
Abstract
Lysosomes are best known for their roles in inflammatory responses by engaging in autophagy to remove inflammasomes. Here, we describe an unrecognized role for the lysosome, showing that it finely controls macrophage inflammatory function by manipulating the lysosomal Fe2+—prolyl hydroxylase domain enzymes (PHDs)—NF-κB—interleukin 1 beta (IL1B) transcription pathway that directly links lysosomes with inflammatory responses. TRPML1, a lysosomal cationic channel, is activated secondarily to ROS elevation upon inflammatory stimuli, which in turn suppresses IL1B transcription, thus limiting the excessive production of IL-1β in macrophages. Mechanistically, the suppression of IL1B transcription caused by TRPML1 activation results from its modulation on the release of lysosomal Fe2+, which subsequently activates PHDs. The activated PHDs then represses transcriptional activity of NF-κB, ultimately resulting in suppressed IL1B transcription. More importantly, in vivo stimulation of TRPML1 ameliorates multiple clinical signs of Dextran sulfate sodium-induced colitis in mice, suggesting TRPML1 has potential in treating inflammatory bowel disease.
Similar content being viewed by others


Introduction
The primary function of the lysosome, a membrane-bound intracellular organelle, is to break down cytosolic molecules with its various digestive hydrolases1. The cytosolic cargo destined for lysosomal degradation are delivered by endosomes or autophagosomes, the latter process is referred to as autophagy2. It is well accepted that the lysosome plays a role in regulating inflammatory responses by engaging in the autophagy process3. For instance, several studies have suggested that autophagy plays a critical role in preventing excessive NLRP3 (NOD-, LRR- and pyrin domain-containing protein 3) inflammasome activation4,5. Timely clearance of inflammasomes by lysosomes via the autophagy process prevents maturation of key proinflammatory cytokines including interleukin-1β (IL-1β) and IL-18, thereby reducing proinflammatory responses. Furthermore, macrophages derived from Atg16L1 knockout mice that are defective in autophagy exhibit enhanced Lipopolysaccharides (LPS)-induced IL-1β production6, suggestive of an anti-inflammatory effect of autophagy. In addition, a variant of the ATG16L1 gene causes increased production of IL-1β in macrophages, thus being strongly associated with the incidence of Crohn’s disease, a type of inflammatory bowel disease (IBD)7.
In addition to the autophagy pathway, studies directly linking lysosomes with inflammation have emerged identifying certain lysosomal enzymes that play a role in autoimmune diseases. For example, defects in α-galactosidase, a lysosomal enzyme, cause aberrant accumulation of lipid antigens and activation of invariant natural killer T cells, which end up in autoimmunity8. Besides, lysosomes regulate innate immune response via the transcription factors EB (TFEB) pathway, a master regulator of lysosome biogenesis, by controlling transcriptional expression of pro-inflammatory cytokines/chemokines involved in antimicrobial host defense or oxidant genes9,10,11,12.
TRPML1, encoded by MCOLN1 gene, is a non-selective cationic channel specifically localized on the lysosome and exerts lysosomal function by maintaining ionic homeostasis in various types of cells that include immune cells such as nature killer cells, memory B cells and macrophages13,14,15,16,17. Functionally, TRPML1 channel engages in a variety of intracellular processes including endocytosis, exocytosis and autophagy by mediating the release of lysosomal Ca2+, Zn2+ and Fe2+’18. In addition, reactive oxygen species (ROS) serve as endogenous activator of the TRPML1 channel19. This implies that TRPML1 may participate in the pathological processes of a range of diseases in which ROS levels are abnormally elevated such as in inflammation responses, neurodegenerative diseases, and myocardial ischemia-reperfusion (I/R) injury20.
In this work, considering the crucial role of TRPML1 channel in maintaining functional lysosomes, we wanted to determine if TRPML1 channel plays a role in regulating inflammation process in macrophages as well as the potential underlying mechanisms. Here, we show that TRPML1 channel prevents sustained inflammation by limiting PHDs-regulated NF-κB activation in macrophages.
Results
The activity of TRPML1 channel finely controls macrophage IL-1β production
To explore whether TRPML1 channel plays a role in regulating inflammatory responses, we began with comparing the expression of MCOLN1 in peripheral blood leukocytes taken from patients diagnosed with Ulcerative Colitis (UC) or Crohn’s disease (CD), two main IBD disorders whose pathogenesis is closely correlated with defective autophagy, and healthy controls. Strikingly, leukocyte expression of MCOLN1 in patients with either UC or CD was significantly down-regulated compared to healthy controls (Fig. 1a). Consistent with this, gene expression profiling of GSE53306 revealed that intestinal MCOLN1 expression in patients with active UC was also down-regulated compared to healthy controls. Notably, when patients with UC were inactive, MCOLN1 expression was not different to that of Healthy controls (Fig. 1b). By contrast, the other two members of TRPML family, TRPML2 (gene name: MCOLN2) or TRPML3 (gene name: MCOLN3) mRNA levels were comparable between UC and Healthy controls (Fig. 1b). Consistently, TRPML1 mRNA expression was significantly down-regulated in BMDMs following 4 hours and longer periods of LPS stimulation (Supplementary Fig. 1A). Furthermore, the intestinal TRPML1 expressions of both mRNA and protein levels negatively correlated with severity of colitis in mice induced by DSS (3%; via drinking water for 6 days21) (Supplementary Fig. 1B, C), whereas protein levels of TRPML2 and TRPML3 in colonic tissues remained unchanged in DSS-treated mice (moderate and severe) (Supplementary Fig. 1D). Collectively, these results demonstrate that TRPML1 channel plays a role in the pathogenesis of IBD.

a MCOLN1 mRNA levels were analyzed in leukocytes harvested from the serum of healthy controls, UC, or CD patients. The line inside the box denotes the median value, while the top and bottom of the box contain the 25th to 75th percentiles of the dataset. The whiskers indicate the minimum and maximum values. b MCOLN1, MCOLN2, and MCOLN3 mRNA levels in colonic tissues collected from healthy controls, active UC, or inactive UC patients were analyzed based on the RNA-seq data. c Volcano plot representing differentially expressed genes in BMDMs treated with LPS (100 ng/mL) vs LPS (100 ng/mL) + ML-SA5 (0.1 μM) conditions. d GSEA enrichment plot displayed IBD signaling pathway (Hsa05321) was enriched within the down-regulated genes in the ML-SA5-treated BMDMs upon LPS stimulation. e IL1B mRNA levels in BMDMs under the indicated conditions. f Relative IL1B mRNA expression in TRPML1-Tet expressed BMDMs under Vehicle or LPS treatment. g Relative mRNA expression of IL1B in BMDMs cells under LPS or LPS + ML-SA5 treatment for 0, 4, 8 or 24 h. h IL1B mRNA levels under LPS or LPS + ML-SA5 treatment in BMDMs collected from WT or MCOLN1 KO mice. i Relative IL1B mRNA levels in NC shRNA or ML1 shRNA expressed BMDMs under the indicated conditions. j Relative IL6 mRNA expression in BMDMs under LPS or LPS + ML-SA5 treatment for 0, 4, 8, or 24 h. k Representative immunoblots and quantitative analysis of pro-IL-1β protein levels in BMDMs under the indicated conditions. l Representative immunoblots and quantitative analysis of pro-IL-1β protein levels in BMDMs collected from MCOLN1 KO mice under the indicated treatments. m, n Secreted IL-1β by WT BMDMs (m) or by MCOLN1-null BMDMs (n) under the indicated conditions was as measured by ELISA assay. ATP (4 mM for 1 h) was added to facilitate the secretion of IL-1β in all groups. The line inside the box denotes the median value and the whiskers indicate the minimum and maximum values. Means ± SEMs were shown in b, e–h, I, j, k. Significant differences were evaluated using one-way ANOVA followed by Tukey’s test.
Levels of the three major pro-inflammatory cytokines, interleukin-6 (IL6), tumor necrosis factor (TNF), and IL-1β, are significantly elevated in both sera collected from IBD patients and in colonic tissues of DSS-treated mice (Supplementary Fig. 1E; GSE148505 gene set). This may positively correlate with poor outcomes of IBD22,23. Our RNA-seq data showed that the IL1B mRNA expression as well as the KEGG IBD pathway (hsa05321) was suppressed following TRPML1 channel activation using its selective agonist, ML-SA5 (0.1 μM)24,25,26, in LPS-stimulated Bone-marrow-derived macrophages (BMDMs; Fig. 1c, d). Given these results, we systematically investigated the role of TRPML1 channel in regulating IL-1β expression.
Stimulating TRPML1 channel with ML-SA5 (0.1 μM) markedly suppressed IL1B mRNA expression in inactive BMDMs (Fig. 1e) and macrophage-like RAW264.7 cells (Supplementary Fig. 1F) in a time-dependent manner (Supplementary Fig. 1G). Alternatively, stimulating TRPML1 channel with treatment of MK6-83 (0.5 μM) or ML-SA1 (2 μM) similarly suppressed the expression of IL-1β mRNA in both inactive and LPS-stimulated BMDMs (Supplementary Fig. 1H). Consistent with this, TRPML1 channel overexpression (TRPML1-Tet on; Supplementary Fig. 2A) in inactive BMDMs or RAW264.7 cells also resulted in significant suppression of IL1B mRNA expression in these cells (Fig. 1f and Supplementary Fig. 2B). Conversely, inhibiting TRPML1 channel with its selective antagonist, ML-SI3 (10 μM)15,27, significantly increased IL1B mRNA expression in inactive BMDMs and RAW264.7 cells (Fig. 1e and Supplementary Fig. 1F). Collectively, these results demonstrate that TRPML1 channel activity bidirectionally regulates transcriptional IL1B expression in macrophages.
We further investigated whether activation of TRPML1 channel also suppresses the enhanced IL1B mRNA expression in pro-inflammatory macrophages stimulated by LPS treatment. The significantly elevated IL1B expression, triggered by LPS treatment (100 ng/ml for 4, 8 and 24 h), was also suppressed by application of ML-SA5 (0.1 μM) (Fig. 1g and Supplementary Fig. 2C, D) or TRPML1 channel overexpression (Fig. 1f and Supplementary Fig. 2E) in both BMDMs and RAW264.7 cells. Co-application of ML-SI3 (10 μM) abolished suppression of IL1B expression resulting from ML-SA5 treatment (0.1 μM) in both inactive and LPS-stimulated BMDMs (Fig. 1e and Supplementary Fig. 2C), as well as RAW264.7 cells (Supplementary Fig. 1F, 2D). These results suggest that the suppressive effect of ML-SA5 treatment on transcriptional IL1B expression is delivered through TRPML1 channel in both inactive and pro-inflammatory macrophages. In agreement with the results of ML-SI3 treatment, ML-SA5 application (0.1 μM for 12 h) had no effects on IL1B expression in both inactive and LPS-stimulated BMDMs collected from MCOLN1 knockout (KO) mice (Fig. 1h and Supplementary Fig. 2F, G). More importantly, inhibiting TRPML1 channel activity, either by the removal of MCOLN1 gene (Fig. 1h) or by ML-SI3 treatment (Supplementary Fig. 2H), significantly facilitated the increase in IL1B transcription induced by LPS treatment. In addition, the KEGG IBD pathway (hsa05321) was significantly up-regulated in the LPS-stimulated MCOLN1-null BMDMs compared to the LPS-stimulated WT BMDMs (Supplementary Fig. 2I). This is indicative of the crucial role of TRPML1 channel in controlling IL1B expression, and consequently, the inflammatory responses elicited by infectious stimulation. Consistent with this, in comparison to lentiviral-negative control (LV-NC) shRNA expressed BMDMs, down-regulating TRPML1 channel with LV-ML1 shRNA (Supplementary Fig. 2J) abolished suppression of IL1B expression following ML-SA5 treatment (0.1 μM) in both inactive and LPS-stimulated BMDMs (Fig. 1i). These results confirmed that activation of TRPML1 channel specifically suppresses transcriptional IL1B expression in both inactive and LPS-stimulated BMDMs.
As demonstrated in IBD pathway, IL1B, IL6 and Tnf are the most common pro-inflammatory genes, regulated by NF-κB signaling pathway, our investigation employed both unbiased RNA-seq analysis and qPCR analysis to carefully measure the changes in expression of IL6 and Tnf, alongside IL1B, in LPS-stimulated BMDMs treated with ML-SA5. With these approaches, we found that the expression levels of Il6 or Tnf remained largely unchanged following activation of TRPML1 channel at 4, 8 and 24 hours compared with the expression of IL1B in LPS-treated BMDMs (Fig. 1c, j and Supplementary Fig. 2K).
Next, we evaluated the changes in pro IL-1β protein expression in response to TRPML1 channel activation. In agreement with changes in IL1B transcription, ML-SA5 application (0.1 μM) abolished the increase in pro IL-1β protein levels triggered by LPS stimulation (100 ng/ml for 24 h) in BMDMs (Fig. 1k). Similarly, treatment of MK6-83 (0.5 μM) or ML-SA1 (2 μM) significantly suppressed pro-IL-1β protein levels in LPS-stimulated BMDMs (Supplementary Fig. 3A), resembling the effects of ML-SA5 treatment. Moreover, overexpression of TRPML1 channel in BMDMs also resulted in a significant suppression on pro IL-1β protein levels in the cells (Supplementary Fig. 3B). The suppression of pro IL-1β protein levels resulting from ML-SA5 treatment was inhibited by co-application of ML-SI3 (10 μM) (Fig. 1k). Furthermore, deletion of TRPML1 channel prevented ML-SA5 treatment from suppressing pro IL-1β protein in LPS-stimulated BMDMs collected from MCOLN1 KO mice (Fig. 1l). Likewise, down-regulating TRPML1 with LV-ML1 shRNA abolished the suppression of pro IL-1β protein following ML-SA5 treatment in LPS-stimulated BMDMs (Supplementary Fig. 3C). In addition to this, ML-SA5 application suppressed pro IL-1β protein levels in both inactive and LPS-stimulated RAW264.7 (Supplementary Fig. 3D).
Finally, enhanced IL-1β secretion from BMDMs, triggered by LPS stimulation, was also suppressed by either ML-SA5 treatment (0.1 μM) (Fig. 1m) or TRPML1 channel overexpression (Supplementary Fig. 3E), as assessed by ELISA assay. In comparison to wild type (WT) mice, IL-1β secretion (Fig. 1n) and pro IL-1β protein levels (Supplementary Fig. 3F) were greatly enhanced in BMDMs collected from MCOLN1 KO mice in response to LPS stimulation, further indicating that TRPML1 channel plays an essential role in restricting excessive production of IL-1β in macrophages in response to inflammatory stimuli.
Much like LPS, Zymosan-A induces pro-inflammatory cytokines in immune cells via Toll-like receptor (TLR) 2 signaling28, while Resiquimod activates TLR7/8 to exert antiviral and antitumoral activity29. We also tested the effect of ML-SA5 on the suppression of expression of IL-1β mRNA and pro Il-1β protein levels in macrophages treated with Zymosan-A or Resiquimod. As observed, the activation of TRPML1 with ML-SA5 significantly suppresses the elevated IL-1β mRNA and pro IL-1β protein levels caused by Zymosan-A (100 μg/mL) or Resiquimod (1 μg/mL) in both BMDMs and RAW264.7 cells (Supplementary Fig. 4A–C) as if by LPS treatment. These experiments suggest that TRPML1 channel regulates the process of inflammation in response to diverse stimuli in general.
TRPML2, located in early/recycling endosomes, has been implicated in the release of chemokine CCL2 from macrophages and in macrophage migration during the innate immune response30. Therefore, we have used genetic depletion techniques (MCOLN2 or MCOLN3 shRNA; Supplementary Fig. 4D) to further exclude the possible involvement of TRPML2 or TRPML3 in the regulation of IL1B expression following ML-SA5 treatment in both inactive and LPS-treated BMDMs. We observed that the knockdown of either TRPML2 or TRPML3 did not alter the suppression of IL-1β mRNA levels induced by ML-SA5 treatment in both inactive or LPS-stimulated BMDMs (Supplementary Fig. 4E). Furthermore, the activation of TRPML1 channel with ML-SA5 treatment suppressed IL-1β mRNA and protein levels in MCOLN2 KO or MCOLN3 KO HEK293T cell lines (Supplementary Fig. 4F–H). The extent of suppressions of IL-1β mRNA and protein levels resulting from ML-SA5 treatment was comparable between WT and MLOLN2 KO or MCOLN3 KO HEK293T cells (Supplementary Fig. 4G, H). These results were contrast to the ones that ML-SA5 treatment failed to induce suppression of IL-1β mRNA and protein levels in both inactive and LPS-stimulated MCOLN1-null BMDMs (Fig. 1h, l and Supplementary Fig. 2G).
When taken together, these results demonstrate that TRPML1 channel activity regulates macrophage inflammatory function by finely controlling pro IL-1β production and subsequent secretion of cleaved IL-1β in both inactive and pro-inflammatory BMDMs.
The activation of TRPML1 channel induces the release of lysosomal Fe2+, which in turn activates PHDs, resulting in suppression of macrophage IL1B transcription
We next sought to explore the mechanisms through which TRPML1 channel activity regulates IL1B transcription. First, we used various chelators to define which cation is the effector of TRPML1 channel and therefore responsible for suppressing IL1B transcription in macrophages. Co-application of ML-SA5 with the iron chelators dexrazoxane (DXZ) (200 μM)31,32,33,34, 2,2’-bipyridyl (150 μM)35,36, deferoxamine (DFX) (200 μM)37, or deferasirox (100 μM)38 abolished the suppression of IL-1β mRNA and protein levels induced by ML-SA5 treatment (0.1 μM) in BMDMs and RAW264.7 cells (Fig. 2a, b and Supplementary Fig. 5A–C). Notably, application of DXZ or 2,2’-bipyridyl completely abolished the suppression of IL-1β mRNA and protein levels induced by ML-SA5 treatment (Fig. 2a, b). By comparison, the application of DFX only partially, but still significantly, reversed the suppression of IL-1β mRNA and protein levels caused by ML-SA5 treatment (Fig. 2a, b). Our reasoning for the partial reversal observed with DFX, less effective than DXZ or 2,2’-bipyridyl, is that DFX may exhibit toxicity to BMDMs, assessed by cell viability assay (Supplementary Fig. 5A). In addition, application of DXZ (200 μM) or DFX (200 μM) alone significantly increased IL1B mRNA levels in both inactive and LPS-treated BMDMs and RAW264.7 cells (Supplementary Fig. 5D). Conversely, application of ferric ammonium citrate (FAC; 50 μM) that increases labile cytoplasmic iron pool, led to the suppression of IL1B mRNA levels in both inactive and LPS-treated BMDMs as well as RAW264.7 cells (Supplementary Fig. 5E). These results suggest a role of iron homeostasis in the regulation of transcriptional expression of IL1B in macrophages. In contrast, neither the Ca2+ chelator Bapta-am (20 μM) nor the zinc chelator TPEN (10 μM)24, altered suppression of IL1B transcription levels and protein levels following ML-SA5 treatment (Fig. 2a and Supplementary Fig. 5F). Furthermore, application of the autophagy inhibitor Wortmannin (100 nM pretreatment for 2 h)20,26 had no effects on the ML-SA5-induced suppression on IL1B transcription in BMDMs, thereby indicating that the suppression on IL1B transcription by ML-SA5 treatment does not result from the effect of TRPML1 channel on regulating autophagy process by mediating the release of lysosomal Zn2+24,25, (Fig. 2a).

a Relative IL1B mRNA levels in BMDMs under control, ML-SA5 (0.1 μM), ML-SA5 + DXZ (200 μM), ML-SA5 + 2, 2’-Bipyridyl (150 μM), ML-SA5 + DFX (200 μM), ML-SA5 + Bapta-am (20 μM), ML-SA5 + TPEN (10 μM), or ML-SA5 + Wortmannin (100 nM pretreatment for 2 h). All treatments were for 24 h. n = 4-16. b Representative immunoblots and quantitative analysis of pro IL-1β protein levels in BMDMs under control, LPS, LPS + ML-SA5, LPS + ML-SA5 + DXZ, LPS + ML-SA5 + 2, 2’-Bipyridyl, LPS + ML-SA5 + DFX, DXZ, 2, 2’-Bipyridyl, or DFX. All treatments were for 24 h. n = 3–6. c Relative ODD-luciferase activity of BMDMs following control, ML-SA5 (0.1 μM), LPS (100 ng/mL), and LPS + ML-SA5 conditions was measured by ODD-luciferase reporter assays. All treatment were for 4 h. n = 4–8. d Relative ODD-luciferase activity of BMDMs following LPS, LPS + ML-SA5, LPS + ML-SA5 + ML-SI3 (10 μM), or LPS + ML-SA5 + 2, 2’-Bipyridyl conditions were measured by ODD-luciferase assays. All treatment were for 4 h. n = 4–8. e Relative IL1B mRNA levels in BMDMs under control, ML-SA5, ML-SA5 + Roxadustat (100 μM), ML-SA5 + Vadadustat (200 μM), or ML-SA5 + GSK1278863 (250 μM). All treatments were for 24 h. n = 4–10. f Representative immunoblots and quantitative analysis of pro IL-1β protein levels in BMDMs under control, LPS, LPS + ML-SA5, LPS + ML-SA5 + Roxadustat, LPS + ML-SA5 + Vadadustat, LPS + ML-SA5 + GSK1278863, Roxadustat, Vadadustat, or GSK1278863. All treatments were for 24 h. n = 3–6. g Relative IL1B mRNA levels under control, ML-SA5, LPS, or LPS + ML-SA5 in NC shRNA, PHD1 shRNA, PHD2 shRNA, PHD3 shRNA, or PHD1 shRNA + PHD2 shRNA (denoted as PHD1 + 2 shRNA) expressed BMDMs. All treatments were for 24 h. n = 4–6. h Representative immunoblots and quantitative analysis of pro IL-1β protein levels under LPS or LPS + ML-SA5 in NC shRNA, PHD1 shRNA, PHD2 shRNA, PHD3 shRNA, or PHD1 + 2 shRNA expressed BMDMs. All treatments were for 24 h. n = 3–12. Means ± SEMs were shown in a–h. Significant differences were evaluated using one-way ANOVA followed by Tukey’s test.
Consistent with the alterations in IL1B transcription, suppression of pro IL-1β protein levels following ML-SA5 treatment was abolished with co-application of either DXZ, 2,2’-bipyridyl, or DFX, in LPS-stimulated BMDMs, whereas these iron chelators alone did not affect pro IL-1β protein levels (Fig. 2b). In contrast to this, co-application of either Bapta-am (20 μM), or TPEN (10 μM), or pretreatment with Wortmannin (100 nM pretreatment for 2 h), did not rescue the ML-SA5-induced suppression of pro IL-1β protein levels in pro-inflammatory BMDM stimulated by LPS treatment (Supplementary Fig. 5F).
To exclude the possible involvement of ML-SA5 treatment in the inflammasome pathway, we have determined that ML-SA5 treatment (0.1 μM) does not alter the LPS-induced activation of either NLRP3 inflammasome or caspase-139 in BMDMs (Supplementary Fig. 6A), which are both required to cleave pro IL-1β for maturation of IL-1β. To exclude its effects on the autophagy process, we have demonstrated that ML-SA5 application (0.1 μM) does not affect autophagic flux in either inactive or LPS-stimulated BMDMs, as assessed by the measurement on lipidated LC3 (LC3-II) and p62 (Supplementary Fig. 6B). In addition to using autophagy inhibitor Wortmannin, we tested the effect of TRPML1 in suppressing IL-1β mRNA and protein levels when inhibiting autophagy initiation with ATG5 shRNA (which affects phagosome expansion)40 in BMDMs (Supplementary Fig. 6C). We found that the inhibition of autophagy by ATG5 shRNA did not affect the suppression of IL-1β mRNA following ML-SA5 treatment in BMDMs (Supplementary Fig. 6D). In addition, we employed WT normal rat kidney epithelial (NRK) cells and the corresponding ATG12 KO NRK cells, in which autophagy is not able to be initiated24. As observed, ML-SA5 treatment suppressed mRNA and protein levels of IL-1β in ATG12 KO cell lines, and the extent of suppressions of IL-1β mRNA and protein levels resulting from ML-SA5 treatment was comparable between WT and ATG12 KO NRK cells (Supplementary Fig. 6E, F). These results further indicate that the TRPML1 channel does not control macrophage inflammation through the regulation of the autophagy process. Instead, it appears to act via the regulation of the “lysosomal Fe2+—PHDs—NF-κB—IL1B transcription” axis.
Furthermore, we have systematically examined the potential involvement of TFEB in the TRPML1-regulated IL1B transcription. Firstly, we synchronized BMDMs by starvation/refeeding (serum starvation overnight and 1-hour amino acid starvation/full nutrient medium for 2 hours)41,42 to reset cytosolic localization of TFEB. With this approach, we observed that nuclear translocation of TFEB was not induced by ML-SA5 treatment at the concentration of 0.1 μM in BMDMs (Supplementary Fig. 7A). Furthermore, ML-SA5 application at the concentration of 0.1 μM did not alter the LPS-induced nuclear translocation of TFEB (Supplementary Fig. 7A). These results confirmed that activation of TRPML1 with ML-SA5 treatment at the concentration of 0.1 μM does not alter TFEB activation in inactive and LPS-stimulated macrophages. Secondly, we compared the suppression of IL-1β mRNA and protein levels resulting from ML-SA5 treatment between BMDMs expressing NC shRNA and those expressing TFEB shRNA (Supplementary Fig. 7B). We found that knockdown of TFEB did not alter the suppression of IL-1β mRNA levels and protein levels caused by ML-SA5 treatment in BMDMs (Supplementary Fig. 7C, D). In light of the knockdown experiments of TFEB performed on BMDMs (Supplementary Fig. 7B–D), we proceeded to investigate the regulation of TRPML1 channel on both IL1β mRNA and protein levels in the TFEB/TFE3 double KO (DKO) Hela cell line (Supplementary Fig. 7E). The elevations in IL-1β mRNA and protein levels following LPS treatment were reduced in TFEB/TFE3 DKO Hela cells compared to those in WT Hela cells treated with LPS (Supplementary Fig. 7F, G). These results indicate a role of TFEB/TFE3 in the transcriptional regulation in the innate immune response consistent with the previous report9. However, the suppression of IL1β mRNA and protein levels resulting from ML-SA5 treatment was not altered in the TFEB/TFE3 DKO Hela cell line compared with the WT Hela cells (Supplementary Fig. 7F, G). These results have comprehensively ruled out the involvement of TFEB in the TRPML1-regulated IL1B transcription in macrophages.
When taken together, these results suggest that the suppression of IL1B transcription following ML-SA5 treatment results from modulation of TRPML1 channel on the release of lysosomal Fe2+ in BMDMs, but not ones of either lysosomal Ca2+, or lysosomal Zn2+ that is a determinant in the TRPML1-regulated autophagy process24,25.
In accordance with the conclusion that lysosomal Fe2+ is the downstream effector of TRPML1 channel suppressing IL1B transcription, gene set enrichment analysis (GSEA) of our RNA-seq data revealed that the iron metabolism signaling pathway was enriched within the up-regulated genes in LPS-stimulated BMDMs when exposing the treatment of ML-SA5 (Supplementary Fig. 8A). Specifically, some iron metabolism-related genes including Slc7a11 (cystine/glutamate antiporter; also see Fig. 1c), Tfrc (transferrin receptor) and Ncoa4 (nuclear receptor coactivator 4) were significantly up-regulated following exposure to ML-SA5 in LPS-stimulated BMDMs (Supplementary Fig. 8A). It is well acknowledged that TRPML1 mediates the release of lysosomal Fe2+43, and our results further prove that activation of TRPML1 channel stimulated by ML-SA5 treatment (0.1 μM) induces the release of lysosomal Fe2+ to the cytosol in BMDMs, as manifested by the reduction in the co-localization of Fe2+ stained by the Labile Fe2+ probe (green) with lysosomes represented by LysoTracker (red) (Supplementary Fig. 8B). Co-application of ML-SI3 (10 μM) abolished the release of lysosomal Fe2+ induced by ML-SA5 in BMDMs (Supplementary Fig. 8B). To further determine the relationship between the expression of TRPML1 channel and its Fe2+ efflux activity of the channel, we monitored the release of lysosomal Fe2+ into the cytosol following the up-regulation of TRPML1 channel. This was achieved by overexpressing the channel in BMDMs (TRPML1-Tet on; Supplementary Fig. 2A). In comparison to naïve BMDMs, the overexpression of TRPML1 channel in BMDMs triggered the release of lysosomal Fe2+ into the cytosol. This was evident from the notable reduction in the co-localization of Fe2+ stained by the Labile Fe2+ probe (green) with lysosomes, represented by LysoTracker (red), in TRPML1-overexpressed BMDMs (Supplementary Fig. 8C). This phenomenon was similar to the one observed in ML-SA5-treated BMDMs (Supplementary Fig. 8B). In contrast, ML-SA5 treatment did not affect the release of lysosomal Fe2+ in MCOLN1-null BMDMs (Supplementary Fig. 8C). In addition, ML-SA5 treatment did not affect the degradation capability, the numbers, or the acidic pH of lysosomes in BMDMs, as assessed by the DQ-BSA assay and LysoTracker staining (Supplementary Fig. 8D, E). When taken together, these results indicate that the activation of TRPML1 channel, whether induced by ML-SA5 treatment or by overexpression of the channel, leads to the release of lysosomal Fe2+ into the cytosol in BMDMs.
Post-transcriptional regulation of intercellular iron homeostasis occurs through the action of iron regulatory proteins, specifically iron regulatory protein (IRP). Alternatively, we utilized the ACO1 (IRP1)/IREB2 (IRP2) RNA binding assay to monitor the release of lysosomal Fe2+ into the cytosol following TRPML1’s opening in BMDMs. In response to TRPML1’s activation with ML-SA5 treatment, lysosomal Fe2+ was released into the cytosol, as manifested by the decrease in ACO1/IREB2 RNA binding in both inactive and LPS-stimulated BMDMs (Supplementary Fig. 8F, G). Likewise, inducing Fe2+ influx by LPS stimulation also decreased the bindings in ACO1/IREB2 RNA in BMDMs (Supplementary Fig. 8F). Furthermore, either inhibiting TRPML1 channel activity with ML-SI3 or chelating Fe2+ with 2-2’ Bipyridyl, abolished the decrease in ACO1/IREB2 RNA binding resulting from ML-SA5 treatment in LPS-treated BMDMs (Supplementary Fig. 8G). When combined, these results indicate that the activation of the TRPML1 channel, whether induced by ML-SA5 treatment or by overexpression of the channel, results in the release of lysosomal Fe2+ into the cytosol in BMDMs.
Further, two approaches to exclude the potential involvement of extracellular iron. Firstly, we used EDTA to chelate iron in the culture medium for BMDMs (iron concentration: 0.25 μM in DMEM medium + 62.5 μM in FBS; Gibco). 0 mM Fe2+ culture medium was achieved by combining medium with 5 mM EDTA (pretreatment for 10 min), as calculated with Maxchelator program (https://somapp.ucdmc.ucdavis.edu/pharmacology/bers/maxchelator/webmaxc/webmaxcS.htm). The suppression of IL1B expression caused by ML-SA5 treatment was not altered in BMDMs cultured in iron-free medium compared to those in normal medium (Supplementary Fig. 8H). This result indicates that the Fe2+ influx into the cytosol mediated by TRPML1 channel stems from the lysosome, rather than from extracellular fluids. Through this mechanism, TRPML1 channel in turn regulates the transcriptional expression of IL1B in macrophages. Secondly, we used Transferrin Receptor Protein 1 (TFR1) shRNA to down-regulate the expression of TFR1 in BMDMs (Supplementary Fig. 8I), thus limiting the extracellular iron transport into the cytosol. We confirmed that the knockdown of TFR1 did not alter lysosomal Fe2+ levels likely through a compensatory mechanism by promoting ferritinophagy, the process of iron-bound ferritin being delivered to lysosomes, replenishing iron levels in the lysosome in BMDMs (Supplementary Fig. 8J), and observed that the suppression of mRNA and protein levels of IL-1β resulting from ML-SA5 treatment did not differ in BMDMs expressing NC shRNA and those expressing TFR1 shRNA (Supplementary Fig. 8K, L). These experiments have sufficiently excluded the potential involvement of extracellular iron in the regulation of IL1B transcription resulting from TRPML1-mediated release of lysosomal Fe2+ in macrophages.
In addition, it has been suggested that DMT1 mediates iron transport across endosomes and lysosomes44, we examined the potential involvement of DMT1 in the release of lysosomal Fe2+ following ML-SA5 treatment. This was achieved by comparing the effects of ML-SA5 treatment on suppressing IL-1β mRNA and protein levels between BMDMs expressing NC (negative control) shRNA and those expressing DMT1 shRNA (Supplementary Fig. 9A). We observed that the knockdown of DMT1 did not alter the suppression of IL-1β mRNA or protein levels due to ML-SA5 treatment in both inactive or LPS-stimulated BMDMs (Supplementary Fig. 9B, C). This stands in sharp contrast to the pronounced reversal effect when TRPML1 channel was completely removed, knocked down, or inhibited using methods such as MCOLN1-deletion, ML1 shRNA or ML-SI3 treatment. By using the DMT1 knockdown approach to exclude its potential involvement, we have confirmed that the activation of TRPML1 channel is indeed responsible for mediating the release of lysosomal Fe2+.
Further, we used Perls’ Prussian blue staining in vivo to detect iron accumulation (ferric iron) in colonic tissues collected from mice administrated with DSS. Once the released lysosomal Fe2+ enters into the cytoplasm, it is ready to be utilized, chelated, or translocated to different cellular compartments45. Considering the 7 days DSS administration period, it is reasonable to be concerned that the influx of Fe2+ from the lysosome through the TRPML1 channel is redistributed to other organelles for storage or bind to iron-binding proteins such as ferritin in colonic tissues in response to inflammatory stimuli. Thus, purposely chose the Perls’ Prussian blue staining to evaluate the changes in cytosolic Fe3+ in macrophages within colonic tissues in response to inflammatory stimuli. This would indirectly visualize the influx of lysosomal Fe2+ in macrophages in colonic tissues in response to inflammatory responses. Iron accumulation in colonic tissues, induced by administration of 3% DSS, was negatively correlated with the severity of colitis in mice, as detected using the Perls’ Prussian blue staining (Blue; Supplementary Fig. 9D), as if the colonic expressions of TRPML1 did (Supplementary Fig. 1B, C). Even more important, iron accumulation in mice colonic tissues induced by treatment with DSS, or in BMDMs treated with LPS (100 ng/mL), was all inhibited by the ablation of MCOLN1 gene, as determined using the Perls’ Prussian blue staining (Supplementary Fig. 9E, F). These results further prove that TRPML1-mediated lysosomal Fe2+ release is indeed involved in the pathogenesis of IBD.
Taken all together, these results support the conclusion that the release of lysosomal Fe2+ following TRPML1’s opening is the effector responsible for suppressing macrophage IL1B transcription.
Next, we investigated the downstream effectors of lysosomal Fe2+ involved in TRPML1-regulated IL1B transcription. Previous studies have determined that Prolyl hydroxylases (PHDs), members of the Fe2+- and α-ketoglutarate (α-KG)-dependent dioxygenase enzyme family, hydroxylate IKKβ to regulate NF-κB activation through their interaction with IkB kinases (IKKs)46,47,48. As such, we sought to determine if PHDs are the downstream effectors of TRPML1-mediated lysosomal Fe2+ release and therefore responsible for suppressing IL1B transcription in macrophages. We found that PHD activity was greatly increased in response to ML-SA5 treatment (0.1 μM) in both inactive and LPS-stimulated BMDMs, as assessed by ODD-luc activity, which is inversely corelated with PHD activity49 (Fig. 2c). On top of that, in comparison with WT BMDMs, PHD activity was suppressed in inactive MCOLN1-null BMDMs, as manifested by the increase in ODD-luc activity in MCOLN1-null BMDMs (Supplementary Fig. 10A). The suppressive effect on PHD activity was further facilitated in MCOLN1-null BMDMs under LPS stimulation (100 ng/mL) compared to WT BMDMs (Supplementary Fig. 10A). Furthermore, co-application of either ML-SI3 (10 μM) or 2,2’-bipyridyl (150 μM) completely abolished ML-SA5-induced PHDs activity (Fig. 2d). These results demonstrate that lysosomal Fe2+ following the opening of TRPML1 channel activates PHDs in macrophages.
Further, co-application with the PHDs inhibitors Roxadustat (100 μM), Vadadustat (200 μM), or GSK1278863 (250 μM)50, completely abolished the suppression resulting from ML-SA5 treatment (0.1 μM) on both IL1B transcription in inactive BMDMs (Fig. 2e) and pro IL-1β protein levels in LPS-stimulated BMDMs (Fig. 2f), as well as in RAW264.7 cells (Supplementary Fig. 10B). When taken together, these results demonstrate that PHDs are activated to suppress IL1B transcription secondary to the release of lysosomal Fe2+ through TRPML1 channel in macrophages.
We also investigated which specific PHDs are the effector molecules for TRPML1 and therefore responsible for suppressing IL1B transcription by using PHD1, PHD2 and PHD3 shRNA (Supplementary Fig. 10C), respectively. Down-regulation of expression of PHD1 or PHD2, but not PHD3, enhanced the increase in IL1B mRNA stimulated by LPS (35 ~ 50-fold) in BMDMs (Fig. 2g), indicating that the activity of PHD1 or PHD2 plays a critical role in regulating IL1B transcription in macrophages. Consistent with this, down-regulation of PHD1 or PHD2 significantly inhibited ML-SA5-induced suppression of IL1B transcription (Fig. 2g) and pro IL-1β protein levels (Fig. 2h) in both inactive and LPS-stimulated BMDMs, as well as RAW264.7 cells (Supplementary Fig. 10D).
Collectively, these results demonstrate that PHD1 and PHD2 are the downstream effectors responsible for suppressing IL1B transcription in response to the release of lysosomal Fe2+ following TRPML1’s opening in BMDMs.
TRPML1-mediated PHDs activity modulates NF-κB signaling pathway, which in turn controls IL1B transcription in macrophages
It is well known that the transcriptional activity of NF-κB controls IL1B transcription and is directly regulated by PHDs51. Thus, it is conceivable that activated PHDs following TRPML1 channel activation modulate NF-κB signaling pathways that suppress macrophage IL1B transcription. To delineate the role of NF-κB signaling involved in the TRPML1-regulated IL1B transcription in BMDMs, we first monitored the changes in NF-κB activity following ML-SA5 treatment (0.1 μM) in BMDMs, as measured by a NF-κB-luciferase assay (see methods). In response to LPS stimulation, NF-κB was swiftly activated, with phosphorylated NF-κB nuclear translocation peaking within 30 minutes (Fig. 3a), as measured by the NF-κB-luciferase assay52. This activation subsequently returns to the basal level after 2 hours of LPS treatment and remains stable for the next 24 hours (Fig. 3a). This rapid rate of phosphorylated NF-κB nuclear translocation (within 30 minutes) is responsible for inducing numerous proinflammatory mediators, which in turn trigger the inflammatory response in the innate immune system. A 30-minutes treatment with ML-SA5 significantly repressed the rapidly elevated NF-κB activity induced by LPS stimulation in BMDMs (Fig. 3a). Furthermore, much like the rapid activation of NF-κB activity in response to LPS stimulation, basal NF-κB activity was also repressed within 30 min following application of ML-SA5 in inactive BMDMs. This repression was gradually followed by a gradual restoration of NF-κB activity, returning to levels close to the basal state after 120 minutes and remaining stable for up to 24 hours of ML-SA5 treatment (Fig. 3b). Therefore, it can be concluded that a 30-minutes treatment with ML-SA5 is sufficient to suppress NF-κB activity, with the peak of this repressive effect occurring within the first 30 minutes and gradually diminishing thereafter. We reasoned that the rapid repressive effect of ML-SA5 treatment is designated to counteract the rapid activation of NF-κB in response to the inflammatory stimuli. Alternatively, NF-κB transcriptional activity was significantly suppressed by ML-SA5 treatment (0.1 μM), as determined by a reduction in p65 phosphorylation, a key transcriptional subunit of NF-κB complexes, in both inactive and LPS-stimulated BMDMs (Fig. 3c) as well as in RAW264.7 cells (Supplementary Fig. 10E). Furthermore, co-application with ML-SI3 (10 μM) rescued the reduction on the phosphorylation of p65 caused by ML-SA5 treatment in BMDMs and RAW264.7 cells (Fig. 3c and Supplementary Fig. 10E). Phosphorylation of IKKα/β and IKBα kinases, two critical upstream kinases of NF-κB signal transduction cascade, were also suppressed by treatment of ML-SA5 in both inactive and LPS-stimulated BMDMs as well as in RAW264.7 cells (Fig. 3c and Supplementary Fig. 10E). Even more, we observed that the increases in phosphorylation on IKKα/β, IKBα, or p65 following 30 minutes of LPS stimulation were facilitated in MCOLN1-null BMDMs compared to the ones in WT BMDMs (Supplementary Fig. 10F). This result provides further support for the conclusion that the TRPML1 channel regulates macrophage NF-κB activity. On the other hand, phosphorylation on IKKα/β, IKBα, or p65 following LPS stimulation for 48 hours were markedly reduced in either WT or MCOLN1-null BMDMs compared to the treatment for 30 minutes (Supplementary Fig. 10F). Again, this result suggests that phosphorylated NF-κB nuclear translocation occurs rapidly within 30 minutes in response to inflammatory stimuli. Collectively, these results indicate that activation of TRPML1 channel suppresses NF-κB transcriptional activity by regulating the canonical NF-κB signal transduction cascade. When taken together, these results confirmed that the activation of TRPML1 with ML-SA5 treatment specifically suppresses NF-κB transcriptional activity in both inactive and pro-inflammatory macrophages.

a Relative NF-κB activity in LPS-stimulated BMDMs treated with either of ML-SA5 (0.1 μM) or Control for different treatment periods as indicated was assessed by a NF-κB reporter assay. n = 3. b Relative NF-κB activity in BMDMs treated with ML-SA5 for different treatment periods as indicated was assessed by a NF-κB reporter assay. n = 4. c Representative immunoblots and quantitative analysis of phosphorylated p65, IKBα and IKKα/β protein levels in BMDMs under control, LPS (100 ng/mL), LPS + ML-SA5 (0.1 μM), ML-SA5, LPS + ML-SA5 + ML-SI3 (10 μM), or ML-SA5 + ML-SI3. All treatments were for 30 min. n = 3–9. d Relative NF-κB activity in BMDMs under control, ML-SA5 (0.1 μM), ML-SA5 + ML-SI3 (10 μM), ML-SA5 + DXZ (200 μM), or ML-SA5 + Roxadustat (100 μM). All treatment were for 30 min. n = 5. e Relative NF-κB activity under control or ML-SA5 (0.1 μM) conditions in NC shRNA or PHD1 shRNA + PHD2 shRNA expressed RAW264.7 cells. All treatment were for 30 min. n = 3. f Relative IL1B luciferase activity in BMDMs under control, ML-SA5 (0.1 μM), LPS (100 ng/mL), LPS + ML-SA5 (0.1 μM), LPS + ML-SA5 + ML-SI3 (10 μM), LPS + ML-SA5 + DXZ (200 μM), or LPS + ML-SA5 + Roxadustat (100 μM) was measured by luciferase assays. All treatment were for 24 h. n = 3. g Relative IL1B luciferase activity under control, ML-SA5 (0.1 μM), LPS (100 ng/mL), or LPS + ML-SA5 (0.1 μM) in NC shRNA or p65 shRNA expressed BMDMs was measured by luciferase assays. All treatments were for 12 h. n = 3–4. h Chromatin immunoprecipitation (ChIP)-qPCR was used to amplify chromatin derived from immunoprecipitations with anti-p65 antibody. Co-precipitating chromatin fragments were analyzed by real time PCR to quantify enrichment of IL1B at the −688 of IL1B position in BMDMs treated with LPS (100 ng/mL), LPS + ML-SA5 (0.1 μM), LPS + ML-SA5 + ML-SI3 (10 μM), LPS + ML-SA5 + Roxadustat (100 μM), or LPS + ML-SA5 + 2,2’ Bipyridyl (150 μM). n = 3. All treatments were for 6 h. Means ± SEMs were shown in a–h. Significant differences were evaluated using one-way ANOVA followed by Tukey’s test.
Next, we wanted to determine if NF-κB is the terminal effector of the “TRPML1—lysosomal Fe2+—PHDs (PHD1 and PHD2)” axis that directly suppresses IL1B transcription. We found that the repression on NF-κB activity resulting from ML-SA5 treatment (0.1 μM) was abolished by co-treatment with either ML-SI3 (10 μM), DXZ (200 μM), or Roxadustat (100 μM) in BMDMs, as assessed by the NF-κB-luciferase reporter assay (Fig. 3d). These results indicate that TRPML1-regulated NF-κB activity is controlled by the “lysosomal Fe2+—PHDs” axis. In addition to this, when PHDs activity was inhibited by down-regulating PHD1 and PHD2 expression (PHD1 + 2 shRNA), the suppression of NF-κB activity following ML-SA5 treatment in RAW264.7 cells was abolished (Fig. 3e). Collectively, these results demonstrate that NF-κB is restrained in the cytosol by the activation of PHDs in response to the release of lysosomal Fe2+ following TRPML1’s opening in BMDMs.
Next, we used a IL1B-luciferase reporter assay (see methods) to determine if NF-κB directly regulates IL1B transcription in response to TRPML1 channel activation. Basal and LPS-induced IL1B luciferase activity was blocked by ML-SA5 treatment (0.1 μM) in BMDMs (Fig. 3f). Furthermore, suppression of IL1B luciferase activity resulting from ML-SA5 treatment was inhibited by the co-application of either ML-SI3, DXZ (200 μM), or Roxadustat (100 μM) (Fig. 3f). These results were consistent with the results of IL1B mRNA and further confirm that the “TRPML1—lysosomal Fe2+—PHDs” axis controls the IL1B transcription in macrophages. Furthermore, the knockdown of p65 (Supplementary Fig. 10G) abolished the repression on both basal and LPS-induced IL1B luciferase activity resulting from ML-SA5 treatment (Fig. 3g). Alternatively, we confirmed the direct regulation of the “TRPML1—lysosomal Fe2+—PHDs” axis on IL1B transcription by using the Chromatin immunoprecipitation (CHIP) assay. ChIP–PCR analysis in LPS-stimulated BMDMs showed p65 bound near the −688 position of IL1B promoter region, which was inhibited by the treatment of ML-SA5 (0.1 μM) (Supplementary Fig. 10H). Furthermore, suppression of the binding of p65 on IL1B promotor caused by ML-SA5 was inhibited by co-application of either ML-SI3 (10 μM), Roxadustat (100 μM), or 2,2’-bipyridyl (150 μM), as measured by CHIP-PCR analysis (Fig. 3h). These results demonstrate that NF-κB, regulated by the “TRPML1—lysosomal Fe2+—PHDs” signaling pathway, is the direct effector responsible for suppressing IL1B transcription in macrophages.
It has been reported that Hypoxia-inducible factor 1α subunit (HIF-1α) plays a role in LPS-induced IL1B expression53,54 and its degradation is closely regulated by PHDs55. As such, we investigated if HIF-1α plays a role in regulating IL1B transcription in response to the activation of PHDs following ML-SA5 treatment. Deletion of HIF1A in RAW264.7 cells (Supplementary Fig. 11A) subtly reduced the suppression of IL1B expression resulting from ML-SA5 treatment (Supplementary Fig. 11B). Alternatively, disrupting HIF-1α function using BAY87-2243, a potent and selective HIF-1 inhibitor56, minorly inhibited the suppression of IL1B expression by ML-SA5 treatment in both inactive and LPS-stimulated BMDMs (Supplementary Fig. 11C). These results indicate that HIF-1α plays a minor role in the regulation on macrophage IL1B transcription manipulated by the “TRPML1—lysosomal Fe2+—PHDs” axis. How PHD1 and PHD2 control the activation of NF-κB without affecting HIF-1α. Previous research has reported that PHD1 or PHD2 also regulates the transcriptional activity of NF-κB independent on HIF-1α function. Mechanistically, PHD1 or PHD2 represses the activation of NF-κB through their hydroxylation on IKKβ, a non-HIF hydroxylation substrate that regulates the phosphorylation of inhibitory IKBα to dissociate from NF-κB complex. The hydroxylation of PHD1 or PHD2 inhibits IKKβ kinase activity (more hydroxylation thus less phosphorylation), thereby reducing the phosphorylation on IKBα. As a result, the inhibited activity of ΙΚΚβ by PHD1 or PHD2 fails to trigger dissociation of the inhibitory IKBα from NF-κB, resulting in the suppressive effect on NF-κB activation47. We then measured the phosphorylation on IKKα/β or ΙΚΒα in BMDMs under control conditions, Roxadustat (Roxa) treatment, LPS stimulation, and LPS + Roxa treatment. We observed that the inhibition of PHDs activity with Rosa treatment significantly promoted the phosphorylation on IKKα/β or ΙΚΒα in LPS-stimulated BMDMs (Supplementary Fig. 11D). Further, we compared phosphorylation of IKKα/β or ΙΚΒα between BMDMs expressing NC shRNA and those expressing PHD1/2 shRNA. We found that down-regulating PHDs expression also facilitated phosphorylation of IKKα/β in LPS-stimulated BMDMs (Supplementary Fig. 11E), similar to the effect observed with Roxa treatment in macrophages (Supplementary Fig. 11D). These results indicate that PHD activation limits the phosphorylation on IKKs-IKBs to suppress NF-κB activation in macrophages.
When taken together, these results indicate that NF-κB is the terminal downstream effector responsible for directly suppressing macrophage IL1B transcription in response to the activation of PHDs following the release of lysosomal Fe2+ through TRPML1 channel.
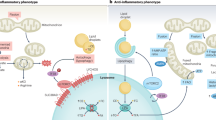
ROS are the upstream effector that activate TRPML1 channel to switch macrophages from a pro-inflammatory to an anti-inflammatory state
We have identified the downstream signaling pathways that are regulated by TRPML1 activation resulting in alterations in macrophage IL1B expression in response to inflammatory stimuli. We next wanted to find out what triggers TRPML1 channel to dampen proinflammatory responses, as represented by the state of macrophages switching from M1 to M257 in the presence of ML-SA5 (Fig. 4a). Reactive oxygen species (ROS) are an endogenous activator of TRPML1 and highly elevated during inflammation process19,58. As such, it is possible that ROS generated during the inflammation process surpass the threshold level required to activate TRPML1 channel, which in turn triggers the release lysosomal Fe2+ to regulates the “PHDs—NF-κB” signaling pathway. This will result in suppressed IL1B expression in macrophages.

a A heat map representation of the relative mRNA expressions of M1 and M2 markers in LPS-stimulated BMDMs treated with either of ML-SA5 (0.1 μM) or control by RNA-seq analysis. b Relative IL1B mRNA expression in WT or MCOLN1-null BMDMs under the treatment of LPS (100 ng/mL), LPS + NAC (3.5 mM), LPS + 2-2’ Bipyridyl (150 μM), or LPS + BAPTA-AM (20 μM). n = 4–5. c Relative mRNA levels of CXCL11, CXCL12, CD74 and CCL22 in BMDMs under LPS (100 ng/mL), LPS + NAC (3.5 mM), LPS + 2-2’ Bipyridyl (150 μM), or LPS + BAPTA-AM (20 μM) treatment. n = 4–7. All treatments were for 6 h. d, e Pro IL-1β protein levels (d) and secreted IL-1β levels (e) in WT or MCOLN1-null BMDMs under LPS (100 ng/mL) or LPS + NAC (3.5 mM) conditions. ATP (4 mM for 1 h) was added to facilitate the secretion of IL-1β in all groups. The line inside the box denotes the median value, while the top and bottom of the box contain the 25th to 75th percentiles of the dataset. The whiskers indicate the minimum and maximum values. n = 3–5. f Representative images of Perls’ Prussian blue staining for WT BMDMs under the treatment of control, LPS (100 ng/mL), or LPS + NAC (3.5 mM). Arrows denote accumulated Fe3+. Scale bar = 50 μm. The average number of Perls’ positive cells per region of interest in BMDMs under the treatments of control, LPS, or LPS + NAC. n = 5. g Representative images of lysosomal Fe2+, stained by the HMRhoNox-M probe, in WT and MCOLN1-null BMDMs under the treatment of control, LPS (100 ng/mL), or LPS + NAC (3.5 mM). Scale bar = 5 μm. Relative fluorescence intensity of HMRhoNox-M was normalized to the control condition in WT or MCOLN1-null BMDMs group. n = 25. Both WT and MCOLN1-null BMDMs were preloaded with 100 μM ferric ammonium citrate (FAC) for 2 h. Means ± SEMs were shown in b–d, f, g. Significant differences were evaluated using one-way ANOVA followed by Tukey’s test.
To test this hypothesis, we first confirmed that ROS levels in BMDMs were significantly elevated following LPS treatment (100 ng/mL), as measured by flow cytometry assay with H2DCFDA probe (Supplementary Fig. 12A), and a commonly-used antioxidant, N-acetylcysteine (NAC), inhibited the elevation of ROS in LPS-stimulated BMDMs (Supplementary Fig. 12A). ML-SA5 treatment alone (0.1 μM) did not alter ROS levels in either inactive or LPS-stimulated BMDMs (Supplementary Fig. 12B). In addition, ROS levels did not differ between WT and MCOLN1-null BMDMs with or without LPS stimulation (Supplementary Fig. 12C). Treatment of NAC (3.5 mM for 6 h) further enhanced the increase in IL1B expression induced by LPS treatment (100 ng/mL) in BMDMs collected from WT mice (Fig. 4b), which was consistent with a previous report59. The same treatment of NAC did not alter the viability of LPS-stimulated BMDMs (Supplementary Fig. 12D). In addition to IL1B, application of NAC (3.5 mM for 6 h) increased expression of the other M1 markers such as CXCL10 and CXCL11 (Fig. 4c) and decreased expression of M2 markers including CD74 and CCL22 (Fig. 4c) in LPS-stimulated BMDMs. More importantly, the same NAC application did not alter IL1B expression in LPS-stimulated BMDMs collected from MCOLN1 KO mice (Fig. 4b). These results indicate that ROS play a role in switching the state of macrophages from M1 to M2 following 6 h of LPS treatment. Similarly, chelating Fe2+ with 2-2 Bipyridyl increased expression of IL1B, CXCL10 and CXCL11 and decreased expression of CD74 and CCL22 in LPS-stimulated BMDMs as if NAC did (Fig. 4c). In contrast, treatment with Bapta-am did not alter the expressions of either IL1B, CXCL10, CXCL11, CD74, or CCL22 in LPS-stimulated BMDMs (Fig. 4c).
Likewise, a similar phenomenon was observed with both pro IL-1β proteins and secreted IL-1β levels in WT and MCOLN1-null BMDMs (Fig. 4d, e). Furthermore, NAC (3.5 μM) application significantly reduced iron accumulation in WT BMDMs, induced by treatment of LPS, as determined by Perls’ Prussian blue staining (Fig. 4f). Alternatively, we directly monitored the changes in lysosomal Fe2+ of BMDMs following LPS or LPS + NAC treatment by using HMRhoNox-M, a Fe2+-selective fluorescent probe localized on the lysosome membrane60. In agreement with the results of Perls’ Prussian blue staining, LPS triggered the release of lysosomal Fe2+ from WT BMDMs (preloaded with 100 μM of ferric ammonium citrate), as manifested by the reduction in fluorescence intensity of HMRhoNox-M. The reduction of fluorescence intensity of HMRhoNox-M resulting from LPS treatment was inhibited by co-application of NAC (Fig. 4g), again suggesting that LPS treatment results in the elevation in ROS, which in turn activate TRPML1 channel to release the lysosomal Fe2+ in macrophages. In contrast, LPS-triggered lysosomal Fe2+ release was inhibited in MCOLN1-null BMDMs, as accessed by the HMRhoNox-M probe (Fig. 4g). In accordance with the in vivo iron staining results of colonic tissues of mice (Supplementary Fig. 9E) and in vitro ones from BMDMs (Supplementary Fig. 9F), these results suggest that ROS are accumulated during the inflammation process and at a level that is sufficient to activate TRPML1, which in turn suppresses IL1B transcription to switch macrophages from a pro-inflammatory to an anti-inflammatory state.
The in vivo stimulation of TRPML1 channel ameliorates clinical signs of DSS-induced colitis in mice by suppressing IL-1β production
Given that stimulating TRPML1 channel with ML-SA5 treatment suppressed IL-1β release from pro-inflammatory BMDMs, we explored the in vivo effects of ML-SA5 administration on DSS-induced colitis in mice, an intestinal inflammation mouse model that has been developed to elucidate the pathogenesis of IBD61. ML-SA5 administration (0.1 mg/kg; intraperitoneal injection (IP)) protected against DSS (3%; via drinking water; Supplementary Fig. 13A)-induced colitis-associated lethality (Fig. 5a) and weight loss (Fig. 5b), decreased the high disease activity index (DAI; a numeric value denoting weight loss, diarrhea, and bleeding of mice) (Fig. 5c), and prevented the shortening of the colon during the course of colitis in mice (Fig. 5d). In line with these observations, colon sections harvested from ML-SA5-treated mice showed less severe mucosal damage and inflammatory cell infiltration (Fig. 5e). Furthermore, the elevated IL-1β levels both in macrophages, stained by CD68, in colonic tissues (Fig. 5f and Supplementary Fig. 13B) and in serum (Fig. 5g) harvested from mice subjected to DSS administration were all markedly reduced by ML-SA5 administration (0.1 mg/kg). Moreover, co-administration of ML-SI3 (0.5 mg/kg) abolished the effect of ML-SA5 on protecting against the DSS-induced colitis (Fig. 5b–g and Supplementary Fig. 13B). Similarly, ML-SA5 administration did not affect DSS-induced colitis-associated lethality (Supplementary Fig. 13C), weight loss (Supplementary Fig. 13D), shortening of the colon (Supplementary Fig. 13E), or elevation on serum IL-1β levels (Supplementary Fig. 13F) during the course of colitis in MCOLN1-KO mice, which sharply contrasted with the protective effect of ML-SA5 administration in WT mice (Fig. 5). These results indicate that the ameliorative effect of ML-SA5 administration on DSS-induced colitis is mediated through TRPML1 channel. Notably, ML-SA5 administration alone did not alter the properties of colon tissues in naïve mice (Fig. 5d, f). Even more important, replenishment of IL-1β (Fig. 6a, b) abolished the effects of ML-SA5 on ameliorating clinical signs of DSS-induced colitis including body weight loss (Fig. 6c), shortening of colon (Fig. 6d), disease activity (Fig. 6e), and mucosal damage (Supplementary Fig. 13G). When combined, by suppressing IL-1β production in intestinal macrophages, the in vivo stimulation of TRPML1 channel greatly improves survival and clinical signs of DSS-induced colitis in mice.

a Comparison of survival rate for C57BL/6 mice treated with 3% DSS or 3% DSS + ML-SA5 administration (0.1 mg/kg; IP injection) for 6 days (D). Mice were then monitored daily until day 12. n = 6. b Percentage of weight changes of mice treated with vehicle (n = 45), 3% DSS administration (n = 28), 3% DSS + ML-SA5 (0.1 mg/kg) administration (n = 5), or 3% DSS + ML-SA5 (0.1 mg/kg) + ML-SI3 (0.5 mg/kg) administration (n = 10) for 6 days. Initial weight of each mouse was defined as 100%. c A time course of disease activity index (DAI) for the mice shown in (b) was monitored daily until day 7. d Representative images and length measurement of colons from mice treated with vehicle, 3% DSS administration, 3% DSS + ML-SA5 (0.1 mg/kg) administration, 3% DSS + ML-SA5 (0.1 mg/kg) + ML-SI3 (0.5 mg/kg) administration, or ML-SA5 (0.1 mg/kg) alone for 6 days. n = 6–35. e Representative H&E staining of colons from mice treated with vehicle, 3% DSS administration, 3% DSS + ML-SA5 (0.1 mg/kg) administration (n = 5), or 3% DSS + ML-SA5 (0.1 mg/kg) + ML-SI3 (0.5 mg/kg) administration for 6 days. Scale bar = 50 μm. Histological analysis of colon tissue from mice under indicated administration at day 7. n = 15–18. f Representative IL-1β staining of colonic tissues collected from mice treated with vehicle, 3% DSS administration, 3% DSS + ML-SA5 (0.1 mg/kg) administration, or ML-SA5 (0.1 mg/kg) administration alone for 6 days. Relative IL-1β fluorescence intensity was normalized to the vehicle group. Scale bar = 50 μm. n = 5–8. g IL-1β levels in serum collected from mice treated with vehicle, 3% DSS administration, 3% DSS + ML-SA5 (0.1 mg/kg) administration, or 3% DSS + ML-SA5 (0.1 mg/kg) + ML-SI3 (0.5 mg/kg) administration for 6 days, as assessed by ELISA assay. The line inside the box denotes the median value, while the top and bottom of the box contain the 25th to 75th percentiles of the dataset. The whiskers indicate the minimum and maximum values. n = 5. Means ± SEMs were shown in a–f. Significant differences were evaluated using one-way ANOVA followed by Tukey’s test.

a C57BL/6 mice were injected with human recombinant IL-1β by intravenous injection (i.v) through tail veins three times during DSS administration as illustrated and administrated with ML-SA5 by intraperitoneal (i.p) injection nine times before and during DSS administration as described in the methods. b–e Serum IL-1β levels (b), percentage of weight changes (c), colon length (d), and DAI index (e) of mice treated with 3% DSS administration (n = 5), 3% DSS + ML-SA5 administration (n = 5), or 3% DSS + ML-SA5 (0.1 mg/kg) + IL-1β (0.01 mg/kg) administration (n = 5) for 6 days (measured at day 7). Initial weight of each mouse was defined as 100%. f Comparison of survival rate for WT and MCOLN1−/− mice treated with 3% DSS for 6 days. The mice were then followed until day 13. n = 6. g Percentage of weight changes in WT, MCOLN1−/−, or MCOLN1−/− mice treated with vehicle or 3% DSS for 6 days. h Colon length of WT, MCOLN1−/−, or MCOLN1−/− mice treated with vehicle or 3% DSS for 6 days. n = 6. i IL-1β levels in serum collected from WT, MCOLN1−/−, or MCOLN1−/− mice treated with vehicle or 3% DSS for 6 days (measured at day 7), as assessed by ELISA assay. The line inside the box denotes the median value, while the top and bottom of the box contain the 25th to 75th percentiles of the dataset. The whiskers indicate the minimum and maximum values. n = 6. j A working model illustrates that TRPML1 channel is activated secondarily to the accumulated ROS at the late stage of the inflammation process in macrophages, which in turn triggers the release of lysosomal Fe2+ to the cytosol, resulting in activation of PHDs. Activated PHDs subsequently restrain NF-κB in the cytosol, leading to suppression of macrophage IL1B transcription and resultant production and secretion of IL-1β from macrophages. As a result, the property of controlling production and subsequent release of IL-1β from macrophages allows the lysosome to finely limit excessive inflammatory responses after an initial insult has been dealt with. Means ± SEMs were shown in a–h. Significant differences were evaluated using one-way ANOVA followed by Tukey’s test.
Conversely, ablation of TRPML1 channel significantly weakened the resistance of mice to DSS treatment, as manifested by the shorter survival in MCOLN1−/− mice subjected to DSS administration compared with WT mice (Fig. 6f). In agreement with the survival data, weight loss (Fig. 6g), shortening of the colon (Fig. 6h) at day 7 in MCOLN1−/− or MCOLN1−/− mice subjected to DSS administration were all worse than that in WT mice. Furthermore, serum IL-1β levels (Fig. 6i) harvested from MCOLN1−/− or MCOLN1−/− mice subjected to DSS administration were elevated much more severely than that from WT mice. In agreement with this, MCOLN1 expression was down-regulated in colonic tissues collected from DSS-induced colitis mice (Supplementary Fig. 1B, C) and from patients with IBD (Fig. 1a, b), these in vivo results indicate that the down-regulated TRPML1 activity contributes to the colitis progression, and thus targeting TRPML1 may represent a promising therapeutic approach for the treatment of IBD.
Discussion
Lysosomes are best known for their roles in the inflammatory response by engaging in the autophagy process. Our work has established a previously unappreciated role of the lysosome in engaging in macrophage inflammatory responses by finely controlling IL-1β production via regulating the lysosomal Fe2+ release through TRPML1 channel. Mechanistically, ROS accumulated during a prolonged inflammatory stimulation in macrophages, cause activation of the TRPML1 channel, which triggers the release of lysosomal Fe2+. The Fe2+ influx to the cytosol in turn activates PHDs, whose activation represses the transcriptional NF-κB activity by restraining p65 in the cytosol, ultimately resulting in suppressed IL1B transcription in macrophages (Fig. 6j). As such, the property of controlling production and subsequent release of IL-1β from macrophages allows the lysosome to finely restrict excessive inflammatory responses after an insult has been dealt with. These findings have demonstrated that apart from relying on its degradative capability, the lysosome restricts excessive inflammatory responses in macrophages by modulating the release of lysosomal Fe2+. This regulation serves to dampen proinflammatory responses and facilitate the restoration of cellular and tissue homeostasis (Fig. 6j).
These findings also uncovered that TRPML1 is required for the lysosome to limit macrophage excessive inflammatory responses to inflammatory stimuli. A certain level of ROS is the integrator connecting TRPML1 and inflammatory responses (Fig. 4). During the inflammation process, ROS are accumulated (Supplementary Fig. 12A). Only in this scenario, TRPML1 is able to be activated and restricts excessive inflammatory responses, allowing healing to commence (Fig. 4). Presumably, the lysosome does not affect the early state of inflammatory responses that is a self-defense to harmful stimuli, as ROS levels are not sufficient for the activation of TRPML1 at this stage. As such, the “ROS-TRPML1-Fe2+” pathway switches macrophages from a pro-inflammatory to an anti-inflammatory state once an inflammatory stimulus has been dealt with (Fig. 6j). Several iron metabolism-related genes, including Slc7a11, TFR1 and Ncoa4, were up-regulated following exposure to ML-SA5 in LPS-stimulated BMDMs (Supplementary Fig. 8A). We propose that the up-regulation of the iron metabolism signaling pathway following TRPML1 channel activation serves as a compensatory mechanism to manage the elevated Fe2+ levels released from the lysosome after opening of the TRPML1 channel. This response is crucial, as excess in labile Fe2+ could otherwise trigger ferroptosis in macrophages. In response to TRPML1 activation, the up-regulation of Slc7a11 promotes glutathione biosynthesis to inhibit ferroptosis. Likewise, the up-regulation of NCOA4 may facilitate ferritinophagy to remove the excessive Fe2+ following TRPML1 activation by carrying more ferritin bound to the released Fe2+ into lysosomes. All these compensatory mechanisms are designed to inhibit ferroptosis in macrophages, along with suppressing inflammation following the release of lysosomal Fe2+ through TRPML1 channel. Additionally, the up-regulation of TFR1 could be a compensatory mechanism to replenish iron to the lysosome. In fact, the up-regulation of iron metabolism signaling pathway following exposure to ML-SA5 in LPS-stimulated BMDMs further supports the notion that TRPML1 channel mediates the release of lysosomal Fe2+ in macrophages in response to inflammatory stimuli. This conclusion could be further proved by the observations that chelating Fe2+ with 2-2 Bipyridyl increased expression of IL1B, CXCL10 and CXCL11 and decreased expression of CD74 and CCL22 in LPS-stimulated BMDMs as if NAC did (Fig. 4c), whereas treatment with Bapta-am (a Ca2+ chelator) did not alter the expressions of either IL1B, CXCL10, CXCL11, CD74, or CCL22 in LPS-stimulated BMDMs (Fig. 4b, c). However, it remains elucidated that how environmental cues such as ROS activate TRPML1 channel to mediate the release of Fe2+ over Ca2+ or Zn2+, which in turn exerts the anti-inflammatory function in macrophages.
As demonstrated in IBD pathway, IL1B, IL6 and Tnf are among the most common recognized pro-inflammatory genes regulated by NF-κB signaling pathway. We were intrigued by the observation that the activation of TRPML1 channel did not significantly affect the expression of either Il6 or Tnf during the inflammatory process in BMDMs. In line with our study, several research groups have reported similar observations. For instance, Tannahill et al. reported that inhibiting glycolysis with 2-deoxyglucose (2DG) suppresses LPS-induced transcriptions of IL1B, but not Tnf or IL6, in mouse macrophages53. In the discussion, the authors discussed the distinct regulation of IL1B over Il6 or Tnf resulting from 2DG in LPS-treated mouse macrophages. They interpreted that in addition to regulating the NF-κB pathway, 2DG affects the “PHD—HIF-1α” pathway to regulate transcription of IL1B through HIF-1α binding site in the promotor region of IL1B. In contrast, there are no canonical HIF-1α binding site upstream of the transcription start site for either Il6 or Tnf. The additional transcriptional regulation of IL1B by 2DG may amplify the effect on IL1B over Il6 or Tnf in LPS-stimulated macrophages. This hypothesis might apply to our observations as well, as the HIF-1α also plays a minor role in the regulation on macrophage IL1B transcription manipulated by the “TRPML1—lysosomal Fe2+—PHDs” axis (Supplementary Fig. 11).
Furthermore, as stated by Fang et al., pretreatment with palmitic acid significantly increased LPS-induced TNF-α production and secretion in macrophages, while suppressing IL-1β production and secretion62. The authors argued that the differential regulation of LPS-induced TNF-α and IL-1β production by PA was associated with cellular metabolism of PA, because inhibiting metabolism of PA with etomoxir or pretreatment with Br-PA, which cannot be metabolized, reversed these effects.
In addition to its direct transcriptional effect, individual co-activators and co-regulatory factors involved in NF-κB-mediated transcription on IL-1β may also contribute to the distinct regulation of IL-1β expression by the TRPML1 channel in macrophages. For instance, NF-κB-mediated transcription of IL-1β involves specific co-activators such as p300, which influence chromatin remodeling and exhibits histone acetyltransferase activities. This recruitment of co-regulators is specific to IL-1β and distinct from those recruited to the Tnf promoter63. Conversely, other transcription factors such as proteins of the bZIP, signal transducer and activator of transcription (STAT), and interferon regulatory factor (IRF) families may contribute to the distinctive role in enhancing Tnf transcription, but not IL1B64. Other than these evidence, the complex feedback inhibition and cross-regulation may also build up another layer of complexity on how TRPML1 affecting IL1B expression outweighs that of Tnf and Il6 expression. Therefore, the regulation of LPS-induced production of TNF-α, IL-6 or IL-1β by different environmental cues does not always appear to be synchronized in macrophages. This may be the case for the distinct regulation by the TRPML1 channel on IL1B, as well as other genes such as MAF, IL10, IL23R, IL18RAP, IL18, SMAD3, JUN, IL21R, IL2RG, and IFNGR1 (IBD pathway hsa05321) but not for Tnf or Il6. In short, our observations suggest that the NF-κB signaling contributes to the reduced expression of IL-1β by the TRPML1 channel; however, the underlying mechanism remains complex.
Zymosan-A induces proinflammatory cytokines release in immune cells via Toll-like receptor (TLR) 2 signaling28, while Resiquimod activates TLR7/8 to exert antiviral and antitumoral activity29. The activation of TRPML1 with ML-SA5 significantly suppresses the elevated IL-1β mRNA and pro IL-1β protein levels caused by Zymosan-A (100 μg/mL) or Resiquimod (1 μg/mL) in both BMDMs and RAW264.7 cells as if by LPS treatment. These experiments suggest that TRPML1 channel regulates the process of inflammation in response to diverse stimuli in general. As demonstrated in this study, ROS accumulation caused by LPS stimulation is the “switch” evoking TRPML1 channel to exert the anti-inflammatory function. Similar to LPS stimulation, it has been widely recognized that TLR agonists trigger a signaling cascade leading to the production of ROS65. For instance, Zymosan-A treatment induces ROS production through a TLR2/MyD88/c-Src/NADPH oxidase pathway66,67 and Resiquimod administration induces priming of neutrophil ROS production68. Therefore, it is conceivable that secondary to ROS accumulation, in response to different stimuli such as Zymosan-A and Resiquimod in macrophages, activation of TRPML1 channel limits the excessive inflammation via restraining IL1B transcriptions.
TFEB, a master regulator of lysosome biogenesis69, regulates inflammatory processes by controlling the expression of pro-inflammatory cytokine and chemokine genes9. Also, the cytosol-to-mitochondria translocation of TFEB has been suggested to regulate inflammation by modulating the electron transport chain complex I in S. Typhimurium-infected cells70. In addition, there are several studies reporting that TRPML1-mediated Ca2+ release from the lysosomes, promotes lysosomal biogenesis and autophagy by regulating the calcineurin-TFEB pathway71. As a result, there are possibilities that TRPML1-mediated Ca2+ release regulates inflammation via the TFEB pathway or autophagic pathway.
By using TFEB/TFE3 double KO Hela or ATG12 KO NRK cells (Supplementary Figs. 6 and 7), we have investigated the potential involvement of TFEB/TFE3 or autophagic pathway in the TRPML1-mediated macrophage inflammation. Under our experimental conditions, our results demonstrate that the TRPML1 channel regulates macrophage inflammation independent on the TFEB/TFE3 signaling pathway. Given the complexity of the TRPML1-autophagy relationship, the variation in interpretation leads to the disparity in conclusions. For instance, the dynamic changes of the autophagic flux might not be adequately captured by monitoring autophagy parameters at the limited observed timing points. Consequently, the prolonged disruption of fusion, leading to accumulation of autophagosomes, could be misinterpreted as changes in autophagy initiation. Furthermore, the variations in observations could be explained by the differences in the choice of cell types selected, experimental conditions applied, or the levels of TRPML1 expressed in the various cell lines used by the different research groups. It is well known that autophagy is a highly context-specific cellular process; these discrepancies highlight the importance of exercising caution when interpreting the changes in autophagic flux. Our conclusion is based on the overall evidence obtained from the specific cell line, treatment doses, and disease model we tested.
The implication of TRPML1 channel in IBD has been also reflected by that expression of TRPML1 in colonic tissues is negatively correlated to the severity of colitis in mice (DSS-induced) (Supplementary Fig. 1B, C) and in patients bearing IBD, a disorder involving chronic inflammation of the gastrointestinal tract (Fig. 1a, b). We reasoned that down-regulated TRPML1 caused a reduction in the release of lysosomal Fe2+ (Fig. 4g and Supplementary Fig. 9E), resulting in limited suppression of IL1B transcription upon infectious stimuli. Consequently, excessive IL-1β is secreted by macrophages, thus causing chronic inflammation in colonic tissues in DSS-treated mice and in IBD patients. In accordance with the correlation between TRPML1 expression and the severity of colitis; survival rate, body weight loss, and shortening of colon, as well as secretion of IL-1β were all worse in MCOLN1 KO mice treated with DSS when compared to WT mice. When combined, these results demonstrate that MCOLN1 is a crucial self-protective gene that limits excessive inflammatory responses in macrophages within the human body. Moreover, the down-regulation of TRPML1 expression appears to underlie the pathogenesis of chronic intestinal inflammation in IBD. Interestingly, expression of TRPML1 was down-regulated in macrophages when exposing to infectious stimuli (i.e., DSS) (Supplementary Fig. 1A–C). Furthermore, intestinal MCOLN1 expression in patients with active UC was also down-regulated compared to healthy controls (Fig. 1a). Notably, MCOLN1 expression in patients with inactive UC was not different to healthy controls (Fig. 1a). To this end, these results may indicate that epigenetic modification may play a role in reducing expression of MCOLN1, consequence of which weakens the capacity of lysosomes to suppress excessive IL-1β secretion during the inflammation process in macrophages within the human body. Moreover, the survival data, weight loss, and shortening of the colon at day 7 in MCOLN1−/− mice subjected to DSS administration were all more severe than those in MCOLN1−/− mice. These animal experimental results imply that a compensatory mechanism, potentially involving other iron release channels, may be initiated when TRPML1 channel is deleted in the MCOLN1 KO mice (Fig. 6f–i). Recent studies have implicated TRPML2 channel, localized in early and recycling endosomes, in chemokine release in response to LPS stimulation30,72 or in immune cell surveillance and viral trafficking72,73. As such, when the TRPML1 channel is compromised, there is a possibility that TRPML2, TRPML3, or another iron release channel compensates for TRPML1 to regulate macrophage inflammatory responses.
Iron supplementation has been suggested as a means to manage iron deficiency, which is a common comorbidity among IBD patients74. However, the safety of oral iron supplementation remains controversial. It has been discovered that iron can modulate bacterial growth and function, potentially exacerbating disease activity in a subset of IBD patients75. Furthermore, several animal studies have indicated that altering dietary iron intake can exacerbate DSS-induced colitis76,77. Therefore, this concern raises questions about the potential benefit effect of iron supplementation therapy to apply in the treatment of IBD. Functionally, the in vivo stimulation of TRPML1 channel, by directly suppressing IL-1β production in intestinal macrophages, greatly improves survival rate and ameliorates clinical symptoms of DSS-induced colitis in mice. Thus, our in vivo research suggests that TRPML1 could be a possible target for the prevention or treatment of IBD. In contrast to supplementation of iron, by directly suppressing IL-1β production, in vivo stimulation of TRPML1 channel ameliorates clinical signs of DSS-induced colitis in mice. These in vivo results not only further prove the molecular mechanism by which TRPML1 finely controls macrophage IL-1β production, but also represent TRPML1 as a possible target for prevention or treatment of IBD.
Methods
Mice
MCOLN1 KO mice on a C57BL/6 J background were purchased from the GemPharmatech Co., Ltd. Experimental mice were housed separately at typically 8–10 weeks of age. Animal experiments were performed at the Experimental Animal Center of Xuzhou Medical University according to the protocols that were approved by the Institute Animal Care Committee of Xuzhou Medical University. To generate experimental colitis, C57BL/6 J mice were treated with Dextran Sulfate Sodium (DSS; 3% wt/vol (w/v), Molecular Weight: 36,000–50,000; MP Biomedicals, Santa Ana, CA) via drinking water ad libitum. After acclimation, mice were randomly assigned to the following groups: Vehicle, 3% (w/v) DSS, 3% DSS + ML-SA5 (0.1 mg/kg; daily; i.p.; plus pre-administration three times (please refer to Fig. 6a)), or 3% DSS + ML-SA5 (0.1 mg/kg; daily; i.p.; plus pre-administration three times) + ML-SI3 (0.5 mg/kg; daily; i.p.). Body weight, stool softness, and blood in the rectum or stool were recorded daily from day 0 to day 7.
Assessment of colitis severity
Body weight loss, stool consistency, and fecal blood were measured daily during DSS administration. A colitis disease activity index (DAI) was calculated daily from general clinical signs including body weight loss, occult blood (no blood, blood visible in stool, or gross bleeding), and stool consistency (normal stool, loose stool or diarrhea). A score for each mouse was given based on the standard described in the paper61.
H&E and Perl’s Prussian blue staining
Proximal and distal colonic sections were fixed in 4% paraformaldehyde and embedded in paraffin. The 5-μm sections were stained with hematoxylin and eosin (H&E) or Prussian blue, respectively. Histological assessment of colitis was performed by a board-certified pathologist, who was blinded to experimental details, as previously described78.
Immunofluorescence staining
Specimens were fixed in 4% paraformaldehyde for 24 h at room temperature, then embedded with paraffin, and sliced into 5-μm sections. The sections were incubated overnight with goat anti-mouse IL-1β antibody (BD Biosciences, San Jose, CA, USA) at a concentration of 1:20 and rat anti-mouse CD68 (Bio-Rad, Hercules, CA, USA) at a concentration of 1:100. The nuclei were stained with DAPI (Invitrogen, Carlsbad, CA, USA).
Isolation, culture, and treatment of BMDMs
Bone marrow was isolated from the femora and tibiae of 6–8 weeks old C57BL/6 J mice. After mice were sacrificed, the femora and tibiae were dissected using scissors, and muscles connected to the bone and the epiphyses were removed using sterile scissors and forceps. Then the bones were flushed with a sterile syringe filled with DMEM (Sigma-Aldrich, St. Louis, MO, USA) to extrude bone marrow cells into a 15 mL sterile polypropylene tube. A 5 mL plastic pipette was used to gently homogenize the bone marrow with DMEM containing 10% FBS (inactivated at 55°C for 30 min; Gibco, Waltham, MA, USA) and supplemented with 20 ng/mL of mouse M-CSF (PEROTECH). The 1×106/mL bone marrow cell suspension was transferred into a 100-mm dish and cultured at 37°C with 5% CO2. Change the fresh culture medium every two days. On day 7, floating cells were discarded and the remaining cells were used for further studies.
Leukocyte isolation
Blood samples of IBD patients were obtained from the Shengjing Hospital under the consent of the patients and all related experiments were approved by the ethics committee of Shengjing Hospital and Xuzhou Medical University. 3 mL whole blood of the patients was drawn into EDTA tubes (BD, Franklin Lakes, NJ, USA) and homogenized with 9 mL red blood cell lysis buffer (Solarbio, Beijing, China), placed at room temperature for 5 min. The supernatant was discarded after centrifugation (1000 × g, 5 min) and sterile PBS (Hyclone, Logan, UT, USA) was used to wash the leukocytes twice gently. All samples were stored at −80 °C for RNA extraction.
Quantitative ELISA assay
The level of IL-1β in mice serum or in supernatant of BMDMs culture was measured by ELISA assay. Suspensions were centrifuged at 2000 × g for 15 min and supernatant was then assayed using an IL-1β ELISA kit (R&D Systems, Minneapolis, MN, USA) according to the manufacturer’s instruction.
Luciferase assays
The luciferase reporter gene assay was measured as described previously79. pNF-κB-luc, ODD-luc and IL1B-luc were inserted into the pGL3-basic lentiviral vector for high efficiency of infection in BMDMs, respectively. After 24 h post-infection, cells were seeded in a 12-well plate and grown for another 16 h. Then, the cells were treated with different chemicals as indicated. Luciferase activities were determined using a ONE-GloTM luciferase Reporter Assay kit (Promega, E6120).
Plasmids and shRNAs
ODD-luc (#18965) was obtained from Addgene. pNF-kB-luc (P0455) was obtained from Miaolingbio.
5’-CTCCCTCAGTTGCCAGGGTCTGTG-3’ and 5’-CTGCATCACCTGTATCTAT TACCTG-3’ were the sequences corresponding to nucleotides 398–412 and 1303–1327 of the mouse EGLN2(PHD1) mRNA (EGLN2 shRNA);
5’-CAAGGTAAGTGGAGGTATTCTTCGA-3′ and 5’-CTCATGAAGTACAGCCAGCATACG-3’ were the sequences corresponding to nucleotides 1139–1163 and 1249–1272 of the mouse EGLN1(PHD2) mRNA (EGLN1 shRNA);
5’-CTACTACCTGAATAAGAATTGGGA-3’ and 5’-CTGGTACTTCGATGCTGAAGAAAGG-3′ were the sequences corresponding to nucleotides 760–783 and 943–967 of the mouse EGLN3(PHD3) mRNA (EGLN3 shRNA);
5’-CTGTGCGACAAGGTGCAGAAAGAA-3’ and 5’-CTTCCCGAAGTGCGTACACATTCTG-3’ were the sequences corresponding to nucleotides 937–960 and 1995–2019 of the mouse p65 mRNA (p65 shRNA);
5’-CCCACATCCAGGAGTGTAA-3’ was the sequence corresponding to nucleotides 963–981 of the mouse TRPML1 mRNA (MCOLN1 or ML1 shRNA).
5’- GCGGTTGAGGCTCACTTTA-3’ was the sequence corresponding to nucleotides of the mouse ATG5 mRNA (ATG5 shRNA)
5’-TGAGACAGACTGCATTCACCTGGAC-3’ and 5’-GGAAAATCAAAATCTATCTCAACAG-3’ were the sequences corresponding to nucleotides of the mouse TRPML2 mRNA (MCOLN2 shRNA)
5’-CTCAACCTGAGCCTGGACTTCCACAG-3’ and 5’-CAACGGGTGGTACATTATGATCATCAT-3’ were the sequences corresponding to nucleotides of the mouse TRPML3 mRNA (MCOLN3 shRNA)
5’- ATAAGCCACGCTAACCACTGCAGCC-3’ was the sequence corresponding to nucleotides of the mouse DMT1 mRNA (DMT1 shRNA)
5’- GCAGCAGGC TGTCATGCATTA-3’ was the sequence corresponding to nucleotides of the mouse TFEB mRNA (TFEB shRNA)
5’-CCGACAATAACATGAAGGCTA-3’ was the sequence corresponding to nucleotides of the mouse TfR1 mRNA (Tfrc shRNA)
All the shRNAs including negative control (NC) shRNA were cloned into lentiviral vector.
CRISPR-mediated genome editing in HEK 293 T cells
Fragment knockout strategy was used with the following gRNAs:
TRPML2 gRNA1: 5’-CTTTGAAAGCAACCACCAGC-3’
TRPML2 gRNA2: 5’- CTGCAGTGTATATACTCAAG-3’
TRPML3 gRNA1: 5’-CAGCTACTCACAACTACCTC-3’
TRPML3 gRNA2: 5’- TCGAGCCCAGAACTTCTCAC -3’.
The gRNA1 was cloned into the pX458 plasmid (Addgene #48138) according to the standard procedures and the gRNA2 carrying U6 promotor was cloned into pX458 with gRNA1 by Gibson assembly. The gRNA sequences were designed to target exon3 of TRPML2 and exon2 of TRPML3, respectively. Freshly plated low passage 293 T cells were transfected with 2 μg pX458 plasmid (with gRNA1 and gRNA2) using 8 μl Lipomaster 3000 (Vazyme, China). We transfected 2×105 293 T cells in a 3.5 cm culture dish and the medium was changed 24 h post-transfection. After 48 h we sorted single 3% GFP+ cells into 96-well plates using FACS. These cells were maintained in DMEM containing 20% FBS with antibiotics at sub-confluent densities. The knock out monoclone was finally verified by sequencing and WB.
Western blotting
Standard western blotting procedures were used. Antibodies used for Western blots include anti-GAPDH (#2118, Cell Signaling Technology), anti-beta Actin (abl1010, Abbkine Scientific), anti-beta Tubulin (#2146, Cell signaling Technology), anti-mouse IL-1β (BD Biosciences, San Jose, CA, USA), anti-NLRP3 (#15101, Cell Signaling Technology), anti-Caspase-1 (AG-20B-0042-C100, AdipoGen), anti-HIF-1α (NB100-449, Novus), anti-PHD1 (NBP2-92974, Novus), anti-PHD2 (NB100-137, Novus), anti-PHD3 (NB100-139, Novus), anti-LC3 (L8918, Sigma-Aldrich), anti-p62 (#5114, Cell Signaling Technology), anti-p-p38 (#4511, Cell Signaling Technology), anti-p38 (#8690, Cell Signaling Technology), anti-p-ERK (#4370, Cell Signaling Technology), anti-ERK (#4695, Cell Signaling Technology), anti-p-JNK (#4668, Cell Signaling Technology), anti-JNK (#9252, Cell Signaling Technology), anti-p-p65 (#3033 Cell Signaling Technology), anti-p65 (#8242, Cell Signaling Technology), anti-p-IKBα (#2859, Cell Signaling Technology), anti-p-IKKα/β (#2697, Cell Signaling Technology), anti-TFEB (#4240, Cell Signaling Technology), anti-TFE3 (#A0548, ABclonal), anti-TRPML1 (ACC-081, Alomone labs), anti-TRPML2 (ACC-082, Alomone labs), anti-TRPML3 (ACC-083, Alomone labs), anti-TFR1 (bsm-52793R, Bioss).
qPCR analysis
Total RNA from BMDMs was isolated using RNAiso Trizol reagent (Sangon Biotech, China), and reverse-transcribed with PrimeScriptTM RT Master Mix (Takara, Japan) according to the manufacturer’s instructions. Then a TB GreenTM Premix EX TaqTM II (Takara, Japan) in a LightCycler 480 qPCR instrument (Roche) was used for qRT-PCR. qRT-PCR was performed in triplicate and results were normalized against GAPDH. Relative fold expressions were calculated with the comparative threshold cycle (2−ΔΔCt) method. The sequences of all primers for qRT-PCR are listed in below:
mIL1B forward: 5’-AGTTGACGGACCCCAAAAG-3’
mIL1B reverse: 5’-AGCTGGATGCTCTCATCAGG-3’
mIL6 forward: 5’-CCAGGTAGCTATGGTACTCCA-3’
mIL6 reverse: 5’-GCTACCAAACTGGCTATAATC-3’
mTNF forward: 5’-CATGAGCACAGAAAGCATGATC-3’
mTNF reverse: 5’-CCTTCTCCAGCTGGAAGACT-3’
mPHD1 forward: 5’-CGCAGCATTCGTGGGGACCAGATT-3’
mPHD1 reverse: 5’-CATCGCCGTGGGGATTGTCAACAT-3’
mPHD2 forward: 5’-AACTCAAGCCCAATTCAGTCAGCA-3’
mPHD2 reverse: 5’-CCCCACAGTACAGTCCCAGCAGAT-3’
mPHD3 forward: 5’-GGCACCTGCGAGGCGACCAGAT-3’
mPHD3 reverse: 5’-TGGCGAACATAACCTGTCCCATTT-3’
mMCOLN1 forward: 5’-AAACACCCCAGTGTCTCCAG-3’
mMCOLN1 reverse: 5’-GAATGACACCGACCCAGACT-3’
hMCOLN1 forward: 5’-TCTTCCAGCACGGAGACAAC-3’
hMCOLN1 reverse: 5’-GCCACATGAACCCCACAAAC-3’
mCXCL10 forward: 5’-TGCCTCATCCTGCTGGGTCTG-3’
mCXCL10 reverse: 5’-TCCCTATGGCCCTCATTCTCACTG-3’
mCXCL11 forward: 5’-GTAACGGCTGCGACAAAGTTGAAG-3’
mCXCL11 reverse: 5’-GAGGCGAGCTTGCTTGGATCTG-3’
mCD74 forward: 5’-GCCAGGAAGAAGTCAGCCACATC-3’
mCD74 reverse: 5’-GGGAACACACACCAGCAGTAGC-3’
mCCL22 forward: 5’-CTCCTGGTGGCTCTCGTCCTTC-3’
mCCL22 reverse: 5’-TCCTGGCAGCAGATACTGTCTTCC-3’
mMCOLN2 forward: 5’-TCTACTCAGAGAAGCACTCACTACC-3’
mMCOLN2 reverse: 5’-CAGCCTCAGAGCAAGAACAATGG-3’
mMCOLN3 forward: 5’-ATGGCAATCTGTCAGCACTTCTAC-3’
mMCOLN3 reverse: 5’-TCTCCAGGCGTTCCAAGGTG-3’
hIL1B forward: 5’-CCGACCACCACTACAGCAAGG-3’
hIL1B reverse: 5’-GGGCAGGGAACCAGCATCTTC-3’
rIL1B forward: 5’-CTCACAGCAGCATCTCGACAAGAG-3’
rIL1B reverse: 5’-TCCACGGGCAAGACATAGGTAGC-3’
hGAPDH forward: 5’-AGCCACATCGCTCAGACAC-3’
hGAPDH reverse: 5’-GCCCAATACGACCAAATCC-3’
rGAPDH forward: 5’-GTGAAGCTCATTTCCTGGTATG-3’
rGAPDH reverse: 5’-GATGGAATTGTGAGGGAGATGC-3’
Chromatin immunoprecipitation (ChIP) assay
ChIP assays were performed using a ChIP kit (P2078, Beyotime, Shanghai, China) according to the manufacturer’s protocol. BMDMs were fixed by adding paraformaldehyde to a final concentration of 1% and then incubated at 37 °C for 10 min to allow cross-linking of endogenous proteins and DNA. After three washes using cold PBS supplemented with 1 mM PMSF, the BMDMs were resuspended in a buffer containing 1% SDS and 1 mM PMSF and then lysed by sonication. Supernatant was collected and chromatin was incubated with anti-p65 (#8242, Cell Signaling Technology), and rotated overnight at 4 °C. Input control and DNA obtained from the immunoprecipitation (IP) were amplified by PCR using primers specific for the mouse IL1B promoter containing the p65 binding site as follows:
(−923/−563) forward: 5’-CAGGAGTGGGTGGGTGAGT-3’
reverse: 5’-GGATGCGCACATTTAGACGCA-3’;
(−301/−292) forward: 5’-GAGGCGAGAGAGGTGACACA -3’
reverse: 5’-TGTCATCGTGGTGGAAATGGG-3’;
(−137/−128) forward: 5’-GCCACCCCTTGACTTCCA-3’
reverse: 5’-AACTGAAGGGTGGGGGAGG-3’
RNA-seq analysis
Total RNA was extracted from BMDMs treated as indicated conditions. Sample libraries were prepared from purified mRNA. Then, paired-end sequencing of the constructed library was carried out at Sangon Biotech. Co. Ltd. (Shanghai, China) on an Illumina HiSeq 2500 system. The quality of raw reads was evaluated and controlled using FastQC and Trimmomatic. Reads were aligned to the human reference genome GRCh37 using HISAT2 (version 1.12.4). Differentially expressed genes were identified using the DESeq2 package with standard settings. Subsequently, reads counts per million (CPM) were applied to perform differential gene expression analysis with DESeq2 package.
Gene set enrichment analysis (GSEA)
According to GSEA User Guide for RNA-seq data, DESeq2 was applied to perform the differential gene expression analysis. Parameters of log Fold change value and p adj value were selected to build pre-ranked list following the ranking method guide provided by Dr. Veronique Voisin (http://www.baderlab.org/CancerStemCellProject/VeroniqueVoisin/AdditionalResources/GSEA. The ferroptosis gene set collections were obtained from the KEGG PATHWAY Database (https://www.kegg.jp/entry/hsa04216). GSEA was performed based on the downloaded gene set collections using GSEA software (v4.1.3, https://www.gsea-msigdb.org/). Significance of the enrichment was calculated based on 1000 cycles of permutations.
Flow cytometry
For flow cytometry analyses, BMDMs were treated with different chemicals as indicated. BMDMs were collected and washed with PBS twice. H2DCFDA (ThermoFisher Scientific, D399) was used for detecting ROS, according to the manufacturer’s instruction. Stained BMDMs were analyzed with a BD FACSAria III flow cytometer and data was processed using FlowJo software.
Fe2+ staining and cell Perl’s Prussian blue staining
To maximally visualize cytosolic Fe2+ and Fe3+, BMDMs were seeded on glass coverslips or 12-well plates and treated with different chemicals as indicated. BMDMs on coverslips were washed 3 times with PBS, and then loaded with 2.5 μM BioTracker™ Far-red Labile Fe2+ Live Cell Dye and 100 nM LysoTrackerTM Red DND-99 for 45 min. Confocal microscopy was performed using Olympus confocal microscopes. BMDMs on 12-well plates were fixed in 4% paraformaldehyde for Perl’s Prussian blue staining by using a commercial kit (G1426, Solarbio, Beijing, China), following the manufacturer’s instructions.
RNA immunoprecipitation (RIP) assay
ACO1 (Iron Regulatory Protein)-IREB2 (Iron Regulatory Element) binding level was detected using RIP-PCR. In brief, ACO1-overexpression BMDMs were harvested and resuspended in freshly prepared nuclear isolation buffer on ice for 20 min. After centrifugation, nuclear pellet was resuspended in freshly prepared RIP buffer containing 0.5% NP40, RNAase inhibitor and protease inhibitors and then lysed by sonication. Supernatant was collected and chromatin was incubated with anti-ACO1 (12406-1-AP, Proteintech), and rotated overnight at 4oC. Input control and RNA obtained from the immunoprecipitation (IP) were purified by TRIzol and amplified by PCR using primers specific for the mouse Ferritin L chain mRNA IREs sequences containing the ACO1 binding site (CAGUGU) as follows:
Fert-L forward: 5’-AACAGTGTTTGAACGGAACAG-3’
Fert-L reverse: 5’-GGTCCCGGAGGCTGCGACTG-3’
CCK8 assay
Freshly detached cells were diluted in a 96-well plate (100 μl/well). Ten μL of the CCK-8 solution (Dojindo, CK04) was added into each well of the plate. Incubate the plate for 1–4 hours in the incubator and the absorbance were measured at 450 nm using a microplate Reader.
DQ-BSA proteolytic assay
DQ-red-BSA was used as an artificial substrate to evaluate lysosomal proteolytic degradation15. Briefly, BMDMs were treated with indicated chemicals, followed by DQ-red-BSA (ThermoFisher Scientific, D12051; 10 μg/ml) staining for 2 h at 37 °C. Cells were starved (amino acid-free and serum withdrawal) to trigger autophagic degradation. Upon cleavage, DQ-red-BSA exhibited red fluorescence, as detected by confocal imaging.
Reagents and chemicals
LysoTracker Red DND-99, and BioTracker™ Far-red Labile Fe2+ Live Cell Dye were from Life Technologies. DXZ, 2,2′-Bipyridyl, DFX, LPS, BAPTA-am, Wortmannin, and TPEN were purchased from Sigma. Roxadustat, Vadadustat, and GSK1278863 were purchased from MedChemExpress. All reagents were dissolved and stored in DMSO or water. ML-SI3 was synthesized by Dr. Huang at the Department of Pharmacology at the Xuzhou Medical University.
Statistical analysis
All experiments are repeated at least three times. Data are presented as the means ± standard errors of the mean. GraphPad Prism 9 Statistics software was unitized for conducting statistical analysis. For multiple groups with equal variances, the statistical significance of differences was evaluated using one-way ANOVA followed by Tukey’s multiple comparisons tests. For multiple groups with unequal variances, Brown-Forsythe and Welch ANOVA tests followed by Dunnett’s tests were adopted. P values < 0.05 were considered statistically significant.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The publicly available datasets can be accessed at the GEO database with accessions GSE53306 and GSE148505. The sequence read data generated in this study have been deposited in the NCBI SRA database under the reference PRJNA1206533. Related main and supplementary source data supporting the findings of this study are available within the Source Data file. Source data are provided with this paper.
Code availability
The RNA-seq data analysis source data and relevant code generated in this study are available at https://github.com/StumbleMQ/NC_2025_SourceData.git.
References
Xu, H. & Ren, D. Lysosomal physiology. Annu. Rev. Physiol. 77, 57–80 (2015).
Klionsky, D. J. et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition)(1). Autophagy 17, 1–382 (2021).
Marchi, S., Guilbaud, E., Tait, S. W. G., Yamazaki, T. & Galluzzi, L. Mitochondrial control of inflammation. Nat. Rev. Immunol. 23, 159–173 (2023).
Zhou, R., Yazdi, A. S., Menu, P. & Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 469, 221–225 (2011).
Nakahira, K. et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 12, 222–230 (2011).
Saitoh, T. et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 456, 264–268 (2008).
Murthy, A. et al. A Crohn’s disease variant in Atg16l1 enhances its degradation by caspase 3. Nature 506, 456–462 (2014).
Darmoise, A. et al. Lysosomal alpha-galactosidase controls the generation of self lipid antigens for natural killer T cells. Immunity 33, 216–228 (2010).
Pastore, N. et al. TFEB and TFE3 cooperate in the regulation of the innate immune response in activated macrophages. Autophagy 12, 1240–1258 (2016).
Lu H. et al. TFEB inhibits endothelial cell inflammation and reduces atherosclerosis. Sci. Signal. 10, eaah4214 (2017).
Visvikis, O. et al. Innate host defense requires TFEB-mediated transcription of cytoprotective and antimicrobial genes. Immunity 40, 896–909 (2014).
Chen, H. D. et al. HLH-30/TFEB-mediated autophagy functions in a cell-autonomous manner for epithelium intrinsic cellular defense against bacterial pore-forming toxin in C. elegans. Autophagy 13, 371–385 (2017).
Wang, W., Zhang, X., Gao, Q. & Xu, H. TRPML1: an ion channel in the lysosome. Handb. Exp. Pharm. 222, 631–645 (2014).
Spix, B., Chao, Y. K., Abrahamian, C., Chen, C. C. & Grimm, C. TRPML cation channels in inflammation and immunity. Front. Immunol. 11, 225 (2020).
Wang, W. et al. Up-regulation of lysosomal TRPML1 channels is essential for lysosomal adaptation to nutrient starvation. Proc. Natl Acad. Sci. USA 112, E1373–E1381 (2015).
Samie, M. et al. A TRP channel in the lysosome regulates large particle phagocytosis via focal exocytosis. Dev. Cell 26, 511–524 (2013).
Qi, J. et al. MCOLN1/TRPML1 in the lysosome: a promising target for autophagy modulation in diverse diseases. Autophagy 20, 1712–1722 (2024).
Qi J. et al. MCOLN1/TRPML1 in the lysosome: a promising target for autophagy modulation in diverse diseases. Autophagy, 1–11 (2024).
Zhang, X. et al. MCOLN1 is a ROS sensor in lysosomes that regulates autophagy. Nat. Commun. 7, 12109 (2016).
Xing, Y. et al. Blunting TRPML1 channels protects myocardial ischemia/reperfusion injury by restoring impaired cardiomyocyte autophagy. Basic Res. Cardiol. 117, 20 (2022).
Aarts, M. et al. A key role for TRPM7 channels in anoxic neuronal death. Cell 115, 863–877 (2003).
Coccia, M. et al. IL-1beta mediates chronic intestinal inflammation by promoting the accumulation of IL-17A secreting innate lymphoid cells and CD4(+) Th17 cells. J. Exp. Med. 209, 1595–1609 (2012).
Ligumsky, M., Simon, P. L., Karmeli, F. & Rachmilewitz, D. Role of interleukin 1 in inflammatory bowel disease–enhanced production during active disease. Gut 31, 686–689 (1990).
Qi, J. et al. MCOLN1/TRPML1 finely controls oncogenic autophagy in cancer by mediating zinc influx. Autophagy 17, 4401–4422 (2021).
Xing Y. et al. Autophagy inhibition mediated by MCOLN1/TRPML1 suppresses cancer metastasis via regulating a ROS-driven TP53/p53 pathway. Autophagy 18, 1932–1954 (2021).
Liu, Y. et al. TRPML1-induced autophagy inhibition triggers mitochondrial mediated apoptosis. Cancer Lett. 541, 215752 (2022).
Qi J. et al. MCOLN1/TRPML1 finely controls oncogenic autophagy in cancer by mediating zinc influx. Autophagy 17, 4401–4422 (2021).
Dillon, S. et al. Yeast zymosan, a stimulus for TLR2 and dectin-1, induces regulatory antigen-presenting cells and immunological tolerance. J. Clin. Invest. 116, 916–928 (2006).
Wilkinson, A. et al. Lipid conjugation of TLR7 agonist resiquimod ensures co-delivery with the liposomal cationic adjuvant formulation 01 (CAF01) but does not enhance immunopotentiation compared to non-conjugated resiquimod+CAF01. J. Control Release 291, 1–10 (2018).
Plesch E. et al. Selective agonist of TRPML2 reveals direct role in chemokine release from innate immune cells. Elife 7, e39720 (2018).
Kwok, J. C. & Richardson, D. R. The cardioprotective effect of the iron chelator dexrazoxane (ICRF-187) on anthracycline-mediated cardiotoxicity. Redox Rep. 5, 317–324 (2000).
Elihu, N., Anandasbapathy, S. & Frishman, W. H. Chelation therapy in cardiovascular disease: ethylenediaminetetraacetic acid, deferoxamine, and dexrazoxane. J. Clin. Pharm. 38, 101–105 (1998).
Hasinoff, B. B., Hellmann, K., Herman, E. H. & Ferrans, V. J. Chemical, biological and clinical aspects of dexrazoxane and other bisdioxopiperazines. Curr. Med. Chem. 5, 1–28 (1998).
Cvetkovic, R. S. & Scott, L. J. Dexrazoxane: a review of its use for cardioprotection during anthracycline chemotherapy. Drugs 65, 1005–1024 (2005).
Breuer, W., Epsztejn, S. & Cabantchik, Z. I. Iron acquired from transferrin by K562 cells is delivered into a cytoplasmic pool of chelatable iron(II). J. Biol. Chem. 270, 24209–24215 (1995).
Kaes, C., Katz, A. & Hosseini, M. W. Bipyridine: the most widely used ligand. A review of molecules comprising at least two 2,2’-bipyridine units. Chem. Rev. 100, 3553–3590 (2000).
Umemura, M. et al. The iron chelating agent, deferoxamine detoxifies Fe(Salen)-induced cytotoxicity. J. Pharm. Sci. 134, 203–210 (2017).
Cappellini, M. D. et al. A phase 3 study of deferasirox (ICL670), a once-daily oral iron chelator, in patients with beta-thalassemia. Blood 107, 3455–3462 (2006).
O’Brien, W. T. et al. The NLRP3 inflammasome in traumatic brain injury: potential as a biomarker and therapeutic target. J. Neuroinflamm. 17, 104 (2020).
Pang, Y., Wu, L., Tang, C., Wang, H. & Wei, Y. Autophagy-inflammation interplay during infection: balancing pathogen clearance and host inflammation. Front. Pharm. 13, 832750 (2022).
Reddy, K. et al. Dysregulation of nutrient sensing and CLEARance in presenilin deficiency. Cell Rep. 14, 2166–2179 (2016).
Curnock, R. et al. TFEB-dependent lysosome biogenesis is required for senescence. EMBO J. 42, e111241 (2023).
Dong, X. P. et al. The type IV mucolipidosis-associated protein TRPML1 is an endolysosomal iron release channel. Nature 455, 992–996 (2008).
Tabuchi, M., Yoshimori, T., Yamaguchi, K., Yoshida, T. & Kishi, F. Human NRAMP2/DMT1, which mediates iron transport across endosomal membranes, is localized to late endosomes and lysosomes in HEp-2 cells. J. Biol. Chem. 275, 22220–22228 (2000).
Rizzollo, F., More, S., Vangheluwe, P. & Agostinis, P. The lysosome as a master regulator of iron metabolism. Trends Biochem. Sci. 46, 960–975 (2021).
Van Welden, S., Selfridge, A. C. & Hindryckx, P. Intestinal hypoxia and hypoxia-induced signalling as therapeutic targets for IBD. Nat. Rev. Gastroenterol. Hepatol. 14, 596–611 (2017).
Cummins, E. P. et al. Prolyl hydroxylase-1 negatively regulates IkappaB kinase-beta, giving insight into hypoxia-induced NFkappaB activity. Proc. Natl. Acad. Sci. USA 103, 18154–18159 (2006).
Takeda, Y. et al. Macrophage skewing by Phd2 haplodeficiency prevents ischaemia by inducing arteriogenesis. Nature 479, 122–126 (2011).
Li, F. et al. Regulation of HIF-1alpha stability through S-nitrosylation. Mol. Cell 26, 63–74 (2007).
Haase, V. H. HIF-prolyl hydroxylases as therapeutic targets in erythropoiesis and iron metabolism. Hemodial. Int. 21, S110–S124 (2017).
Liu T., Zhang L., Joo D. & Sun S.C. NF-kappaB signaling in inflammation. Signal Transduct. Target. Ther. 2, 17023 (2017).
Bagaev, A. V. et al. Elevated pre-activation basal level of nuclear NF-kappaB in native macrophages accelerates LPS-induced translocation of cytosolic NF-kappaB into the cell nucleus. Sci. Rep. 9, 4563 (2019).
Tannahill, G. M. et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature 496, 238–242 (2013).
Wen, H. & Ting, J. P. Agitation by suffocation: how hypoxia activates innate immunity via the Warburg effect. Cell Metab. 17, 814–815 (2013).
Watts, E. R. & Walmsley, S. R. Inflammation and hypoxia: HIF and PHD isoform selectivity. Trends Mol. Med. 25, 33–46 (2019).
Ellinghaus, P. et al. BAY 87-2243, a highly potent and selective inhibitor of hypoxia-induced gene activation has antitumor activities by inhibition of mitochondrial complex I. Cancer Med. 2, 611–624 (2013).
Ahamada, M. M., Jia, Y. & Wu, X. Macrophage polarization and plasticity in systemic lupus erythematosus. Front. Immunol. 12, 734008 (2021).
Mittal, M., Siddiqui, M. R., Tran, K., Reddy, S. P. & Malik, A. B. Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal. 20, 1126–1167 (2014).
Ohnishi, T., Bandow, K., Kakimoto, K., Kusuyama, J. & Matsuguchi, T. Long-time treatment by low-dose N-acetyl-L-cysteine enhances proinflammatory cytokine expressions in LPS-stimulated macrophages. PLoS One 9, e87229 (2014).
Niwa, M., Hirayama, T., Okuda, K. & Nagasawa, H. A new class of high-contrast Fe(II) selective fluorescent probes based on spirocyclized scaffolds for visualization of intracellular labile iron delivered by transferrin. Org. Biomol. Chem. 12, 6590–6597 (2014).
Neudecker, V. et al. Myeloid-derived miR-223 regulates intestinal inflammation via repression of the NLRP3 inflammasome. J. Exp. Med. 214, 1737–1752 (2017).
Fang, C. et al. Differential regulation of lipopolysaccharide-induced IL-1beta and TNF-alpha production in macrophages by palmitate via modulating TLR4 downstream signaling. Int. Immunopharmacol. 103, 108456 (2022).
Talreja J. et al. HIF-1alpha regulates IL-1beta and IL-17 in sarcoidosis. Elife 8, e44519 (2019).
Falvo, J. V., Tsytsykova, A. V. & Goldfeld, A. E. Transcriptional control of the TNF gene. Curr. Dir. Autoimmun. 11, 27–60 (2010).
Werling, D. & Jungi, T. W. TOLL-like receptors linking innate and adaptive immune response. Vet. Immunol. Immunopathol. 91, 1–12 (2003).
Li, X. J. et al. Protein-tyrosine phosphatase Shp2 positively regulates macrophage oxidative burst. J. Biol. Chem. 290, 3894–3909 (2015).
Li, J. T. et al. Subanesthetic isoflurane relieves zymosan-induced neutrophil inflammatory response by targeting NMDA glutamate receptor and Toll-like receptor 2 signaling. Oncotarget 7, 31772–31789 (2016).
Wang, Y. Y., Liu, S., Lian, F., Yang, W. G. & Xue, S. Toll-like receptor 7/8 agonist resiquimod induces late preconditioning in neonatal cardiac myocytes. Acta Pharm. Sin. 32, 565–572 (2011).
Sardiello, M. et al. A gene network regulating lysosomal biogenesis and function. Science 325, 473–477 (2009).
Calabrese, C. et al. Mitochondrial translocation of TFEB regulates complex I and inflammation. EMBO Rep. 25, 704–724 (2024).
Medina, D. L. et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 17, 288–299 (2015).
Sun, L., Hua, Y., Vergarajauregui, S., Diab, H. I. & Puertollano, R. Novel role of TRPML2 in the regulation of the innate immune response. J. Immunol. 195, 4922–4932 (2015).
Chen C.C. et al. TRPML2 is an osmo/mechanosensitive cation channel in endolysosomal organelles. Sci. Adv. 6, eabb5064 (2020).
Kumar A. & Brookes M.J. Iron therapy in inflammatory bowel disease. Nutrients 12, 3478 (2020).
Ellermann, M. et al. Dietary iron variably modulates assembly of the intestinal microbiota in colitis-resistant and colitis-susceptible mice. Gut Microbes 11, 32–50 (2020).
Mahalhal, A. et al. Oral iron exacerbates colitis and influences the intestinal microbiome. PLoS One 13, e0202460 (2018).
Reifen, R., Matas, Z., Zeidel, L., Berkovitch, Z. & Bujanover, Y. Iron supplementation may aggravate inflammatory status of colitis in a rat model. Dig. Dis. Sci. 45, 394–397 (2000).
McNamee, E. N. et al. Novel model of TH2-polarized chronic ileitis: the SAMP1 mouse. Inflamm. Bowel Dis. 16, 743–752 (2010).
Dar, A. A., Belkhiri, A., Ecsedy, J., Zaika, A. & El-Rifai, W. Aurora kinase A inhibition leads to p73-dependent apoptosis in p53-deficient cancer cells. Cancer Res. 68, 8998–9004 (2008).
Acknowledgements
This work was supported by National Natural Science Foundation of China (NSFC) grants (82101314 to Y.X., 82200107 to M.W. and 82372205 to D.C), Key University Science Research Project of Jiangsu Province (20KJA310001 to W.W.), Major Projects of Jiangsu Provincial Health Commission (ZD2022051 to W.W.); Jiangsu Specially-Appointed Professor award to W.W., Jiangsu Province Innovative and Entrepreneurial Talent program to W.W and Jiangsu Province Innovative and Entrepreneurial Team program to W.W. Natural Science Foundation of Liaoning Province (2021-MS-161 to M.M.W.), Natural Science Foundation of Shanghai (23ZR1447900 to D.C.), Shanghai Jiao Tong University “Jiao Tong University Star” Plan Medical-Industrial Interdisciplinary Research Fund (YG2021GD03 to D.C.). We are grateful to Dr. Haoxing Xu (Liangzhu Laboratory and School of Basic Medical Sciences, Zhejiang University) for sharing the TFEB/TFE3 double KO Hela cell line, Ms. Josée Normand (Department of Pharmacology, Dalhousie University, Canada) for proofreading the manuscript, and appreciate the encouragement and helpful comments from other members of the Wang laboratory.
Author information
Authors and Affiliations
Contributions
W.W. and J.Q. conceived of the presented idea. Y.X., M.W., F.Z., T.X., X.W., R.C., Z.S., Y.D., D.X., X.Q., Q.L. (Lu), Q.L. (Li), Y.W., and J.Q. performed experiments and contributed to the interpretation of the results. F.Z., W.C., and M.H. made the K.O. cell lines used in this study. W.W., J.Q., D.C., and J.C. supervised the findings of this work. W.W. and J.Q. wrote the manuscript. All authors discussed the results and contributed to the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Osamu Takeuchi and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xing, Y., Wang, Mm., Zhang, F. et al. Lysosomes finely control macrophage inflammatory function via regulating the release of lysosomal Fe2+ through TRPML1 channel. Nat Commun 16, 985 (2025). https://doi.org/10.1038/s41467-025-56403-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-56403-x
