
Abstract
Programmed-cell death is an antimicrobial defense mechanism that promotes clearance of intracellular pathogens. Toxoplasma counteracts host immune defenses by secreting effector proteins into host cells; however, how the parasite evades lytic cell death and the effectors involved remain poorly characterized. We identified ROP55, a rhoptry protein that promotes parasite survival by preventing lytic cell death in absence of IFN-γ stimulation. RNA-Seq analysis revealed that ROP55 acts as a repressor of host pro-inflammatory responses. In THP-1 monocytes ΔROP55 infection increased NF-κB p65 nuclear translocation, IL-1β production, and GSDMD cleavage compared to wild type or complemented parasites. ΔROP55 infection also induced RIPK3-dependent necroptosis in human and mouse primary macrophages. Moreover, ΔROP55 parasites were significantly impaired in virulence in female mice and prevented NF-κB activation and parasite clearance in mBMDM. These findings place ROP55 as a major virulence factor, dampening lytic cell death and enabling Toxoplasma to evade clearance from infected cells.
Similar content being viewed by others
Introduction
Toxoplasma gondii is an obligate intracellular parasite that is estimated to infect a third of the world human population, making it one of the most successful apicomplexan parasites1,2. Although most infections are generally asymptomatic, toxoplasmosis poses a serious health threat to immunocompromised individuals and to the developing fetus3. The recent emergence of highly virulent strains causing diseases in immunocompetent individuals also reinforces its global importance4,5. Toxoplasma can establish a chronic infection that persists for the lifetime of the host. To do so, the parasite has evolved several mechanisms to subvert the host’s immune response. These strategies depend on an arsenal of parasite effector proteins that are secreted into the host cell from two distinct intracellular compartments: the rhoptries and the dense granules6,7. These parasite effectors counteract host signaling pathways and dictate the variations in the virulence between Toxoplasma strains, the most common of which in Europe and North America are referred to as Type I, II and III6,7,8.
Programmed cell death is a critical aspect of the innate immune response and controls infections by killing and clearing infected cells, thereby depriving intracellular pathogens of their replicative niche9. Cell death pathways can either induce cell lysis and inflammation, as observed in both pyroptosis and necroptosis, or engage a nonlytic death program that does not induce inflammation, such as apoptosis10. Pyroptosis occurs through the formation of multiprotein complexes called inflammasomes, in which inflammatory caspases play a central role for the maturation of IL-1 family cytokines and the initiation of cell death11. The activation of NLRP3, which is the most well-studied inflammasome, is a tightly regulated process requiring two distinct signals11. The first signal arises from PAMPs and DAMPs that activate pattern recognition receptors and NF-κB signaling, leading to a priming stage in which the inactive forms of NLRP3 and IL-1β are synthesized12. A second signal is then necessary for the maturation of IL-1β by caspase-1. This signal is triggered by disturbances to cellular homeostasis caused by multiple agonists; among them, ionic flux, reactive oxygen species, and mitochondrial dysfunction, inducing NLRP3 inflammasome assembly to promote caspase-1 activation13. Caspase-1 activity is also required for cleaving Gasdermin D (GSDMD), which, upon insertion in the plasma membrane, forms pores that induces cell swelling and osmotic rupture, resulting in pyroptotic cell death. Necroptotic cell death, on the other hand, is a caspase-independent program that can be initiated by diverse triggers including ligation of death receptors, Toll-like receptors (TLRs) or cytosolic nucleic acid sensors10,14. These diverse stimuli converge to activate the serine/threonine protein kinase RIPK3, which, in turn, phosphorylates the pseudokinase MLKL. Active MLKL anchors in the plasma membrane and forms pores, leading to the release of damage-associated molecular patterns (DAMPs), cytokines and chemokines, and the lytic death of the cell10,14. Depending on the trigger that initiates necroptosis, RIPK3 undergoes oligomerization with MLKL with or without the involvement of RIPK1. In either scenario, necroptosis can lead to NF-kB signaling and transcriptional activation15,16,17,18 and can only proceed when caspase-8 is inhibited or insufficiently activated, since caspase-8 inhibits necroptosis by cleaving RIPK1 and RIPK310,19.
Toxoplasma repression of lytic cell death pathways have emerged as an important parasite strategy to favor survival, assuring long term persistence. For example, Toxoplasma bradyzoites block interferon-induced necroptosis by the combined function of two dense granule proteins (TgNSM and TgIST)20, contributing to survival of intracellular cysts. These findings suggest that necroptosis might be regulated in Toxoplasma-infected cells, but the extent to which these effectors or others play a role in vivo or during acute infection, remain unknown. In contrast, pyroptosis has been identified as the pathway associated with the resistance to chronic Toxoplasma infection in Lewis rats21. This resistance mechanism is linked to the activation of the NLRP1 sensor, resulting in host cell lysis and parasite clearance in vivo22. However, across different host species, Toxoplasma infection does not consistently lead to pyroptotic cell death. Indeed, inflammasome activation relies on the specific combination of host cell type and Toxoplasma strain. For instance, whereas infection of human THP-1 cells with type II parasites induces the nuclear translocation of NF-κB, production of IL-1β and activation of NLRP3 inflammasome23,24,25, infection with type I parasites does not23,26. This difference is in part due to allelic differences in the gene encoding GRA15, between the type I RH strain and type II strains23,26. Additionally, type I RH parasites actively employ counter-measures to block the NF-κB/IL-1β signaling axis, as type I Toxoplasma infection inhibits LPS-induced NF-κB nuclear translocation and IL-1β production, dampening host inflammatory responses27,28,29. In neutrophils, this strategy of immune evasion was shown to be dependent on actively invading parasites27,30. However, the identity of the parasite effectors involved in this suppression remains poorly characterized.
Here we identify a new Toxoplasma rhoptry bulb protein, ROP55, that prevents the induction of host lytic cell death through the repression of NF-κB signaling. The absence of ROP55 triggers both pyroptosis and RIPK3-dependent necroptosis of infected cells. Furthermore, we show that deletion of the ROP55 gene leads to a significant decrease in parasite virulence in vivo. Our findings reveal the role of ROP55 as a major virulence factor in Toxoplasma, preventing lytic host cell death as a conserved mechanism of evading parasite clearance in human and mouse cells.
Results
TGGT1_300220 is a new rhoptry protein, named ROP55
Rhoptry genes display a characteristic expression pattern and can be identified through bioinformatic analysis31. Using this approach, we identified TGGT1_300220, encoding a hypothetical protein of 1823aa, with a predicted signal sequence and a mRNA periodicity typical of rhoptry proteins (Fig. 1a). TGGT1_300220 is not predicted to be essential for growth in fibroblasts (fitness score of –0.1532), has no characterized domains using PROSITE33, HHpred34 or Phyre-235 searches, and appears to be restricted to coccidian parasites (Supplementary Fig. 1).

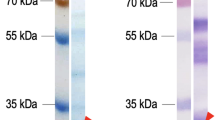
a Expression pattern of known rhoptry proteins and TGGT1_300220 in green during the tachyzoite cell cycle. Data obtained from ToxoDB (Behnke et al. 2010). b Immunoblot of lysates from parental (RH Δku80) and Tg300220-HA3 lines using anti-HA antibodies. ROP55 migrates at the expected size of 206 kDa. Loading control using anti-ROP5 antibodies. n = 4 experiments (c) Colocalization of TGGT1_300220-HA3 (Tg300220) (green) with rhoptry neck RON2 or rhoptry bulb ROP5 (red). Inset: higher magnification. n = 6 experiments. Source data are provided as a Source Data file.
To determine its localization, we endogenously tagged TGGT1_300220 with a triple HA tag at its C-terminus in the type I background strain RH Δku80 (Tg300220-HA3 line) (Supplementary Fig. 2a, b) confirming its expected size of 206 kDa (Fig. 1b). Immunofluorescence analysis (IFA) revealed a clear apical dot, suggestive of rhoptry localization that partially colocalized with the rhoptry neck protein RON2, but to a greater degree with ROP5, a rhoptry bulb protein (Fig. 1c). This rhoptry bulb localization fits with the hyperLOPIT (spatial proteomics method hyperplexed Localisation of Organelle Proteins by Isotopic Tagging) analysis36 which predicted TGGT1_300220 to be associated with rhoptry fraction 1, which contains luminal proteins that are secreted and injected into the host cell upon invasion.
These data show that TGGT1_300220 is a new rhoptry bulb protein, named hereafter ROP55, for rhoptry protein 55.
ROP55 is a major virulence factor
In order to study the function of ROP55, we generated a knock-out in the ROP55-HA3 line (ΔROP55) (Supplementary Fig. 2c, d) and a complemented line (compl.) by reintroducing the HA-tagged rop55 gene under its own promotor in the UPRT locus of the ΔROP55 line (Supplementary Fig. 2e, f). Western blot and IFA analyses confirmed the deletion and the re-expression of ROP55 in the complemented cell line and its targeting to the correct location (Fig. 2a, b).

a Western blot showing the absence of ROP55 in the ΔROP55 line and its re-expression in complemented strain (Compl). ROP55 was detected with an αHA antibody. n = 4 experiments. b Absence of ROP55 in ΔROP55 strain and correct localization of ROP55 (red) at the bulb of the rhoptries in the complemented strain. ROP5 (green), n = 4 experiments. c, d Plaque assay of RH Δku80 parental line (WT), ΔROP55 and complemented (compl.) lines on HFF monolayers after 6 days. Values are normalised relative to the percentage of lysis in the control strain and reported as mean ± SEM. c Lysis plaque areas of one representative experiment showing 3 technical replicates. Replicated experiments are shown in Supplementary Fig 2g, h. d Number of lysis plaques showing 3 independent experiments each having 3 technical replicates. e BALB/c mice were intraperitoneally infected with 100 tachyzoites of the WT and the complemented strains (5 mice per condition, n = 3), or with increasing doses of 104 tachyzoites (5 mice, n = 3), 105 tachyzoites (5 mice, n = 2) or 106 tachyzoites (10 mice, n = 1) of ΔROP55 knock-out parasites. Survival curves were compared using two-sided Log-rank (Mantel-Cox) test, p < 0,0001 for WT versus 106 ΔROP55 infected mice (f) Expression of TgSAG1 measured by qRT-PCR in spleens of BALB/c mice 4 days after infection. Representative graph of 3 independent experiments. Bars represent means ± SEM of three to five animals in each group. Replicated experiments are shown in Supplementary Fig. 2j, k. Statistical significance was determined using one-way ANOVA followed by Tukey’s post hoc test for (c). d, f Source data are provided as a Source Data file.
To test the ability of ΔROP55 to complete the lytic cycle in human foreskin fibroblasts (HFFs) we performed plaque assays over 6 days. We observed a trending reduction in both size and number of plaques (Fig. 2c, d), when compared with the wild type line (WT), and this phenotype was fully rescued in the complemented strain (Fig. 2c, d). We next evaluated the role of ROP55 in vivo during acute infection of BALB/c mice injected intraperitoneally (i.p.) with increasing doses of ΔROP55 tachyzoites. All mice died by day 8 following injection of 100 WT or complemented tachyzoites (Fig. 2e). Conversely, all mice infected with 104 and 105 ∆ROP55 tachyzoites survived, and only the injection of 1 million tachyzoites resulted in the death of 50% of mice (Fig. 2e). Moreover, when we assessed parasite burden at 4 days post-infection (dpi), by quantitative RT-PCR (qPCR), we observed substantially decreased TgSAG1 expression in the spleens of mice infected with 1000 ∆ROP55 tachyzoites, suggesting that the survival of mice infected with ΔROP55 is associated with a reduction in parasite burden (Fig. 2f and Supplementary 2i, j).
Altogether, these results show that ROP55, although not essential for Toxoplasma growth in vitro, has a prominent role in vivo and contributes to the virulence of the RH strain in female mice.
ROP55 controls parasite and host cell survival
To determine which stage of the lytic cycle was impaired in ΔROP55 parasites, we analyzed the individual steps involved in tachyzoite propagation in vitro. We observed a modest increase (25%) in the percentage of intracellular ΔROP55 parasites relative to the WT strain after 5 min of contact with HFF cells (Fig. 3a). When we counted the number of parasites per vacuole at 24 h-post infection (hpi), ΔROP55 parasites were not affected in their capacity to replicate (Fig. 3b). By triggering egress from infected host cells with the Ca++ ionophore A23187, ΔROP55 parasites exhibited similar egress to the WT line (Fig. 3c). However, when we measured parasite burden by qRT-PCR at 24 hpi, we detected a significant reduction in parasite load in cells infected with ΔROP55 parasites compared to WT or compl. parasites (Fig. 3d and Supplementary 3a, b). To test whether this reduction was due to an elimination of parasite vacuoles, we counted the vacuoles at 4 and 24 hpi. We observed a marked reduction in the number of vacuoles containing ΔROP55 parasites at 4 hpi, which persisted at 24 hpi, suggesting that the elimination occurred shortly after invasion (Fig. 3e and Supplementary 3c).

a Percentage of invaded HFFs quantified after 5 min of invasion. Values are normalized relative to the percentage of cells invaded by the control strain and are reported as mean ± SEM (n = 6 biological replicates, each with 3 technical replicates). b Number of parasites per vacuole at 24 hpi. Plotted are the mean values of 6 independent experiments and bars represent SEM. c Percentage of egress events following A23187 treatment at 30 hpi. 200 vacuoles were counted per coverslip. Values are reported as mean ± SEM (n = 3 biological replicates, each with 3 technical replicates). d Parasite burden measured by qRT-PCR in HFFs infected for 24 hpi. One representative experiment out of 3 is plotted, replicated experiments are shown in Supplementary Fig. 3a, b. Bars represent mean ± SEM (n = 3 technical replicates). e Number of vacuoles per field in infected HFFs. Representative graph of 2 independent experiments with 2 technical replicates counting 48 microscopic fields in WT, 52 in ΔROP55 and 36 in complemented infected cells at 4hpi and 50 microscopic fields in WT, 49 in ΔROP55 and 40 in complemented infected cells at 24hpi. Values are absolute numbers. Replicated experiment is shown in Supplementary Fig. 3c. f Percentage of HFF nuclei staining positive for propidium iodide (PI) per field at 6 hpi. Graph showing one independent experiment counting 35 microscopic fields for WT and ΔROP55, and 20 microscopic fields for compl. coverslips, replicated independent experiment is shown in Supplementary Fig. 3f. g Quantification of LDH released into the medium of HFFs infected for 6 or 24 h. Representative graph of 3 independent experiments. Values were normalized to 100% of lysis control (with addition of supplier lysis buffer) and are reported as mean ± SEM (n = 3 technical replicates). Replicated experiments are shown in Supplementary Fig. 3g, h. Statistical significance was determined using a two-tailed paired t test for (a, c), one-way ANOVA for (d, f) and two-way ANOVA (b, g). followed by Tukey’s pos-hoc test. Source data are provided as a Source Data file.
When culturing ΔROP55 parasites, we noticed the presence of abnormal host cells with elongated and condensed nuclei (Supplementary Fig. 3d). In addition, the cells appeared to be detaching and dying. The elongated nucleus phenotype was detectable within 2 hpi and increased to 6 hpi (Supplementary Fig. 3e). To assess host cell membrane integrity and cell viability, infected fibroblasts were stained with propidium iodide (PI), a non-permeant fluorescent dye commonly used to detect dead cells. At 6 dpi, the number of cells staining with PI was significantly increased when infected with ∆ROP55 parasites compared to WT and complemented lines (Fig. 3f and Supplementary 3f). This was concomitant with an increased level of lactate dehydrogenase (LDH) release into the culture medium of ΔROP55 infected cells (Fig. 3g and Supplementary 3g, h), which is indicative of lytic cell death37. These data show that infection with RH tachyzoites lacking ROP55 leads to an increase in host cell mortality, accompanied by the elimination of parasite vacuoles and a decrease in parasite load. Importantly, the cell death was observed in the absence of any stimulation, such as IFN-γ or LPS. However, ROP55 deficiency did not induce the death of all infected HFFs, and when the parasites were found within a viable host cell, they grew normally, as shown in Fig. 3b. Accordingly, electron microscopy images did not reveal any ultra-structural defects of the host cell, the parasites, or the PV at 24 hpi (Supplementary Fig. 3i, j). These images also showed normal rhoptry shape.
Altogether, these results suggest a role for ROP55 in controlling host cell viability, which may contribute to maintaining the parasite’s replicative niche.
ROP55 is a repressor of inflammatory host cell death
The ability of ROP55 to affect host cell fate prompted us to investigate the host transcriptional response during infection with ∆ROP55 parasites. At 6 hpi, a substantially different gene expression profile was displayed in HFFs infected with ∆ROP55 relative to WT (Fig. 4a). We found 388 differentially expressed genes (DEGs), with an adjusted p value < 0.05, of which 283 were up-regulated and 105 were down-regulated. Interestingly, many of these DEGs were associated with cell death and the inflammatory response of the host cell (Fig. 4a and Supplementary data 1). This profile was similar at 24 hpi, with 125 differentially expressed genes, of which 44 were up-regulated and 81 were down-regulated (Supplementary Fig. 4a, b and Supplementary data 2). When focusing on the set of genes showing the greatest level of change (Log2 fold change above 2 (Log2FC > 2)) at 6 hpi, we found a group of 62 genes specifically overexpressed after infection with ΔROP55 (Fig. 4b). Among them was a set of inflammatory mediators known to be up-regulated during inflammation in fibroblasts38. These genes included IL6, LIF, IL23 and CSF3 and their related transcription factors ELF3 and STAT4, as well as IL1A, IL1B and IL1RN (encoding the IL1 receptor antagonist), three important members of the IL1 family (IL1F). The complete list of DEGs is presented in the Supplementary data files, and are accessible through NCBI SRA (PRJNA881613). Importantly, the complementation of ∆ROP55 parasites broadly reverted the expression profile to that of the WT strain (Fig. 4a, Supplementary Fig. 4c), in strong support of the ROP55-dependent regulation of host gene expression.

a Heatmap of HFFs infected with RH ∆ku80 parental strain (WT), ΔROP55 or complemented (compl.) strains for 6 h, or left non-infected (NI). b Volcano plot highlighting in black the genes that passed the cut off of a differential expression higher than a log2 fold change of 2 and an adjusted p value lower than 0.05. Genes labeled in red are amongst the top 25 DEGs. c Bar plot representing selected list of biological processes enriched after gene ontology (GO) analysis for the upregulated genes with an adjusted p value lower than 0.05. The number of genes in each category is indicated after the bars, and the scale shows the weighted Fisher p value. d IPA analysis representing top upstream regulators of the overexpressed DEGs. Numbers after the bars indicate the activation z-score and the scale shows the Benjamini-Hochberg (BH) corrected p value. All data represents analysis at 6 hpi. Statistical test used for data analysis were for (b). Two-sided Wald test using DESeq2; BH-adjusted p < 0.05 and |log2FoldChange | > 2, (c) Two-sided Fisher’s exact test and weight01 algorithm using topGO; BH-adjusted p < 0.05, and for (d). Two-sided Fisher’s exact test and Z-score algorithm using IPA; BH-adjusted p < 0.05 and |Z-score | >2.
A Subsequent Gene Ontology (GO) enrichment analysis on the 283 specifically up-regulated genes at 6 hpi showed that the biological processes associated with ΔROP55 infection involved a range of stress-related responses, such as regulation of cell proliferation and the induction of immune responses (Fig. 4c, Supplementary data 3). Ingenuity Pathway Analysis (IPA) performed on the overexpressed genes revealed proinflammatory stimuli, such as pathogen/damage-associated molecular patterns (PAMPs/DAMPs) and TLR ligands as potential up-stream regulators of these cellular responses (TNF, LPS, IL1B, poly rI:rC-RNA (Fig. 4d)). All of these stimuli converge on NF-κB and MAPK signaling activation39, which are critical hubs of inflammatory responses and cell death programming. Accordingly, RELA (NF-κB p65 subunit), p38 MAPK, and ERK1/2 were also found in the IPA analysis (Fig. 4d).
Altogether, these findings indicate a potential role for ROP55 as a suppressor of proinflammatory pathways triggered during infection with the RH Toxoplasma strain.
ROP55 suppresses activation of the NF-κB/IL-1β signaling axis in fibroblasts
The inhibition of NF-κB activation is responsible for the repression of cellular inflammatory responses during infection by the RH Toxoplasma strain27,28,30,40. As transcriptomic analysis of ΔROP55- infected cells revealed the upregulation of several mediators of the NF-κB/IL-1β axis (Fig. 4), we hypothesized that ROP55 might control this signaling pathway. To validate the observed increase in IL1B expression (Fig. 4b), we conducted qPCR in cells infected for 6 and 24 h. A significant increase in IL1B mRNA was observed at 6 hpi that further increased at 24 hpi in ΔROP55-infected HFFs (Fig. 5a and Supplementary Fig. 5a-d). This resulted in increased levels of IL-1β detected in the cell supernatant by ELISA (Fig. 5b).

a Expression of IL1B measured by qRT-PCR in HFFs infected for 6 h and 24 h or non-infected cells (NI). Representative graph from 3 independent experiments. Values are reported as mean ± SEM (n = 3 technical replicates), replicated experiments are shown in Supplementary Fig. 5a–d. b IL-1β secretion from HFFs infected for 6 or 24 h measured by ELISA. Values are reported as mean ± SEM (n = 3 independent biological replicates). c Representative immunofluorescence image of NF-κB p65 subunit nuclear translocation (red) in HFFs infected for 2 h with the indicated strains. Intracellular parasites are staining with anti-SAG1 antibody (green), nuclei are staining with DAPI (blue). Quantification of p65 nuclear signal intensity in HFFs infected for 2 (d) and 6hpi (e). Horizontal line represents the mean fluorescence of p65 staining in cells infected with the WT strain. (d) n = 210 nuclei in WT infected cells, 227 in ΔROP55, 227 in complemented and 205 in Pru. (e) nuclei n = 204 in WT infected cells 217 in ΔROP55, 194 in complemented and 228 in Pru. Representative violin plots from 2 independent experiments, replicated experiments are shown in Supplementary Fig. 5f, g. f Immunofluorescence image of GFP expression in HEK 293 NF-κB reporter cells, infected for 24 h with the indicated strains. Intracellular parasites are stained with anti-GAP45 antibody (red), nuclei are staining with DAPI (blue). Scale bar represents 10 µm. Quantification of GFP intensity of HEK 293 NF-κB reporter cells infected for 6 h (g) or 24 h (h) with the indicated strains. Horizontal line represents the mean GFP fluorescence of cells infected with the WT strain. (g) n = 426 nuclei in WT, 408 in ΔROP55, 403 in complemented and 406 in Pru infected cells. h nuclei n = 394 in WT, 396 in ΔROP55, 408 in complemented and 398 in Pru infected cells. Representative graphs of 2 independent experiments. Replicated experiments are shown in Supplementary Fig. 5h. Statistical significance was determined using a two-way ANOVA followed by Tukey’s post hoc test. Source data are provided as a Source Data file.
Since NF-κB is the main transcriptional activator of IL-1β, we tested whether ROP55 might inhibit NF-κB activation. We infected HFFs and measured the nuclear translocation of the NF-κB p65 subunit by IFA at 2 and 6 hpi (Fig. 5c). The type II Pru strain, known to induce p65 NF-κB nuclear translocation in a GRA15II-dependent manner was used as a positive control26. A significant increase in p65 nuclear translocation was observed in cells infected with ΔROP55 (Fig. 5d, e and Supplementary Fig. 5e, f), supporting a model in which ROP55 inhibits NF-κB p65 activation shortly after invasion.
To test whether the NF-κB translocation was coupled with a downstream transcriptional activity, we infected the human embryonic kidney HEK293 reporter cell line that expresses GFP under the control of NF-κB responsive elements26 and quantified the GFP expression at 6 and 24 hpi (Fig. 5f). Negligible levels of GFP fluorescence were observed following infection with the WT or complemented strains at both timepoints (Fig. 5g, h and Supplementary Fig. 5g). In contrast, reporter cells infected with ∆ROP55 displayed sustained GFP expression, corroborating the p65 nuclear translocation observed in HFFs.
Overall, these findings suggest a regulatory role of ROP55 in controlling NF-κB activation and the subsequent proinflammatory response in human fibroblasts.
ROP55 function is conserved in human and murine immune cells
We next sought to determine if ROP55 also plays a role in controlling host cell death and inflammation in immune cells. Consistent with the findings in human fibroblasts, the absence of ROP55 induced significant lytic cell death of primary murine bone-marrow-derived macrophages (mBMDMs) (Fig. 6a. and Supplementary Fig. 6a). This was accompanied by a decrease in parasite load (Fig. 6b and Supplementary Fig. 6b–d), that was not due to a defect of invasion, since ∆ROP55 invaded mBMDM at the same level as WT parasites (Fig. 6c and Supplementary Fig. 6e). Furthermore, a significant increase in the nuclear translocation of p65 NF-κB subunit was observed upon infection of mBMDMs with ΔROP55 parasites (Fig. 6d, e and Supplementary Fig. 6f, g).

a Quantification of LDH released into the medium of mBMDM at 2, 6 or 24 hpi. Representative graph of 2 independent experiments. Values were normalized to 100% of lysis control and are reported as mean ± SEM of 3 technical replicates. Replicated experiments are shown in Supplementary Fig. 6a. b Expression of TgGADPH in mBMDM at 3, 6 or 24 hpi measured by qRT-PCR. Representative graph of 4 independent experiments. Bars represent means ± SEM of three technical replicates. Replicated experiments are shown in Supplementary Fig. 6b, c, d. c Percentage of invaded mBMDM quantified after 1 h. Values are normalized relative to the percentage of cells invaded by the control strain and are reported as mean ± SEM of 3 technical replicates. Representative graph of 2 independent experiments. Replicated experiments are shown in Supplementary Fig. 6e. Quantification of p65 nuclear signal intensity in mBMDMs infected for 2 h (d) and 6 h. (e). Representative violin plots from 2 independent experiments quantifying in (d) 310 nuclei for WT and 336 for ΔROP55 infected BMDM, and in (e) n = 221 nuclei for WT and 201 for ΔROP55. Horizontal line represents the mean fluorescence of p65 staining in cells infected with the WT strain. Replicated experiments are shown in Supplementary Fig. 6f, g. Statistical significance was determined using one-way ANOVA for (c, d, e) and two-way ANOVA for (a, b). followed by Tukey’s post hoc test. Source data are provided as a Source Data file.
As the effectors shaping immune responses against Toxoplasma might differ between murine and human hosts, we also evaluate ΔROP55 infection in human immune cells. Monocytes play a central role in initiating innate defenses against Toxoplasma41,42 and the THP-1 cell line recapitulates many features of primary human monocytes. ΔROP55-infected THP-1 monocytes also displayed increased mortality, as assessed by LDH release (Fig. 7a and Supplementary Fig. 7a, b) and PI incorporation (Supplementary Fig. 7c). Whereas THP-1 were equally infected by WT and ΔROP55 parasites at 2 hpi, significantly reduced parasite load was observed at 24 hpi in ΔROP55-infected cells (Fig. 7b and Supplementary Fig. 7d, e). Accordingly, at 16 hpi, we observed a higher proportion of PI positive cells infected with CFSE-labeled ΔROP55 parasites versus CFSE-labeled WT or complemented parasites (Supplementary Fig. 7c). Since CFSE-parasites enables to discern infected from uninfected cells, we observed that a high proportion of non-infected cells were also dying in ΔROP55 THP-1 cultures (Supplementary Fig. 7c).

a LDH released into the medium of THP-1 cells at 2 or 6 hpi or non-infected cells (NI). Representative graph of 3 independent experiments. Values normalized against 100% of lysis control showing means ± SEM of three technical replicates. Replicated experiments shown in Supplementary Fig. 7a, b. b TgSAG1 levels measured by qPCR in THP-1 at 2 and 24hpi. Bars represent means ± SEM of three technical replicates. Replicated experiments shown in Supplementary Fig. 7c, d. c Immunofluorescence assay showing NF-κB p65 nuclear translocation (red) in THP-1 infected for 2 h with WT, ΔROP55 and complemented parasites stained with anti-SAG1 antibody (green). Nuclei are labeled with DAPI (blue). d Quantification of p65 nuclear intensity in THP-1 from (c) counting 119 nuclei per condition. Representative violin plot from 2 independent experiments. Replicated experiment is shown in Supplementary Fig. 7f. e WB of IκBα levels in THP-1 lysates 16 hpi or non-infected (NI). f Densitometry quantification of IκBα bands observed in (c). Arbitrary units obtained after normalization to the βactin signal of each corresponding line. Pool of 3 independent experiments. g Expression of hIL1B in THP-1 at 2 and 6 hpi measured by qRT-PCR. Representative graph of 3 independent experiments. Bars represent means ± SEM of three technical replicates. Replicated experiments shown in Supplementary Fig. 7g, h. h ELISA measuring IL-1β secretion from supernatants of THP-1 after 2 and 6 hpi. Pooled results of 3 independent experiments. Bars represent means ± SEM of three biological replicates. i WB analysis mature (IL-1β) and immature (pro-IL-1β) forms of IL-1β present in cell lysates (cells) or supernatants of THP-1 cells infected for 6 h or left non-infected (NI). As a control, THP-1 were stimulated with LPS for 6 h, and ATP was added the last 30 min before harvesting. Loading control: human HSC70, representative result of three independent experiments. Statistical significance was determined using one-way ANOVA for (d, f) and two-way ANOVA (a, b, g, h). followed by Tukey’s or Sidak’s (b) post hoc test. Source data are provided as a Source Data file.
As previously described23,26, nuclear translocation of the p65 subunit of NF-κB was not detected in THP-1 cells infected with WT RH parasites (Fig. 7c, d)23,26. In contrast, and consistent with our findings in human fibroblasts and murine mBMDMs, unstimulated THP-1 monocytes infected with ΔROP55 parasites displayed a significant increase in the nuclear intensity of p65 NF-κB (Fig. 7c, d and Supplementary Fig. 7f). In the canonical NF-κB signaling pathway, p65 is maintained in the cytoplasm by the inhibitor of NF-kB, IκBα, and activation of the pathway results in ubiquitination and proteasomal degradation of IκBα, releasing p65 to enter the nucleus. Consistent with the increase in p65 nuclear translocation, we observed a significant reduction in IκBα protein levels in ΔROP55-infected THP-1 (Fig. 7e, f). Consequently, the induction of IL-1β transcripts and the release of IL-1β protein in the culture media were observed in ΔROP55-infected THP-1 (Fig. 7g, h and Supplementary Fig. 7g, h). Furthermore, we detected both immature and cleaved forms of the cytokine in the cellular lysates as well as in the supernatants of ΔROP55- infected THP-1 (Fig. 7i).
Collectively, these data indicate that infection of THP-1 monocytes with ΔROP55 parasites induces an early NF-κB signaling response that activates IL-1β transcription, maturation and secretion, coupled with lytic cell death. These observations suggest a role for ROP55 in controlling upstream NF-κB signaling to escape host clearance by inflammatory cell death.
We noticed that ∆ROP55 parasites cultured continuously for several months showed a consistent decline in their ability to induce IL-1β secretion in THP-1 cells, eventually reaching almost undetectable levels of the cytokine (Supplementary Fig. 7i). However, the increased lytic cell death and NF-κB translocation in cells infected with ∆ROP55 parasites were sustained over the time (Supplementary Fig. 7j, k), indicating that the mutant line was able to adapt in vitro to avoid specifically the induction of IL-1β. Given this adaptation, for all the following experiments described here, unless otherwise stated, freshly thawed ∆ROP55 parasites capable of inducing IL-1β secretion were utilized.
Activation of the NLRP3 inflammasome and pyroptotic cell death in THP-1 cells infected by ΔROP55 parasites
IL-1β production is regulated by the NLRP3 inflammasome in human monocytes infected by Prugniaud strain Toxoplasma24,25. To assess the involvement of the NLRP3 inflammasome during ∆ROP55 infection, we first assessed inflammasome priming by evaluating the transcriptional upregulation of NLRP3 in THP-1 cells. While the transcript levels of NLRP3 remained low in non-infected cells and those infected with control parasites, ∆ROP55-infected THP-1 cells showed increased NLRP3 transcripts (Fig. 8a and Supplementary Fig. 8a, b). Then, to test the requirement of NLRP3 inflammasome activation for IL-1β secretion, we pre-treated THP-1 cells with MCC950, a specific NLRP3 inhibitor that prevents inflammasome oligomerization and activation43. In THP-1 infected with ΔROP55 parasites, MCC950 treatment resulted in decreased IL-1β secretion (Fig. 8b). As a control, THP-1 were treated with canonical NLRP3 inflammasome activators LPS and ATP, and MCC950 also reduced IL-1β release in these conditions. Since the classical NLRP3 inflammasome recruits caspase-1 to process pro-IL-1β into mature IL-1β11, we evaluated the levels of IL-1β secretion in caspase-1 knockout THP-1 cells (ΔCASP1)44 and observed significantly reduced IL-1β secretion (Fig. 8c). These data show the involvement of the canonical NLRP3 inflammasome pathway for IL-1β production in response to infection with ΔROP55 parasites. This also provide evidence for the involvement of ROP55 in hindering the initial cascade leading to NLRP3 priming in THP-1 cells infected with Toxoplasma.

a Expression of NLRP3 measured by qRT-PCR in THP-1 cells infected for 2 and 6 h or non-infected cells (NI). Representative graphs of 3 independent experiments. Bars represent means ± SEM of three technical replicates. Replicated experiments are shown in Supplementary Fig. 8a, b. b Quantification by ELISA of IL-1β secreted in the culture supernatants of THP-1 cells pretreated with 5 µM MCC950 or DMSO 40 min before infection with the corresponding strains or left non-infected (NI). Pooled results of 2 independent experiments each one including 3 technical replicates. Bars represent means. c Quantification by ELISA of IL-1β secreted in the media of Δcaspase1 THP-1 cells (ΔCASP1) at 2hpi with the respective strains. Pooled results of 3 independent experiments. Bars represent means ± SEM three biological replicates, each with 2 technical replicates. d Western blot showing the full length-GSDMD (FL-GSDMD) and N-term-GSDMD (N-GSDMD) forms of gasdermin D in the cell lysates of THP-1 cells infected for 14 h with the respective strains. β-actin was used as loading control n = 4 experiments. e LDH released of Δcaspase1 THP-1 cells (ΔCASP1) 2hpi with the respective strains. Values represent means ± SEM of three technical replicates from one representative experiment. Replicated experiment in Supplementary Fig. 8d. f Fold change increase in PI positive cells after infection with the respective strains. Values of infected THP-1 WT and ΔGSDMD THP-1 cells (ΔGSDMD) were normalized to non-infected cells. Values are reported as mean ± SEM of 4 biological replicates. Statistical significance was determined using two-way ANOVA for (a, c, f), and one-way ANOVA for (e) followed by Tukey’s or Sidak post hoc for (c, f). Source data are provided as a Source Data file.
To next address if NLRP3 inflammasome activation induced pyroptotic cell death upon infection with ΔROP55 parasites, we analyzed Gasdermin D (GSDMD) cleavage, which is a hallmark of pyroptosis. We detected the 30 kDa cleaved N-terminal fragment of GSDMD only in cells infected with ∆ROP55 (Fig. 8d and Supplementary Fig. 8c). We then compared the mortality of ΔCASP1 and ΔGSDMD25 THP-1 against wild-type THP-1 cells upon infection with ∆ROP55. Interestingly, a significant but only partial reduction in mortality was observed in these two pyroptosis-deficient cell lines (Fig. 8e and f and Supplementary Fig. 8d). These findings indicate that ∆ROP55-infected THP-1 are dying, in part, through GSDMD-dependent pyroptosis. On the other hand, these results also highlight the triggering of a pyroptosis-independent cell death mechanism in THP-1 cells infected with parasites lacking ROP55.
The absence of ROP55 triggers necroptosis of infected cells
Since only partial rescue of the THP-1 cell death was observed in absence of caspase 1 or GSDMD, these findings suggested the activation of at least two different death programs in cells infected with ∆ROP55. We thus explored alternative cell death pathways that could account for the residual mortality not associated with the pyroptotic cascade. The subsequent experiments were performed with adapted-ΔROP55 parasites that did not trigger IL-1β induction in THP-1 cells, but that still induced lytic host cell death.
GSDME is another pore forming gasdermin involved in a secondary lytic death pathway that is activated by apoptotic caspases. In this process, named secondary necrosis45, caspase-3 is responsible for GSDME cleavage, releasing its N-terminal fragment and causing pore formation and loss of membrane integrity in a similar manner to GSDMD. To test if secondary necrosis occurs upon infection with ∆ROP55, we evaluated the activation state of caspase-3 using an intrinsically non-fluorescent probe, that produces a fluorogenic response upon caspase-3 and caspase-7 cleavage. As positive control of caspase-3/7 cleavage, THP-1 cells were treated with staurosporine (Fig. 9a). A slight but not significant increase in caspase-3/7 activity was observed 2 hpi with ΔROP55 (Fig. 9a). This result prompted us to investigate the pore forming activity of GSDME. However, we did not detect any GSDME cleaved N-terminal fragment under infection with ΔROP55 or control parasites (Fig. 9b). This was consistent with the fact that GSDME knockout THP-1 cells did not display a reduction in the mortality after infection with ΔROP55 (Fig. 9c), excluding secondary necrosis as a death mechanism in ΔROP55-infected cells.

a Percentage of Caspase 3/7 positive THP-1 cells infected for 2 or non-infected cells (NI), Staurosporine (Stau). Pooled results of 4 independent experiments. Bars represent means ± SEM. b Western blot showing the full length-GSDME (FL-GSDME) 55 kDa and N-term-GSDME (N-GSDME) 30 kDa forms in the cell lysates of THP-1 16hpi. β-actin as loading control. NS: nonspecific band at 25 kDa. One representative gel of 3 independent experiments, (quantification in Supplementary Fig. 8d). c Fold change in PI positive cells after infection with the respective strains. Values of infected THP-1 WT and ΔGSDME THP-1 cells (ΔGSDME) were normalized to non-infected cells and are reported as mean ± SEM of 3 biological replicates. LDH released of THP-1 cells pretreated with 40 µM Nec1, 5 µM GSK872, 40 µM Necrosulfonamide or DMSO 45 min before 2 hpi (d) or 6 hpi (e) with the corresponding strains or left non-infected (NI). Representative graph of 3 independent experiments. Values were normalized against 100% of lysis control and represent means ± SEM of three technical replicates. Replicated experiments are shown in Supplementary Fig. 9a–d. f Western blot of THP-1 WT (left side lines) and ΔCASP1 THP-1(right side lines) cell lysates 14 hpi showing full length pro-caspase8 (FL) and p43/41 and p18 subunits after TNFα and ciclohexamide (CHX) treatment. One representative of 3 independent experiments. g Fold change in PI positive hMDM 18hpi with the respective strains. Data were normalized to non-infected cells. hMDM were pretreated or not with 10 µM necrosulfonamide 40 min before infection. Values are reported as mean ± SEM of 3 biological replicates using primary hMDM from two different donors. Statistical significance was determined using one-way ANOVA for (a) and two-way ANOVA (c, d, e, g) or Sidak post hoc for (b, g). Source data are provided as a Source Data file.
We next investigated the occurrence of necroptosis, which along with pyroptosis, is a major mechanism of defense against intracellular pathogens. For this, we performed pharmacological inhibition of RIPK1 and RIPK3, the two kinases involved in necroptotic pathways14, as well as MLKL, which is a critical substrate of RIPK3 for necroptosis execution. THP-1 cells were pre-incubated either with Nec1, an allosteric inhibitor of RIPK146; GSK872, a competitive inhibitor of RIPK347; or Necrosulfonamide48, an inhibitor of MLKL polymerization. The cells were then infected or left untreated and the mortality was assessed by measuring LDH release after 2h and 6 h. Whereas RIPK1 inhibition had no major effect on the mortality of ΔROP55-infected THP-1, RIPK3 and MLKL inhibition significantly decreased the percentage of LDH release from ΔROP55-infected THP-1 (Fig. 9d, e, Supplementary Fig. 9a–d). A requirement for the triggering of the necroptotic cascade is the lack of caspase-8 activity19. We thus assessed the activation state of caspase-8, by evaluating its auto-proteolysis into p18 caspase-8 by western blot. As control for caspase-8 processing, THP-1 cells were treated with TNF-α and cyclohexamide, resulting in generation of the p18 subunit. In ΔROP55-infected THP-1, only the 58 kDa form of pro-caspase 8 was detected (Fig. 9f, left side lanes), consistent with uncleaved caspase-8 promoting necroptosis14. In addition, we found that in ΔROP55-infected ΔCASP-1 THP-1 cells, caspase-8 is also not active (Fig. 9f, right side lanes). ΔCASP-1 THP-1 also display a rescue of ΔROP55-induced mortality under RIPK3 and MLKL inhibition, reinforcing the involvement of necroptosis in pyroptosis-deficient cells (Supplementary Fig. 9e).
In order to evaluate if ΔROP55 infection also triggers necroptosis in human primary immune cells, we isolated monocytes from healthy adult donor peripheral blood mononuclear cells (PBMCs), and differentiated them into macrophages. We found that infection of human monocyte-derived macrophages (hMDM) with ΔROP55 also induced death of host cells at 18 hpi (Fig. 9g). Furthermore ΔROP55-induced hMDM mortality was rescued by inhibiting the necroptotic executioner pseudokinase MLKL with necrosulfonamide (Fig. 9g). In mBMDM, RIPK3 inhibition also suppressed ΔROP55-induced mortality at 2 hpi (Supplementary Fig. 9f), whereas RIPK1 inhibition did not induce changes at this time point. At 6hpi, a significant reduction in mBMDM mortality was detected with both RIPK1 and RIPK3 inhibitors (Supplementary Fig. 9g). Besides, we observed that the vast majority of nuclei of mBMDM infected with ∆ROP55 appeared swollen or disrupted at 6hpi and this phenotype was rescued under the treatment with necroptosis inhibitors (Supplementary Fig. 9h).
Overall, these results show a conserved role for ROP55 in the subversion of a RIPK3-dependent cell death pathway in mouse and human cells.
Discussion
During infection with intracellular pathogens, regulated cell death pathways are key components of innate immunity that sense danger signals and activate effector mechanisms critical for the subsequent establishment of adaptative immunity9. As a result of successful coevolution with the host, several intracellular pathogens have developed diverse strategies to suppress the activation of cell death pathways10, and Toxoplasma is not an exception. Both, avirulent and highly virulent Toxoplasma strains have been shown able to block apoptosis49, pyroptosis25 and necroptosis20. By these means, the parasite maintains its replicative niche, which is essential for intracellular survival, host propagation and chronic persistence. However, how Toxoplasma circumvents inflammatory cell death, and how the virulent RH strain counteracts the activation of NF-kB, the central transcription factor involved in all these responses, remains poorly characterized. In the present study we have identified a new rhoptry protein, ROP55, that, when removed, induces significant NF-kB activation and lytic death of infected host cells. Consequently, a decrease of parasite load is observed in cells infected with ROP55-deficient parasites, supporting a role for ROP55 in the maintenance of the parasite niche. Importantly, ROP55 appears to be one of the major virulence factors required for the establishment of acute toxoplasmosis in mice.
Transcriptomic analysis of HFFs infected with ΔROP55 parasites revealed an increased inflammatory response, that we further characterized in THP-1 monocytes and found that priming of the NLRP3 inflammasome was responsible for IL-1β production and secretion. Previous studies using the RH strain have suggested the existence of parasite factors able to block NF-κB signaling and host cell inflammatory responses. These were based on the fact that: (i) infection of macrophages, monocytes, neutrophils and fibroblasts by RH tachyzoites does not result in the activation of NF-κB27,28,29,30, and (ii) the RH strain is able to block NLRP3 inflammasome activation and IL-1β production during activation of human peripheral blood neutrophils with LPS27, by suppressing the activation of NF-κB signaling. This suppression was also observed in macrophages and fibroblasts28,30; however, the parasite effectors involved in this effect were unknown. Here, we show that upon infection with RH parasites deficient in ROP55, this repression is released in three human cell lines (HFF, THP-1 and HEK293) as well as in primary mouse macrophages. ROP55 acts during the first two hours after infection, which suggests an early role for ROP55 in preventing the transduction of parasite sensing through NF-κB signaling, potentially hindering NLRP3 priming. It is worth noting that ROP55 effects were observed in the absence of exogenous IFN-γ or LPS stimulation, indicating that the triggers of these responses arise from molecular patterns directly linked to Toxoplasma infection.
In contrast to RH, the Type II Pru strain constitutively activates IL-1β expression and release from infected cells, but, without inducing cell death. Two counterbalancing mechanisms for these inflammatory responses arise from dense granule effectors: (i) TEEGR50, that regulates EZH2 to antagonize NF-κB-induced cytokines; and (ii) MAG1 that inhibits IL-1β induction51. Here, we show, in RH parasites, another level of control involving the rhoptries. Dense granule and rhoptry effectors might cooperate to modulate host inflammation and cell death. Lima et al.27 used mycalolide B to block parasite invasion and dense granule release, but not rhoptry secretion in neutrophils, showing that rhoptry secretion alone doesn’t suppress LPS-induced IL-1β production. Unlike Lima’s study, we used different cell types without LPS stimulation. Toxoplasma PAMPs may activate different pathways than LPS for IL-1β production. Further studies will be necessary to address whether ROP55 may have additive contribution or synergize with dense granule effectors in different Toxoplasma strains.
ΔROP55 infection induced pyroptotic lytic death of THP-1 monocytes that was significantly dependent on caspase-1 and on the executioner protein GSDMD. However, approximately 50% of cells deficient in either caspase-1 or GSDMD survived infection with ΔROP55 parasites, suggesting that an additional, pyroptosis-independent mechanism of death occurs. We then assessed apoptotic caspases and necroptosis, which are also known to play a role during Toxoplasma infection, particularly in IFN-γ-activated cells20,52. A crosstalk between cell death programs has been documented10,53, showing that they do not operate in a mutually exclusive manner, but rather, they are interconnected signaling processes. Although we found a slight but not significant increase in caspase-3/7 activation, our data demonstrate that secondary necrosis was not occurring in ΔROP55-infected THP-1 cells. Using pharmacological inhibition, we demonstrated that RIPK3- and MLKL-dependent necroptosis was responsible for the death of cells infected with ΔROP55 parasites. This was demonstrated in human THP-1 monocytes as well as in primary human and mouse macrophages. The enzymatic activity of RIPK1 was not involved in ΔROP55-induced necroptosis in THP-1 cells at either time point, nor in mBMDMs at 2 hpi. Interestingly, at a later time point (6 hpi), RIPK1 inhibition significantly reduced the mortality of mBMDMs. A role for RIPK1 in THP-1 cells or in mBMDMs at 2 hpi cannot be excluded, because Nec1 does not inhibit RIPK1-scaffolding function, which is important contributing to NF-κB signal transduction14. Altogether these results suggest that ROP55 may interfere with a necroptotic signaling cascade, which eventually intersects with the NLRP3 priming pathway through NF-κB transduction, protecting infected cells from both necroptosis and pyroptosis.
We were surprised to observe that ∆ROP55 parasites cultured for several months consistently showed a gradual decline in their ability to induce IL-1β secretion in cells, eventually reaching undetectable levels of the cytokine. In this adapted-∆ROP55 mutant, NF-κB signal transduction was maintained. Interestingly, NLRP3 transcriptional induction was lost. These findings suggest that the mutant adapted by blocking the initial inflammasome priming signal to prevent inflammasome-induced pyroptosis. This also implies that the signal initiating inflammasome activation may differ from the signal triggering necroptosis, because necroptosis was maintained in the adapted-∆ROP55 mutant. It might be possible that, due to the pressure from the inflammatory environment in the culture, ∆ROP55 parasites adapted by downregulating effectors that prime the inflammasome response. How the ∆ROP55 parasites adapted remains an open question, which in the future might help to understand the cross-talk between necroptosis and pyroptosis and the characterization of potential new parasite cell death and inflammasome regulators.
Preventing both necroptosis and NF-κB-dependent inflammatory signaling is not unique to Toxoplasma. Viruses and pathogenic bacteria counteract these cellular responses as a strategy to maintain their replicative niche54,55. RIPK3-dependent necroptosis has been largely characterized as an antiviral defense mechanism triggered by TLR316 and ZBP1 sensors56 after detection of viral genomes. In these pathways, RIPK3 activation induces both necroptotic cell death and NF-κB mediated cytokine production15,57. Interestingly, RIPK1-scaffolding activity is crucial for mediating NF-κB activation in RIPK3-dependent necroptosis16,17,58 whereas its kinase activity is dispensable57. One fascinating viral protein subverting both necroptosis59 and inflammatory signaling is the mouse cytomegalovirus M45 protein. This protein interacts with RIP homotypic interaction motifs (RHIM) present in RIPKs and in ZBP1, and induces degradation of NF-κB essential modulator (NEMO)60 of infected cells. As a result, M45 blocks both TLR and IL-1R signaling pathways, which are initiated by two different triggers that eventually converge to induce NF-κB translocation.
Few studies have examined necroptosis as a defense against Toxoplasma. The dense granule proteins TgNSM and TgIST block interferon-regulated necroptotic gene expression20,61, preserving intracellular cysts, but their role in tachyzoite survival is unstudied, and no clear evidence shows that necroptosis kills tachyzoites or controls acute infection. For example, RIPK3-deficient macrophages do not affect Toxoplasma survival. RIPK3-knockout mice resist intraperitoneal infection normally but show better survival in oral infections due to increased immune cell infiltration and edema in the intestinal lining, not to reduced parasite numbers62. Additionally, Mlkl−/− mice are similar susceptibility to oral Toxoplasma infection suggesting that RIPK3 impact on host survival may be due to its pyroptotic, not necroptotic, activity62. Using intraperitoneal infection of RH ΔROP55 parasites, we found improved survival of mice, which is correlated with a decrease in parasite burden in the spleen at day 4 post-infection. Studies examining the infection of Ripk3−/− or Mlkl−/− mice by ΔROP55 parasites are still needed to evaluate the contribution of RIPK3-dependent necroptotic cell death to the loss of ΔROP55 virulence in vivo.
The sensors for RIPK3 activation upon ΔROP55 infection are unknown. Interestingly, Influenza A virus is sensed by ZBP1 and this can induce RIPK3 necroptosis within the nucleus, resulting in nuclear envelope disruption63, a feature that might be reminiscent of the nuclei morphology in mBMDMs infected by ΔROP55. It has been proposed that ZBP1 can assist host control during Toxoplasma infection in vitro and in vivo, because Zbp1−/− mice had increased parasitemia and inflammatory cytokines compared to wild-type mice64. There is no direct evidence of Toxoplasma sensing by ZBP1; however, it has been shown that ZBP1 can also bind endogenous nucleic acids, acting as a sensor for excessive transcription states generated during infection65,66. The detection of Toxoplasma DNA through AIM2 is involved in atypical apoptosis mediated by caspase-8 in IFN-γ-primed macrophages52. It is worth noting that this and previously reported cell death mechanisms upon infection with Toxoplasma20,67 differ greatly from the death triggered by ΔROP55 in naive HFF, mBMDM, hMDM and undifferentiated THP-1 monocytes. The lytic cell death observed upon ΔROP55 infection is unique in the sense that it is observed in the absence of exogenous stimulation, owing to the detection of Toxoplasma molecular patterns or/and the sensing of danger signals coming directly from the infection. Although direct evidence for ROP55 inhibition of NF-kB is not show in this study, its potential to modulate host cell signaling and viability, warrants further investigation. ROP55 may hypothetically interfere with host proteins within NF-kB pathway or may also interact with parasite effectors to suppress effector-triggered immunity.
ROP55 is crucial in orchestrating an early repression mechanism that allows the escape of lytic cell death and ensures parasite survival within the host. The lytic cell death was observed in two human cell lines and in primary mouse and human macrophages, revealing the control of conserved pathways in different cell types and species by ROP55. Among the Toxoplasma mutants for GRAs and ROPs effectors that have been characterized, ΔROP55 appears to be one of the most attenuated parasites in a mouse model of acute toxoplasmosis. Our work reveals a previously unappreciated rhoptry function in the complex mechanisms developed by Toxoplasma to escape inflammatory cell death and opens new avenues to understand how this parasite avoids elimination by lytic host cell-death.
Methods
Ethics statement
All mice protocols were approved by the Institutional Animal Care and Utilization Committee (IACUC) of the American University of Beirut (AUB). Animals were obtained from Jackson laboratory USA, and housed in specific pathogen free facility with a 12 h ON/OFF light cycle, 50–60% humidity and 21–24 °C ambient temperature. Humane endpoints were fully respected as per AUB IACUC following AAALAC (Association for Assessment and Accreditation of Laboratory Animal Care International) guidelines and guide of animal care use book (Guide, NRC 2011). Mice were monitored on a daily basis. To verify the acute phase of the infection, blood was drawn following anesthesia with isoflurane by inhalation, and sera were probed against tachyzoite extracts by Western blot. Mice were sacrificed if any abnormal health features were noticed. Animals were anesthetized before cervical dislocation.
Cell lines and maintenance and treatments
Human Foreskin Fibroblasts (HFF) (ATCC CRL 1634) and HEK 293 NF-κB reporter cell line (System Biosciences, Cat. # TR860A-1), were cultured in Dulbecco’s Modified Eagle’s Medium (Gibco) supplemented with 5% or 10% (v/v) of heat inactivated Fetal Bovine Serum (FBS), 2 mM Glutamine (Gibco) and 50 µg/mL Penicillin/Streptomycin at 37 °C with 5% CO2. THP-1 WT, ΔGSDMD THP-125 Δcaspase-1 THP-1, and ΔGSDME THP-1 cells44 were cultured in RPMI 1640 (Gibco) supplemented with 100 U/mL penicillin, 100 ug/mL streptomycin, 2 mM L-glutamine and 10% (v/v) heat inactivated FBS and 1 µg/ml of puromycin. THP-1 cells were treated with a final concentration of 5 µM MCC950 (Sigma, 5381200001) as described by Gov et al.24. Mouse BMDM were isolated from BALB/c mice tibias and differentiated for 7 days following standard procedures68.
Type I RH parasite strains lacking the ku80 gene (RH Δku80) were used for all experiments unless stated otherwise and routinely passed in HFF cells. Type II Prugniaud (Pru) were cultured in the same conditions as RH parasites.
Human monocyte-derived macrophage differentiation
Human whole blood was obtained from healthy adult donors at the University of California, under the auspices of the Institute for Clinical and Translational Science, with donors providing written informed consent. The collection followed the ethical guidelines approved by the Institutional Review Board at the University of California, Irvine. Peripheral blood mononuclear cells (PBMCs) were separated from the whole blood using density gradient centrifugation with lymphocyte separation media (MP Biomedicals, Santa Ana, CA). Monocytes were subsequently isolated from the PBMCs through counterflow elutriation, as previously described69, and assessed for purity after each isolation. Typically, this method yielded highly pure monocyte cultures (resulted in >93% pure monocytes), based on CD11b+ and CD3- CD20- CD56- marker staining. After isolation, monocytes were differentiated into macrophages using 100 ng/ml recombinant human macrophage colony-stimulating factor (M-CSF; Gemini Bio-Products, West Sacramento, CA) over 6 to 7 days. Fresh media with M-CSF was supplemented on day 3. On days 6 to 7, the monocyte-derived macrophages were replated and allowed 4 h to adhere before being used in infection experiments with a multiplicity of infection (MOI) of 2.
Parasite transfections
For each transfection 20 × 106 freshly lysed parasites were electroporated at 2.02 kV, 50Ω, 25μF in an Electro Cell Manipulator 630 (BTX) with 5 μg of PCR products obtained with the KOD polymerase in addition to 30 μg CRISPR/Cas9 plasmid or with the linearized pLIC-HA3-CAT plasmid. Transgenic parasites were selected with 1 μM pyrimethamine, 20 μM Chloramphenicol or 5 μM FUDR. Clones were isolated through limiting dilution and correct integrations were verified by diagnostic PCR.
Plasmid constructs
The HA3-tagging at the endogenous locus of ROP55 was performed by single homologous recombination using the ligation-independent-cloning strategy. The last 2 Kb of the rop55 genomic sequence were amplified with primers ML2258 and ML2259 and inserted in frame with the sequence coding for HA3 tag in the vector pLIC-HA3-CAT. After linearization with SfuI and transfection, the integration of the construct at rop55 locus was verified with primers ML2363 and ML1476.
For the generation of ∆ROP55 line, two different RNA guides (gRNAs)s were cloned into the pU6-Cas9-YFP plasmid in order to target the 5ʹ and 3ʹ UTR of rop55 in the ROP55-HA3-tagged line. Briefly, gRNAs were generated by annealing primers ML2258/ML2259 and primers ML2831/ML2832, respectively. The annealed DNA fragments were cloned in the BsaI sites of vector pU6-Cas9 to obtain plasmids p5’ROP55 and p3’ROP55. A donor DNA fragment containing the DHFR resistance cassette flanked by 30 bp homology regions to the 5ʹ UTR and 3ʹ UTR of rop55 locus was amplified with primers ML2829 and ML2828 from pLIC-HA3DHFR vector. The double homologous recombination events were verified by PCR using primers ML2636 and ML2454 for 5’ integration and ML2601 and ML2830 for 3ʹ integration.
To complement the ∆ROP55 line, an extra copy of the rop55 gene, driven by its own promotor and fused with a C-terminal sequence encoding for a triple HA tag, was inserted at the uprt locus. Briefly, a 7 kb DNA fragment was synthetized containing the following sequences: 50 bp of homology region with the 5ʹUTR of the uprt locus, the rop55 promotor region (927 pb), the full cDNA sequence of rop55 fused to a triple HA tag sequence, the 3ʹUTR of the hxgprt gene and 50 bp of homology regions with the 3ʹUTR of the uprt locus. After cloning in pUC vector, digestion with PstI and NaeI, the 7 kb excised fragment was co-transfected with two gRNAs targeting the 5ʹand 3ʹUTR of the uprt locus. The plasmid guide p5’UPRT was constructed by annealing primers ML3445 and ML3446 and the plasmid guide p3’UPRT by annealing primers ML2087 and ML2088. The integration of the exogenous copy of rop55 at the uprt locus was verified with primers ML3187 and ML2081 and ML2384 and ML3190. Primer sequences are in Supplementary Table 1.
Plaque assay
Cells were seeded on 24-well plates and grown to 100% confluency and infected with 7000 tachyzoites per well. Four-fold dilutions were then performed from the top row to the next row. The plate was incubated at 37 °C, 5% CO2 and left untouched for 6 days. On day 6 post-infection, cells were fixed with 4% paraformaldehyde (PFA) (w/v) for 20 min, and stained with Diff-Quik dyes (Medion Diagnostics). Images of the lower row of the plate were acquired with an Olympus MVX10 stereo microscope. Lysis plaque size was measured using the Zen software, Blue edition (Zeiss). Graphs shows a representative assay out of 3 experiments.
Intracellular replication and number of vacuoles
Confluent cells on coverslips were infected with 2 × 105 freshly released parasites for one hour, and then the remaining extracellular parasites were washed away. 24 h later, cells were fixed in 4% PFA for 20 min, and immunofluorescence staining (IFA) of the inner membrane complex (using αGAP45) was used to count the number of parasites per vacuole and the number of vacuoles per field. The number of vacuoles containing either 2, 4, 6, 8 or 16 parasites were counted using a Leica DM2500, 100x oil objective NA = 1.4, microscope (Leica Biosystems), and expressed as percentage. 200 vacuoles/coverslips (n = 3 coverslips/experiment) were analyzed. Graphs show means of 6 independent experiments. For the number of vacuoles per fiel, 20 fields per coverslips were counted, in duplicates. Graphs show means of 2 independent experiments
Invasion
Confluent cells on coverslips were put on ice. Cells were infected with 3 × 106 freshly lysed parasites in 300 μl of complete medium. Parasites were let to set for 20 min with the plate on ice. The plate was then moved to a water bath at 38 °C for 5 min. After 5 min, invasion was blocked by fixing infected cells with 4% PFA (w/v) in HBSS, (Gibco). Differential IFA was then performed to distinguish intracellular from extracellular parasites. Not-permeabilized samples were saturated with HBSS 10% FBS and then probed with anti-SAG1 antibodies to stain extracellular parasites. Samples were then permeabilized with 0.1% saponin for 15 min and with anti-ROP1 antibodies to detect the PVM of intracellular parasites. Intracellular and extracellular parasites were counted in 20 fields per coverslips in triplicates. The values were expressed as the number of intracellular parasites per field and normalized to that of the control line (RH ΔKu80) arbitrarily fixed to 100%. Graphs shows the mean of 6 independent experiments, including three technical replicates each. For mBMDM 5 × 105 cells seeded on coverslips where infected with 1 × 106 tachyzoites for 1 h at 37 °C in triplicate for each strain. Not-permeabilized samples were probed with anti-SAG1 antibodies and then permeabilized with 0.2% Triton 15 min to stain extracellular parasites with anti-GAP45 antibodies. The values were expressed as the number of intracellular parasites per field and normalized to that of the control line (RH ΔKu80).
Egress
HFFs grown on coverslips in a 24-well plate were infected with 1 × 105 tachyzoites and washed after 2 h. Following ∼30 h of infection, parasite egress was stimulated with calcium ionophore A23187 (1 μM) for 8 min and then samples were fixed with 4% PFA and processed for IFA with an antibody recognizing the dense granule protein GRA3. Egress events (GRA3 released in the PV upon egress) were quantified by analysing 200 vacuoles per coverslip (n = 3 coverslips). Values were normalized to that of the control line (RH ΔKu80) arbitrarily fixed to 100%. Graphs show means of three independent experiments, including three technical replicates each.
Immunofluorescence analysis
To determine the localization of ROP55 in intracellular parasites, HFF cells were infected with ROP55-HA3 tachyzoites for 24 h and fixed with 100% cold methanol for 8 min at –20 °C. After three washes, infected cells were then blocked with PBS 10% FBS with Triton X-100 0.3%, incubated with primary antibody (anti-HA, anti-ROP55, anti-ROP2 antibodies) for 1 h in PBS 2% FBS plus Triton X-100 0.3%, washed three times and then incubated with secondary antibodies (Alexa Fluor 488 and 594- or 647-conjugated antibodies against mouse, rat or rabbit IgG, Molecular Probes). Coverslips were mounted in Immu-Mount (ThermoScientific). For intracellular replication and egress quantifications, cells were permeabilized with Triton X-100 0.1% in PBS for 10 min, staining was performed as described above.
Immunoblots
Infected HFF cells were trypsinised at the intended time-point, washed in PBS and the total number of cells was counted. The cell pellet was then resuspended in Laemmli buffer containing 10 % (v/v) of β-mercaptoethanol and protease inhibitors in order to have the same number of cells per mL. For the analysis of IL-1β in HFF cells, cells were grown in serum free media and 5 mL of supernatant were concentrated with Amicon filters (with a cut off of 10 kDa, Millipore) and then precipitated over-night with ice-cold acetone at –20 °C. For IL-1β immunoblot from THP-1 cells, samples were treated as described by Pandori et al.25. For GSDMD immunoblot, THP-1 cells were solubilized with 1% NP-40, and the insoluble fraction, was treated for 30 min with DNAse I in 1% NP-40 buffer and then subjected to SDS-PAGE and western blotting. A wet transfer was performed for 1 h at room-temperature to PVDF membranes. Membranes were blocked for 1 h at RT with 5% non-fat dry milk in TNT buffer (140 mM NaCl, 15 mM Tris, 0.05% Tween 20). The primary antibodies used were: rat anti-HA (3F10, Roche, 1:1000); mouse anti-ROP5; mouse anti-IL-1β (1:5000) (3ZD clone); rabbit anti-Gasdermin D (E8G3F Cell Signaling); mouse anti-Hsc70 (1:1000) (clone 1B5 from Stressgen); rabbit anti-DFNA5/GSDME EPR19859 N-terminal (ab215191 from abcam); rabbit anti-IκBα 44D4 (#4812 from Cell Signaling); mouse anti-caspase-8 1C12 (#9746 from Cell Signaling). After washes with TNT buffer, the membranes were incubated with alkaline phosphatase conjugated secondary antibodies or with horseradish peroxidase (HRP)-conjugated goat anti-mouse or anti rabbit antibodies (Jackson ImmunoResearch). Uncropped scans of the blots are in the Source Data file.
Quantitative real time-PCRs
At time of sample collection, cells were washed three times to remove extracellular parasites, centrifuged for 5 min at 1800 RPM, and the pellets were resuspended in RNA lysis buffer RA1 (NucleoSpin, Macherey-Nagel) containing 1% of β-mercaptoethanol. RNA extractions were performed according to the manufacturer’s protocol, which includes a DNase treatment to remove contaminating DNA (NucleoSpin, Macherey-Nagel). cDNA synthesis was done with a normalized quantity of RNA between all samples and performed according to manufacturer’s instructions (Superscript III First-Strand Synthesis kit (Invitrogen)). As controls, samples containing water or lacking the reverse transcriptase were used in the cDNA synthesis. cDNA was used as a template in qRT-PCR, performed in triplicates using the SensiFAST™ SYBR No-ROX Kit (Bioline) and a LightCycler 480 Real-Time PCR System (Roche). Primers were designed using the LightCycler Probe Design Software 2.0. The primers used are listed in Supplementary table 1. Data were analysed using the LightCycler 480 program with a relative quantification measurement (ΔCT), normalized to the expression of the housekeeping gene, mouse or human β-actin. For measuring parasitaemia, TgGAPDH or TgSAG1 values were normalized to the mouse or human β-actin of the samples infected with the control strain. Absence of amplification was confirmed in the negative controls with water and no reverse transcriptase. For absolute quantification of TgSAG1 from DNA extracted from infected THP-1 cells. A standard curve was set up with known concentrations of parasite DNA using automatic efficiency calculation from LightCycler® 480 Software, which also determine the concentration of target DNA in the sample using the second derivative maximum method.
LDH assay
Cell viability was determined with the Pierce LDH Cytotoxicity Assay according to manufacturer’s protocol (Thermo scientific). Cells were washed with PBS to remove any phenol red contamination from the culture medium, and infected with freshly lysed tachyzoites at a MOI = 2 in DMEM without phenol red (Gibco). At required time-points, the supernatants were harvested, LDH detection assay was performed, and 490 nm and 680 nm absorbances were measured with a plate reader (TECAN). Lysis Buffer-treated cells were considered as the maximum LDH activity control. Each value was normalized to that of the control arbitrarily fixed to 100%. Graphs shows the mean of three independent experiments, including three technical replicates each.
Propidium Iodide staining for microscopy analysis
To analyse cell death, confluent HFF cells were infected with freshly lysed parasites, the plate was centrifuged for 5 min at 1200 RPM, and cells were washed one hour later to remove extracellular parasites. One hour before the intended time-point, propidium iodide (Molecular probes, Life Technologies) at 10 μM and DAPI (1 µg/ml) was added to the wells and incubated for 1 h at 37 °C, and protected from light. Live imaging of infected cells was performed using an inverted Zeiss microscope and images were acquired with the ZEN software. At least 15 fields were quantified by counting the total number of cells and the nuclei staining positive for propidium iodide. The values were expressed as the % of PI positive cells per field. Graphs shows one independent experiment out of two, including two technical replicates each.
ELISA
Tachyzoites were washed in PBS before use in infection experiments, in order to avoid transferring any IL-1β from the parasite cultures. Confluent HFFs in 24 well plates were then infected with 5 × 105 freshly lysed tachyzoites. THP-1 cells were plated in 24-well plates at 5 × 105 cells/well and infected at a MOI = 2 or stimulated at the same time with 100 ng/mL of E. coli LPS (List Biological Laboratories, Campbell, CA) and with 5 mM ATP (Sigma-Aldrich, St. Louis, MO) added in the 30 min prior to harvest. Harvested supernatants were centrifuged for 5 min at 2000 RPM. ELISA was then performed according to manufacturer’s protocol (IL-1β ELISA Kit, Thermofisher). Absorbance at 450 nm was measured with the Magellan software in a plate reader (TECAN). A standard curve of known concentrations of human IL-1β was prepared in each experiment to convert the absorbance to IL-1β concentrations in pg/ml. Graphs shows the mean of three independent experiments, including three technical replicates each.
NF-κB activation measurements
For GFP reporter quantifications, glass coverslips were incubated in 24-well plates at 37 °C overnight with 20 µg/mL Poly-D lysine prepared in PBS. Reporter cells were then plated at a concentration of 5 × 105 into each well and 24 h later cells were infected with 1 million parasites/well. After 6 or 24 h post infection, coverslips were fixed with 4% PFA and probed whit anti-GAP45 antibody followed by Alexa Fluor 594-conjugated anti-rabbit IgG (Molecular Probes) to visualize infected cells. For quantification of NF-κB activation in HFFs, a confluent monolayer of cells grown on coverslips in 24-well plates were infected with 1 million parasites/well and then fixed and probed with anti NF-κB p65 (D14E12) Rabbit mAb (1:800) (Cell Signaling) and anti-SAG1 mouse antibodies (1:100) followed by Alexa Fluor 594-conjugated anti-rabbit IgG and Alexa Fluor 488 anti-mouse IgG (Molecular Probes). For quantification of NF-κB activation in THP-1 cells, cells were plated at a concentration of 5×105 in 24-well plates and infected with 1 million parasites/well. After 2 or 6 h of infection, the cells were fixed with 4% PFA and probed with antibodies as HFF cells in 96 round-bottomed well plates. Image acquisitions were performed with a Leica Thunder microscope and the mean intensity of the fluorescence of the red channel was measured for each nucleus using the quantification tool in the Leica Application Suite X program. The mean value of the fluorescence from at least 100 nuclei for each condition was plotted in GraphPad.
Flow cytometry
THP-1 cells were plated at 5×105 cells/well and infected with the indicated strain at a MOI = 2. Cells were harvested at 2 or 16 h after infection and resuspended in binding buffer (10 mM HEPES, 140 mM NaCl, and 2.5 mM CaCl2, pH 7.4). Propidium Iodide (Life Technologies, Eugene, OR) was used at 1 µg/mL per 100 µL of resuspended cells. Cells were stained for 15 min at room temperature, protected from light. 400 µL of buffer was then added to the cells. The cells were immediately analysed using a NovoCyte flow cytometer with NovoExpress software (Agilent) or a FACSCalibur flow cytometer with CellQuest Software (BD Biosciences). FCS files were analysed using FlowJo software (TreeStar, Ashland, OR). PI values were normalized to the non-infected controls considered as 1 for each cell line. For Caspase 3/7 activity assay, cells were labeled with the CellEvent™ probe C10423 from Invitrogen during 30 min according with the manufacturer instructions.
Total RNA isolation for RNA sequencing analysis
Total RNA from cell pellets was resuspended in RNA lysis buffer RA1 (NucleoSpin, Macherey-Nagel) containing 1% of β-mercaptoethanol and extracted according to the manufacturer’s protocol (NucleoSpin, Macherey-Nagel). To remove potential DNA contamination, RNA samples were treated twice with 1 U DNase I (Life Technologies) per 10 μg of RNA for 30 min at 37 °C, followed by inactivation of the DNase I enzyme.
mRNA isolation and cDNA synthesis
mRNA was isolated using the NEBNext Ultra II Directional RNA Library Prep Kit for Illumina (NEB #E7420L) per the manufacturer instructions. Briefly, 8 μl NEBNext first strand synthesis reaction buffer, 2 μl NEBNext primers, and 10 μl nuclease-free buffer, were mixed and placed in ice. 20 μl of NEBNext oligo d(T)-25 beads were washed with 100 μl NEBNext RNA binding buffer (2X) in a 1.5 ml microcentrifuge tube and placed on a magnetic rack at room temperature for 2 min. The supernatant was removed, and beads were washed with another 100 μl of RNA binding buffer (2X). The beads were resuspended in a volume of 50 μl RNA binding buffer (2X), transferred to 0.2 ml PCR tubes, and 50 μl of total RNA was added. To denature the RNA and facilitate binding to the beads, tubes were heated in a thermocycler at 65 °C for 5 min with the lid set to 75 °C. To assist further binding of mRNA, beads were resuspended, and samples let sit at room temperature for 5 min for two cycles. To remove contaminants, we placed the samples on the magnetic rack for 2 min and removed the supernatant and washed in 200 μl of RNA wash buffer twice. 50 μl of NEBNext Tris buffer was added and tubes were placed in a thermocycler at 80 °C for 2 min with the lid set to 90 °C, then cooled to 25 °C. The beads were mixed with 50 μl of RNA binding buffer (2X) at room temperature for 5 min. The supernatant was removed and 200 μl of NEBNext wash buffer was added to each tube. Samples were mixed, placed back on the rack for 2 min at room temperature, and the supernatant removed. After spinning down and removing any additional wash buffer, the RNA was eluted by adding 15.5 μl of the prepared first strand reaction buffer and incubating at 94 °C for 15 min. Using the magnet, 13.5 μl of supernatant from each sample was transferred to clean PCR tubes and placed on ice.
First strand cDNA was synthetized also using the NEBNext Ultra II Directional RNA Library Prep per the manufacturer instructions. RNA polymerase inhibitor, Actinomycin D, was dissolved in nuclease-free water to 0.1 μg/μl and 5 μl was added to each sample along with 0.5 μl murine RNAse inhibitor and 1 μl ProtoScript II reverse transcriptase. Samples were then incubated in a thermocycler at 25 °C for 10 min, 42 °C for 15 min, and 70 °C for 15 min. The Second strand was synthesized by adding 48 μl of nuclease-free water, 8 μl NEBNext second strand reaction buffer, and 4 μl of second strand synthesis enzyme to each sample, and incubating in a thermocycler for 1 h at 16 °C with the lid set to 40 °C. cDNA was then purified using 1.8 volumes (144 μl) of AMPure XP magnetic beads (ref# A63881) to cDNA samples and incubated at room temperature for 5 min. cDNA was separated using a magnet rack and the supernatant removed. The beads were washed twice with 500 μl of 80% ethanol, the supernatant removed, and beads were spun down one more time to remove any excess ethanol. Beads were dried for 5 min on the magnet and resuspended in 60 μl of 0.1X TE buffer and incubated at room temperature for 2 min. The supernatant was collected and transferred to a new PCR tube. Ends were repaired using 6.5 μl of NEBNext end repair reaction buffer (10X) and 3 μl of NEBnext end prep enzyme mix and incubated in a thermocycler at 20 °C for 30 min and at 65 °C for 30 min with the lid set to 75 °C.
Adapter ligation for next generation sequencing and PCR enrichment
Adapters were ligated using 15 μl of NEB blunt/TA ligase master mix, 1 μl of 1.5 μM NEBNext multiplexing Illumina adaptors, and 2.5 μl nuclease-free water to each sample. Samples were mixed and incubated at 20 °C for 15 min and an additional 16.5 μl of nuclease-free water added. The ligation reactions were purified on DNA beads using using 100 μl of AMPure beads and eluted in 52 μl of 0.1X TE buffer. 50 μl of supernatant was transferred to a new tube and purified again, this time using 50 μl of AMPure beads and eluted in 19 μl of 0.1X TE buffer. 17 μl of the supernatant was mixed with 10 μl of universal barcode/primer, 25 μl KAPA HiFi HotStart Ready Mix (2X), and 3 μl of NEBNext USER enzyme. cDNA was amplified in a thermocycler at 37 °C for 15 min, 12 cycles of 98 °C for 30 s, 55 °C for 10 s, and 62 °C for 75 s, then 62 °C for 5 min. cDNA was purified using 45 μl of AMPure beads and eluted with 23 μl of 0.1X TE buffer. Libraries were sequenced using the Novaseq 6000 sequencing system from Illumina in order to obtain at least 50 million reads per sample.
RNAseq pipeline
The quality of the fastq sequences were checked using FastQC and the per base sequence quality and content were used to determine the number of nucleotides to trim from the ends using Trimmomatic3. Sequences were aligned to the most recent version of the human genome (GRCh38.p13) using Hisat24. Samtools5 was used to convert the files from.sam to.bam, which were then sorted and indexed. Using Bedtools6, we converted the files from.bam to.bed then visualized the alignments with IGV7. We utilized HTSeq8 and the human genome feature file (.gff) to count the number of uniquely mapped reads for each gene, then performed differential expression analysis using DESeq29. Using the differential expression data from DESeq2, we performed GO term enrichment using the topGO10.
IPA analysis
Using the Ingenuity Pathway Analysis (IPA) software (QIAGEN Inc., https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis), the lists of differentially expressed genes output from DESeq2 were analysed more deeply. Tab-delimited matrices of the differentially expressed genes, including -log10 p value and log2 fold change, were loaded into IPA and an initial core analysis was performed by the software11. The core analysis provides summarized data about the listed genes regarding canonical pathways involved, upstream regulators, diseases and disorders, molecular and cellular functions, physiological and system development functions, functions in toxicity, regulator effect networks, and associated network functions. Additionally, Fig.s were generated from regulatory networks and associated molecular and cellular functions to visualize the data.
In vivo studies
Eight to ten weeks old female BALB/c mice were intraperitoneally injected with wild type (WT), knock-out ROP55 (∆ROP55) or complemented parasites. To assess the ∆ROP55 virulence in vivo, freshly harvested tachyzoites (102, 104, 105) of WT or ∆ROP55 parasites, or lethal dose (106) ∆ROP55 parasites were intraperitoneally injected into 5-10 mice per condition and monitored for survival. To study parasite load, mice were infected with 103 tachyzoites and 4 dpi the animals were sacrificed and spleens were collected in order to perform RT-qPCR.
Statistical analysis
GraphPad was used for all statistical analysis other than for RNAseq data. A normality test was performed to analyse sample distribution. When data was normally distributed and had equal variance, a two-tailed unpaired t test was used. When data was normally distributed but sample size and variance were unequal, a Welch t test was used. For non-normally distributed data, a two-tailed Mann–Whitney U test was used. Group analysis were performed with Tukey’s or Sidaks’s multiple comparison tests. Statistical significance was considered for p values below 0.05. Values in bar graphs are means ± SEM.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data are present in the main text and the supplementary materials. All biological materials and data are available from the authors upon request. Source data are provided with this paper. RNAseq data is available in NCBI SRA https://www.ncbi.nlm.nih.gov/sra (code PRJNA881613). Source data are provided with this paper.
References
Flegr, J., Prandota, J., Sovičková, M. & Israili, Z. H. Toxoplasmosis–a global threat. Correlation of latent toxoplasmosis with specific disease burden in a set of 88 countries. PloS One 9, e90203 (2014).
Pappas, G., Roussos, N. & Falagas, M. E. Toxoplasmosis snapshots: global status of Toxoplasma gondii seroprevalence and implications for pregnancy and congenital toxoplasmosis. Int. J. Parasitol. 39, 1385–1394 (2009).
Montoya, J. G. & Liesenfeld, O. Toxoplasmosis. Lancet (Lond., Engl.) 363, 1965–1976 (2004).
Demar, M. et al. Fatal outbreak of human toxoplasmosis along the Maroni River: epidemiological, clinical, and parasitological aspects. Clin. Infect. Dis.: Off. Publ. Infect. Dis. Soc. Am. 45, e88–e95 (2007).
Blaizot, R. et al. Outbreak of amazonian toxoplasmosis: A one health investigation in a remote amerindian community. Front Cell Infect. Microbiol 10, 401 (2020).
Hunter, C. A. & Sibley, L. D. Modulation of innate immunity by Toxoplasma gondii virulence effectors. Nat. Rev. Microbiol. 10, 766–778 (2012).
Boothroyd, J.C. & Hakimi, M.-A. Chapter 17 - Effectors produced by rhoptries and dense granules: an intense conversation between parasite and host in many languages, in Toxoplasma gondii (Third Edition). (eds. L.M. Weiss & K. Kim) 789-806 (Academic Press, 2020).
Barragan, A. & Sibley, L. D. Transepithelial migration of Toxoplasma gondii is linked to parasite motility and virulence. J. Exp. Med. 195, 1625–1633 (2002).
Jorgensen, I., Rayamajhi, M. & Miao, E. A. Programmed cell death as a defence against infection. Nat. Rev. Immunol. 17, 151–164 (2017).
Tummers, B. & Green, D. R. The evolution of regulated cell death pathways in animals and their evasion by pathogens. Physiol. Rev. 102, 411–454 (2022).
Broz, P. & Dixit, V. M. Inflammasomes: mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 16, 407–420 (2016).
Bauernfeind, F. G. et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 183, 787–791 (2009).
Groslambert, M. & Py, B. F. Spotlight on the NLRP3 inflammasome pathway. J. Inflamm. Res. 11, 359–374 (2018).
Grootjans, S., Vanden Berghe, T. & Vandenabeele, P. Initiation and execution mechanisms of necroptosis: an overview. Cell death Differ. 24, 1184–1195 (2017).
Orozco, S. L. et al. RIPK3 activation leads to cytokine synthesis that continues after loss of cell membrane integrity. Cell Rep. 28, 2275–2287.e2275 (2019).
Meylan, E. et al. RIP1 is an essential mediator of toll-like receptor 3-induced NF-kappa B activation. Nat. Immunol. 5, 503–507 (2004).
Kaiser, W. J., Upton, J. W. & Mocarski, E. S. Receptor-interacting protein homotypic interaction motif-dependent control of NF-κB activation via the DNA-dependent activator of IFN regulatory factors1. J. Immunol. 181, 6427–6434 (2008).
Moriwaki, K. et al. The necroptosis adaptor RIPK3 promotes injury-induced cytokine expression and tissue repair. Immunity 41, 567–578 (2014).
Mocarski, E. S., Guo, H. & Kaiser, W. J. Necroptosis: The Trojan horse in cell autonomous antiviral host defense. Virology 479-480, 160–166 (2015).
Rosenberg, A. & Sibley, L. D. Toxoplasma gondii secreted effectors co-opt host repressor complexes to inhibit necroptosis. Cell Host Microbe 29, 1186–1198.e1188 (2021).
Cavaillès, P. et al. The rat Toxo1 locus directs toxoplasmosis outcome and controls parasite proliferation and spreading by macrophage-dependent mechanisms. Proc. Natl Acad. Sci. USA 103, 744–749 (2006).
Cirelli, K. M. et al. Inflammasome sensor NLRP1 controls rat macrophage susceptibility to Toxoplasma gondii. PLoS Pathog. 10, e1003927 (2014).
Gov, L., Karimzadeh, A., Ueno, N. & Lodoen, M.B. Human innate immunity to Toxoplasma gondii is mediated by host caspase-1 and ASC and parasite GRA15. mBio 4, https://doi.org/10.1128/mbio.00255-13 (2013).
Gov, L., Schneider, C. A., Lima, T. S., Pandori, W. & Lodoen, M. B. NLRP3 and potassium efflux drive rapid IL-1β release from primary human monocytes during Toxoplasma gondii infection. J. Immunol. 199, 2855–2864 (2017).
Pandori, W. J. et al. Toxoplasma gondii activates a Syk-CARD9-NF-κB signaling axis and gasdermin D-independent release of IL-1β during infection of primary human monocytes. PLOS Pathog. 15, e1007923 (2019).
Rosowski, E. E. et al. Strain-specific activation of the NF-κB pathway by GRA15, a novel Toxoplasma gondii dense granule protein. J. Exp. Med. 208, 195–212 (2011).
Lima, T.S., Gov, L. & Lodoen, M.B. Evasion of Human Neutrophil-Mediated Host Defense during Toxoplasma gondii Infection. mBio 9, https://doi.org/10.1128/mbio.02027-17 (2018).
Butcher, B. A., Kim, L., Johnson, P. F. & Denkers, E. Y. Toxoplasma gondii tachyzoites inhibit proinflammatory cytokine induction in infected macrophages by preventing nuclear translocation of the transcription factor NF-kappa B. J. Immunol. 167, 2193–2201 (2001).
Shapira, S. et al. Initiation and termination of NF-kappaB signaling by the intracellular protozoan parasite Toxoplasma gondii. J. cell Sci. 118, 3501–3508 (2005).
Butcher, B. A. & Denkers, E. Y. Mechanism of entry determines the ability of Toxoplasma gondii to inhibit macrophage proinflammatory cytokine production. Infect. Immun. 70, 5216–5224 (2002).
Camejo, A. et al. Identification of three novel Toxoplasma gondii rhoptry proteins. Int. J. Parasitol. 44, 147–160 (2014).
Sidik, S. M. et al. A Genome-wide CRISPR screen in toxoplasma identifies essential apicomplexan genes. Cell 166, 1423–1435 e1412 (2016).
Sigrist, C. J. et al. New and continuing developments at PROSITE. Nucleic Acids Res. 41, D344–D347 (2013).
Zimmermann, L. et al. A completely reimplemented mpi bioinformatics toolkit with a new HHpred server at its core. J. Mol. Biol. 430, 2237–2243 (2018).
Kelley, L. A., Mezulis, S., Yates, C. M., Wass, M. N. & Sternberg, M. J. E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858 (2015).
Barylyuk, K. et al. A comprehensive subcellular atlas of the toxoplasma proteome via hyperLOPIT provides spatial context for protein functions. Cell Host Microbe 28, 752–766.e759 (2020).
Kovacs, S. B. & Miao, E. A. Gasdermins: Effectors of pyroptosis. Trends Cell Biol. 27, 673–684 (2017).
Nguyen, H. N. et al. Autocrine loop involving IL-6 family member LIF, LIF receptor, and STAT4 drives sustained fibroblast production of inflammatory mediators. Immunity 46, 220–232 (2017).
Broz, P. & Monack, D. M. Newly described pattern recognition receptors team up against intracellular pathogens. Nat. Rev. Immunol. 13, 551–565 (2013).
Robben, P. M. et al. Production of IL-12 by macrophages infected with Toxoplasma gondii depends on the parasite genotype. J. Immunol. 172, 3686–3694 (2004).
Dunay, I. R. et al. Gr1(+) inflammatory monocytes are required for mucosal resistance to the pathogen Toxoplasma gondii. Immunity 29, 306–317 (2008).
Robben, P. M., LaRegina, M., Kuziel, W. A. & Sibley, L. D. Recruitment of Gr-1+ monocytes is essential for control of acute toxoplasmosis. J. Exp. Med. 201, 1761–1769 (2005).
Coll, R. C. et al. MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nat. Chem. Biol. 15, 556–559 (2019).
Pandori, W. J. et al. Role for Caspase-8 in the Release of IL-1β and Active Caspase-1 from Viable Human Monocytes during Toxoplasma gondii Infection. J Immunol 212, 1161–1171 (2024).
Rogers, C. et al. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat. Commun. 8, 14128 (2017).
Degterev, A. et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 4, 313–321 (2008).
Zhuang, C. & Chen, F. Small-molecule inhibitors of necroptosis: Current status and perspectives. J. Med. Chem. 63, 1490–1510 (2020).
Sun, L. et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148, 213–227 (2012).
Matsuno, S. Y., Pandori, W. J. & Lodoen, M. B. Capers with caspases: Toxoplasma gondii tales of inflammation and survival. Curr. Opin. Microbiol. 72, 102264 (2023).
Braun, L. et al. The Toxoplasma effector TEEGR promotes parasite persistence by modulating NF-κB signalling via EZH2. Nat. Microbiol 4, 1208–1220 (2019).
Tomita, T. et al. Toxoplasma gondii Matrix Antigen 1 is a secreted immunomodulatory effector. mBio 12, https://doi.org/10.1128/mbio.00603-21 (2021).
Fisch, D. et al. Human GBP1 is a microbe-specific gatekeeper of macrophage apoptosis and pyroptosis. EMBO J. 38, e100926 (2019).
Chen, K.W. et al. Extrinsic and intrinsic apoptosis activate pannexin-1 to drive NLRP3 inflammasome assembly. EMBO J. 38, e101638 (2019).
Koehler, H. et al. Inhibition of DAI-dependent necroptosis by the Z-DNA binding domain of the vaccinia virus innate immune evasion protein, E3. Proc. Natl Acad. Sci. USA 114, 11506–11511 (2017).
Ashida, H., Sasakawa, C. & Suzuki, T. A unique bacterial tactic to circumvent the cell death crosstalk induced by blockade of caspase-8. EMBO J. 39, e104469 (2020).
Thapa, R. J. et al. DAI Senses Influenza A Virus Genomic RNA and Activates RIPK3-Dependent Cell Death. Cell Host Microbe 20, 674–681 (2016).
Peng, R. et al. Human ZBP1 induces cell death-independent inflammatory signaling via RIPK3 and RIPK1. EMBO Rep. 23, e55839 (2022).
Rebsamen, M. et al. DAI/ZBP1 recruits RIP1 and RIP3 through RIP homotypic interaction motifs to activate NF-κB. EMBO Rep. 10, 916–922 (2009).
Upton, J. W., Kaiser, W. J. & Mocarski, E. S. Cytomegalovirus M45 cell death suppression requires receptor-interacting protein (RIP) homotypic interaction motif (RHIM)-dependent interaction with RIP1. J. Biol. Chem. 283, 16966–16970 (2008).
Fliss, P. M. et al. Viral mediated redirection of NEMO/IKKγ to autophagosomes curtails the inflammatory cascade. PLoS Pathog. 8, e1002517 (2012).
Henry, B., Phillips, A.J., Sibley, L.D. & Rosenberg, A. A combination of four Toxoplasma gondii nuclear-targeted effectors protects against interferon gamma-driven human host cell death. mBio 15, https://doi.org/10.1128/mbio.02124-24 (2024).
Cervantes, P.W., Martorelli Di Genova, B., Erazo Flores, B.J. & Knoll, L.J. RIPK3 facilitates host resistance to oral toxoplasma gondii Infection. Infect. immunity 89, e00021–21 (2021).
Zhang, T. et al. Influenza Virus Z-RNAs induce ZBP1-mediated necroptosis. Cell 180, 1115–1129.e1113 (2020).
Pittman, K. J., Cervantes, P. W. & Knoll, L. J. Z-DNA binding protein mediates host control of toxoplasma gondii infection. Infect. Immun. 84, 3063–3070 (2016).
Nozaki, K., Li, L. & Miao, E. A. Innate sensors trigger regulated cell death to combat intracellular infection. Annu. Rev. Immunol. 40, 469–498 (2022).
Maelfait, J. et al. Sensing of viral and endogenous RNA by ZBP1/DAI induces necroptosis. EMBO J. 36, 2529–2543 (2017).
Niedelman, W., Sprokholt, J. K., Clough, B., Frickel, E.-M. & Saeij, J. P. J. Cell death of gamma interferon-stimulated human fibroblasts upon Toxoplasma gondii infection induces early parasite egress and limits parasite replication. Infect. Immun. 81, 4341–4349 (2013).
Gonçalves, R. & Mosser, D. M. The isolation and characterization of murine macrophages. Curr. Protoc. Immunol. 111, 14.11.11–14.11.16 (2015).
Bobak, D. A., Frank, M. M. & Tenner, A. J. Characterization of C1q receptor expression on human phagocytic cells: Effects of PDBu and fMLP. J. Immunol. 136, 4604–4610 (1986).
Acknowledgements
We thank Sebastian Lourido fot the the pU6-Universal plasmid and Dominique Soldati-Favre for anti-GAP45 antibodies, we also thank J. Saeij for kindly gift of HEK 293 NF-κB reporter cell line (System Biosciences, Cat. # TR860A-1), we are also grateful to Derek Abbott for sharing the ΔGSDMD THP-1 line; we are also grateful to Elodie Jublanc and Vicky Diakou at the imaging facility MRI at the University of Montpellier, part of the national infrastructure France-BioImaging supported by the French National Research Agency (ANR-10-INBS-04, «Investments for the future»). Dr Maryse Lebrun is an INSERM researcher. This work was supported by the Laboratoire d’Excellence (LabEx) (ParaFrap ANR-11-LABX-0024) and Fondation pour la Recherche Médicale (Equipe FRM DEQ20130326508) to M.L., the National Institutes of Allergy and Infectious Diseases and the National Institutes of Health (R01 AI136511 and R21 AI142506) and the University of California, Riverside (NIFA-Hatch-225935) to K.L.R, NIH (R01AI120846 and R21AI156452) to M.B.L. M.T.G.R. was funded by the French Parasitology consortium ParaFrap (ANR-11-LABX-0024) and was a recipient of FRM (award no. FDT20190400862).
Author information
Authors and Affiliations
Contributions
M.T.G.R. and D.M.P.V performed the experiments and analysed the data, they generated parasite lines and performed measurements in HFF and THP-1 cells; M.H., H.E.H. and M. G. performed the mouse experiments; T.L. and K.L.R generated cDNA, performed and analyzed RNAseq experiments. J.S., K.O.Y., S.M., and M.B.L. performed western blot of GSDMD, GSDME, Caspase 8, IκBα and quantification of cell mortality in THP-1 cell lines by cell cytometry. J.S performed human MDM experients. D.M.P.V performed mouse mBMDM experiments and NF-κB translocations, A.G. Performed IL-1β ELISA and LDH in THP-1 cells with the help of D.M.P.V; LBS performed electron microscopy images; M.T.G.R., D.M.P.V., and M.L. conceived, directed the project and wrote the manuscript with input from all the authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Antonio Barragan, Sarah Ewald and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Grilo Ruivo, M.T., Shin, Jh., Lenz, T. et al. The Toxoplasma rhoptry protein ROP55 is a major virulence factor that prevents lytic host cell death. Nat Commun 16, 709 (2025). https://doi.org/10.1038/s41467-025-56128-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-56128-x
