
Abstract
Secondary organic aerosols (SOAs) significantly impact Earth’s climate and human health. Although the oxidation of volatile organic compounds (VOCs) has been recognized as the major contributor to the atmospheric SOA budget, the mechanisms by which this process produces SOA-forming highly oxygenated organic molecules (HOMs) remain unclear. A major challenge is navigating the complex chemical landscape of these transformations, which traditional hypothesis-driven methods fail to thoroughly investigate. Here, we explore the oxidation of α-pinene, a critical atmospheric biogenic VOC, using a novel reaction discovery approach based on molecular dynamics and state-of-the-art enhanced sampling techniques. Our approach successfully identifies all established reaction pathways of α-pinene ozonolysis, as well as discovers multiple novel species and pathways without relying on a priori chemical knowledge. In particular, we unveil a key branching point that leads to the rapid formation of alkoxy radicals, whose high and diverse reactivity help to explain hitherto unexplained oxidation pathways suggested by mass spectral peaks observed in α-pinene ozonolysis experiments. This branching point is likely prevalent across a variety of atmospheric VOCs and could be crucial in establishing the missing link to SOA-forming HOMs.
Similar content being viewed by others


Introduction
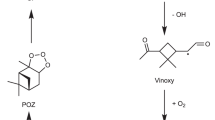
Organic aerosols are ubiquitous in the Earth’s atmosphere and have significant impacts on human welfare1,2,3. On a regional level, they contribute to the formation of urban haze4,5, which reduces visibility and endangers human health by increasing the risk of respiratory and cardiovascular diseases6,7. At the global scale, they affect the Earth’s radiative balance through both direct and indirect mechanisms3,8, with the former involving the scattering and absorption of solar radiation while the latter altering cloud properties. Model studies indicate that organic aerosol in general, and secondary organic aerosol (SOA) in particular9,10,11, constitute a substantial fraction of the total number of aerosol particles in the atmosphere3,12. Unlike primary organic aerosol (POA) that are directly emitted in particulate form, SOAs form through the clustering of low-volatility compounds generated by the oxidation of gas-phase parent volatile organic compounds (VOCs), which in turn originate from both biogenic and anthropogenic sources such as vegetation or traffic. Some of these oxidation reactions can lead to highly oxygenated organic molecules (HOMs) with extremely low volatility, that can either partition into preexisting aerosols, thus increasing their size and mass, or even foster the formation of entirely new aerosol particles (Fig. 1)13,14,15,16,17,18.

The intricate potential energy landscape arising from the degradation of atmospheric volatile organic compounds (VOCs) is systematically explored through the enhanced sampling aided approach, facilitating the enumeration of potential reaction pathways for subsequent high-level quantum chemical calculations.
Traditionally, the oxidation of atmospheric parental VOCs was believed to proceed in a stepwise manner, with one oxidant (such as ozone and OH radical) attack adding at most 2 to 3 new oxygen-containing functional groups to the carbon backbone before forming closed-shell products. These products typically require further oxidation cycles to generate HOMs. This conventional perspective, however, strongly limits the potential yields of low-volatility HOMs, as the intermediate products are prone to being scavenged by partitioning into condensed-phase before undergoing another oxidation cycle. Recent research15 has demonstrated that under suitable conditions, certain VOCs can undergo autoxidation, wherein a single oxidant attack leads (via sequential unimolecular isomerization and O2 addition reactions) to products containing as many as 10 new oxygen atoms. These findings underscore the need for a more comprehensive understanding of parent VOC reactivities and their fates, especially the competition between autoxidation and sequential oxidation. Such insights are crucial for accurately assessing regional and global aerosol budgets and quantifying their consequential impacts on climate and health.
Except for polluted urban regions19,20,21, biogenic VOCs have been identified as the main precursors for the global SOA budget, and total emissions of known biogenic SOA precursors are estimated to be up to 10 times larger than their anthropogenic counterpart3. Monoterpenes, a family of C10H16 compounds, comprise around 11% of global biogenic VOC emissions by mass12. They are considered as a crucial class of precursors for the global SOA budget12,22,23, primarily owing to their high reactivity (in particular, their tendency to undergo autoxidation) and the low volatility of the numerous oxidation products. α-Pinene is the most emitted monoterpene, accounting for approximately one third of the total emissions24. Its ozonolysis reaction (α-pinene + O3) is one of the most efficient systems for SOA generation in the atmosphere25,26. Though significant efforts have been made in understanding this important reaction over the past decades13,16,26,27,28,29,30,31, an explicit chemical mechanism that links the α-pinene ozonolysis to the formation of various lowest-volatility HOMs, which are SOA precursors, has not yet been established32.
The initial steps of α-pinene ozonolysis reaction, and the formation of closed-shell oxidation products with up to 4 to 5 oxygen atoms, are well documented and included, for example, in the widely used Master Chemical Mechanism33 (MCM) for tropospheric degradation of VOCs. Further sequential oxidation of α-pinene is also reasonably well described by automated reaction mechanism generators such as the Generator of Explicit Chemistry and Kinetics of Organics in the Atmosphere34,35 (GECKO-A). In contrast, the autoxidation pathways observed to lead to substantial yields of aerosol-forming HOMs13,36 on a sub-second timescale are still poorly understood despite some promising recent advances26. An issue complicating the experimental analysis is that especially these autoxidation channels are only competitive in relatively clean conditions (low reactant concentrations) – at the high concentrations required for the application of spectroscopic techniques, for example, they are overwhelmed by termination reactions. Currently, mass spectrometry is the principal analytical technique in identifying and characterizing autoxidation pathways driving HOMs formation. However, without an added dimension of separation provided by, for example, ion mobility or chromatographic analysis, mass spectrometric techniques cannot distinguish different molecules with the same mass37. Computational methods complement mass spectrometry by identifying the actual molecular-level reaction steps behind the observed autoxidation process. In the recent decades, computations have become efficient and accurate enough that theoretical studies are sometimes even able to guide experiments, rather than the other way around. However, except for a few very recent studies38,39, both experimental and theoretical studies in the field of atmospheric reaction mechanisms have so far relied heavily on chemical expertise and intuition to identify, for example, autoxidation pathways. This limits the exploration of complex reaction networks characterized by multiple competitive reaction pathways.
In contrast to the hypothesis-driven strategy, several discovery-based approaches have recently been proposed to characterize the reaction space based on ab initio molecular dynamics (MD)40,41,42,43,44,45,46. Some of these methods employ high temperatures and pressures to induce chemical reactions40,45,46, whereas others accelerate the sampling of reactive events by applying an external bias potential to a set of collective variables (CVs) that encode the generally slow degrees of freedom of a system41,42. In this context, Raucci et al.42 proposed a workflow for reaction discovery based on a CV derived from spectral graph theory47 and the explore version of the on-the-fly probability enhanced sampling method48 (OPESE). In this approach, a molecule is represented as a graph whose vertices and edges are its atoms and chemical bonds, respectively. The maximum eigenvalue of the symmetric adjacency matrix associated with the graph is then used as CV in discovery simulations (Fig. 1). This approach is suitable for blind sampling of reactive events because it is based on a generic CV that does not require any a priori knowledge about the reactivity of the system, allowing molecular dynamics simulations to freely explore new chemical pathways42,49,50. Furthermore, the dynamics of the system is perturbed in a controlled way by a barrier cutoff in OPESE, which sets a limit on the maximum value of the bias, and grants control over the exploratory phase.
In this study, we unravel the complexity of α-pinene ozonolysis using this enhanced sampling aided reaction discovery approach. In a three-step strategy, we first performed extensive MD guided reaction discovery simulations, and then characterized the energetics of the newly discovered pathways with high-level ab initio electronic structure calculations (up to CCSD(T) level). Finally, a kinetic master equation approach was set up to understand the significance of the newly observed reaction routes.
Without relying on human heuristic, we were able to uncover all the known reaction pathways of the α-pinene ozonolysis reaction, including the ring-opening channel and the OH roaming channels reported very recently by Iyer et al.26 and Klippenstein and Elliott51, respectively, as well as discover several new species and reaction pathways that are of high atmospheric relevance. In particular, our approach revealed the importance of a new reaction class in α-pinene ozonolysis: the unimolecular rearrangement of alkyl radicals containing endoperoxide functionalities, resulting in the formation of alkoxy radicals. This reaction type was investigated in aromatic oxidation but found to be negligible for benzene52. In contrast, within the α-pinene ozonolysis system, this reaction type becomes activated due to the excess energy retained from preceding reaction steps. In addition, using the analogy between this reaction and a previously reported epoxidation reaction53, we have identified both anthropogenic and biogenic model systems in which the endoperoxy-alkyl radical rearrangement is competitive with O2 addition even as a thermal reaction, i.e., without the chemical activation driven by the excess energy. A key feature of this reaction is that it unimolecularly converts alkyl radicals into alkoxy radicals, and thus – due to the versatility of alkoxy radical atmospheric chemistry – in effect opens new and unanticipated branching points in atmospheric oxidation sequences. Considering the ubiquity of endoperoxide functionalities in aromatic compounds and terpene-derived intermediates, such branching points will offer crucial evidence in explaining the extensive array of as-yet unexplained molecular compositions observed in mass spectrometric autoxidation experiments, and in delineating their role in atmospheric aerosol formation.
Results
Thorough exploration of known α-pinene ozonolysis pathways via MD guided reaction discovery
The initial steps of the α-pinene ozonolysis reaction follows the general mechanism of cyclic alkene ozonolysis. The ozone molecule adds to the double bond, forming a primary ozonide, which rapidly decomposes into a carbonyl-substituted Criegee Intermediate (CI; carbonyl oxide) (Fig. 2a). Due to the asymmetry of the addition site, as well as the high barrier for rotation around the O-O bond in the CI, there are four distinct CI isomers. According to MCM33, the branching ratios for their formation are reasonably similar33 (between 0.33 and 0.2). Three of the four CI isomers (as shown in Fig. 2a) have rapid 1,4-hydrogen shift channels, leading to the formation of three vinyl hydroperoxides (VHPs). These VHPs subsequently release an OH radical, producing vinoxy radicals—resonance-stabilized species with both alkyl and alkoxy radical characteristics. The vinoxy radicals then add O₂, forming four distinct types of peroxy radicals (RO₂), commonly referred to as the first-generation RO₂ in α-pinene ozonolysis. The remaining CI isomer (shown in Supplementary Fig. 1a) cannot undergo H-shifts, and thus either reacts bimolecularly (for example with water vapor) or isomerizes into a dioxirane which then rearranges or decomposes further to a variety of closed-shell products including carboxylic acids. The “textbook” reaction sequence, depicted in gray in Fig. 2a, cannot explain the experimentally observed HOMs with more than 6 O atoms in α-pinene ozonolysis. This is because the H-shift reactions of the first-generation RO₂ are generally too slow to compete with potential bimolecular termination reactions (with NOₓ or HO₂) in the atmosphere. Even under the cleanest conditions, these bimolecular termination reactions typically exhibit pseudo-first-order reaction rates exceeding 10⁻² s⁻¹ (10⁻² s⁻¹ to 10² s⁻¹ in typical atmospheric conditions). Kurtén et al.30 calculated all possible H-shift reactions for the first-generation RO₂ and found that each of the four RO₂ can, at most, undergo one H-shift reaction (with a rate at around 10⁻² s⁻¹ to 10⁻¹ s⁻¹), while all other H-shifts are significantly slower (ranging from 10⁻²⁶ s⁻¹ to 10⁻³ s⁻¹). Consequently, under atmospheric conditions, such H-shifts can only lead to the addition of one more O₂ to the first-generation RO₂. In 2021, Iyer et al.26 partially addressed this issue by proposing an alternative reaction pathway, in which one of the vinoxy radicals undergoes a chemically activated ring-opening, followed by O₂ addition to form a new RO₂ radical. This RO₂ is capable of rapid unimolecular isomerization, outcompeting bimolecular termination reactions and facilitating several more steps of autoxidation. Experiments on selectively deuterated α-pinene isomers32 have later confirmed that this reaction channel is indeed competitive – but also demonstrate the existence of additional, yet undiscovered reaction channels. Specifically, while the reaction route forming closed-shell products with 7 or more O atoms has been identified in Iyer et al.26, such routes inevitably involve H-shifts of the aldehydic H atom attached to the carbon that was originally the secondary C atom in the double bond (carbon 3 in Meder et al.32). But the deuteration experiments indicate that almost half of these products have not undergone such a reaction. This highlights the existence of unaccounted processes essential for explaining the α-pinene ozonolysis experiments.

a The known and newly discovered pathways. The gray region represents the conventional perception of the α-pinene ozonolysis mechanisms. The ring-breaking mechanism proposed by Iyer et al.26 is marked orange. The OH roaming mechanism leading to a closed-shell product with two double bonds found in this work is marked green. The endoperoxy-alkyl radical rearrangement mechanism found in this work is marked blue. b Complete set of products from the OH roaming mechanism.
The “textbook” α-pinene ozonolysis mechanism33 described above is reproduced in full by the MD reaction discovery procedure, illustrated in gray in Fig. 2a. Moreover, the reaction discovery engine independently discovered the ring-breaking mechanism (illustrated in orange in Fig. 2a) and its main subsequent steps proposed by Iyer et al.26. In addition to these already-known reaction channels, the sampling procedure produced a vast number of additional reaction mechanisms following the ring-breaking of Vinoxy-c, summarized in Supplementary Fig. 1b. While these reaction channels may represent actual reaction pathways (minimum-energy paths connecting potential energy surface minima) that could be accessible under suitable conditions, the vast majority of them are not competitive with bimolecular sinks in the atmosphere, which typically constrain the lifetimes of alkyl radicals to less than 10−6 s (due to the reaction with O2) and peroxy radicals to less than 10−2 s to 102 s (due to reactions with either NOx or HO2, depending on the conditions). The lifetimes and fates of alkoxy radicals are more structure-dependent54, but inevitably less than 10−3 s.
Notably, the reaction discovery engine also independently identified the OH roaming pathways (Fig. 2b), which were recently proposed by Klippenstein and Elliott based on systematic theoretical calculations51. The OH roaming was thought to be responsible for the production of hydroxycarbonyl products during the dissociation of Criegee intermediates, as observed in the pioneering experimental work55 and several recent works56,57 by Lester and co-workers. In the context of α-pinene ozonolysis, Klippenstein and Elliott’s theoretical calculations51 indicate that the OH roaming channels are kinetically significant, with a branching ratio of around 20% at room temperature for stabilized VHPs, and this ratio can further increase at the lower temperatures typical of the troposphere. However, the overall impact of the OH roaming channels is constrained by the branching of stabilized VHPs. The initial step of OH roaming produces addition or abstraction products with intact cyclobutane rings, which could possess energy exceeding the dissociation threshold of the cyclobutane ring and then lead to ring-breaking products51. We found a new ring-breaking product with two carbon-carbon double bonds, highlighted in green in Fig. 2a. This product forms when the dissociated OH abstracts a hydrogen atom from one of the two spectator methyl groups, leading to cyclobutane ring cleavage. Such unsaturated products have a high potential for subsequent oxidation reactions and could thereby contribute to the formation of HOMs. Their oxidation paths and related products merit in-depth studies in the future.
New branching point in α-pinene ozonolysis: endoperoxy-alkyl radical rearrangement
One recurring structural motif in the novel channels discovered by the reaction discovery engine was the high reactivity of alkyl radicals with endoperoxide (cyclic peroxide) substituents. These were observed to undergo a variety of different isomerization reactions, typically resulting in the conversion of an alkyl radical with an endoperoxide group into an alkoxy radical with an epoxide or ether group. Due to lifetime constraint discussed above, most of these reaction channels are unlikely to be competitive in the atmosphere. Similar rearrangement mechanisms have been reported for other organic compounds. For instance, Zador et al.38 reported this mechanism in a selected intermediate from the limonene + OH system. Additionally, calculations by Xu et al.52 suggested that this rearrangement mechanism is excessively slow in the oxidation of benzene. Nevertheless, for the radical depicted in blue in Fig. 2a, this reaction channel may have significant yields due to large amount of excess energy retained from preceding reaction steps. Indeed, this novel reaction channel occurs downstream of the ring-breaking mechanism proposed by Iyer et al.26, and involves two new isomerization steps for the RB-RO2 species. The first step, with a barrier of 16.3 kcal/mol at the CCSD(T) level, yields an alkyl radical with an endoperoxide group (abbreviated EPO in Fig. 2a). The second step, with a CCSD(T) barrier of 17.9 kcal/mol, then rearranges the EPO into an alkoxy radical with an epoxide group (abbreviated AOE).
To quantify the impact of excess energy in overcoming the above rearrangement channel barriers, we conducted comprehensive kinetic master equation simulations covering the entire potential energy surface of α-pinene ozonolysis along the targeted Criegee pathway (CI-a), depicted in Fig. 3a. Within these simulations, the dissipation of excess energy was addressed using a carefully evaluated molecular heat transfer model (Supplementary Note 2). Our simulations unveiled that over 75% of RB species was formed within 2 ns after the initial ozone attack on α-pinene (Supplementary Fig. 4), suggesting that RB species should retain a significant portion of the 80 kcal/mol excess energy acquired in preceding reactions. Traditionally, it has been assumed that such excess energy dissipates during the next O2 addition step (given its relatively lengthy timescale of approximately 100 ns), thereby exerting minimal influence on subsequent reaction steps. However, this assumption is challenged by our current simulation results: even though excess energy may dissipate after the completion of O2 addition, the early-formed addition product can retain sufficient energy to surmount the transition state (TS5 and TS6) barriers, leading to the substantial formation of AOE within nanoseconds. The SimpleRRKM method in the kinetic master equation simulation used to treat the O2 addition step requires a transition state, but these are difficult to optimize. Therefore, we used a model transition state with the correct number of rovibrational modes but with an energy that was varied to provide a pseudo-first-order O2 addition rate within the atmospherically relevant53,58,59 range of 107 s−1 to 108 s−1. The yield of this endoperoxy-alkyl radical rearrangement channel exhibits a high sensitivity to the O2 addition rate. We found that, at an effective pseudo-first-order O2 addition rate of 1.9 × 107 s⁻¹, the yield is approximately 9.0%, while a rate of 9 × 107 s⁻¹ elevates the yield to 31.5%, as is depicted in Fig. 3b. Assuming roughly equal yields for the four Criegee-forming channels of α-pinene ozonolysis, the 31.5% yield from the studied single channel translates into an overall yield of approximately 7.9% for the complete α-pinene ozonolysis reaction (this percentage roughly equals to the full HOMs yield from α-pinene ozonolysis). It is noteworthy that, although previous results30 indicate that the fastest H-shift reaction rates for first-generation RO₂ range from 10⁻² to 10⁻¹ s⁻¹ under thermalized conditions, these rates could be significantly enhanced by the excess energy discussed in this paper, potentially allowing for nanosecond-scale formation of certain H-shift products (see Supplementary Note 4). However, the excess energy likely affects only one H-shift cycle and is unlikely to persist into subsequent cycles due to rapid dissipation.

a Potential energy surface of α-pinene ozonolysis for the CI-a isomeric pathway. Numerical values are zero-point corrected energies as explained in the Methods section. b Yield of the alkoxy radical (AOE) from the endoperoxy-alkyl radical rearrangement reaction along the CI-a isomeric pathway compared to its competing channels. The gray shaded area represents the atmospherically relevant pseudo-first-order O2 addition rate range.
Moreover, the reactant alkyl radical EPO is resonance-stabilized, indicating potential reversibility of the competing O2 addition reaction, and a further enhancement of the endoperoxy-alkyl radical rearrangement channel. However, additional kinetic master equation simulations suggest that the impact of this back-reaction becomes significant only when the lifetime of the O2 addition product (denoted EPO-RO2 in Fig. 2) is unrealistically long, exceeding 105 s (Supplementary Note 5). Lowering the rearrangement reaction barrier to approximately 14 kcal/mol is necessary for the back reaction to exert influence under realistic RO2 lifetimes of 1 s to 100 s. The dependence of the endoperoxy-alkyl radical rearrangement channel on the O2 addition product lifetime provides a typical example of the competition between kinetic and thermodynamic control in O2 addition to resonance-stabilized alkyl radicals, as discussed for example by the work of Wennberg and co-authors60 in the context of isoprene oxidation. While not applicable to the α-pinene case, the O2 addition back-reaction may play a role in other atmospherically relevant systems discussed in the subsequent section, for which the endoperoxy-alkyl radical rearrangement reaction barriers are in the range of 11 kcal/mol to 14 kcal/mol.
The substantial yield observed within the plausible pseudo-first-order O2 addition rate range53,58,59 of 107 s−1 to 108 s−1 suggests that the newly identified endoperoxy-alkyl radical rearrangement mechanism could be a major channel in α-pinene ozonolysis. The existence of this rearrangement channel is supported by recent flow reactor experiments61 which show that the yield of 8-oxygen containing acyl peroxy radical is likely lower than estimated by Iyer et al.26. This indicates an alternative fate of one or more of the preceding intermediates, such as the rearrangement of the endoperoxy-alkyl radical reported here. We note that in contrast to the peroxide product from the O2 addition, the alkoxy radical product formed from the rearrangement reaction is likely to have a large number of rapid reaction channels available that could help explain the hitherto unexplained oxidation pathways suggested by the mass spectral peaks.
Potential routes to highly oxygenated organic molecules
Potential follow-up reaction channels for the endoperoxy-alkyl radical rearrangement product AOE were explored (Supplementary Note 9). While the scission reaction leading to acetone and a C7 product (AE-C7 in Fig. 4a) is the fastest (this reaction was also observed more frequently than others in the MD reaction discovery simulations), several H-shift channels are likely to have non-negligible yields. In particular, kinetic master equation simulations using the DFT barriers in Supplementary Table S4 suggest that at 300 K the 1,7 H-shift of AOE (producing AE-C10) has a rate roughly one tenth that of its scission reaction (producing AE-C7), converting to a branching ratio of about 0.1 v.s. 0.9 for the two paths. O2 addition products (RO2) following AE-C7 and AE-C10 contain multiple active sites for H-shifts (e.g., H atoms on the carbonyl carbon of the aldehyde and on the carbons of the epoxide ring) and hence have a strong potential for further autoxidation. According to the recently published structure activity relationship by Vereecken and Noziere62, H-shifts on the aldehyde exhibit rates exceeding 1 s−1. We are unaware of any specific structure activity relationship for H-shifts of RO2 containing an epoxide group. As a rough estimate, we can assume that H atoms on cyclic ether (epoxide) carbons have similar reactivity as H atoms on carbons with an acyclic ether substituent. For these, the structure activity relationship63 predicts H-shift rates up to 0.1 s−1. Our DFT calculations suggest that for AE-C7-RO2, the H-shifts from the aldehyde and epoxide carbons have barriers of around 22–25 kcal/mol, corresponding to rates exceeding 0.01 s−1 at the low barrier end. These rates align with the assumption that H-shifts from cyclic (epoxide) and acyclic ether carbons have similar rates. However, our calculations predict somewhat lower rates than the structure activity relationship62 for H-shifts from the aldehyde carbon. This is likely due to the steric hindrance caused by the epoxide ring on the carbon chain in the current case. Nevertheless, these rates suggest that H-shift reactions following AE-C7 and AE-C10 could be competitive with their bimolecular sinks. Fig. 4a shows potential routes for autoxidation leading to HOMs with up to 10 or 11 oxygen atoms. Note that these routes are only illustrative, as the order of H-shifts may vary due to their similar rates. Additionally, some of the H-shift products from the epoxide (e.g., the 1,4 H-shift of AE-C10-RO2) will likely not retain the epoxide ring intact, but instead form a carbonyl with O2 adding to the adjacent carbon. Many of these routes likely contribute to the mass spectrometry peaks for C7 and C10 HOMs observed in our flow reactor experiments (Fig. 4b, c).

a Potential autoxidation routes for AE-C7 and AE-C10, where reactions after AE-C7-RO2 and AE-C10-RO2 are based on structure activity relationship and chemical intuition. b Mass Spectrometry for C10 highly oxygenated organic molecules (HOMs) observed at the reaction time of 600 ms. c Mass Spectrometry for C7 highly oxygenated organic molecule (HOM) observed at the reaction time of 600 ms.
Endoperoxy-alkyl radical rearrangement for other atmospheric compounds
The endoperoxy-alkyl radical rearrangement mechanism discovered here can be thought of as an extension of the epoxide-forming channel investigated early by Møller et al.53. Direct experimental observation of that reaction was also achieved in the notable work of Klippenstein, Lester, and their co-workers64. In that reaction, an alkyl radical adjacent to a hydroperoxy group reacts to form a closed-shell epoxide and a OH radical. Møller et al.53 found that the thermal reaction rate is competitive with O2 addition when the alkyl radical carbon has an OH substituent, which can form a H-bond to an acceptor substituent on or adjacent to the other C atom in the nascent epoxide ring. We carried out test calculations on a variety of systems (replacing the OOH group with a cyclic peroxide) and found that the presence of the OH substituent on the alkyl radical carbon can indeed significantly reduce the barrier of the newly discovered endoperoxy-alkyl radical rearrangement channel. The corresponding reaction barriers for most of these tailored molecules are approximately 11 kcal/mol to 14 kcal/mol at the CCSD(T) level (Supplementary Note 7), in contrast to the 17.9 kcal/mol at the CCSD(T) level observed in the case of α-pinene. While even these reduced barrier heights still do not correspond to competitive reaction rates against O2 addition under thermalized conditions, simulations on model systems (Supplementary Note 6) indicate that the reactions could have non-negligible yields when considering a modest and plausible amount of excess energy (e.g., 20 kcal/mol) from preceding steps. Despite not identifying such an OH substituent on the alkyl radical carbon in the known or sampled α-pinene ozonolysis system, this structural motif is likely present in the atmospheric oxidation of a diverse array of anthropogenic and biogenic compounds.
We identified three representative examples with endoperoxy-alkyl radical rearrangement reaction rates potentially competitive against O2 addition: the naturally emitted monoterpene α-ocimene (Fig. 5), the industrial chemical 1,4 hexadiene (Supplementary Fig. 11a), and the terpenoid alcohol geraniol, which is both naturally emitted and used in fragrances (Supplementary Fig. 11b). For the 1,4 hexadiene + OH and geraniol + OH systems, the corresponding endoperoxy-alkyl radical rearrangement reaction barriers are 13.4 kcal/mol and 11 kcal/mol at the CCSD(T) level, respectively. Simulations on model systems suggest that such barrier heights could result in about 10% and 40% yields, respectively, when factoring in 20 kcal/mol of excess energy from preceding reaction steps. Considering the potential reversibility of O2 addition, this yield could likely be increased further. For the α-ocimene + OH system, the endoperoxy-alkyl radical rearrangement reaction barrier is only 3.1 kcal/mol at the CCSD(T) level, indicating a 100% yield even under thermalized conditions without any excess energy. This typical example in the α-ocimene + OH system highlights the importance of the new mechanism. Conventionally, hydroxyalkyl radicals are expected to terminate the oxidation propagation by releasing the H atom to an O2, resulting in the formation of closed-shell carbonyl co-products. However, the new mechanism suggests that these intermediates do not necessarily conclude the oxidation propagation or the formation of HOMs.

The barrier for this endoperoxy-alkyl radical rearrangement reaction is 3.1 kcal/mol at the CCSD(T) level, indicating a much faster reaction rate and 100% yield against its competing O2 addition reaction.
Discussion
These selected examples demonstrate that the endoperoxy-alkyl radical rearrangement mechanism presented here is likely to be relatively common in atmospheric autoxidation processes. OH groups, alkyl radical sites, endoperoxides and H-bond acceptor groups are ubiquitous functionalities – the specific structural combination required to make the proposed reaction pathway competitive is not implausibly restrictive. The main atmospheric implication of the reaction is that it opens new and unexpected branching points in oxidation mechanisms: while alkyl radicals, with a few exceptions, have traditionally been assumed to exclusively react with O2, alkoxy radicals are arguably the most versatile species in atmospheric chemistry, and often have multiple competing reaction channels available. A recurring theme of both smog chamber and field studies of atmospheric oxidation has been the surprisingly large numbers of different progressively oxygenated product compounds (e.g., in terms of elemental compositions in mass spectra). The unexpected branching points revealed by our new mechanism helps explain this diversity.
Methods
Enhanced sampling aided reaction discovery
Our approach for conducting reaction discovery simulations combines a collective variable (CV) derived from spectral graph theory42 and the explore variant of the On-the-fly Probability Enhanced Sampling (OPESE) method48. This workflow42 is briefly resumed here. The molecule is represented as a graph where vertices and edges represent atoms and chemical bonds, respectively. The adjacency matrix A is associated to the graph, and its elements aij indicates whether atoms i and j are connected by a chemical bond:
where σij represent the typical bond lengths between atoms of types i and j. We use the largest eigenvalue of the adjacency matrix A (denoted as λmax) as collective variable in the enhanced sampling simulations. λmax is real, positive, and non-degenerate, and its choice as a CV guarantees translational, rotational and permutational invariance while preserving the chemical information encoded in the molecular graph (λmax grows with the number of bonds and its value lies between the average and maximum coordination number47).
The fluctuations of λmax are enhanced using the OPESE method, which builds the bias by estimating the probability distribution of the CV on-the-fly. In particular, the bias is built as48:
where Zn is a normalization factor, γ > 1 is the bias factor, and \({p}^{{WT}}\left(s\right)\) represents the well-tempered distribution. An important feature of OPESE is the presence of a regularization term ε (\(\varepsilon=\,{e}^{-\beta \Delta E/(1-1/\gamma )}\)) which is related to the maximum value of the bias that can be deposited. This allows limiting the extent of exploration by preventing high energy transition states from being visited, and thus, represents a key parameter to control the discovery phase.
The discovery engine is complemented by a reaction analysis module that enables detection of new chemical species. We use the Open Babel65 program to identify chemical species in every trajectory snapshot, converting the Cartesian coordinates of each frame into their corresponding SMILES representations. The adjacency matrix A includes all the atoms present in the system. Its elements (aij) are computed using PLUMED66, with the σij parameters of Eq. (1) being equal to 1.6 Å for O-H, 1.9 Å for O-O, C-C, and C-O, and 1.2 Å for C-H and H-H. The exponents of the switching functions are chosen as n = 6 and m = 10 to enforce a smooth behavior over a wide range of distances. In the case of the VHP species, defining the graph with all the atoms in the system predominantly resulted in sampling various conformers. Therefore, we conducted simulations using a CV more tailored on a specific functional group that undergoes the OH decomposition (i.e., the -COOH group in the VHP), observing several reactive pathways involving the formation of OH radicals. Notably, even with this focused CV, we still observed unexpected products, such as those from the roaming OH radical pathway, demonstrating the tool’s capability to discover reaction pathways beyond those predicted by conventional chemical intuition. To explore these channels, we treated the system as an unrestricted singlet state in the dynamics. Additionally, we have performed MD discovery simulations at the unrestricted wB97xd/6-31 G level, starting from VHP-a in Fig. 2. Using the same CV (including the COOH group in the graph), we have identified the same products as those obtained with the PM6 level of theory.
We started the discovery phase from α-pinene and ozone. We performed multiple molecular dynamics (0.5 ns each) setting the barrier value in OPES explore at 100, 200, 300, 400, and 500 kJ/mol. Whenever a new species was detected, we initiated new simulations from that structure, again with multiple values of the ε parameter in OPESE. O2 was not included in the simulation until the formation of vinoxy. Starting from vinoxy, each new simulation was carried out both in the presence and absence of O2. Each discovered species was first optimized at DFT level (B3LYP/6-31 + G(d)) to assess its stability. Then, the most relevant pathways were characterized by computing transition states (TS) energies at a high level of theory (CCSD(T)), as explained in the following section. Molecular dynamics simulations were performed using the CP2K 8.1167 software, combined with a development version of PLUMED 2.8.266. All simulations of the discovery phase were conducted at the PM668 level of theory, with an integration time step of 0.5 fs. We sampled the NVT ensemble using the velocity rescaling thermostat69, maintaining a temperature of 300 K and a time constant of 100 fs. The computational cost of these MD-based discovery simulations is comparable to that of regular MD simulations, and it is primarily determined by the electronic level of theory. From this perspective, this methodology is highly generalizable and can be implemented using both GPU and CPU codes for running molecular dynamics. In our particular case, we used the CPU version of CP2K, and with 4 cores on a desktop workstation, we were able to perform each step in an average of 0.02 s. As a result, we were able to complete 500 ps of simulation in approximately 6 h.
Electronic structure calculations
A thorough exploration of the reactants, intermediates, transition states, and products on the discovered reaction pathways was conducted by sampling their conformers using the Spartan’18 (Wavefunction, Inc) program. The conformer sampling procedure was carried out using the MMFF method70. All conformers were first optimized at the B3LYP/6-31 + G(d) level of theory71,72,73,74, and those within 2 kcal/mol in relative electronic energies of the lowest energy conformer were chosen to be optimized at the ωB97X-D/aug-cc-pVTZ level of theory75,76,77. Finally, only the lowest energy conformer was chosen for the final single-point electronic energy calculation at the ROHF-ROCCSD(T)-F12a/VDZ-F12 level78,79,80,81,82 to correct the final energies. These calculations were performed using the Molpro 2022.2.2 program83. To obtain transition state (TS) structures, initial guesses were generated in Spartan'18 and subsequently optimized at the B3LYP/6-31 + G(d) level of theory using Gaussian 09, with relevant TS bond distances constrained. The selection of the B3LYP functional for the initial TS calculations was based on its established capability to determine TS structures. Unconstrained TS optimization at the same level of theory was then performed immediately after the constrained optimization of the initial guess TS structures to precisely locate the TS structures. Once the TS structures were identified, they underwent conformer sampling in Spartan with relevant bonds being constrained. The resulting TS conformers were optimized once again under constraint and subsequently subjected to unconstrained TS optimization at the B3LYP/6-31 + G(d) level in Gaussian 16. Finally, conformers with relative electronic energies within 2 kcal/mol were optimized at the higher ωB97X-D/aug-cc-pVTZ level of theory. The lowest energy conformer was selected for the final single-point electronic energy calculation at the ROHF-ROCCSD(T)-F12a/VDZ-F12 level using the Molpro program83.
Kinetic master equation calculations
The RRKM simulations were carried out using the Master equation solver for multi-energy well reactions (MESMER) program84. The potential energy surface and the methods used until the formation of the ring broken RO2 are identical to those used in our previous work26. Briefly, the SimpleRRKM method in MESMER with Eckart tunneling was used to treat the initial association reaction. Similar treatment was applied to the other intermediate complexes separated by transition states. Approximations of the nascent energy allocation to both vinoxy and OH products resulting from VHP decomposition were tackled employing the pseudo-isomerization methodology85. The parameter m value, which controls the fraction of the density of states of the vinoxy radical to spread the excess energy over, was set to 0.5. The O2 addition step of the ring-broken product RB was treated using the SimpleRRKM method, employing a model transition state with variable energies (while O2 concentration was fixed at 5 × 1018 cm⁻³) to modulate the effective pseudo-first-order addition rate within the range of about 107 s−1 to 108 s−1, a plausible rate range for O2 addition to vinoxy/alkyl radicals under atmospheric conditions53,58,59 (T = 298.15 K and P = 1 atm). The O2 addition step for all other vinoxy/alkyl radicals was managed using the “Simple Bimolecular Sink” method, wherein the rate is governed by an adjustable bimolecular loss rate coefficient and an O2 “excess reactant” concentration of 5 × 1018 cm⁻³. The dissipation of the excess energy through interaction with bath gases was regulated by the ΔEdown parameter in MESMER and was set to be 175 cm−1. This selection was guided by a molecular heat dissipation model discussed in the Supplementary Note 2. All intermediates were designated as “modeled” throughout the simulations, with corresponding Lennard–Jones potentials assigned as sigma = 6.5 Å and epsilon = 600 K. The simulation employed a grain size of 120 cm−1, with the energy spanned by the grains of 60 kBT. The initial ozone concentration was set identical to our prior work, at 1018 molecules cm⁻³, to minimize the overall simulation time. Previous test calculations26 run with the more ambiently relevant ozone concentration (1012 molecules cm⁻³) have shown that the initial ozone concentration did not influence results. This rationale aligns with the observation that, beyond the initial association reaction, subsequent ozonolysis steps exhibit independence from the initial ozone concentration. Although our MESMER simulations did not account for multi-conformer effects, we have included a discussion of these effects in the Supplementary Note 13.
Mass spectrometry experiments
A time-of-flight chemical ionization mass spectrometer with nitrate charger ions (NO3-CIMS) was used to detect the oxidation products of α-pinene ozonolysis at 600 ms reaction time. The experiments were carried out in a quartz tube flow reactor. Liquid α-pinene (Sigma-Aldrich, purity 91%) was bubbled from a reservoir using a pure N2 flow. This was allowed to interact with ozone produced by flowing synthetic air through an ozone generator equipped with a 184.9 nm (Hg PenRay) lamp. The reaction time between α-pinene and ozone was controlled in two ways, 1) by introducing the ozone into the flow reactor through an injector tube and consequently limiting the α-pinene and ozone interaction time, and 2) by increasing the inlet flow through the reactor to the mass spectrometer. High inlet flow rates of 20–30 liters per minute to achieve sub-second reaction times were made possible by the use of the Multi-scheme chemical ionization inlet86 (MION). The experiments were conducted under atmospheric conditions.
Data availability
The authors declare that the primary data supporting the findings of this study are available within the paper and supplementary information, as well as in the Zenodo repository [https://doi.org/10.5281/zenodo.14006848]. Additional data can be obtained from the corresponding author upon request. Source data are provided with this paper.
Code availability
The input scripts developed in this study for molecular dynamics reaction discovery, electronic structure calculations, and kinetic master equation simulations are available in the Zenodo repository [https://doi.org/10.5281/zenodo.14006848].
References
Jimenez, J. L. et al. Evolution of organic aerosols in the atmosphere. Science 326, 1525–1529 (2009).
Pöschl, U. Atmospheric aerosols: composition, transformation, climate and health effects. Angew. Chem. Int. Ed. 44, 7520–7540 (2005).
Kanakidou, M. et al. Organic aerosol and global climate modelling: a review. Atmos. Chem. Phys. 5, 1053–1123 (2005).
Guo, S. et al. Elucidating severe urban haze formation in China. Proc. Natl Acad. Sci. 111, 17373–17378 (2014).
An, Z. et al. Severe haze in northern China: A synergy of anthropogenic emissions and atmospheric processes. Proc. Natl Acad. Sci. 116, 8657–8666 (2019).
Al-Kindi, S. G., Brook, R. D., Biswal, S. & Rajagopalan, S. Environmental determinants of cardiovascular disease: lessons learned from air pollution. Nat. Rev. Cardiol. 17, 656–672 (2020).
Pope, C. A. III & Dockery, D. W. Health effects of fine particulate air pollution: lines that connect. J. Air Waste Manag. Assoc. 56, 709–742 (2006).
Kiehl, J. Aerosols, climate, and the hydrological cycle. Science 294, 2119–2124 (2001).
Hallquist, M. et al. The formation, properties and impact of secondary organic aerosol: current and emerging issues. Atmos. Chem. Phys. 9, 5155–5236 (2009).
Shrivastava, M. et al. Recent advances in understanding secondary organic aerosol: Implications for global climate forcing. Rev. Geophys. 55, 509–559 (2017).
Ziemann, P. J. & Atkinson, R. Kinetics, products, and mechanisms of secondary organic aerosol formation. Chem. Soc. Rev. 41, 6582–6605 (2012).
Guenther, A. et al. A global model of natural volatile organic compound emissions. J. Geophys. Res. Atmos. 100, 8873–8892 (1995).
Ehn, M. et al. A large source of low-volatility secondary organic aerosol. Nature 506, 476–479 (2014).
Bianchi, F. et al. New particle formation in the free troposphere: A question of chemistry and timing. Science 352, 1109–1112 (2016).
Bianchi, F. et al. Highly oxygenated organic molecules (HOM) from gas-phase autoxidation involving peroxy radicals: A key contributor to atmospheric aerosol. Chem. Rev. 119, 3472–3509 (2019).
Tröstl, J. et al. The role of low-volatility organic compounds in initial particle growth in the atmosphere. Nature 533, 527–531 (2016).
Vereecken, L., Müller, J. F. & Peeters, J. Low-volatility poly-oxygenates in the OH-initiated atmospheric oxidation of α-pinene: impact of non-traditional peroxyl radical chemistry. Phys. Chem. Chem. Phys. 9, 5241–5248 (2007).
Vereecken, L. & Peeters, J. Nontraditional (per) oxy ring-closure paths in the atmospheric oxidation of isoprene and monoterpenes. J. Phys. Chem. A 108, 5197–5204 (2004).
Yao, L. et al. Atmospheric new particle formation from sulfuric acid and amines in a Chinese megacity. Science 361, 278–281 (2018).
Nie, W. et al. Secondary organic aerosol formed by condensing anthropogenic vapours over China’s megacities. Nat. Geosci. 15, 255–261 (2022).
Guo, S. et al. Remarkable nucleation and growth of ultrafine particles from vehicular exhaust. Proc. Natl Acad. Sci. 117, 3427–3432 (2020).
Hoffmann, T. et al. Formation of organic aerosols from the oxidation of biogenic hydrocarbons. J. Atmos. Chem. 26, 189–222 (1997).
Atkinson, R. & Arey, J. Gas-phase tropospheric chemistry of biogenic volatile organic compounds: a review. Atmos. Environ. 37, 197–219 (2003).
Sindelarova, K. et al. Global data set of biogenic VOC emissions calculated by the MEGAN model over the last 30 years. Atmos. Chem. Phys. 14, 9317–9341 (2014).
Lee, A. et al. Gas‐phase products and secondary aerosol yields from the ozonolysis of ten different terpenes. J. Geophys. Res. Atmos. 111, D07302–D07319 (2006).
Iyer, S. et al. Molecular mechanism for rapid autoxidation in α-pinene ozonolysis. Nat. Commun. 12, 878 (2021).
Zhang, X. et al. Formation and evolution of molecular products in α-pinene secondary organic aerosol. Proc. Natl Acad. Sci. 112, 14168–14173 (2015).
Quéléver, L. L. et al. Effect of temperature on the formation of highly oxygenated organic molecules (HOMs) from alpha-pinene ozonolysis. Atmos. Chem. Phys. 19, 7609–7625 (2019).
Rissanen, M. P. et al. Effects of chemical complexity on the autoxidation mechanisms of endocyclic alkene ozonolysis products: From methylcyclohexenes toward understanding α-pinene. J. Phys. Chem. A 119, 4633–4650 (2015).
Kurten, T. et al. Computational study of hydrogen shifts and ring-opening mechanisms in α-pinene ozonolysis products. J. Phys. Chem. A 119, 11366–11375 (2015).
Kristensen, K. et al. High-molecular weight dimer esters are major products in aerosols from α-pinene ozonolysis and the boreal forest. Environ. Sci. Technol. Lett. 3, 280–285 (2016).
Meder, M. et al. Selective deuteration as a tool for resolving autoxidation mechanisms in α-pinene ozonolysis. Atmos. Chem. Phys. 23, 4373–4390 (2023).
Saunders, S. M., Jenkin, M. E., Derwent, R. & Pilling, M. Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part A): tropospheric degradation of non-aromatic volatile organic compounds. Atmos. Chem. Phys. 3, 161–180 (2003).
Aumont, B., Szopa, S. & Madronich, S. Modelling the evolution of organic carbon during its gas-phase tropospheric oxidation: development of an explicit model based on a self generating approach. Atmos. Chem. Phys. 5, 2497–2517 (2005).
Camredon, M., Aumont, B., Lee-Taylor, J. & Madronich, S. The SOA/VOC/NO x system: an explicit model of secondary organic aerosol formation. Atmos. Chem. Phys. 7, 5599–5610 (2007).
Rissanen, M. P. et al. The formation of highly oxidized multifunctional products in the ozonolysis of cyclohexene. J. Am. Chem. Soc. 136, 15596–15606 (2014).
Sandström, H., Rissanen, M., Rousu, J. & Rinke, P. Data‐Driven Compound Identification in Atmospheric Mass Spectrometry. Adv. Sci. 11, 2306235 (2024).
Zádor, J. et al. Automated reaction kinetics of gas-phase organic species over multiwell potential energy surfaces. J. Phys. Chem. A 127, 565–588 (2023).
Barber, V. P., Green, W. H. & Kroll, J. H. Screening for new pathways in atmospheric oxidation chemistry with automated mechanism generation. J. Phys. Chem. A 125, 6772–6788 (2021).
Wang, L. et al. Discovering chemistry with an ab initio nanoreactor. Nat. Chem. 6, 1044–1048 (2014).
Grimme, S. Exploration of chemical compound, conformer, and reaction space with meta-dynamics simulations based on tight-binding quantum chemical calculations. J. Chem. Theory Comput. 15, 2847–2862 (2019).
Raucci, U., Rizzi, V. & Parrinello, M. Discover, sample, and refine: Exploring chemistry with enhanced sampling techniques. J. Phys. Chem. Lett. 13, 1424–1430 (2022).
Yang, M. et al. Combined molecular dynamics and coordinate driving method for automatic reaction pathway search of reactions in solution. J. Chem. Theory Comput. 14, 5787–5796 (2018).
Unsleber, J. P. & Reiher, M. The exploration of chemical reaction networks. Annu. Rev. Phys. Chem. 71, 121–142 (2020).
Xu, R., Meisner, J., Chang, A. M., Thompson, K. C. & Martínez, T. J. First principles reaction discovery: from the Schrodinger equation to experimental prediction for methane pyrolysis. Chem. Sci. 14, 7447–7464 (2023).
Chang, A. M., Meisner, J., Xu, R. & Martínez, T. J. Efficient Acceleration of Reaction Discovery in the Ab Initio Nanoreactor: Phenyl Radical Oxidation Chemistry. J. Phys. Chem. A 127, 9580–9589 (2023).
Pietrucci, F. & Andreoni, W. Graph theory meets ab initio molecular dynamics: atomic structures and transformations at the nanoscale. Phys. Rev. Lett. 107, 085504 (2011).
Invernizzi, M. & Parrinello, M. Exploration vs convergence speed in adaptive-bias enhanced sampling. J. Chem. Theory Comput. 18, 3988–3996 (2022).
Das, S., Raucci, U., Neves, R. P., Ramos, M. J. & Parrinello, M. How and when does an enzyme react? Unraveling α-Amylase catalytic activity with enhanced sampling techniques. ACS Catal. 13, 8092–8098 (2023).
Raucci, U., Sanchez, D. M., Martínez, T. J. & Parrinello, M. Enhanced sampling aided design of molecular photoswitches. J. Am. Chem. Soc. 144, 19265–19271 (2022).
Klippenstein, S. J. & Elliott, S. N. OH Roaming during the Ozonolysis of α-Pinene: A New Route to Highly Oxygenated Molecules? J. Phys. Chem. A 127, 10647–10662 (2023).
Xu, L., Møller, K. H., Crounse, J. D., Kjaergaard, H. G. & Wennberg, P. O. New insights into the radical chemistry and product distribution in the OH-initiated oxidation of benzene. Environ. Sci. Technol. 54, 13467–13477 (2020).
Møller, K. H., Kurten, T., Bates, K. H., Thornton, J. A. & Kjaergaard, H. G. Thermalized epoxide formation in the atmosphere. J. Phys. Chem. A 123, 10620–10630 (2019).
Iyer, S., Rissanen, M. P. & Kurtén, T. Reaction between peroxy and alkoxy radicals can form stable adducts. J. Phys. Chem. Lett. 10, 2051–2057 (2019).
Taatjes, C. A. et al. Hydroxyacetone production from C3 Criegee intermediates. J. Phys. Chem. A 121, 16–23 (2017).
Liu, T. et al. OH roaming and beyond in the unimolecular decay of the methyl-ethyl-substituted Criegee intermediate: observations and predictions. J. Am. Chem. Soc. 145, 19405–19420 (2023).
Liu, T. & Lester, M. I. Roaming in the Unimolecular Decay of syn-Methyl-Substituted Criegee Intermediates. J. Phys. Chem. A 127, 10817–10827 (2023).
Wu, D. & Bayes, K. D. Rate constants for the reactions of isobutyl, neopentyl, cyclopentyl, and cyclohexyl radicals with molecular oxygen. Int. J. Chem. Kinet. 18, 547–554 (1986).
Atkinson, R. Gas-phase tropospheric chemistry of volatile organic compounds: 1. Alkanes and alkenes. J. Phys. Chem. Ref. Data 26, 215–290 (1997).
Teng, A. P., Crounse, J. D. & Wennberg, P. O. Isoprene peroxy radical dynamics. J. Am. Chem. Soc. 139, 5367–5377 (2017).
Zang, H. et al. Direct probing of acylperoxy radicals during ozonolysis of α-pinene: constraints on radical chemistry and production of highly oxygenated organic molecules. Atmos. Chem. Phys. 23, 12691–12705 (2023).
Vereecken, L. & Nozière, B. H migration in peroxy radicals under atmospheric conditions. Atmos. Chem. Phys. 20, 7429–7458 (2020).
Otkjær, R. V., Jakobsen, H. H., Tram, C. M. & Kjaergaard, H. G. Calculated hydrogen shift rate constants in substituted alkyl peroxy radicals. J. Phys. Chem. A 122, 8665–8673 (2018).
Hansen, A. S. et al. Watching a hydroperoxyalkyl radical (• QOOH) dissociate. Science 373, 679–682 (2021).
O’Boyle, N. M. et al. Open Babel: An open chemical toolbox. J. Cheminformatics 3, 1–14 (2011).
Bonomi, M. et al. Promoting transparency and reproducibility in enhanced molecular simulations. Nat. Methods 16, 670–673 (2019).
Kühne, T. D. et al. CP2K: An electronic structure and molecular dynamics software package-Quickstep: Efficient and accurate electronic structure calculations. J. Chem. Phys. 152, 194103 (2020).
Stewart, J. J. Optimization of parameters for semiempirical methods V: Modification of NDDO approximations and application to 70 elements. J. Mol. Model. 13, 1173–1213 (2007).
Bussi, G., Donadio, D. & Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 014101 (2007).
Halgren, T. A. MMFF VII. Characterization of MMFF94, MMFF94s, and other widely available force fields for conformational energies and for intermolecular‐interaction energies and geometries. J. Computational Chem. 20, 730–748 (1999).
Beck, A. D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 98, 5648–5646 (1993).
Lee, C., Yang, W. & Parr, R. G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 37, 785 (1988).
Hehre, W. J., Ditchfield, R. & Pople, J. A. Self-consistent molecular orbital methods. XII. Further extensions of Gaussian-type basis sets for use in molecular orbital studies of organic molecules. J. Chem. Phys. 56, 2257–2261 (1972).
Frisch, M. J., Pople, J. A. & Binkley, J. S. Self-consistent molecular orbital methods 25. Supplementary functions for Gaussian basis sets. J. Chem. Phys. 80, 3265–3269 (1984).
Chai, J. D. & Head Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 10, 6615–6620 (2008).
Dunning, T. H. Jr Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 90, 1007–1023 (1989).
Kendall, R. A., Dunning, T. H. & Harrison, R. J. Electron affinities of the first‐row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 96, 6796–6806 (1992).
Watts, J. D., Gauss, J. & Bartlett, R. J. Coupled-cluster methods with noniterative triple excitations for restricted open-shell Hartree-Fock and other general single determinant reference functions. Energies and analytical gradients. J. Chem. Phys. 98, 8718–8733 (1993).
Adler, T. B., Knizia, G. & Werner, H. J. A simple and efficient CCSD (T)-F12 approximation. J. Chem. Phys. 127, 221106 (2007).
Knizia, G., Adler, T. B. & Werner, H.-J. Simplified CCSD (T)-F12 methods: Theory and benchmarks. J. Chem. Phys. 130, 054104 (2009).
Werner, H. J., Knizia, G. & Manby, F. R. Explicitly correlated coupled cluster methods with pair-specific geminals. Mol. Phys. 109, 407–417 (2011).
Peterson, K. A., Adler, T. B. & Werner, H. J. Systematically convergent basis sets for explicitly correlated wavefunctions: The atoms H, He, B–Ne, and Al–Ar. J. Chem. Phys. 128, 084102 (2008).
Werner, H. J. et al. The Molpro quantum chemistry package. J. Chem. Phys. 152, 144107 (2020).
Glowacki, D. R., Liang, C. H., Morley, C., Pilling, M. J. & Robertson, S. H. MESMER: an open-source master equation solver for multi-energy well reactions. J. Phys. Chem. A 116, 9545–9560 (2012).
Shannon, R. J., Robertson, S. H., Blitz, M. A. & Seakins, P. W. Bimolecular reactions of activated species: An analysis of problematic HC (O) C (O) chemistry. Chem. Phys. Lett. 661, 58–64 (2016).
Rissanen, M. P., Mikkilä, J., Iyer, S. & Hakala, J. Multi-scheme chemical ionization inlet (MION) for fast switching of reagent ion chemistry in atmospheric pressure chemical ionization mass spectrometry (CIMS) applications. Atmos. Meas. Tech. 12, 6635–6646 (2019).
Acknowledgements
We thank Prof. Michele Parrinello for reading a preliminary version of the paper and for his encouraging remarks and support. We also thank Valerio Rizzi, Axel Tosello Gardini, and Enrico Trizio for helpful discussions. T.K. acknowledges the support from The Jane and Aatos Erkko Foundation (JAES) and the Research Council of Finland Center of Excellence VILMA under Grant No. 346369. H.V. acknowledges the support from Research Council of Finland Center of Excellence VILMA under Grant No. 346368. S.I. acknowledges the funding support from the Research Council of Finland funding number 355966. M.R. acknowledges the funding support from the European Research Council under the European Union’s Horizon 2020 research and innovation program under Grant No. 101002728. We thank the CSC IT center for science for computing resources.
Author information
Authors and Affiliations
Contributions
H.Y., U.R., S.I., and T.K. conceived the study and designed the research; H.Y., U.R., S.I., and T.K. performed the research with input from G.H., T.G.A., S.B., and A.S.; M.R., J.K., and H.V. provided scientific input; H.Y., U.R., S.I., and T.K. analyzed the data and wrote the manuscript; All authors reviewed and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yang, H., Raucci, U., Iyer, S. et al. Molecular dynamics-guided reaction discovery reveals endoperoxide-to-alkoxy radical isomerization as key branching point in α-pinene ozonolysis. Nat Commun 16, 661 (2025). https://doi.org/10.1038/s41467-025-55985-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-55985-w
