
Abstract
Hypoxic tumors present a significant challenge in cancer therapy due to their ability to adaptation in low-oxygen environments, which supports tumor survival and resistance to treatment. Enhanced mitophagy, the selective degradation of mitochondria by autophagy, is a crucial mechanism that helps sustain cellular homeostasis in hypoxic tumors. In this study, we develop an azocalix[4]arene-modified supramolecular albumin nanoparticle, that co-delivers hydroxychloroquine and a mitochondria-targeting photosensitizer, designed to induce cascaded oxidative stress by regulating mitophagy for the treatment of hypoxic tumors. These nanoparticles are hypoxia-responsive and release loaded guest molecules in hypoxic tumor cells. The released hydroxychloroquine disrupts the mitophagy process, thereby increasing oxidative stress and further weakening the tumor cells. Additionally, upon laser irradiation, the photosensitizer generates reactive oxygen species independent of oxygen, inducing mitochondria damage and mitophagy activation. The dual action of simultaneous spatiotemporal mitophagy activation and mitophagy flux blockade results in enhanced autophagic and oxidative stress, ultimately driving tumor cell death. Our work highlights the effectiveness of hydroxychloroquine-mediated mitophagy blockade combined with mitochondria-targeted photosensitizer for cascade-amplified oxidative stress against hypoxic tumors.
Similar content being viewed by others

Introduction
Hypoxia (pO2 ≤ 2.5 mmHg) is a characteristic feature of 50–60% of solid tumors, primarily attributed to the formation of irregular tumor micro-vessels caused by the rapid proliferation of tumor cells1,2. The formation of a hypoxic microenvironment is closely associated with tumors’ proliferation, differentiation, angiogenesis, and metabolic adaptations3,4. However, this complex microenvironment creates formidable obstacles in the treatment of hypoxic tumors, including challenges such as drug delivery, suboptimal efficacy of aerobic treatments, and drug resistance5,6,7. Therefore, it is crucial to develop therapeutic strategies specifically tailored to the intricate microenvironment of hypoxic tumors.
Hypoxia imposes several significant pressures on tumor cells, affecting their metabolic, oxidative, and nutrient-dependent processes8,9. In hypoxic tumors, metabolic adaptations include lactic acid fermentation, resulting in low pH and elevated oxidative stress10,11,12. This oxidative stress renders the mitochondria vulnerable to damage, jeopardizing cellular survival by increasing reactive oxygen species (ROS) production13,14,15. Enhanced mitophagy—a process that selectively removes damaged mitochondria—was observed in hypoxic tumors as a protective mechanism to ensure cellular survival16,17,18. This phenomenon is primarily mediated by pathways involving hypoxia-inducible factor 1-α (HIF-1α)/BCL2-interacting protein (Beclin 1) or PTEN-induced putative kinase 1 (PINK1) and Parkinson protein 2 (Parkin)19,20,21,22,23. However, the strategy of mitophagy inhibition to enhance oxidative stress and combat hypoxic tumors has not been explored before, making mitophagy inhibition a potential strategy to hinder hypoxic tumor progression. Furthermore, due to the intricate regulatory networks within the tumor cellular environment, directly modulating mitophagy-related proteins does not always guarantee consistent therapeutic efficacy24,25. Hydroxychloroquine (HCQ), a clinically approved autophagy and mitophagy inhibitor, works by alkalinizing lysosomes, thus impeding autophagosome-lysosome fusion26,27,28,29. Nevertheless, mitophagy inhibition therapy with HCQ alone may not be sufficient to completely eradicate tumor cells in hypoxic tumors.
Similar to autophagy, mitophagy can act as a double-edged sword in hypoxic tumors22. Excessive mitophagy can lead to the clearance of healthy mitochondria, compromising normal cellular functions and survival23. Hence, we hypothesize that modulating the inhibition and activation of mitophagy at different stages could significantly amplify the disruption of hypoxic tumors. Mitochondria-targeting photodynamic therapy (M-PDT) is a light-controlled, selective therapeutic strategy that induces mitochondrial dysfunction and activation through ROS generation30,31,32,33,34. However, mitophagy can eliminate pro-apoptotic mitochondria35, potentially diminishing M-PDT therapeutic efficacy. Thus, M-PDT could be a promising component of mitophagy inhibition therapy for hypoxic tumors. However, most reported mitochondria-targeting photosensitizers36,37, such as triphenylphosphonium (TPP)-modified photosensitizers (PSs)38, are highly dependent on oxygen, which limits their effectiveness against hypoxic tumors. Alternatively, Type I M-PDT, produces cytotoxic ROS such as superoxide anion radicals (O2•-) and hydroxyl radicals (•OH) through electron transfer, with minimal oxygen dependence39,40,41,42,43, making it more suitable for hypoxic tumors. Therefore, the combined application of the mitophagy blockade and Type I M-PDT, aimed at mitochondrial damage and mitophagy activation, may represent an effective therapeutic strategy for hypoxic tumors.
The impact of mitophagy on tumors is complex. The survival or death of hypoxic tumor cells is intricately linked to the concentration and interactions of mitophagy modulators44,45. Previously, we reported a series of hypoxia-responsive supramolecular therapeutic platforms based on azocalix[4]arene (AC4A)46,47,48,49,50,51. Unlike conventional hypoxia-responsive nanomaterials, AC4A-based supramolecular nanoparticles have cavities with high binding affinity and precise stoichiometry for drug molecules52,53,54. Furthermore, they can release the drug molecules through the cleavage of azobenzene bonds in response to hypoxia stimulus55,56. Despite these advantages, the rapid clearance of AC4A from the body due to its small size and biocompatibility needs further improvement. The human serum albumin (HSA)-based platform has been shown to enhance biocompatibility, extend circulation time, achieve accurate targeting (via Gp60-mediated active targeting and passive targeting mechanisms of HSA), and enable stimuli-responsive drug release behavior57. HSA provides multiple sites for surface modification, such as the thiol group in cysteine58. Therefore, by designing appropriate chemical bonds, AC4A can be modified onto the surface of HSA to obtain HSA-AC4A. HSA-AC4A retains the original biocompatibility and tumor-targeting specificity of HSA, while enabling quantitative and ratiometric drug encapsulation due to the modification of macrocyclic calixarenes.
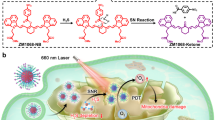
In this study, we engineer an AC4A-modified supramolecular albumin nanoparticle (SHC4H) to treat hypoxic tumors. As illustrated in Fig. 1a, SHC4H is prepared through the self-assembly of AC4A-modified reduced HSA chains (HSA-AC4A), the mitophagy inhibitor (HCQ), and the mitochondrial-targeting Type І photosensitizer (SMNB). Under hypoxia, the host molecule (AC4A) on SHC4H is reduced to aminocalix[4]arene (NH2C4A), which exhibits low drug affinity, consequently releasing the guest molecules (HCQ and SMNB). HCQ blocks hypoxia-induced mitophagy by alkalinizing lysosomes, disrupting pro-survival metabolic adaptations, and increasing oxidative stress in tumor cells. Furthermore, the released SMNB restores its original fluorescence, enabling real-time monitoring of the treatment process. Subsequently, upon exposure to 660 nm light, SMNB generates ROS through the electron transfer mechanism, damaging mitochondria and further enhancing oxidative stress. At the same time, the tumor cells undergo a dual attack of ROS-induced mitophagy and lysosomal destruction caused by autophagic flux blockade, inducing mitophagy stress, autophagosome accumulation, amplifying oxidative stress, and ultimately cell death (Fig. 1b). Due to the lack of studies on mitophagy-related therapy in hypoxic tumor treatment, we further explore the effectiveness of this strategy across different cell types. Our results indicate that the SHC4H+hv treatment shows excellent therapeutic effects in B16, 4T1, PANC1 (caspase-3 expressing), and MCF-7 (caspase-3-deficient) hypoxic tumors via different cell death pathways. In summary, SHC4H provides an effective strategy for hypoxic tumor treatment by orchestrating mitochondrial disruption and mitophagy regulation to cascade-amplify oxidative stress.

a The conjugation of HSA with free thiol groups to methacrylamide-monomodification sulfonate modified azocalix[4]arene (MAM-SAC4A) through a photoclick chemistry reaction. SHC4H was constructed by self-assembly process after encapsulating SMNB and HCQ. b i-ii. Hypoxia-responsive drug release and fluorescence imaging capabilities of SHC4H. Before laser irradiation, HCQ alkalized lysosomes to block hypoxia-induced pro-survival mitophagy, resulting in an imbalance and increased oxidative stress. iii. Upon laser irradiation, SMNB underwent O2•--mediated type I M-PDT, and the continuous accumulation of mitophagy flux further induced an intracellular ROS storm and amplified oxidative stress. The ROS storm within the mitochondria led to two distinct modes of cell death in different tumor cells.
Results
Effect of pro-survival mitophagy inhibition on hypoxic tumor cells
B16 and MCF-7 cells were cultured in a hypoxic chamber (1% O2) to simulate the conditions found in hypoxic tumors. Various concentrations of mitophagy inhibitor (HCQ) were added to tumor cells (B16, MCF-7, A431, and CT26 cells). As shown in Supplementary Fig. 1, B16 and MCF-7 cells demonstrated greater sensitivity to HCQ under hypoxic conditions compared to normoxic ones. Notably, the heightened sensitivity was more pronounced in B16 and MCF-7 cells than in A431 and CT26 cells (Supplementary Table 1). Thus, B16 and MCF-7 cell models were chosen as the primary models for further studies. Furthermore, compared to normoxic conditions, exposure to hypoxia resulted in elevated expression of HIF-1α and mitophagy-related markers (Beclin, LC3II/І) in B16 and MCF-7 tumor cells, indicating the activation of pro-survival mitophagy. Following HCQ treatment, oxidative stress levels increased, and ATP production decreased in B16 and MCF-7 tumor cells, indicating mitophagy inhibition (Fig. 2a–c, Supplementary Fig. 2, 3a). Crystal violet staining showed a decreased survival rate of hypoxic B16 and MCF-7 cells after HCQ treatment (Fig. 2d, Supplementary Fig. 3b). These observations suggest that HCQ-mediated mitophagy inhibition is an effective strategy for combatting hypoxic tumors.

Western blot analysis to reflect the impact of hypoxia and HCQ on HIF-1α, Beclin1, LC3 II/I expression in (a) B16 and (b) MCF-7 cells. c Detection of intracellular ATP after HCQ treatment under hypoxia (n = 3 independent experiments; mean values ± SD). Statistical differences were determined by One-way ANOVA with a Tukey post-hoc test. d Crystal violet staining to visualize the growth-inhibitory effect in B16 and MCF-7 cells incubated with 10 μM of HCQ (n = 4 independent experiments; scale bar = 5 mm). e Competitive fluorescence titration of MAM-SAC4A•RhB (0.5/0.5 μM) with SMNB (up to 3.33 μM) in PBS. Inset in (e): the associated titration curve at λem = 578 nm was fitted according to a 1:1 binding stoichiometry. f Competitive fluorescence titration of MAM-SAC4A•RhB (0.5/0.5 μM) with HCQ (up to 1.18 μM) in PBS. Inset in (f): the associated titration curve at λem = 578 nm was fitted according to a 1:1 binding stoichiometry. g Molecular structure of HSA-AC4A chain. h TEM images and particle sizes of (h1) HC4, (h2) HC4H, (h3) SHC4, (h4) SHC4H I (SMNB: HCQ = 1: 40), (h5) SHC4H II (SMNB: HCQ = 1: 100). Three independent experiments were performed and representative results are shown. Scale bar = 100 nm. i Fluorescence spectra of SMNB (10 μM) and MAM-SAC4A•SMNB complexes (1/10, 2/10, 3/10, 4/10, 5/10 μM) in PBS. j Fluorescence spectra of SMNB (10 μM), SHC4 (5/10 μM), SHNH2C4 (5/10 μM) in PBS. k Relative fluorescence intensity at 527 nm of MAM-SAC4A–RHo123 (30/10 mM) at different times after adding SDT (1.0 mM). Inset: fluorescence spectra of MAM-SAC4A-RHo123 (30/10 mM) before and after reduction by SDT. Source data are provided as a Source Data file.
However, mitophagy inhibition alone was inadequate for complete tumor eradication, highlighting the need for synergistic integration of diverse treatment modalities to optimize therapeutic outcomes. Our synthesized SMNB acts as a mitochondria-targeted Type I photosensitizer, capable of inducing mitochondrial oxidative damage and mitophagy activation. The synthesis details of SMNB were provided in the supporting information (Supplementary Figs. 4–7). SMNB demonstrated excellent photostability and robust ROS production under both normoxic (21% O2) and hypoxic (1% O2) conditions (Supplementary Fig. 8). SMNB exhibited similar cytotoxicity under both normoxic and hypoxic conditions when exposed to 660 nm laser irradiation, which suggests that its ability to generate ROS was unaffected by oxygen concentration. Additionally, SMNB showed minimal impact on B16 and MCF-7 cells proliferation in the absence of irradiation, indicating its excellent cellular safety profile (Supplementary Fig. 9, Supplementary Table 2). Hence, SMNB has great potential when combined with mitophagy inhibitors (HCQ) to treat hypoxic tumors.
The optimal ratio of SMNB /HCQ for synergistic anti-tumor cells in hypoxia
In combinatorial therapy, HCQ functions as a mitochondrial autophagy inhibitor, whereas SMNB serves as both a mitochondrial disruptor and a mitophagy inducer, allowing for the spatiotemporal regulation of mitophagy. Since modulating mitophagy can either synergistically disrupt cellular homeostasis in hypoxic tumors or contribute to therapeutic resistance, determining the optimal SMNB-to-HCQ ratio is critical for effective treatment.
The range of synergistic ratio between SMNB and HCQ was screened by a cytotoxicity assay in B16 and MCF-7 cells (Supplementary Fig. 10a, b). The combination index (Q-value) was calculated to assess the synergistic effect. Concomitant administration of SMNB (0.25 μM) and HCQ (10 μM) in B16 cells exhibited a potent synergistic effect (Q > 1.4) (Supplementary Fig. 10c). Similarly, co-administration of SMNB (0.1 μM) and HCQ (10 μM, 20 μM) in MCF-7 cells resulted in a significant synergistic response (Q > 1.4) (Supplementary Fig. 10d). In general, a synergistic molar ratio of SMNB and HCQ at 1:40 (SMNB: 0.25 μM; HCQ: 10 μM) was optimal for killing B16 cell, whereas a synergistic molar ratio of SMNB and HCQ at 1:100 (SMNB: 0.10 μM; HCQ: 10 μM) was found to be the most effective in MCF-7 cells.
Development of a ratiometric co-delivery albumin nanocarrier
After determining the optimal drug combination ratio, it is imperative to ensure precise delivery of SMNB and HCQ to the hypoxic tumors. To achieve it, a hypoxia-responsive compound (MAM-SAC4A) was designed and synthesized as the fundamental component (for detailed synthetic procedures refer to the Supplementary Fig. 11–14). The binding constants between the drugs and MAM-SAC4A were measured to quantitatively assess the interaction between drugs and carriers with Rhodamine B (RhB) serving as the reporter dye. Competitive fluorescent titrations were adopted to determine the binding affinities (Ka) of HCQ and SMNB to MAM-SAC4A. The affinity of RhB to MAM-SAC4A was (3.42 ± 0.24) × 105 M−1 (Supplementary Fig. 15). The RhB fluorescence gradually recovered following the titration of guest molecules into the RhB and MAM-SAC4A mixed solution. The Ka values were (1.32 ± 0.64) × 107 M−1 and (1.69 ± 0.58) × 106 M−1 for the MAM-SAC4A-HCQ and MAM-SAC4A-SMNB by fitting with a 1:1 competitive titration model (Fig. 2e, f). The strong binding affinities between drugs and MAM-SAC4A enabled the precise loading of SMNB and HCQ without the need for additional binding sites through covalent modification.
To improve biocompatibility and tumor-targeting capability, MAM-SAC4A was modified with human serum albumin (HSA). By cleaving the disulfide bonds in HSA to obtain thiol groups, the modification sites for MAM-SAC4A conjugation increased. The double bonds on MAM-SAC4A were conjugated with thiol groups through a mild and efficient photo-click reaction to obtain the carrier scaffold HSA-AC4A. Moreover, the encapsulation of drugs into the cavities of MAM-SAC4A facilitated the self-assembly of HSA-AC4A, resulting in the formation of nanoparticles (Fig. 2g, Supplementary Fig. 16). MAM-SAC4A displayed a characteristic UV absorption peak at 362 nm, with its absorbance increasing linearly with concentration (Supplementary Fig. 17a, b). After conjugation, the absorption spectrum of HSA-AC4A showed a prominent peak at 363 nm, corresponding to the absorbance of the MAM-SAC4A component. The degree of MAM-SAC4A modification in HSA-AC4A was 14.30%, indicating that each HSA molecule was on average conjugated with five MAM-SAC4A molecules (Supplementary Fig. 17c). The nanoparticles formed from the carrier skeleton alone were named HC4. The nanoparticles loaded only with HCQ were named HC4H. The nanoparticles loaded only with SMNB were named SHC4. For B16 cells, the optimal loading ratio of SMNB to HCQ was 1:40; thus, SHC4H prepared at this ratio was named SHC4H-I. For MCF-7 cells, the optimal loading ratio of SMNB to HCQ was 1:100; the corresponding nanoparticles were named SHC4H-II. SHC4H-I and SHC4H-II are collectively referred to as SHC4H in all subsequent experiments.
Morphology, particle size, and zeta potential are critical parameters affecting nanoparticle pharmacokinetics, biodistribution, and cellular uptake. Transmission electron microscope (TEM) results revealed that all the albumin-based nanoparticles exhibited a spherical morphology with a particle size of approximately 20 nm (Fig. 2h). Dynamic light scattering (DLS) analysis showed that the particle sizes of nanoparticles slightly decreased after drug loading, likely due to the electrostatic interaction between the components. Furthermore, all albumin-based nanoparticles showed excellent long-term storage stability at 4 °C for 35 days (Supplementary Fig. 18a-e). These nanoparticles exhibited a negatively charged surface originating from the negative charge of HSA (Supplementary Fig. 18f). Upon drug loading, the zeta potential increases, which can be attributed to the positive charge of the loaded HCQ and SMNB.
SMNB displayed a distinct near-infrared fluorescence emission at 700 nm. Due to the photoinduced electron transfer mechanism of MAM-SAC4A towards SMNB, the fluorescence of SMNB gradually quenched upon the addition of HSA-AC4A solution (Fig. 2i). A substantial reduction in SMNB fluorescence was observed when the concentration of HSA-AC4A reached 5 μM, suggesting the complete loading of SMNB (Fig. 2i, inset). Interestingly, significant fluorescence recovery could be observed from supramolecular albumin nanoparticles loaded with SMNB (SHC4) after reducing MAM-SAC4A to NH2C4A to simulate conditions in hypoxic tumor cells (Fig. 2j). This intriguing property shows excellent potential for hypoxia-responsive fluorescence imaging. To further substantiate this hypothesis, the MAM-SAC4A•Rhodamine 123 (RHo123) complex was used to simulate hypoxia-responsive drug release and subsequent fluorescence recovery. The addition of sodium dithionite (SDT), mimicking the action of azo reductase overexpressed in hypoxic tumor cells, restored RhB fluorescence in the MAM-SAC4A•RHo123 complex solution (Fig. 2k). Rat liver microsomes and NADPH are commonly used to study the activation of hypoxia-responsive probes and prodrugs. In our in vitro drug release tests, the formulations were incubated with rat liver microsomes (40 μg/mL) and NADPH (40 μM) under either hypoxic or normoxic conditions. As depicted in Supplementary Fig. 19, the cumulative release of HCQ and SMNB from the formulations under hypoxic conditions exceeded 75% within 15 min, while almost no drug release was observed under normoxic conditions. These findings suggested that SHC4 is a promising platform for hypoxia-responsive drug delivery and imaging.
Intracellular hypoxia imaging and mitochondrial targeting proficiency
Various upregulated reductases in hypoxic cancer cells could efficiently reduce and cleave azobenzene bonds in SHC4H to release drugs (Fig. 3a). To assess the performance of hypoxia imaging, B16 and MCF-7 cells were treated with SHC4H in hypoxia (1% O2) or normoxia (21% O2). When cells were co-cultured with SHC4H under normoxic conditions, a weak red fluorescence was observed in the cytoplasm after 12 h. On the contrary, cells co-cultured with SHC4H under hypoxic conditions displayed bright fluorescence at 6 h, which further intensified after 12 h, suggesting the release of SMNB and the fluorescence restoration (Fig. 3b, Supplementary Fig. 20). These findings proved the effectiveness of SHC4H in accurately detecting hypoxia, providing a potential approach for diagnosis and real-time monitoring during treatment in hypoxic tumors.

a Schematic diagram of the hypoxic imaging mechanism. b Confocal laser scanning microscopy (CLSM) images of B16, MCF-7 cells incubated with SHC4H under hypoxic (1% O2) or normoxic (21 % O2) conditions for 12 h (Blue: DAPI; Red: SMNB). Scale bar = 50 μm. Three independent experiments were performed and representative results are shown. c CLSM images showing the co-localization of mitochondrial and SMNB in B16, MCF-7 cells after different treatments under hypoxic (1% O2) for 6 h, along with corresponding fluorescence curves (Blue: DAPI, Green: mitochondrial/lysosome tracker, Red: SMNB, Yellow: merge). Scale bar = 25 μm. Three independent experiments were performed and representative results are shown. d Cell viability of B16 and MCF-7 cells after treatment with varying concentrations of HC4 for 48 h (n = 3 independent experiments; mean values ± SD). Cell viability of e B16 and f MCF-7 after different treatments under normoxic (21% O2) or hypoxic (1% O2) conditions, respectively (n = 4 independent experiments; mean values ± SD). Statistical differences were determined by Two-way ANOVA with Tukey’s multiple comparisons test. Source data are provided as a Source Data file.
The fluorescence property of SMNB allowed for precise tracking of its cellular uptake and intracellular distribution. Positively charged photosensitizers have been reported to exhibit specificity for targeting cancerous mitochondria (-220 mV) due to their hyperpolarization compared to normal mitochondria (-157 mV)59. The distribution of SHC4H in major intracellular organelles was thus evaluated. After treating B16 and MCF-7 cells with SMNB, SHC4, and SHC4H, a pronounced co-localization of green mitochondrial fluorescence and red SMNB fluorescence was observed. Pearson’s R values of 0.85 and 0.93 were observed in the SHC4H groups of B16 and MCF-7 cells, respectively, indicating the mitochondrial targeting proficiency of SMNB due to its positive charge (Fig. 3c, Supplementary Fig. 21, 22). In comparison, after incubation for 6 h, the red fluorescence of SMNB and the green fluorescence of lysosome only partially overlapped in MCF-7 cells (Pearson’s R values of 0.39). These outcomes revealed that SHC4H could selectively release SMNB in response to hypoxic conditions for fluorescence imaging, and SMNB was ultimately localized to the mitochondria.
Synergistic anti-hypoxic tumor effects of SHC4H under irradiation
As depicted in Fig. 3d, the viability of B16 and MCF-7 cells co-incubated with HSA-AC4A (MAM-SAC4A equivalent concentration: 0-32 μM) remained above 90%, indicating excellent biocompatibility. Similarly, no significant dark toxicity was observed when cells were incubated with SHC4 (SMNB equivalent concentration: 0-8 μM) in the absence of light irradiation (Supplementary Fig. 23). Our hypothesis suggests that successful inhibition of mitophagy is crucial for amplifying intracellular oxidative stress through SHC4H+hv treatment. Lysosomes play a pivotal role in the mitophagy pathway, selectively engulfing and degrading damaged mitochondria. Previous studies26,27,28,29 indicate significant lysosomal alkalization ~24 h after treatment after HCQ at a concentration of 10 µM. Thus, in our prior screening of pure drug combinations, light exposure was initiated after 24 h of HCQ treatment. To further validate this approach, we assessed lysosomal alkalinization levels after treating cells with SHC4H (HCQ: 10 μM) for varying durations (6, 12, and 24 h). Our findings revealed a reduction in green fluorescence after 24 h of incubation with SHC4H, indicating lysosomal alkalinization (Supplementary Fig. 24). Moreover, the SHC4H-24 h+hv group exhibited the highest ROS production and most pronounced inhibition of cell growth compared to the SHC4H-6 h+hv and SHC4H-12 h+hv groups (Supplementary Fig. 25). These results underscore the critical role of lysosomal alkalinization before light exposure, confirming its necessity in our experimental strategy.
The cytotoxicity of different albumin supramolecular nanoparticles was validated using Cell Counting Kit-8 (CCK8) and Calcein-AM and Propidium Iodide staining assays. Under hypoxic conditions (1% O2), the viabilities of B16 and MCF-7 cells treated with SMNB+hv were 48.27% (0.25 μM of SMNB) and 52.66% (0.10 μM of SMNB), respectively, with no significant difference compared to normoxic conditions (21% O2). These results suggest that the ROS production by the SMNB+hv group was oxygen-independent (Fig. 3e, f). SHC4H+hv group displayed significant cytotoxicity against B16 and MCF-7 cells under hypoxia, but did not affect cell proliferation under normoxia, due to its hypoxia-responsive drug release properties. Notably, the SHC4H+hv group displayed a more potent inhibition of B16 and MCF-7 cells growth than the SHC4+hv group. The Q values for the SHC4H+hv group, calculated using formula (1), exceeded the threshold value of 1.15 for both QMCF-7 (1.29) and QB16 (1.23), confirming a significant synergistic inhibitory effect on MCF-7 and B16 cells. The excellent phototoxicity and synergistic inhibitory effect of the SHC4H+hv group were further validated by live/dead cell co-staining assays. The fluorescence images in Supplementary Fig. 26 showed the highest proportion of red/green fluorescence when cells were treated with the SHC4H+hv group under hypoxia, consistent with the CCK8 results. These results demonstrated a significant synergistic effect through HCQ-mediated mitophagy inhibition and mitochondria-targeted SMNB+hv-induced ROS against hypoxic B16 and MCF-7 cells.
ROS storm trigged by SHC4H+hv within hypoxic tumor cells
Under irradiation, SMNB generates ROS through electron transfer, including O2•-, •OH, and H2O2, independently of oxygen. (Fig. 4a). The fluorescence probe DHR123 was used to detect the O2•- production in the solution. The fluorescence intensity of SHNH2C4H-DHR123 progressively increased during irradiation, indicating continuous O2•- production (Fig. 4b, c, Supplementary Fig. 27). To simulate hypoxic conditions, the aqueous solution containing supramolecular nanoparticles was purged with oxygen-depleted gas for 5 min and maintained under hypoxic conditions during irradiation. The amount of O2•- generated by SHNH2C4H in hypoxia showed no significant deviation from that produced in normoxia, with 83.5% of the level observed in normoxia. These findings prove that the oxygen level had minimal impact on O2•- production.

a Schematic diagram of O2•- mediated Type I PDT mechanism. b, c Detection of O2•- of SMNB+hv, SNH2C4+hv, SNH2C4H+hv and dihydrorhodamine 123 (DHR123) in normoxia (N) and hypoxia (H) (n = 3 independent experiments; mean values ± SD). d ROS detection in B16 and MCF-7 cells using DCFH-DA, DHR123, and hydroxyphenyl fluorescein (HPF) as total ROS, O2•-, and •OH fluorescence indicator, respectively (Blue: DAPI, Green: DCFH-DA/HPF ROS tracker; Red: DHR123 ROS tracker). Three independent experiments were performed and representative results are shown. Scale bar = 40 μm. e The CLSM images of mitochondrial membrane potential (MMP) in B16 and MCF-7 cells subjected to different treatments assessed by JC-10 assays (Green: J-monomer; Red: J-aggregates). Three independent experiments were performed and representative results are shown. Scale bar = 40 μm. f Schematic illustration of sequential oxidative impairment of biomacromolecules. g Lipid peroxidation products (Malondialdehyde, MDA) detection (n = 4 independent experiments; mean values ± SD). h The level of 8-hydroxy-2-deoxyguanosine (8-OHdG) was measured by Elisa kit assay after different treatments (n = 4 independent experiments; mean values ± SD). i Intracellular glutathione in B16 and MCF-7 cells treated with SHC4+hv, SHC4H, SHC4H+hv. Reduced glutathione (GSH)/oxidized glutathione (GSSG) was reported as the ratio of GSH to GSSG (n = 6 independent experiments; mean values ± SD). Statistical differences were determined by One-way ANOVA with a Tukey post-hoc test. Source data are provided as a Source Data file.
Silicon phthalocyanine dichloride (SIPN2), a type II photosensitizer whose efficacy depends on oxygen, was chosen as the control group for further verification. Under hypoxic conditions and light irradiation, bright red fluorescence of DHR123 was found in cells upon irradiation, indicating excellent in vitro O2•- photoactivity of SHC4H+hv treatment (Fig. 4d). Furthermore, O2•- is converted to •OH through an intracellular cascade reaction. Under hypoxic conditions, SHC4H exhibited excellent potential as a type I photodynamic nanosystem under irradiation, as evidenced by the remarkable green fluorescence of HPF, indicating the presence of •OH upon irradiation. Nevertheless, the SIPN2+hv group exhibited minimal presence of O2•- and •OH. The nonspecific ROS probe DCFH-DA was used to test the intracellular ROS generation of SHC4H. A significantly reduced green fluorescence of DCFH was observed in cells treated with SIPN2+hv under hypoxic conditions than normoxic conditions. In contrast, cells showed an intense green fluorescence of DCFH after treating SMNB+hv under both normoxia and hypoxia.
Furthermore, the ROS production by SHC4H can be precisely induced under controlled conditions of hypoxia and irradiation, as evidenced by the absence of green fluorescence in cells subjected to hypoxia or irradiation alone. Notably, the fluorescence intensity of the SHC4H+hv exhibited an increase compared to the SHC4+hv group. A flow cytometric assay for ROS quantitative measurement further confirmed the significant ROS increase in the SHC4H+hv group compared with the SHC4+hv group (Supplementary Fig. 28, 29). These observations suggest that mitophagy inhibition in the SHC4H+hv group enhances intracellular ROS levels compared to the SHC4+hv group, warranting further investigation into the underlying mechanisms.
Enhanced oxidative stress induced by a dual attack
Oxidative stress disrupts the cellular redox balance, resulting in the overproduction and accumulation of excessive ROS. Compared to SMNB-mediated M-PDT monotherapy, mitophagy inhibition combined with SMNB induced elevated ROS levels, amplifying oxidative stress. Oxidative stress triggers oxidation reactions and damages crucial intracellular organelles and biomolecules, such as DNA, lipids, and other biomacromolecules. Cellular oxidative stress was thus assessed from damaged mitochondria, reducing substances, and oxidative products to further study the heightened intracellular oxidative stress.
Maintaining mitochondrial membrane potential (MMP) was essential for normal mitochondrial physiological functions. JC-10, a fluorescent probe, confirmed the decline of MMP by showing a fluorescence switch from red to green. Under hypoxic conditions, the SHC4H+hv group exhibited significant green fluorescence, indicating reduced MMP, mitochondrial damage, and early-stage apoptosis (Fig. 4e). The malondialdehyde (MDA) level, an ultimate byproduct of the lipid oxidation stress process, was elevated in SHC4+hv and SHC4H+hv compared to PBS group (Fig. 4f, g, Supplementary Fig. 30a, b). 8-Hydroxy-2’-deoxyguanosine (8-OHdG) was an oxidative adduct formed as a result of ROS attacking the eighth carbon atom of the guanine base within DNA molecules. Figure 4h showed a significant increase in the 8-OHdG amount in the SHC4H+hv group compared with the SHC4+hv group, indicating an elevated ROS level (Supplementary Fig. 30c). Under normal conditions, intracellular glutathione predominantly exists in its reduced state. However, in the presence of oxidative stress, it undergoes oxidation to form GSSG (oxidized glutathione). A notable decline in the GSH/GSSG ratio was explicitly observed in the group treated with M-PDT, indicating an increased conversion of GSH to GSSG. Remarkably, the SHC4H+hv group exhibited the lowest GSH/GSSG ratio to other groups (Fig. 4i, Supplementary Fig. 30d, 31). Furthermore, intracellular hypoxia and oxidative stress levels were tested using ROS-ID Hypoxia/Oxidative Stress Detection Kit. The highest level of oxidative stress of SHC4H+hv was obtained in B16 and MCF-7 cells (Supplementary Fig. 32).
In summary, these findings suggest that synergistic therapy involving HCQ-mediated mitophagy inhibition and SMNB+hv-induced mitochondrial oxidative damage, along with mitophagy activation, led to a ROS storm and cellular amplified oxidative stress in hypoxic tumors, damaging crucial cell components, including mitochondria, DNA, and lipids. This phenomenon may be attributed to the regulation of mitophagy, where continuous ROS-induced mitochondrial damage and mitophagy activation occur. HCQ-mediated mitophagy inhibition leads to the accumulation of damaged mitochondria, increasing ROS levels, and further amplifying oxidative stress. Further investigations are needed to explore the underpinning hypothesis.
Evaluation of SHC4H+hv-induced mitophagy regulation and mitophagy stress
Hypoxia and elevated ROS levels in mitochondria trigger cellular mitophagy, where HCQ disrupts lysosomal function by raising lysosomal pH, hindering enzymatic activity, thus blocking autophagosome degradation and inducing mitophagy stress (Fig. 5a). Co-localization analysis was used to investigate mitophagy initiation and inhibition by SHC4H+hv. Carbonyl cyanide m-chloro phenylhydrazone (CCCP), a mitophagy agonist, was used as a positive control. Compared to normoxic conditions, B16 and MCF-7 cells exhibited a greater degree of co-localization between mitochondria (marked with red mito-tracker) and autophagosomes (marked with green-LC3), as well as between mitochondria and lysosomes (marked with green lyso-tracker) in hypoxic conditions (Fig. 5b and Supplementary Fig. 33, 34). This observation suggests heightened activation of mitophagy in hypoxic conditions. Furthermore, results revealed an excellent co-localization effect between the autophagosome and mitochondria in B16 and MCF-7 cells after CCCP, SHC4+hv, and SHC4H+hv treatments, as shown by the Pearson Correlation Coefficient (PCC). This finding indicated that SMNB could induce mitophagy under light conditions. However, following SHC4H+hv treatment, there was limited co-localization between intracellular mitochondria and lysosomes, indicating that HCQ disrupted the degradation pathway of damaged mitochondria in lysosomes (Supplementary Fig. 35). The perturbation would then seriously disrupt the cellular machinery responsible for recycling damaged mitochondria and wasted cellular material.

a Schematic diagram of HCQ blocking mitophagic flux. b CLSM images of fluorescent co-localization of mitochondria (Mito-Tracker Red) and autophagosomes (Green-LC3II). PCC, Pearson Correlation Coefficient. Scale bar = 40 μm. TEM images of c B16 and d MCF-7 cells after different treatments under hypoxia (Yellow arrow: autophagosomes; Red arrow: autolysosomes). Scale bar = 4 μm or 1 μm. Immunofluorescence images of the key protein change in e B16 and f MCF-7 cells for mitophagy, including PINK1 and Parkin, after different treatments under hypoxia. PBS-N represents the PBS group cultured under normoxic conditions (Blue: DAPI; Green: Mito-Tracker; Orange: PINK1; Red: Parkin). Scale bar = 20 or 4 μm. Three independent experiments in b–f were performed with similar results. g Intracellular ATP levels in B16 and MCF-7 cells following treatment with PBS, HCQ, HC4H, SHC4+hv, and SHC4H+hv (HCQ: 10 μM) treatments (n = 3 independent experiments; mean values ± SD). Statistical differences were determined by One-way ANOVA with a Tukey post-hoc test. h The expression of key proteins for mitophagy in B16 and MCF-7 cells, including LC3II/I, p62, after different treatments. NAC, N-acetylcysteine (5 mM). At least four independent experiments were performed and representative results are shown. After different treatments, apoptosis analysis by flow cytometry in i B16, j MCF-7 cells. Three independent experiments were performed and representative results are shown. Source data are provided as a Source Data file.
Adenovirus transfection and the cell TEM sections experiments were conducted to visualize the autophagic stress and autophagosome accumulation following treatment with SHC4H+hv. The autophagic flux within cells from different groups was measured by the transfection of cells with AdPlus-mCherry-GFP-LC3B adenovirus. Under hypoxia, the control group showed a diffused yellow fluorescence pattern, with only a few red/yellow spots observed, indicating a mild induction of autophagy. The appearance of red dots in MCF-7 cells treated with SHC4+hv implied the fusion of autophagosomes and lysosomes. Distinct and highly overlapping yellow spots were observed in MCF-7 cells treated with SHC4H+hv, suggesting the accumulation of autophagic flux (Supplementary Fig. 36). Furthermore, TEM images revealed that the SHC4H+hv group significantly promoted numerous double-membrane vesicles (autophagosomes) and concomitantly decreased unilamellar vesicles (autolysosomes) compared to the SHC4+hv group (Fig. 5c, d), resulting from the autophagic stress. Both autophagosomes and autolysosomes were observed in the PBS and SHC4+hv groups, indicating that hypoxia and SMNB+hv spatiotemporally triggered mitophagy. Meanwhile, the intracellular ATP level declined after HCQ treatment, demonstrating that the mitophagy inhibition decreased the cellular energy supply (Fig. 5g).
To further elucidate the mitophagy stress through the activation of mitophagy and the blockade of autophagic flux, vital markers associated with the mitophagy cascade were examined. The PINK1/Parkin pathway was involved in mitophagy induced by ROS or hypoxia, with activation occurring upon mitochondrial membrane potential disruption. In the SHC4+hv group, the expressions of PINK1 were significantly increased compared to the PBS group. Furthermore, confocal microscopy images showed a significant increase in Parkin (Red) co-localization with the mitochondrial (Green-MitoTracker) post-SHC4H+hv treatment, indicating effective recruitment of Parkin to the mitochondrial (Fig. 5e, f, Supplementary Fig. 37).
The primary function of Parkin protein is to regulate mitochondrial membrane proteins through ubiquitination, thereby tagging damaged mitochondria for autophagic clearance. Translocase of outer mitochondrial membrane 20 (TOMM20) and mitofusin-2 (MFN2), key proteins on the outer mitochondrial membrane, were observed to undergo a significant reduction in expression levels as shown in confocal microscopy images and western blot analysis results post SHC4+hv treatment, indicative of their degrade by ubiquitination (Supplementary Fig. 38, 39). In contrast, the SHC4H+hv group exhibited elevated expression levels of TOMM20 and MFN2 compared to the SHC4+hv group, attributed to the inhibition of mitochondrial degradation by HCQ. These results indicate the involvement of the PINK1/PRKN pathway in SHC4+hv-induced mitophagy in B16 and MCF-7 cells, while simultaneously inhibiting the degradation of damaged mitochondria mediated by mitophagy. There may be other mitochondrial autophagy pathways involved, which merit further exploration in the future.
During the process of mitophagy, the cytoplasmic protein LC3-I was converted into membrane-type LC3-II by enzymatic hydrolysis. The presence of autophagic flux blockade was investigated. A heightened expression of p62 (sequestosome-1) is a marker for autophagic flux blockade, and based on the western blot results, the increased ratio of autophagosome-bound lipidated LC3II/ LC3I and p62 levels in both B16 and MCF-7 cells after SHC4H+hv treatment further confirmed the activation of mitophagy and the blockade of autophagic flow. To further substantiate that the activation of mitophagy is attributed to the generation of ROS, N-acetylcysteine (NAC), as a model antioxidant, was used to pretreat tumor cells to alleviate the oxidative stress induced by SHC4+hv. Results showed that NAC partially reversed changes seen with SHC4+hv, as evidenced by the SHC4+hv-induced LC3-II accumulation could be neutralized by the presence of NAC. These results suggested ROS were responsible for mitophagy activation (Fig. 5h, Supplementary Fig. 40).
In summary, hypoxia and increased SMNB+hv-induced ROS levels were proven to damage mitochondria and trigger cellular mitophagy. HCQ induced perturbation in lysosomal function by alkalinizing lysosomes and impairing their enzymatic activity, effectively blocking damaged mitochondria degradation and inducing mitophagy stress. These findings indicate that the spatiotemporal regulation of mitophagy caused by SHC4H+hv induced mitochondrial damage and blocked the degradation of damaged mitochondria, contributing to the cascade amplification of oxidative stress in hypoxic tumors.
Cell demise mechanisms and mitophagy regulation measured by RNA sequencing
The role of mitophagy regulation varies across different types of tumors, with the intricate interplay or crosstalk between mitophagy and cell death remaining complex and not fully understood. Traditionally, PDT primarily triggers apoptosis as the principal mode of cell death. However, tumor cells may also undergo alternative forms of cell death, such as pyroptosis, necroptosis, and ferroptosis. Thus, a comprehensive exploration is imperative to understand the consequences of mitophagy regulation on cell death in diverse hypoxic cellular environments. The Annexin V-FITC assay results revealed that apoptosis was the primary mode of cell death in B16 cells after various treatments. In the SHC4H+hv group, the late-stage apoptosis ratio was 70.86% (Fig. 5i, Supplementary Fig. 41). However, in MCF-7 cells, both apoptosis and necroptosis were observed following SHC4H+hv treatment (Fig. 5j). As shown in Supplementary Fig. 42, the typical morphology of cells in the PBS group (untreated) differed from the apoptotic cells in the SHC4H+hv group. The presence of apoptotic bodies and the intact plasma membrane displaying blistering morphology were observed in B16 and MCF-7 cells. Apart from apoptosis, the SHC4H+hv treated MCF-7 cells exhibited necroptotic characteristic features, including enlarged nuclei, disrupted plasma membrane, and subsequent release of cellular contents.
To further investigate the impact of SHC4H+hv on B16 and MCF-7 cells, RNA-sequencing was performed. The correlation heatmap and principal component analysis (PCA) provided an overview of the differentially expressed genes between the PBS and SHC4H+hv groups (Supplementary Fig. 43–45, Supplementary Table 3, 4). The volcano plot (SHC4H+hv vs. PBS) revealed 1861 downregulated, 1954 upregulated genes in B16 cells and 1566 down-regulated, 2268 upregulated genes in MCF-7 cells (Supplementary Fig. 46), respectively (|log2fold change | >1 and p value < 0.05). Gene ontology (GO) enrichment analysis and Kyoto encyclopedia of genes and genomes (KEGG) analysis in B16 and MCF-7 cells (PBS vs. SHC4H+hv groups) exhibited the same enrichment pathway, including mitophagy regulation, cytochrome-c (cyt c) oxidase activity, intrinsic apoptosis, and mitochondrial outer membrane permeabilization pathway, reactive oxygen species, oxidative stress-induced cell death (Fig. 6a, Supplementary Fig. 47–50). However, compared to B16 cells, the significantly enriched pathways in MCF-7 cells after SHC4H+hv treatment included necroptosis pathway and signal pathways related to necroptosis, including tumor necrosis factor (TNF) signal pathway, NF-κappaB signal pathway, mitochondrial DNA metabolic process. Gene set enrichment analysis (GSEA) further substantiated the divergence in cell demise modalities between B16 and MCF-7 cells after SHC4H+hv treatment (Fig. 6b, c, Supplementary Fig. 51–53). Previous reports have demonstrated that in caspase-deficient cells, the accumulation of ROS and mitochondrial outer membrane permeabilization could activate the TNF-dependent RIP3 (Receptor-interacting protein kinase 3)/MLKL (Mixed lineage kinase domain-like pseudokinase) signaling pathway, leading to necroptosis60. Thus, the initiation of necroptosis in MCF-7 cells following SHC4H+hv treatment may be ascribed to caspase-3-deficient and ROS-induced mitochondrial outer membrane permeabilization (MOMP). To further confirm it, we performed RNA sequencing and western blot analysis.

a Bubble diagram of the DEGs enriched in the GO pathway (False Discovery Rate, FDR < 0.05), n = 3 independent experiments. b GSEA of the DEGs in B16 cells after treatment with SHC4H followed by irradiation (p-val<0.05), n = 3 independent experiments. A: Positive regulation of the execution phase of apoptosis; B: Regulation of cyt (c) release from mitochondria. C: Regulation of mitochondrial membrane potential. D: Cytochrome c oxidase activity. E: Positive regulation of intrinsic apoptotic signaling pathway. NES: normalized enrichment score. c GSEA of the DEGs in MCF-7 cells after treatment with SHC4H followed by irradiation (p-val<0.05), n = 3 independent experiments. A: DNA adduct; B: Glutathione metabolism; C: Reactive oxygen species; D: Mitophagy; E: Necroptosis. d Heatmap of genes associated with the mitophagy and cell death mode (Apoptosis or Necroptosis) in B16 and MCF-7 after different treatments. The red color represents upregulation, and the blue color indicates downregulation, n = 3 independent experiments. e PPI (protein-protein interaction) network of identified differentially expressed genes after treating SHC4H+hv in B16 and MCF-7 cells (n = 3 independent experiments). f The amount of intracellular cytochrome-c after different treatments in B16 and MCF-7 cells (n = 5 independent experiments; mean values ± SD). g The expression of key proteins for apoptosis in B16 cells, including cleaved-Caspase-3 (cCasp3), Bcl-2, after different treatments. Four independent experiments were performed and representative results are shown. h After different treatments, the expression of key proteins for necroptosis in MCF-7 cells, including p-Mlkl and p-Ripk3. Three independent experiments were performed and representative results are shown. i The levels of intracellular TNF after different treatments in MCF-7 cells (n = 4 independent experiments; mean values ± SD). Statistical differences were determined by One-way ANOVA with a Tukey post-hoc test. Source data are provided as a Source Data file.
The essential genes associated with cell apoptosis, necroptosis, mitophagy, and MOMP were further analyzed (Fig. 6d, e). Several intrinsic apoptosis-related and mitophagy-related genes, such as CYTP, CASP3 or CASP7, CASP6, BCL-2, PRKN, and PINK1, were distinctly upregulated or downregulated in B16 and MCF-7 cells. Necroptosis-related genes, including PTPN6, RIPK3, RIPK1, MLKL, and TNF, were distinctly upregulated or downregulated in MCF-7 cells. Subsequently, Elisa and western blot assays were also conducted to reveal the cell demise mechanisms induced by SH4C+hv in B16 and MCF-7 cells. The expression levels of pro-apoptotic proteins (Cyc-c, cCasp3) exhibited an obvious reduction. In contrast, the expression of Bcl-2, a potent inhibitor of apoptosis, demonstrated a decrease specifically in SHC4H+hv-treated B16 cells compared to those in other groups (Fig. 6f, g). Meanwhile, the expression of TNF, p-Ripk3, and p-Mlkl (a necroptosis executive protein) was significantly upregulated in SHC4H+hv-treated MCF-7 cells, suggesting the induction of TNF-dependent necroptosis (Fig. 6h, i, Supplementary Fig. 54). However, the expression of p-Mlkl and p-Ripk3 in B16 cells treated with SHC4H+hv was similar to that in the PBS group, suggesting that B16 cells did not undergo necroptosis (Supplementary Fig. 55). In summary, these studies discussed the diverse modes of cell death induced by SHC4H+hv treatment in B16 and MCF-7 cells. Our findings showed that while apoptosis was the primary mode of cell death in B16 cells following various interventions, MCF-7 cells exhibited both apoptosis and necroptosis after SHC4H+hv treatment. RNA-sequencing analysis revealed differential gene expression patterns, with pathways related to mitophagy, apoptosis, and necroptosis being influenced. The study indicated that caspase-3-deficient MCF-7 cells may undergo TNF-dependent necroptosis in response to SHC4H+hv treatment due to mitochondrial membrane permeabilization.
Tumor hypoxia imaging and anti-tumor efficacy in B16 tumor-bearing mice model
A mouse melanoma cancer model was established to investigate the anti-tumor efficacy of SHC4H (Fig. 7a). To investigate the potential of the SHC4H as a fluorescent off-on NIR nanoprobe, PBS, SMNB, and SHC4H were administered intravenously in B16 tumor-bearing nude mice (150 mm3). Remarkably, even after 48 h, the tumor tissue in the SMNB group exhibited no signs of fluorescence. In contrast, the SHC4H group showed a marked increase in fluorescence within the tumor tissue after injection, peaking at 24 h post-injection (Fig. 7b, d). These results demonstrated the SHC4H’s ability to release SMNB in hypoxic conditions, enabling in vivo hypoxic tumor detection and imaging. For further analysis, tumor tissues and major organs were excised for ex vivo fluorescence imaging after 24 h of injection. As shown in Fig. 7c, the SMNB group displayed prominent fluorescence in the kidney and liver, with minimal fluorescence detected in the tumor. This suggests that SMNB was primarily metabolized by the liver and kidneys. The fluorescence intensity in tumor tissues of mice from the SHC4H group was 3.5 times higher than that in the SMNB group, consistent with in vivo imaging results, indicating that loading SMNB onto the supramolecular albumin nanocarriers enhanced its ability to enter tumor tissues.

a Schematic illustration of the treatment schedule for the anti-tumor efficacy study. The green circle represents intravenous drug administration (n = 6 mice per group). b In vivo fluorescence imaging of B16 tumor-bearing mice at indicated time points after intravenous injection of PBS, SMNB, and SHC4H. c Ex vivo images of tumors and other significant organs at 24 h post-injection, with corresponding mean fluorescence intensity analysis (n = 3 independent experiments; mean values ± SD). d Mean fluorescence intensity (MFI) of tumors in different groups with elapsing time (n = 3 independent experiments; mean values ± SD). p value represents SHC4H group versus SMNB group at 24 h. e Tumor growth curves during the treatment (n = 6 mice per group; mean values ± SD). f Tumor weights of different groups at the experimental endpoint (n = 5 mice per group; mean values ± SD). g Image of the harvested tumors at the end of treatment (n = 5 mice per group). h Body weight changes of mice during the treatment (n = 6 mice per group; mean values ± SD). i Western blot analysis (left) and relative ratio (right) of LC3II/LC3I, p62, and β-actin. Parallel processing of samples from the same experiment and blots (n = 4 independent experiments; mean values ± SD). j Representative immunofluorescence images of Parkin and p62 in the B16 tumor tissues after different treatments (n = 4 independent experiments; mean values ± SD). Scale bar = 100 μm. k TUNEL immunofluorescence staining images of tumor tissues to determine the level of apoptosis (n = 4 independent experiments; mean values ± SD). Scale bar = 100 μm. l Apoptotic protein (cCasp3) and inhibiting apoptotic protein (Bcl-2) immunofluorescence staining images of the tumor tissues in each group. Blue fluorescence represents DAPI-stained nuclei (n = 4 independent experiments; mean values ± SD). Scale bar = 100 μm. Statistical differences were determined by One-way ANOVA with a Tukey post-hoc test. Source data are provided as a Source Data file.
After the treatment period, the PBS group exhibited the most rapid tumor proliferation rate, with the average tumor volume reaching 1553 mm3 on the 12th day, close to the critical threshold of 1600 mm3, necessitating euthanasia promptly (Fig. 7e, Supplementary Fig. 56). The HC4H and SHC4H groups showed similar tumor growth rates to the PBS group, with an average tumor volume of ~1300 mm3 by the 14th day, indicating limited therapeutic efficacy of HCQ alone. The HCQ + SMNB+hv group exhibited a tumor suppression rate of 37.03%, likely due to minimal SMNB accumulation in tumor carriers caused by the lack of carrier. Compared to the HCQ + SMNB+hv group, the SHC4+hv, and the SHC4H+hv groups showed significantly enhanced tumor growth inhibition, highlighting the superior targeted efficacy of the HSA-AC4A carrier. In comparison to the SHC4+hv group, the SHC4H+hv group displayed superior anti-tumor efficacy, exhibiting almost complete inhibition of tumor growth. Consistent findings were also noted in assessing tumor weight and observing tumor sizes among all experimental groups (Fig. 7f, g). Furthermore, the treatment of B16 tumor-bearing mice with SHC4H+hv significantly improved survival (Supplementary Fig. 57). These remarkable outcomes could be ascribed to the efficient targeting ability of tumors and the synergistic effects of HCQ-mediated mitophagy inhibition and mitochondria-targeted SMNB+hv-induced ROS. Ensuring the biocompatibility of therapeutic agents is of paramount importance in biomedical research and clinical applications. No noticeable body weight changes were observed during the treatment process, which preliminarily confirmed the safety profile of these nanoparticles. In addition, negligible hemolysis and the absence of noteworthy pathological abnormalities in major organs further emphasized the remarkable biosafety of our nanoparticles (Fig. 7h, Supplementary Fig. 58, 59).
Consistent with the previous assessment of mitophagy levels, the expression of Parkin, LC3II, and p62 was upregulated in tumors obtained from the HC4H, SHC4H, and SHC4H+hv groups, compared to the other groups (Fig. 7i, j, Supplementary Fig. 60). These findings elucidated the suppression of mitophagy through the interception of autophagic flux and the hindered degradation of substrates within the autophagosomes. Tumor section analysis was conducted to evaluate the comprehensive anti-tumor effects further. Hematoxylin and Eosin (H&E) and Terminal-Deoxynucleotidyl Transferase Mediated Nick End Labeling (TUNEL) staining results of tumor tissues in the SHC4H+hv group revealed a pronounced apoptotic feature (Fig. 7k, Supplementary Fig. 61). The SHC4H+hv group also exhibited the highest expression of cleaved Caspase-3 (cCasp 3) levels, consistent with TUNEL results. Bcl-2, an anti-apoptotic protein, in SHC4H+hv-treated mice also decreased significantly (Fig. 7l). As shown in Supplementary Fig. 62, the lowest expression of the HIF-1ɑ in the SHC4H+hv-treated group was observed. The aforementioned findings underscored the superior anti-tumor efficacy of SHC4H+hv, predominantly driven by the induction of apoptosis in B16 tumor cells.
Tumor hypoxia imaging and anti-tumor efficacy in MCF-7 tumor-bearing mice model
The in vivo tumor hypoxia imaging and anti-tumor efficacy performance of SHC4H was also confirmed in the MCF-7 tumor-bearing mice model (Fig. 8a). Upon intravenous drug administration, the SHC4H group exhibited strong and time-dependent fluorescence signals within the tumors, distinguishing them from the surrounding tissue (Fig. 8b). The SHC4H group also exhibited a more pronounced tumor accumulation compared to the SMNB group, consistent with the experimental results in the B16 tumor model. Quantitative analysis showed that 24 h post-injection was the appropriate time point for achieving maximal therapeutic efficacy in the hypoxic tumor regions as the MFI values reached the maximum level at this time point (Fig. 8d). The fluorescence intensity after 24 h declined, indicating the gradual clearance of SHC4H at later stages. To assess the distribution of the probes, mice were sacrificed 24 h after injection. The major organs, including the heart, liver, spleen, lung, kidney, and tumor tissues, were collected for analysis (Fig. 8c). The experimental findings revealed a predominant distribution of SMNB within the tumors in the SHC4H group.

a Schematic representation of the anti-tumor efficacy study process. The green circle emblemizes intravenous drug administration (n = 6 mice per group). b In vivo fluorescence imaging of MCF-7 tumor-bearing mice following intravenous administration of PBS, SMNB, and SHC4H. c Ex vivo images of tumors and major organs captured 24 h after injection and d a corresponding analysis of MFI (n = 3 independent experiments; mean values ± SD). p value represents SHC4H group versus SMNB group at 24 h. e Tumor growth curves in each group during treatment (n = 6 mice per group; mean values ± SD). f Tumor weights of different groups at the experimental endpoint (n = 5 mice per group; mean values ± SD). g Images of the tumors obtained from each experimental group (n = 5 mice per group). h Body weight changes in different groups during the experimental period (n = 6 mice per group; mean values ± SD). i Expression of LC3 and p62 of MCF-7 tumor in BALB/c-nu mice via Western Blot analysis (left) and the quantification of the ratio of LC3-II to LC3I and p62 to β-actin expression (right), n = 4 independent experiments, mean values ± SD. j Immunofluorescence staining for evaluating the expression levels of mitophagy-related proteins (Parkin) and anti-apoptotic protein (Bcl-2) in tumor tissues from each experimental group. Blue fluorescence represents DAPI-stained nuclei (n = 4 independent experiments; mean values ± SD). Scale bar = 100 μm. k Immunofluorescence TUNEL staining for assessing the apoptosis of tumors (n = 4 independent experiments; mean values ± SD). Scale bar = 100 μm. l p-Ripk3 and p-Mlkl immunohistochemical staining in the tumor tissues after different treatments (n = 4 independent experiments; mean values ± SD). Scale bar = 100 μm. Statistical differences were assessed using one-way ANOVA followed by a Tukey post-hoc test. Source data are provided as a Source Data file.
Mice-bearing MCF-7 tumors were divided into six groups once the tumor size reached ~150 mm3. As shown in Fig. 8e, the HC4H group and SHC4H group only showed a moderate tumor growth inhibition effect compared to the PBS-treated group, demonstrating that HCQ treatment alone was not effective enough to suppress tumor growth. The SHC4H+hv group demonstrated the most pronounced anti-tumor efficacy (Fig. 8f, g, Supplementary Fig. 63). Furthermore, the Kaplan–Meier survival curves (Supplementary Fig. 64) showed that the lifespan of mice in the SHC4H+hv group was prolonged compared with those in other groups, owing to the remarkable tumor growth inhibition.
During the treatment process, no pronounced loss of body weight was observed, implying that there was no severe systemic toxicity (Fig. 8h). Furthermore, no obvious abnormity was found in H&E staining results, indicating the excellent biocompatibility of SHC4H (Supplementary Fig. 65).
Western blot analysis and immunofluorescence staining of tumor sections were performed to evaluate autophagy levels after treatment. Analogous outcomes were observed in the MCF-7 tumors. Alterations in the expression of mitophagy-related proteins substantiated the effective mitophagy inhibition (Fig. 8i, j, Supplementary Fig. 66). In the B16 model, SHC4H+hv treatment also induced the most prominent level of cell apoptosis, as evidenced by the results of TUNEL and H&E staining (Fig. 8k, Supplementary Fig. 67). The expression of HIF-1ɑ was correlated with tumor size, serving as an indicative marker of tumor hypoxia (Supplementary Fig. 68). Immunofluorescence images showed a significant increase in the expression of caspase7, an essential protein associated with programmed cell death, in the tumor tissues of the SHC4H+hv group (Supplementary Fig. 69). This indicates that caspase-3-deficient MCF-7 tumors may undergo endogenous cell apoptosis through caspase7 activity. As shown in Fig. 8l and Supplementary Fig. 70, the tumor tissue from the SHC4H+hv-treated group exhibited elevated levels of TNF and crucial necrosis-associated proteins, namely p-Ripk3 and p-Mlkl, suggesting the occurrence of necroptosis. In short, these experiments confirmed that SHC4H+hv treatment could significantly suppress tumor growth through mitochondria-targeted SMNB+hv-induced and HCQ-mediated mitophagy inhibition in the MCF-7 tumor-bearing mice model. These studies have also demonstrated the induction of apoptosis and the potential occurrence of necroptosis in tumor tissues following SHC4H+hv treatment.
Anti-tumor efficacy in PANC1 subcutaneous tumor and 4T1 orthotopic breast cancer models
To validate the clinical application prospects of SHC4H+hv treatment, we conducted additional experiments in other types of hypoxic tumors. As shown in Supplementary Fig. 71, 72, the SHC4H+hv treatment demonstrates excellent cytotoxic effects against 4T1, PANC1, CT6, and A431 cell lines under hypoxic conditions in vitro. Furthermore, the cell death mechanisms in 4T1 and PANC1 cells following SHC4H+hv treatment are predominantly mediated through mitochondria-dependent apoptosis (Supplementary Fig. 73). Based on these results, the PANC1 subcutaneous and the 4T1 orthotopic breast tumor models were established for further investigation (Supplementary Fig. 74a, 76a). Supplementary Fig. 74b–e demonstrated that the SHC4H+hv group exhibited significant capability to inhibit tumor growth, which almost ablated the whole tumors with slight body weight variation. The Kaplan–Meier survival curves of mice bearing PANC1 also indicated that the SHC4H+hv group possessed the highest average life span over the therapy episode as compared to those from other treatment groups, owing to the remarkable growth inhibition of PANC1 tumors after SHC4H+hv treatment (Supplementary Fig. 74f, 75). Moreover, Supplementary Fig. 74g showed that tumor sections from mice treated with SHC4H+hv had the highest percentage of dead cells compared to the other groups.
As depicted in Supplementary Fig. 76b–d, both the SHC4+hv and SHC4H+hv treatment groups exhibited improved 4T1 tumor suppression without significant changes in mice body weight. Notably, SHC4H+hv treatment demonstrated superior tumor inhibition compared to SHC4+hv alone, as evidenced by Supplementary Fig. 76e. Moreover, the survival rate of mice treated with SHC4H+hv was notably higher, reaching 80% on day 60 (Supplementary Fig. 76f). H&E-stained tumor sections (Supplementary Fig. 76g) further validated the efficacy of SHC4H+hv in eradicating orthotopic 4T1 tumors. Additionally, H&E staining (Supplementary Fig. 77) revealed no significant pathological damage to major organs following SHC4H+hv treatment. The blood routine and biochemistry analyzes of the healthy mice treated with PBS, healthy mice treated with SHC4H, and tumor-bearing mice cured via SHC4H+hv (Supplementary Fig. 78) showed no detectable differences, highlighting the excellent biosafety of our formulation.
These findings underscore the robust anti-tumor efficacy and excellent biosafety profile of SHC4H+hv, supporting its potential clinical utility in hypoxic tumor therapy.
Discussion
Hypoxic tumors present significant therapeutic challenges due to their complex microenvironment and increased adaptability, necessitating the development of effective strategies61,62,63. Recent research has emphasized the crucial role of mitophagy in maintaining redox and metabolic homeostasis in hypoxic tumors11,12. However, research into the impact of mitophagy-regulating treatment and oxidative stress on hypoxic tumors remains largely unexplored. This prompted a comprehensive investigation into their effect on hypoxic tumors. Our initial studies revealed that the mitophagy inhibitor HCQ significantly suppressed the growth of hypoxic tumor cells (B16, MCF-7), with heightened sensitivity under hypoxic conditions (1% O2) to HCQ, compared to under normoxic conditions (21% O2). However, as expected, HCQ-mediated singular mitophagy inhibition therapy was inadequate in completely eradicating hypoxic tumor cells, possibly due to insufficient levels of damaged mitochondria and oxidative stress. To address this, we synthesized SMNB, a mitochondrial-targeted type I photosensitizer, which offers promise as a combinational therapeutic, serving as both a mitochondrial disruptor and a stimulator of mitophagy.
Unlike conventional photosensitizers36,37, our SMNB demonstrated efficient mitochondrial targeting and generated significant levels of ROS under hypoxic conditions. As a result, the combination of mitophagy inhibition and type I M-PDT treatment emerged as a promising approach to damaging mitochondria and modulating mitophagy in hypoxic tumors. As delivering SMNB and HCQ to hypoxic tumors in precise proportions while ensuring responsive drug release was critical, the optimal ratio of the two drugs was determined to achieve optimal therapeutic outcomes.
In this study, we report a hypoxia-responsive supramolecular albumin-based nanoparticle, SHC4H, for treating hypoxic tumors. The HSA-AC4A can effectively load equal proportions of SMNB and HCQ due to the strong binding affinities between MAM-SAC4A and SMNB, as well as HCQ. This design allows for precise delivery of drugs to hypoxic tumors and ensures the hypoxia-response release of therapeutic agents. The hypoxia-responsive imaging capability of SHC4H was demonstrated in vitro and in vivo, facilitating the detection of hypoxic tumors and real-time monitoring of the treatment process. This aligns with recent research showing that C4A-loaded fluorescent agents can release and restore fluorescence under hypoxic conditions, making them valuable for image-guided therapy55,56.
Our study aims to achieve a cascade-amplification of oxidative stress through the spatiotemporal regulation of mitophagy, combining mitochondria-targeted SMNB-induced ROS and mitophagy inhibition in hypoxic tumors. Our study demonstrates that SHC4+hv treatment specifically targets mitochondria and induces significant ROS generation under hypoxic conditions in B16 and MCF-7 cells, leading to decreased ATP levels and mitochondrial membrane potential, which are indicative of mitophagy initiation. This conclusion is supported by several key findings: including the presence of prominent autophagosomes in TEM images, strong colocalization of mitochondria and LC3 in fluorescence images, significant changes in autophagy-related proteins as shown by Western blot analysis, and activation of mitophagy pathways as confirmed by enrichment analysis. Additionally, SHC4+hv treatment led to increased PINK1 levels, recruitment of Parkin to the mitochondrial membrane, and decreased levels of TOMM20 and MFN2, emphasizing the involvement of the PINK1/Parkin pathway in mitophagy induction. The use of the antioxidant N-acetylcysteine (NAC) inhibited SHC4+hv-induced mitophagy, suggesting that ROS is a trigger for this process, consistent with previous findings on oxidative stress-induced mitophagy. SHC4H+hv treatment resulted in lysosomal alkalization, a blockage in autophagic flux, autophagosome accumulation, increased levels of LC3 II and P62, and impaired degradation of mitochondrial proteins, indicating mitophagy inhibition. These findings confirmed that SHC4H+hv treatment induces mitophagy and disrupts the downstream autophagic pathway, leading to the accumulation of damaged mitochondria and causing mitophagy stress.
After 24 h of light exposure, the SHC4H+hv group produced higher levels of ROS and oxidative stress in hypoxic B16 and MCF-7 cells compared to the SHC4+hv group. This was characterized by intensified oxidation of critical biomolecules, such as 8-OHdG and MDA. An explanation could be that the tumor cells suffered a dual assault from ROS-triggered mitophagy and autophagic flux impedance, resulting in autophagic stress and substantial accumulation of autophagosomes. These autophagosomes, harboring impaired mitochondria, continuously contribute to ROS generation, culminating in a significant increase in oxidative stress. Cellular toxicity and in vivo tumor inhibition experiments validated the synergistic therapeutic effect of the HCQ and SMNB combination in B16 and MCF-7 hypoxic tumors.
Notably, unlike the common apoptosis-driven mechanism associated with PDT64, our SHC4H+hv treatment strategy induced distinct modes of cell death across different cell types (B16, 4T1, and PANC1 cells: caspase-3-expressing and MCF-7 cells: caspase-3-deficient types). After SHC4H+hv treatment, B16, 4T1, and PANC1 cells primarily exhibited mitochondria-mediated cell apoptosis, characterized by the release of cytochrome c from mitochondria and subsequent activation of the caspase cascade, ultimately leading to cellular apoptosis. In addition to mitochondria-mediated apoptosis, MCF-7 cells also exhibited necrotic apoptosis. The whole transcriptome sequencing results revealed significant differences in mitochondrial membrane potential, permeability, and increased TNF expression compared to the PBS group. These findings were consistent with previous studies by Tait60, suggesting that mitochondrial outer membrane permeabilization and intracellular damaged DNA can trigger TNF-dependent necrotic cell death in the absence of caspase-3. Furthermore, as reported65,66 caspase-3 could regulate autophagy by cleaving the autophagy regulator Beclin1. However, our findings showed that the differential expression of caspase-3 in B16 and MCF-7 cells does not significantly affect mitophagy regulation within cells treated with SHC4H+hv. This discrepancy may arise from the complex interplay of various autophagy regulatory beyond caspase-3 within B16 and MCF-7 cells.
To assess the clinical potential of SHC4H+hv treatment, we conducted additional experiments in other types of hypoxic tumors. The SHC4H+hv treatment demonstrates excellent cytotoxic effects against 4T1, PANC1, CT26, and A431 cell lines under hypoxic conditions in vitro. Furthermore, SHC4H+hv treatments significantly inhibited tumor growth in the PANC1 subcutaneous and the 4T1 orthotopic breast tumor models. Although we have verified that SHC4H+hv treatment is effective in a variety of hypoxic tumors, it requires adjusting the ratio of SMNB to HCQ for different cell types. Optimizing this drug ratio for each patient may complicate the treatment process. In the future, we plan to further study the SHC4H concentration requirements of different cell lines and in vitro tumor models and integrate these data into a medication guide library to increase the versatility of our formulation in the treatment of hypoxic tumors.
In summary, our study demonstrated that SHC4H+hv treatment effectively suppresses hypoxic tumors through cascade-amplification of oxidative stress-mediated by spatiotemporal regulation of mitophagy. Before light exposure, HCQ disrupts lysosomal function to inhibit mitophagy, leading to increased ROS and oxidative stress. Subsequent irradiation further induces a substantial production of ROS by SMNB, triggering mitochondrial apoptosis and mitophagy. At this stage, the hypoxia-induced and ROS-induced mitophagy led to the accumulation of autophagosomes, triggering intracellular autophagic stress and resulting in an oxidative stress storm. Furthermore, this strategy exhibited excellent efficacy in B16, 4T1, PANC1, and MCF-7 tumors in vitro and in vivo via different modes of cell death. Our findings outline a versatile strategy based on supramolecular albumin nanoparticles, potentially applicable to diverse therapeutic agents for hypoxic tumors. However, our study also has limitations. Our current work focuses on the combined effects of SMNB-mediated mitochondrial disruption and mitophagy activation, alongside HCQ-mediated mitophagy inhibition, leading to enhanced oxidative stress in some hypoxic tumors. These findings are limited to the combined treatment of SMNB and HCQ.
In future, we plan to further investigate the therapeutic effects of SMNB in combination with other autophagy inhibitors or by knocking out key molecules (such as ATG5, PINK1, PRKN, and others) to inhibit mitophagy. Such studies will broaden the scope of our research, providing a deeper understanding of the specific mechanisms of mitophagy in oxidative stress and cell apoptosis. This will not only enhance our overall knowledge of cellular autophagy processes but may also reveal effective therapeutic strategies to improve cancer treatment outcomes.
Methods
Ethical statement
All animal experiments were performed according to protocols approved by the Animal Care and Use Committee at the South China University of Technology (SCUT) (official approval number: 2021047) and the Institutional Animal Care and Use Committee (IACUC) of Sun Yat-sen University (protocol number of SYSU-IACUC-2024-000868). Following Institutional Animal Care and Use Committee (IACUC) guidelines and local authorities (the Animal Care and Use Committee at SCUT), the tumor size in any single mouse does not exceed 2000 mm3, and the average tumor size within the group remains below 1600 mm3.
Eukaryotic cell lines
MCF-7 (HTB-22 BCRC), B16 (JCRB0202), CT26 (CRL-2638), 4T1 (CRL-2539), PANC1(CRL-1469) cells were purchased from Procell Life Science&Technology Co.,Ltd., Wuhan. A431(CRL-1555) cells were purchased from GAINING BIOLOGICAL, Shanghai. All cell lines were routinely tested with mycoplasma free by Mycoplasma Stain Assay Kit (C0296, Beyotime) and authenticated by Short Tandem Repeat(STR) profiling.
Synthesis of SMNB
Synthesis of 2-amino-5-diethylaminobenzene thiosulfate sodium (Bunte salt)41. The crude product was further purified by refluxing in methanol for 3 h and filtered to obtain a dark gray solid with a yield of 33.23%. [M-H]-:m/z calcd. for (C10H16N2S2O3-): 276.06, found: 276.06.
Synthesis of Sulfursubstituted Methyl Nile Blue analog (SMNB). Firstly, Sodium 2-amino-5-diethylaminophenylthiosulfate (1.02 g, 3.70 mmol) and naphthylamine (0.80 g, 5.07 mmol) were dissolved in DMSO, followed by the addition of potassium dichromate (1.20 g, 4.05 mmol) and the mixture was stirred at room temperature for 20 min. Next, the reaction mixture was transferred to 200 mL of methanol, and 20 mL of 2 M hydrochloric acid was added. The mixture was stirred at room temperature for 40 min until the reaction was complete. The solvent was then removed under reduced pressure, and the remaining solution was slowly poured into 100 mL of saturated sodium chloride to precipitate the dark blue solid. The crude product was purified by silica gel column chromatography using a mixture of CH2Cl2/CH3OH (10:1, v/v) as the eluent. The dark blue solid SMNB was obtained with a yield of 21.46%. 1H NMR (400 MHz, Chloroform-d) δ 11.70 (s, 1H), 9.23 (d, J = 8.0 Hz, 1H), 8.89 (dd, J = 7.9, 1.6 Hz, 1H), 7.90 (d, J = 9.3 Hz, 1H), 7.83–7.69 (m, 2H), 7.09 (dd, J = 9.4, 2.6 Hz, 1H), 6.84 (s, 1H), 6.78 (d, J = 2.7 Hz, 1H), 3.61 (q, J = 7.1 Hz, 4H), 3.40 (d, J = 4.7 Hz, 3H), 1.35 (t, J = 7.1 Hz, 6H). [M-H]-:m/z calcd. For (C21H21N3S+): 348.15, found: 348.
Synthesis of MAM-SAC4A
Synthesis of MAM-SAC4A50: Compound 1-mono-carboxytrisulfonic acid azocalix[4]arene (MCASAC4A) (200 mg, 0.18 mmol) was dissolved in DMF (20 mL), DIPEA (44 mg, 0.34 mmol) and HATU (78 mg, 0.21 mmol) were sequentially added to the DMF solution, and activated for 30 min. Compound 2 - PEG3-monomethacrylamide (MAM-PEG3) (90 mg, 0.35 mmol) was added to the reaction system, and the mixture was stirred at room temperature for 12 h. After vacuum concentration, the crude product was recrystallized with water and methanol to obtain 205 mg of brick-red powder with a yield of 83% (Compound 3).
1H NMR (400 MHz, DMSO-d6) δ 7.95 (d, J = 8.2 Hz, 2H), 7.89 – 7.76 (m, 10H), 7.76 – 7.66 (m, 12H), 5.63 (s, 1H), 5.29 (s, 1H), 4.14 (s, 4H), 3.82 (s, 4H),3.65 – 3.32 (m, 14H), 3.24 (d, J = 5.6 Hz, 2H), 1.82 (s, 3H) ppm. 13C NMR (100 MHz, DMSO-d6) δ 167.68, 165.86, 158.58, 153.66, 152.13, 149.06, 144.87, 139.87, 135.18, 130.39, 128.48, 126.65, 123.97, 121.79, 121.68, 119.26, 70.14, 69.64, 69.02, 31.78, 18.70 ppm. Mass spectrum (ESI): [M-3Na + 2H]-:m/z calcd. For (C65H61N10O18S3-): 1365.34, found: 1365.33.
Basic characterization
A UV spectrophotometer (Implen N60 Touch) was used to measure the degree of substitution of AC4A and assess the photostability of the photosensitizer. Particle size, zeta potential analyzes, and stability analysis on different albumin-based nanoparticles were performed using a Malvern particle analyzer (Zetasizer Pro). The morphology of the nanoparticles was visualized using a Transmission Electron Microscope (HT7800, 120 V). A fluorescence spectrophotometer (PerkinElmer LS55) was employed to meticulously assess the intricate phenomenon of fluorescence quenching exhibited by SMNB post its encapsulation within HC4, with subsequent fluorescence recovery manifesting upon vector perturbation.
Hypoxia-induced mitophagy by western blot analysis
The expressions of HIF-1ɑ, Beclin1, and LC3B were determined by western blot analysis. B16 and MCF-7 cells were treated with PBS (21% O2), PBS (1% O2), and HCQ (1% O2, 10 μM) for 24 h. Protein extraction was performed using a robust protocol involving RIPA buffer supplemented with a potent cocktail of proteinase and phosphatase inhibitors (Leagene). Subsequently, the protein samples were subjected to electrophoresis on a high-quality 15% polyacrylamide gel using the advanced PAGE Gel Fast Preparation Kit (EpiZyme). The primary antibodies and secondary antibodies, including anti-HIF-1ɑ (ab179483, Abcam, WB: 1:1000), anti-Beclin (AF5128, Affinity, WB: 1:1000), anti-LC3B (L754, Sigma, WB: 1:1000), β-actin (AF7018, Affinity, WB: 1:5000), and anti-rabbit HRP-linked IgG (S0010, Affinity, WB: 1:5000) were applied. The blots were developed using an enhanced chemiluminescence (ECL) solution. The intensity of blots was quantified by the Image J software (flimj-plugin).
In vitro cytotoxicity assay of SMNB and HCQ
B16 and MCF-7 cells were treated with SMNB (0, 0.05, 0.1, 0.25, 0.5, 1.0, 2.0 μM) and HCQ (0, 5, 10, 20, 40 μM) for 24 or 48 h in a normoxic incubator (21% O2) or a hypoxic incubator chamber (1% O2). For the light irradiation group, cells were irradiated with a 660 nm laser (100 mW/cm2; 5 min) after 12 h of co-culturing with the drug. Cell viability was evaluated using a CCK8 assay kit, and the UV absorption (405 nm) was measured using a SpectraMax® i3x spectrophotometer.
In vitro cytotoxicity assay for screening SMNB/HCQ ratio
MCF-7 and B16 cells were treated with free SMNB/HCQ pair at various combinations of molar ratios for 24 h, under normoxic (21% O2) or a hypoxic (1% O2) condition. For the light group, cells were irradiated with a 660 nm laser (100 mW/cm2; 5 min) after 12 h of co-culturing with the drug. Cell viability was evaluated using a cck8 assay, and measured using a SpectraMax® i3x spectrophotometer at 405 nm. The results were analyzed using the Q-value method to obtain a combination index (CI) value.
V(a + b) represents the inhibition rate of the combination therapy of A and B, while Va and Vb represent the inhibition rate of A and B as monotherapies (Q < 0.85 indicates antagonism, 0.85 ≤ Q < 1.15 indicates additivity and Q ≥ 1.15 indicates synergy).
Preparation of HC4
To prepare HSA with free thiol groups, glutathione (33.50 mg) and HSA (100 mg) were dissolved in 10 mL of deionized water and stirred at room temperature for 12 h. After completion of the reaction, the resulting solution was dialyzed using a dialysis membrane (MWCO = 2000) for 24 h to remove any unreacted reagents and impurities. Finally, the dialyzed solution was freeze-dried to obtain the purified HSA with free sulfhydryl groups.
To conduct a click reaction between thiol groups and double bonds, 24 mg of reduced HSA, 20 mg of MAM-SAC4A, and 20 mg of 2,2-bis(hydroxymethyl)propionic acid were dissolved in 20 mL of ultra-pure water. The reaction mixture was exposed to a purple light at 365 nm and allowed to react for 24 h, followed by dialysis for 24 h through a dialysis membrane (MWCO = 2000). The purified solution was then freeze-dried to obtain a red solid powder (25 mg, 69.10% yield).
Binding affinities of MAM-SAC4A with guests
Rhodamine B (RhB) was used as a fluorescent probe, and the binding constants (Ka) of RhB and MAM-SAC4A were measured by fluorescence direct titration. The direct fluorescent titration method’s fluorescence intensity (PerkinElmer LS55) of 0.5 μM RhB was measured first. Then, different concentrations of MAM-SAC4A were added to it, and the fluorescence intensity decreased with the increase of MAM-SAC4A concentration. Host and guest bonding constants (Ka) were determined according to the 1:1 bonding model. The binding constants of MAM-SAC4A to SMNB and HCQ were measured by fluorescence competitive titration, respectively. The competitive fluorescent titration method added different concentrations of competitors into the solution of host-guest dyes with known binding constants. The fluorescence intensity of the fluorescent dye and the change in the concentration of the guest were tested to obtain the fluorescence intensity change curve. The binding constants (Ka) of the host and the guest to be tested were determined according to the 1:1 competitive bonding model.
Data analysis of fluorescence titrations. Data fitting from direct host-guest titrations and competitive titrations was performed nonlinearly. The fitting modules were downloaded from the website of Prof. Nau’s group (http://www.jacobs-university.de/ses/wnau) under the column “Fitting Functions.”
Preparation of SHC4H
To obtain SHC4H, HSA-AC4A was dissolved in a PBS solution and treated with SMNB and HCQ separately. The reaction mixture was stirred for 2 h to allow for the conjugation of the SMNB and HCQ to the HSA-AC4A conjugate. The hydrodynamic diameter and zeta potential were measured by dynamic light scattering using a Malvern Mastersizer 2000 instrument. The TEM images of nanoparticles were collected by JEM-ARM200P.
Evaluation of drug release behavior
The drug release behavior of SHC4H was detected by UV-Vis spectroscopy (LAMBDA365 Uv-vis). Rat liver microsomes (40 μg/mL) and NADPH (40 μM) were added to a mixed solution of HSA-AC4A (10 μM) and HCQ (10 μM) and incubated in hypoxic conditions at 37 °C. At various time intervals, collect 0.5 mL of the solutions, centrifuge for 5 min using an ultrafiltration tube (MWCO 3000), assess the absorption at 342 nm of the concentrated solution, and quantify the release of HCQ based on the HCQ standard curve. Repeat this process three times at each time point. The release behavior evaluation of SMNB is also conducted following the above experimental method.
The hypoxia imaging capability of SHC4H in vitro analysis
MCF-7 and B16 cells were treated with SHC4H (SMNB concentration: 50 nM) and exposed to either normoxic (21% O2) or hypoxic (1% O2) conditions for 6 or 12 h. Following treatment, the cells were stained with DAPI. Confocal Laser Scanning Microscopy (LSN 880 microscope with the ZEN 3.2 software) was employed for detailed observation and documentation of the cells, capturing intricate features (Excitation wavelengths of 633/700 nm for SMNB).
Photosensitizers located at lysosomes and mitochondria analysis
MCF-7 and B16 cells were treated with SMNB, SHC4, or SHC4H (SMNB concentration: 50 nM) and cultured under hypoxic conditions (1% O2) for 12 h. After treatment, the cells were incubated with Mito-Tracker Green (100 nM) or Lyso-Tracker Green (50 nM) pre-incubated at 37 °C for 30 min. Following a thorough triple wash with PBS, the cells were stained with DAPI for 10 min. Cellular observations were carried out using CLSM (LSN 880 microscope with the ZEN 3.2 software), with excitation/emission wavelengths of 490/516 nm for Mito-Tracker Green, 504 /511 nm for Lyso-Tracker Green, 340/488 nm for DAPI, and 633/650-700 nm for SMNB. The Image J software (Fiji) analyzed the Pearson’s R-value.
Optimizing incubation time analysis of SHC4H
To determine the optimal incubation time of SHC4H with cells to ensure sufficient lysosomal alkalinization and achieve maximum cytotoxicity and ROS generation upon light exposure. Cells were treated with SHC4H (HCQ: 10 μM) and incubated for 6, 12, and 24 h, and then co-incubated with Lyso-Tracker Green (Beyotime, C1047S) for 30 min. The cell samples were observed under a CLSM (LSN 880 microscope with the ZEN 3.2 software). Post-incubation with SHC4H for different times, cells were exposed to light specific for SMNB activation. Cell viability was evaluated post-light exposure using a cytotoxicity assay kit. Intracellular ROS levels were measured immediately using a ROS detection assay kit.
The cytotoxicity analysis of SHC4H+hv treatment
MCF-7, B16, CT26, and 4T1 cells were seeded in 96-well plates (5000 cells per well) and incubated for 12 h. Similarly, A431 and PANC1 cells were seeded in 96-well plates (10000 cells per well) and incubated for 12 h. The original cell medium was then replaced with a novel medium containing PBS, HCQ, SMNB, SHC4, HC4H, and SHC4H (at a ratio of SMNB: HCQ = 0.1:10 μM for MCF-7, CT26, A431, PANC1 cells and 0.25:10 μM for B16 cells). The cells were cultured in either a normoxic cell incubator (21% O2) or a hypoxia incubator chamber (1% O2) for 24 h. In light-treated groups, cells were exposed to a 660 nm laser (100 mW/cm2; 5 min) and cultured for 12 h. After these treatments, the cell medium was replaced with fresh medium containing 10% CCK8 and incubated for 2 h. Finally, cell viability was evaluated by measuring absorbance at 450 nm using a Microplate Reader. For calcein/PI staining analyzes, MCF-7 and B16 cells were seeded in 12-well plates and incubated for 12 h. Different formulations were used after replacing the culture medium. Calcein AM/PI solution was added and incubated for 30 min, followed by CLSM (LSN 880 microscope with the ZEN 3.2 software) using excitation wavelengths of 494/517 nm for Calcein and 535/617 nm for PI. Cell apoptosis assay data were collected using Beckman cytoflex with flowJo (Vision 10.6.2) and analyzed using Flowjo FlowJo 10.8.1 software.
ROS generation capability analysis of SHC4H
MCF-7 and B16 cells were then treated with PBS, HCQ, SMNB, SIPN2, SHC4, or SHC4H and cultured under either normoxic (21% O2) or hypoxic (1% O2) conditions for 24 h. Following treatment, cells were co-incubated with DCFH-DA (10 μM), dihydrorhodamine 123 (DHR123), or hydroxyphenyl fluorescein (HPF) at 37°C for 30 min. Subsequently, the cells were irradiated with a 660 nm laser (100 mW/cm2; 5 min) and stained with DAPI for 10 min. Utilizing CLSM (LSN 880 microscope with the ZEN 3.2 software), meticulous cellular observation and image capture were conducted, with excitation/emission wavelengths of 358/461 nm for DAPI and 481/535 nm for DCFH-DA, DHR123, and HPF. Furthermore, flow cytometry analysis was employed for subsequent quantitative assessment of O2•-, •OH, and total ROS generation.
Detection of mitochondrial membrane potential
After different groups of treatments, the cells were incubated with JC-10 staining solution (Mitochondrial Membrane Potential Kit, Solarbio, M8650) for 20 min and observed under a CLSM (LSN 880 microscope with the ZEN 3.2 software). The fluorescence signals of JC-10 monomer and JC-10 aggregate were detected at 490/530 nm and 525 /590 nm, respectively.
Enhanced oxidative stress detection
Following PBS, SHC4, SHC4H, and SHC4H treatments for 24 h under hypoxia, the cells were exposed to 660 nm laser irradiation (100 mW/cm2; 5 min). After the treatment, cells were harvested for lysis to obtain intracellular lysate. Subsequently, the GSH and GSSG Assay Kit (Beyotime, S0053), Human/Mouse 8-Hydroxydeoxyguanosine Elisa Detection Kit (Mlbio, ml057433), and Lipid Peroxidation MDA Assay Kit (Beyotime, S0131S) were employed for the quantitative assessment of GSSG, 8-OHdG, and MDA levels, respectively. Furthermore, the enhanced oxidative stress was detected by a ROS-ID Hypoxia/ Oxidative Stress Detection Kit (ENZO, ENZ-51042-0125). The cells were then observed by CLSM (LSN 880 microscope with the ZEN 3.2 software), with excitation wavelengths of 490/525 nm for oxidative stress detection and 596/670 nm for hypoxia detection.
Analysis of the regulation of mitophagy
The cells were treated with PBS (21% O2), PBS, SHC4+hv, SHC4H, and SHC4H+hv. To analyze the co-localization of mitochondria and lysosomes, the cells were co-incubated with Lyso-Tracker Green (50 nM) and Mito-Tracker Red stain working medium (30 nM) at 37°C for 90 min. To investigate the occurrence and blockage of mitochondrial autophagy, the cells were co-incubated with anti-LC3B (Sigma, L754, IF: 1:100) for 12 h and stained with Alexa Fluor® 488 Conjugate anti-rabbit IgG (4412S, Cell Signaling Technology, IF: 1:500). The cells were observed and imaged using a CLSM (LSN 880 microscope with the ZEN 3.2 software), with excitation wavelengths of 504/511 nm for Lyso-Tracker Green, 579/599 nm for Mito-Tracker Red, and 488/525 nm for anti-LC3B. Furthermore, the TEM images of tumor cells with different treatments were collected by HITACHI HT7700 for the detection of autophagosome aggregation and cell death mode.
AdPlus-mCherry-GFPLC3B adenovirus was added to the cells at a concentration of 10 μL (10 MOI) to detect the autophagy flow. After incubating for 24 h, the cells were treated with different treatments and observed using a CLSM. The fluorescence signals of mCherry and GFP were detected at 587/610 nm and 488/507 nm, respectively.
B16 and MCF-7 cells were prepared on a confocal dish and fixed with 4% paraformaldehyde to preserve protein localization. Subsequently, permeabilization was carried out through 0.1% Triton X-100 in PBS, facilitating antibody penetration. Blocking with 5% BSA in PBS followed, reducing nonspecific binding. Primary antibodies anti-PINK1 (DF7742S, Abmart, WB:1:1000, IF: 1:100) and anti-Parkin (AF5128, Affinity, WB:1:1000, IF: 1:100), anti-TOMM20 (T55527S, Abmart, WB:1:1000, IF: 1:100), and anti-MFN2 (12186-1-AP, Proteintech, WB:1:1000, IF: 1:200) were then applied, and cells were incubated according to manufacturer recommendations. After thorough washing, secondary antibodies Alexa Fluor® 488 Conjugate anti-rabbit IgG (4412S, Cell Signaling Technology, IF: 1:500) were used for visualization. CLSM was used to capture fluorescence images. Moreover, the western blot analysis was also conducted by the above antibodies. Co-localization quantification and the western blot analysis results were analyzed using Image J software (Fiji).
ATP levels detection
After co-culturing with the different drugs in an anaerobic incubator (1%O2) for 24 h, the light group was irradiated (100 mW/cm2, 5 min). Following an additional 12 h incubation, the cell culture supernatant and cell lysates were harvested. The ATP level of the sample was detected by an Enhanced ATP Assay Kit (S0027, Beyotime). The chemiluminescence was measured using an enzyme labeling instrument (SpectraMax®i3x).
Detection of mitophagy-related proteins
The cells were treated with PBS (21% O2), PBS, SHC4+hv, SHC4H, and SHC4H+hv. The mitophagy-related protein expression (LC3, SQSTM1) was determined. The primary antibodies and secondary antibodies included anti-LC3B (L754, Sigma, WB: 1:1000), anti-SQSTM1 (Abcam, ab56416, WB: 1:1000), β-actin (AF7018, Affinity, WB: 1:1000), Goat Anti-Mouse IgG (H + L) HRP (S0002, Affinity, WB: 1:5000) and anti-rabbit HRP-linked IgG (S0010, Affinity, WB: 1:5000). Western blotting strips were obtained by Mini-PROTEAN Tetra, UVP ChemStudio 815 and BG-GDAAUTO730. The blots were developed using an enhanced chemiluminescence (ECL) solution. The intensity of blots was quantified by the Image J software (Fiji). The expression of PIKN1 and Parkin was also measured by the immunofluorescence staining and observed by CLSM.
RNA sequencing and analysis
B16 and MCF-7 cells were seeded in 6-well culture plates and cultured for 24 h. Then, the cells received different treatments, including PBS, SHC4+hv, SHC4H, and SHC4H+hv. Irradiation was applied at a 100 mW/cm2 power density for 5 min. The cells were incubated for another 6 h, and the total RNA was extracted from the treated cells using a Trizol reagent (Ambion, 260808). High-throughput sequencing was performed by Shanghai Personal Biotechnology Co., Ltd. (3 sets of PBS, 3 sets of SHC4+hv, and 3 sets of SHC4H+hv, both in B16 and MCF-7 cell line). Sequencing libraries were created using the NEBNext Ultra II RNA Library Prep Kit and sequenced on the Illumina novaseq 6000 platform. A differential expression analysis was carried out between two sets of comparisons using the DESeq (v1.38.3) software. Gene Ontology (GO) enrichment analysis was performed with TopGO (v2.50.0), and p-values were determined using the hypergeometric distribution method. GO terms significantly enriched among the differentially expressed genes (DEGs), whether upregulated or downregulated, were identified based on a p-value threshold of <0.05. The hypergeometric distribution test was also utilized for the analysis of pathways from the Kyoto Encyclopedia of Genes and Genomes (KEGG).
Animal tumor models
BALB/c nude mice (Female; 5–6 weeks; 18–22 g) were used as the experimental model. B16 (5 × 105 cells in 100 μL PBS), MCF-7 (1 × 106 cells in 100 μL PBS), or PANC1 (1 × 106 cells in 100 μL PBS) cells were subcutaneously implanted into the thighs of each nude mouse. To establish an orthotopic homologous breast tumor model, PBS (100 μL) containing 1 × 106 4T1 cells was subcutaneously injected into the fat pat under the fourth mammary gland of the right side. The tumors were allowed to grow until their size reached 150 mm3 for subsequent experiments. BALB/c nude mice were bred and housed in a standard barrier animal facility at a temperature of 23 ± 2°C, relative humidity range between 40–70 % environment with a 12 h light/dark cycle.
Hypoxia-responsive fluorescence imaging
The MCF-7 or B16 tumor-bearing mice were randomized into three groups: PBS group, SMNB group, and SHC4H group. The mice were evaluated and imaged using a mouse imaging apparatus with an excitation wavelength of 680 nm and an emission wavelength of 750 nm at 0, 6, 12, 24 and 48 h post-administration via the caudal vein. At 24 h post-administration, the mice were humanely sacrificed, and their tumors, heart, liver, spleen, lung, and kidney were excised. The tumors were rinsed with normal saline and dried with filter paper before fluorescence imaging using the IVIS Spectrum system (Lumina XR Series III).
In vivo efficacy of anti-MCF-7 and B16 tumor models
The tumor-bearing mice were randomly allocated into six groups (n = 6 mice per group): PBS, SMNB + HCQ+hv, HC4H, SHC4+hv, SHC4H, and SHC4H+hv group. The tumor-bearing mice were intravenously injected on days 0, 3, 6, and 9 in the MCF-7 model and on days 0, 2, 4, and 6 in the B16 model, while their tumor sites were irradiated with a 660 nm laser (100 mW/cm², 5 min) on various days. The body weight and tumor volume were measured and recorded every two or three days during the experiment. After the treatments, the tumors obtained from each treatment group were analyzed by H&E, immunofluorescence, and western blot analysis. The tumor image of the mice at the end of the experiment was taken by Xiaomi Civi 3. Immunofluorescent staining images were acquired with the ZEN 3.2 software on a LSN 880 microscope. Histology images were processed and analyzed using NDP. View 2.
In vivo efficacy of anti-PANC1 and 4T1 orthotopic breast tumor models
The mice with an average tumor volume of about 150 mm3 were randomly allocated to 5 treatment groups (n = 6 mice for each group), which were PBS, HC4H, SHC4+hv, SHC4H, and SHC4H+hv. The drugs were injected via tail vein administration, and the 660 nm laser irradiation (100 mW/cm², 5 min) was performed at 24 h after days 0 and day 3 administration. The body weight and tumor volume were measured and recorded every three days during the experiment. After the treatments, the tumors and the major organs of the represented mice were harvested for H&E staining to evaluate the effect and the safety of our formulations.
Blood routine and biochemical analysis
To further investigate the biosafety of the nanoparticles, we collected blood samples from healthy mice (n = 3 mice) without treatment and healthy mice treated with SHC4H (n = 3 mice) four times. Furthermore, at the end of the SHC4H+hv treatments, the blood samples of each mouse (n = 3 mice) were also collected for blood routine analysis and blood biochemistry determination.
Statistical and data reproducibility
Quantitative data supporting our study are presented as mean ± standard deviation (SD) and were analyzed using GraphPad Prism 8.0. The schemes and illustrations were produced by Adobe Illustrator 2022. Statistical analyzes were performed using unpaired, one-way or two-way ANOVA, followed by Tukey’s multiple comparison test for multiple group comparisons. A p-value of less than 0.05 was considered statistically significant. As for the choice of sample size, we reviewed the sample size of similar studies and conducted preliminary experiments to estimate effect size and data variability. The experiments were repeated independently at least three times with similar results unless otherwise mentioned, and the micrographs were the representative data from individual samples (the n was given in figure captions).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The RNA sequencing data generated in this study have been deposited in the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) database at https://www.ncbi.nlm.nih.gov/sra with accession codes PRJNA1153548 and PRJNA1152985. NMR spectrum data of SMNB and MAM-SAC4A are included in Supplementary Fig. 6, 7, 12, 13. Mass spectrum data of SMNB and MAM-SAC4A are summarized in Supplementary Fig. 5, 14. All other data supporting this study are available in the main article, Supplementary Information or Source Data file. Source data file is provided with this paper. Source data are provided with this paper.
Change history
28 February 2025
A Correction to this paper has been published: https://doi.org/10.1038/s41467-025-57018-y
References
Helmlinger, G. et al. Interstitial pH and pO2 gradients in solid tumors in vivo: high-resolution measurements reveal a lack of correlation. Nat. Med. 3, 177–182 (1997).
Wei, X. et al. Mechanisms of vasculogenic mimicry in hypoxic tumor microenvironments. Mol. Cancer 20, 7–24 (2021).
Chen, Z. et al. Hypoxic microenvironment in cancer: molecular mechanisms and therapeutic interventions. Signal Transduction Targeted Ther. 8, 70–92 (2023).
Blouw, B. et al. The hypoxic response of tumors is dependent on their microenvironment. Cancer Cell 4, 133–146 (2003).
Liu, J.-N. et al. Chemical design and synthesis of functionalized probes for imaging and treating tumor hypoxia. Chem. Rev. 117, 6160–6224 (2017).
Zou, M.-Z. et al. Advances in nanomaterials for treatment of hypoxic tumor. Natl. Sci. Rev. 8, nwaa160 (2021).
Singleton, D. C. et al. Therapeutic targeting of the hypoxic tumour microenvironment. Nat. Rev. Clin. Oncol. 18, 751–772 (2021).
Meng, W., Hao, Y., He, C., Li, L. & Zhu, G. Exosome-orchestrated hypoxic tumor microenvironment. Mol. Cancer 18, 1–14 (2019).
Bhandari, V. et al. Molecular landmarks of tumor hypoxia across cancer types. Nat. genet. 51, 308–318 (2019).
Jin, M.-Z. & Jin, W.-L. The updated landscape of tumor microenvironment and drug repurposing. Signal Transduction Targeted Ther. 5, 166 (2020).
Semenza, G. L. et al. Tumor metabolism: cancer cells give and take lactate. J. Clin. Invest. 118, 3835–3837 (2008).
Liu, L. et al. Hypoxia-induced energy stress regulates mRNA translation and cell growth. Mol. Cell 21, 521–531 (2006).
Bader, S. B. et al. Replication catastrophe induced by cyclic hypoxia leads to increased APOBEC3B activity. Nucleic Acids Res. 49, 7492–7506 (2021).
Taylor, C. T. & Scholz, C. C. The effect of HIF on metabolism and immunity. Nat. Rev. Nephrol. 18, 573–587 (2022).
Murphy, M. P. et al. Mitochondrial dysfunction indirectly elevates ROS production by the endoplasmic reticulum. Cell Metab. 18, 145–146 (2013).
Gustafsson, Å. B. & Dorn, G. W. Evolving and expanding the roles of mitophagy as a homeostatic and pathogenic process. Physiol. Rev. 99, 853–892 (2019).
Wang, S. et al. The mitophagy pathway and its implications in human diseases. Signal Transduction Targeted Ther. 8, 304 (2023).
Youle, R. J. & Narendra, D. P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 12, 9–14 (2011).
Assi, M. & Kimmelman, A. C. Impact of context-dependent autophagy states on tumor progression. Nat. Cancer 4, 596–607 (2023).
Fay, H. R. S. et al. Mitophagy plays a key role in the anti-leukemic activity of autophagy inhibitors under hypoxia in acute myeloid leukemia. Blood 134, 1278 (2019).
Vyas, S., Zaganjor, E. & Haigis, M. C. Mitochondria and cancer. Cell 166, 555–566 (2016).
Xue, Q. et al. Copper metabolism in cell death and autophagy. Autophagy 19, 2175–2195 (2023).
Xie, X.-Q. et al. Targeting ATAD3A-PINK1-mitophagy axis overcomes chemoimmunotherapy resistance by redirecting PD-L1 to mitochondria. Cell Res. 33, 215–228 (2023).
Zuo, L. et al. Biomimetic nanovesicle with mitochondria-synthesized sonosensitizer and mitophagy inhibition for cancer sono-immunotherapy. Nano Lett. 23, 3005–3013 (2023).
Gong, X. et al. Enhanced chemodynamic therapy mediated by a tumor-specific catalyst in synergy with mitophagy inhibition improves the efficacy for endometrial cancer. Small 19, e2301497 (2023).
Xu, F. et al. Development of a pH-responsive polymersome inducing endoplasmic reticulum stress and autophagy blockade. Sci. Adv. 6, eabb8725 (2020).
Li, Y. et al. PINK1-mediated mitophagy promotes oxidative phosphorylation and redox homeostasis to induce drug-tolerant persister cancer cells. Cancer Res. 83, 398–413 (2023).
Qu, F. et al. Manipulation of mitophagy by “all-in-one” nanosensitizer augments sonodynamic glioma therapy. Autophagy 16, 1413–1435 (2020).
Deng, Y. et al. Biomimetic nanoparticle synchronizing pyroptosis induction and mitophagy inhibition for anti-tumor therapy. Biomaterials 301, 122293 (2023).
Li, Z. et al. Activatable nano-photosensitizers for precise photodynamic cancer therapy. Coord. Chem. Rev. 493, 215324 (2023).
Chen, W. et al. Type I photosensitizer targeting G-quadruplex RNA elicits augmented immunity for cancer ablation. Angew. Chem. Int. Ed. 62, e202300162 (2023).
Yao, Q. et al. The concept and examples of type-III photosensitizers for cancer photodynamic therapy. Chem 8, 197–209 (2022).
Shin, J. et al. Mitochondria-targeted nanotheranostic: harnessing single-laser-activated dual phototherapeutic processing for hypoxic tumor treatment. Matter 4, 2508–2521 (2021).
Son, S. et al. Cancer therapeutics based on diverse energy sources. Chem. Soc. Rev. 51, 8201–8215 (2022).
Ma, Z. et al. A pH-driven small-molecule nanotransformer hijacks lysosomes and overcomes autophagy-induced resistance in cancer. Angew. Chem. Int. Ed. 61, e202204567 (2022).
Nash, G. T. et al. Nanoscale metal-organic layer isolates phthalocyanines for efficient mitochondria-targeted photodynamic therapy. J. Am. Chem. Soc. 143, 2194–2199 (2021).
Li, H. et al. Mitochondria-targeted smart AIEgens: imaging and therapeutics. Coord. Chem. Rev. 473, 214818 (2022).
Wang, D. et al. Microbial synthesis of Prussian blue for potentiating checkpoint blockade immunotherapy. Nat. Commun. 14, 2943 (2023).
An, J. et al. An unexpected strategy to alleviate hypoxia limitation of photodynamic therapy by biotinylation of photosensitizers. Nat. Commun. 13, 2225 (2022).
Teng, K.-X., Niu, L.-Y. & Yang, Q.-Z. Supramolecular photosensitizer enables oxygen-independent generation of hydroxyl radicals for photodynamic therapy. J. Am. Chem. Soc. 145, 4081–4087 (2023).
Li, M. et al. Unimolecular photodynamic O2-economizer to overcome hypoxia resistance in phototherapeutics. J. Am. Chem. Soc. 142, 5380–5388 (2020).
Kim, J. H. et al. A small molecule strategy for targeting cancer stem cells in hypoxic microenvironments and preventing tumorigenesis. J. Am. Chem. Soc. 143, 14115–14124 (2021).
Won, M. et al. An ethacrynic acid-brominated BODIPY photosensitizer (EA-BPS) construct enhances the lethality of reactive oxygen species in hypoxic tumor‐targeted photodynamic therapy. Angew. Chem. Int. Ed. 60, 3196–3204 (2021).
Ji, Y. et al. Use of ratiometrically designed nanocarrier targeting CDK4/6 and autophagy pathways for effective pancreatic cancer treatment. Nat. Commun. 11, 4249 (2020).
Georgakopoulos, N. D., Wells, G. & Campanella, M. The pharmacological regulation of cellular mitophagy. Nat. Chem. Biol. 13, 136–146 (2017).
Han, H. et al. Corralarenes: a family of conjugated tubular hosts. J. Am. Chem. Soc. 144, 20351–20362 (2022).
Li, J.-J. et al. Lactose azocalixarene drug delivery system for the treatment of multidrug-resistant pseudomonas aeruginosa infected diabetic ulcer. Nat. Commun. 13, 6279 (2022).
Guo, J.-S. et al. Dual hypoxia-responsive supramolecular complex for cancer target therapy. Nat. Commun. 14, 5634 (2023).
Hu, X. Y. et al. Hypoxia-responsive host-guest drug delivery system. Acc. Mater. Res. 4, 925–938 (2023).
Yao, S. Y. et al. An antitumor dual-responsive host-guest supramolecular polymer based on hypoxia-cleavable azocalix[4] arene. Angew. Chem. Int. Ed. 135, e202213578 (2023).
Li, S. et al. Drug in drug: A host-guest formulation of azocalixarene with hydroxychloroquine for synergistic anti-inflammation. Adv. Mater. 34, 2203765 (2022).
Zhang, T.-X. et al. A general hypoxia-responsive molecular container for tumor-targeted therapy. Adv. Mater. 32, e1908435 (2020).
Yue, Y. X. et al. Promoting tumor accumulation of anticancer drugs by hierarchical carrying of exogenous and endogenous vehicles. Small Struct. 3, 2200067 (2022).
Huang, Q. et al. Coupling an artificial receptor with macrophage membrane for targeted and synergistic treatment of cholestasis. Supramol. Mater. 1, 100020 (2022).
Yao, S.-Y. et al. Single molecular nanomedicines based on macrocyclic carrier-drug conjugates for concentration-independent encapsulation and precise activation of drugs. J. Am. Chem. Soc. 146, 14203–14212 (2024).
Chen, F. Y. et al. Molecular recognition of cyclophanes in water. Chin. Chem. Lett. 35, 109161 (2023).
Wang, Y. et al. Calixarene-modified albumin for stoichiometric delivery of multiple drugs in combination-chemotherapy. Theranostics 12, 3747–3757 (2022).
Shi, Z. et al. NIR-dye bridged human serum albumin reassemblies for effective photothermal therapy of tumor. Nat. Commun. 14, 6567 (2023).
Guo, X. et al. Mito-bomb: targeting mitochondria for cancer therapy. Adv. Mater. 33, e2007778 (2021).
Giampazolias, E. et al. Mitochondrial permeabilization engages NF-κB-dependent anti-tumour activity under caspase deficiency. Nat. Cell Biol. 19, 1116–1129 (2017).
Zhang, C. et al. Bypassing the immunosuppression of myeloid-derived suppressor cells by reversing tumor hypoxia using a platelet-inspired platform. Adv. Funct. Mater. 30, 2000189 (2020).
Zhou, S. et al. Acid and hypoxia tandem-activatable deep near-infrared nanoprobe for two-step signal amplification and early detection of cancer. Adv. Mater. 35, 2212231 (2023).
Ge, L. et al. Light-activated hypoxia-sensitive covalent organic framework for tandem-responsive drug delivery. Nano Lett. 21, 3218–3224 (2021).
Li, X. et al. Impairing tumor metabolic plasticity via a stable metal-phenolic-based polymeric nanomedicine to suppress colorectal cancer. Adv. Mater. 35, e2300548 (2023).
Mariño, G. et al. Baehrecke self-consumption: the interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell Bio. 15, 81–94 (2014).
Tsapras, P. et al. Caspase involvement in autophagy. Cell Death Differ. 24, 1369–1379 (2017).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (32101065, 32471390, and U20A20259), the Science, Technology & Innovation Commission of Shenzhen Municipality (JCYJ20220818102810023), and the Natural Science Foundation of Guangdong Province (2023A1515012015).
Author information
Authors and Affiliations
Contributions
H. Chen, D. Guo, X. Zeng, and W. Wang conceived the project. W. Wang performed the experiments, analyzed the results, and wrote the manuscript. S. Yao completed the synthesis of MAM-SAC4A and the detection of the interaction between drugs and carriers. J. Luo, Q. Huang, Y Yang, Z Shi, J Lin helped in some cell and animal experiments. C. Ding contributed to crafting the schematic diagram and the refinement of the manuscript. Y. Pan contributed to the refinement of the manuscript. All other authors revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, W., Yao, SY., Luo, J. et al. Engineered hypoxia-responsive albumin nanoparticles mediating mitophagy regulation for cancer therapy. Nat Commun 16, 596 (2025). https://doi.org/10.1038/s41467-025-55905-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-55905-y
