
Abstract
Eukaryotic translation elongation factor 1A (eEF1A) is a highly abundant, multi-domain GTPase. Post-translational steps essential for eEF1A biogenesis are carried out by bespoke chaperones but co-translational mechanisms tailored to eEF1A folding remain unexplored. Here, we use AlphaPulldown to identify Ypl225w (also known as Chp1, Chaperone 1 for eEF1A) as a conserved yeast protein predicted to stabilize the N-terminal, GTP-binding (G) domain of eEF1A against its misfolding propensity, as predicted by computational simulations and validated by microscopy analysis of ypl225wΔ cells. Proteomics and biochemical reconstitution reveal that Ypl225w functions as a co-translational chaperone by forming dual interactions with the eEF1A G domain nascent chain and the UBA domain of ribosome-bound nascent polypeptide-associated complex (NAC). Lastly, we show that Ypl225w primes eEF1A nascent chains for binding to GTP as part of a folding mechanism tightly coupled to chaperone recycling. Our work shows that an ATP-independent chaperone can drive vectorial folding of nascent chains by co-opting G protein nucleotide binding.
Similar content being viewed by others


Introduction
Protein folding across all three kingdoms of life is guided by a variety of chaperones, many of which recognize their clients as nascent chains emerging from the ribosome exit tunnel1. In eukaryotes, the major burden of co-translational folding falls on heat shock protein 70 (Hsp70) paralogs and their co-chaperones; additional contributions critical to general protein folding come from the Hsp90 system, as well as the TRiC/CCT chaperonin which works to fold tubulin, actin, and many other proteins with complex topologies2.
How does the eukaryotic ribosome choreograph co-translational chaperones and nascent chain targeting/processing factors as they all vie for the limited surface area surrounding the nascent chain exit site3? One body of work has shown that the ribosome-associated complex (RAC) facilitates Hsp70 activation and binding to specific nascent chain sites soon after they emerge from the exit tunnel4,5,6,7. The translating ribosome can also dynamically recruit the highly abundant nascent polypeptide-associated complex (NAC) to several distinct regions surrounding the exit site. Here, NAC functions as a gatekeeper for sorting nascent chains towards the signal recognition particle (SRP) or a peptidase for N-terminal methionine excision8,9. By contrast, the mechanism by which NAC assists co-translational protein folding remains poorly understood10.
Recently, we found that Zpr1 and its co-chaperone Aim29 mediate de novo folding of the highly abundant and essential eukaryotic translation elongation factor 1A (eEF1A)11,12. eEF1A, a GTPase, delivers amino acid-charged tRNAs to the translating ribosome and is recycled by a regulated G protein cycle13. A unique aspect of the Zpr1-Aim29 folding mechanism is its reliance on conformational changes driven by GTP hydrolysis of its client. This process occurs post-translationally and is likely preceded by individual folding of the three eEF1A domains: the N-terminal GTP-binding domain (domain I, DI) and the two C-terminal β-barrel domains (DII and DIII). Structure-guided loss-of-function mutations in Zpr1 or Aim29 lead to growth arrest or slow growth dependent on the degree to which eEF1A misfolding drives induction of the heat shock response (HSR) or activation of the integrated stress response (ISR) via reduction in eEF1A levels due to proteasomal degradation.
We considered the possibility of additional eEF1A folding factors restricted to co-translational stages of eEF1A biogenesis. This notion came partially from a single-molecule analysis of co-translational folding of EF-G, a prokaryotic GTPase that mediates ribosome translocation during elongation14,15. In this study, co-translational misfolding between the nascent N-terminal GTP-binding domain (DI) and the ensuing β-barrel domain II (DII) was suppressed by Trigger Factor, a ribosome-associating, ATP-independent chaperone unique to prokaryotes. Owing to the sequence and structural similarity between EF-G DI/DII and the corresponding eEF1A domains, we wondered if protein folding factors specific to eukaryotes play an analogous role during eEF1A biogenesis.
Indeed, in this study, we discover that the conserved protein Ypl225w acts as a chaperone tailored to the co-translational folding of eEF1A DI. We show that ribosomal recruitment of Ypl225w by NAC enables Ypl225w to associate with eEF1A nascent chains and thereby prime DI for GTP binding. The subsequent GTP-binding step enables eEF1A DI folding concomitant with Ypl225w recycling. Our work reveals how an ATP-independent chaperone system on the ribosome co-opts the nucleotide binding of a nascent G protein client into a folding switch. In line with our results, an independent study found a similar body of evidence while our work was being considered for publication, supporting a largely similar working model for Ypl225w’s mechanism of action16.
Results
Computational modeling identifies Ypl225w as an eEF1A chaperone candidate
To search for new eEF1A chaperones, we used a version of AlphaPulldown17 (a derivative of AlphaFold) specific for the budding yeast Saccharomyces cerevisiae. In this screening approach, eEF1A’s sequence served as the bait against a library of “prey” sequences consisting of yeast proteins conserved across eukaryotes. We filtered interactions based on structures with an ipTM (interface pTM, a measure of structure confidence) greater than 0.8 (see “Methods”) (Fig. 1a). The top hits were two bona fide eEF1A physical and functional interactors (Efb1 and Efm4)18,19, as well as the conserved protein Ypl225w. The structural model of the Ypl225w•eEF1A interaction predicts a high confidence interface (ipTM = 0.82) in which the N-terminal α-helix (NaH) of Ypl225w stabilizes a non-native arrangement of DI’s switch regions that are normally involved in guanyl nucleotide binding (Fig. 1b).

a Table showing the top three hits (ipTM > 0.8) from an AlphaPulldown screen that utilized eEF1A’s sequence as bait. Bona fide eEF1A interactors are highlighted in blue. b ColabFold model showing eEF1A DI (light blue) bound to Ypl225w (light purple). The Switch I (SwI), Switch II (SwII), and P-loop regions of eEF1A DI are outlined in black. c The indicated strains carrying the Hsf1 reporter (4xHSE-YFP) were analyzed by flow cytometry. Bar graphs show median YFP intensity values normalized to both cell size (side scatter area) and the average median YFP intensity of wild-type (YPL225W) cells. Error bars represent the standard deviation from three biological replicates. d Wild-type (YPL225W) and ypl225wΔ strains expressing endogenously tagged eEF1A-GFP were imaged by confocal microscopy. Shown are representative micrographs normalized to the same intensity, with cells outlined. Scale bar represents 2.5 µm. 100 cells were quantified for each sample (right panel). The data shown is representative of three independently performed experiments. e Wild-type (YPL225W) and ypl225wΔ strains expressing endogenously tagged eEF1A-GFP were analyzed by flow cytometry as described in panel (c). Shown are bar graphs of median GFP intensity values normalized to both cell size and the average median GFP intensity of wild-type cells. Measurements from three replicates were compared by an unpaired, two-tailed t-test and yielded a p-value of 0.0000076. ****p < 0.0001. f Strains bearing the indicated genotypes were grown to mid-log phase (OD600 = 0.4) in YPD media, serially diluted by a factor of 10, and spotted onto YPD plates prior to being incubated at the indicated temperatures. Plates were imaged after 48 h.
We looked for additional computational evidence that Ypl225w recognizes eEF1A in the process of folding by testing whether DI is prone to forming non-native structures at various nascent chain lengths (see “Methods”) using enhanced sampling Monte Carlo simulations with DBFOLD, a computational Monte Carlo-based algorithm that predicts conformational free energy landscapes of proteins at various stages of translation20,21,22,23. This approach revealed that the region of eEF1A predicted to interact with Ypl225w (amino acids 1–72) is indeed prone to forming kinetically trapped, misfolded intermediates (Supplementary Fig. 1a). Specifically, our simulations of eEF1A (amino acids 1–72) populated four major conformational states with the propensity to form non-native β-strands (Supplementary Fig. 1a; ES1-ES4). ES1, the lowest free energy state of eEF1A at the length of 72 amino acids, possesses 4.2% ɑ-helices and 30.6% β-strands, as opposed to the native structure’s 54.2% ɑ-helices and absence of β-strands (Supplementary Fig. 1a). To define the sequence specificity of these findings, we analyzed a highly similar region in Sup35, a nascent chain release factor in S. cerevisiae (eRF3) and a closely related paralog of eEF1A24. Strikingly, the predicted co-translational folding pathway of this Sup35 region (amino acids 254–325, following the N-terminal prion domain that is unique to the fungal eRF3 lineage) was dominated by a native-like folding intermediate (SS1), exhibiting 55.6% ɑ-helical content and lacking β-strands, which closely resembled its native secondary structure (54.2% ɑ-helices and absence of β-strands) (Supplementary Fig. 1a). Additionally, ColabFold modeling found no strong evidence of an interaction between Ypl225w and Sup35, supporting the hypothesis that Ypl225w and eEF1A are a highly selective chaperone-client pair (ipTM = 0.177, Supplementary Fig. 2c).
To predict the nascent chain length at which Ypl225w engages eEF1A, we examined longer chain lengths of both eEF1A and Sup35 and found that at the emergence of the 95th amino acid, the folding landscapes of both paralogs converge to native-like intermediates (EL1 and SL1-SL2; Supplementary Fig. 1b). These data suggest that Ypl225w associates with eEF1A nascent chains in the window of ribosomal emergence of amino acids 72–95.
From an evolutionary perspective, Ypl225w belongs to a highly conserved protein family (PF04669) found across eukaryotes. We were thus intrigued by the apparent absence of PF04669 family members in plant species, including Arabidopsis thaliana. This is notable because the predicted free energy landscape for folding of A. thaliana eEF1A (amino acids 1–72) revealed two, well-populated species with native-like structures (A1 and A4, comprising ~42% of the snapshots, Supplementary Fig. 1c), despite its lack of global resemblance to the Sup35 landscape. By comparison, our simulations of eEF1A folding in humans, a species with a clear Ypl225w ortholog (PBDC1), predicted the formation of only non-native intermediates rich in β-strands (H1-H3, Supplementary Fig. 1c) similar to the yeast eEF1A. Finally, the local DI interactions we observed in our Ypl225w•eEF1A structural model remained largely unaffected in a Ypl225w•eEF1A DI ColabFold model lacking DII and DIII altogether, suggesting eEF1A may not yet possess these domains when Ypl225w first associates (Supplementary Fig. 2a, b). Collectively, these predictions led us to experimentally test Ypl225w as a candidate chaperone that staves off the potential of eEF1A’s G domain to form kinetically trapped, non-native β-strands during co-translational folding.
ypl225wΔ cells display the hallmarks of defective eEF1A biogenesis
A genome-wide screen for mutations that chronically induce an HSR reporter had already implicated Ypl225w in some major aspect of protein folding in the cell by ranking ypl225wΔ as the 12th highest hit25. When the latter study was completed, deletion of AIM29, a gene now known to encode a co-chaperone for eEF1A-mediated folding by Zpr1, was the only other uncharacterized gene at that time that ranked higher than ypl225wΔ. Thus, we began by examining ypl225wΔ cells for signs of disrupted eEF1A biogenesis that we saw previously in cells lacking Zpr1 or Aim2911,12. First, we confirmed that loss of Ypl225w resulted in the expected elevation of basal Hsf1 activity by utilizing the HSR reporter from the genome-wide study (Fig. 1c). Second, we looked for evidence of eEF1A-GFP aggregates in ypl225wΔ cells and observed fluorescent punctae in nearly half of the imaged cells (44%; Fig. 1d). Third, we detected a small but significant reduction in eEF1A protein levels (12–15% relative to WT cells) by flow cytometry analysis of eEF1A-GFP fluorescence (Fig. 1e). These latter phenotypes—aggregation and reduced abundance—are the expected outcomes of eEF1A misfolding leading to protein aggregation or degradation12.
ypl225wΔ cells grow normally under standard conditions and the eEF1A biogenesis defects reported above are milder than those caused by disruption of Zpr1 or Aim29 function (Supplementary Fig. 2d). However, when we replaced wild-type Hsf1 with a C-terminally truncated hsf1 allele that is non-inducible at elevated temperatures26, we observed a strong synthetic growth defect with ypl225wΔ (Fig. 1f). These data suggest that an adaptive HSR in ypl225wΔ cells partially restores eEF1A proteostasis towards its original set-point. Finally, we tested whether the function of Ypl225w in maintaining proteostasis was conserved across two diverged yeast species (>500 MYA) and found that heterologous expression of the Schizosaccharomyces pombe Ypl225w ortholog (SPBC3E7.07c) fully suppressed the HSR in ypl225wΔ cells (Supplementary Fig. 2e, f). Collectively, these findings reveal that Ypl225w has a conserved but poorly defined function in eEF1A biogenesis.
Ypl225w is a bona fide eEF1A chaperone
To test Ypl225w’s biochemical activity as an eEF1A chaperone, we turned to our in vitro translation system for studying eEF1A biogenesis and found several lines of supporting evidence11,12. First, eEF1A synthesized in ypl225wΔ extracts acquired only ~20–30% of its trypsin resistance relative to the wild-type and Zpr1-depleted extract controls (which exhibits ~0% trypsin resistance relative to WT extracts) (Fig. 2a and Supplementary Fig. 3a). Second, pure, recombinant Ypl225w-3xFLAG restored eEF1A folding in ypl225wΔ extracts to wild-type levels when added back co-translationally (Fig. 2b), as well as post-translationally but to a lesser extent (Fig. 2c). To control for potential effects of altered ypl225wΔ cell physiology on eEF1A folding in our extracts, we immunodepleted extracts of endogenously expressed Ypl225w-3xFLAG but observed a comparable folding defect that could be complemented biochemically (Supplementary Fig. 3b). Finally, in agreement with our computational modeling, newly synthesized Sup35 acquired trypsin resistance independently of Ypl225w (Supplementary Fig. 3c).

a Trypsin resistance of 35S-methionine-radiolabeled eEF1A was determined following in vitro translation (IVT) for 45 min in wild-type (YPL225W) or ypl225w∆ extracts and addition of 100 µg/mL cycloheximide (CHX) to terminate further protein synthesis. The “0” time point represents the undigested sample. Following digestion for the indicated times, samples were resolved by SDS-PAGE and visualized by autoradiography. “Fraction resistant” is the signal of near full-length, trypsin-resistant (Tryp.-Res.) eEF1A fragment divided by the total synthesized eEF1A in the undigested “0” time point. Samples were also analyzed by immunoblotting (IB) with the indicated antibody. The data shown is representative of three independently performed experiments. b Trypsin resistance of 35S-methionine-radiolabeled eEF1A (45-min translation time) was assessed after 5 min of trypsin digestion and with addition of the indicated amounts of recombinant Ypl225w-3xFLAG protein prior to IVT. Samples were also analyzed by IB with the indicated antibodies. “Fraction resistant” is the signal of near full-length, trypsin-resistant (Tryp.-Res.) eEF1A fragment divided by the total synthesized eEF1A in the corresponding undigested lane normalized to the WT YPL225W signal. The data shown is representative of three independently performed experiments. c Trypsin resistance of 35S-methionine-radiolabeled eEF1A (20-min translation time) was assessed after 5 min of trypsin digestion. “Co-translational addback” reactions were supplemented with Ypl225w-3xFLAG protein prior to translation initiation and immediately treated with trypsin post-CHX addition. “Post-translational addback” reactions were only supplemented with Ypl225w-3xFLAG protein after CHX treatment and then incubated at room temperature for 20 min before trypsin treatment. The data shown is representative of two independently performed experiments. d Affinity-purified samples from one replicate of the untagged (YPL225W) and YPL225W-3xFLAG samples that were used for mass spectrometry analysis shown here after SDS-PAGE followed by silver staining. The data shown is representative of two independently performed experiments. e Mass spectrometry analysis of samples similar to the ones in panel (d). The dotted line represents a false discovery rate at the p-value of 0.05. The FDR was calculated on the Empirical Bayes moderated t-statistic (see “Methods”). The inset within the volcano plot represents various categories of the enriched Ypl225w-3xFLAG interactors. The black dots represent interactors outside of the indicated categories.
Ypl225w is associated with translating ribosomes
We next hypothesized that Ypl225w’s previously noted ribosome association27 might explain its apparently stronger biochemical activity as a co-translational eEF1A chaperone. To first corroborate the ribosomal association, we immunoprecipitated Ypl225w-3xFLAG from cell extracts and quantitatively analyzed its interactors by SDS-PAGE and mass spectrometry (Fig. 2d, e and Supplementary Data 1: IP-MS data). Besides eEF1A, we detected numerous large and small ribosomal subunit proteins among the top hits (Fig. 2e and Supplementary Data 1: IP-MS data). In a complementary approach, we subjected extracts to sucrose gradient fractionation. Western blotting revealed a population of Ypl225w-3xFLAG in the heavy fractions containing polysomes, which we could eliminate by extract pre-treatment with EDTA, a condition that dissociates ribosomal subunits (Supplementary Fig. 3d).
Ypl225w is a co-translational chaperone for the GTP-binding domain of eEF1A
Our ColabFold model of Ypl225w•eEF1A suggested that Ypl225w specifically chaperones the DI portion of eEF1A nascent chains. We found two additional lines of evidence supporting this hypothesis. In the first approach, we compared wild-type and ypl225wΔ cells expressing eEF1A DI fused to GFP and observed fluorescent puncta in the majority of mutant cells (a sign of DI aggregation), as well as a ~12% reduction in fluorescence by flow cytometry (a sign of DI degradation) (Fig. 3a, b and Supplementary Fig. 4a). Despite its sequence and structural similarity to eEF1A DI, Sup35 DI-GFP fluorescence remained diffuse (a sign of DI solubility) in ypl225wΔ cells (Supplementary Fig. 4a). Further testifying to the specificity of this eEF1A DI aggregation phenotype in ypl225wΔ cells, we have previously shown that eEF1A DI-GFP remains soluble following engineered depletion of Zpr112.

a WT (YPL225W) and ypl225wΔ strains expressing eEF1A DI-GFP driven by the GAL1 promoter (integrated at the URA3 locus) were imaged by confocal microscopy and analyzed as in Fig. 1d. Scale bar represents 2.5 µm. 100 cells were quantified for each sample (right panel). See “Methods” for details of how expression of GAL1 was induced. The data shown is representative of three independently performed experiments. b Strains expressing eEF1A DI-GFP from panel (a) were analyzed via flow cytometry as described in Fig. 1e. Measurements were compared by an unpaired, two-tailed t-test and yielded a p-value of 0.0000077. ****p < 0.0001. c Cartoon schematic of ribosome-nascent chain complexes (RNCs) and sample processing for RNC-crosslinking experiment in panel (d). V160/V332 represent stalled RNCs of 160 or 332 amino acids in length stalled at a valine (V) codon. Cartoon templates have been used in our previous work11,12. d IVT reactions from ypl225wΔ extracts supplemented with 1 µM Ypl225w-3xFLAG and nonstop eEF1A mRNAs (V160 or V332 RNCs) were translated for 15 min prior to crosslinking with 1 mM BS3. Samples were then subjected to denaturing FLAG immunoprecipitations (IPs) and analyzed by SDS-PAGE followed by autoradiography. The data shown is representative of three independently performed experiments. Filled arrow: V160-NC, asterisk: V160-tRNA, open arrow: V160-tRNA x Ypl225w-3xFLAG, filled circle: V160-NC x Ypl225w-3xFLAG, open square: V332-NC, open circle: V332-tRNA.
In the second approach, we examined the “timing” of Ypl225w’s association with ribosome-nascent chain complexes (RNCs) using cell-free translation28. Here, we stabilized RNCs at the 3’ ends of two mRNAs encoding truncated versions of eEF1A but lacking the terminal stop codon (Fig. 3c). The shorter truncation, V160-RNC, represents a translation elongation intermediate in which only a portion of DI has been synthesized, while the longer truncation, V332-RNC proceeds to additionally synthesize DII. To specifically monitor Ypl225w-3xFLAG interaction with these RNCs, we used chemical crosslinking followed by denaturation and immunoprecipitation (Fig. 3c). This approach allowed us to detect cross-linked adducts between Ypl225w and V160-nascent chain (NC) but not V332-NC (Fig. 3d). We used RNase digestion to confirm that the higher molecular weight adduct corresponded to tRNA-conjugated V160-NC while the other species are deacylated products thereof, likely formed during sample processing (Supplementary Fig. 4b). These in vitro data strongly support a model in which Ypl225w interacts with nascent chains of eEF1A’s DI but completes its chaperone function as elongation proceeds further.
NAC facilitates eEF1A folding in vitro by ribosomal recruitment of Ypl225w
In considering how Ypl225w might gain access to eEF1A RNCs, we noted the presence of the NAC on our list of interactors (Fig. 2e). The major form of yeast NAC comprises the Egd2 α subunit in complex with the Egd1 β subunit29. Our ColabFold modeling of Ypl225w’s interaction with NAC revealed a high confidence interface with the C-terminal UBA domain of Egd2, which could accommodate the nearby eEF1A DI interaction (Fig. 4a). Intriguingly, a similar interface between NAC UBA and the signal recognition particle (SRP) was recently resolved by CryoEM analysis of RNCs with an exposed ER signal sequence (Supplementary Fig. 5a)9. This interaction is critical for tethering SRP to the ribosome prior to receiving the ER signal sequence from NAC. Since NAC, to our knowledge, has not been previously shown to recruit folding factors to the ribosome, we decided to explore this possibility in the context of Ypl225w-mediated eEF1A folding.

a ColabFold models of eEF1A DI (light blue) bound to Ypl225w (light purple) and NAC (Egd1: light green, Egd2: dark green). Model on the right was rotated ~50° and highlights the interaction of the ubiquitin-associated (UBA) domain of Egd2 (outlined in gold) with Ypl225w. b Extracts derived from the untagged strain (EGD2, YPL225W) or those tagged at the indicated endogenous loci (EGD2-13xMyc, YPL225W-3xFLAG) were subjected to FLAG IP. Elutions were resolved by SDS-PAGE followed by immunoblotting (IB) with the indicated antibodies. The data shown is representative of two independently performed experiments. c The indicated proteins were prepared and subject to FLAG IP as described in “Methods”. Total and eluted (IP) fractions were analyzed by SDS-PAGE and Coomassie staining or immunoblotting (IB) for the Myc tag on Egd2. Ypl225w-3xFLAG was quantitatively captured under these conditions. Dashes indicate cropping from the same gel to remove irrelevant lanes. NAC refers to recombinant Myc-Egd2•10xHis-Egd1. The data shown is representative of two independently performed experiments. d FLAG IP of the indicated proteins was carried out as in panel (c). Dashes indicate cropping from the same Coomassie-stained gel to remove irrelevant lanes. The data shown is representative of two independently performed experiments. e Trypsin resistance of 35S-methionine-radiolabeled eEF1A (45-min in vitro translation) in wild-type (EGD2) or egd2∆ extracts and addition of 100 µg/mL cycloheximide (CHX) to terminate translation. f Trypsin resistance (5-min trypsin digestion time) of 35S-methionine-radiolabeled eEF1A produced from IVT reactions (45-min translation time) in EGD2 or egd2Δ extracts supplemented with the indicated concentrations of recombinant NAC proteins. Dashes indicate cropping from the same gel to remove irrelevant lanes. The data shown is representative of two independently performed experiments. g Trypsin resistance of 35S-methionine-radiolabeled eEF1A was assessed as in panel (f) but with egd2Δ extracts supplemented with the indicated concentrations of Ypl225w-3xFLAG. The data shown is representative of two independently performed experiments.
First, we observed that Ypl225w-3xFLAG co-immunoprecipitated a C-terminally Myc-tagged version of Egd2 (Fig. 4b). Next, we attempted to pull down recombinant NAC with recombinant Ypl225w-3xFLAG using anti-FLAG magnetic beads but found no evidence of a stable interaction (Fig. 4c). We observed a similar negative result when we incubated Ypl225w-3xFLAG with salt-washed ribosomes purified from yeast cell extracts, which notably lacked stoichiometric amounts of NAC (Fig. 4c). Strikingly, Ypl225w-3xFLAG co-immunoprecipitated near quantitative amounts of both NAC and ribosomes following incubation of all components together (Fig. 4c). This interaction was abolished when we replaced wild-type NAC (NACWT) with a NAC mutant lacking the UBA domain (NACubaΔ) altogether or containing point mutations within the UBA domain (NACL165A and NACV166A) that our ColabFold model predicted would disrupt the interaction with Ypl225w (Fig. 4d and Supplementary Fig. 5a).
To examine the requirement for NAC during eEF1A biogenesis in our cell-free system, we generated egd2Δ cell extracts and found evidence of severe eEF1A misfolding relative to wild-type extracts but no discernible effect on Sup35 folding (Fig. 4e and Supplementary Fig. 5b). Importantly, co-translational addback of recombinant NACWT but not NACubaΔ or NACL165A + V166A mutants restored folding to normal levels (Fig. 4f). Next, we reasoned that if NAC facilitates timely co-translational capture of eEF1A nascent chains, then supraphysiological concentrations of Ypl225w-3xFLAG might bypass the need for NAC in vitro. Consistently, titration of egd2Δ cell extracts with additional recombinant Ypl225w-3xFLAG progressively suppressed their eEF1A folding defect (Fig. 4g). Lastly, we found that Ypl225w-3xFLAG crosslinking to V160 RNCs was diminished in egd2Δ extracts in a manner that was rescued by addback of NACWT but not NACubaΔ (Supplementary Fig. 5c). In sum, these findings strongly argue that NAC facilitates co-translational eEF1A folding via an SRP-like tethering mechanism directed at Ypl225w.
Ypl225w mediates eEF1A folding using an N-terminal α-helix with a hydrophobic patch
To address how Ypl225w facilitates eEF1A folding, we examined Ypl225w’s long NaH, which is predicted to insert itself into the switch helical region of eEF1A’s DI (Fig. 1b). We focused specifically on a hydrophobic patch on NaH comprising several highly conserved residues, which we rendered more hydrophilic by mutating phenylalanine at position 19 to alanine (F19A; Fig. 5a and Supplementary Fig. 6a). We found multiple lines of evidence that F19A ablates Ypl225w’s chaperone function. First, replacing wild-type Ypl225w-3xFLAG at the endogenous locus with the stably expressed F19A mutant allele resulted in a ypl225wΔ-like HSR phenotype (Fig. 5b and Supplementary Fig. 6b). Second, we analyzed Ypl225wF19A-3xFLAG interactors via SDS-PAGE, Western blotting, and negative stain electron microscopy and found a strong reduction in Ypl225w-bound eEF1A, ribosomal proteins, and ribosomal particles (Fig. 5c and Supplementary Fig. 6c). Third, co-translational addback of Ypl225wF19A-3xFLAG to ypl225wΔ extracts, even at supraphysiological concentrations, failed to restore trypsin resistance to eEF1A (Fig. 5d and Supplementary Fig. 6d). Fourth, Ypl225wF19A-3xFLAG yielded a much weaker interaction with V160 RNCs by our crosslinking assay (Fig. 5e). Lastly, we examined the effect of F19A under fully defined, in vitro conditions and observed that Ypl225w-3xFLAG quantitatively pulls down recombinant eEF1A DI, as well as full-length eEF1A, whereas Ypl225wF19A-3xFLAG does not interact with either (Fig. 5f). As further evidence of pull-down specificity, we observed no stable interaction between Ypl225w-3xFLAG and Sup35 DI (Supplementary Fig. 6e). Taken together, these data demonstrate that NaH mediates Ypl225w’s interaction with its folding client using a selective, hydrophobic interface.

a ColabFold models of eEF1A DI in surface representation bound to Ypl225w (light purple ribbon). Hydrophobicity was defined using ChimeraX molecular lipophilicity potential (“mlp”) command with default settings. Inset on the right was rotated ~150° and illustrates the hydrophobic interaction of the eEF1A DI SwI region with the NaH of Ypl225w. The F19 residue on the helix is highlighted in black. b Indicated strains carrying the Hsf1 reporter (4xHSE-YFP) were analyzed by flow cytometry as in Fig. 1c and normalized to YPL225WWT-3xFLAG values. c FLAG IPs of untagged (YPL225W), YPL225WWT-3xFLAG, and YPL225WF19A-3xFLAG samples. Samples were analyzed with Coomassie staining (left), as well as immunoblotting (IB, right) with the indicated antibodies. The data shown is representative of two independently performed experiments. d Trypsin resistance of 35S-methionine-radiolabeled eEF1A (45-min translations) in ypl225wΔ extracts supplemented with the indicated concentrations of Ypl225wWT-3xFLAG or Ypl225wF19A-3xFLAG proteins. Samples were digested with trypsin for 5 min. The data shown is representative of two independently performed experiments. e IVT reactions were used to generate V160-RNCs in ypl225wΔ extracts supplemented with Ypl225wWT-3xFLAG or Ypl225wF19A-3xFLAG proteins, and were cross-linked where indicated (lanes with BS3) before denaturing FLAG IPs. Filled arrow: V160-NC, asterisk: V160-tRNA, filled circle: V160-NC x Ypl225w-3xFLAG, open arrow: V160-tRNA x Ypl225w-3xFLAG. The data shown is representative of two independently performed experiments. f FLAG IPs of the indicated proteins (see “Methods”). eEF1A indicates the full-length protein. Dashes indicate cropping from the same Coomassie-stained gel to remove irrelevant lanes. The data shown is representative of two independently performed experiments.
GTP binding to eEF1A drives Ypl225w recycling
Ypl225w is predicted to interact with DI switch regions in their nucleotide-free state raising the intriguing possibility that GTP binding drives release of Ypl225w from eEF1A nascent chains. We found initial support for this idea by observing that inclusion of excess GTP during our isolation of endogenous Ypl225w-3xFLAG reduced co-IP of full-length eEF1A but not ribosomes (Supplementary Fig. 7a). We observed no effect when we further included an energy regenerating system (Supplementary Fig. 7a) but the addition of puromycin led to significantly reduced ribosome co-IP (Supplementary Fig. 7a). Collectively, these data suggest that endogenous Ypl225w (re)binds full-length eEF1A in extracts that have depleted GTP but remains stably bound to relatively short eEF1A nascent chains (i.e., not yet capable of interacting with GTP) regardless of nucleotide depletion (see “Discussion”). Next, we examined binding of recombinant Ypl225w-3xFLAG to eEF1A DI and found strong evidence of GTP-mediated disruption (Supplementary Fig. 7b). To confirm that this effect is driven by formation of a GTP-bound eEF1A conformation, we used a GTP-binding defective version of eEF1A DI (DID156N)30,31. As expected, Ypl225w-3xFLAG interaction with DID156N was resistant to disruption by GTP (Supplementary Fig. 7b). Next, we developed a bead-based assay for monitoring the kinetics of GTP-driven release of DI-immobilized Ypl225w-3xFLAG. We pre-formed protein complexes in the absence of nucleotides and bound them to anti-FLAG magnetic beads. Following GTP addition, we observed fast release of DIWT which occurred in a concentration-dependent manner (Fig. 6a–c). The addition of GDP and non-hydrolyzable GTP analogs, but not GMP or the guanosine base alone also stimulated release of DIWT, suggesting that the negatively charged β-phosphate group of GTP is critical for driving release but that GTP hydrolysis is not required (Supplementary Fig. 7c). To additionally probe the base specificity of these nucleotide effects, we monitored the effect of the xanthosine triphosphate (XTP) analog on DI release. In earlier work, eEF1AWT was shown to have little affinity for XTP relative to GTP, whereas eEF1AD156N had the opposite specificity for these two nucleotides30,31. As expected, GTP but not XTP caused rapid release of DIWT from Ypl225w-3xFLAG, whereas the reverse was true for DID156N (Fig. 6b, c).

a eEF1A DI•Ypl225w-3xFLAG complexes were first immobilized on beads (~300 nM Ypl225w and ~150 nM eEF1A DI), which were subsequently incubated with or without 100 µM GTP. After 10 min, samples were subjected to magnetic separation and removal of the supernatant fraction, followed by elution of the beads with FLAG peptide. Samples were resolved by SDS-PAGE and visualized by immunoblotting (IB). The “Input” lane represents beads that were directly eluted with FLAG peptide after the initial immobilization of eEF1A DI•Ypl225w-3xFLAG. “Fraction released” is the eEF1A DI signal in each supernatant over the input signal. *Asterisk indicates FLAG bleed-through signal in anti-eEF1A immunoblot. The data shown is representative of three independently performed experiments. b Line plot showing quantification of DIWT or DID156N released from Ypl225w-3xFLAG beads as in panel (a) but in the presence or absence of 100 µM GTP or XTP for the indicated times. Data represents mean ± standard deviation of three replicates. c Line plot showing quantification of DIWT or DID156N released from Ypl225w-3xFLAG beads at different concentrations of nucleotide as in panel (b) but with either GTP or XTP. Data represents mean ± standard deviation of three replicates. A sigmoidal (4PL) curve was fitted to the data in GraphPad Prism. d The association (“Assoc.”) of DIWT or DID156N to Ypl225w-3xFLAG-coated BLI tips and subsequent dissociation (“Dissoc.”) in 1 mM GTP, 1 mM XTP, or buffer (“-GTP”) was measured in triplicate and plotted as shown. The dissociation rate constants (koff) for the different reactions are indicated on the plot. e IVT reactions were used to generate V160- and V332-RNCs in ypl225wΔ extracts supplemented with 1 µM Ypl225w-3xFLAG but with either eEF1AWT or eEF1AD156N mRNA templates. Samples were cross-linked with BS3 where indicated and subsequently used for denaturing FLAG IPs. The data shown is representative of two independently performed experiments. Filled arrow: V160-NC, asterisk: V160-tRNA, filled circle: V160-NC x Ypl225w-3xFLAG, open arrow: V160-tRNA x Ypl225w-3xFLAG, open square: V332-NC, open circle: V332-tRNA, filled square: V332-NC x Ypl225w-3xFLAG, open star: V332-tRNA x Ypl225w-3xFLAG. f As in panel (e) but 3 min prior to crosslinking, reactions were supplemented with 1 mM XTP where indicated. The data shown is representative of two independently performed experiments.
The bead release data above are consistent with two competing models. In the first, GTP actively promotes dissociation of Ypl225w from nucleotide-free DI. Alternatively, DI might be capable of rapidly dissociating and rebinding from immobilized Ypl225w-3xFLAG; in that view, by blocking rebinding, GTP would lead to an apparently rapid release from the beads. To distinguish between these possibilities, we used bio-layer interferometry (BLI), a technique for measuring the kinetics of molecular interactions in real time32. First, we immobilized Ypl225w-3xFLAG on BLI protein G tips that we loaded previously with anti-FLAG antibody. Next, we monitored DIWT and DID156N binding to the tips before shifting them into a dissociation regime where we monitored the kinetic effects of nucleotide addition. GTP induced almost instantaneous dissociation of DIWT, while XTP was only marginally faster than spontaneous dissociation (Fig. 6d). In the case of DID156N, we observed the anticipated reversal of these effects (Fig. 6d). These data establish that GTP actively dissociates Ypl225w from DI rather than causing free client “trapping.”
The above data suggest that GTP facilitates Ypl225w recycling during co-translational eEF1A folding. We tested two predictions of this model using our in vitro translation system. First, reduced binding of GTP to growing D156N RNCs should interfere with Ypl225w recycling. Second, the above defect should be suppressed in the presence of XTP. We validated the first prediction by repeating our crosslinking assay using D156N V160-RNC and V332-RNCs. In contrast to the wild-type control in which only the V160-RNC interacted with Ypl225w-3xFLAG, we observed strong cross-links to both D156N V160- and V332-RNCs (Fig. 6e). To test the second prediction, we included XTP either during D156N RNC translation or following inhibition of further protein synthesis by cycloheximide. In both instances, we observed the expected outcome in which XTP selectively disrupted Ypl225w’s interaction with V332-RNCs (Fig. 6f and Supplementary Fig. 7d). Collectively, these data demonstrate that GTP binding to DI nascent chains drives recycling of Ypl225w during co-translational folding of eEF1A.
Discussion
Co-translational protein folding in prokaryotes depends on Trigger Factor (TF), an ATP-independent chaperone unique to prokaryotes33. TF is also the only known chaperone in prokaryotes that binds ribosomes even though other ATPase chaperones, including Hsp70, can associate with nascent chains33. By contrast, studies of co-translational protein folding in eukaryotes have shown that RAC exerts control over the Hsp70 ATPase cycle by a complex and still poorly understood mechanism5,6,10,34. In addition, eukaryotic ribosomes are bound to stoichiometric amounts of NAC3. The precise role of NAC in folding has remained unclear, but recent work has clearly illustrated NAC’s versatility in mediating other kinds of protein biogenesis decisions, ranging from ER targeting to enzymatic processing of nascent N-termini3,8,9. In this study, we provide evidence for a mechanistic model (Fig. 7), in which NAC facilitates ribosomal recruitment of Ypl225w, an ATP-independent chaperone, to mediate co-translational folding of eEF1A, one of the most abundant proteins in eukaryotes.

Cartoon model emphasizing the GTP-driven chaperone mechanisms of the eEF1A folding pathway en route to the formation of eEF1A ternary complexes competent for translation elongation. The sequence begins with NAC UBA domain-dependent recruitment of Ypl225w upon eEF1A nascent chain emergence. Ypl225w uses its N-terminal ɑ-helix (NaH) to stabilize DI of eEF1A nascent chain until DI is able to bind GTP, at which point Ypl225w is released. Full-length eEF1A is subsequently chaperoned by Zpr1 using its ɑ-helical hairpin (aHH) and zinc finger domain (ZnF), before complex disassembly via the Zpr1 co-chaperone Aim29 and GTP hydrolysis. The eEF1A nucleotide exchange factor eEF1B promotes GDP dissociation. GTP-bound eEF1A can then associate with charged tRNAs for delivery to ribosomes for translation elongation. See “Discussion” for further details. Cartoon templates have been used in our previous work11,12.
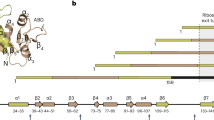
In the initial stages, we propose that the UBA domain of Egd2 (the NAC ɑ subunit in budding yeast) facilitates Ypl225w recruitment to ribosomes with partially synthesized DI nascent chains. The timing of these co-translational events in vivo was established in a parallel study using selective ribosome profiling, which revealed that Ypl225w becomes stably associated in a NAC-dependent manner with the ribosome following emergence of the DI switch regions16. This is consistent with both our folding simulations showing that these regions are prone to forming a kinetically trapped, misfolded intermediate, as well as our analysis of Ypl225w crosslinking to relatively short eEF1A RNCs in vitro. In the ensuing steps, Ypl225w maintains the nascent chain in a state primed for GTP binding upon the emergence of the remaining DI structural elements needed to form a bona fide GTP-binding pocket. Finally, in the recycling stage, GTP binding induces a switch-like, DI folding mechanism concomitant with Ypl225w release from the ribosome.
The very first step of our working model resembles the dynamic interaction between the NAC UBA domain and the SRP in metazoans, which a recent study showed was critical for ER protein targeting9. Our in vitro pull-down data also imply that, similar to SRP, Ypl225w has an additional affinity for the ribosome. In that regard, it is notable that ER signal sequences emerging from the ribosome first interact with NAC to “make room” for SRP docking to the ribosome. It will be interesting to test if the highly conserved N-terminal sequence of eEF1A can similarly prime NAC for a subsequent substrate hand-off to Ypl225w.
By contrast to the essential function of NAC in metazoans, yeast nac mutants grow normally under optimal conditions suggesting that a unique but still mysterious aspect of yeast cell biology bypasses NAC’s otherwise essential role. Indeed, our evidence for NAC’s involvement in eEF1A folding comes only from analyzing eEF1A folding in yeast cell-free extracts. A similar situation has been noted by studies of how NAC potently modulates SRP targeting to nascent chains with ER signal sequences in yeast extracts despite only mild effects of nac mutations on ER protein targeting in vivo35. Nonetheless, we posit that our work has uncovered a broadly conserved chaperone system. First, we have shown by a cross-species complementation experiment that expression of a Ypl225w ortholog from a distantly related fission yeast restores proteostasis to ypl225wΔ cells (Supplementary Fig. 2e). Second, an interaction between the C. elegans orthologs of Egd2 (icd-2, worm NACA) and Ypl225w (Pbdc1) has been previously reported36, as have those between the human orthologs of eEF1A (EEF1A) and Ypl225w (PBDC1)37,38,39. An important future goal will be to measure what portion of NAC’s essential function in metazoans is to maintain proteostasis by facilitating co-translational eEF1A folding.
Our work sets the stage for further exploring the evolutionary origins behind eEF1A’s dependence on a dedicated, co-translational chaperone system. Specifically, we have found computational and experimental evidence that the highly related DI of Sup35 folds independently of Ypl225w. Thus, one future strategy would be to screen for eEF1A DI mutant sequences that fold along a smoother, Sup35-like folding pathway. Such chaperone bypass might have naturally taken place in plant species such as Arabidopsis thaliana, which lacks a discernible Ypl225w homolog. While speculative, this intriguing possibility is consistent with our folding simulations of A. thaliana eEF1A predicting the existence of native-like intermediates reminiscent of Sup35 rather than full dominance by non-native structures rich in β-strands as in yeast eEF1A (Supplementary Fig. 1a–c).
Our model resembles the mechanism by which Ric8 mediates folding of the Ras-like domain of G alpha proteins, despite uncertain timing of those folding events relative to translation40,41,42,43. Namely, Ric8 maintains its client in a near-native state that is primed for GTP binding, which leads to folding concomitant with chaperone recycling. Together with our previous study of how eEF1A GTP hydrolysis drives the post-translational folding steps mediated by Zpr1 and Aim2911,12, this collective body of work illustrates the mechanistic versatility with which ATP-independent chaperones manipulate the nucleotide cycles of their clients to facilitate G protein folding.
Methods
Yeast strains
All strains used in this study are listed in Supplementary Table 1. Cells were grown or cultured at 30 °C unless otherwise indicated.
Plasmids
All plasmids used in this study are listed in Supplementary Table 2.
Oligos
All oligos used in this study are listed in Supplementary Table 3.
Antibodies
For immunoblotting, primary antibodies were used at the indicated dilution and fluorescent secondary antibodies were used at a 1:10,000 dilution. Primary antibodies include the following: rabbit-anti-eEF1A ([1:10,000 - IB], ED7001, Kerafast Inc., Boston, MA), mouse-anti-FLAG M2 ([1:2000–1:5000 - IB], F3165, MilliporeSigma, Burlington, MA), mouse-anti-Pgk1 ([1:2000 - IB], 459250, ThermoFisher Scientific, Waltham, MA), mouse-anti-Myc 9E10 ([1:10,000 - IB], 13-2500, ThermoFisher Scientific, Waltham, MA), and rabbit-anti-hexokinase ([1:2000 - IB], U.S. Biological Life Sciences, Swampscott, MA).
Immunoblotting
Samples were analyzed via SDS-PAGE, transferred to a nitrocellulose membrane, and blocked using 5% skim milk in TBST (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 0.1% Tween-20, 0.25 mM EDTA) for 1 h. Primary antibodies at the indicated dilutions (see “Antibodies” section) were used to probe membranes overnight at 4 °C. Prior to secondary antibody incubation (see “Antibodies” section), membranes were washed three times using TBST (10 min per wash). Secondary antibodies were visualized using a LI-COR Odyssey scanner.
Autoradiography
After separation via SDS-PAGE, gels were soaked in a fixing solution consisting of 50% methanol and 10% acetic acid for 30 min before being rinsed in a drying solution consisting of 30% methanol and 5% glycerol for 30 min. Gels were then dried under a vacuum for 2 h. Next, gels were exposed using phosphor-screens (GE Healthcare) for 1–3 days and subsequently scanned on a Sapphire Biomolecular Imager (Azure Biosciences).
Microscopy
VDY6176 and VDY6206 cells were grown to saturation overnight in synthetic complete (SC) media containing 2% glucose. After back-dilution to an OD600 of 0.1 in SC + 2% glucose, cells were grown for 3–4 h until they reached mid-log phase. Cells were then imaged using a Nikon TI inverted microscope with a 100× oil-immersion objective (1.45 NA), a Yokogawa dual spinning disk confocal unit, and a Hamamatsu ImagEM EM-CCD camera with a 488 nm laser (GFP). Images were captured with MetaMorph. Maximum intensity z-projections were used to generate the final images from collected Z stacks.
VDY6205 and VDY6240 cells expressing pGAL1-eEF1A DI-GFP (pVD2659) were grown overnight in SC + 2% raffinose. After back-dilution to an OD600 = 0.1 in SC + 2% galactose, cells were grown for another 4 h and imaged as described above.
VDY6374 and VDY6375 cells were transformed with pVD3006/3007. Transformants were grown overnight in S-Trp (synthetic media lacking tryptophan) + 2% glucose. Cells were next back-diluted to an OD600 of 0.1 and grown for another 4 h and subsequently imaged as described above.
Flow cytometry
For flow cytometry, experiments were performed using the FACSymphony A3 analyzer (BD Biosciences) with the 488 nm laser (FITC). 10,000 events were collected for every sample.
VDY3334 and VDY6165 cells were grown to saturation overnight in SC + 2% glucose. After back-dilution to an OD600 of 0.1, cells were grown for 4–5 h until OD600 ~ 0.5. Samples were then measured, and medians were collected using the Bioconductor packages flowCore and flowViz.
To determine the HSR of Ypl225wF19A-3xFLAG-expressing cells relative to Ypl225wWT-3xFLAG-expressing cells and ypl225wΔ cells, VDY6292, VDY6334, and VDY6341 cells were grown to saturation overnight in SC + 2% glucose. Subsequent dilution, growth, measurement, and analysis was performed as described above.
VDY6176 and VDY6206 cells were grown to saturation in SC + 2% glucose media. Cells were back-diluted to an OD600 of 0.1 in SC + 2% glucose and grown until OD600 = 0.5. Samples were immediately measured. 10,000 cells were measured for each sample and medians were collected as described above.
VDY6205 and VDY6240 cells were grown overnight in SC + 2% raffinose media. Cells were back-diluted to an OD600 = 0.1 in SC + 2% galactose and grown for 4 h. Samples were measured and sample medians were analyzed as described above.
Growth assays
VDY6285, VDY6287, VDY6335, and VDY6336 cells were grown to mid-log phase and back-diluted to an OD600 of 0.4. The first spot of each row represents the undiluted culture and subsequent spots were taken from serial dilutions (1:10).
VDY3334 and VDY6165 cells were grown to mid-log phase before back-dilution to an OD600 of 0.05 in 200 µL of YPD rich media in a 96-well plate. Cells were grown with shaking for 24 h at 30 °C with OD600 measurements collected at 10-min intervals. Three replicates were measured for each sample.
Polysome profiling
VDY6217 cells were grown logarithmically in YPD before back-dilution to reach an OD600 of 0.5. Next, cells were harvested by rapid vacuum filtration and snap freezing in liquid nitrogen. The frozen paste was mixed with frozen polysome lysis buffer (20 mM Tris pH 8.0, 70 mM KCl, 5 mM MgCl2, 100 µg/mL cycloheximide, 1 mM DTT, 1% Triton X-100, 0.025 U/µL Turbo DNAse [AM2238, ThermoFisher Scientific]) with 1 mL lysis buffer added per 500 OD600 units. The frozen mix was then cryogenically lysed using the Retsch MM 400 mixer mill and the resultant grindate was thawed for 2 min at 30 °C with intermittent flicking and placed on ice. 40 mM EDTA was added during this thawing step for samples that received this treatment. Samples were clarified via centrifugation for 10 min at 16,000 × g and 4 °C and the RNA concentration was normalized across samples. 1 mg of RNA was loaded onto linear 10–50% sucrose gradients and centrifuged in a Beckmann Coulter SW-41 rotor for 3 h at 154,000 × g and 4 °C. Gradients were fractionated using a BioComp Gradient Station and the absorbance at 260 nm was measured during fractionation. Trichloroacetic acid (TCA) was added to fractionated samples to a final concentration of 20%, and samples were stored at −20 °C overnight. The next day, samples were spun down for 30 min at 4 °C, washed three times with ice-cold acetone (where each wash consisted of a 10-min 4 °C centrifugation step), and vacuum-dried. The protein pellet was solubilized in SDS loading buffer and separated via SDS-PAGE before immunoblotting with the indicated antibodies.
Immunoprecipitations
Cells containing endogenously tagged Ypl225wWT-3xFLAG or Ypl225wF19A-3xFLAG were grown in 1.5 L cultures to an OD600 of 1.8 in liquid YPD media. Cells were spun down at 3500 × g for 15 min at 4 °C, washed in 50 mL double-distilled water twice, and resuspended in 1 mL (per gram of pellet) resuspension buffer (1.2% PVP-40, 20 mM HEPES pH 7.4, 1x protease inhibitor cocktail [Roche], 1% solution P [2 mg pepstatin A, 90 mg PMSF, 5 mL 100% ethanol], 1 mM DTT). Cells were spun down at 3500 × g for 15 min to remove all traces of buffer. The cell paste was placed into a syringe and was pushed out to be frozen as “noodles” in a 50 mL conical tube filled with liquid nitrogen. Liquid nitrogen was decanted from the tube and frozen “noodles” were stored at −80 °C until cryogenic lysis using a Retsch PM100 ball mill. Next, 250 mg of grindate powder was thawed at 4 °C followed by resuspension in 1 mL of HIP buffer (40 mM HEPES-KOH pH 7.5, 110 mM KOAc, 2 mM MgCl2, 1% Triton X-100, 0.1% Tween, 1x protease inhibitor cocktail [Roche], 1% solution P, 1 mM DTT). The lysate was next passed through a Whatman 25 mm GD/X disposable filter (Cat No. 6888-2527) and equal volumes of the filtered lysates were added to 62.5 μL protein G Dynabeads pre-bound to 3.75 μL of anti-FLAG M2 antibody. Samples were subsequently agitated for 30 min at 4 °C. The beads were then washed twice with 750 µL of HIP buffer. After the final wash, proteins were eluted from beads with 40 μL of 1 mg/mL 3xFLAG peptide in HIP buffer at room temperature for 30 min. Samples were analyzed via SDS-PAGE followed by immunoblotting with the indicated antibodies.
For Supplementary Fig. 7, 1 mM GTP and the regeneration system (1 mM ATP, 10 mM creatine phosphate, 0.04 mg/mL creatine kinase) were supplemented as indicated to all HIP wash buffer steps.
Pure eEF1A, eEF1A DI, or Sup35 DI was diluted in Buffer A (20 mM HEPES pH 7.4, 100 mM KOAc, 2 mM Mg(OAc)2, 2 mM DTT) with 5% glycerol at 2× the final concentration. Ypl225w-3xFLAG and nucleotide (if present) were added for 30 min so that the final concentration of each protein was 3 µM and the nucleotide concentration was 100 µM or 1 mM. An aliquot was removed as the total. Samples were added to protein G Dynabeads pre-conjugated to anti-FLAG antibody (~1 µL antibody per 16.6 µL beads) resuspended in 1 reaction volume of Buffer A with 5% glycerol. Samples were next agitated at 4 °C for 30 min before the unbound material was removed and samples were washed 3x in 1 mL cold wash buffer (Buffer A with 5% glycerol and 0.05% Tween). Samples were eluted after agitating for 30 min at RT with 1 mg/mL 3xFLAG peptide in Buffer A with 5% glycerol. Equal amounts of totals and IPs were added to 2x SDS/urea sample buffer (20 mM Tris-HCl pH 6.8, 4 M urea, 2.5% SDS, 0.05 mM EDTA, 1% β-mercaptoethanol, 10% glycerol), heated at 80 °C, and analyzed by SDS-PAGE and Coomassie staining.
A final concentration of 125 A260 units of high salt-washed ribosomes, 7.5 µM NAC or NAC mutant protein, and 3 µM Ypl225w-3xFLAG protein were incubated as indicated together for 30 min at room temperature. An aliquot was removed as the total. Samples were added to protein G Dynabeads pre-conjugated to anti-FLAG antibody (~1 µL antibody per 16.6 µL beads) resuspended in 1 reaction volume of Buffer A with 5% glycerol. Samples were next agitated at 4 °C for 30 min, the unbound material was removed, and samples were washed 3x in 1 mL cold wash buffer (Buffer A with 5% glycerol and 0.05% Tween) before elution via agitation for 30 min at room temperature with 1 mg/mL 3xFLAG peptide in Buffer A with 5% glycerol. Equal amounts of totals and IPs were added to 2x SDS/urea sample buffer, and analyzed by SDS-PAGE and Coomassie staining or immunoblotting with the indicated antibodies.
For release assays to monitor eEF1A DI dissociation from Ypl225w-3xFLAG, the following changes were made: reactions were set up so that the final concentration of eEF1A and Ypl225w would be ~150 nM and 300 nM, respectively, in 24 µL after splitting the washed beads into 13 tubes. One sample was set aside, and the remaining 12 were incubated in 24 µL Buffer A + 5% glycerol with the indicated added components for the indicated times at room temperature with agitation. The supernatant was removed, and the beads for all 13 samples were eluted with 24 µL of 1 mg/mL 3xFLAG peptide in Buffer A + 5% glycerol as before. Equal amounts of supernatant and eluate from the beads were added to 2x SDS/urea sample buffer and analyzed by SDS-PAGE and immunoblotting.
Yeast in vitro transcription and translation
The mMESSAGE mMACHINE T7 kit (Invitrogen) was used as described previously to generate capped mRNAs from PCR templates44. PCR products encoding TEF2 were generated from pVD2609 using primers oVD13102 and oVD13103. Nonstop mRNAs were generated from pVD2609 using primers oVD13102 and oVD13432 (V160-RNCs) or oVD13603 (V332-RNCs). D156N versions of eEF1A V160/V332 nonstop mRNAs were generated using pVD2860 as a template. PCR products encoding SUP35 were generated from genomic S. cerevisiae DNA.
Yeast in vitro translation (IVT) extracts were prepared as described previously12. Briefly, 1.5 L YPD media of indicated strains were grown to an OD600 ~ 1.8–2.0. Cells were centrifuged at 2556 × g for 15 min in a JS-4.2 rotor at 4 °C. Next, pellets were first resuspended in DEPC-treated water, centrifuged, and then resuspended in Buffer A (30 mM HEPES pH 7.4, 100 mM KOAc, 2 mM Mg(OAc)2) with 2 mM DTT. Finally, 1 mL Buffer A with 2 mM DTT, 14% glycerol (Buffer ADG), and 2x protease inhibitor cocktail (Roche) was added per 6 grams of cell weight and frozen via a “popcorn” method (frozen drop by drop in liquid nitrogen) before being lysed in a Retsch PM100 ball mill. The powder was thawed at 4 °C and centrifuged at 13,336 × g for 10 min at 4 °C. The resultant supernatant was transferred to an SW-55 rotor and centrifuged at 291,000 × g for 30 min at 4 °C with no brake. The middle two layers were removed for subsequent desalting (using five connected Hi-Trap desalting columns (GE Healthcare, Chicago, IL, USA) on an AKTA Pure FPLC system). Fractions with A260 > 60 were pooled, RNase-treated (and subsequently deactivated using EGTA), and snap-frozen.
Each single IVT reaction consisted of 7.5 µL of nuclease-treated extracts, 2 µL Buffer ADG, 2.5 µL of 6x translation buffer (132 mM HEPES-KOH, pH 7.4, 720 mM KOAc, 9 mM Mg(OAc)2, 4.5 mM ATP, 0.6 mM GTP, 150 mM creatine phosphate [Roche], 0.24 mM of each amino acid but lacking methionine [Promega, Madison, WI], 10.2 mM DTT), 0.5 µL creatine kinase [20 mg/mL in 50% glycerol, Roche], 0.5 µL RiboGuard [Lucigen, Middleton, WI, USA], 1 µL 35S-Methionine [PerkinElmer, Waltham, MA, USA], and 1 µL of 200 ng/µL capped mRNA. Briefly, IVT reactions (once translation had initiated) were incubated for 45 min at room temperature (unless otherwise stated). Cycloheximide was added to 100 µg/mL to arrest translation and an aliquot was taken for “Before Trypsin” gel samples. The remainder of the reaction was treated with 50 µg/mL trypsin (Promega) and incubated at room temperature for the indicated times. Samples were quenched using an equal volume of 2x SDS/urea sample buffer before being heated for 3 min at 70 °C. Samples were analyzed via SDS-PAGE, autoradiography, and immunoblotting.
For the generation of RNCs, four IVT reactions were used per sample, and each sample was programmed with nonstop eEF1A mRNA encoding a truncation of either 160 or 332 amino acids. Translation reactions were incubated for 15 min at room temperature prior to arrest with cycloheximide. Next, samples were then treated with either water or BS3 to 1 mM final concentration. Crosslinking samples were incubated on ice for 90 min in the dark. After 90 min, 20 mM Tris pH 6.8 was added to quench the crosslinking reagent. SDS and DTT were then added to each sample to 1% SDS and 0.5 mM DTT final concentration to denature non-covalent interactions, and samples were heated subsequently for 10 min at 65 °C. Next, cold IP buffer (buffer ADG + 0.1% Triton X-100) was added to dilute denaturant 1:10 (e.g., 450 µL IP buffer added to 50 µL IVT reaction) and samples were added to protein G Dynabeads pre-conjugated to anti-FLAG antibody (0.5 µL antibody per 10 µL beads). Samples were immunoprecipitated by gentle nutation for 30 min at 4 °C and Dynabeads were then washed twice in cold IP buffer. Samples were eluted using 70 °C-heated 1x SDS sample buffer (40 mM Tris-HCl pH 6.8, 5% SDS, 0.1 mM EDTA, 1% β-mercaptoethanol, 0.1 mg/mL bromophenol blue, 20% glycerol). Samples were analyzed via SDS-PAGE, autoradiography, and immunoblotting.
Recombinant protein expression and purification
eEF1A DI or Sup35 DI constructs were cloned into pET29b vectors with a Myc-6xHis C-terminal tag. Rosetta (DE3) cells were transformed with these vectors and grown at 37 °C in Terrific Broth (TB) media until OD600 of 0.5. The cultures were moved to 4 °C for 1 h and then supplemented with 0.4 mM IPTG (US Biological Life Sciences) to induce protein expression at 16 °C for 16 h. Cells were harvested at 3500 × g for 20 min and resuspended in PBS. After a second spin, cell pellets were frozen in liquid nitrogen and stored at −80 °C until purification.
Cell pellets were thawed at 4 °C and resuspended in lysis buffer (50 mM Tris pH 7.5, 150 mM NaCl, 2% glycerol, 2 mM MgCl2) supplemented with 2 mM β-mercaptoethanol, protease inhibitor cocktail [Roche], and 1 mM PMSF. Cells were lysed using a sonicator set to 40% power for 5 × 30-s intervals on ice with 1 min on ice in between. Lysed cells were spun down at 20,000 × g for 15 min at 4 °C in a JLA 16.25 rotor. Supernatants were loaded on 1 mL equilibrated HisPur Ni-NTA resin (ThermoFisher Scientific) in gravity flow columns twice. The resin was washed sequentially with 20 mL wash buffer 1 (50 mM Tris pH 7.5, 500 mM NaCl, 2% glycerol, 2 mM MgCl2, 2 mM β-mercaptoethanol), 20 mL wash buffer 2 (50 mM Tris pH 7.5, 150 mM NaCl, 2% glycerol, 2 mM MgCl2, 10 mM imidazole, 2 mM β-mercaptoethanol), and 20 mL wash buffer 3 (50 mM Tris pH 7.5, 150 mM NaCl, 2% glycerol, 2 mM MgCl2, 50 mM imidazole, 2 mM β-mercaptoethanol). The resin was then incubated with 3 mL of elution buffer (50 mM Tris pH 7.5, 150 mM NaCl, 2% glycerol, 2 mM MgCl2, 250 mM imidazole, 2 mM β-mercaptoethanol) for 5 min before eluting.
Eluate was concentrated to <1 mL and applied to an equilibrated Superdex 200 10/300 column for gel filtration in SEC buffer (40 mM HEPES pH 7.5, 110 mM KOAc, 2 mM MgCl2, 2% glycerol, 0.5 mM TCEP). Peak fractions were aliquoted, frozen in liquid nitrogen, and stored at −80 °C.
Ypl225w-3xFLAG constructs were cloned into pET29b vectors with a 3xFLAG-3c-6xHis C-terminal tag. Rosetta (DE3) cells were transformed with these vectors and grown at 37 °C in TB media until OD600 of 0.5. The cultures were then supplemented with 1 mM IPTG to induce protein expression at 37 °C for 4 h. Cells were harvested at 3500 × g for 20 min and resuspended in PBS. After a second spin, cell pellets were frozen in liquid nitrogen and stored at −80 °C until purification.
Cell pellets were thawed at 4 °C, resuspended in B1 Lysis Buffer (20 mM sodium phosphate pH 8.0, 300 mM NaCl, 20 mM imidazole) supplemented with benzonase (MilliporeSigma), 5 mM β-mercaptoethanol, protease inhibitor cocktail [Roche], and 0.2 mM PMSF. Cells were lysed with 5 passes on an Emulsiflex cell disruptor at ∼12–15k PSI. Lysed cells were spun down at 28,000 × g for 30 min at 4 °C in a JLA 16.25 rotor. The resultant supernatant was loaded onto an equilibrated 5 mL HisTrap HP column. The column was washed with 10 column volumes of B1 buffer before eluting with a linear gradient of B2 elution buffer (20 mM sodium phosphate pH 8.0, 300 mM NaCl, 500 mM imidazole, 5 mM β-mercaptoethanol) over 5 column volumes. Peak elution fractions were pooled and concentrated to 2.5 mL with a 10 kDa cutoff filter before desalting with a PD10 desalting column into B5 SEC buffer (50 mM Tris pH 8.0, 200 mM NaCl, 10% glycerol, 0.5 mM TCEP). 1 mL of desalted sample was applied to an equilibrated Superdex 200 10/300 column for gel filtration in B5 SEC buffer. Peak fractions were aliquoted, frozen in liquid nitrogen, and stored at −80 °C.
NAC constructs were cloned into pETDuet vectors with N-terminal Myc-Egd2 and 10xHis-Egd1 tags. Rosetta (DE3) cells were transformed with these vectors and grown at 37 °C in Luria (LB) media until OD600 of 0.6. The cultures were then supplemented with 1 mM IPTG to induce protein expression at 37 °C for 3 h. Cells were harvested at 3500 × g for 20 min and resuspended in IMAC-A buffer (50 mM HEPES pH 7.5, 300 mM NaCl, 20 mM imidazole, 1 mM DTT, 5% glycerol) supplemented with 1 mM PMSF. After a second spin, cell pellets were frozen in liquid nitrogen and stored at −80 °C until purification.
Cell pellets were thawed at 4 °C and resuspended in IMAC-A buffer supplemented with 1 mM PMSF. Next, cells were lysed using a sonicator set to 40% power for 3 × 30-s intervals on ice. Lysed cells were spun down at 40,000 × g for 30 min at 4 °C in a JA 25.5 rotor. Resultant supernatants were loaded on 1 mL equilibrated HisPur Ni-NTA resin (ThermoFisher Scientific) in gravity flow columns and washed with 15 mL of IMAC-A buffer. The protein was eluted in 3 mL IMAC-B buffer (50 mM HEPES pH 7.5, 300 mM NaCl, 500 mM imidazole, 1 mM DTT, 5% glycerol). The eluate was concentrated to 0.5 mL with a 10 kDa cutoff filter before being applied to an equilibrated Superdex 200 10/300 column for gel filtration in SEC buffer (50 mM HEPES pH 7.5, 150 mM NaCl, 0.5 mM TCEP). Peak fractions were combined, concentrated, aliquoted, frozen in liquid nitrogen, and stored at −80 °C.
Purification of native eEF1A from yeast
Native eEF1A was purified from S. cerevisiae as previously described12. VDY465 (WT) cells were grown in large cultures to an OD600 of 1.8 in liquid YPD media. 3 L of cells were spun down at 3500 × g for 20 min at 4 °C, washed in 50 mL double-distilled water twice, and resuspended in 1 mL per gram of pellet of resuspension buffer (1.2% PVP-40, 20 mM HEPES pH 7.4, 1x protease inhibitor cocktail [Roche], 1% solution P [2 mg pepstatin A, 90 mg PMSF, 5 mL 100% ethanol], 1 mM DTT). Cells were spun down twice at 3500 × g for 10 min to remove all traces of buffer. The resultant cell paste was placed into a syringe and was pushed out to be frozen as “noodles” in a 50 mL conical tube filled with liquid nitrogen. Liquid nitrogen was decanted from the tube and frozen “noodles” were stored at −80 °C until cryogenic lysis using a Retsch PM100 ball mill.
Next, 8 g of frozen grindate was thawed in 80 mL I-100 buffer (20 mM Tris pH 7.5, 0.1 mM EDTA, 100 mM KCl, 25% glycerol, 1 mM DTT) supplemented with 1x protease inhibitor cocktail (Roche). The lysate was spun at 8000 × g for 5 min at 4 °C in a JA 25.5 rotor, and the supernatant was transferred into new tubes and spun at 20,000 × g for 15 min at 4 °C in a JA 25.5 rotor. The supernatant was then spun at 300,000 × g for 106 min at 4 °C in a Ti-70 ultracentrifuge rotor. Next, the supernatant was incubated with 10 g of pre-equilibrated DEAE resin (in buffer I-100) for 1 h while agitated on a tilt table at 4 °C. The unbound fraction was removed by centrifugation. The resin was washed with an additional 20 mL I-100 buffer, spun down, and the wash was added to the unbound fraction. The unbound fraction and wash were incubated with 60 mL pre-equilibrated CM-sepharose slurry (in buffer I-100) for 1 h while agitated on a tilt table at 4 °C. A Buchner funnel with Whatman type 1 paper (∼11 µm pore size) was used to separate the unbound fraction. The resin was washed with 40 mL I-100 before removing from the Buchner funnel, resuspended in 60 mL I-100 with solid KCl supplemented to 500 mM final concentration of KCl, and incubated for 1 h while agitated on a tilt table at 4 °C. Eluate was collected using the Buchner funnel, and resin was washed with an additional 20 mL buffer I-100 supplemented with solid KCl for a final concentration of 500 mM KCl, which was combined with the eluate. Next, the eluate was concentrated with a 30 kDa cutoff concentrator to ∼5 mL, filtered with a 0.22 µm filter and desalted into I-50 buffer (20 mM Tris pH 7.5, 0.1 mM EDTA, 50 mM KCl, 25% glycerol, 1 mM DTT) using 4 × 5 mL sequential HiTrap desalting columns. ∼8 mL of flow-through was collected and applied onto a Source 15S 4.6/100 column. The column was washed with 40 column volumes of I-50 buffer before eluting with a linear gradient of I-300 buffer (20 mM Tris pH 7.5, 0.1 mM EDTA, 300 mM KCl, 25% glycerol, 1 mM DTT) over 20 column volumes. Peak fractions containing eEF1A were pooled and concentrated to ∼2 mL with a 30 kDa cutoff concentrator. The sample was next applied onto a HiPrep 16/60 Sephacryl S-100 column. Native eEF1A eluted as the second peak. Fractions were pooled and concentrated with a 30 kDa cutoff concentrator. Aliquots were frozen in liquid nitrogen and stored at −80 °C.
Purification of high-salt-washed ribosomes
VDY6292 cells were grown in large cultures to an OD600 1.8 in liquid YPD media. 4.5 L of cells were spun down at 3500 × g for 20 min at 4 °C, washed in 50 mL double-distilled water twice, and resuspended in 1 mL per gram of pellet of resuspension buffer. Cells were spun down twice at 3500 × g for 10 min to remove the buffer. The cell paste was frozen as “noodles” and cryogenically lysed using a Retsch PM100 ball mill.
Next, the powder was thawed in 25 mL of ribosome lysis buffer (1x Buffer A, 1 mg/mL heparin sodium salt [Sigma]) and supplemented with 2 mM DTT, 1x protease inhibitor cocktail (Roche), and 0.1 mM PMSF at 4 °C. The lysate was cleared by centrifugation at 20,000 × g for 30 min at 4 °C in a JA 25.5 rotor. Next, the supernatant was brought up to 35 mL with a ribosome lysis buffer supplemented with 2 mM DTT, 1x protease inhibitor cocktail (Roche), and 0.1 mM PMSF. ~17.5 mL of the solution was layered onto two 3 mL low-salt sucrose cushions (1x Buffer A, 100 mM KCl, 500 mM sucrose) before centrifugation for 106 min at 370,000 × g in a Ti-70 rotor at 4 °C. The supernatant was removed carefully, and each pellet was resuspended carefully in 1.5 mL high salt wash buffer (1x Buffer A, 600 mM KCl, 1 mg/mL heparin, 2 mM DTT). The volumes were combined, brought up to 9 mL in a 50 mL falcon tube and left overnight at 4 °C to fully resuspend. 3 mL of the sample was layered onto 3 × 250 µL high-salt sucrose cushions (1x Buffer A, 600 mM KCl, 500 mM sucrose) before centrifugation at 100,000 × g for 30 min at 4 °C. Pellets were rinsed once in freezing buffer (1x Buffer A, 10% glycerol, 2 mM DTT) before resuspending in 100 µL freezing buffer. Aliquots were frozen in liquid nitrogen and stored at −80 °C.
Bio-layer interferometry
Biolayer interferometry was performed on an Octet RED384 instrument (Sartorius). Anti-FLAG M2 was immobilized on protein G biosensors (Sartorius Cat. No. 18-5082) at 5 µg/mL (in binding buffer: Buffer A with 5% glycerol, 2 mM DTT, and 0.05% Tween-20) for 120 s, after which a baseline measurement was performed in a new well with the same buffer for 30 s. Sensors were then moved into 1 µM Ypl225w-FLAG for 30 s to capture Ypl225w-FLAG. After Ypl225w-FLAG binding, sensors were moved into a binding buffer with 1 mM GTP (Roche), 1 mM XTP (TriLink Biotechnologies), or no nucleotide to acquire baseline measurement for 30 s. Then sensors were moved into 300 nM eEF1A DI (DIWT or DID156N) domain in binding buffer without nucleotide for the association phase. After 100 s, the sensors were moved back into baseline wells with or without nucleotides to measure eEF1A DI domain dissociation in the presence of different nucleotides. Probes were regenerated in 0.1 mM glycine pH 1.5 and neutralized in binding buffer before rebinding anti-FLAG M2 and reusing for subsequent measurements. Measurements were obtained in quadruplicate or triplicate for each condition. Data were analyzed in the included Octet RED384 data analysis software. Briefly, each sensor was subjected to double reference subtraction. First, a reference sensor with no Ypl225w-3xFLAG was subtracted from each sensor. Then, a reference well with no eEF1A DI domain was subtracted from sensors subjected to eEF1A DI domain binding. The data were corrected to adjust signal to the same value between the association and dissociation steps to correct for optical artifacts caused by moving sensors between wells.
Negative stain of YPL225W, YPL225WWT-3xFLAG, and YPL225WF19A-3xFLAG strains
Samples were stained using a 1.5% (w/v) uranyl formate solution and imaged on a ThermoFisher Scientific Tecnai T12 electron microscope equipped with a LaB6 filament, operated at 120 kV, and at 59,000× magnification. The microscope was equipped with a Gatan UltraScan 895 (4k x 4k) CCD camera.
Ypl225w-3xFLAG immunoprecipitation mass spectrometry and analysis
Three technical replicates of immunoprecipitations from either untagged cells or cells expressing Ypl225w-3xFLAG protein were used for downstream mass spectrometry and analysis. The samples were prepared using filter-aided sample preparation (FASP). Protein samples were loaded onto the 10k filter and centrifuged at 13,500 × g for 10 min followed by a wash with 100 mM triethylammonium bicarbonate (TEAB). Proteins were reduced by 20 mM tris(2-carboxyethyl)phosphine (TCEP) for 1 h at 37 °C followed by centrifugation as described above and then alkylated in 40 mM iodoacetamide for 1 h in darkness at room temperature followed by centrifugation as described above. Protein digestion was then performed using trypsin with 1:50 ratio (w:w, substrate: enzyme) and incubating overnight at 37 °C. Digested filtrate samples were cleaned up with C18 Spin Tips (Pierce, ThermoFisher Scientific) and reconstituted with 0.1% formic acid prior to LC-MS/MS analysis.
Samples were analyzed by Lumos Tribrid Orbitrap Mass Spectrometer coupled with Ultimate 3000 nano-HPLC (ThermoFisher Scientific) at the Harvard Center for Mass Spectrometry. Peptides were separated onto a 150 μm inner diameter microcapillary trapping column packed with approximately 2 cm of C18 Reprosil resin (5 μm, 100 Å, Dr. Maisch GmbH, Germany) followed by an analytical column (µPAC, C18 pillar surface, 50 cm bed, ThermoFisher Scientific). Separation was achieved by applying a gradient from 7–37% acetonitrile in 0.1% formic acid over 90 min at 500 nL/min. Electrospray ionization was enabled by applying a voltage of 2.2 kV using a homemade electrode junction at the end of the microcapillary column and sprayed from metal tips (PepSep, Denmark). The mass spectrometry survey scan was performed in the Orbitrap in the range of 400–1800 m/z at a resolution of 12 × 104, followed by the selection of the twenty most intense ions (TOP20) for CID-MS2 fragmentation in the Ion trap using a precursor isolation width window of 2 m/z, AGC setting of 10,000, and a maximum ion accumulation of 100 ms. Singly charged ion species were not subjected to CID fragmentation. The normalized collision energy was set to 35 V and an activation time of 10 ms. Ions in a 10 ppm m/z window around ions selected for MS2 were excluded from further selection for fragmentation for 90 s.
Raw data was then submitted for analysis in Proteome Discoverer 2.5 software (ThermoFisher Scientific). The mass spectrometry data was searched against the Uniprot-reviewed Saccharomyces cerevisiae database along with known contaminants such as human keratins and common lab contaminants. Quantitative analysis between samples was performed by label-free quantitation (LFQ). Sequest HT searches were performed using the following guidelines: a 10 ppm MS tolerance and 0.6 Da MS/MS tolerance; trypsin digestion with up to two missed cleavages; carbamidomethylation (57.021 Da) on cysteine were set as static modification; oxidation (+15.995 Da) of methionine and protein N-terminus acetylation set as dynamic modification; minimum required peptide length set to ≥6 amino acids. At least one unique peptide per protein group is required for identifying proteins. All MS2 spectra assignment FDR of 1% on both protein and peptide level was achieved by applying the target-decoy database search by Percolator.
Computational screening for interactors of eEF1A
AlphaPulldown in the “pulldown” mode with default parameters was used to screen 3769 yeast proteins conserved in humans (source: DIOPT Ortholog Finder) for candidates forming a binary complex with eEF1A17,45. MSAs were generated using ColabFold/MMSeqs2 prior to running the structure predictions in parallel as a HPC job array. Complexes were filtered for an inter-chain PAE < 5 and a Protein Interaction score based on structural properties of the complex (PI_score > 0.1)46. The full filtered list of eEF1A predicted interactions can be found in Supplementary Data 2.
ColabFold modeling of Ypl225w•eEF1A and NAC•Ypl225w•eEF1A complexes
ColabFold version 1.5.2 was used with 5 models, 20 seeds, 20 recycles and the default parameters with msa_mode: “MMseqs2 (UniRef+Environmental)” and model_type: “AlphaFold2-multimer-v3” to model the S. cerevisiae eEF1A•Ypl225w and eEF1A DI•Ypl225w complexes. For the S. cerevisiae eEF1A DI•Ypl225w•Egd2•Egd1 complex, ColabFold version 1.5.2 was run with the same parameters but with 1 seed. Sequences were obtained from Uniprot. eEF1A DI contains residues 1–239. Figure 1b highlighted residues on eEF1A are: 36–75 (Switch I), 95–110 (Switch II), and 14–21 (P-loop). Models are provided in Supplementary Data 3. Consurf was used to determine conservation scores for Ypl225w residues, and CLUSTAL Omega was used for sequence alignment, as shown in Supplementary Fig. 6. Molecular graphics and analyses for all models were performed with UCSF ChimeraX.
Co-translational folding simulations of eEF1A and Sup35
For the simulations of eEF1A and Sup35, we employed Monte Carlo Protein Unfolding (MCPU), an all-atom protein Markov Chain Monte Carlo (MCMC) simulation, using the MCPU software21. This software serves as the core protein Monte Carlo (MC) simulation engine with knowledge-based potential of the DBFOLD Python package20. The version of the software used is available at https://github.com/amirbitran/dbfold. MCMC simulations were chosen for their efficiency in exploring the rugged energy landscapes, allowing for the capture of non-native but significant conformational states. To further enhance the sampling of the free energy and to search for any non-native structures, we utilized temperature replica exchange and umbrella sampling as described and implemented in the DBFOLD package20.
In this study, two truncated chains for each protein were selected to capture snapshots of the co-translational folding landscape at early folding stages by looking at the free energy landscape of varying chain lengths22,23. The truncation points were strategically chosen to be at 72 and 95 amino acids from the N-terminus to ensure that the secondary structures of interest were retained. These specific truncation points were selected based on the interaction pattern between eEF1A and Ypl225w predicted by ColabFold, as well as experimental insights from cell-free translation experiments that investigated “timing” of Ypl225w association.
The initial structures for these simulations were predicted by ColabFold and processed using a minimization process that ran for 50 million steps at a temperature T = 0.1. This low temperature was chosen to refine the initial structure by minimizing the thermal noise and relax the structure in the forcefield so that we could use it as a more accurate starting point for the production simulations. In MCPU software, this temperature scales the difference in potential energy during the MC simulation, where lower values correspond to lower physical temperatures, thereby facilitating structure stabilization.
After minimization, a total N = (number of temperatures) × (number of setpoints) replicas were initiated and simulated using replica exchange at every 10,000th step. Temperatures for production simulation ranged from T = 0.4 to T = 1.0 in increments of ∆T = 0.025. For umbrella sampling, a harmonic bias potential was applied along the number of native contacts as the reaction coordinates, as implemented in the DBFOLD. A grid of setpoints was established from 0 to the total number of native contacts, rounded to the nearest 10 with increments of 10. This ensured sufficient overlap between replicas, enabling the statistical reweighting to unbias the system during analysis. The bias in the potential is applied as follows:
where for conformation, \(({x}_{n})\), \(E({x}_{n})\) is the biased potential energy, \(E{({x}_{n})}^{0}\) is the unbiased potential energy, \({k}_{{bias}}\) is the constant that controls the magnitude of the bias, \(N({x}_{n})\) is the number of native contacts, and \(S\) is the set-point.
After performing 600 million MC steps and once the simulation reached equilibrium, analyses were conducted to characterize the sampled ensembles. To ensure the Markov Chain had equilibrated, the first 200 million steps were discarded as a burn-in stage. This threshold was selected based on the point where the moving average energy of each replica reached a plateau. From the remaining 400 million samples, every 500,000th samples were kept for analysis, ensuring that the samples were independent and identically distributed.
For each conformation sampled, the proportions of secondary structures (helices and strands) were computed based on the DSSP (Dictionary of Secondary Structure of Proteins) definitions using the Python package MDTraj47. MDTraj is also used to compute the Radius of gyration (Rg) of each sample. Using helical content and Rg as features, we conducted free energy calculations using the Multistate Bennett Acceptance Ratio (MBAR) method48, which reweights the data from biased trajectories to estimate observables under the unbiased condition. This step was essential to correct for biases introduced during enhanced sampling, thereby ensuring unbiased free energy estimations from the simulated trajectories.
To construct smoother free energy landscapes and estimate the underlying probability density function (PDF) of samples, Kernel Density Estimation49 is performed with Python package Scipy. After obtaining the PDF, probabilities were computed for the conformational states and converted into free energy using \(F=-\log (P)\), where \(F\) is in units of \({k}_{B}T\). For the representation, free energy values were evaluated with respect to the lowest free energy conformation and colored accordingly. To highlight the low-energy conformations, free energy differences greater than 5 kT were masked in dark red. The input files to reproduce simulations can be found in Supplementary Data 4.
Quantification and statistical analysis
Using the BioConductor packages flowCore and flowViz, flow cytometry samples were gated for live, single cells. Samples were normalized to cell size by dividing the raw GFP/YFP intensity (FITC.A) by side scatter (SSC.A). Data was log-transformed for histograms and bar graphs.
To detect statistically significant differences in protein abundance, the R package limma (v.3.50.0) was used to calculate the FDR on the Empirical Bayes moderated t-statistic44. A protein was defined as significant if FDR < 0.05. The results of this analysis are provided in Supplementary Data 1.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Further information, resources or reagents are available upon request. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE50 partner repository with the dataset identifier PXD058955. Data generated in this study are provided in the Supplementary Information and Source Data files. Source data are provided with this paper.
Code availability
The version of the software used for MC simulations of eEF1A and Sup35 is available at https://github.com/amirbitran/dbfold.
References
Kramer, G., Shiber, A. & Bukau, B. Mechanisms of cotranslational maturation of newly synthesized proteins. Annu. Rev. Biochem. 88, 337–364 (2019).
Balchin, D., Hayer-Hartl, M. & Hartl, F. U. In vivo aspects of protein folding and quality control. Science 353, aac4354 (2016).
Gamerdinger, M. & Deuerling, E. Cotranslational sorting and processing of newly synthesized proteins in eukaryotes. Trends Biochem. Sci. S0968-0004, 00258 (2023).
Chen, Y., Tsai, B., Li, N. & Gao, N. Structural remodeling of ribosome associated Hsp40-Hsp70 chaperones during co-translational folding. Nat. Commun. 13, 3410 (2022).
Zhang, Y. et al. The ribosome-associated complex RAC serves in a relay that directs nascent chains to Ssb. Nat. Commun. 11, 1504 (2020).
Kišonaitė, M. et al. Structural inventory of cotranslational protein folding by the eukaryotic RAC complex. Nat. Struct. Mol. Biol. 30, 670–677 (2023).
Lee, K. et al. Pathway of Hsp70 interactions at the ribosome. Nat. Commun. 12, 5666 (2021).
Gamerdinger, M. et al. NAC controls cotranslational N-terminal methionine excision in eukaryotes. Science 380, 1238–1243 (2023).
Jomaa, A. et al. Mechanism of signal sequence handover from NAC to SRP on ribosomes during ER-protein targeting. Science 375, 839–844 (2022).
Deuerling, E., Gamerdinger, M. & Kreft, S. G. Chaperone interactions at the ribosome. Cold Spring Harb. Perspect. Biol. 11, a033977 (2019).
McQuown, A. J. et al. A Zpr1 co-chaperone mediates folding of eukaryotic translation elongation factor 1A via a GTPase cycle. Mol. Cell 83, 3108–3122.e13 (2023).
Sabbarini, I. M. et al. Zinc finger protein Zpr1 is a bespoke chaperone essential for eEF1A biogenesis. Mol. Cell 83, 252–265.e13 (2023).
Dever, T. E., Dinman, J. D. & Green, R. Translation elongation and recoding in eukaryotes. Cold Spring Harb. Perspect. Biol. 10, a032649 (2018).
Liu, K., Maciuba, K. & Kaiser, C. M. The ribosome cooperates with a chaperone to guide multi-domain protein folding. Mol. Cell 74, 310–319.e7 (2019).
Prabhakar, A., Puglisi, E. V. & Puglisi, J. D. Single-molecule fluorescence applied to translation. Cold Spring Harb. Perspect. Biol. 11, a032714 (2019).
Minoia, M. et al. Chp1 is a dedicated chaperone at the ribosome that safeguards eEF1A biogenesis. Nat. Commun. 15, 1382 (2024).
Yu, D., Chojnowski, G., Rosenthal, M. & Kosinski, J. AlphaPulldown—a Python package for protein-protein interaction screens using AlphaFold-Multimer. Bioinformatics 39, btac749 (2023).
Andersen, G. R. et al. Structural basis for nucleotide exchange and competition with tRNA in the yeast elongation factor complex eEF1A:eEF1Bα. Mol. Cell 6, 1261–1266 (2000).
White, J. T. et al. Protein methylation and translation: role of lysine modification on the function of yeast elongation factor 1A. Biochemistry 58, 4997–5010 (2019).
Bitran, A., Jacobs, W. M. & Shakhnovich, E. Validation of DBFOLD: an efficient algorithm for computing folding pathways of complex proteins. PLoS Comput. Biol. 16, e1008323 (2020).
Yang, J. S., Chen, W. W., Skolnick, J. & Shakhnovich, E. I. All-atom ab initio folding of a diverse set of proteins. Structure 15, 53–63 (2007).
Bitran, A., Jacobs, W. M., Zhai, X. & Shakhnovich, E. Cotranslational folding allows misfolding-prone proteins to circumvent deep kinetic traps. Proc. Natl Acad. Sci. USA 117, 1485–1495 (2020).
Bitran, A., Park, K., Serebryany, E. & Shakhnovich, E. I. Co-translational formation of disulfides guides folding of the SARS-CoV-2 receptor binding domain. Biophys. J. 122, 3238–3253 (2023).
Inagaki, Y., Blouin, C., Susko, E. & Roger, A. J. Assessing functional divergence in EF-1α and its paralogs in eukaryotes and archaebacteria. Nucleic Acids Res. 31, 4227–4237 (2003).
Brandman, O. et al. A ribosome-bound quality control complex triggers degradation of nascent peptides and signals translation stress. Cell 151, 1042–1054 (2012).
Krakowiak, J. et al. Hsf1 and Hsp70 constitute a two-component feedback loop that regulates the yeast heat shock response. eLife 7, e31668 (2018).
Fleischer, T. C., Weaver, C. M., McAfee, K. J., Jennings, J. L. & Link, A. J. Systematic identification and functional screens of uncharacterized proteins associated with eukaryotic ribosomal complexes. Genes Dev. 20, 1294–1307 (2006).
Perara, E., Rothman, R. E. & Lingappa, V. R. Uncoupling translocation from translation: implications for transport of proteins across membranes. Science 232, 348–352 (1986).
Reimann, B. et al. Initial characterization of the nascent polypeptide-associated complex in yeast. Yeast 15, 397–407 (1999).
Ozturk, S. B. et al. Unique classes of mutations in the Saccharomyces cerevisiae G-protein translation elongation factor 1A suppress the requirement for guanine nucleotide exchange. Genetics 174, 651–663 (2006).
Cavallius, J. & Merrick, W. C. Site-directed mutagenesis of yeast eEF1A: viable mutants with altered nucleotide specificity. J. Biol. Chem. 273, 28752–28758 (1998).
Shah, N. B. & Duncan, T. M. Bio-layer interferometry for measuring kinetics of protein-protein interactions and allosteric ligand effects. J. Vis. Exp. 18, e51383 (2014).
Hoffmann, A., Bukau, B. & Kramer, G. Structure and function of the molecular chaperone Trigger Factor. Biochim. Biophys. Acta 1803, 650–661 (2010).
Döring, K. et al. Profiling Ssb-nascent chain interactions reveals principles of Hsp70-assisted folding. Cell 170, 298–311.e20 (2017).
Zhang, Y. et al. NAC functions as a modulator of SRP during the early steps of protein targeting to the endoplasmic reticulum. Mol. Biol. Cell 23, 3027–3040 (2012).
Simonis, N. et al. Empirically controlled mapping of the Caenorhabditis elegans protein-protein interactome network. Nat. Methods 6, 47–54 (2009).
Huttlin, E. L. et al. The BioPlex network: a systematic exploration of the human interactome. Cell 162, 425–440 (2015).
Huttlin, E. L. et al. Architecture of the human interactome defines protein communities and disease networks. Nature 545, 505–509 (2017).
Huttlin, E. L. et al. Dual proteome-scale networks reveal cell-specific remodeling of the human interactome. Cell 184, 3022–3040.e28 (2021).
Papasergi-Scott, M. M. et al. Structures of Ric-8B in complex with Gα protein folding clients reveal isoform specificity mechanisms. Structure 31, 553–564.e7 (2023).
Seven, A. B. et al. Structures of Gα proteins in complex with their chaperone reveal quality control mechanisms. Cell Rep. 30, 3699–3709.e6 (2020).
Tall, G. G., Krumins, A. M. & Gilman, A. G. Mammalian Ric-8A (synembryn) is a heterotrimeric Gα protein guanine nucleotide exchange factor. J. Biol. Chem. 278, 8356–8362 (2003).
Tall, G. G. & Gilman, A. G. Purification and functional analysis of Ric-8A: a guanine nucleotide exchange factor for G-protein alpha subunits. Methods Enzymol. 390, 377–388 (2004).
Schuldiner, M. et al. The GET complex mediates insertion of tail-anchored proteins into the ER membrane. Cell 134, 634–645 (2008).
Hu, Y. et al. An integrative approach to ortholog prediction for disease-focused and other functional studies. BMC Bioinformatics 12, 357 (2011).
Malhotra, S., Joseph, A. P., Thiyagalingam, J. & Topf, M. Assessment of protein-protein interfaces in cryo-EM derived assemblies. Nat. Commun. 12, 3399 (2021).
McGibbon, R. T. et al. MDTraj: a modern open library for the analysis of molecular dynamics trajectories. Biophys. J. 109, 1528–1532 (2015).
Shirts, M. R. & Chodera, J. D. Statistically optimal analysis of samples from multiple equilibrium states. J. Chem. Phys. 129, 124105 (2008).
Scott, D. W. Multivariate Density Estimation: Theory, Practice and Visualization (Wiley, 1992).
Vizcaíno, J. A. et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 44, D447–D456 (2016).
Acknowledgements
We thank Jeffrey Prince, Kate Guerin, and Oliver Rancu for their help with the initial characterization of Ypl225w. We would like to thank Mei Chen and Steven Kowalski of the Harvard Center for Mass Spectrometry for their help with mass spectrometry analysis and preparation. We also would like to thank the Harvard Center for Macromolecular Interactions for their help with bio-layer interferometry. This work was supported by the National Institutes of Health (R35-GM127136 to V.D. and R35-GM139571 to E.I.S.), DFG (DO 545/28-1 to V. Dötsch), and the National Science Foundation Graduate Research Fellowships Program (DGE1745303 to I.M.S.).
Author information
Authors and Affiliations
Contributions
Conceptualization, I.M.S. and V.D.; methodology, I.M.S., D.R., K.P., A.J.M., A.R.N., C.T., and V.D.; formal analysis, I.M.S., D.R., K.P., A.R.N.; investigation, I.M.S., D.R., K.P., A.J.M., and A.R.N.; resources, A.W.M. and V.D.; writing—original draft, I.M.S., D.R., and V.D.; writing—reviewing and editing, all authors; supervision, V.D., E.I.S., A.W.M., and V.D.; funding acquisition, I.M.S. and V.D.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Fabio Trovato and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s Note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Sabbarini, I.M., Reif, D., Park, K. et al. A ribosome-associating chaperone mediates GTP-driven vectorial folding of nascent eEF1A. Nat Commun 16, 1277 (2025). https://doi.org/10.1038/s41467-025-56489-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-56489-3
