
Abstract
Zinc (Zn)-based batteries have been persistently challenged by the critical issue of inhomogeneous zinc deposition/stripping process on substrate surface. Herein, we reveal that zinc electrodeposition behaviors dramatically improved through the introduction of highly zincophilic copper oxide nanoparticles (CuO NPs). Strong electronic redistribution between Zn and CuO explains the high Zn affinity on CuO, with negligible nucleation overpotential. Additionally, CuO exhibits remarkable electron-accepting and -donating capabilities in electron-rich and electron-deficient environments, resembling a sponge. This ‘Electron Sponge’ effect emerges from stable Zn-O bonding in CuO, enhancing electron duality in the Zn-O bond region. This unique strategy is pivotal in mitigating dendritic growth, fostering dendrite-free zinc-based flow batteries with enhanced rate performance and cyclability. It presents significant performance with not only high energy density (180 Wh L−1) but also the long cycle stability (> 2500 cycles) at high current density (140 mA cm−2).
Similar content being viewed by others

Introduction
Over the past decades, owing to high energy density and reliable rechargeability, Li-ion batteries (LIBs) have been attracting attention as preferred candidates for the electrical energy storage system (ESS) market1. However, their practical application to grid-scale energy storage is seriously hindered by explosion risk of flammable organic electrolytes and the high cost of limited lithium resources2,3. Alternatively, Zn-based batteries have been evaluated for use as next generation safe and inexpensive ESS devices. Zn-based batteries can provide the intrinsic safety of aqueous electrolyte, material/fabrication cost benefits, and competitive volumetric capacity (5854 mAh cm−3)4,5,6. To date, there has been extensive research related to aqueous Zn-based batteries such as Zn-metal (Fe and Mn)7,8, Zn-halide (Cl, Br, and I)9,10, and Zn-organic (TEMPO and quinone)11 redox flow batteries (RFBs), as well as static Zn-ion batteries. Among the various Zn-based batteries, Zn-polyiodide flow batteries (ZIFBs) are promising candidates owing to their attractive features of high reversibility, energy density, and cost-effectiveness10,12,13,14,15,16,17,18,19,20,21,22. They employ redox couples of I−/I3− and Zn0/Zn2+ in the cathode and anode, respectively, based on the following reactions:
In the ZIFB system, an iodide cathode with a high standard rate constant of k = 1.3 × 10−5 m s−1 (versus bromide, k = ~ 4 × 10−7 m s−1) can improve the current and power densities in comparison with those of commercial Zn-bromine RFBs23,24. Also, iodine is much less toxic and corrosive than bromine. Moreover, ZIFBs exhibit an attractive theoretical energy density of 322 Wh L−1 at the solubility limit of ZnI2 electrolyte in water (7 mol L−1)10. Their electrolyte cost ($41 KWh−1) is much lower than that of commercial all-vanadium RFBs (VRFB, $87 KWh−1) and recently progressive all-organic RFBs (Viologen-TEMPO, $92 KWh−1)25.
However, as with other conventional batteries adopting Zn anodes, ZIFBs have faced critical issues with the inhomogeneous Zn deposition/stripping process on the substrate surface20,21. This inhomogeneous deposition can easily evolve and accelerate via trivial changes in the microenvironment, such as electrode roughness, Zn2+ ion distribution, and electric field around the electrode. It leads to undesirable Zn dendrite formation in a direction perpendicular to the electrode. A small dendrite can rapidly develop into a large one, pierce the separator, and ultimately cause a short circuit18,24. The formation of Zn dendrites worsens under experimental conditions of high areal capacity and high current density. Thus, to guarantee cyclic stability, ZIFBs are typically operated at limited levels of areal capacity and current density.
To inhibit dendrite formation, much research has been devoted to nucleation adjustment, 2D diffusion suppression, and polarization mitigation. First, the energy barrier for Zn nucleation has been strategically controlled to strengthen interaction between zinc nuclei and electrode substrate, because initial homogeneous Zn nucleation plays a significant role in suppressing dendrite formation. By pre-positioning nucleation seed materials with strong zincophilic characteristics onto the electrode, uniform electrodeposition can be induced even at low nucleation overpotential (NOP)26. Recent studies have attempted to lower NOP by decorating electrode surface with a series of metal-based seeds (Au, Ag, Cu, and Sn) that have good affinity for Zn and can act as nucleation sites27,28,29,30. In addition to metal species, carbon structures with various defects and impurities have been widely employed to enhance Zn affinity and suppress dendrite formation31. Non-conductive coating layers such as oxides (ZnO, ZrO2, Al2O3, and TiO2) and sulfides (ZnS) have also been applied to inhibit dendrite formation by utilizing the strong Zn affinity of oxygen- or sulfur-containing groups32,33,34,35,36. Second, adsorbed Zn2+ ions diffuse on the surface to locate the most energetically favorable sites for charge transfer; this process is defined as 2D diffusion. Facile 2D diffusion of Zn2+ ions on the electrode surface can facilitate a biased electrodeposition around a specific electrode area. Furthermore, they aggregate and grow into Zn dendrites, which stabilize the surface energy of the exposed active areas. Recently, various approaches have been reported to control 2D diffusion and fasten Zn ions around the nucleation sites using several organic (polyamide and polyethylene glycol) or inorganic species (CaCO3 and Kailon) with high affinity for Zn2+ ions37,38,39,40. Also, an electrostatic shield effect using co-cations (Li+, Na+, Mn2+, and Co2+) can effectively suppress 2D diffusion41,42,43,44. Third, the reduction of polarization, including concentration polarization and electrochemical polarization, is one of the main factors in preventing dendrite formation; this reduction can be achieved using a 3D porous substrate with high specific surface area45.
Herein, we report CuO NPs as unique nucleation seeds, demonstrating simultaneous nucleation adjustment, 2D diffusion suppression, control of Zn crystallinity, and efficient stripping with minimal residual Zn (Fig. 1). The electrodeposition behavior of atomic zinc on a substrate with CuO NPs is thoroughly investigated by a comprehensive analysis of the series of surface reactions including the nucleation and growth steps. CuO NPs have a strong zincophilic property that facilitates homogenous Zn nuclei formation around uniformly pre-distributed CuO sites. NOP and 2D-surface diffusion of initial nuclei were investigated by combining electrochemical analysis and computational approaches. The strong binding energy between CuO and Zn nuclei plays an important role in determining the growth direction of the subsequent accumulation of Zn electrocrystal by epitaxial mechanism. As the areal capacity increased during the charging process, variation of the Zn crystallinity was monitored by surface atomic structure characterization, backed up by density functional theory (DFT) calculations. The densely electrodeposited Zn layer with (002)-preferred surface plane provided remarkably high areal capacity of 463 mAh cm−2, the highest value among all reported Zn-based batteries, including ZIFBs. Furthermore, CuO NPs contribute to effective stripping of Zn metal during the discharge process. A series of positive electrochemical effects of CuO NPs on the nucleation, growth, and dissolution of Zn can be explained by the concept of the electron sponge. Consequently, ZIFB with CuO NPs electrodes exhibited an extended lifespan exceeding 2500 cycles, even at a high current density of 140 mA cm−2.

CuO NPs act as unique nucleation seeds, adjusting nucleation, suppressing 2D diffusion, controlling Zn crystallinity, and enabling efficient stripping with minimal Zn residue.
Results
CuO NPs as efficient Zn nucleation seed
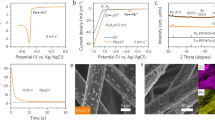
Nucleation seeds can help Zn nuclei to anchor homogeneously on the electrode surface, can stabilize them at the seed site and inhibit their 2D-diffusion, and furthermore can induce crystal growth parallel to the electrode surface. The NOP is a critical parameter to evaluate the ability of the nucleation seed to attract the Zn species to the electrode surface and stabilize Zn nuclei; NOP is defined as the difference between initial voltage dip and later voltage plateau during galvanostatic Zn deposition26,28. To allow a comparison of Zn NOP values, Fig. 2a and Supplementary Fig. 1 indicate the voltage profiles of galvanostatic Zn deposition on several metal or metal oxide substrates. As expected, because Zn metal does not itself require a nucleation step, Zn metal substrate did not show any initial overpotential stage and immediately exhibited a constant steady-state voltage. On the other hand, many other metal substrates including Sn, Fe, and Ni were characterized and found to have large NOPs, which may be ascribed to a mismatch of lattice structures between metal substrates and Zn. For example, Sn had an NOP of 95.8 mV, the minimum level required to overcome the heterogeneous nucleation barrier formed by the large thermodynamic mismatch between Zn and Sn. Metal oxides such as ZnO and SnO displayed similarly large NOPs of 67.63 and 135.36 mV, respectively. Carbon is the most commonly adopted electrode material in zinc-based flow batteries12,13,14,15,16,17; it exhibited a much larger NOP of 155.2 mV. In contrast, the electrodeposition of Zn on Cu or CuO substrates led to very different nucleation behavior, with negative NOP, indicating the strong affinity of Zn species for those substrates and their potential use as nucleation seeds. It is notable Zn started to be electrodeposited on CuO substrate at a more anodic potential by 195.5 mV than the standard reduction potential of Zn/Zn2+ reaction.

a Voltage profiles of galvanostatic Zn deposition on various substrates at a current density of 20 mA cm−2. b Adsorption energy of single Zn atom on various substrates. The inset image is a charge density difference map comparing CuO (001) and adsorbed Zn atom to their preadsorption states. The depletion and accumulation of electron charge are indicated by cyan and yellow, respectively (isosurface level: 0.005 e Å−3). c Variation of nucleation overpotential as a function of current density on b-graphite and CuO-graphite substrates.
DFT calculations were carried out to investigate the adsorption energy of atomic Zn (single Zn adsorbate, \({{{\rm{Zn}}}}_{1}^{*}\)) for various substrates (Fig. 2b). Based on our XRD data for each substrate, multiple crystal surfaces were considered. The Zn adsorption energy shows a strong correlation with the above NOP values, except for that of Cu, which was reported to form Cu-Zn alloys29,46. The binding energy between Zn and CuO was significantly high at −5.96 eV, obviously larger than that of graphitic carbon (−0.35 eV). The strong adsorption of Zn on CuO was likely induced by a significant electronic redistribution between Zn and CuO, as shown in the inset of Fig. 2b.
To investigate in more detail the effect of CuO NPs as Zn nucleation seeds, we decorated the graphite surface with CuO NPs with sizes of less than 50 nm (CuO-graphite) and compared the resulting electrochemical behavior to that of bare graphite electrode (b-graphite). These CuO NPs distributed uniformly to form homogeneous Zn electrodeposition on CuO-graphite (Supplementary Fig. 2). Since the reduction in NOP is more pronounced with smaller particle sizes, i.e., with an increase in the surface-active area of CuO, this study adopted CuO-NPs below 50 nm (Supplementary Fig. 3). NOP is strongly dependent on the applied current density as well as the type of substrate. Figure 2c and Supplementary Fig. 4 show the variation of NOP as a function of current density on b-graphite and CuO-graphite substrates. NOP is proportional to the current density; it shows a considerably large overpotential, exceeding 300 mV, at 60 mA cm−2 for b-graphite. This is one of the main causes of cell failure: the overpotential causes dendrite issues when zinc-based batteries are operated at high current densities. In contrast, CuO-graphite exhibited values of NOP of less than zero for all current densities, providing its reliable performance as a nucleation seed even at high current density.
In Fig. 3a, b, electron redistribution between single Zn adsorbate (\({{{\rm{Zn}}}}_{1}^{*}\)) and substrates (CuO and graphite) is indicated according to Bader charge values. CIF files used for electronic structure calculations are included in supplementary data files 1–4. Specifically, using the Bader charges of the Zn atom and the pristine surface models as reference states (Supplementary Figs. 5–7), the electron redistribution by Zn adsorption was calculated. At the neutral charge state (\(\Delta\)ne = 0), the condition under which adsorption energy \({{{\rm{Zn}}}}_{1}^{*}\) was calculated (Fig. 2b), CuO actively accepted electrons from \({{{\rm{Zn}}}}_{1}^{*}\) (\(\Delta\)TECuO = 1.22). Here, \(\Delta\)TE is defined as electron transfer relative to a reference system; it is calculated by measuring the deviation of the Bader atomic charge due to Zn adsorption. Positive \(\Delta\)TE values imply that more electrons are distributed by adsorption with respect to the reference systems. Electron redistribution between graphite and \({{{\rm{Zn}}}}_{1}^{*}\) is negligible (\(\Delta\)TEGR = 0.02). With this strong electronic redistribution, \({{{\rm{Zn}}}}_{1}^{*}\) it can be favorably anchored to the CuO substrate, explaining the low NOP during galvanostatic Zn deposition.

a, b Bader charge analysis of single Zn atom adsorbed on substrates from Δne = −2.0 to Δne = +2.0 [(a) CuO (001), (b) graphite]. c-f Charge density difference maps of discharged state (Δne = −2.0) and charged state (Δne = +2.0) [discharged: (c) CuO (001) and (d) graphite; charged: (e) CuO (001) and (f) graphite]. Depletion and accumulation of electron charge are indicated by cyan and yellow, respectively (isosurface levels: 0.005 e Å−3 for CuO (001), 0.001 e Å−3 for graphite). g-j Density of states before Zn adsorption for CuO (001) (g) and graphite (h). The density of states after Zn adsorption for CuO (001) (i) and graphite (j). For visibility, the states are normalized to the number of atoms and then multiplied by 1, 1, 3, and 10 for zinc, copper, oxygen, and carbon.
To examine the electronic behavior of CuO and graphite substrates in electron-rich and electron-deficient environments, we added up to two electrons to the model system (\(\Delta\)ne = +2) at 0.5 increments and removed up to two electrons from the model system (\(\Delta\)ne = −2) at 0.5 increments to mimic the charging and discharging conditions of the Zn/Zn2+ redox reaction, respectively. The gradient of \(\Delta\)TE (\(\Delta \Delta\)TE, Supplementary Table 1) represents the sensitivity of each substrate to change in the charge state of the electrode. A higher \(\Delta \Delta\)TE means that a substrate can take and release electrons more easily than its surroundings, thus functioning as an electron sponge. Both CuO and graphite substrates respond more actively to charging and discharging electrons than does \({{{\rm{Zn}}}}_{1}^{*}\). Comparing substrates, CuO shows a higher \(\Delta \Delta\)TE value than that of graphite, indicating that CuO accepts more electrons in an electron-rich environment and releases more electrons in an electron-deficient environment than other substrates. Simulated charge-discharge tests thus predict that CuO will become strongly negatively polarized in the charging condition, electrostatically attracting additional Zn2+ to the CuO substrate rather than to the already adsorbed Zn* and leading to homogeneous lateral growth of Zn (vide infra). In the discharging condition, CuO is converted to a positively polarized state, while Zn* is maintained in a much high oxidation state (partial atomic charge derived by Bader analysis, δ ~ +1.36), which promotes electro-dissolution of Zn/Zn2+ and its hydration by the surrounding water. Furthermore, there is a repulsive force between the positively charged CuO surface and Zn*, facilitating Zn dissolution.
Charge distribution changes of \({{{\rm{Zn}}}}_{1}^{*}\) on CuO and graphite substrates in charge (\(\Delta\)ne = +2.0) and discharge (\(\Delta\)ne = −2.0) conditions are visualized with regard to the neutral charge state (\(\Delta\)ne = 0.0) in Fig. 3c–f. Figure 3e, f show changes in charge distribution of Zn* on CuO and graphite substrates during electron addition (\(\Delta\)ne = +2.0). Similarly, Fig. 3c, d indicate changes when removing electrons (\(\Delta\)ne = −2.0). Electrons are highly localized on the surface region of CuO. In graphite, electrons are widely dispersed on the entire surface model, with mild localization on the surface region. This trend is consistent with previous observations that the \(\Delta \Delta\)TE of \({{{\rm{Zn}}}}_{1}^{*}\)@CuO is lower than that of \({{{\rm{Zn}}}}_{1}^{*}\)@graphite.
Figure 3g–j show density of state values (DOS) of surface layers of CuO and graphite with and without the single Zn adsorbent, \({{{\rm{Zn}}}}_{1}^{*}\). When Zn is adsorbed on the CuO substrate (Fig. 3g, i), the energy level of the O(2p) state shifts down as Zn(3 d) and O(2p) states are hybridized, with a main peak at approximately −6.0 eV below the main peak of Cu(3 d) near −4.0 eV (Zn and OZn states are shown in Supplementary Fig. 8). In contrast to the significant redistribution of CuO orbital states, the orbital states of graphite undergo only minor changes by\(\,{{{\rm{Zn}}}}_{1}^{*}\). The orbital shape of \({{{\rm{Zn}}}}_{1}^{*}\) on graphite closely resembles that of a Zn atom.
Our DOS analysis clearly elucidated the operating mechanism of the electron sponge effect in Zn*@CuO, the aim of this study. Zn(3 d)–O(2p) hybridization, which is relatively more stable than Cu(3 d)–O(2p) hybridization, allows the formation of a surface Zn-O layer that is desensitized to electron polarization (either addition or removal of electrons). Thus, with Zn*-O bonds, which are relatively more durable than Cu-O bonds, the Zn* becomes resistant to electron polarization; this phenomenon was also observed by Bader charge analysis (\(\Delta \Delta\)TEZn*@CuO < \(\Delta \Delta\)TEZn*@GR in Supplementary Table 1). In place of the resistant Zn, CuO acts as an electron sponge and is sensitive to electron transfer, which is also verified by the value of \(\Delta \Delta\)TECuO > \(\Delta \Delta\)TEGR (Supplementary Table 1).
Electrochemical effect of CuO NPs on Zn nucleation/growth
Electrochemical characterization was performed to study the deposition/stripping behavior of Zn on CuO-graphite substrate in an electrolyte of 2.5 M ZnI2; results are shown in Fig. 4a. shows cyclic voltammetry (CV) curves for the redox reactions of Zn/Zn2+ at a scan rate of 20 mV s-1. While the deposition potential is defined as the point at which Zn2+ ions begin to be reduced on the substrate (A, B) during the forward potential scan, the crossover potential can be observed as the intersection point at which the current switches from cathodic to anodic during the reverse potential scan (A’, B’). The difference between these two potentials (|A − A’| or |B − B’|) indicates the extent of polarization due to Zn nucleation47. For b-graphite, |A − A’| was very large at 26.2 mV, showing the large nucleation energy barrier on the carbon substrate (Supplementary Table 2). In contrast, |B − B’| was close to zero, with a negligible nucleation energy barrier for CuO-graphite; this is consistent with the aforementioned galvanostatic overpotential. The negligible nucleation energy can be ascribed to the decorated CuO NPs, which provide strong adsorption sites at which Zn nuclei can stably anchor to the substrate with improved affinity. Homogeneous nucleation on CuO-graphite promoted the reversible redox reaction of Zn/Zn2+, producing the higher anodic and cathodic peak current densities. By replacing ZnI2 with Na2SO4 in the electrolyte for CV measurements, we confirmed that the influence of hydrogen evolution reaction (HER) in the corresponding potential range can be disregarded (Supplementary Fig. 9).

a Cyclic voltammograms for Zn/Zn2+ redox reaction at scan rate of 20 mV s−1. b Tafel polarization curves at scan rate of 2 mV s−1 under OCV condition. c Nyquist plots at −1.0 V versus Ag/AgCl reference in a frequency range of 30 mHz – 300 kHz before (top) and after (bottom) electrodeposition of a thin Zn layer of 1.67 mAh cm−2. d CA curves under constant overpotential of 150 mV for 140 s. e Comparison of 2D diffusion times at applied overpotentials of 100 – 300 mV. f Reaction paths of Zn atom and energy barriers on graphite and CuO (001), derived by DFT calculations. The inset is a magnified image of the energy barrier of graphite.
The effect of CuO NPs on the Zn nucleation behavior was further investigated by Tafel polarization analysis at a scan rate of 2 mV s−1 under open circuit voltage (OCV) (Fig. 4b). The b-graphite followed the typical Butler-Volmer relation, with negligible mass-transfer effect at high concentration of Zn2+ ion. In contrast, CuO-graphite gave a distorted cathodic curve. Notably, during the nucleation period, an early cathodic current plateau was observed at the more positive potential, −0.91 V, than at the Zn/Zn2+ redox potential. This high and constant nucleation current can be explained by the interaction between CuO NPs and Zn2+ ion. After zinc coverage of nearly all CuO NPs at around −0.94 V, the current resembled that of b-graphite and followed a similar polarization curve.
To investigate the kinetic effect of CuO NPs on the Zn/Zn2+ redox reaction, we performed electrochemical impedance spectroscopy (EIS) during both nucleation and growth stages (Fig. 4c). EIS measurements were conducted at −1.0 V versus Ag/AgCl reference in a frequency range of 30 mHz – 300 kHz. Pristine surfaces of b-graphite and CuO-graphite substrates showed different charge-transfer behaviors. While b-graphite yielded a large charge-transfer resistance of 1.13 ohm‧cm2, CuO-graphite was characterized by a remarkably smaller charge-transfer resistance of 0.32 ohm‧cm2 due to the high Zn affinity of the CuO surface during the nucleation stage (Supplementary Table 3). To compare the charge-transfer resistance during the Zn growth stage, a constant amount of thin Zn layer was electrodeposited on the electrode substrates at 20 mA cm−2 for 300 sec. During the growth stage, the charge-transfer resistance of CuO-graphite (0.12 ohm‧cm2) became slightly smaller than that of b-graphite (0.17 ohm‧cm2). The difference in the resistance is the result of different Zn crystallites formed on the b-graphite and CuO-graphite substrates; this will be discussed in more detail below.
During nucleation and the early growth period, adsorbed Zn2+ ions move laterally along the electrode surface to locate, through 2D diffusion, the most energetically favorable sites. However, this 2D diffusion should be suppressed for the homogeneous Zn deposition because it causes nuclei aggregation and uneven dendrite growth. The extent of 2D diffusion can be identified simply by looking at current variation during chronoamperometry (CA) because this variation reflects changes in surface morphology due to 2D diffusion48. The current-time curve was obtained under a constant overpotential of 150 mV during CA (Fig. 4d). The current density for b-graphite showed a sharp increase during initial electrodeposition; current density became stable at −48 mA cm−2 after the long 2D diffusion period of over 17 s, indicating a rampant 2D diffusion process. On the other hand, for CuO-graphite, the 2D diffusion time was very short, within 0.7 s. Figure 4e compares the period of 2D diffusion as a function of the applied overpotential. As the overpotential increased from 100 mV to 300 mV, the 2D diffusion time decreased from 19 s to 11 s but remained high for b-graphite. For CuO-graphite, the 2D diffusion time showed negligible differences regardless of applied overpotential (Supplementary Fig. 10). This result indicates that 2D diffusion was effectively inhibited by the intrinsically strong Zn-affinity of CuO NPs, leading to even Zn deposition. To show how CuO prevents 2D diffusion of Zn atoms, calculations for diffusion energy barrier were performed (Fig. 4f). CIF files used for diffusion energy barrier calculations are included in supplementary data files 5–8. While only a negligible energy barrier (0.01 eV) was required for Zn atom to move by 2D diffusion on graphite, a much higher energy barrier (0.86 eV) must be overcome for such movement on CuO; this is because the weak bonding between carbon and Zn atoms facilitates surface movement of Zn atoms, while hybridized Zn-O bonds for Zn*@CuO are hard to break (Fig. 3g–j and Supplementary Figs. 11-16).
Zn atomic structure on CuO-graphite electrode
The surface atomic structure of electrodeposited Zn is an important characteristic in determining the overall surface morphology and, thus, the maximum available areal capacity of ZIFB. As the electrodeposited material becomes denser, more Zn2+ ions can be stably charged in the limited anode space and more battery capacity becomes available. Furthermore, a smooth and dense electrodeposit creates a homogeneous electric field around it, which inhibits dendrite growth by inducing uniform mass transfer of Zn2+ ions from the bulk solution.
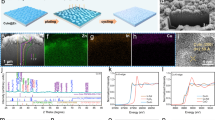
To compare the maximum available capacity of b-graphite and CuO-graphite as an anode, ZIFB was charged at a constant current density of 80 mA cm−2 and its cell voltage was monitored (Fig. 5a). When Zn electrodeposits or dendrites contact and pierce the membrane, short circuits occur between anode and cathode and cell voltage starts to drop; this is accompanied by strong voltage fluctuation18,19,20,21. Therefore, we can clearly identify the maximum areal capacity from the voltage profile. While the short circuit occurred at an areal capacity of 276.5 mAh cm−2 for b-graphite, the short circuit of CuO-graphite took place at an even higher areal capacity of 378.0 mAh cm−2, an improvement of 36.7%.

a Voltage profile during charging at a constant current density of 80 mA cm−2. b-e Surface analysis of Zn electrodeposits on b-graphite and CuO-graphite electrodes after charging to 160 mAh cm−2, of which position is marked by a star symbol in the voltage profile in (a). b XRD patterns of zinc electrodeposit. c Distribution diagram of zinc crystal planes. d SEM images of surface structure and e SEM cross-section images to identify surface roughness and packing density of Zn electrodeposits: top (low magnification) and bottom (high magnification) images. f Comparison of areal capacities demonstrated from previous Zn-based batteries. g, h Side and top views of charge density difference maps of Zn clusters with vertical (3D) and lateral (2D) growths and their relative energies between 2D and 3D [(g) graphite; (h) CuO (001)]. The charge density difference map was created by measuring the change in charge density from Zn3* to Zn4*. The depletion and accumulation of electron charges are colored cyan and yellow, respectively [isosurface levels: 0.0004 e Å−3 for graphite and 0.004 e Å−3 for CuO (001)]. i Energetically favored sequential growth of Zn clusters on graphite and CuO (001).
To scrutinize the extension effect of areal capacity, we investigated the Zn crystal orientation on the two electrode substrates by X-ray diffraction (XRD) after charging to 160 mAh cm−2 (Fig. 5b). By switching the b-graphite anode with the CuO-graphite anode, the occupying ratio of (002) and (004) crystal planes, in which the Zn electrodeposits grow parallel to the electrode surface and have the highest atomic packing density, sharply increased from 30.8% to 57.2%; these values were determined by the XRD intensity ratio (Fig. 5c and Supplementary Table 4). On the other hand, the occupying ratio of (101), (102), and (103) crystal planes, tilted in a range of 15 ~ 70 degrees with respect to the electrode surface and prone to dendrite growth, significantly decreased from 62.9% to 35.6%. Therefore, the higher areal capacity of CuO-graphite was induced by the change in Zn crystallite formed during Zn growth stage. Consistent results can be observed in the scanning electron microscopy (SEM) images (Fig. 5d and Supplementary Fig. 17). Zn electrodeposition typically grows in the form of crystalline aggregates, as commonly observed in Zn-based flow batteries. Comparing the crystallinity of zinc within the aggregates, the difference is evident from the high-magnification SEM images (Fig. 5d). For the Zn electrodeposit formed on the b-graphite, Zn layers grown obliquely in various directions constituted a unit structure. On the other hand, for CuO-graphite, the Zn layer grew in one direction parallel to the electrode surface, corresponding to the (002)/(004) crystal planes. Figure 5e demonstrates the difference in the arrangement of Zn aggregates accumulating on the electrode surface through a comparison of electrode cross-sections. In CuO-graphite, the aggregates stacked regularly, forming a relatively flat electrode surface, whereas in bare graphite, the aggregates densely concentrated in specific areas, resulting in the formation of protrusions. As a result, the maximum height of the Zn electrodeposit for CuO-graphite was smaller at 530 µm than that for b-graphite (683 µm), preventing the electrodeposit from touching the membrane. By closely examining the cross-sectional image, the Zn electrodeposit on b-graphite and CuO-graphite electrodes occupied 62.8% and 55.2% of the entire anode space, respectively. This means that the packing of the Zn electrodeposit on CuO-graphite was 14% denser than that on b-graphite.
The areal capacity is affected by the applied current density as well as the electrode substrate. We performed charge-discharge testing in the ZIFB single cell to identify the maximum areal capacities as functions of current density. At a current density of 20 mA cm−2, the areal discharge capacity of 463.7 mAh cm−2 was stably maintained, with a high energy efficiency of 89%; this is the highest areal capacity ever reported for all Zn-based batteries (Fig. 5f and Supplementary Fig. 18)10,12,13,14,15,16,17,18,19,20,21,22,30,49,50,51,52,53,54,55,56,57,58,59,60. This high areal capacity opens a new horizon for realizing the applicability of Zn-based batteries as long-period ESSs.
Figure 5g, h show charge density differences when \({{{\rm{Zn}}}}_{3}^{*}\) clusters become \({{{\rm{Zn}}}}_{4}^{*}\) following either vertical or lateral growth modes (Supplementary data files 9-12). Here, using DFT, Elateral – Evertical was calculated as the energy difference between a laterally grown \({{{\rm{Zn}}}}_{4}^{*}\) cluster and a vertically grown one. A positive value indicates that the vertically grown \({{{\rm{Zn}}}}_{4}^{*}\) cluster is more stable than the laterally grown cluster. For the graphite substrate, Zn atoms prefer to adsorb on top of pre-adsorbed Zn clusters, resulting in a vertical growth mode, while Zn* on CuO (001) exhibits lateral growth mode in which Zn atoms adsorb to the sides of pre-adsorbed Zn clusters, so that they have direct interaction with the substrate. The charge density difference maps show that Zn atoms adsorbed on graphite share more electrons with pre-existing Zn* clusters than with graphite during its vertical growth. On the other hand, the Zn atoms on CuO (001), with its lateral growth, have more active electronic redistribution with the substrate than pre-existing Zn* clusters. Figure 5i shows snapshots of Zn clusters grown sequentially along the stable energy path on graphite (upper panel) and CuO (lower panel) (Supplementary Figs. 19 and 20 for more details). This illustrates a clearer preference for Zn lateral growth on CuO substrate. CIF files used to simulate growth mode are included in supplementary data files 13–24.
ZIFB performance with CuO-graphite anode
ZIFB performance was compared under a current density of 20 mA cm−2 using an electrolyte compromising 3.0 M ZnI2 and 4.0 M NH4I, with a theoretical volumetric capacity of 160.8 Ah L−1 (Fig. 6a). The experiment was repeated three times under the same conditions (Supplementary Fig. 21). The CuO-graphite demonstrated a discharge capacity of 154.9 ± 1.4 Ah L−1 with a high capacity utilization of 96.3 ± 0.8%, while the b-graphite had an even smaller capacity of 134.6 ± 4.6 Ah L−1 due to the crossover of I3− ions. For the b-graphite, the loose zinc electrodeposits or dendrites touched and pierced the ion exchange membrane, creating porous defects. I3− ions easily transferred from the cathode to the anode side through the porous defects, causing Zn corrosion and self-discharge. The anolyte color changed from colorless to brown due to I3− crossover (Fig. 6a inset). For the CuO-graphite, the full capacity was stably operated without any color change in the anolyte. The discharge energy density reached 180.3 ± 3.5 Wh L−1 with an energy efficiency of 86.9 ± 2.2 %, which is among the highest performances reported to date for ZIFBs at the current density of 20 mA cm−210,12,13,14,15,16,17,18,19,20,21,22. The energy density was calculated only based on the volume of the catholyte due to the ambipolar characteristics of ZnI2. It can be converted to 90.2 ± 1.3 Wh L−1 based on the total volume of anolyte and catholyte. The NOP reduction effect of CuO NPs was also observed during cell operation. The voltage profile during the initial charging time was magnified and is plotted in Fig. 6b. The initial voltage overshoot is closely related to the Zn NOP and decreased by more than half in the CuO-graphite. This can be explained by the decrease in NOP on CuO NPs. The average charge and discharge voltages were 1.31 V and 1.15 V, respectively.

a Charge-discharge voltage profile with electrolyte of 3.0 M ZnI2 and 4.0 M NH4I at a constant current density of 20 mA cm−2. b High-magnification of initial charging voltage curves. c Variation of coulombic and energy efficiencies at different current densities in range of 40–400 mA cm−2. d Cycling stability of ZIFBs at constant current density of 140 mA cm−2. e Performance comparison of ZIFBs demonstrated in previous studies. f SEM images of Zn electrodeposits on b-graphite (top) and CuO-graphite (bottom) electrodes at 100th, 500th, and 1000th cycles during cycling test. g Change in particle size distribution of deposited Zn clusters. h Zinc crystal distribution calculated from XRD analysis for Zn electrodeposits at each cycle.
Rate performance analysis was conducted for the 1.67 M ZnI2 and 4.0 M NH4I electrolyte, which demonstrated dendrite-free ZIFB operation with CuO-graphite at high current density (Fig. 6c). ZIFB with CuO-graphite delivered satisfactory cell performance with high coulombic efficiency (97.6%) and discharge specific capacity (61.3 Ah L−1, corresponding to a capacity utilization of 68.5%) at 400 mA cm−2. At the same current density, b-graphite suffered from a low coulombic efficiency of 92.6% due to dendrite formation. For b-graphite, dendrite formation, crossover, and self-discharge began to deteriorate cell stability above 240 mA cm−2 (Supplementary Fig. 22). As previously shown in Fig. 2, higher current density correlates with increased NOP, fostering dendrite formation and eventually resulting in cell failure. However, the CuO-graphite effectively inhibited dendrite formation based on the negligible NOP and the (002)-preferred Zn electrocrystallization, even at the high current density of 400 mA cm−2. Furthermore, the ZIFB with CuO-graphite at 70% SoC (State-of-Charge) delivered a maximum power density of 317.2 mW cm−2 with a corresponding current density of 500 mA cm−2 (Supplementary Fig. 23), a superior rate performance compared to reported ZIFBs13,16,18.
Dendrite-free stability was also observed in the long-term charge-discharge operation under current density of 140 mA cm−2 and areal capacity of 40 mAh cm−2 (Fig. 6d and Supplementary Fig. 24). The ZIFB with b-graphite anode deteriorated severely with the sharp drop of coulombic efficiency at approximately 1000th cycle. On the other hand, the CuO-graphite anode exhibited extended stability to 2500 cycles with average coulombic efficiency of 98.7%. Therefore, compared to previously reported ZIFB studies, the CuO-graphite in this study enabled significant performance with not only high energy density (180 Wh L−1) but also very long cycle stability (> 2500 cycles) at high current density (140 mA cm−2). (Fig. 6e). In particular, achieving a cumulative capacity of 100 Ah cm−2 over 2500 cycles, the CuO-graphite exhibited remarkably superior stability characteristics compared to other zinc-based batteries (Supplementary Table 5). From the analysis results of FE-SEM, (FE-TEM), and XRD conducted on the electrode after cycling, it can be concluded that CuO NPs performed their function reliably and remained physicochemically stable in the ZIFB electrolyte over 2500 cycles (equivalent to 2.2 months) (Supplementary Figs. 25–27).
The surface morphology of the anodes during the 100th, 500th, and 1000th cycles was investigated by FE-SEM (Fig. 6f). For b-graphite, agglomerations of unit crystals growing in various directions were observed; these agglomerates became more prominent as the cycle number increased. Notably, dendritic growth with Zn particles larger than 500 µm was seen during the 1000th cycle. In the case of CuO-graphite, uniform surface morphology without agglomeration was maintained for all cycles. The particle size distribution of the Zn crystals was measured by analyzing multiple SEM images (Fig. 6g). The b-graphite yielded a broad size distribution ranging from 60 µm to 520 µm. As the cycle number increased, the particle size gradually increased. In contrast, a constant size distribution with average particle size of about 150 µm was observed for all cycles for CuO-graphite. From the overall surface images of b-graphite and CuO-graphite electrodes after charging during the 1000th cycle, it was obvious that CuO nucleation seeds resulted in homogeneous Zn deposition (Supplementary Fig. 28). It can be observed that the (002)/(004)-preferred Zn basal planes on CuO-graphite dominantly developed at a ratio of approximately 42.0%, regardless of the cycle number (Fig. 6h and Supplementary Fig. 29). For b-graphite, as the cycle number increased, those basal planes sharply decreased from 25.7% to 13.5%; instead, vertical planes such as (110) and (100) planes increased from 10.1% to 19.2% (Supplementary Table 6). Therefore, we can conclude that the (002)/(004)-preferred Zn crystallinity on the CuO nucleation seeds inhibited growth and proliferation of Zn dendrites, prolonging the cyclability of the ZIFB system.
Dendrite-free stability can also be enhanced by efficient Zn dissolution on CuO-graphite during discharge. After discharge, the residual amount of Zn on CuO-graphite was even less than that on b-graphite, as shown by EDX analysis (Supplementary Fig. 30) and XRD analysis (Supplementary Fig. 31). To quantitatively investigate the residual Zn, it was completely oxidized using 0.14 M VOSO4 solution and the change in valence state of vanadium ions was monitored by potentiometric titration (Supplementary Fig. 32). The residual amount of Zn on CuO-graphite was 78 mmol cm−2, which was about 4.5 times lower than that on b-graphite (353 mmol cm−2). This is in good agreement with the above computational analysis, indicating that CuO NPs with electron sponge function promote the dissolution behavior of Zn. Efficient Zn dissolution during discharge contributes to preventing dendritic Zn growth in subsequent charging cycles.
Discussion
In this study, we demonstrated dendrite-free Zn electrodeposition on CuO-graphite with an remarkably low NOP. The strong electronic redistribution between Zn and CuO can explain the high Zn affinity on CuO-graphite, which is supported by large predicted binding energy. Furthermore, through computational analysis, it was shown that CuO exhibits remarkable electron-accepting and -donating capabilities in electron-rich and electron-deficient environments.. The establishment of stable Zn-O bonding within CuO gives rise to electron duality, indicating distinct electronic structures between the CuO matrix and the Zn-O bond region. This electron sponge-like feature facilitates plating/stripping behavior of Zn under charging and discharging conditions. The highly negatively polarized CuO and strong binding energy between Zn and CuO induced lateral growth of Zn on the CuO-graphite electrode, leading to even Zn deposition with (002)-preferred crystal planes. As a result, the battery, with notably high areal capacity of 463.7 mAh cm−2, was stably charged and discharged without dendrite issues. Based on densely packed Zn electrodeposits, the discharge energy density reached 180 Wh L−1 with the energy efficiency of 86.9% at 20 mA cm−2 in the ZIFB full cell, which is among the highest performances reported to date at this current density. Furthermore, dendrite-free stability was also demonstrated during long-term charge-discharge operation under a current density of 140 mA cm−2. The CuO-graphite anode exhibited an extended lifespan, enhancing the cell stability to 2500 cycles, with average coulombic efficiency of 98.7%.
Methods
Preparation of b-graphite and CuO-graphite
For b-graphite, a mixture of conductive carbon (Super-P) and polyvinylidene fluoride (PVDF, 99%, Sigma-Aldrich) binder was stirred into a slurry at a mass ratio of 8:2. The slurry was coated to a thickness of 50 µm on a bipolar carbon plate (Sigracell TF6, SGL Carbon, thickness: 0.6 mm) and was then dried at 80 °C for 24 h. The as-obtained sample was compressed to a thickness of 0.605 mm at 45 °C using a heating roll press. For CuO-graphite, Super-P, PVDF, and CuO NPs (≤ 50 nm particle size, Sigma-Aldrich) were mixed into a slurry at a mass ratio of 7:2:1. SEM images and XRD patterns of commercial CuO NPs are shown in Supplementary Fig. 33. The areal loading amount of CuO NPs was 0.4 mg cm−2. In addition, the graphite surface decorated with SnO2 NPs (≤ 100 nm particle size, Sigma-Aldrich) and ZnO NPs (≤ 50 nm particle size, Sigma-Aldrich) was prepared using the same methods.
Material characterization
Field emission scanning electron microscopy (FE-SEM, S-4800, Hitachi) at an accelerating voltage of 5 kV was performed to both examine the surfaces and determine the cross-sectional morphologies of electrodes. To investigate the elemental distribution on the electrode surface, energy dispersive X-ray spectroscopy (EDX, Unti® Max, Oxford Instruments) was carried out. Samples were cut with an ion milling system (IMS, E-3500, Hitachi) prior to cross-sectional analysis. High-resolution TEM images were obtained using a F200 (HR-TEM, JEOL) at an accelerating voltage of 200 kV. The crystal planes of Zn were investigated using a high-resolution powder X-ray diffractometer (XRD, SmartLab, Rigaku), from 2Ө = 30°–80° at a scan rate of 5° min−1.
Electrochemical analyses
Electrochemical measurements were performed on a VSP Potentiostat (Biologic) using a three-electrode system consisting of b-/CuO-graphite (or metallic) substrates, Zn plate, and Ag/AgCl corresponding to working, counter, and reference electrode, respectively, in an electrolyte of 2.5 M ZnI2 at room temperature. The working electrode area was applied 0.19625 cm2. All metallic substrates were purchased from Sigma-Aldrich. These metallic substrates were carefully polished with sandpaper (Cw 600-2c ~ 2000-2c) to eliminate corrosion products formed due to air exposure. After polishing, the substrates were washed with DI water before use. CV curves were obtained at a scan rate of 20 mV s−1 within a potential range of −0.6 to −1.1 V. To quantify NOP on several electrode substrates, CP tests were conducted at various current densities from 10 to 60 mA cm−2. NOP was calculated as the difference between the nucleation potential within the initial 0.5 s and the growth potential between 10 s and 15 s. CA data were recorded under constant overpotential of the Zn2+/Zn redox reaction so as to monitor current variation as a function of time. The 2D diffusion time was determined as follows for ease of comparative analysis: (1) We plotted a regression line (gray line) based on CA current values between 50 and 100 s, a duration considered sufficiently long for the predominant occurrence of the 3D diffusion process. (2) The 2D diffusion time was quantified by defining it as the point at which the difference between the actual current value and the regression value begins to decrease below 1 mA cm−2. For EIS measurement, an AC voltage with 10 mV amplitude was applied over a frequency range of 30 mHz – 300 kHz. EIS analysis was performed on each electrode before and after a constant amount of Zn layer had been galvanostatically electrodeposited at a current density of 20 mA cm−2 for 5 min. Tafel polarization curves were obtained at scan rate of 2 mV s−1 within the overpotential range of ±150 mV under OCV. The exchange current density was calculated from the anodic/cathodic Tafel slopes of a linear region within the plot.
Potentiometric titration (Tiamo, Metrohm, Switzerland) experiments were performed to measure amounts of residual Zn on electrode substrates. The ZIFB single cell was charged to an areal capacity of 160 mAh cm−2 and discharged to voltage cut-off of 0.1 V at current density of 80 mA cm−2. Residual Zn was completely dissolved and oxidized by circulating 50 mL of 0.14 M VOSO4 + 0.5 M H2SO4 solution as an oxidizing agent. Then, the change in valence state of vanadium ions was monitored by potentiometric titration using 0.1 M KMnO4 solution.
Flow battery tests
Configurations of ZIFB single cells are shown in Supplementary Fig. 34. b-/CuO-graphite electrodes and pristine carbon plates were used as negative and positive electrodes, respectively. Two types of ZIFB single cells were fabricated with different electrode areas: an electrode area of 9 cm2 was applied for analysis of zinc growth behavior, while an electrode area of 35 cm2 was adopted for the electrochemical battery test, which included cycling and rate performance. A copper plate was used as current collector. To allow electrolyte circulation, 2.0 mm and 3.0 mm thick flow frames were placed next to the negative and positive electrodes, respectively. 0.7 mm thick polypropylene mesh was positioned in the negative flow frame. This non-conductive mesh functioned as both a spacer for Zn electrodeposition and a turbulent flow promoter for the negative electrolyte, improving the mass-transfer characteristics of Zn2+ ion. A piece of carbon felt (XF-30A, Toyobo) with thickness of 4.3 mm was placed in the positive flow frame; this fabric played a significant role as an electrode, leading to enlarged interfacial area between the electrolyte and electrode. A Nafion membrane (N-211, Dupont, USA) with thickness of 25.4 µm was employed as a cation exchange membrane sandwiched between the negative and positive flow frames. It was soaked in deionized (DI) water for 24 h prior to use. ZIFB electrolytes with different concentrations were prepared by dissolving ZnI2 (98%, Sigma-Aldrich) and NH4I (99%, Kanto chemical) in DI water. The battery tests have been conducted at room temperature.
To investigate the extent to which CuO-graphite improved the energy density of the ZIFBs, charge-discharge tests were conducted at a constant current density of 20 mA cm−2 using an anolyte and a catholyte with the same composition: 3.0 M ZnI2 and 4.0 M NH4I. They were charged to 100% SoC, equivalent to an areal capacity of 183.8 mAh cm−2, and then discharged to 0.1 V using battery test equipment (Maccor Series 4000). Reflecting the ambipolar characteristics of ZnI2, unless otherwise specified, all capacity and energy densities are calculated only based on the volume of the catholyte. Energy densities are calculated by multiplying discharge-specific capacity and average voltage. Power densities are determined by dividing the discharge output by the unit area. For the rate performance test, ZIFBs were operated at different current densities ranging from 40 to 400 mA cm−2 using an anolyte and a catholyte with the same composition: 1.67 M ZnI2 and 4.0 M NH4I. The current density was incrementally increased by 40 mA cm−2 every 5 cycles. Cycling tests were performed at constant current density of 140 mA cm−2 with 40% SoC. The anolyte and catholyte shared identical compositions: 1.67 M ZnI2 and 4.0 M NH4I.
In the all the battery tests, the volumes of anolyte and catholyte were the same at 40 ml. Using a peristaltic pump, the electrolytes were circulated at constant flow rate of 75 mL min−1 in each half-cell (Masterflex pump, Cole-Parmer).
Density functional theory (DFT) calculations
DFT calculations were conducted using the Vienna Ab initio Simulation Package (VASP)61, along with the projector-augmented wave (PAW) method and the Perdew-Burke-Ernzerhof (PBE) functional62. A convergence test was conducted with a criterion of 0.01 eV atom−1 to determine the cutoff energy and Monkhorst-Pack grids63. To ensure accuracy, denser grids (four times for both a and b-axes) were used to analyze the density of states (DOS). The electronic and total ground state energies were converged with the criteria of 10-6 eV and 0.01 eV Å−1, respectively. Hubbard U correction was adopted for Cu 3 d of CuO (C2/c), with a value of \({U}_{{{\rm{eff}}}}\) of 5.5 eV64, which provided a good overall representation of experimental values (Supplementary Fig. 35)65.
Both 4×3 and 1×1 CuO surface models were constructed to describe each Zn-atom-and-cluster adsorbed surface and the fully Zn covered surface. The asymmetric CuO slab models were passivated with hydrogen atoms to minimize the influence of dangling bonds present on the opposite side of the Zn adsorption surface66. A nudged elastic band approach was used to calculate the diffusion barrier of the fixed CuO and the graphite surface67.
Values of NOP were evaluated by calculating the adsorption energy of Zn atoms, (\({E}_{{{\rm{Zn}}}}^{{{\rm{ad}}}}\)):
where \({E}_{{{{\rm{Zn}}}}^{*}}\), \({E}_{*}\), and \({E}_{{{\rm{Zn}}}}\) are DFT total energies of adsorbed Zn (Zn*), substrate, and Zn atoms in a box (20×20×20 Å3), respectively. To quantify the change in energy level of the oxygen 2p state upon Zn adsorption, we evaluated the oxygen p-band centers in the following way68,69:
where EF, E, and ρ(E) are Fermi energy, p-band energy, and number of states, respectively.
To understand the growth behavior of Zn on the substrates, we assumed sequential adsorption of Zn atoms. The process of sequential Zn adsorption begins with a cluster of three Zn atoms (Zn3*). Then, adsorption calculations of Zn were performed on adjacent (lateral) and top (vertical) sites of the previously adsorbed Zn*. The adsorption process was then repeated based on the energetically most stable structures. (Supplementary Figs. 19 and 20).
We used both relaxed and fixed surfaces for the electron sponge calculations to investigate the relationship between electron sponge behavior and surface relaxation. To prevent artificial charge transfer to hydrogen when the total number of electrons in the system was controlled, symmetric CuO models were adopted. Considering the experimental conditions in which Zn2+ ions in solution are converted to Zn metal with two electrons, we set the maximum number of electrons, which varies from plus two to minus two, to simulate charge and discharge conditions. The VESTA software is employed to visualize the crystal structures and charge density difference maps in this paper (Figs.1, 2b, 3c–f, 4f and 5g–i)70.
DFT calculations of hydration effects on zinc adsorption and growth on CuO and graphite surfaces
For all calculations in this study (Figs. 2b, 3, 4f, and 5g–i), the contribution of H2O (H2Osolvation for the hydration of Zn, and H2Osurface for H2O adsorption on the electrode surfaces) have been explicitly investigated by calculating Zn adsorption, electron transfer behavior in electrode surfaces, Zn diffusion, and growth mode of Zn cluster. (Supplementary Figs. 36–41).
Our calculations confirm the minor contribution of H2O to the overall behavior of Zn ions on CuO and graphite surfaces in aqueous zinc batteries. Despite the inherent presence of water, where water solvation (H2Osolvation) and surface adsorption (H2Osurface) could potentially impact Zn adsorption energetics, electron transfer, and growth mechanisms, our findings demonstrate that these hydration effects do not significantly alter the key behaviors or trends. Specifically, while the presence of H2O introduces slight changes in adsorption energy and electron exchange dynamics, it does not substantially affect the overall preference for two-dimensional zinc growth on CuO or the minor energy differences between two-dimensional and three-dimensional growth modes on graphite. Thus, our results align with our hypothesis that the contribution of H2O to adsorption, electron transfer, and growth processes is negligible, affirming the robustness of our theoretical models and the conclusions derived from them (see Section S1 in supplementary for more details).
Data availability
All data provide to ensure transparency and validity of conclusions in this study are present in the manuscript or the supplemental information. Source data are provided with this paper.
References
Assat, G. & Tarascon, J.-M. Fundamental understanding and practical challenges of anionic redox activity in Li-ion batteries. Nat. Energy 3, 373–386 (2018).
Shin, J. & Choi, J. W. Opportunities and reality of aqueous rechargeable batteries. Adv. Energy Mater. 10, 2001386 (2020).
Ding, M. et al. A stable and energy-dense polysulfide/permanganate flow battery. ACS nano 17, 16252–16263 (2023).
Yin, J., Zhang, W., Alhebshi, N. A., Salah, N. & Alshareef, H. N. Electrochemical zinc ion capacitors: fundamentals, materials, and systems. Adv. Energy Mater. 11, 2100201 (2021).
Lu, B. et al. Catholyte engineering to release the capacity of iodide for high‐energy‐density iodine‐based redox flow batteries. SusMat 3, 522–532 (2023).
Wang, G. et al. Unlocking the solubility limit of ferrocyanide for high energy density redox flow batteries. Mater. Today Energy 28, 101061 (2022).
Yang, M. et al. High performance and long cycle life neutral zinc-iron flow batteries enabled by zinc-bromide complexation. Energy Storage Mater. 44, 433–440 (2022).
Xiang, W. et al. Alkaline Zn-Mn aqueous flow batteries with ultrahigh voltage and energy density. Energy Storage Mater. 61, 102894 (2023).
Lee, J.-H. et al. Dendrite-free Zn electrodeposition triggered by interatomic orbital hybridization of Zn and single vacancy carbon defects for aqueous Zn-based flow batteries. Energy Environ. Sci. 13, 2839–2848 (2020).
Li, B. et al. Ambipolar zinc-polyiodide electrolyte for a high-energy density aqueous redox flow battery. Nat. Commum. 6, 6303 (2015).
Winsberg, J. et al. Aqueous 2, 2, 6, 6-tetramethylpiperidine-N-oxyl catholytes for a high-capacity and high current density oxygen-insensitive hybrid-flow battery. ACS Energy Lett. 2, 411–416 (2017).
Li, B. et al. Metal–organic frameworks as highly active electrocatalysts for high-energy density, aqueous zinc-polyiodide redox flow batteries. Nano Lett. 16, 4335–4340 (2016).
Weng, G.-M., Li, Z., Cong, G., Zhou, Y. & Lu, Y.-C. Unlocking the capacity of iodide for high-energy-density zinc/polyiodide and lithium/polyiodide redox flow batteries. Energy Environ. Sci. 10, 735–741 (2017).
Xie, C., Zhang, H., Xu, W., Wang, W. & Li, X. A long cycle life, self‐healing zinc–iodine flow battery with high power density. Angew. Chem. Int. Ed. 130, 11341–11346 (2018).
Liu, J. et al. MoS2-modified graphite felt as a high performance electrode material for zinc–polyiodide redox flow batteries. Inorg. Chem. Front. 6, 731–735 (2019).
Mousavi, M. et al. Decoupled low-cost ammonium-based electrolyte design for highly stable zinc–iodine redox flow batteries. Energy Storage Mater. 32, 465–476 (2020).
Yang, J., Song, Y., Liu, Q. & Tang, A. High-capacity zinc–iodine flow batteries enabled by a polymer–polyiodide complex cathode. J. Mater. Chem. A 9, 16093–16098 (2021).
ShakeriHosseinabad, F. et al. Influence of flow field design on zinc deposition and performance in a zinc-iodide flow battery. ACS Appl. Mater. Interfaces 13, 41563–41572 (2021).
Jian, Q., Wu, M., Jiang, H., Lin, Y. & Zhao, T. A trifunctional electrolyte for high-performance zinc-iodine flow batteries. J. Power Sources 484, 229238 (2021).
Jin, S. et al. Stabilizing zinc electrodeposition in a battery anode by controlling crystal growth. Small 17, 2101798 (2021).
Wang, S. et al. Act in contravention: A non-planar coupled electrode design utilizing “tip effect” for ultra-high areal capacity, long cycle life zinc-based batteries. Sci. Bull. 66, 889–896 (2021).
Mousavi, M. et al. Elucidating and tackling capacity fading of zinc-iodine redox flow batteries. Chem. Eng. J. 412, 128499 (2021).
Jang, W. J., Cha, J. S., Kim, H. & Yang, J. H. Effect of an iodine film on charge-transfer resistance during the electro-oxidation of iodide in redox flow batteries. ACS Appl. Mater. Interfaces 13, 6385–6393 (2021).
Xu, Z., Fan, Q., Li, Y., Wang, J. & Lund, P. D. Review of zinc dendrite formation in zinc bromine redox flow battery. Renew. Sustain. Energy Rev. 127, 109838 (2020).
Khor, A. et al. Review of zinc-based hybrid flow batteries: From fundamentals to applications. Mater. Today Energy 8, 80–108 (2018).
Foroozan, T. et al. Non-dendritic Zn electrodeposition enabled by zincophilic graphene substrates. ACS Appl. Mater. Interfaces 11, 44077–44089 (2019).
Cui, M. et al. Quasi-isolated Au particles as heterogeneous seeds to guide uniform Zn deposition for aqueous zinc-ion batteries. ACS Appl. Energy Mater. 2, 6490–6496 (2019).
Zhang, Y., Howe, J. D., Ben-Yoseph, S., Wu, Y. & Liu, N. Unveiling the origin of alloy-seeded and nondendritic growth of Zn for rechargeable aqueous Zn batteries. ACS Energy Lett. 6, 404–412 (2021).
Huang, Y. et al. Layer-by-layer zinc metal anodes to achieve long-life zinc-ion batteries. Chem. Eng. J. 431, 133902 (2022).
Yin, Y. et al. Dendrite‐free zinc deposition induced by tin‐modified multifunctional 3D host for stable zinc‐based flow battery. Adv. Mater. 32, 1906803 (2020).
Liu, P. et al. Mesoporous hollow carbon spheres boosted, integrated high performance aqueous Zn-ion energy storage. Energy Storage Mater. 25, 858–865 (2020).
Kim, J. Y., Liu, G., Shim, G. Y., Kim, H. & Lee, J. K. Functionalized Zn@ ZnO hexagonal pyramid array for dendrite‐free and ultrastable zinc metal anodes. Adv. Funct. Mater. 30, 2004210 (2020).
Liang, P. et al. Highly reversible Zn anode enabled by controllable formation of nucleation sites for Zn‐based batteries. Adv. Funct. Mater. 30, 1908528 (2020).
He, H., Tong, H., Song, X., Song, X. & Liu, J. Highly stable Zn metal anodes enabled by atomic layer deposited Al2O3 coating for aqueous zinc-ion batteries. J. Mater. Chem. A 8, 7836–7846 (2020).
Zhao, K. et al. Ultrathin surface coating enables stabilized zinc metal anode. Adv. Mater. Interfaces 5, 1800848 (2018).
Hao, J. et al. An in‐depth study of Zn metal surface chemistry for advanced aqueous Zn‐ion batteries. Adv. Mater. 32, 2003021 (2020).
Zhao, Z. et al. Long-life and deeply rechargeable aqueous Zn anodes enabled by a multifunctional brightener-inspired interphase. Energy Environ. Sci. 12, 1938–1949 (2019).
Mitha, A., Yazdi, A. Z., Ahmed, M. & Chen, P. Surface adsorption of polyethylene glycol to suppress dendrite formation on zinc anodes in rechargeable aqueous batteries. ChemElectroChem 5, 2409–2418 (2018).
Kang, L. et al. Nanoporous CaCO3 coatings enabled uniform Zn stripping/plating for long‐life zinc rechargeable aqueous batteries. Adv. Energy Mater. 8, 1801090 (2018).
Deng, C. et al. A sieve‐functional and uniform‐porous kaolin layer toward stable zinc metal anode. Adv. Funct. Mater. 30, 2000599 (2020).
Ding, F. et al. Dendrite-free lithium deposition via self-healing electrostatic shield mechanism. J. Am. Chem. Soc. 135, 4450–4456 (2013).
Wan, F. et al. Aqueous rechargeable zinc/sodium vanadate batteries with enhanced performance from simultaneous insertion of dual carriers. Nat. Commum. 9, 1656 (2018).
Soundharrajan, V. et al. The dominant role of Mn2+ additive on the electrochemical reaction in ZnMn2O4 cathode for aqueous zinc-ion batteries. Energy Storage Mater. 28, 407–417 (2020).
Ma, L. et al. Initiating a mild aqueous electrolyte Co3O4/Zn battery with 2.2 V-high voltage and 5000-cycle lifespan by a Co (III) rich-electrode. Energy Environ. Sci. 11, 2521–2530 (2018).
Guo, W. et al. Dendrite-free Zn anode with dual channel 3D porous frameworks for rechargeable Zn batteries. Energy Storage Mater. 30, 104–112 (2020).
Zhang, Q. et al. The three‐dimensional dendrite‐free zinc anode on a copper mesh with a zinc‐oriented polyacrylamide electrolyte additive. Angew. Chem. Int. Ed. Engl. 58, 15841–15847 (2019).
Wang, S. et al. A highly reversible zinc deposition for flow batteries regulated by critical concentration induced nucleation. Energy Environ. Sci. 14, 4077–4084 (2021).
Zhou, M. et al. Surface‐preferred crystal plane for a stable and reversible zinc anode. Adv. Mater. 33, 2100187 (2021).
Xiang, H. X., Tan, A. D., Piao, J. H., Fu, Z. Y. & Liang, Z. X. Efficient nitrogen‐doped carbon for zinc–bromine flow battery. Small 15, 1901848 (2019).
Suresh, S., Ulaganathan, M. & Pitchai, R. Realizing highly efficient energy retention of Zn–Br2 redox flow battery using rGO supported 3D carbon network as a superior electrode. J. Power Sources 438, 226998 (2019).
Li, X., Xie, C., Li, T., Zhang, Y. & Li, X. Low‐cost titanium–bromine flow battery with ultrahigh cycle stability for grid‐scale energy storage. Adv. Mater. 32, 2005036 (2020).
Li, X. et al. A complexing agent to enable a wide‐temperature range bromine‐based flow battery for stationary energy storage. Adv. Funct. Mater. 31, 2100133 (2021).
Lee, H. J., Kim, D.-W. & Yang, J. H. Estimation of state-of-charge for zinc-bromine flow batteries by in situ Raman spectroscopy. J. Electrochem. Soc. 164, A754 (2017).
Wu, M., Zhao, T., Jiang, H., Zeng, Y. & Ren, Y. High-performance zinc bromine flow battery via improved design of electrolyte and electrode. J. Power Sources 355, 62–68 (2017).
Naresh, R. P., Ragupathy, P. & Ulaganathan, M. Carbon nanotube scaffolds entrapped in a gel matrix for realizing the improved cycle life of zinc bromine redox flow batteries. ACS Appl. Mater. Interfaces 13, 48110–48118 (2021).
Shi, X. et al. Homogeneous deposition of zinc on three-dimensional porous copper foam as a superior zinc metal anode. ACS Sustain. Chem. Eng. 7, 17737–17746 (2019).
Kang, Z. et al. 3D porous copper skeleton supported zinc anode toward high capacity and long cycle life zinc ion batteries. ACS Sustain. Chem. Eng. 7, 3364–3371 (2019).
Cai, Z. et al. Chemically resistant Cu–Zn/Zn composite anode for long cycling aqueous batteries. Energy Storage Mater. 27, 205–211 (2020).
Chen, Y. et al. A dendrite-free Zn@ CuxZny composite anode for rechargeable aqueous batteries. Electrochim. Acta 399, 139334 (2021).
Zhou, J. et al. Encapsulation of metallic Zn in a hybrid MXene/Graphene aerogel as a stable zn anode for foldable Zn‐ion batteries. Adv. Mater. 34, 2106897 (2022).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169 (1996).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865 (1996).
Monkhorst, H. J. & Pack, J. D. Special points for Brillouin-zone integrations. Phys. Rev. B 13, 5188 (1976).
Dudarev, S. L., Botton, G. A., Savrasov, S. Y., Humphreys, C. J. & Sutton, A. P. Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA+ U study. Phys. Rev. B 57, 1505 (1998).
Chase, M. W. NIST-JANAF thermochemical tables and pp. 1−1951 (National Information Standards Organization, 1998).
Yoo, S. H., Lymperakis, L. & Neugebauer, J. Efficient electronic passivation scheme for computing low-symmetry compound semiconductor surfaces in density-functional theory slab calculations. Phys. Rev. Mater. 5, 044605 (2021).
Sheppard, D., Terrell, R. & Henkelman, G. Optimization methods for finding minimum energy paths. J. Chem. Phys. 128, 13 (2008).
Paßens, M. et al. Tuning the surface electronic structure of a Pt3Ti (111) electro catalyst. Nanoscale 8, 13924–13933 (2016).
Wang, L. et al. Tunable intrinsic strain in two-dimensional transition metal electrocatalysts. Science 363, 870–874 (2019).
Momma, K. & Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Cryst. 44, 1272–1276 (2011).
Acknowledgements
This work was supported by the Samsung Research Funding & Incubation Center of Samsung Electronics under Project Number SRFC-MA2101-04 and the National Supercomputing Center with supercomputing resources including technical support (KSC-2021-CRE-0357).
Author information
Authors and Affiliations
Contributions
J.S.C., C.W.L. and J.H.Y. conceived and designed the research. J.S.C. and N.S. conducted characterizations and electrochemical measurements. J.S.C. and J.H.Y. performed the spectroscopic and electrochemical analyses. S.P. and C.W.L. performed the DFT calculation analysis. Y.C.K. facilitated the collaboration and supervised the research. All authors contributed to the data analysis and discussion. J.S.C., S.P., C.W.L., and J.H. Y. wrote the manuscript with input from all the coauthors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Cha, J.S., Park, S., Seo, NU. et al. Zincophilic CuO as electron sponge to facilitate dendrite-free zinc-based flow battery. Nat Commun 16, 844 (2025). https://doi.org/10.1038/s41467-025-56011-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-56011-9
