
Abstract
The introduction of fluoroalkyl groups into pharmaceutical compounds has the potential to enhance their therapeutic properties. Nevertheless, the synthesis of enantiomerically pure C(sp³)–CF₃ compounds poses a significant challenge. Biocatalysis offers precise stereochemical control, however, the scarcity of fluorine-containing natural products makes it difficult to find enzymes capable of incorporating fluoroalkyl groups. Herein, we develop a ground-state flavin-dependent enzyme-catalyzed strategy for the radical-mediated enantioselective trifluoromethylation. Two engineered flavin-dependent enzymes are successfully developed to catalyze stereoselective hydrotrifluoromethylation and trifluoromethyl-alkyl cross-electrophile coupling reactions using trifluoromethyl thianthrenium triflate as a radical donor. Experimental investigations and computational simulations demonstrate that the reaction is initiated through single-electron transfer from the ground state flavin hydroquinone (FMNhq) and quenched through hydrogen atom transfer by flavin semiquinone (FMNsq). This strategy provides an opportunity to bridge the gap between biocatalysis and organic fluorides but also introduces an alternative approach to address challenging stereoselective fluoroalkylation reactions in organic synthesis.
Similar content being viewed by others

Introduction
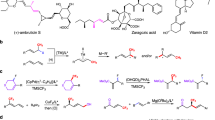
The incorporation of fluoroalkyl motifs has emerged as an important tool in medicinal chemistry, enhancing the properties of drug molecules such as lipophilicity, bioavailability, binding selectivity, and metabolic stability, ultimately exerting a significant impact on treatment outcomes1,2. Among the fluoroalkyl motifs, the perfluoroalkyl groups, especially trifluoromethyl (CF3) groups have garnered growing interest in recent decades because of their crucial role in the synthesis of bioactive molecules. (representative examples are shown in Fig. 1a)3,4,5. As such, the development of general technologies for constructing CF3-containing organofluorines has become a significant focus of research for chemists. To date, significant progress has been made in the formation of C(sp2)–CF3 bonds6. However, the synthesis of C(sp3)–CF3 motifs, especially in the production of optically pure C(sp3)–CF3 containing compounds, are relatively limited. (Fig. 1b)7,8,9. Given the well-established importance of chirality in pharmaceutical research and development, there is a growing demand for the development of a general and practical strategy to generate optically pure CF3–containing compounds.

a Representative CF3–containing bioactive molecules. b The production of optically pure C(sp3)–CF3 containing compounds. c Previous photo-induced flavin-dependent enzymes catalyzed reaction d Proposed ground state flavin-dependent enzyme catalyzed enantioselective trifluoroalkylation in this work. e Challenges in flavin-dependent enzyme catalyzed enantioselective trifluoroalkylation.
Recent advancements in the enantioselective synthesis of CF₃–containing substrates can be categorized into two primary approaches. The first involves the asymmetric functionalization of prochiral CF3–containing substrates10, necessitating the synthesis of precursors with diverse fluorine-containing building blocks. Another strategy involves the direct enantioselective installation of a CF3 group to prochiral substrates11, which poses a more significant challenge in stereochemical control11,12,13. In recent years, biocatalysis has played an increasingly significant role in facilitating the enantioselective synthesis of high-value compounds14,15,16,17,18,19,20. However, the scarcity of fluorine-containing natural products creates a gap between organofluorine chemistry and biocatalysis21. To date, only one type of natural enzyme (S‐adenosylmethionin-dependent) has been identified that is capable of forming C–F bonds22,23. Furthermore, naturally occurring CF3–containing compounds remain undiscovered, indicating the difficulty of locating natural enzymes capable of catalyzing the formation of C–CF3 bonds. To bridge the gap between organofluorine chemistry and biocatalysis, researchers have developed some elegant methods to incorporate fluorine-containing substrates into complex functional molecules. These methods involve installing fluoroalkyl groups using aldolases24, methyltransferases25,26, polyketide synthases27,28,29, and iron-containing proteins30,31, as well as the direct incorporation of trifluoromethyl groups through laccases32 or the natural cofactor pyridoxal-5’-phosphate33 (PLP). Among limited enantioselective examples, heme enzymes successfully synthesize CF3-containing compounds by employing 2,2,2-trifluoro-1-diazoethane as the carbene precursor34,35,36,37. However, there are still limited examples of utilizing a biocatalytic strategy for the direct enantioselective installation of a CF3 group onto prochiral substrates. Very recently, during the review of our work, Zhao’ group reported an asymmetric photoenzymatic process for the incorporation of fluorinated motifs into olefins38.
Excited-state-mediated biocatalysis39,40,41, particularly those involving flavin-dependent enzymes have emerged as an important strategy for the development of new-to-nature reactions, enriching the diversity of biosynthesis. This strategy involves generating radicals by harnessing the photoinduced capabilities of enzyme cofactors42,43,44,45,46,47,48,49,50,51,52,53,54 or photocatalysts55,56,57,58,59, while leveraging the enzyme’s pocket to precisely control stereoselectivity. We proposed that CF3 radicals could be formed in the active site of flavin-dependent enzymes and then captured by radical acceptors, enabling an enantioselective trifluoromethylation reaction. To achieve this these flavin-dependent enzymes catalyzed processes typically rely on the formation of an Electron Donor-Acceptor (EDA) complex by the interaction of flavin with a substrate, enabling the absorption of visible light to initiate the reaction (Fig. 1c). However, predicting these complexes with specific substrates is difficult because of the elusive electrostatic attraction and electron affinity that dictate their formation. In addition, the degradation of flavin-containing cofactors under visible light irradiation affects the efficiency of photoenzymatic processes60. Utilizing ground-state electron transfer to initiate these reactions offers an alternative approach to photoenzymatic processes (Fig. 1d)41. This method depends on the reducing capability of flavin hydroquinone (FMNhq, E1/2 = –0.45 V vs. saturated calomel electrode) and does not require the presence of light for activation. So far, this method has only been utilized to generate α-carbonyl carbon-centered radicals61,62. Considering the varying oxidation-reduction potentials of CF3 radical donors, we aim to identify a reagent capable of facilitating ground-state electron transfer for flavin-dependent enzymes, further enabling the enantioselective radical trifluoromethylation reaction. Another challenge stems from the high electrophilicity of CF3 radicals, which makes them prone to being quenched by electrons and limits their capture by other radical acceptors (Fig. 1e)63. In this article, we describe a flavin-dependent enzymes catalyzed method for enantioselective trifluoromethylation using trifluoromethyl thianthrenium triflate (TT–CF3+OTf–)64 as a radical donor and either styrenes or nitroalkanes as acceptors. By employing protein engineering, two engineered flavin-dependent enzymes effectively catalyze stereoselective hydrotrifluoromethylation and trifluoromethyl-alkyl cross-electrophile coupling reactions with up to 98% yield and 99:1 e.r. This study not only establishes a connection between biocatalysis and organofluorine chemistry but also presents an alternate solution to the challenge of stereochemical control in radical fluoroalkylation reactions.
Results
Enantioselective hydrotrifluoromethylation
We investigated the enantioselective hydrotrifluoromethylation with α-methyl styrene (1a) and CF3 radical donors in the presence of 1 mol% flavin-dependent enzymes. Considering the limited single-electron reduction ability of the ground-state FMNhq, we assessed the reduction potentials of typical CF3 radical donors (Fig. S12). Following this, we conducted a simultaneous screening of flavin-dependent enzymes and CF3 radical donors, considering both the effects of reduction potentials and protein structure on the binding of CF3 radical donors. As shown in Fig. 2b and Table S2, when trifluoromethylsulfonylbenzene (CF3–1) was employed as a CF3 radical donor, only a modest yield of the desired product was observed. The results obtained with Togni’s reagent I (CF3–3) were significantly superior to those achieved with Togni’s reagent II (CF3–2). Diphenyl(trifluoromethyl)sulfonium trifluoromethanesulfonate (CF3–4) yielded positive results, producing (S)-2a in 60% yield and 94:6 e.r. with old yellow enzyme 1 from Saccharomyces cerevisiae (OYE1) (Fig. 2b, entry 2). The best results were achieved with TT–CF3+OTf– (CF3-5) in combination with OYE1, resulting in a 72% yield and 94:6 e.r. of (S)-2a (Fig. 2b, entry 3). It is noteworthy that the most readily reducible Umemoto reagents (CF3-6) did not yield the optimal results, reflecting the influence of protein structure on the radical donor. Decreasing the enzyme concentration to 0.1 mol% also led to a 50% yield (Fig. 2b, entry 4). Control experiments validated crucial aspects of this reaction. The exclusion of OYE1 and the NADPH recycling system (Fig. 2b, entry 5-6) resulted in no product formation. The hydrotrifluoromethylation proceeded in 8% yield as a racemate with free flavin mononucleotide (FMN, 1 mol%). (Fig. 2b, entry 7), indicating the important role of the protein scaffold. There was a slight decrease in yield upon the introduction of an additional light source, (Fig. 2b, entry 8), possibly due to the photodegradation of FMN60.

a The rder of reduction potentials for typical trifluoromethyl (CF3) radical donors. b Development of the flavin-dependent enzymes catalyzed enantioselective hydrotrifluoromethylation. Reaction conditions: 1a (5 mM), CF3 donor (10 mM), OYE1 (1 mol%), NADP+ (0.1 μmol), GDH (0.1 mg, about 3 U), glucose (100 μmol), solvent (960 μL 50 mM tris buffer, pH 8.0 and 40 μL DMSO), reaction for 12 h under N2 atmosphere. Yield and enantiomeric ratio (e.r.) were determined by GC. n.d. = not determined, OYE1 = old yellow enzyme 1 from Saccharomyces cerevisiae. GDH Glucose dehydrogenase, FMN flavin mononucleotide. c Structural representation of the active site of OYE1.d Engineering of OYE1 for enantioselective hydrotrifluoromethylation.
To further improve the catalytic efficiency of OYE1, the active site residues closest to the flavin cofactor (Tyr82, Asn194, Tyr196, Phe250, Asn251, Phe 296, Tyr375) were selected for mutation. Through screening (Table S3), we found that the majority of the mutants examined exhibited decreased enantioselectivity, except for N251M, which showed 81% yield and 98:2 e.r. and 550 total turnover number (TTN) for this enzymatic hydrotrifluoromethylation.
Enantioselective trifluoromethyl-alkyl cross-electrophile coupling
Encouraged by these results, we aimed to apply our strategy to the trifluoromethyl-alkyl cross-electrophile coupling using nitroalkanes as radical acceptors. This approach involves the use of nitroalkanes to produce nitronates under basic conditions to capture radicals47,65. The elimination of the nitro group during the radical addition process facilitates the construction of C(sp3)–CF3 bond bearing a CF3–substituted stereogenic carbon. We sought to perform this reaction using CF3–5 and 1-methoxy-4-(1-nitroethyl)benzene (3a) in tris buffer (pH = 9.0) with OYE1. However, the reaction proceeded inefficiently, yielding 20% of the desired product with 62:38 e.r. (Table S4). Therefore, a set of flavin-dependent enzymes was further employed for screening (Table S4). The promising enzymes were old yellow enzyme 3 from Saccharomyces cerevisiae (OYE3) and GluER. OYE3 yielded product (S)-5a with 40% yield and 74:26 e.r., while GluER achieved a higher yield of 60% with moderate enantioselectivity (69:31 e.r.) (Fig. 3a, entry 1, 2). Taking into consideration both yield and stereoselectivity, we chose GluER as the template for further engineering. Following the screening of mutations in active site residues (Table S5), the mutant Y177F displayed improved enantioselectivity of 95:5 e.r. with a yield of 71% (Fig. 3a, entry 3). Decreasing the enzyme concentration to 0.1 mol% resulted in a 30% yield with an unchanged e.r. value (Fig. 3a, entry 4). The TTN of the Y177F variant was also measured to be 320. To investigate the influence of pH on the production of nitronates from nitroalkanes, we conducted the model experiment at lower pH levels. It is worth noting that the decrease in yield (Fig. 3a, entry 5) at lower pH levels, which was due to the increased difficulty in forming nitronate 4a under these conditions. Control reactions were also implemented and confirmed that the enzyme scaffold and NADPH recycling system are essential for this reaction (Fig. 3a, entry 7–8).

a Development of the flavin-dependent enzymes catalyzed enantioselective trifluoromethyl-alkyl cross-electrophile coupling. Reaction conditions: 1a (5 mM), CF3–5 (10 mM), GluER (1 mol%), NADP+ (0.1 μmol), GDH (0.1 mg, about 3 U), glucose (100 μmol), solvent (960 μL tris buffer, pH 9.0 and 40 μL DMSO), reaction for 12 h under N2 atmosphere. Yield and e.r. were determined by GC. OYE3 old yellow enzyme 3 from Saccharomyces cerevisiae, GluER = enoate reductase from Gluconobacter oxydans. b Structural representation of the active site of GluER. c, Engineering of GluER for enantioselective trifluoromethyl-alkyl cross-electrophile coupling.
Substrate scope
With the best mutant in hand, we investigated the substrate scope of these stereoselective trifluoromethylation reaction (Fig. 4). For hydrotrifluoromethylation reaction, most tested styrene derivatives act as radical acceptors were compatible with standard reaction conditions, leading to the formation of corresponding products with moderate to excellent yields and good enantioselectivities, showcasing the good functional group tolerance of this reaction. Among them, styrenes bearing electron-donating or withdrawing groups at the ortho-, para- and meta-position of phenyl group were well-tolerated, yielding corresponding products with up to 98% yields and 99:1 e.r. values (2b–2 h). The heterocyclic substrate also delivered the desired product (S)-2i (62% yield), although with lower enantioselectivity (82:18 e.r.). The utilization of the mutant F296A in the library led to an improved result (80% yield, 92:8 e.r.). The disubstituted derivative substrate was also accommodated without issue (2j, 68% yield, 97:3 e.r.). In contrast, the activity of α-phenyl and ethyl styrene decreased significantly (2k, 2l). It is noteworthy that the phenyl group (2k) generated a higher yield than the ethyl group, probably due to the combined effects of steric hindrance and the electron-donating properties of the benzene ring. Considering the significance of further elongating the perfluoroalkyl chain in pharmaceutical compounds, we proceeded to extend this reaction to perfluorobutylation using the TT–C4F9+X− (X = OSO2C4F9). Gratifyingly, styrenes bearing electron-donating or withdrawing groups could effectively incorporate perfluorobutyl groups with high yields and excellent stereoselectivity (2m–2o). Additionally, the reaction was scaled up to 20 mg (0.2 mmol 1a) with a 70% yield (51% isolated yield, the product is highly volatile) and no loss in stereoselectivity.

a Hydrotrifluoromethylation, b Trifluoromethyl-alkyl cross-electrophile coupling. Yields and e.r. (S: R) determined by GC or HPLC.
We next examined the substrate scope of the trifluoromethyl-alkyl cross-electrophile coupling reaction. α-Aryl nitroalkanes featuring electron-donating substituents, such as methoxyl (5a, 5d), and methyl (5c) as well as electron-withdrawing substituents such as ethoxycarbonyl (5e), halogens (5f, 5g) on phenyl group were all compatible in this reaction. Among them, there was no significant difference in the yields of nitroalkanes with electron-donating and electron-withdrawing groups on the phenyl group, highlighting the compatibility of this process. Due to steric hindrance, (1-nitropropyl)benzene could not undergo this reaction (5i). Furthermore, by conducting the reaction with bulky naphthyl substrate was also afforded in 27% yield and 97:3 e.r. (5h). A scaled-up experiment was also performed with 3a (0.2 mmol), affording the desired product (S)-5a in 62% yield (44% isolated yield) with 95:5 e.r.
Mechanistic investigations
To investigate the mechanism of this reaction, ultraviolet-visible spectrum experiments were first conducted, as shown in Fig. 5a66. No significant absorption was detected in the visible light spectrum for TT–CF3+OTf– (CF3–5) or the enzyme solution of OYE1–N251M with flavin hydroquinone (FMNhq) (Fig. 5a, Fig S8). Upon the addition of CF3–5 to the enzyme solution, a slight absorption band in the 400–450 nm range was observed (Fig. 5a, red trace). Extending the time by ten minutes resulted in the observation of a significant oxidation peak in the range of 400-–450 nm, suggesting that the ground state FMNhq was oxidized by CF3–5 (Fig. 5a, green trace). Similar results were observed with GluER-Y177F (Fig. 5b). In the radical capture experiments, diethyl 2,2-diallylmalonate and 2,2,6,6-Tetramethylpiperidinooxy (TEMPO) were used as radical acceptors, respectively. The adducts served to confirm the formation of the trifluoromethyl radical (Figs. S6, 7). Isotope incorporation experiments were conducted to investigate the mechanism of radical termination (Fig. 5c). Initially, we performed the model reaction in a deuterated buffer using the NADPH/glucose system, which is responsible for generating FMNsq. A low level of deuterium incorporation was observed in the resulting products 3a and 5a. (Fig. 5c, eq. 1 and 3). In contrast, when employing deuterated glucose to produce deuterated flavin semiquinone (FMNsq) in the reaction, we detected a significant enhancement in the deuterium incorporation rate of the products (Fig. 5c, eq. 2 and 4). These results suggested that the reaction tends to be terminated through hydrogen atom transfer (HAT) by FMNsq. Based on these experiments, we propose the following reaction mechanism: TT–CF3+OTf– undergoes ground state electron transfer from FMNhq to generate the CF3 radical, which is subsequently captured by the radical acceptor. The final product is formed through stereoselective quenching by FMNsq (Fig. 5d). Finally, quantum-mechanics/molecular-mechanics (QM/MM) calculations were conducted to confirm the energy feasibility of both hydrotrifluoromethylation and trifluoromethyl-alkyl cross-electrophile coupling (Figs. S16–20). The results characterized a thermodynamically favorable and kinetically feasible process for both reactions.

a Ultraviolet-visible spectrum experiments of OYE1-N251M. Source Data are provided as a Source Data file. b Ultraviolet-visible spectrum experiments of GluER-Y177F. c Isotope incorporation experiments. d Proposed reaction mechanism.
To gain insight into the origin of reactivity and stereoselectivity for these stereoselective trifluoromethylation reactions, we performed molecular dynamics simulation studies. The typical snapshots were chosen in Fig. 6 for structural analysis. The N251M mutation in OYE1 and the Y177F mutation in GluER alter the structure of the substrate-binding pocket, resulting in a reduced quenching distance between FMNsq and the product radical intermediate. The binding energies obtained through Molecular Mechanics/Generalized Born Surface Area (MM/GBSA) calculations show that the N251M exhibits a tendency to slightly increase the binding affinity to the radical intermediate compared to the WT-OYE1, and such tendency is much more distinct when comparing the binding free energies of WT and Y177F GluER. (WT-OYE1 vs. N251M, ΔG = −26.90 ± 0.81 vs. −27.30 ± 0.71 kcal/mol, WT-GluER vs. Y177F, ΔG = −22.87 ± 1.52 vs. −26.86 ± 1.94, Table S9). Per-residue decomposition of the binding free energies indicates that N251M mutation of OYE1 leads to increased contributions from F374 and Y177F mutation of GluER leads to significantly increased contribution from R261 and thianthrenium triflate (TT) (Fig. S23i & S25i). For enantioselectivity analysis, the improper dihedral torsion angle formed between the coplanar carbon atoms (C3, C7, and C8 for 2a and C3, C8, and C10 for 5a) of product radical intermediates 2a/5a and the hydrogen atom at N5 of FMNsq (C3–C8–C7–H) was measured. The positive and negative dihedral torsions corresponded to the pro-(R) and pro-(S) forms of the product. The changes in improper dihedral torsion angles during the molecular dynamics simulations are illustrated in Fig. S22–25. A comparison of the distributions of improper dihedral torsion angles indicate that MD reproduces the improved enantioselectivity for Y177F relative to the WT-GluER and confirms that N215M allows the maintenance of the selectivity of OYE1.

a WT-OYE1, b N251M-OYE1, c, WT-GluER and d Y177F-GluER. Product radical intermediate of 2a in a and b, product radical intermediate of 5a in c and d are shown in wheat.
Discussion
In this study, we have developed flavin-dependent enzyme-catalyzed stereoselective trifluoromethylation reactions involving both hydrotrifluoromethylation and trifluoromethyl-alkyl cross-electrophile reactions, granting access to essential chiral fluoroalkyl motifs. This biocatalytic approach uses readily available CF3 reagents to enable the direct enantioselective incorporation of a CF3 group onto prochiral substrates. This approach involves the utilization of the ground state FMNhq to catalyze electron transfer, resulting in the generation of CF3 radicals without the requirement of light irradiation. Two engineered enzymes facilitate the efficient capture of highly reactive CF3 radicals by radical acceptors and precisely control enantioselectivity. This method not only creates opportunities to bridge biocatalysis and organic fluorides but also provides a new strategy for addressing challenging stereoselective fluoroalkylation reactions in organic synthesis.
Methods
General procedure for the flavin-dependent enzyme-catalyzed stereoselective trifluoromethylation reactions
960 μL Enzyme solution (0.025- 0.05 μmol pure enzyme, tris buffer, pH = 8.0 for hydrotrifluoromethylation, pH = 9.0 for trifluoromethyl-alkyl cross-electrophile coupling) containing NADP+ (0.5 μmol), GDH (0.1 mg, about 3U), glucose (100 μmol) was added to a 10 mL Schlenk tube containing a magnetic stir bar. 40 μL stock solutions (DMSO) with styrene 1 or nitroalkane 3 (5 μmol) and TT–CF3+OTf− or TT–C4F9+OSO2C4F9− (10 μmol) were following added into the system. The mixture was then degassed by freeze-pump-thaw three times under N2 atmosphere. The reaction was stirred for 12 h at room temperature. Then the reaction was extracted by ethyl acetate three times. The yield and enantioselectivity was determined by GC or HPLC.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The X-ray crystallographic coordinates for structures reported in this study have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition number 2339888. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif. Data relating to the materials and methods, experimental procedures, mechanistic studies and computational calculations, gas chromatography, high-performance liquid chromatography spectra, and NMR spectra are available in the Supplementary Information and from the corresponding author(s) upon request. Source data are provided in this paper.
References
Mueller, K., Faeh, C. & Diederich, F. Fluorine in pharmaceuticals: Looking beyond intuition. Science 317, 1881–1886 (2007).
Shabir, G. et al. Chemistry and pharmacology of fluorinated drugs approved by the FDA (2016–2022). Pharmaceuticals 16, 1162–1211 (2023).
Jeffrey, W. C. et al. Inhibition of clinically relevant mutant variants of HIV-1 by quinazolinone Non-nucleoside reverse transcriptase inhibitors. J. Med. Chem. 43, 2019–2030 (2000).
Magueur, G. et al. Fluoroartemisinin: Trifluoromethyl analogues of artemether and artesunate. J. Med. Chem. 47, 2694–2699 (2004).
Keam, S. J. Pirtobrutinib: First approval. Drugs 83, 547–553 (2023).
Tomashenko, O. A. & Grushin, V. V. Aromatic trifluoromethylation with metal complexes. Chem. Rev. 111, 4475–4521 (2011).
Sarver, P. J. et al. The merger of decatungstate and copper catalysis to enable aliphatic C(sp3)–H trifluoromethylation. Nat. Chem. 12, 459–467 (2020).
Bian, K.-J. et al. Photocatalytic hydrofluoroalkylation of alkenes with carboxylic acids. Nat. Chem. 15, 1683–1692 (2023).
Xu, P. et al. Enantioselective radical trifluoromethylation of benzylic C−H bonds via cooperative photoredox and copper catalysis. J. Am. Chem. Soc. 144, 13468–13474 (2022).
Nie, J. et al. Asymmetric construction of stereogenic carbon centers featuring a trifluoromethyl group from prochiral trifluoromethylated substrates. Chem. Rev. 111, 455–539 (2011).
Yang, X., Wu, T., Phipps, R. J. & Toste, F. D. Advances in catalytic enantioselective fluorination, mono-, di-, and trifluoromethylation, and trifluoromethylthiolation reactions. Chem. Rev. 115, 826–870 (2015).
Ma, J.-A. & Cahard, D. Update 1 of: Asymmetric fluorination, trifluoromethylation, and perfluoroalkylation reactions. Chem. Rev. 108, PR1–PR43 (2008).
Wu, B. B. et al. Enantioselective synthesis of secondary β‑Trifluoromethyl alcohols via catalytic asymmetric reductive trifluoroalkylation and diastereoselective reduction. J. Am. Chem. Soc. 144, 6543–6550 (2022).
Reetz, M. T. Biocatalysis in organic chemistry and biotechnology: Past, Present, and Future. J. Am. Chem. Soc. 135, 12480–12496 (2013).
Schwizer, F. et al. Artificial metalloenzymes: Reaction scope and optimization strategies. Chem. Rev. 118, 142–231 (2018).
Zeymer, C. & Hilvert, D. Directed evolution of protein catalysts. Annu. Rev. Biochem. 87, 131–157 (2018).
Devine, P. N. et al. Extending the application of biocatalysis to meet the challenges of drug development. Nat. Rev. Chem. 2, 409–421 (2018).
Chen, K. & Arnold, F. H. Engineering new catalytic activities in enzymes. Nat. Catal. 3, 203–213 (2020).
Hollmann, F., Opperman, D. J. & Paul, C. E. Biocatalytic reduction reactions from a chemist’s perspective. Angew. Chem.-Int. Ed. 60, 5644–5665 (2021).
Buller, R. et al. From nature to industry: Harnessing enzymes for biocatalysis. Science 382, eadh8615 (2023).
Carvalho, M. F. & Oliveira, R. S. Natural production of fluorinated compounds and biotechnological prospects of the fluorinase enzyme. Crit. Rev. Biotechnol. 37, 880–897 (2017).
O’Hagan, D., Schaffrath, C., Cobb, S. L., Hamilton, J. T. G. & Murphy, C. D. Biosynthesis of an organofluorine molecule - A fluorinase enzyme has been discovered that catalyses carbon-fluorine bond formation. Nature 416, 279–279 (2002).
Sun, H. H. et al. Directed evolution of a fluorinase for improved fluorination efficiency with a non-native substrate. Angew. Chem.-Int. Ed. 55, 14275–14278 (2016).
Fang, J., Hait, D., Head-Gordon, M. & Chang, M. C. Y. Chemoenzymatic platform for synthesis of chiral organofluorines based on Type II Aldolases. Angew. Chem.-Int. Ed. 58, 11841–11845 (2019).
Peng, J. et al. Adenosylmethionine as a reagent for enzyme-catalyzed fluoromethylation. Angew. Chem.-Int. Ed. 60, 27178–27183 (2021).
Wang, W. R., Zhao, H. M., Yu, N. H., Chen, F. & Dong, M. Stable S-Adenosylmethionine analogue for enzymatic fluoromethylation. ACS Catal. 13, 13729–13734 (2023).
Walker, M. C. et al. Expanding the fluorine chemistry of living systems using engineered polyketide synthase pathways. Science 341, 1089–1094 (2013).
Rittner, A. et al. Chemoenzymatic synthesis of fluorinated polyketides. Nat. Chem. 14, 1000–1006 (2022).
Sirirungruang, S. et al. Engineering site-selective incorporation of fluorine into polyketides. Nat. Chem. Biol. 18, 886–893 (2022).
Zhao, Q. et al. Engineering non-haem iron enzymes for enantioselective C(sp3)–F bond formation via radical fluorine transfer. Nat. Synth. 3, 958–966 (2024).
Zhao, L.-P. et al. Biocatalytic enantioselective C(sp3)–H fluorination enabled by directed evolution of non-haem iron enzymes. Nat. Synth. 3, 967–975 (2024).
Simon, R. C. et al. Biocatalytic trifluoromethylation of unprotected phenols. Nat. Commun. 7, 13323–11328 (2016).
Wang, Y. et al. Nature-inspired radical pyridoxal-mediated C–C bond formation. J. Am. Chem. Soc. 146, 23321–23329 (2024).
Tinoco, A. et al. Highly diastereo- and enantioselective synthesis of trifluoromethyl-substituted cyclopropanes via Myoglobin-catalyzed transfer of trifluoromethylcarbene. J. Am. Chem. Soc. 139, 5293–5296 (2017).
Zhang, J. E., Huang, X. Y., Zhang, R. J. K. & Arnold, F. H. Enantiodivergent α-Amino C-H fluoroalkylation catalyzed by engineered cytochrome P450s. J. Am. Chem. Soc. 141, 9798–9802 (2019).
Huang, X. Y. et al. A biocatalytic platform for synthesis of chiral α-trifluoromethylated organoborons. ACS Cent. Sci. 5, 270–276 (2019).
Schaus, L. et al. Protoglobin-catalyzed formation of cis-trifluoromethyl-substituted cyclopropanes by carbene transfer. Angew. Chem.-Int. Ed. 62, e202208936 (2023).
Li, M. et al. Asymmetric photoenzymatic incorporation of fluorinated motifs into olefins. Science 385, 416–421 (2024).
Harrison, W., Huang, X. & Zhao, H. Photobiocatalysis for abiological transformations. Acc. Chem. Res. 55, 1087–1096 (2022).
Emmanuel, M. A. et al. Photobiocatalytic strategies for organic synthesis. Chem. Rev. 123, 5459–5520 (2023).
Fu, H. & Hyster, T. K. From ground-state to excited-state activation modes: Flavin-dependent “ene”-reductases catalyzed non-natural radical reactions. Acc. Chem. Res. 57, 1446–1457 (2024).
Biegasiewicz, K. F. et al. Photoexcitation of flavoenzymes enables a stereoselective radical cyclization. Science 364, 1166–1169 (2019).
Huang, X. et al. Photoenzymatic enantioselective intermolecular radical hydroalkylation. Nature 584, 69–74 (2020).
Trimble, J. S. et al. A designed photoenzyme for enantioselective [2+2] cycloadditions. Nature 611, 709–714 (2022).
Sun, N. et al. Enantioselective [2+2]-cycloadditions with triplet photoenzymes. Nature 611, 715–720 (2022).
Zhang, Z. et al. Photoenzymatic enantioselective intermolecular radical hydroamination. Nat. Catal. 6, 687–694 (2023).
Fu, H. et al. An asymmetric sp3–sp3 cross-electrophile coupling using ‘ene’-reductases. Nature 610, 302–307 (2022).
Zhao, B. et al. Direct visible-light-excited flavoproteins for redox-neutral asymmetric radical hydroarylation. Nat. Catal. 6, 996–1004 (2023).
Duan, X. et al. A Photoenzymatic strategy for radical‐mediated stereoselective hydroalkylation with diazo compounds. Angew. Chem.-Int. Ed. 62, e202214135 (2023).
Chen, X. et al. Photoenzymatic hydrosulfonylation for the stereoselective synthesis of chiral sulfones. Angew. Chem.-Int. Ed. 62, e202218140 (2023).
Li, M. et al. Remote stereocontrol with azaarenes via enzymatic hydrogen atom transfer. Nat. Chem. 16, 277–284 (2024).
Ju, S. et. al. Stereodivergent photobiocatalytic radical cyclization through the repurposing and directed evolution of fatty acid photodecarboxylases. Nat. Chem. 16, 1339–1347 (2024).
Shi, Q. et al. Single-electron oxidation-initiated enantioselective hydrosulfonylation of olefins enabled by photoenzymatic catalysis. J. Am. Chem. Soc. 146, 2748–2756 (2024).
Mou, K. et al. Stereodivergent protein engineering of fatty acid photodecarboxylase for light-driven kinetic resolution of sec-alcohol oxalates. Angew. Chem.-Int. Ed. 63, e202318374 (2024).
Cheng, L. et al. Stereoselective amino acid synthesis by synergistic photoredox-pyridoxal radical biocatalysis. Science 381, 444–451 (2022).
Liu, Y. et al. Photoredox/Enzymatic catalysis enabling redox-neutral decarboxylative asymmetric C−C Coupling for asymmetric synthesis of chiral 1,2-amino alcohols. JACS Au 3, 3005–3013 (2023).
Sun, S. et al. Enantioselective decarboxylative alkylation using synergistic photoenzymatic catalysis. Nat. Catal. 7, 35–42 (2024).
Xu, Y. et al. A light-driven enzymatic enantioselective radical acylation. Nature 625, 74–78 (2024).
Ouyang, Y. et al. Synergistic Photoenzymatic catalysis enables synthesis of a‑tertiary amino acids using threonine aldolases. J. Am. Chem. Soc. 146, 13754–13759 (2024).
Moore, W. M. et al. The photochemistry of riboflavin. I. The hydrogen transfer process in the anaerobic photobleaching of Flavins. J. Am. Chem. Soc. 85, 3367–3372 (1963).
Sandoval, B. A., Meichan, A. J. & Hyster, T. K. Enantioselective hydrogen atom transfer: discovery of catalytic promiscuity in flavin-dependent ‘ene’-reductases. J. Am. Chem. Soc. 139, 11313–11316 (2017).
Fu, H. et al. Ground-State electron transfer as an initiation mechanism for biocatalytic C–C bond forming reactions. J. Am. Chem. Soc. 143, 9622–9629 (2021).
Studer, A. A “renaissance” in radical trifluoromethylation. Angew. Chem.-Int. Ed. 51, 8950–8958 (2012).
Jia, H., Häring, A. P., Berger, F., Zhang, L. & Ritter, T. Trifluoromethyl thianthrenium triflate: A readily available trifluoromethylating reagent with formal CF3+, CF3•, and CF3− reactivity. J. Am. Chem. Soc. 143, 7623–7628 (2021).
Fu, H. et al. Asymmetric C‑alkylation of nitroalkanes via enzymatic photoredox catalysis. J. Am. Chem. Soc. 145, 787–793 (2023).
Page, C. G. et al. Regioselective radical alkylation of arenes using evolved photoenzymes. J. Am. Chem. Soc. 145, 11866–11874 (2023).
Acknowledgements
This research was funded by National Natural Science Foundation of China (No. 22322705, 22171243, 22301276), National Key Research and Development Program of China (No. 2021YFC2102000), Zhejiang Provincial Natural Science Foundation of China (LQ24B020009) and Scientific Research Starting Foundation of Zhejiang University of Technology (No. 2020105009029). We thank Dr. Zejie Zhu for her assistance in the electrochemical experiments.
Author information
Authors and Affiliations
Contributions
J.X. conceived and directed the project. X.D., D.C., and M.W. developed the reactions and performed the majority of synthetic experiments. C.J. and X.C. assisted with synthetic experiments. Z.W. conducted computational studies. J.X. and X.D. wrote the paper with input from all authors
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Hans Bunzel and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Duan, X., Cui, D., Wang, M. et al. Ground-state flavin-dependent enzymes catalyzed enantioselective radical trifluoromethylation. Nat Commun 16, 1225 (2025). https://doi.org/10.1038/s41467-025-56437-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-56437-1
This article is cited by
-
Applications of thianthrene chemistry in organic synthesis
Nature Synthesis (2025)
-
Steering oxygen-centred radicals with ground-state ene-reductases for enantioselective intermolecular hydroalkoxylations
Nature Catalysis (2025)
