
Abstract
Allylic ethers and alcohols are essential structural motifs commonly present in natural products and pharmaceuticals. Direct allylic C–H oxygenation of internal alkenes is one of the most direct methods, bypassing the necessity for an allylic leaving group that is needed in the traditional Tsuji–Trost reaction. Herein, we develop an efficient and practical method for synthesizing (E)-allyl ethers from readily available internal alkenes and alcohols or phenols via selective allylic C–H oxidation. Key advances include the use of a Cu/Azodiformate catalyst system to facilitate remote allylic C–H activation and the achievement of excellent chemoselectivity through a dynamic ligand exchange strategy using a bis(sulfonamide) ligand. This method features a broad substrate scope and functional group tolerance, successfully applied to the synthesis of various challenging medium-sized cyclic ethers (7-10 members) and large-ring lactones (14-20 members), with high regioselectivity and stereoselectivity.
Similar content being viewed by others
Introduction
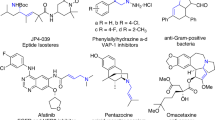
Allylic alcohols and ethers are key functional motifs widely found in various bioactive molecules, pharmaceuticals, and natural products, such as aigialomycin, rosuvastatin, and pacritinib (Fig. 1)1,2,3. An attractive alternative involves the direct oxidative coupling of an allylic C–H bond with alcohols or phenols to forge the desired allylic ethers. Compared to the classical Tsuji–Trost substitution reaction, it avoids the prefunctionalization of alkene substrates and exhibits typical features of simplicity in steps, atom economy, and high reaction efficiency4,5,6. However, oxidative allylic etherification is significantly more challenging, as both the substrate alcohols and phenols, as well as the allylic ethers, are prone to competitive oxidation.

Selected bioactive compounds containing allylic alcohols (Aigialomycin D and Rosuvastatin) and allylic ethers (Herboxidiene, (−)-Zampanolide, (+)-Brasilenyne and Pacritinib).
In this field, the White group has made significant contributions to both inter- and intramolecular allylic C–H functionalization by developing a Pd(II)/bis-sulfoxide catalytic system7,8,9,10,11,12,13,14. This system facilitates the broad cross-coupling of primary, secondary, and tertiary aliphatic alcohols with terminal olefins and can also be employed in the synthesis of various oxygen-containing heterocycles15,16. Since then, catalytic systems for oxidative allylic functionalization have developed rapidly. For instance, Gong and co-workers established a Pd(0)/BQ-PIII catalytic system to promote allylic C–H activation, enabling the synthesis of chiral chromans by using a chiral phosphoramidite ligand17,18,19. Recently, the Gevorgyan group reported a photocatalytic platform that operates through a blue light-induced Pd(0/I/II) manifold with a mild aryl bromide oxidant. This method achieves regio- and diastereoselective allylic C–H oxygenation20,21,22. In addition to palladium-based systems, the Blakey group also discovered regioselective intermolecular allylic etherification reactions based on a Rh(III)Cp*/AgOAc-catalyzed platform (Fig. 2a)23,24. Despite the significant advances in these methods, they remain limited to simple alkenes with cyclic or terminal double bonds and a narrow range of oxygen nucleophile substrates. Therefore, the development of new catalyst systems capable of promoting regioselective allylic functionalization of internal olefins and compatible with a wide range of nucleophiles is highly desirable.

a Transition metal-catalyzed allylic C–H etherification. b Our Cu(I)/azodiformate catalyst system. c Our strategy. d Synthesis of structurally diverse medium-sized and macrocyclic rings.
Recently, we developed an intermolecular allylic C–H amination reaction of internal alkenes, where a π-allyl-copper species (Int A) was involved as a key intermediate25. Inspired by this study, we turned our attention to whether this intermediate could be intercepted by a range of stabilized carbon, nitrogen, and oxygen nucleophiles (Fig. 2b). To test the above hypothesis, we unexpectedly found that this catalytic system is suitable for oxygen nucleophiles, with the only drawback being the formation of an allylamine by-product (see the Supplementary Information Table S1 for details). This phenomenon may be attributed to high-basicity nucleophiles, with pKa’s ranging from ~10 for phenol to >15 for aliphatic alcohols, which may affect the ligand exchange process and disrupt the catalytic cycle. To overcome this issue, our laboratory has recently disclosed a dynamic ligand exchange strategy to suppress C–N bond reductive elimination (Fig. 2c)26,27,28,29,30,31. Specifically, we introduced the bis(sulfonamide) ligand (IBSI) into the reaction system, which rapidly underwent N-ligand exchange with Int A to form the new π-allyl-Cu complex Int B; Subsequently, the hydrogen atom transfers directly from phenol to the N-ligand via ligand-to-ligand hydrogen transfer (LLHT)32,33,34,35,36, forming Int C, thereby enabling the formation of mono-allylic etherification product.
Medium-sized cyclic ethers and macrolides are present in numerous biologically important natural products37,38. Due to the unfavorable entropic and enthalpic factors, as well as transannular interactions associated with these ring systems, their efficient synthesis remains a formidable challenge39,40,41,42,43,44,45. Encouraged by intermolecular etherification reactions, we envisioned that the π-allyl-copper intermediates could be captured by an appropriate intramolecular oxygen nucleophile. As expected, we successfully synthesized a diverse array of medium-sized cyclic ethers (7–10 members) and macrolactones (14–20 members) using our allylic C–H oxidation strategy (Fig. 2d).
Results
Reaction condition optimization
After a short survey of potential oxygen nucleophiles, we selected phenol as a suitable starting substrate for our study. We initially tested the standard conditions developed for our recently reported allylic C–H amination reaction but observed an unexpectedly low yield of the desired allylic ether 3a, along with significant amounts of by-product 3b (3a/3b = 23/68). Next, various copper catalysts were examined, including CuCl, CuTc, Cu2O, and CuCl2, and they were less effective than Cu2O (Table 1, entries 1–4). We also investigated polar solvents such as THF, DMF, and the aromatic solvent toluene, which showed no significant impact on both yield and chemoselectivity (Table 1, entries 5–7). Inspired by ligand exchange strategies, we hypothesized that introducing a protonic acid into the reaction system might undergo proton transfer with azodicarboxylate, thereby facilitating its dissociation from the copper catalyst as hydrazinedicarboxylate. Subsequently, various protonic acids were screened as ligands (see the Supplementary Information Table S4 for details). The results indicate that as the acidity of the ligand decreased, only the desired compound 3a was produced, with the yield gradually increasing from 0% to 68% (Table 1, entries 8–11). However, further reduction in ligand acidity resulted in a decrease in the chemoselectivity of the product, approaching a ratio of 3:4 (Table 1, entries 12–14). To our delight, the choice of ligand BBSI had a remarkable impact on the chemoselectivity and exhibited much better performance, giving 3a in 68% yield, while the side product 3b was observed in 12% yield (3a/3b = 68/12) (Table 1, entry 11). It can be inferred that the acidity of the ligand significantly influences the chemoselectivity of the product. Weaker acidity affects the N-ligand exchange process, while stronger acidity impacts the LLHT process. Encouraged by these results, we evaluated BBSI with different substituents (Table 1, entries 15–18) and found that iPr-L9 (IBSI) exhibited the best performance, furnishing 3a in 88% yield with complete inhibition of the side product 3b. Notably, L11, which has methyl groups at positions 1, 3, and 5 of the benzene ring, exhibited no reactivity, potentially attributed to steric hindrance preventing the LLHT process (more details, see Supplementary Information Table S5).
Substrate scope
Under optimized conditions, the substrate scope of the δ-etherification reaction was investigated (Fig. 3). In all cases, the reaction proceeded with high regioselectivity and excellent E/Z selectivity (>20:1). Firstly, we explored the scope of alkene substrates using phenol and found that the reaction was compatible with a variety of substituted internal alkenes. δ-Etherification with linear, branched, and cyclic substituents resulted in moderate to excellent yields (3a–9). Alkene substrates bearing a di-α-substituent were suitable for this reaction, while mono-α-substituted substrate underwent this transformation with good yield and a 1:1 dr (4, 5). Interestingly, we further evaluated the impact of long alkyl chain substituents with various functional groups. Substituents including chlorine, iodine, ether, ester, azide, and amide were all well tolerated (10–17). In addition, substrates containing terminal alkyne or terminal alkene moieties were also applicable to this reaction (15, 16). This insight was then implemented in reactions with numerous phenol coupling partners. A diverse range of electron-withdrawing and electron-donating groups at various positions on the aryl units were found to be well tolerated, resulting in the corresponding products with yields ranging from 53% to 94% (18–28). The molecular structure of compound 28 has been confirmed by X-ray crystallography. For multisubstituted phenols (29–34), the reactions also proceeded smoothly to afford the corresponding products in satisfactory yields. Additionally, a variety of functional groups, including halogen (18, 28, 34), nitro (19, 31, 32), aldehyde (22), trifluoromethyl (20, 30), sulfonyl (24), carboxylic ester (21, 33), ketone (23, 26, 29), and nitrile (25, 30), were well tolerated on the aryl ring under standard conditions. The etherification reaction also proceeded effectively in the late-stage functionalization of several pharmaceuticals and related bioactive molecules (35–39), including the cosmetic ingredient Birch-Me, the pharmaceutical molecule Farnesane Impurity 37, the natural product estrone, as well as analogs of ibuprofen and naproxen.

a Reaction conditions: 1a (0.2 mmol, 1.0 equiv), 2 (0.6 mmol, 3.0 equiv), DEAD (0.4 mmol, 2.0 equiv), Cu2O (10 mol%) and L9 (20 mol%) in toluene (2.0 mL) at the 80 °C for 10 h. b Isolated yield.
To further demonstrate the synthetic potential of this transformation, various alcohols were subsequently introduced into the etherification reaction (Fig. 4). Under the optimal conditions for phenol, a small amount of compound 41 was detected. By referencing Stahl’s work, modifying the reaction conditions to the CuCl/(EtO)2P(O)H catalytic system significantly improved the yield of the etherification product46. The substrates of methanol (40), ethanol (41), cyclopropylmethanol (42), benzylalcohol (43, 44), and 3-phenylpropanol (45) were all allowed to react with 1a, furnishing the corresponding products in moderate yields. Several ethanol derivatives with various substituents, including bromine, ester, thiophene, alkynyl, vinyl, and naphthyl groups (46–53), exhibit favorable performance in this reaction, affording satisfactory yields in most cases. From products 54–57, it can be observed that alkyl alcohols with trifluoromethyl substitution, as the chain length increases, exhibit a corresponding decrease in product yields. This suggests that the reaction rate may be related to the ease of deprotonation of the O-nucleophilic reagent.

a Reaction conditions: 1a (0.2 mmol, 1.0 equiv), 2 (0.6 mmol, 3.0 equiv), DEAD (0.4 mmol, 2.0 equiv), CuCl (10 mol%) and L12 (20 mol%) in toluene (2.0 mL) at the 80 °C for 10 h. b Isolated yield.
Having successfully achieved intermolecular etherification, we hypothesized whether this catalytic mode could be extended to intramolecular C–H oxidation reactions for the construction of cyclic compounds (Fig. 5). Initially, we attempted the synthesis of more synthetically favorable 5-membered and 6-membered ring compounds. The results indicated that we could obtain tetrahydrofuran (58), tetrahydropyran (59, 60), benzofuran (61), and benzopyran (62, 63) derivatives in moderate yields. It is gratifying to note that the current strategy enables the successful synthesis of medium-sized rings, which are typically considered difficult to prepare. Substrates bearing trifluoromethyl electron-withdrawing groups smoothly obtained the corresponding 7- to 10-membered cyclic ether products (64–67). Subsequently, by employing the different chain lengths of alkyl groups, all 14- to 20-membered macrolides (68–74) can be smoothly obtained in 53–78% yields.

a Reaction conditions: 1 (0.2 mmol, 1.0 equiv), DEAD (0.4 mmol, 2.0 equiv), Cu2O (10 mol%) and L9 (20 mol%) in toluene (2.0 mL) at the 80 °C for 10 h. b Isolated yield.
Synthetic applicability
To demonstrate the applicability and effectiveness of our strategy, both intermolecular and intramolecular gram-scale reactions were conducted under standard conditions. The reactions afforded product 3a in 82% yield (2.72 g) and product 70 in 62% yield (1.51 g) (Fig. 6a). The internal alkenes in the products can be reduced to form 75 or oxidized to generate 76 using K2OsO4. Reduction with LiAlH4 transformed the amide moiety into the corresponding secondary amine to furnish 77, while the 8-aminoquinoline moiety could be conveniently removed by treatment with IBX, affording the primary amine 78 (Fig. 6b). Subsequently, compound 19 was subjected to indium-mediated reduction of the nitro group, followed by CAN oxidative cleavage to afford the linear allylic alcohol 79 (Fig. 6c), which can serve as an important synthetic intermediate.

a Gram-scale reaction. b Demonstration of utility. c Synthesis of allylic alcohol.
Mechanistic study
To investigate the underlying mechanism of the allylic C–H etherification process, a series of control and isotope labeling experiments were conducted. Initially, it was observed that both trans- and cis-hexenamides (1a and 80) produce the same product 3a, indicating that the reaction proceeds via a π-allyl-Cu intermediate (Fig. 7a). No reaction was observed with the N-methyl protected enamide substrate 81, highlighting the critical chelating role of the directing group in the reaction (Fig. 7b). In the presence of two equivalents of TEMPO or BHT as radical scavengers, 3a could still be isolated in 82% and 78% yields, respectively. These results suggest that the reaction may not proceed via a radical pathway (Fig. 7c). Next, the by-product 3b was selected as the substrate and reacted under standard conditions, but no etherification product was obtained. This suggests that the reaction is unlikely to proceed via a nucleophilic substitution mechanism (Fig. 7d). In addition, a deuterium labeling experiment with 83-d2 (59% D) produced 86-d2 (59% D) in an 83% yield without deuterium scrambling and the deuteration at the internal alkene of 84-d2 (97% D) turned out to be identical to that of the corresponding product 87-d2 (97% D), which strongly supported that the alkene isomerization of 1a would not occur during the reaction process. Moreover, the reaction of 85-d3 (>99% D) and 4-nitrophenol smoothly afforded the product 88-d2 with 98% deuteration at the allylic position, suggesting that the allylic C–H cleavage process was most likely to be irreversible (Fig. 7e). Furthermore, the parallel and intermolecular kinetic isotope effects were determined to be 2.5 and 2.3, respectively (Fig. 7f, g). All these results are consistent with the allylic C–H activation pathway, indicating that the allylic C–H cleavage likely involves the rate-limiting step.

a Z-alkene experiment. b N-Me AQ control experiment. c Radical trapping experiments. d Replacing experiment. e D-labeling experiment. f Parellel kinetic isotope effect. g Intermolecular kinetic isotope effect.
DFT calculations
To gain a better understanding of the reaction mechanism, particularly the crucial role of ligand exchange strategies in determining the reaction’s chemoselectivity, density functional theory (DFT) calculations were conducted (Fig. 8). The computational study on the Cu/azodiformate system-catalyzed allylic C–H activation, leading to the formation of the π-allyl-Cu complex A, can be referenced from our previous work25. From the complex A, in the absence of a protic acid ligand, the reaction more readily proceeds through TS1-1 to undergo C–N bond reductive elimination, forming intermediate B-1 with a Gibbs free-energy barrier of 7.6 kcal/mol. However, upon the introduction of the IBSI, intermediate A rapidly undergoes ligand exchange with IBSI via TS1, forming intermediate C with a Gibbs free-energy barrier of 5.9 kcal/mol. For acetic acid and phenol, the protonation transition states TS-1′ and TS-1′′ exhibit relatively high energies, indicating that these ligands are less prone to undergo proton transfer. This suggests that the driving force for ligand exchange is related to the acidity of the ligands. Moreover, intermediate C faces challenges with C–N bond reductive elimination due to the high energy barrier of TS-2′. Instead, it undergoes LLHT with phenol, transitioning via TS2 and TS3 successively, forming intermediate F. This intermediate then dissociates the IBSI to produce the new π-allyl-copper species G, which is capable of undergoing C–O bond reductive elimination (TS4) to generate the desired product. Regarding regioselectivity, DFT calculations were employed to investigate two competing transition states in the inner-sphere pathway. The δ-amination pathway through TS4 was found to be at least 8.8 kcal/mol more favorable than the β-amination pathway.

The DFT-computed free-energy changes of the favorable reaction pathway and the Gibbs free energies of the key transition states.
Discussion
In summary, we have developed a general Cu/azodiformate system-catalyzed allylic oxidation method that efficiently affords (E)-allylethers from readily available internal olefins and alcohols or phenols with various functional groups, exhibiting high regioselectivity and stereoselectivity. This approach allows for the efficient synthesis of a variety of medium-sized cyclic ethers (7–10 members) and large-ring lactones (14–20 members). Experimental and computational studies indicated how an unusual mechanism involving dynamic ligand exchange can be used to control the chemoselectivity and ultimately expand C–O bond-forming methodologies. Insights from this study will help broaden the scope of pro-nucleophiles used in allylic C–H functionalization reactions.
Methods
General procedure
A mixture of amide (0.20 mmol, 1.0 equiv), Cu2O (0.02 mmol, 0.1 equiv), L (0.04 mmol, 0.2 equiv), DEAD (0.40 mmol, 2.0 equiv) and alcohol/phenol (0.60 mmol, 3.0 equiv) in Toluene (2 mL) in a 10 mL glass vial (sealed with PTFE cap) was heated at 80 °C for indicated time. The reaction progress was monitored by thin-layer chromatography. Upon completion, the reaction mixture was concentrated in vacuo and purified by silica gel column chromatography to afford the desired products.
Data availability
The data generated in this study are provided in the Supplementary Information file. The experimental procedures, data of NMR, and HRMS have been deposited in the Supplementary Information file. The X-ray crystallographic coordinates for structures reported in this study have been deposited at the Cambridge Crystallographic Data Centre (CCDC: 2322949) and (CCDC: 2322950). These data could be obtained free of charge from The Cambridge Crystallographic Data Centre (https://www.ccdc.cam.ac.uk/data_request/cif). All data are available from the corresponding author upon request. Source data are provided with this paper.
References
Roughley, S. D. & Jordan, A. M. The medicinal chemist’s toolbox: an analysis of reactions used in the pursuit of drug candidates. J. Med. Chem. 54, 3451–3479 (2011).
Trost, B. M. & Crawley, M. L. Asymmetric transition-metal-catalyzed allylic alkylations: applications in total synthesis. Chem. Rev. 103, 2921–2944 (2003).
Ghosh, A. K. & Cheng, X. Enantioselective total synthesis of (−)-zampanolide, a potent microtubule-stabilizing agent. Org. Lett. 13, 4108–4111 (2011).
Trost, B. M. & Van Vranken, D. L. Asymmetric transition metal-catalyzed allylic alkylations. Chem. Rev. 96, 395–422 (1996).
Trost, B. M., Machacek, M. R. & Aponick, A. Predicting the stereochemistry of diphenylphosphino benzoic acid (DPPBA)-based palladium-catalyzed asymmetric allylic alkylation reactions: a working model. Acc. Chem. Res. 39, 747–760 (2006).
Trost, B. M. & Crawley, M. L. Enantioselective allylic substitutions in natural product synthesis. Top. Organomet. Chem. 38, 321–340 (2011).
Chen, M. S. & White, M. C. A sulfoxide-promoted, catalytic method for the regioselective synthesis of allylic acetates from monosubstituted olefins via C–H oxidation. J. Am. Chem. Soc. 126, 1346–1347 (2004).
Chen, M. S., Prabagaran, N., Labenz, N. A. & White, M. C. Serial ligand catalysis: a highly selective allylic C–H oxidation. J. Am. Chem. Soc. 127, 6970–6971 (2005).
Fraunhoffer, K. J. & White, M. C. syn-1,2-amino alcohols via diastereoselective allylic C–H amination. J. Am. Chem. Soc. 129, 7274–7276 (2007).
Young, A. J. & White, M. C. Catalytic intermolecular allylic C–H alkylation. J. Am. Chem. Soc. 130, 14090–14091 (2008).
Reed, S. A., Mazzotti, A. R. & White, M. C. A catalytic, Brønsted base strategy for intermolecular allylic C–H amination. J. Am. Chem. Soc. 131, 11701–11706 (2009).
Pattillo, C. C. et al. Aerobic linear allylic C–H amination: overcoming benzoquinone inhibition. J. Am. Chem. Soc. 138, 1265–1272 (2016).
Ma, R. & White, M. C. C–H to C–N cross-coupling of sulfonamides with olefins. J. Am. Chem. Soc. 140, 3202–3205 (2018).
Ali, S. Z. et al. Allylic C–H amination cross-coupling furnishes tertiary amines by electrophilic metal catalysis. Science 376, 276–283 (2022).
Ammann, S. E., Rice, G. T. & White, M. C. Terminal olefins to chromans, isochromans, and pyrans via allylic C–H oxidation. J. Am. Chem. Soc. 136, 10834–10837 (2014).
Kaster, S. H. M. et al. Palladium-catalyzed cross-coupling of alcohols with olefins by positional tuning of a counteranion. Science 385, 1067–1076 (2024).
Wang, P. S. et al. Asymmetric allylic C–H oxidation for the synthesis of chromans. J. Am. Chem. Soc. 137, 12732–12735 (2015).
Wang, P. S., Shen, M. L., Wang, T. C., Lin, H. C. & Gong, L. Z. Access to chiral hydropyrimidines through palladium-catalyzed asymmetric allylic C–H amination. Angew. Chem. Int. Ed. 56, 16032–16036 (2017).
Lin, H. C. et al. Nucleophile-dependent Z/E- and regioselectivity in the palladium-catalyzed asymmetric allylic C–H alkylation of 1,4-dienes. J. Am. Chem. Soc. 141, 5824–5834 (2019).
Yamada, K., Cheung, K. P. S. & Gevorgyan, V. General regio- and diastereoselective allylic C–H oxygenation of internal alkenes. J. Am. Chem. Soc. 146, 18218–18223 (2024).
Cheung, K. P. S., Fang, J., Mukherjee, K., Mihranyan, A. & Gevorgyan, V. Asymmetric intermolecular allylic C–H amination of alkenes with aliphatic amines. Science 378, 1207–1213 (2022).
Mukherjee, K., Cheung, K. P. S. & Gevorgyan, V. Photoinduced Pd-catalyzed direct sulfonylation of allylic C–H bonds. Angew. Chem. Int. Ed. 64, e202413646 (2025).
Nelson, T. A. F. & Blakey, S. B. Intermolecular allylic C–H etherification of internal olefins. Angew. Chem. Int. Ed. 57, 14911–14915 (2018).
Burman, J. S. & Blakey, S. B. Regioselective intermolecular allylic C–H amination of disubstituted olefins via rhodium/π-allyl intermediates. Angew. Chem. Int. Ed. 56, 13666–13669 (2017).
Wang, L. et al. Cooperative Cu/azodiformate system-catalyzed allylic C–H amination of unactivated internal alkenes directed by aminoquinoline. Nat. Commun. 15, 1483 (2024).
Huang, W.-Y. et al. Palladium-catalyzed enantioselective multicomponent cross-coupling of trisubstituted olefins. J. Am. Chem. Soc. 146, 16892–16901 (2024).
Jiang, X.-L., Sheng, F.-T., Zhang, Y., Deng, G. & Zhu, S.-L. Ligand relay catalysis enables asymmetric migratory reductive acylation of olefins or alkyl halides. J. Am. Chem. Soc. 144, 21448–21456 (2022).
King, R. P., Krska, S. W. & Buchwald, S. L. A ligand exchange process for the diversification of palladium oxidative addition complexes. Org. Lett. 23, 6030–6034 (2021).
Hamby, T. B., LaLama, M. J. & Sevov, C. S. Controlling Ni redox states by dynamic ligand exchange for electroreductive Csp3–Csp2 coupling. Science 376, 410–416 (2022).
Wang, G. et al. Ligand-exchange-enabled axially chiral recognition in asymmetric aminopalladation/olefination of 2‑alkynylanilides. ACS Catal. 14, 4053–4065 (2024).
Sahoo, H., Zhang, L., Cheng, J. H., Nishiura, M. & Hou, Z. M. Auto-tandem copper-catalyzed carboxylation of undirected alkenyl C–H bonds with CO2 by harnessing β‑hydride elimination. J. Am. Chem. Soc. 144, 23585–23594 (2022).
Zhang, Q.-L., Dong, D.-F. & Zi, W.-W. Palladium-catalyzed regio- and enantioselective hydrosulfonylation of 1,3-dienes with sulfinic acids: scope, mechanism, and origin of selectivity. J. Am. Chem. Soc. 142, 15860–15869 (2022).
Cheng, L. et al. Nickel-catalyzed regio- and enantioselective hydroarylation of 1,3-dienes with indoles. CCS Chem. 4, 2612–2619 (2022).
Li, Q., Wang, Z., Dong, V. M. & Yang, X.-H. Enantioselective hydroalkoxylation of 1,3-dienes via Ni-catalysis. J. Am. Chem. Soc. 145, 3909–3914 (2023).
Sansores-Paredes, M. L. G., Lutz, M. & Moret, M. E. Cooperative H2 activation at a nickel(0)-olefin centre. Nat. Chem. 16, 417–425 (2024).
Tang, S.-E., Eisenstein, O., Nakao, Y. & Sakaki, S. Aromatic C–H σ‑bond activation by Ni0, Pd0, and Pt0 alkene complexes: concerted oxidative addition to metal vs ligand-to-ligand H transfer mechanism. Organometallics 36, 2761–2771 (2017).
Hussain, A., Yousuf, S. K. & Mukherjee, D. Importance and synthesis of benzannulated medium-sized and macrocyclic rings (BMRs). RSC Adv. 4, 43241–43257 (2014).
Marsault, E. & Peterson, M. L. Macrocycles are great cycles: applications, opportunities, and challenges of synthetic macrocycles in drug discovery. J. Med. Chem. 54, 1961–2004 (2011).
Song, B.-C. et al. Rhodium(III)-catalyzed C–H/O2 dual activation and macrocyclization: synthesis and evaluation of pyrido[2,1-a]isoindole grafted macrocyclic inhibitors for influenza H1N1. Angew. Chem. Int. Ed. 62, e202218886 (2023).
Zhang, P.-F. et al. Macrocyclization via remote meta-selective C–H olefination using a practical indolyl template. Chem. Sci. 14, 8279–8287 (2023).
Ding, L., Song, H., Zheng, C. & You, S.-L. Enantioselective synthesis of medium-sized-ring lactones via iridium-catalyzed Z‑retentive asymmetric allylic substitution reaction. J. Am. Chem. Soc. 144, 4770–4775 (2022).
Li, B. et al. Construction of natural-product-like cyclophane-braced peptide macrocycles via sp3 C–H arylation. J. Am. Chem. Soc. 141, 9401–9407 (2019).
Kim, H. & Chang, S. Intramolecular amido transfer leading to structurally diverse nitrogen-containing macrocycles. Angew. Chem. Int. Ed. 56, 3344–3348 (2017).
Li, Y., Hu, M. & Li, J.-H. Silver-catalyzed intermolecular [3 + 2]/[5 + 2] annulation of N‑arylpropiolamides with vinyl acids: facile synthesis of fused 2H‑benzo[b]azepin-2-ones. ACS Catal. 7, 6757–6761 (2017).
Hou, S.-H., Yu, X., Zhang, R., Wagner, C. & Dong, G. B. Rhodium-catalyzed diastereo- and enantioselective divergent annulations between cyclobutanones and 1,5-enynes: rapid construction of complex C(sp3)‑rich scaffolds. J. Am. Chem. Soc. 144, 22159–22169 (2022).
Hu, H. et al. Copper-catalysed benzylic C–H coupling with alcohols via radical relay enabled by redox buffering. Nat. Catal. 3, 358–367 (2020).
Acknowledgements
This work was supported by the National Key Research & Development Program of China (2021YFF0701800, S.-Y.Z. and 2022YFC2703400, S.-Y.Z.), the Fundamental Research Funds for the Central Universities (YG2022ZD021, S.-Y.Z.), and STCSM (22ZR1435100, S.-Y.Z.).
Author information
Authors and Affiliations
Contributions
The project was conceived and directed by S.-Y.Z. (Shu-Yu Zhang), L.W. designed the experiments and analyzed the data. S.-Y.Z. (Shen-Yuan Zhang) and Z.-H.L. performed the DFT calculations. L.W., Y.S., and J.X. performed the experiments. L.W., P.-F. L., and T.-M. D. prepared the manuscript. All authors discussed the results and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Xinyao Li and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, L., She, Y., Xiao, J. et al. Allylic C–H oxygenation of unactivated internal olefins by the Cu/azodiformate catalyst system. Nat Commun 16, 870 (2025). https://doi.org/10.1038/s41467-025-56230-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-56230-0
