation and local electric field enhancement by co-doping Fe and F in CoO nanoneedle arrays for industrial electrocatalytic water oxidation. Nat. Commun. 15, 1012 (2024).
Wang, S. et al. Identifying the geometric catalytic active sites of crystalline cobalt oxyhydroxides for oxygen evolution reaction. Nat. Commun. 13, 6650 (2022).
Chen, Z. et al. Reversible structural evolution of NiCoOxHy during the oxygen evolution reaction and identification of the catalytically active phase. ACS Catal. 8, 1238–1247 (2018).
Yao, N. et al. Intermolecular energy gap-induced formation of high-valent cobalt species in CoOOH surface layer on cobalt sulfides for efficient water oxidation. Angew. Chem. Int. Ed. 61, e202117178 (2022).
Wu, Q., Xiao, M., Wang, W. & Cui, C. In situ coordination environment tuning of cobalt sites for efficient water oxidation. ACS Catal. 9, 11734–11742 (2019).
Risch, M. et al. Water oxidation by amorphous cobalt-based oxides: in situ tracking of redox transitions and mode of catalysis. Energy Environ. Sci. 8, 661–674 (2015).
Görlin, M. et al. Tracking catalyst redox states and reaction dynamics in Ni–Fe oxyhydroxide oxygen evolution reaction electrocatalysts: the role of catalyst support and electrolyte pH. J. Am. Chem. Soc. 139, 2070–2082 (2017).
Zhu, W. et al. Direct dioxygen radical coupling driven by octahedral ruthenium–oxygen–cobalt collaborative coordination for acidic oxygen evolution reaction. J. Am. Chem. Soc. 145, 17995–18006 (2023).
Yang, C., Fontaine, O., Tarascon, J.-M. & Grimaud, A. Chemical recognition of active oxygen species on the surface of oxygen evolution reaction electrocatalysts. Angew. Chem. Int. Ed. 56, 8652–8656 (2017).
Grimaud, A. et al. Activating lattice oxygen redox reactions in metal oxides to catalyse oxygen evolution. Nat. Chem. 9, 457–465 (2017).
Pan, Y. et al. Direct evidence of boosted oxygen evolution over perovskite by enhanced lattice oxygen participation. Nat. Commun. 11, 2002 (2020).
Haschke, S. et al. Direct oxygen isotope effect identifies the rate-determining step of electrocatalytic OER at an oxidic surface. Nat. Commun. 9, 4565 (2018).
Lin, Y. et al. In situ identification and time-resolved observation of the interfacial state and reactive intermediates on a cobalt oxide nanocatalyst for the oxygen evolution reaction. ACS Catal. 12, 5345–5355 (2022).
Yang, H. et al. Intramolecular hydroxyl nucleophilic attack pathway by a polymeric water oxidation catalyst with single cobalt sites. Nat. Catal. 5, 414–429 (2022).
Wang, B. et al. In situ structural evolution of the multi-site alloy electrocatalyst to manipulate the intermediate for enhanced water oxidation reaction. Energy Environ. Sci. 13, 2200–2208 (2020).
VandeVondele, J. & Hutter, J. Gaussian basis sets for accurate calculations on molecular systems in gas and condensed phases. J. Chem. Phys. 127, 114105 (2007).
Moellmann, J. & Stefan, G. DFT-D3 study of some molecular crystals. J. Phys. Chem. C. 118, 7615 (2014).
Acknowledgements
Y. G. Zhao and G. R. Patzke thank the University of Zurich Research Priority Program Solar Light to Chemical Energy Conversion (URPP LightChEC) for financial support. The authors thank the Center for Microscopy and Image Analysis (UZH) for assistance and support in performing scanning electron microscopy characterizations. We are grateful to PSI-SuperXAS-X10DA and HZB-BESSY II-KMC-2 for the allocation of synchrotron radiation beamtime and to Dr. Maarten Nachtegaal and Dr. Götz Schuck for providing assistance in using the SuperXAS-X10DA and the KMC-2 beamlines, respectively, and for their help in setting up the operando XAS tests.
Author information
Authors and Affiliations
Contributions
G. R. Patzke supervised this project. Y. G. Zhao and G. R. Patzke conceived the original concept and contributed to manuscript writing. Y. G. Zhao performed all syntheses, materials characterizations, data analyses, and prepared the manuscript. N. C. Dongfang, and M. Iannuzzi performed the DFT calculations and data analyses. C. Huang and H. Zhao helped with the operando XAS experiments. R. Erni conducted HR-TEM and EELS characterizations. J. G. Li and L. Pan assisted with the electrochemical data analyses. All authors reviewed and commented on the manuscript before submission.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhao, Y., Dongfang, N., Huang, C. et al. Operando monitoring of the functional role of tetrahedral cobalt centers for the oxygen evolution reaction. Nat Commun 16, 580 (2025). https://doi.org/10.1038/s41467-025-55857-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-55857-3
This article is cited by
-
Microbial-vulcanized organic-inorganic dual-modulated cobalt hydroxide for oxygen evolution reaction
Nature Communications (2025)
-
Achievements and challenges in deciphering heterogeneous catalytic reaction mechanisms using operando infrared spectroscopies
Nature Communications (2025)
-
Connecting the dots in a pH-dependent oxygen evolution reaction
Nature Chemistry (2025)
