
Abstract
Structural information on channelrhodopsins’ mechanism of light-gated ion conductance is scarce, limiting its engineering as optogenetic tools. Here, we use single-particle cryo-electron microscopy of peptidisc-incorporated protein samples to determine the structures of the slow-cycling mutant C110A of kalium channelrhodopsin 1 from Hyphochytrium catenoides (HcKCR1) in the dark and upon laser flash excitation. Upon photoisomerization of the retinal chromophore, the retinylidene Schiff base NH-bond reorients from the extracellular to the cytoplasmic side. This switch triggers a series of side chain reorientations and merges intramolecular cavities into a transmembrane K+ conduction pathway. Molecular dynamics simulations confirm K+ flux through the illuminated state but not through the resting state. The overall displacement between the closed and the open structure is small, involving mainly side chain rearrangements. Asp105 and Asp116 play a key role in K+ conductance. Structure-guided mutagenesis and patch-clamp analysis reveal the roles of the pathway-forming residues in channel gating and selectivity.
Similar content being viewed by others


Introduction
Channelrhodopsins (ChRs) are light-gated ion channels from protists and their viruses, and are widely used for optogenetic control of neurons and other excitable cells1,2,3. Unlike all other known ion channels, ChRs are oligomeric retinylidene proteins using photon energy to create a pathway for ion conductance in each protomer. A ChR protomer consists of a bundle of seven transmembrane helices (7TM domain) that form a binding pocket for the retinal and a cytoplasmic domain not required for the channel function. The all-trans-retinal chromophore is attached to a conserved lysine residue in the transmembrane helix 7 (TM7) via a protonated retinylidene Schiff base (RSB). Photon absorption induces isomerization of the all-trans chromophore to the 13-cis form, followed by a series of thermal conformational changes, eventually leading to the recovery of the unphotolyzed state (the photochemical reaction cycle). ChRs’ unitary conductances estimated by noise analysis range from tens to hundreds of fS4,5, placing ChRs in the transporters category rather than that of most canonical channels with their three orders of magnitude higher conductance in the range of 5–500 pS6. The mechanistic relationship between the photochemical conversions of the chromophore and conformational changes in the protein moiety of ChR (i.e., channel gating) remains elusive.
Kalium channelrhodopsins (KCRs) from stramenopile microbes7 are a functional class of ChRs related to “bacteriorhodopsin-like cation channelrhodopsins (BCCRs)”8. Unlike canonical ChRs such as Chlamydomonas reinhardtii ChR2 (CrChR2), KCRs, and other BCCRs show protein sequence homology and share some functional aspects with haloarchaeal outward H+ pumps. In particular, they exhibit three highly conserved residues in TM3 (the DTD motif) required for the outwardly directed proton pumping in haloarchaeal H+ pumps9. Upon expression of their genes in mammalian cells and illumination, KCRs generate K+ efflux, leading to membrane hyperpolarization and inhibition of action potentials in neurons and cardiomyocytes7,10,11,12,13, which makes them promising optogenetic tools. At low cytoplasmic K+ concentration, as in the guard cells of higher plants, KCRs generate H+ influx, leading to transient acidification that induces Ca2+ release from the endoplasmic reticulum14.
Using single-particle cryo-electron microscopy (cryo-EM), we and others have obtained high-resolution structures of the dark-adapted (closed) state of Hyphochytrium catenoides KCR1 (HcKCR1)12,15,16. Compared with most other known K+ channels, which have a tetrameric or pseudotetrameric arrangement and a single pore in the center of the tetramer, HcKCR1 forms a homotrimer, in which each protomer mediates cation flux across the cell membrane. Trimeric assemblies, characteristic of haloarchaeal H+ pumps, are also found in nanodisc-, peptidisc- and detergent-reconstituted HcKCRs12,15,16 and the Na+-selective BCCR from the cryptophyte Rhodomonas lens, known as ChRmine17,18. In contrast, canonical ChRs such as CrChR2 form homodimers19,20.
A key question is how photoisomerization of the retinal chromophore triggers the opening of the ion conduction pathway across the membrane. However, structural analysis of photoinduced channel opening is challenging. Two-dimensional CrChR2 crystals illuminated on an electron microscopy grid and rapidly frozen showed light-induced CrChR2 conformational changes, but the 6 Å resolution was insufficient to resolve side chains of individual amino acid residues21. Time-resolved serial femtosecond crystallography captured the initial stages of a dimeric ChR photocycle, but no open state of the channel was produced due to crystal packing interactions22. The wild-type HcKCR1 channel opens on the submillisecond time scale and closes within ~25 ms under single-turnover photoexcitation conditions7, which makes it difficult to trap the open state for cryo-EM.
In this work, we use the replacement of the highly conserved Cys110 in the middle of TM3 with alanine that results in an >1000-fold decrease in the channel closing rate23, facilitating freeze-trapping. We present cryo-EM structures of the dark-adapted and illuminated states of the HcKCR1_C110A mutant.
Results
HcKCR1_C110A embedded in peptidiscs
For cryo-EM analysis, we reconstituted purified HcKCR1_C110A into peptidiscs and took images before and after photoactivation by laser flashes. This preparation exhibited fast flash-induced absorption changes as detected in detergent-purified protein (Supplementary Fig. 1a). The thermal recovery of the dark-adapted state was slower in peptidiscs than in detergent, with time constants similar to those of channel closing in HEK293 cells23, indicating that peptidiscs provide HcKCR1_C110A an environment similar to that in biological membranes. We subjected the sample to a laser-flash illumination and plunge-freezing protocol that maximized the fraction of protomers in the open state (detected by patch-clamp recording) before thermal recovery (Methods, Supplementary Fig. 1b–f, and Supplementary Discussion).
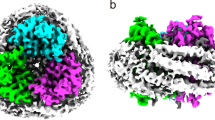
We collected cryo-EM images before and after illumination. Single-particle analysis allowed us to obtain density maps at 3.08 Å resolution for dark-adapted and 3.03 Å for laser-flash-illuminated HcKCR1_C110A, respectively, and to determine high-resolution structures; the resolution of the maps within the 7TM domain reached 2.6 Å (laser-flash-illuminated HcKCR1_C110A) and 2.8 Å (dark-adapted HcKCR1_C110A) (Fig. 1, Table 1, Supplementary Fig. 2). The three-cornered shape of the particles was similar to what we obtained previously with dark-adapted HcKCR1 and HcCCR in peptidiscs15. The same residue pairs as reported for wild-type HcKCR115 stabilized the trimer, including Asp48 (TM1) and Arg128 (TM4), Thr79 (TM2) and Trp161 (TM5), and Asp90 (TM2) and Ser148 (TM4-TM5 loop). This demonstrates that the C110A mutation does not affect the trimeric assembly. The space between the three protomers was filled with six phospholipid molecules carried over from the membranes of the Pichia pastoris expression host, which would prevent ion translocation through it. We modeled these lipids as dioleoylphosphatidylethanolamine (DOPE), three in each bilayer leaflet. Sterol lipids originating from the host membranes were modeled as cholesterol. Cholesteryl hemisuccinate (CHS) was added during purification, but the density of hemisuccinate was weak or missing for clear identification.

a Peptidisc comprising a C3-symmetric HcKCR1_C110A trimer in the dark (blue, magenta, and orange) and upon laser-flash illumination (green, pink, and yellow), lipids and cholesterols (gray), and multiple surrounding 37-residue amphipathic peptides (white). View from the intracellular side. b Structure models of dark-adapted state (left) and illuminated state (right) HcKCR1_C110A viewed from the intracellular side (upper) and the membrane plane (lower) with all-trans-retinal (yellow) and 13-cis retinal (red), respectively. In one protomer of the trimer, the helices are labeled to show the order of transmembrane helices.
The backbone RMSD between the HcKCR1_C110A and the wild-type HcKCR1 dark-adapted state structures was 0.318 Å (Fig. 2a). The chromophore density in the dark-adapted HcKCR1_C110A structure could be modeled with all-trans-retinal and the β-ionone ring in the coplanar (6-s-trans) configuration with respect to the polyene chain, as in the wild-type HcKCR1 structure15. In HcKCR1_C110A, the position of the retinal polyene chain relative to the β-ionone headgroup was slightly shifted towards residue 110 compared with the wild type (Fig. 2b). The replacement of cysteine with alanine allows the RSB to move farther away from its counterion Asp229 (from 3.4 to 4.6 Å), the homolog of Asp212 in bacteriorhodopsin (BR). In contrast to wild-type HcKCR1, which has a direct H-bond between the RSB and Asp229, the H-bond interaction in HcKCR1_C110A involves a bridging water molecule, which explains the observed ~10 nm blue shift of the absorption maximum in the mutant23.

a An overlay of structures of dark-adapted C110A mutant (shown in blue, retinal in yellow) and wild-type (WT, shown in gray). The helices are shown as cartoons, the chromophore and the residue 110 side chains, as sticks. b A zoom-in into the residue 110 region. c Representative photocurrent traces recorded at 0 mV in response to 200-ms light pulses (green bar) normalized at the peak value. d, e Photocurrent rise and decay time constants. The symbols are individual-cell data; the lines are mean ± sem values. *p = 3.7E-4 (n = 10, 26, and 23 cells for the WT, C110A, and C110T, respectively); **p = 4.3E-5 (n = 16, 10, and 17 cells for the WT, C110A, and C110T, respectively) by the two-tailed Mann–Whitney test. f Current-voltage dependencies (mean ± sem, n = 25, 37, and 40 cells for the WT, C110A, and C110T, respectively). Source data are provided as a Source Data file.
Planar automated patch-clamp recording confirmed that the channel in HcKCR1_C110A was closed in the dark, as the membrane resistance of human embryonic kidney (HEK293) cells expressing HcKCR1_C110A and wild-type HcKCR1 was not significantly different (1.92 ± 0.19 and 1.85 ± 0.19 GΩ, respectively; n = 37 and 41 cells, respectively; p = 0.90 by the two-tailed Mann–Whitney test). The photocurrent rise and decay were slower in C110A than in C110T (Fig. 2c–e), and neither mutation compromised K+ selectivity (Fig. 2f). Taken together with the small structural differences from the wild type (Fig. 2a, b), these properties make HcKCR1_C110A a favorable choice for a structural study of the gating mechanisms in ChRs.
Formation of a K+ conduction pathway
The HOLLOW software24 detected a series of internal cavities in the structures of HcKCR1_C110A in the dark-adapted state and upon laser-flash illumination (Fig. 3a). In the structural models, all spherical densities within the cavities were interpreted as water molecules, although some of them could be potassium ions (see below). It is worth to note that stable K+ binding could not be found for dark state HcKCRs12. The dark-adapted HcKCR1_C110A structure contains 20 water molecules per protomer (compared to 15 water molecules and a sodium ion in the wild-type structure15), whereas the laser-flash-illuminated HcKCR1_C110A structure contains 27 water molecules. Illumination connected the separated cavities observed in the dark-adapted state structure into a continuous cation conduction pathway, which can be divided into the intracellular segment, the photoactive site, and the extracellular segment (Fig. 3a). A CAVER25 search with the probe radius 1 Å revealed a continuous intramolecular tunnel in the laser-flash-illuminated but not the dark-adapted HcKCR1_C110A structure (Fig. 3b). Molecular dynamics (MD) simulations revealed that water molecules could fill the continuous tunnel in the laser-flash-illuminated state. In contrast, water penetration into the RSB region is blocked in the dark-adapted state (Fig. 3c).

a Cavities in HcKCR1_C110A protomer modeled with the HOLLOW software. The cavity color shows the electrostatic potential from −5 (red) to +5 (blue). The helices are shown as cartoons; the chromophore, as a stick model. b The CAVER software detects an intramolecular tunnel (yellow) only in the laser-flash-illuminated HcKCR1_C110A structure, not in the dark-adapted state structure. c MD simulations show a water chain connecting the RSB with the intracellular aqueous phase in the structure of the laser-flash-illuminated HcKCR1_C110A trimer.
Photoisomerization unblocks the central gate
Comparison of the dark-adapted and laser-flash-activated HcKCR1_C110A structures revealed a distinct photoinduced change in the retinal chromophore from the all-trans to the 13-cis configuration (Fig. 4). The chromophore density in the illuminated structure was modeled with the RSB in the 13-cis, 15-anti configuration. However, the density allowed for the 13-cis, 15-anti isomer and the 13-cis, 15-syn isomer to potentially fit, suggesting a heterogeneous mixture of states could exist. When symmetry relaxation was applied during map refinement with CryoSPARC software26, individual protomers revealed differences in the RSB densities that could be fit with 13-cis, 15-anti and 13-cis, 15-syn isomers as well as the 11-cis, 15-anti isomer (Supplementary Fig. 3). Retinal extraction and HPLC analysis confirmed a substantial decrease in all-trans-retinal and a corresponding increase in the cis isomers upon laser-flash excitation (Supplementary Fig. 1g, h).

Structures of dark-adapted and laser-flash-illuminated HcKCR1_C110A containing cavities modeled with HOLLOW; the helices are shown as cartoons; the chromophore and key residues, as stick models; water molecules, as small spheres; and the potassium ions transported in molecular dynamics simulations (right panel), as large green spheres.
In the dark, the RSB separates the internal cavities facing inside and outside the cell, thus representing the key structural element constricting the K+ conduction pathway. The pronounced change of the RSB position upon the photoinduced all-trans to 13-cis isomerization altered the conformation of the Lys233 side chain to which the retinal is attached (Supplementary Fig. 3f, g), merging the separated cavities into a continuous K+ conduction pathway (Fig. 4). A slight rearrangement of the Asp105, Tyr106, and Thr109 side chains also assisted this merging (Fig. 4, center). The photoinduced movement of Asp105 pulled the Lys84 side chain out of the conduction path, thereby widening it. The abolishment of photocurrents by the D105N mutation and a slower photocurrent decay rate in the T109A and T109V mutants corroborates the role of these residues in channel gating12,15,16. Kinetic analysis of photocurrents and transient absorption changes in HcKCR1_C110A23 suggested that the RSB was already deprotonated by the time of freezing and imaging the laser-flash-illuminated sample, which is consistent with the RSB NH-bond changing its orientation from facing the extracellular side in the dark-adapted state structure to the cytoplasmic side in the laser-flash-illuminated structure (Fig. 4).
The Leu113 side chain flanks the conduction pathway near Ile73 (Supplementary Fig. 4a), which we earlier identified as required for K+ selectivity7. Replacement of Leu113 with Ala, but not with Ile, caused a 25-nm blue shift of the photocurrent action spectrum (Supplementary Fig. 4b). Both mutations reduced photocurrent desensitization and slowed the photocurrent decay (Supplementary Fig. 4c, d), which explained a higher light sensitivity observed in the L113I mutant27. Neither mutation changed the reversal potential (Vr) (Supplementary Fig. 4e, f), indicating no role of Leu113 in K+ selectivity. The absorption spectrum of L113A showed a similar blue shift compared to the wild type (Supplementary Fig. 4g). Slower recovery of the unphotolyzed state probed by flash photolysis (Supplementary Fig. 4h) correlated with slower channel closing under single-turnover conditions (Supplementary Fig. 4i).
Flipping of the Asp116 side chain
Structural changes caused by light-induced chromophore isomerization propagate towards the intracellular side of the molecule via the hydrophobic side chain of Trp199. Its light-induced movement perturbs the hydrophobic cluster consisting of Phe236, Phe195, and Phe117 (Supplementary Fig. 5a), which may trigger reorientation of the Asp116 carboxylic acid side chain (Fig. 4 top panel). In the dark-adapted state, Asp116 faces the intracellular side to form a salt bridge with Arg244 and a hydrogen bond (H-bond) to Ser70. In contrast, the salt bridge is broken in the laser-flash-illuminated state, and the Asp116 side chain faces away from Arg244 towards the RSB. The Asp116 side chain orientation and the interhelical H-bond to the Ser70 hydroxyl group are reminiscent of Asp96 H-bonded to Thr46 in BR28 and suggest a proton-binding site29. Asp96 corresponds to Asp116 in HcKCR1 and is the internal proton donor to the RSB during BR’s proton pumping cycle. The breakage of the Asp116-Arg244 salt bridge and flipping of the Asp116 side chain in HcKCR1_C110A connect the internal cavities to provide a continuous cation pathway.
In addition to laser-flash-illuminated HcKCR1_C110A, we performed cryo-EM single-particlet analysis of HcKCR1_C110A illuminated with continuous green light before cryo plunging (Supplementary Fig. 6, Table 1). The density map had a global resolution of 3.44 Å and up to 3.2 Å within the 7TM domain allowing us to model the isomerized retinal in the predominant 13-cis, 15-syn configuration. The internal cavities were connected in the RSB region. However, the cavities in the intracellular segment were still separated because the orientation of the Asp116 side chain was similar to that in the dark-adapted state (Supplementary Fig. 5b). Given the weak density of the Asp116 side chain in the laser-flash-illuminated structure, we conclude that the flexible Asp116 side chain regulates cation flow. The model obtained after continuous illumination gives structural insight into a desensitized KCR with no or weak conductance and might comprise a mixture of states as concluded earlier23. The presence of multiple states could also be an explanation for the lower resolution of the map obtained upon continuous illumination of HcKCR1_C110A. Similarly, a complexity of protein conformations and retinal binding was observed in the cryo-EM structures of HcKCR1 and HcKCR1_C110T, respectively, prepared under continuous dim white light16.
Asp116 is critical for K+ selectivity7,10 and shows transient binding of partially dehydrated K+ upon breakage of the Asp116-Arg244 salt bridge in MD simulations using the dark-adapted state wild-type structure12. While the Asp116 side chain flips upon laser-flash illumination of HcKCR1_C110A as seen by cryo-EM, the Arg244 side chain shows only a small movement to connect more tightly to Ser70 (Fig. 4). However, we observed reorientation of the Arg244 side chain towards the intracellular solvent in MD simulations using the laser-flash-illuminated HcKCR1_C110A structure, described in the next section.
Formation of a transient water chain
We performed MD simulations starting from the dark-adapted state and laser-flash-illuminated HcKCR1_C110A trimer structures. The transmembrane region of each protomer was structurally stable during the simulations (Supplementary Figs. 7–9). To evaluate H-bonding at the Asp116/Ser70 site and the RSB (Lys233), we used the Bridge/Bridge2 graph-based algorithm and graphical user interface30,31 to compute H-bond networks of the direct and water-mediated H-bonds between side chains of HcKCR1_C110A; we extracted from these graphs the H-bond clusters of Lys233 and Ser70 (see Methods for details). In the dark-adapted state structure simulations, the RSB connected to the extracellular aqueous phase via an extended water-mediated H-bond network of ~20 side chains, including Tyr81, Lys84, Asp87, Asn99, Trp102, Asp105, Tyr106, Thr109, Gln218, Tyr222, and Asp229 (Supplementary Figs. 10–13). Many of this network’s H-bond connections were frequently sampled in all three protomers (Supplementary Figs. 11–13). At the cytoplasmic side, Asp116 H-bonded mostly directly to Ser70 and Arg244, and the extracellular and cytoplasmic water-mediated H-bond networks of Lys233 and Ser70 remained separated.
The change in the orientation of the RSB NH-bond in the laser-flash-illuminated structure had drastic consequences for the H-bond network of the cytoplasm-oriented 13-cis, 15-anti RSB (Supplementary Figs. 8, 9, 14–21). Within this network, Lys233 bridges in two of the three protomers to the H-bonded Ser70/Asp116 cluster in the intracellular segment via a chain of three H-bonded water molecules (Fig. 3c), similar to a water chain connecting the RSB to Asp96 in BR32,33. In analogy to the role of the H-bonded water chain in BR proton transfer, we hypothesize that the H-bonded water chain that assembles in HcKCR1 could facilitate the H+ conductance of KCRs7,14. Compared with the dark-adapted structure, the connection between Ser70 and Asp116 in the laser-flash-illuminated structure was more frequently mediated by water molecules. Likewise, connections between Arg244 and Ser70 in the laser-flash-illuminated structure are mostly water-mediated. Arg244 can additionally water bridge to Glu248, whereas in the dark-adapted trimer the shortest-distance connection between Arg244 and Glu248 is via Asn67 or Tyr58. These rearrangements of the cytoplasmic H-bond network largely replaced direct H-bonds at the Asp116 cluster with water-mediated bridges, which potentially facilitated the partial dehydration of K+ by its transient binding to Asp116 and Thr120 observed in MD simulations12.
Rearrangement of the aromatic cluster
In the extracellular segment of the laser-flash-illuminated HcKCR1_C110A, the signal propagation from the RSB via Tyr106 results in the perturbation of the cluster of aromatic residues consisting of Trp102, Phe221, Tyr222, and His225 (Fig. 4), which has been suggested to form the K+ selectivity filter7,10,12,15. Trp102 displays an inward movement towards the RSB upon illumination, with His225 moving away from the cavity to compensate (Fig. 4). A cation-π interaction with Trp102 was proposed12 and may allow for the preferential passage of K+ over Na+ ions. While Tyr222 is essential for K+ selectivity7,10, it cannot contribute to cation-π interactions as the aromatic ring’s plane does not face the cavity.
The MD simulations and graph-based analyses confirm that the dynamics of the aromatic cluster are markedly different in the dark-adapted vs. laser-flash-illuminated HcKCR1_C110A trimers. In the dark, Tyr81, Trp102, Tyr106, and Tyr222 are part of the H-bond cluster of the all-trans RSB; the shortest-distance connection between Tyr106 and Tyr222 is via 2–3 H-bonded water molecules, or via water, Tyr81, and Lys84 (Supplementary Figs. 11d, 12d, 13d). By contrast, in the laser-flash-illuminated trimer, the shortest-distance connections between Tyr106 and Tyr222 are via both side chains and water bridges, and the side chains mediating the connection between Tyr106 and Tyr222 can be different than in the dark-adapted state (Supplementary Figs. 11–13 and 15–17).
The K+ flow through the channel
Fig. 5a shows the profile of the intramolecular tunnel that is found in the laser-flash-illuminated HcKCR1_C110A structure, exhibiting four constrictions (C1–C4) with a smaller radius than a bare potassium ion (1.49 Å34). We used a MD-based computational electrophysiology (CompEL) protocol35,36 to determine if the channel is sufficiently open to allow K+ passage under transmembrane voltages (Supplementary Fig. 22).

a A tunnel profile detected by CAVER using the probe radius 1 Å. C1-C4 are the four constrictions. The lines show the positions of the tested residues along the tunnel. b, Photocurrent traces recorded from the wild-type (WT) and Y222T mutant at incremental voltages. c The current-voltage relationships of the WT and Y222T mutant. The symbols are mean ± sem (n = 19 and 17 cells, respectively). d The Vr values are derived by analysis of the current-voltage dependencies (see Methods for solution compositions). e Photocurrent amplitudes normalized to the WT recorded in the same experiment. d, e The symbols are individual-cell data recorded at the time of peak (empty circles) and the end of illumination (filled circles); the lines are mean ± sem values. *p < 0.05; **p < 0.001; ***p < E-5 compared to the WT by the one-way ANOVA with the Tukey means comparison. The numbers of the cells sampled for each variant and the exact p values are provided in the Source Data file.
To accelerate the rate of K+ passage through the channel during the limited simulation time (500 ns), we chose a high transmembrane voltage of 400 to 900 mV (Supplementary Fig. 22b, c). We sampled five independent trajectories for the dark-adapted and laser-flash-illuminated structures. We monitored the root mean square deviations (RMSD) of the protein backbone atoms and observed that the structures remained stable throughout the simulation time (Supplementary Fig. 23). Across the trajectories, we observed altogether 10 K+ ions permeating through the channels in the laser-flash-illuminated structure whereas no cation permeation was observed in the case of the dark-adapted structure. The low simulated conduction rate aligns well with the generally low conductance characteristic of HcKCR1. The 10 permeating events in the illuminated structure were equally distributed among inward and outward permeations under negative and positive transmembrane voltages, respectively (Supplementary Movies 1 and 2). This was facilitated by the pore opening between TM1-3 and TM7 and was largely assisted by the aspartate residues in TM3 and TM7 (Fig. 4, right). During the outward permeation, the cation entered from the intracellular side by binding to Asp116. The cation then migrated toward the extracellular site by interacting with Asp229 followed by binding to Asp105. An inverted sequence of events was observed during the inward permeation where the potassium ion moved towards the intracellular site via Asp105 to Asp229 to Asp116. Strong inhibition of photocurrents in the HcKCR1_D229N mutant corroborates the role of Asp229 in channel gating10,12,15. Apart from the negatively charged aspartate residues, the permeating ion also binds favorably to the polar residues containing hydroxyl groups. Ser70, Thr120, and Tyr222 act as active binding sites for the permeating ion and direct its motion through the channel (Supplementary Movie 3). In addition, we observe rearrangement of the Trp102 residue at the extracellular site, wherein it orients itself towards RSB. It binds the permeating ion via a cation-π interaction and directs it toward the aspartate residues (Asp105 and Asp229) during inward permeation.
As the potassium ions are seldom free and have a coordinated hydration sphere around it, we also monitored the hydration changes. We observed that during permeation in both inward and outward directions, there is at least one coordinated water molecule, even when the potassium ion interacts with the aspartates (Supplementary Fig. 24).
These results qualitatively show that HcKCR1_C110A is conductive for K+ upon photoillumination, with the movement through the channel guided by the aspartate residues. Due to the small number of ions permeating over the limited simulation period, a quantitative analysis of the conductance cannot be performed. A longer simulation time revealing significantly higher permeation events for deriving statistically meaningful single-channel conductance will be the scope of future studies.
Mutagenetic analysis of the K+ pathway
Many residues forming the constrictions of the intramolecular tunnel detected in the laser-flash-illuminated structure (Supplementary Table 1) have been shown earlier to contribute to channel gating and/or selectivity (Asn67, Leu69, Ser70, and Asp116 in C1; Ile73, Cys77, and Thr109 in C2; Tyr81, Lys84, Asp105, Tyr106, and Asp229 in C3; Phe88 and Tyr222 in C4)7,10,12,15,37. Tyr222 is replaced with Thr in Na+-selective HcCCR, and the T222Y mutation converted HcCCR into a K+ channel7. Figure 5b, c shows photocurrent traces and the current-voltage relationships (IV curves) of the opposite mutant, Y222T, compared to the wild-type HcKCR1. The Y222T mutation caused a large curve shift to more positive voltages, which confirmed the importance of Tyr222 for K+ selectivity. The H-bond graph analyses support the key role of Tyr222, as this residue is part of the Lys233 H-bond network in both the structures of dark-adapted and laser-flash-illuminated HcKCR1_C110A (Supplementary Figs. 7–18).
The functional role of several other constriction-forming residues has not yet been tested by mutagenesis. Thr120, Met240, and Arg244 contribute to C1 in the intracellular segment, and Met20, Tyr22, and Leu25 form C4 together with Tyr222 in the extracellular segment. We have also included Val36 and Phe117 in our analysis because these residues are replaced with Leu in WiChR, a KCR variant from Wobblia lunata that shows an increased K+/Na+ permeability ratio compared to HcKCR110. Supplementary Fig. 25 shows the IV curves of the tested mutants, and Fig. 5d, e shows their reversal potentials (Vr) and photocurrent amplitudes at the time of the peak and the end of illumination. M20F, as Y222T, changed only selectivity, causing a parallel shift of the IV curve along the X axis; R244A and R244Q reduced only conductance (the slope of the curve) without the curve shift; most other mutations changed both these parameters. Some mutations, such as R244K, more strongly reduced photocurrents at the voltages above than below the Vr, i.e., caused inward rectification. Of the four tested Arg244 substitutions, Ala and Gln caused the strongest inhibition of photocurrents but did not significantly change the Vr, suggesting that Arg244 is important for channel gating but not selectivity, which is controlled by the Ser70-Asp116 interhelical H-bond. L25A showed a more depolarized Vr compared to the wild type, indicating that Leu25 contributes to the K+ selectivity of the wild type. Surprisingly, all tested substitutions of Met20 showed a more negative Vr than the wild type at the end of illumination and M20A, M20L, M20V, and M20G at the peak time, indicating an increased K+ selectivity. The Vr shift to more depolarized values is likely caused by the accumulation of a less K+-selective photocycle intermediate upon prolonged illumination7. All Met20 mutants showed a reduced Vr shift compared to the wild-type by the two-tailed Mann-Whitney test (Source Data file), suggesting that Met20 contributes to the formation of this intermediate in the wild-type. M20A, M20L, and M20V showed a significantly more negative Vr of the end current (one-way ANOVA followed by the Tukey test for means comparison; Source Data file) than the previously reported mutant H225F (KALI-1)12, included in our study for comparison. The results on Met20 and Leu25 mutants show that the TM1 residues also contribute to selectivity in addition to the TM2 and TM7 residues identified by comparison of HcKCR1 and HcCCR7. Met20 is conserved in Na+-selective HcCCR, which suggests that this residue was evolutionarily selected for a channel property other than selectivity. However, Met20 is located at a 5.4 Å distance from Tyr222, the key determinant of K+ selectivity (Fig. 4), and might influence its H-bonding network and/or hydrophobic interactions.
Discussion
KCRs are unique among K+ channels because of their light-gated conductance. Our structural study of the HcKCR1_C110A mutant revealed that the retinal isomerization changed the orientation of the RSB NH-bond from extracellular to cytoplasmic and directly opened the central gate in the photoactive site of the KCR protomer. The RSB connectivity switch is the key element of active ion transport and sensory signaling in haloarchaeal rhodopsins38,39, and its occurrence in KCRs confirms the similarity between these two protein families. Retinal extraction and HPLC analysis showed a photoinduced increase in 11-cis isomers besides the predominant 13-cis isomers (Supplementary Fig. 1g, h). Although the all-trans to 11-cis photoisomerization is not typical of microbial rhodopsins40, it has been found in middle rhodopsin of Haloquadratum walsbyi41, bestrhodopsin42, and wild-type HcKCR112. Further studies are needed to clarify the functional importance of 11-cis retinal in HcKCR1.
The retinal isomerization signal is propagated through changes in the hydrophobic/aromatic residue clusters and the rearrangement of the two Asp residues (Asp105 and Asp116), which are part of the local H-bond networks. These light-induced changes connect the internal cavities and broaden the putative K+ efflux pathway predicted from the dark-adapted state structures of HcKCRs and HcCCR12,15. Asp105 and Asp116 correspond to Asp85 and Asp96 in BR, which are the key elements in the latter protein’s outward H+-pumping activity. Remarkably, the same residue pair was recruited to regulate photoinduced passive K+ flow in KCRs (Supplementary Fig. 26). A delayed RSB deprotonation in the D105N mutant suggests that Asp105 is an acceptor of the RSB proton in HcKCR112. The Asp116 homolog in a related protein, Guillardia theta cation channelrhodopsin 2 (GtCCR2), undergoes deprotonation directly coupled to channel gating8, which also likely occurs in HcKCR1.
The backbone RMSD between dark-adapted and laser-flash-illuminated HcKCR1_C110A structures is only 0.348 Å, whereas the RMSD between ground state and light-activated bovine rhodopsin structures (PDB IDs 1HZX and 3PQR, respectively) is 1.552 Å. The latter is caused by major conformational changes due to helix movements upon light-activation of rhodopsin43,44. Canonical ChRs such as CrChR2 also undergo large photoinduced changes in the secondary structure involving TM2, TM6, and TM7 detected by Fourier-transform infrared (FTIR) spectroscopy45, 2D crystallography21, and double electron–electron resonance spectroscopy46,47. We did not observe large photoinduced helical movements in HcKCR1_C110A and propose that subtle local changes are sufficient for light-gated K+ conductance in KCRs, as for active H+ translocation by a BR triple mutant48, although the extent of global light-induced conformational changes in haloarchaeal proton pumps is a subject of discussion49. An FTIR observation of only limited protein structural changes upon retinal photoisomerization in HcKCR1 and a homologous protein GtCCR4 supports our hypothesis12,50. In HcKCR1, TM2 interacts with TM4 of the adjacent protomer to form trimers, which makes its photoinduced movement difficult. However, given the presence of a small fraction of apoprotein and a mixture of retinal isomers in our illuminated sample (see Supplementary Discussion), we cannot exclude that some HcKCR1_C110A protomers may show a larger global light-induced conformational change. Further experiments with complementary spectroscopic techniques are needed to test this hypothesis.
The extracellular cavity provides enough space for a hydrated cation. However, constrictions in the pathway would require the cation to be partially dehydrated to pass through the rest of the channel. Since the channel pore features several narrow stretches that fit only the size of a bare K+, it appears that the K+ is partly or fully dehydrated during most of its journey through the channel (Supplementary Fig. 26), potentially interacting with the hydroxyl groups of Ser, Thr, and Tyr side chains, carboxylates of Asp side chains, and the backbone carbonyl groups. K+ ions following the concentration gradient pass the constrictions stepwise by moving from one cavity to the next using local fluctuations of the restricting amino acid side chains to allow cation passage, as we observed in our MD simulations. The constricting residues we identified by structure-guided mutational analysis contribute to the selectivity filter based on the easier dehydration of K+ than Na+, which reduces the cation size, and the cation-π interactions.
The advent of all-optical electrophysiology11 requires spectrally separated optogenetic tools, which are rarely found in nature. Our structural analysis of photoinduced changes in a ChR molecule will facilitate the molecular engineering of color-tuned variants with intact channel function. A better understanding of channel gating and selectivity mechanisms will eventually enable the design of optogenetic tools with a desired combination of biophysical properties.
Our results reveal molecular mechanisms of light-gated K+ and H+ conductance and selectivity of HcKCR1. The structures showed subtle but informative rearrangements of HcKCR1 upon laser-flash-illumination, which are sufficient for ion permeation as confirmed by MD simulations. These insights will facilitate KCR engineering for optogenetic needs. However, further studies using MD simulations of the potassium channel gating, as well as time-resolved vibrational spectroscopy and serial crystallography are needed to further elucidate the chemistry of their unique function.
Methods
Molecular biology
The polynucleotides encoding the amino acid residues 1–265 of HcKCR1 (Genbank accession number MZ826861) were fused with the C-terminal 8His-tag and cloned in the pPICZalpha-A vector (Invitrogen, Cat. # V19520) for expression in Pichia pastoris, or fused with the C-terminal mCherry tag and cloned in the pcDNA3.1(+) vector (Invitrogen, Cat. #V79020) for expression in HEK293 (human embryonic kidney) cells. A QuikChange XL site-directed mutagenesis kit (Agilent Technologies, Cat. #200516) was used to introduce point mutations.
Expression and purification of HcKCR1 mutants from Pichia pastoris
The plasmids encoding the expression constructs were linearized with SacI and used to transform P. pastoris strain SMD1168 (his4, pep4) by electroporation according to the manufacturer’s instructions. Resistant transformants were selected on 0.5 mg ml−1 zeocin (Gibco, Cat. # R25001). The next step involved the induction of transgene expression, which was achieved by the addition of 0.5% methanol in the presence of 5 µM all-trans-retinal (Millipore-Sigma, Cat. # 116-31-4). After 24 h, the pink-colored cells were harvested by centrifugation at 5000 × g for 10 min, and the cell pellets were resuspended in 100 ml ice-cold buffer A (20 mM HEPES, pH 7.4, 150 mM NaCl, 1 mM EDTA, 5% glycerol) and lysed by either French press or bead beater (BioSpec Products, Cat. #607). Membrane fragments were collected by ultracentrifugation and solubilized by incubation with 1% dodecyl maltoside (DDM, Anatrace) for 1.5 h at 4 °C. The supernatant was mixed with nickel-nitrilotriacetic acid agarose beads (Thermofisher, Cat. # R90101) and loaded on a column. The protein was eluted with buffer containing 20 mM HEPES pH 7.5, 300 mM NaCl, 300 mM imidazole, and 0.05% DDM. The protein was further purified by size exclusion chromatography in the elution buffer containing 20 mM HEPES pH 7.5, 300 mM KCl, 1 mM sodium azide, 5% glycerol, and 0.05% DDM/ 0.005% CHS (cholesteryl hemisuccinate, Anatrace), and concentrated for peptidiscs reconstitution.
Absorption spectra of detergent-purified and peptidisc-incorporated protein samples were recorded using a Cary 4000 spectrophotometer (Varian). Hydroxylamine (Millipore-Sigma, Cat. # 5470-11-1) was added to the protein samples under ambient illumination at the final concentration of 100 mM, after which the samples were kept for 20 min in the dark spectrophotometer chamber to avoid the accumulation of intermediates. The difference spectrum exhibited a positive peak at 365 nm, reflecting the retinal oxime formation. Its ε value of 33,900 M−1 cm−1 was used for calculations. The ε values of BR and HcKCR1_C110A at 280 nm were calculated using ExPASy ProtParam tool51.
Photoinduced absorption changes were measured with a laboratory-constructed crossbeam apparatus. Excitation flashes were provided by a Minilite II Nd:YAG laser (532 nm, pulse width 6 ns, energy 5 mJ; Continuum). Measuring light was from a 250-W incandescent tungsten lamp and a McPherson monochromator (model 272, Acton). Absorption changes were detected with a Hamamatsu Photonics photomultiplier tube (model R928) combined with a second monochromator of the same type. Signals were amplified by a low noise current amplifier (model SR445A; Stanford Research Systems) and digitized with a GaGe Octopus digitizer board (model CS8327, DynamicSignals LLC), with a maximal sampling rate of 50 MHz. Logarithmic data filtration was performed using the GageCon program52.
Electron microscopy of HcKCR1_C110A mutant in peptidiscs
Purified HcKCR1_C110A was mixed with a 2:1 excess (w/w) of nanodisc scaffolding peptide NSPr (Nter-DWLKAFYDKVAEKLKEAAPDWFKAFYDKVAEKFKEAF-Cter, purity >80%, A+ peptide Co. Ltd., Shanghai) and diluted to 1/14 of initial concentration before applying to size exclusion chromatography on a Superdex 200 Increase 10/300 GL column in buffer containing 20 mM HEPES pH 7.5 and 150 mM KCl. The peak fraction was diluted to 0.02 mg mL−1 and evaluated by negative staining electron microscopy with uranyl formate15. For cryo-EM, 4 µL of dark-adapted HcKCR1_C110A reconstituted into peptidiscs at 0.4 mg/mL concentration were applied onto homemade holey gold grids. Excess protein was blotted away using a Vitrobot Mark IV set to 277 K, 100% relative humidity, with 2.5 seconds blot time and blot force 1. Grids were plunge-frozen in liquid ethane and stored under liquid nitrogen prior to imaging. To trap the laser-illuminated structure, the grids were illuminated by nanosecond laser pulses at a 10 Hz repetition rate provided by a Minilite II Nd:YAG laser (532 nm, pulse width 6 ns, energy 0.72 mJ per pulse; Continuum) within the ~750 ms time duration between blotting and plunge freezing. For the continuous illumination, a band-pass optical filter (520 ± 20 nm) was attached to a Fiber-Lite Mi-150 illuminator (Dolan-Jenner), and the blotted grids were illuminated continuously (~750 ms, 13.3 mJ/cm2) until plunge freezing.
Cryo-EM image acquisition and data processing
Cryo-EM movies were acquired at 300 kV on a Titan Krios transmission electron microscope equipped with a Falcon 4i detector. Movies were collected in the EER format with a pixel size of 1.03 Å and total exposure of 40 e−/Å2. Data was collected at both 0° and 30° stage tilt. Automated data collection was performed with the EPU (v3.3) software package. A total of 5483 movies were collected for the dark-adapted HcKCR1_C110A samples, and 7683 movies were collected for the laser pulse-illuminated HcKCR1_C110A samples.
Image processing was performed using CryoSPARC v4.4 software package26. All movies were initially preprocessed for patch motion correction and CTF estimation. A subset of particles was selected by blob picking to generate 2D templates, which were then used for template picking across the full datasets.
For the dark-adapted HcKCR1_C110A dataset, 1,320,264 particles were initially selected for 2D classification, and 527,087 particles were selected for 3D ab initio reconstruction. The best class was selected for multiple rounds of heterogenous and non-uniform refinement, followed by reference-based motion correction, and local refinement53. The final map at 3.08 Å resolution was obtained with 86,551 particles and C3 symmetry imposed.
For the laser pulse illuminated HcKCR1_C110A dataset, 5,144,654 particles were selected for 2D classification, of which 1,589,866 were selected for 3D ab initio reconstruction. The best map was selected for the same steps of refinement as the dark-adapted HcKCR1_C110A map. The final map at 3.05 Å resolution was obtained with 230,267 particles and C3 symmetry imposed.
For the continuous light illuminated HcKCR1_C110A dataset, 1,454,190 particles were selected for 2D classification, and 367,702 particles were selected for 3D ab-initio reconstruction, the best map was selected for refinement in the same process as the previous datasets. The final map at 3.44 Å resolution was obtained with 55,716 particles and C3 symmetry imposed.
Model building and refinement
Sharpened density maps prepared in CryoSPARC (v. 4.4) were used to build models in Coot (v. 0.9.8.9)54. Models for dark-adapted and illuminated HcKCR1_C110A generated from wild-type HcKCR1 structure (PDBID:8GI8) were fit to the map in Coot; then the models were further refined in Phenix (v. 1.20.1)55. Structural analysis and figure preparation were performed with UCSF ChimeraX (v. 1.7)56, PyMOL57 (v. 2.5.5), and HOLLOW24 (v. 1.3) software. The structures of the dark-adapted and laser-flash-illuminated HcKCR1 C110A were refined using the hybrid quantum mechanics/molecular mechanics (QM/MM) method58. The QM part included the retinal-lysine (Lys233) protonated Schiff base. The QM/MM boundary was placed between Cδ and Cε of the lysine side chain using the hydrogen link atom scheme. The QM part was described using the CAM-B3LYP-D3(BJ) functional59 in combination with the cc-pVDZ basis set60. The protein within the MM region was treated using the Amber ff14SB force field61 while the water molecules were described using the TIP3P model62. The QM/MM structure optimizations were performed using ORCA 5.0 program63 interfaced with Chemshell software64.
Cavity and tunnel analyses
Intramolecular cavities were identified using HOLLOW (v. 1.3)24 with a grid spacing of 0.3 Å and a surface probe of 0.95 Å in radius. The internal surfaces shown in Figs. 3 and 4 were created by manually selecting the internal spherical probes generated by HOLLOW. Electrostatic potentials were calculated using APBS (v. 3.4.1)65. The intramolecular tunnel in the laser-flash-illuminated structure was detected using CAVER Analyst v. 2.0 Beta25 with a probe radius of 1 Å.
Retinal extraction and high-performance liquid chromatography (HPLC) analysis
The HPLC analysis of retinal isomers was conducted as described elsewhere12. Before extraction, the purified HcKCR1_C110A sample was incubated overnight in the dark at 4 °C. Half of the sample was subjected to laser-flash excitation as described in the “Electron microscopy of HcKCR1_C110A mutant in peptidiscs” section. 120 μL of 90% (v/v) aqueous methanol solution and 10 μL of 2 M hydroxylamine (pH 6.0) (Millipore-Sigma, Cat. # 5470-11-1) were added to 30-μL protein aliquots (concentration ~2 mg mL−1). Retinal oximes were extracted with 500 μL of n-hexane. Extracts (150 μL) were injected into an HPLC system equipped with a silica column (particle size 3 μm, 150 × 6.0 mm; Pack SIL, YMC), a pump (model 1525, Waters), and a UV-Visible detector (model 2489, Waters). As the mobile-phase solvent, n-hexane containing 15% ethyl acetate and 0.15% ethanol was used at a flow rate of 1.5 mL min−1.
Graph-based analyses of the protein-water H-bond networks
The structures of the dark-adapted and laser-flash-illuminated trimers were aligned along the membrane normal using the PPM web server66. All protein residues had standard protonation states, i.e., all Asp/Glu were negatively charged, all Arg/Lys, positively charged, and all His residues, neutral; the RSB was considered protonated in all trimer simulations. Using CHARMM-GUI and CHARMM, the proteins were embedded in a DOPE lipid membrane by preserving the water molecules and the six intra-trimer lipids present in the starting experimental structures. The simulation systems contained ~450 DOPE lipid molecules, ~35.870–37.120 bulk water molecules, and 130 mM neutralizing KCl salt, for a total of ~177.950−181.520 atoms. Atomic-level simulations were performed using CHARMM3667,68,69, the TIP3P water model62, and retinal parameters70,71,72 using the supercomputer JURECA at Forschungszentrum Jülich73. The starting box size was 140.0 Å × 139.2 Å × 113.5 Å for the simulations of the dark-adapted trimer, and 140.2 Å × 141.0 Å × 110.9 Å for the two simulations of the laser-illuminated trimer. All MD simulations were performed using NAMD74. Equilibration protocol: The simulation systems were geometry optimized and heated to 300 K, followed by equilibration in six steps, 1 ns each, in which harmonic constraints placed on selected hetero-atoms were progressively released. Heating and equilibration step #1 used harmonic constraints of 4 kcal mol−1 Å−2 on the backbone, 2 kcal mol−1 Å−2 on the side chains, retinal hetero-atoms, and the oxygen atoms of the crystallographic water molecules, 1 kcal mol−1 Å−2 on ions, bulk water oxygen atoms, and hetero-atoms of the lipid headgroups, and 0.5 kcal mol−1 Å−2 on the hetero-atoms of the lipid acyl chains; equilibration step #2, constraints on the lipid acyl chains were switched off; step #3, harmonic constraints on the backbone were halved, to 2 kcal mol−1 Å−2; step 4, all remaining constraints were halved; step #5, harmonic constraints on the lipid headgroups were removed, and all remaining constraints reduced to 0.5 kcal mol−1 Å−2. This completed the equilibration, all harmonic constraints were removed, and simulations proceeded with production runs. During equilibration and the first 1 ns of the production run, we used an integration step of 1 fs. For the remainder of the production runs we used a reversible multiple timestep algorithm with 1 fs for the bonded forces, 2 fs for the short-range non-bonded, and 4 fs for the long-range electrostatics. Replicates performed: For the dark-adapted trimer, as shown in Supplementary Figs., the H-bond networks of the RSB are very similar in the three protomers; the H-bonds of the Schiff base cluster have high occupancies and similar values in each of the three monomers. Consequently, only one simulation of the dark trimer was performed. For the laser-flash-illuminated trimer, for which the cytoplasmic Schiff base cluster is more dynamic, and there is more variability of the H-bond cluster than in the dark counterpart, we performed one independent simulation. This independent repeat simulation of the illuminated trimer was started from the same starting coordinates as the main simulation, but with a prior geometry optimization of the retinal molecule and Lys233 while keeping the remaining protein fixed; the protocol for geometry minimization of the entire simulation system, equilibration and dynamics, was the same as described above for the main simulation. Length of the simulations: The simulations we prolonged to 484.0 ns for the dark-adapted trimer, 424.0 ns for the main 13-cis-retinal-bound trimer, and 457.3 ns for the repeat 13-cis-retinal-bound trimer. The simulations were then subjected to H-bond network computations using the graph-based algorithm and graphical user interface Bridge/Bridge230,31. An H-bond graph consists of nodes (the H-bonding residues) and edges (here, direct or water-mediated bridges between the side chains). For clarity, H-bonds are computed residue-wise, such that each residue is represented by a single node. A cluster of H-bonds consists of a collection of nodes and edges that are all interconnected to each other. We considered two groups H-bonded when the distance between the donor and acceptor hetero-atoms was within 3.5 Å, and the H-bond angle was within 60°. The H-bond graph computations included direct H-bonds between the protein side chains and water bridges of up to three H-bonding water molecules; for the retinal chromophore, the Schiff base nitrogen atom and C15 were included in the H-bond graph computation. Graph computations used ~20,000 coordinate sets from the last ~200 ns of each simulation. To clarify the H-bond graph representation, we extracted from the graph only H-bonds present during at least 30% of the coordinate sets and only the H-bond clusters of Lys233 and Ser70.
Computational electrophysiology
MD simulations were performed to probe the potassium ion flow through the protein in both dark-adapted and laser-flash-illuminated structures. The 15-anti RSB was modeled in its protonated form in both the isomers: all-trans configuration in the former and 13-cis form in the latter. The simulations were performed through the computational electrophysiology (CompEL) protocol35,36 using one protomer for each structure. In this approach, the separation of two aqueous compartments is obtained by duplicating the membrane patch in the direction of the membrane normal. The transmembrane voltage results from a constant ion concentration difference between the two compartments.
The respective ion channel is embedded in a lipid bilayer of POPC (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine) and solvated in water with 600 mM of K+ and Cl− ions on both sides of the bilayer. The embedding was done with CHARMM-GUI75,76,77. The protonation states of the residues were modeled at pH 7. The single-membrane system was first energy minimized to stabilize the system, thereby avoiding steric clashes. This is followed by equilibration in a stepwise manner through seven NPT cycles where the number of particles (N), pressure (P) and temperature (T) were kept constant. During the seven equilibration steps, positional restraints are applied on the heavy atoms to first equilibrate the solvent and ions around the lipids and protein using the LINCS algorithm78. The positional and torsional restraints are then gradually relaxed to equilibrate the whole system. In the first three equilibration steps the timestep was 1 fs, afterwards it was set to 2 fs. The pressure was maintained at 1 bar using the Berendsen coupling scheme79 while the temperature was kept constant at 303 K by utilizing the velocity-rescaling thermostat80. The equilibrated single-membrane setup is then duplicated and stacked parallel to form the double-membrane configuration (Supplementary Fig. 22). The advantage of using the double-membrane setup in parallel orientation is that it allows simultaneous sampling of the ion movement under the effect of both positive and negative transmembrane voltages.
Periodic boundary conditions were applied, forming two distinct compartments (A and B). To generate transmembrane voltages, an ion imbalance was created between the two compartments. This was achieved by varying the concentration of potassium ions where compartment A had 6 K+ ions less than compartment B while the chloride ion concentration was maintained the same in both compartments. The GROMACS tool gmx potential was used to quantify the transmembrane voltages during the simulation. To keep the number of ions constant in the compartments throughout the simulation time, after ion permeation, ion/water exchanges are carried out. The ion count is checked at every 100 time steps, with the threshold set to 1 and steps set to 10.
The double-membrane setup dimensions for the dark-adapted and laser-flash-illuminated models were 72 × 72 × 192 Å3 and 72 × 72 × 186 Å3, respectively. Each setup consisted of ~90,000 atoms (~16,000 water molecules, 250 lipid molecules, and ~160 potassium and chloride ions). Five replicates of the setup were generated for each model and production runs were simulated independently for 500 ns. For these runs, the temperature was maintained at 303 K using the velocity-rescaling thermostat while the Parrinello-Rahman pressure coupling scheme was employed to maintain the pressure at 1 bar81. All simulations were run using GROMACS (v. 2023.3)82,83. Force field parameters for the retinal were taken from references71,72,84,85,86,87 and used in CHARMM format. TIP3P waters62 and CHARMM36m force field parameters were used68,69. The particle-mesh Ewald method (PME) was used for long-range electrostatics interactions88 with a cutoff of 1.2 nm for Coulomb and Lennard-Jones interactions.
HEK293 cell culture and transfection
No cell lines from the list of known misidentified cell lines maintained by the International Cell Line Authentication Committee were used in this study. HEK293 cells were obtained from the American Type Culture Collection (ATCC; catalog #CRL-1573) and grown as described earlier7. The cells were plated on 2-cm diameter plastic dishes 48–72 hrs before experiments, grown for 24 hrs, and transfected with 10 μl of Lipofectamine LTX with Plus Reagent (ThermoFisher, Cat. #15338100) using 3 μg DNA per dish. All-trans-retinal (Millipore-Sigma, Cat. # 116-31-4) was added immediately after transfection at the final concentration of 5 µM.
Automated patch-clamp recording
Automated whole-cell patch-clamp recording was conducted at room temperature (21 °C) 48–72 h after transfection with a SyncroPatch 384 (Nanion Technologies) based on a Biomek i5 automated liquid handler (Beckman Coulter), using NPC-384T S-type chips (Nanion, Cat. # 222101) with one hole per well. Before measurements, transfected cells were dissociated using TrypLE Express, diluted with CHO-S-SFM-II medium (both from ThermoFisher, Cat.# 12604013 and 31033020, respectively) and resuspended in External Physiological solution (Nation, Cat.# 08 3001), which contained (in mM): NaCl 140, KCl 4, CaCl2 2, MgCl2 1, HEPES 10, glucose 5, pH 7.4. The chips were filled with 80 μL of the same solution without the divalent cations (Nanion ChipFill solution, Cat.# 08 3003). After that, 20 μL of this solution was replaced with 20 μL of the cell suspension, and suction was applied to draw the cells into the holes. After that, 20 μL of the medium was replaced with 40 μL of the Seal Enhancer solution (Nanion External Physiological solution with ten mM CaCl2, Cat.# 08 3012). Finally, 40 μL of the resultant medium was replaced with 40 μL of External Physiological solution to bring the CaCl2 concentration to ~2.6 mM. The KF 110 Internal solution contained (in mM): NaCl 10, KCl 10, KF 110, EGTA 10, HEPES 10, pH 7.2 (Nanion, Cat.# 08 3008). Illumination was provided with LUXEON Z Color Line light-emitting diodes (LEDs) Cat.# LXZ1-PM01 (530 ± 30 nm) arranged in a 6×16 matrix that covered a quarter of the 384-well chip; four quarters were illuminated successively. A thin class IP68 light guide with a counterbore round head (Mentor, Cat. #1292.1601; length 11.5 mm, diameter 2.2 mm) was attached to each LED to bring the light closer to the cells. The forward LED current was 900 mA (which corresponded to the irradiance 2.6 mW mm−2 at the cell level), the illumination duration was 200 ms, and the interval between successive light pulses was 30 s, except in experiments with the C110A and C110T mutants, in which it was 5 min. The LEDs were driven by a derivative of CardioExcyte 96 SOL (Nanion, Cat. #191003) and controlled by Biomek commands. PatchControl384 v. 2.3.0 (Nanion Technologies) software was used for data acquisition at a 5 kHz sampling rate (200 μs per point), except for recording the slow photocurrent decay of the C110A and C110T mutants, which was conducted at 200 Hz (5 ms per point). The photocurrent amplitude at the end of illumination and τ of photocurrent rise and decay were calculated using DataControl384 software v. 2.3.0 (Nanion Technologies). Further analysis was performed using the Origin Pro 2016 software (OriginLab Corporation).
Manual patch-clamp recording
Manual patch-clamp recordings were performed with an Axopatch 200B amplifier (Molecular Devices). The pipette solution contained (in mM) KCl 130, MgCl2 2, HEPES 10 pH 7.4, and the bath solution contained (in mM) NaCl 130, CaCl2 2, MgCl2 2, glucose 10, HEPES 10 pH 7.4. The low-pass filter of the amplifier output was set to 2 kHz. The signals were digitized with a Digidata 1440 A (Molecular Devices) at a 5 kHz sampling rate (200 μs per point) using pClamp 10.7. Patch pipettes with 2–3 MΩ resistances were fabricated from borosilicate glass. Laser excitation was provided by a Minilite Nd:YAG laser (532 nm, pulse width 6 ns, energy 5 mJ; Continuum). The current traces were logarithmically filtered using Logpro software89. Curve fitting was performed using Origin Pro software. Continuous light pulses were provided by a Polychrome V light source (T.I.L.L. Photonics GMBH) in combination with a mechanical shutter (Uniblitz Model LS6, Vincent Associates; half-opening time 0.5 ms). The action spectra of photocurrents were constructed by calculating the initial slope of photocurrent recorded in response to 15-ms light pulses at the intensity <25 µW mm−2, corrected for the quantum density measured at each wavelength, and normalized to the maximal value.
Statistics and reproducibility
The graphs for protein-water H-bond networks were computed separately for each simulation. From each graph, we extracted the H-bond clusters of the RSB and S70 of each protomer with Connected Component Analyses. The H-bond occupancy threshold of 30% was chosen because it gives similar H-bond networks in the three protomers of each simulation. An additional independent simulation performed for the laser-flash-illuminated structure confirmed our findings that a water-mediated network can transiently assemble at the cytoplasmic side of the RSB.
Plasmids encoding different channel variants were randomly assigned to transfect identical cell batches. M20V, M20Y, and K84R were tested upon four independent transfections, and all other mutants upon three independent transfections on different experimental days; the data obtained were pooled together. In automated patch-clamp studies, cells were randomly drawn into the wells. Cells transfected with the wild-type HcKCR1 were included in each experiment as a control. For measurements of the membrane (seal) resistance in the dark in the C110 mutants, wells with photocurrents <20 pA at 0 mV (i.e., those that captured non-expressing cells) were excluded from the analysis. For an unbiased estimation of the photocurrent amplitude, the data from wells that formed seals with a resistance <500 MΩ were excluded. For a more accurate estimation of the Vr values by plotting the IV curves, wells with a seal resistance <500 MΩ and photocurrents of the absolute magnitude <20 pA at −100 mV were excluded. To estimate the decay τ values, wells with a seal resistance <500 MΩ and photocurrents of the absolute magnitude <100 pA at −100 mV were excluded. In manual patch-clamp experiments, the cells were selected for patching by inspecting their tag fluorescence; non-fluorescent cells and cells in which no GΩ seal was established or lost during recording were excluded from the analysis. In automated and manual patch-clamp experiments, the photocurrent traces recorded from different cells transfected with the same construct were considered biological replicates (reported as n values). These values indicate how often the experiments were performed independently.
Statistical analysis of the patch-clamp data was performed using Origin Pro 2016 software. The Kolmogorov–Smirnov test at the 0.05 level was used to determine the data distribution’s normality. For normally distributed data, 1-way ANOVA followed by comparisons of the means by the Tukey test was used. If the normal distribution of the data was not assumed or rejected, the non-parametric two-tailed Mann–Whitney test was used to compare the means. No statistical methods were used to pre-determine sample sizes, but our sample sizes are similar to those reported in the previous publication15.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The cryo-EM maps have been deposited in the Electron Microscopy Data Bank (EMDB) under accession codes EMD-45467 (dark-adapted HcKCR1_C110A), EMD-45468 (laser-flash-illuminated HcKCR1_C110A), and EMD-45469 (continuous green light illuminated HcKCR1_C110A). The coordinates have been deposited in the RCSB Protein Data Bank (PDB) under accession codes 9CDC (dark-adapted HcKCR1_C110A), 9CDD (laser-flash-illuminated HcKCR1_C110A), and 9CDE (continuous green light illuminated HcKCR1). Coordinates of the dark-adapted and laser-flash-illuminated structures along with the input files used for the computational electrophysiology simulations, have been uploaded to the Figshare repository and can be accessed at https://doi.org/10.6084/m9.figshare.27292005. Coordinates at the starting and final points of the trimer simulations, along with corresponding protein structure files, have been uploaded to https://github.com/anb2022/KCRopenTrimerSimulations. Source data are provided with this paper.
References
Deisseroth, K. Optogenetics: 10 years of microbial opsins in neuroscience. Nat. Neurosci. 18, 1213–1225 (2015).
Emiliani, V. et al. Optogenetics for light control of biological systems. Nat. Rev. Method Primers 2, 55 (2022).
Piatkevich, K. D. & Boyden, E. S. Optogenetic control of neural activity: the biophysics of microbial rhodopsins in neuroscience. Q. Rev. Biophys. 57, e1 (2023).
Feldbauer, K. et al. Channelrhodopsin-2 is a leaky proton pump. Proc. Natl. Acad. Sci. USA 106, 12317–12322 (2009).
Govorunova, E. G., Sineshchekov, O. A., Janz, R., Liu, X. & Spudich, J. L. Natural light-gated anion channels: A family of microbial rhodopsins for advanced optogenetics. Science 349, 647–650 (2015).
Dawson D. C. Permeability and conductance of ion channels A primer. In: Molecular Biology of Membrane Transport Disorders (eds. Schultz SG, et al.). Springer USA (1996).
Govorunova, E. G. et al. Kalium channelrhodopsins are natural light-gated potassium channels that mediate optogenetic inhibition. Nat. Neurosci. 25, 967–974 (2022).
Sineshchekov, O. A., Govorunova, E. G., Li, H. & Spudich, J. L. Bacteriorhodopsin-like channelrhodopsins: Alternative mechanism for control of cation conductance. Proc. Natl Acad. Sci. USA 114, E9512–E9519 (2017).
Govorunova, E. G., Sineshchekov, O. A. & Spudich, J. L. Potassium-selective channelrhodopsins. Biophys. Physicobiol. 20, e201011 (2023).
Vierock, J. et al. WiChR, a highly potassium-selective channelrhodopsin for low-light one- and two-photon inhibition of excitable cells. Sci. Adv. 8, eadd7729 (2022).
Fan, L. Z. et al. All-optical physiology resolves a synaptic basis for behavioral timescale plasticity. Cell 186, 543–559.e19 (2023).
Tajima, S. et al. Structural basis for ion selectivity in potassium-selective channelrhodopsins. Cell 186, 4325–4344.e26 (2023).
Ott, S. et al. Kalium channelrhodopsins effectively inhibit neurons. Nat. Commun. 15, 3480 (2024).
Huang, S., Shen, L., Roelfsema, M. R. G., Becker, D. & Hedrich, R. Light-gated channelrhodopsin sparks proton-induced calcium release in guard cells. Science 382, 1314–1318 (2023).
Morizumi, T. et al. Structures of channelrhodopsin paralogs in peptidiscs explain their contrasting K(+) and Na(+) selectivities. Nat. Commun. 14, 4365 (2023).
Shan, Y. et al. Channelrhodopsins with distinct chromophores and binding patterns. Nat. Commun. 15, 7292 (2024).
Tucker, K., Sridharan, S., Adesnik, H. & Brohawn, S. G. Cryo-EM structures of the channelrhodopsin ChRmine in lipid nanodiscs. Nat. Commun. 13, 4842 (2022).
Kishi, K. E. et al. Structural basis for channel conduction in the pump-like channelrhodopsin ChRmine. Cell 185, 672–689.e23 (2022).
Volkov, O. et al. Structural insights into ion conduction by channelrhodopsin 2. Science 358, eaan8862 (2017).
Li, H. et al. Crystal structure of a natural light-gated anion channelrhodopsin. Elife 8, e41741 (2019).
Muller, M., Bamann, C., Bamberg, E. & Kuhlbrandt, W. Light-induced helix movements in channelrhodopsin-2. J. Mol. Biol. 427, 341–349 (2015).
Oda, K. et al. Time-resolved serial femtosecond crystallography reveals early structural changes in channelrhodopsin. Elife 10, e62389 (2021).
Sineshchekov, O. A., Govorunova, E. G., Li, H., Wang, Y. & Spudich, J. L. Channel gating in kalium channelrhodopsin slow mutants. J. Mol. Biol. 436, 168298 (2024).
Ho, B. K. & Gruswitz, F. HOLLOW: generating accurate representations of channel and interior surfaces in molecular structures. BMC Struct. Biol. 8, 49 (2008).
Brezovsky, J., Kozlikova, B. & Damborsky, J. Computational analysis of protein tunnels and channels. Methods Mol. Biol. 1685, 25–42 (2018).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Duan X. et al. Suppression of epileptic seizures by transcranial activation of K+-selective channelrhodopsin. bioRxiv, https://www.biorxiv.org/content/10.1101/2024.01.03.573747v1 (2024).
Luecke, H., Schobert, B., Richter, H. T., Cartailler, J. P. & Lanyi, J. K. Structure of bacteriorhodopsin at 1.55 A resolution. J. Mol. Biol. 291, 899–911 (1999).
Lazaratos, M., Siemers, M., Brown, L. S. & Bondar, A. N. Conserved hydrogen-bond motifs of membrane transporters and receptors. Biochim. Biophys. Acta Biomemb. 1864, 183896 (2022).
Siemers, M. et al. Bridge: a graph-based algorithm to analyze dynamic H-bond networks in membrane proteins. J. Chem. Theory Comput. 15, 6781–6798 (2019).
Siemers, M. & Bondar, A. N. Interactive interface for graph-based analyses of dynamic h-bond networks: application to spike protein S. J. Chem. Inf. Model. 61, 2998–3014 (2021).
Roux, B., Nina, M., Pomes, R. & Smith, J. C. Thermodynamic stability of water molecules in the bacteriorhodopsin proton channel: a molecular dynamics free energy perturbation study. Biophys. J. 71, 670–681 (1996).
Freier, E., Wolf, S. & Gerwert, K. Proton transfer via a transient linear water-molecule chain in a membrane protein. Proc. Natl. Acad. Sci. USA 108, 11435–11439 (2011).
Volkov, A. G., Paula, S. & Deamer, D. W. Two mechanisms of permeation of small neutral molecules and hydrated ions across phospholipid bilayers. Bioelectrochem. Bioenerg. 42, 153–160 (1997).
Kutzner, C., Grubmuller, H., de Groot, B. L. & Zachariae, U. Computational electrophysiology: the molecular dynamics of ion channel permeation and selectivity in atomistic detail. Biophys. J. 101, 809–817 (2011).
Kutzner, C. et al. Insights into the function of ion channels by computational electrophysiology simulations. Biochim. Biophys. Acta 1858, 1741–1752 (2016).
Govorunova, E. G., Sineshchekov, O. A., Brown, L. S. & Spudich, J. L. Biophysical characterization of light-gated ion channels using planar automated patch clamp. Front. Mol. Neurosci. 15, 976910 (2022).
Spudich, J. L., Sineshchekov, O. A. & Govorunova, E. G. Mechanism divergence in microbial rhodopsins. Biochim. Biophys. Acta 1837, 546–552 (2014).
Haupts, U., Tittor, J., Bamberg, E. & Oesterhelt, D. General concept for ion translocation by halobacterial retinal proteins: the isomerization/switch/transfer (IST) model. Biochemistry 36, 2–7 (1997).
Ernst, O. P. et al. Microbial and animal rhodopsins: structures, functions, and molecular mechanisms. Chem. Rev. 114, 126–163 (2014).
Sudo, Y. et al. A microbial rhodopsin with a unique retinal composition shows both sensory rhodopsin II and bacteriorhodopsin-like properties. J. Biol. Chem. 286, 5967–5976 (2011).
Kaziannis, S. et al. Multiple retinal isomerizations during the early phase of the best rhodopsin photoreaction. Proc. Natl Acad. Sci. USA 121, e2318996121 (2024).
Choe, H. W. et al. Crystal structure of metarhodopsin II. Nature 471, 651–655 (2011).
Standfuss, J. et al. The structural basis of agonist-induced activation in constitutively active rhodopsin. Nature 471, 656–660 (2011).
Ritter, E., Stehfest, K., Berndt, A., Hegemann, P. & Bartl, F. J. Monitoring light-induced structural changes of Channelrhodopsin-2 by UV-visible and Fourier transform infrared spectroscopy. J. Biol. Chem. 283, 35033–35041 (2008).
Krause, N., Engelhard, C., Heberle, J., Schlesinger, R. & Bittl, R. Structural differences between the closed and open states of channelrhodopsin-2 as observed by EPR spectroscopy. FEBS Lett. 587, 3309–3313 (2013).
Sattig, T., Rickert, C., Bamberg, E., Steinhoff, H. J. & Bamann, C. Light-induced movement of the transmembrane helix B in channelrhodopsin-2. Angew. Chem. Int. Ed. Engl. 52, 9705–9708 (2013).
Tittor, J. et al. Proton translocation by bacteriorhodopsin in the absence of substantial conformational changes. J. Mol. Biol. 319, 555–565 (2002).
Hirai, T., Subramaniam, S. & Lanyi, J. K. Structural snapshots of conformational changes in a seven-helix membrane protein: lessons from bacteriorhodopsin. Curr. Opin. Struct. Biol. 19, 433–439 (2009).
Yamauchi, Y. et al. Molecular properties of a DTD channelrhodopsin from Guillardia theta. Biophys. Physicobiol. 14, 57–66 (2017).
Gasteiger, E. et al. ExPASy: The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 31, 3784–3788 (2003).
Waschuk, S. A., Bezerra, A. G. Jr, Shi, L. & Brown, L. S. Leptosphaeria rhodopsin: bacteriorhodopsin-like proton pump from a eukaryote. Proc. Natl. Acad. Sci. USA 102, 6879–6883 (2005).
Punjani, A., Zhang, H. & Fleet, D. J. Non-uniform refinement: adaptive regularization improves single-particle cryo-EM reconstruction. Nat. Methods 17, 1214–1221 (2020).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr D. Biol. Crystallogr. 66, 486–501 (2010).
Liebschner, D. et al. Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D. Struct. Biol. 75, 861–877 (2019).
Goddard, T. D. et al. UCSF chimeraX: meeting modern challenges in visualization and analysis. Protein Sci. 27, 14–25 (2018).
Schrodinger L. The PyMOL molecular graphics system, version 2.5.5. (2010).
Field, M. J., Bash, P. A. & Karplus, M. A combined quantum-mechanical and molecular mechanical potential for molecular-dynamics simulations. J. Comput. Chem. 11, 700–733 (1990).
Yanai, T., Tew, D. P. & Handy, N. C. A new hybrid exchange-correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 393, 51–57 (2004).
Dunning, T. H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 90, 1007–1023 (1989).
Maier, J. A. et al. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput 11, 3696–3713 (2015).
Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W. & Klein, M. L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 (1983).
Neese, F. Software update: the ORCA program system-Version 5.0. Wiley Interdiscip. Rev. Comput. Mol. Sci. 12, e1606 (2022).
Metz, S., Kästner, J., Sokol, A. A., Keal, T. W. & Sherwood, P. ChemShell-a modular software package for QM/MM simulations. Wiley Interdiscip. Rev. Comput. Mol. Sci. 4, 101–110 (2014).
Jurrus, E. et al. Improvements to the APBS biomolecular solvation software suite. Protein Sci. 27, 112–128 (2018).
Lomize, M. A., Pogozheva, I. D., Joo, H., Mosberg, H. I. & Lomize, A. L. OPM database and PPM web server: resources for positioning of proteins in membranes. Nucleic Acids Res. 40, D370–D376 (2012).
Huang, J. & MacKerell, A. D. Jr CHARMM36 all-atom additive protein force field: validation based on comparison to NMR data. J. Comput. Chem. 34, 2135–2145 (2013).
MacKerell, A. D. et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 102, 3586–3616 (1998).
Mackerell, A. D. Jr., Feig, M. & Brooks, C. L. 3rd Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 25, 1400–1415 (2004).
Gruia, A. D., Bondar, A. N., Smith, J. C. & Fischer, S. Mechanism of a molecular valve in the halorhodopsin chloride pump. Structure 13, 617–627 (2005).
Nina, M., Roux, B. & Smith, J. C. Functional interactions in bacteriorhodopsin: a theoretical analysis of retinal hydrogen bonding with water. Biophys. J. 68, 25–39 (1995).
Tajkhorshid, E., Baudry, J., Schulten, K. & Suhai, S. Molecular dynamics study of the nature and origin of retinal’s twisted structure in bacteriorhodopsin. Biophys. J. 78, 683–693 (2000).
Thörnig P. JURECA: data centric and booster modules implementing the modular supercomputing architecture at Jülich supercomputing centre. J. Large-Scale Res. Facil. JLSRF 7 (2021).
Phillips, J. C. et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 153, 044130 (2020).
Jo, S., Kim, T., Iyer, V. G. & Im, W. CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput. Chem. 29, 1859–1865 (2008).
Wu, E. L. et al. CHARMM-GUI Membrane Builder toward realistic biological membrane simulations. J. Comput. Chem. 35, 1997–2004 (2014).
Lee, J. et al. CHARMM-GUI input generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM simulations using the CHARMM36 additive force field. J. Chem. Theory Comput 12, 405–413 (2016).
Hess, B., Bekker, H., Berendsen, H. J. C. & Fraaije, J. G. E. M. LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 18, 1463–1472 (1997).
Berendsen, H. J. C., Postma, J. P. M., Vangunsteren, W. F., Dinola, A. & Haak, J. R. Molecular-dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690 (1984).
Bussi, G., Donadio, D. & Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 014101 (2007).
Parrinello, M. & Rahman, A. Polymorphic transitions in single-crystals - a new molecular-dynamics method. J. Appl. Phys. 52, 7182–7190 (1981).
Abraham, M. J. et al. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1, 19–25 (2015).
Abraham M. et al. GROMACS 2023.3 Source code (2023.3) Zenodo https://manual.gromacs.org/2023.3/download.html (2023).
Adam, S. & Bondar, A. N. Mechanism by which water and protein electrostatic interactions control proton transfer at the active site of channelrhodopsin. PLoS One 13, e0201298 (2018).
Tajkhorshid, E., Paizs, B. & Suhai, S. Conformational effects on the proton affinity of the Schiff base in bacteriorhodopsin: a density functional study. J. Phys. Chem. B 101, 8021–8028 (1997).
Tajkhorshid, E. & Suhai, S. Influence of the methyl groups on the structure, charge distribution, and proton affinity of the retinal Schiff base. J. Phys. Chem. B 103, 5581–5590 (1999).
Baudry, J., Crouzy, S., Roux, B. & Smith, J. C. Quantum chemical and free energy simulation analysis of retinal conformational energetics. J. Chem. Inf. Comput. Sci. 37, 1018–1024 (1997).
Essmann, U. et al. A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577–8593 (1995).
Spudich J. L. LogPro. v1 edn. Zenodo https://zenodo.org/records/4264796 (2022).
Acknowledgements
We thank Dr. Tingting W. Mills (UTHealth) for access to and help with the HPLC facility and Dr. Yuki Sudo (Okayama University) for advice on retinal extraction. We thank U. Benjamin Kaupp for critically reading the manuscript. This work was supported by the National Institutes of Health Grants R35GM140838 (J.L.S.), 1S10OD032293-01 (J.L.S.), and RF1NS133657 (J.L.S.), the Robert A. Welch Foundation Endowed Chair AU-0009 (J.L.S.), and in part by the Natural Sciences and Engineering Research Council of Canada Discovery Grant RGPIN-2023-05764 (O.P.E.) and the Canadian Institutes of Health Research (CIHR) Operating Grants PJT-159464 (O.P.E.) and PJT-520404 (O.P.E. and T.M.). I.S. acknowledges funding from the German Research Foundation (CRC 1078—Protonation Dynamics in Protein Function, project C6) and from the Israel Science Foundation (Research Center, grant no. 3131/20). A.-N.B. was supported in part by the National Institutes of Health Grant R01 GM151326. We thank Dwayne Miller and Jessica Besaw for their helpful discussions. We thank Samir Benlekbir and Zhijie Li of the Toronto High-Resolution High Throughput cryo-EM facility, supported by the Canada Foundation for Innovation and Ontario Research Fund, for data acquisition. We gratefully acknowledge computing time on the supercomputer JURECA at Forschungszentrum Jülich73 under grant number DYNAMICNETWORKS.
Author information
Authors and Affiliations
Contributions
T.M. and K.K. reconstituted HcKCR1_C110A into peptidiscs and performed cryo-EM sample preparation, data acquisition and processing, structure modeling, and refinement. H.L. cloned, expressed, and purified HcKCR1_C110A. Y.W. helped with protein production and cloning. E.G.G. performed electrophysiology experiments. P.N. and I.S. did the refinement of the retinal Schiff base in the cryo-EM structures using hybrid quantum mechanics/molecular mechanics (QM/MM). H.L., E.G.G., I.S., and A.N.B. helped with structure interpretation. H.L. performed HPLC experiments, and O.A.S. performed the HPLC data analysis. O.A.S. performed spectroscopic studies, L.S.B. helped with laser experiments, P.N. performed, and I.S. supervised computational electrophysiology, T.D., S.H., and H.S. helped with computational electrophysiology, A.N.B. performed water dynamics MD simulations and did the hydrogen bond analysis, J.L.S. and O.P.E. supervised the project. T.M., K.K., O.A.S., E.G.G., P.N., and A.N.B. prepared the figures, and T.M., E.G.G., and O.P.E. prepared the manuscript with contributions from all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Stephan Hirschi, Tsutomu Kouyama, Thomas Mager, Andrea Pasquadibisceglie, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Morizumi, T., Kim, K., Li, H. et al. Structural insights into light-gating of potassium-selective channelrhodopsin. Nat Commun 16, 1283 (2025). https://doi.org/10.1038/s41467-025-56491-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-56491-9
