
Abstract
Electrocatalytic NO reduction reaction (NORR) offers a promising route for sustainable NH3 synthesis along with removal of NO pollutant. However, it remains a great challenge to accomplish both high NH3 production rate and long duration to satisfy industrial application demands. Here, we report an in situ-formed hierarchical porous Cu nanowire array monolithic electrode ensembled in a pressurized electrolyzer to regulate NORR reaction kinetics and thermodynamics, which delivers an industrial-level NH3 partial current density of 1007 mA cm–2 with Faradaic efficiency of 96.1% and remains stable at 1000 mA cm–2 for 100 hours. Integrating the Cu nanowire array monolithic electrode with pressurized electrolyzer boosts the NH3 production rate to 10.5 mmol h–1 cm–2, which is over tenfold that using commercial Cu foam at 1 atm. The NORR performance can be attributed to the promoted NO mass transfer to the enriched Cu surface, which could increase the NO coverage on Cu and then destabilize adsorbed NO and weaken hydrogen adsorption, thereby facilitating NO hydrogenation to NH3 while suppressing the competing hydrogen evolution.
Similar content being viewed by others


Introduction
Ammonia (NH3) is one of the most important commodity chemicals in modern society, which is widely used in the manufacture of fertilizers and almost all nitrogenous chemicals1,2,3,4,5. To date, global NH3 production is up to 180 million tons per year via the Haber-Bosch process, which is conducted at severe operation conditions i.e., high temperatures (~ 500 °C) and high pressures (> 100 atm), resulting in 1-2% of world’s energy consumption and massive CO2 emission6,7,8. Electrochemical ammonia synthesis, powered by green and economical renewable electricity under mild conditions, has drawn increasing attention and is considered as a hopeful substitutable route to the Haber-Bosch process9,10,11,12,13,14. Electrocatalytic NO reduction reaction (NORR) not only offers a fascinating route for sustainable NH3 synthesis but also provides an effective method for the removal of NO pollutants. Nearly tens of million tons of exhaust NO are emitted globally each year, in which the concentration of NO pollutants generated during the synthesis processes of adipic acid and nitric acid is approximately 10% to 20%15. In addition, NO serves as a key intermediate in the feasible N2-NOx-NH3 reaction process, which can be obtained with high purity and large quantities through practicable nonthermal plasma processes16,17. Much effort has been focused on enhancing the current density and Faradaic efficiency (FE) of NH3 via exploring efficient catalysts to facilitate the activation and hydrogenation of NO18,19,20,21,22,23,24. Encouragingly, copper (Cu) is screened out to be the optimal transition metal catalyst for NORR to NH3 owing to its moderate reactivity20,25,26. Nevertheless, the limited active sites of Cu particles, poor solubility of NO in water (~ 1.92 mM) and the undesirable hydrogen evolution reaction (HER) greatly hinder the NH3 production, resulting in insufficient current density and FE far below the demand from industrial application. Improving NO mass transfer and promoting the NO hydrogenation while synchronously inhibiting the competing HER and N-N coupling are of great significance to facilitate the NO electroreduction to NH3, yet remain a great challenge owing to the aforementioned issues.
Herein, we present that the in situ-formed hierarchical porous Cu nanowire array (Cu NWA) monolithic electrode can significantly boost the NORR to NH3 activity in a pressurized electrolyzer via facilitating both the reaction kinetics and thermodynamics. The Cu NWA monolithic electrode exhibits an industrial-level NH3 partial current density of 1007 mA cm–2 with an FE of 96.1% under 5 atm NO conditions. Remarkably, an NH3 yield rate of 10.5 mmol h–1 cm–2 can be accomplished at 10 atm of NO, which is over tenfold that using commercial Cu foam at 1 atm. Moreover, this system exhibits excellent stability with negligible attenuation in the NH3 production rate and FE in over 100 hours of electrolysis at 1000 mA cm–2. Multiple characterizations and theoretical studies reveal that the outstanding NORR performance benefits from both the in situ-formed hierarchical porous structure of the Cu NWA electrode, which maximizes the exposure of Cu active sites and enhances internal mass transfer and the pressurized electrocatalytic system, which improves NO solvation and external mass transfer, thereby promoting the adsorption of NO on Cu. A high NO coverage on the Cu surface could destabilize the adsorbed NO and weaken the hydrogen adsorption, thereby facilitating the NO hydrogenation to NH3 while inhibiting the competing hydrogen evolution. These aspects realize the superior FE and larger current density for NH3 production from NORR.
Results
Catalyst synthesis and characterizations
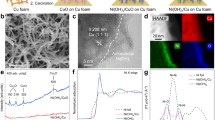
The monolithic Cu NWA electrode was prepared by treating commercial Cu foam with chemical etching in a mixed solution of NaOH and (NH4)2S2O8 followed by electrochemical reduction (Fig. 1a; see “Methods”). X-ray diffraction (XRD) analysis and scanning electron microscope (SEM) were utilized to investigate the structural variation of Cu foam, which presents that the chemical etching creates CuOx nanowire arrays (CuOx NWA) which were then electro-reduced to Cu nanowire arrays with lengths of ~ 10 μm (Fig. 1b–d and Supplementary Fig. 1). In situ Raman measurements were conducted to monitor the electrochemical reduction step. The chemically etched sample shows the typical Raman peaks of CuO and Cu2O at 150, 216, 298, and 652 cm−1, respectively27,28,29, which all disappeared during the electrochemical reduction process (Supplementary Fig. 2), indicating the complete reduction of surface oxide phase to metallic Cu. Further high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) and high-magnification SEM images demonstrate the formation of hierarchical pores on the surface of Cu foam skeleton, built with stacked arrays of mesoporous Cu nanowires with a diameter of 100 ~ 150 nm (Fig. 1e and Supplementary Fig. 3). To be specific, the monolithic hierarchical porous Cu nanowire array (NWA) structure possesses a three-level distribution of pores, i.e., the first-level micrometer-scale pores of skeleton, the second-level macropores of stacked arrays, and the third-level mesopores of nanowires (Fig. 1a–e). Such an architecture not only maximizes the exposure of Cu active sites but also are beneficial for improving the internal mass transfer of NO to access the active sites.

a Schematic illustration of the Cu NWA synthesis process. b XRD patterns of CuOx NWA and Cu NWA with standard PDF cards of Cu, CuO, and Cu2O. c SEM image of Cu NWA. d Enlarged SEM image of the zone marked in (c). e HAADF-STEM image of Cu NWA. f Double layer capacitance for the Cu NWA and Cu foam electrodes in 1 M KOH electrolyte at 25 oC. g EIS measurements of Cu NWA and Cu foam in 5 atm NO-saturated 1 M KOH solution at 25 oC and corresponding equivalent circuit diagram, showing electron transfer resistance of Cu NWA with 10.9 ± 0.1 Ω cm−2 and of Cu foam with 68.4 ± 0.2 Ω cm−2. Source data are provided as a Source Data file.
High-resolution HAADF-STEM image of Cu NWA present the lattice distances of 0.18 and 0.21 nm, which corresponds to the Cu(100) and Cu(111) facets (Supplementary Fig. 4), respectively, which matches well with the XRD result. Further analysis by Krypton adsorption shows that the Cu NWA possesses Brunauer–Emmett–Teller (BET) specific surface area of 17000 cm2 g−1, which is over 18 times that of the commercial Cu foam (Supplementary Fig. 5) due to the in situ-formed hierarchical porous Cu nanowire array. This indicates the substantially enlarged Cu surface of the Cu NWA catalyst, which is further evidenced by its remarkably increased electrochemical active surface area (ECSA) and corresponding double layer capacitance (Cdl) compared with those of commercial Cu foam (Fig. 1f and Supplementary Fig. 6). The enriched Cu surface with abundant active site in Cu NWA monolithic electrode is beneficial for the adsorption and catalytic conversion of NO. In addition, electrochemical impedance spectroscopy (EIS) characterizations show that the interfacial electron transfer resistance of the Cu NWA (10.9 ± 0.1 Ω cm−2) is notably lower than that of the Cu foam (68.4 ± 0.2 Ω cm−2) (Fig. 1g), suggesting a faster electron transfer rate of the Cu NWA, which is favorable for elevating the current density.
Electrocatalytic NORR performance
The electrochemical NORR performance metrics of the Cu NWA monolithic electrode were evaluated in an H-type pressurized electrolyzer, which were conducted at different partial pressures of NO (pNO). Firstly, the pressure-dependent performance of NORR was investigated over the Cu NWA catalyst, which presents that under identical cathodic potential of – 0.4 V versus reversible hydrogen electrode (RHE), the current density enhances significantly from 299 to 1047.4 mA cm–2 with the rise of pNO from 1 to 5 atm and then becomes steady at higher pNO (Fig. 2a, Supplementary Table 1 and Supplementary Fig. 7). The remarkably increased current density at elevated pressures could be due to the enhanced NO solvation and mass transfer in the electrolyte which markedly boost the NORR. The steadied current density at pNO of above 5 atm may be attributed to the limited size of the Cu NWA electrode reaching its full catalytic capacity.

a The effect of NO pressure on NORR product distribution and total current density over Cu NWA at – 0.4 V versus RHE at 25 oC. b, c NH3 FE (b) and NH3 partial current density (c) over Cu NWA and commercial Cu foam at different applied potentials at 25 oC under 1 atm, 5 atm NO, respectively. d The NH3 partial current densities and corresponding NH3 yield rate of Cu NWA and Cu foam under 5 and 10 atm NO at 25 oC, respectively. e Long-term chronopotentiometry stability test of Cu NWA at 500 and 1000 mA cm–2 in 5 atm NO-saturated 1 M KOH electrolyte at 25 oC, and the electrolyte were refreshed every 10 h and 4 h. a–d Error bars were gained after at least three times independent measurements. All of the potentials were reported without iR corrected. Source data are provided as a Source Data file.
Moreover, the pNO has a notable effect on the FE for NH3 production (\({\rm{FE}}_{{\rm{NH}}_3}\)), in which the \({\rm{FE}}_{{\rm{NH}}_3}\) increases gradually from 82.1% to 96.1% with the increase of the NO pressure from 1 to 5 atm (Fig. 2a), which can be attributed to the inhibition of competitive HER caused by an increase in NO coverage. Compared with the case under 1 atm, the Cu NWA electrode always exhibits considerably higher \({\rm{FE}}_{{\rm{NH}}_3}\) and partial current density under 5 atm at all applied potentials (Fig. 2b, c and Supplementary Tables 2, 3). Notably, the NH3 partial current density can reach up to 1415.4 mA cm–2 at – 1.0 V versus RHE when further increasing the pNO to 10 atm (Fig. 2d). The competitive HER is effectively suppressed in a wide potential range at the pressurized electrocatalytic system (Supplementary Fig. 8). Above results show the positive effect of high pNO on the NORR to NH3. Nevertheless, it is worth mentioning that an optimal pNO exists for \({\rm{FE}}_{{\rm{NH}}_3}\) since excessive NO coverage on the Cu surface will promote the N − N coupling for N2 formation, leading to a decrease in the \({\rm{FE}}_{{\rm{NH}}_3}\) (Fig. 2a).
Furthermore, the NH3 partial current densities on the Cu NWA are always notably higher than those on the Cu foam under 1 atm NO at all applied potentials (Fig. 2c). The advantages of Cu NWA on NORR performance are more pronounced at pNO of 5 atm with enhanced external mass transfer (Fig. 2c). These results indicate that the Cu NWA possess more active sites for the NORR to NH3. Only a slight promotion effect on the NH3 partial current density was observed for the Cu foam under 5 atm, which could be due to its insufficient number of active sites (Supplementary Tables 4, 5). Moreover, the ECSA-normalized NH3 partial current densities of Cu NWA increased significantly with the rise of NO pressure from 1 to 10 atm across a wide potential range from 0.2 to – 1.0 V versus RHE, suggesting an increase in reaction pressure can significantly enhance mass transfer and promote NORR to NH3 (Supplementary Fig. 9 and Supplementary Tables 6, 7). By applying 5 atm pNO over the Cu NWA cathode, an industrial-level partial current density of 1007 mA cm–2 for NH3 electrosynthesis can be achieved with \({\rm{FE}}_{{\rm{NH}}_3}\) of 96.1% at – 0.4 V versus RHE, which dominates over the performances of previous reports (Supplementary Table 8). At the NH3 partial current density of 1415.3 mA cm–2, an unprecedented NH3 yield rate of 10.5 mmol cm–2 h–1 is reached at – 1.0 V versus RHE under 10 atm NO, which is over tenfold that using commercial Cu foam at 1 atm and even two orders of magnitude higher than already reported NORR systems (Fig. 2d and Supplementary Table 8). The Cu NWA electrode, which possesses abundant active sites, combined with high-pressure NO that enhances NO mass transfer, effectively regulates the reaction kinetics and thermodynamics of NORR. This synergy promotes the electrocatalytic conversion of NO to NH3, resulting in an exceptionally high NH3 partial current density.
The NORR stability tests were carried out by using chronoamperometry under 5 atm pNO at constant current densities of 1000 and 500 mA cm−2, respectively, during which the electrolyte and reaction gas was replaced every 4 and 10 hours accordingly to remove the products and resupply the reactants. The results show that the \({\rm{FE}}_{{\rm{NH}}_3}\) and applied potential can be well maintained for over 100 h under 1000 mA cm−2 and over 200 h under 500 mA cm−2 (Fig. 2e). The pH of the electrolytes after the long-term stability test remains relatively stable compared to fresh 1 M KOH, indicating that the reaction potential is unlikely affected by changes in hydroxide ion concentration (Supplementary Fig. 10). SEM and XRD analyses show that the morphology and crystal structure of Cu NWA have no visible changes after the durability test (Supplementary Figs. 11 and 12). X-ray photoelectron spectroscopy (XPS) characterization reveals the maintained metallic state of Cu after the 100 h stability test under 1000 mA cm−2 (Fig. 3a, b). Inductively coupled plasma-optical emission spectrometer (ICP-OES) analysis of the electrolyte reveals that the Cu content is below the detection limit, suggesting that there is no significant Cu detachment from the surface of the monolithic Cu NWA electrode (Supplementary Fig. 13). These results indicate the Cu NWA, benefiting from its in situ-formed hierarchical porous monolithic structure, possesses a high structural stability towards efficient NORR to NH3.

a, b, XPS of Cu NWA and Cu NWA after the stability test of 100 h under 1000 mA cm−2 for the Cu 2p orbitals (a) and Cu LMM (b). c The 1H NMR spectra obtained from 15NO, NO, and Ar as feed gas for electroreduction at – 0.4 V versus RHE, respectively. d Pressure-dependent ATR-SEIRAS of NOad on the Cu NWA at − 0.4 V versus RHE. e, f Potential-dependent in situ ATR-SEIRAS of the Cu NWA from 0 V to − 0.6 V versus RHE under NO pressure of 1 atm (e) and 5 atm (f), respectively. c–e All of the potentials were reported without iR corrected. Source data are provided as a Source Data file.
Moreover, NORR conducted with low concentrations of NO demonstrates that our pressurized electrolyzer system can effectively convert NO into NH3 even at concentrations as low as 1% (Supplementary Fig. 14a). In addition, we evaluated the NORR performance in the presence of CO impurity, which are commonly found in emitted NO exhaust gas and can lead to catalyst poisoning in many catalytic processes. No carbon-containing products such as methane, ethylene, acetic acid, and ethanol are detected by gas chromatography and nuclear magnetic resonance spectroscopy, which should be attributed to the lower adsorption-free energy and more positive electroreduction equilibrium potential of NO than that of CO (Supplementary Fig. 14b). Moreover, the faradaic efficiency and yield rate of NH3 during a chronopotentiometry stability test at 1000 mA cm–2 for 4 h at the cathode from NORR remain nearly independent of CO concentration, indicating that CO impurity has a negligible effect on both the electrode and the anion exchange membrane (Supplementary Fig. 14c).
Identifying the active sites and reaction mechanism
In order to investigate active sites and the mechanism of NORR, we performed in situ spectroscopic measurements to study the chemical state of Cu and reaction intermediates for Cu NWA during the NORR. In situ Raman spectroscopy measurements present that no characteristic peaks of surface Cu oxides were detected at the voltage range from 0.2 to – 0.6 V versus RHE (Supplementary Fig. 15). In addition, the quasi in situ X-ray absorption spectra of Cu K-edge show that the valence state and coordination of Cu in the Cu NWA catalyst before and after the NORR at – 0.4 V versus RHE, are all in line with those of the reference Cu foil (Supplementary Fig. 16). These results prove that the metallic Cu is the active site for the NORR. Isotope-labeling experiments using 15NO combined with 1H NMR spectroscopy show that only 15NH4+ was detected in the product, confirming that all the NH3 were generated from the NORR without NH3 formation from possible N-containing contaminant in the system (Fig. 3c). In addition, ultraviolet-visible spectrophotometry shows no signal of hydroxylamine in the electrolyte after the NORR process, which indicates that hydroxylamine is not likely an intermediate in electrochemical reduction of NO to NH3 for Cu NWA catalyst (Supplementary Fig. 17).
In situ attenuated total reflection surface-enhanced infrared absorption spectroscopy (ATR-SEIRAS) was further performed to reveal the effects of pNO on NO coverage and NORR over Cu NWA catalyst (Supplementary Fig. 18). As shown in Fig. 3d, the peak intensity of adsorbed NO (NOad at 1523 cm−1)30,31 gradually increases with the increase of NO pressure from 1 to 6.5 atm, which demonstrates that high pNO can promote the mass transfer and coverage of NO on the surface of Cu NWA. Moreover, the enhanced NO protonation on the electrode surface induces reorientation of H2O molecules surrounding the NOad species, leading to an increased peak intensity of O-H stretching and H-O-H bending at higher pNO32,33. Furthermore, potential-dependent in situ ATR-SEIRAS measurements were conducted under 1 and 5 atm NO pressures, respectively (Fig. 3e, f). On the one hand, a slight increase in the peak intensity of adsorbed NO (NOad) at 1 atm pressure was observed as the applied potential varied from 0 to − 0.6 V versus RHE (Fig. 3e and Supplementary Fig. 19), suggesting a stronger NO bonding on the more negatively charged Cu surface owing to the promoted π-back donation34. On the other hand, with the increase of NO pressure to 5 atm, notably increased peak intensities of NOad and ammonium ion (NH4+)35,36,37 were observed compared with that at 1 atm, indicating an improved NO coverage on the Cu surface and boosted NORR activity for NH3 production at a higher NO pressure (Fig. 3f).
DFT calculation
To get an in-depth understanding of the pressure effect of NO in promoting NORR to NH3 over the Cu NWA, we investigated the reaction process by density functional theory (DFT) calculations. On the basis of a series of structural characterizations, the abundant and the most thermodynamically Cu(111) surface was employed to calculate the reaction mechanism. Though NO can be strongly adsorbed on the Cu(111) surface to reach a high coverage, the practical NO coverage under reaction conditions depends on the equilibrium between the NO mass transfer and NO consumption rates. Since the high pressure-promoted NO mass transfer could facilitate NO adsorption on Cu, the effect of NO coverage on the reaction thermodynamics was studied at increasing NO coverages of 1/9, 2/9, and 4/9 monolayer (ML) (Supplementary Data 1 and Supplementary Fig. 20). The results show that among the NORR reaction steps, hydrogenation of adsorbed NO (NO*) to NOH* is the potential determining step (PDS) for NH3 formation (Fig. 4a). With the increase of NO coverage from 1/9 to 4/9 ML, the reaction free energy of the PDS notably decreases from 0.22 to 0.10 eV, indicating that the increased NO coverage can promote the NO hydrogenation. In addition, the reaction-free energies for the hydrogenation of N* to NH* and NH2* to NH3* are decreased with the increase of NO coverage. Projected density of states (PDOS) of NO* and its O and N atoms show that their p-band centers all shift up to higher energy levels with the increase of NO coverage from 1/9 to 4/9 ML (Fig. 4b and Supplementary Fig. 21), suggesting that the interaction between Cu and NO* is moderately weakened at higher NO coverage, leading to destabilized NO* in favor of subsequent NO* hydrogenation.

a Reaction pathway for the formation of NH3 on Cu(111) at different NO coverage (1/9, 2/9 and 4/9 ML) and key intermediate structures inserted, respectively. b Projected density of states of the NO* species at different NO coverage (1/9, 2/9 and 4/9 ML). c Free energy of the N* protonation and N–N coupling steps at 1/9 and 4/9 ML NO coverage, respectively. d The H* adsorption free energy at 1/9 and 4/9 ML NO coverage. e Atomic configurations, corresponding differential charge density, and the average number of per NO* obtained from Cu of 1/9 and 4/9 ML NO coverage, respectively. Cu, N, O, and H atoms were presented in rose, blue, red, and white spheres, respectively. Source data are provided as a Source Data file.
The competitive reaction for N-N coupling and H2 formation was further studied to understand the high FE for NH3. Compared with N-N coupling between N* and NO*, the hydrogenation of N* is energetically more favorable with a notably lower free energy at both 1/9 and 4/9 ML NO coverage (Fig. 4c). More importantly, the increase of NO coverage can efficiently weaken the hydrogen adsorption on Cu(111), resulting in suppressed HER for H2 formation (Fig. 4d). Analyses of Bader charge and differential charge densities of NO* show that the number of electrons obtained from Cu decreases as the NO coverage increases from 1/9 to 4/9 ML, which presents the similar result from PDOS analysis that the increased NO coverage can effectively weaken the Cu–NO* interaction (Fig. 4e). Consequently, we propose that an increase in NO coverage could enhance the hydrogenation of NO* while simultaneously suppressing the competing HER, thereby boosting the NORR to produce NH3.
Discussion
In this study, we present a promising strategy to achieve sustainable electrosynthesis of NH3 from NO with ampere-level current density by using a monolithic hierarchical porous Cu NWA electrode with abundant active sites combined with pressurizing the NO to enhance both the internal and external mass transfer. This system delivers an ampere-level NH3 partial current density of 1007 mA cm–2 with a high NH3 FE of 96.1%, an NH3 yield rate of 10.5 mmol h–1 cm–2, and excellent stability of over 100 h electrolysis at 1000 mA cm–2, exhibiting great potential for practical application. The Cu NWA monolithic electrode possesses an enriched Cu surface with abundant active sites, porous interconnected nanowire structure, and high conductivity, which are beneficial for improving the reaction rate of NO electroreduction to NH3. The high NO pressure can not only enhance the mass transfer but also increase the NO coverage on the Cu surface, which could weaken the Cu-NO* interaction and the hydrogen adsorption, therefore facilitating NO hydrogenation to NH3 while suppressing the competing hydrogen evolution.
Methods
Materials and chemicals
Cu foam (> 99.99%, 0.15 mm thickness, 57 mg cm−2) was purchased from Kunshan Guangjiayuan new material Co., Ltd. Hydrochloric acid (36 ~ 38%), sulfuric acid (96 ~ 98%), KOH (AR), NaOH (AR), (NH4)2S2O8 (AR), and dimethyl sulfoxide (AR) were supplied by Sinopharm Chemical Reagent Co., Ltd. The anion exchange membrane (Fumasep FAB-PK-130, 130 μm) was purchased from SCI Materials Hub. Ar (99.99%), NO (99.9%) and He (99.99%) were purchased from Dalian Special Gases Co., Ltd. D2O (99.9%) was purchased from Energy Chemical, SAAN Chemical Technology Co., Ltd. Cu foil (> 99.99%, 0.05 mm thickness) was purchased from Canrd limited company. Sodium nitrite-15N (> 98.5%, 99 atom%) was purchased from Aladdin. The Pt mesh (1 × 1 cm2), Ag/AgCl (saturated KCl solution), Hg/HgO (1 M KOH solution), and glassy carbon electrode were purchased from Gaoshi Ruilian (Tianjin) Optoelectronic Technology Co., Ltd.
Preparation of Cu NWA
The Cu NWA was achieved by combining chemical etching and electrochemical reduction methods38. Typically, a 1 cm2 Cu foam was firstly washed using hydrochloric acid for 30 min, and the cleaned Cu foam was etched at 10 oC by a mix solution containing 1.2 M NaOH and 0.06 M (NH4)2S2O8 for 30 min. Then, it was dried at 60 oC and heat treated at 400 oC in Air conditions for 1 h to gain CuOx NWA. Finally, an electrochemical reduction step was conducted, which was carried out using the cyclic voltammetry method (0 to − 1 V versus RHE, 0.1 V s−1) for 30 cycles in 1 M KOH to obtain the Cu NWA. The Cu NWA electrode with an area of 1 cm2 and a thickness of 0.15 mm was used directly as the cathode.
Structural characterization
XRD patterns were collected by an Empyrean-100 diffractometer equipped Cu Kα radiation source. SEM images were obtained on a Quanta 200 F worked at 20 kV. HRSEM images were acquired on a Hitachi S-5500 microscope worked at 30 kV. HAADF-STEM images were gained by a JEOL ARM300F worked at 300 kV. BET specific surface area was analyzed using Krypton as analysis gas by an Autosorb-iQ. EIS measurement was probed under OCV with the frequency of 106 to 0.1 Hz in 5 atm NO-saturated 1 M KOH solution. ICP-OES was carried out on Avio 550 Max.
Electrochemical measurements
We conducted the electrocatalytic measurements in an H-type high-pressure electrolyzer with three electrodes (Supplementary Fig. 22). The volumes of the cathode and anode compartments were 225 cm3 and 30 cm3, respectively. Electrochemical signals were reported by a CHI 660E electrochemical workstation. The Cu NWA monolithic electrode (1 × 1 cm2) was used as the working electrode. The catalyst loading was calculated based on the percentage of the volume of the active Cu nanowire array relative to the total volume of the Cu nanowire array electrode, resulting in an estimated loading of approximately 3.8 mg cm‒2. The Pt mesh and Ag/AgCl were utilized as the counter and reference electrode, respectively. 1 M KOH (pH 14.02 ± 0.01) was utilized as the electrolyte for NORR. The cathode and anode chambers contain 100 mL and 20 mL electrolytes, respectively. An anion exchange membrane was used to balance and transfer the hydroxide ions between the cathode and the anode, ensuring a constant hydroxide concentration at both electrodes. The anion exchange membrane was immersed in a 1 M KOH solution for 12 hours prior to use. A small hole in the Ag/AgCl reference electrode was designed to balance the internal and external pressure, which can significantly minimize the effect of reaction pressure on the calibration of the reference electrode to RHE under pressurized conditions (Supplementary Fig. 23). To prevent the leakage of toxic mercury from the Hg/HgO electrode, we used an Ag/AgCl reference electrode instead of the commonly used Hg/HgO electrode under alkaline conditions. To assess the impact of using Ag/AgCl, we have tested the performance of the Cu NWA electrode in 1 M KOH solution under 1 atm NO with Ag/AgCl and Hg/HgO as reference electrodes, respectively. The Cu NWA electrode exhibits comparable current densities and NH3 production with both types of reference electrodes (Supplementary Fig. 24). This indicates that the chlorine leaching from the Ag/AgCl reference electrode has a negligible effect on performance due to the relatively short reaction time. Different pressures of NO were introduced to the electrolyzer for 15 min to achieve equilibrium. Under each condition, the test was conducted for 10 min using the i-t technic before detecting the products. The electrolyte was refreshed every 10 h at 500 mA cm–2 and every 4 h at 1000 mA cm–2 during the long-term stability test. Fluctuations of potential after refreshing the electrolyte could be due to the low concentration of NO in the refreshed electrolyte in the initial stage. To mitigate any potential impact of chlorine leaching during long-term stability tests, we replaced the saturated KCl solution in the Ag/AgCl reference electrode with a fresh electrolyte for each cycle, aiming to minimize chlorine leaching effects. Linear sweep voltammetry (LSV) was executed at the scan rate of 0.05 V s−1.
The reference electrode calibration was performed in H2-saturated 1 M KOH at 25 oC. The pH of the solution was recorded when the display value remained steady using a Leici PHS-3G pH meter. All of the potentials were reported versus Ag/AgCl and further switched to the RHE without iR compensation on the basis of the following equation, E (versus RHE) = E (versus Ag/AgCl) + 0.197 V + 0.059 × pH (pH 14.02 ± 0.01).
Isotope labeling experiment with 15NO
The 15NO was prepared in lab by using Sodium nitrite-15N following the reaction.
\(3{N}{a}^{15}N{O}_{2}\,+\,{H}_{2}S{O}_{4}\to {2}^{15}{NO}\uparrow+\,N{a}^{15}N{O}_{3}\,+\,{{Na}}_{2}S{O}_{4}\,+\,{H}_{2}O\)
In detail, 1 g Na15NO2 was first dissolved in 5 mL water and then a clean Cu foil was introduced as the catalyst. Next, 5 mL 3 M H2SO4 was added into the above Na15NO2 solution for reaction, and then the produced 15NO was collected in a gas sample bag for isotope labeling NORR experiment.
Products analysis
The as-produced NH3 concentration was quantified by Ion chromatography based on the standard curve (Supplementary Fig. 25). After the electrochemical measurements, the electrolyte was diluted by water in a certain proportion to obtain a suitable NH3 concentration for detection.
The FE of NH3 was calculated on the basis of the following equation:
n: electron transfer numbers
c: the concentration of NH3 (mol L–1)
F: 96485 C mol–1
Velectrolyte: the volume of cathodic electrolyte (L)
Q: the number of charges transferred (C)
The NH3 partial current density was calculated according to the following equation:
j: the total current density
The yield rate of NH3 was calculated on the basis of the following equation:
S: the geometrical area of the electrode in centimeter (cm2)
t: the reaction time in hours (h)
1H NMR spectroscopy was utilized to analyze NH3 in the isotope-labeling experiment. Firstly, the sample after the electrochemical measurements was regulate to pH 2.0 using 1 M HCl. Subsequently, 0.48 mL sample was mixed with 0.02 mL 10 mM DMSO and 0.1 mL D2O, where DMSO serves as the internal standard and D2O was used for shimming. Finally, the prepared sample was measured by a Bruker Avance 400 M NMR spectrometer.
The possible gaseous products of NORR (such as N2 and H2) were analyzed using a Shimadzu GC-2014 gas chromatography (GC). The gaseous products concentration was quantified by a thermal conductivity detector.
FEs of gaseous products were calculated using the following equation:
n: electron transfer numbers
Peak area: the area of products on GC
A: a conversion factor based on calibration of the GC
P: the partial pressure of NO (atm)
Vgas: the volume of gas (L)
F: = 96485 C mol–1
Po: = 1 atm
Vm: = 24 L mol–1
Q: the number of charges transferred (C)
ECSA measurement
The ECSA was obtained by the double layer capacitance (Cdl) method. The CV curves in Cdl determination were recorded within the non-Faradaic potential range at scan rates of 200, 300, 400, 500, 600, and 700 mV s–1 in 1 M KOH.
The Cdl, and ECSA were calculated using the following equation:
jcapacitance: capacitive current at different scan rates
jcapacitance = (janode – jcathode)/2 at the open-circuit voltage
V: scan rates
Cs: specific capacitance for a smooth metallic surface, a general value of Cs = 0.029 mF cm–2
EIS measurement
Test the open-circuit potential and cycle the CV curves within the range of ± 0.1 V of the open-circuit potential for 10 times to clean the electrode surface in 5 atm NO-saturated 1 M KOH solution. EIS was probed under open-circuit potential by the A.C. Impendence technic with the frequency of 106 Hz to 0.1 Hz. Each sample was tested three times to gain charge transfer resistance.
In situ Raman measurement
A Renishaw inVia Raman device was utilized for in situ Raman tests. The spectra were obtained using a 785 nm laser as the excitation source. A typical H-type electrochemical Raman cell was used for the tests (Supplementary Fig. 26). A Corundum (Al2O3) optical window between the sample and the objective was utilized for the measurements. 1 M KOH was used to electrolysis. A Pt wire and an Ag/AgCl was utilized for the counter and reference electrode, respectively. CuOx NWA served as a working electrode to probe the in situ CV electrochemical reduction process and the surface chemical state of the electrode during the subsequent NORR.
Quasi in situ XPS measurement
XPS measurements were carried out on a SPECS near ambient pressure XPS (NAP-XPS) device with Al Kα (1486.6 eV) as the excitation source. Quasi in situ XPS samples were measured after the electrochemical treatment without exposure to air atmosphere.
Quasi in situ XAS measurement
XAS was collected using the Si (311) monochromator in the transmission mode at the BL14W1 beamline in the Shanghai Synchrotron Radiation Facility (SSRF). Quasi in situ XAS samples were measured after the electrochemical treatment without exposure to an air atmosphere.
In situ ATR-SEIRAS measurement
A Bruker Invenio FT-IR spectrometer equipped with a two-compartment high-pressure three-electrode cell was utilized for in situ ATR-SEIRAS measurement. Cu NWA catalyst dropped onto the Au/Si prism acted as the working electrode. IrO2 loaded on carbon felt and Ag/AgCl were utilized as the counter and reference electrode, respectively. All spectra were shown in absorbance, calculated using the following equation, A = − log(R/R0), where R and R0 is the reflectance of the sample spectrum and the background spectrum, respectively.
Computational method
DFT calculations were conducted using the Vienna Ab-initio Simulation Package wherein the projector augmented-wave pseudo-potential39,40,41. All calculations were carried out on the generalized gradient approximation method with the Perdew-Burke-Ernzerhof functional for the exchange-correlation term42,43,44. DFT-D3 method by Grimme and co-workers was applied45,46. Geometry optimization was performed until the atomic forces converged to values smaller than 0.05 eV Å–1. The four-layer Cu(111) surface was built in a 3 × 3 supercell to simulate the catalyst surface. The vacuum thicknesses were set to 15 Å between the layers. The surface Brillouin zone was sampled with a (4 × 4 × 1) Monkhorst-Pack k-points grid mesh for surfaces. The free energies of NO molecules (GNO(g)) were calculated on the basis of the following equation: \({{E}_{{total}}+{ZPE}+H-{TS}+k}_{B}T{\mathrm{ln}}\frac{P}{{P}_{0}}\), where \({E}_{{total}}\) is total energy, \({ZPE}\) is the zero-point energy, \(T\) is 298.15 K, \(H\) and \(S\) are enthalpy and entropy based on ideal gas approximation, \({k}_{B}\) is ideal Boltzmann constant, \(P\) is 5 atm of NO partial pressure, and \({P}_{0}\) is standard atmosphere pressure of 1 atm.
Data availability
All the data that support the findings of this study are available within the paper and its Supplementary Information files. Source data are provided in this paper.
References
Suryanto, B. H. R. et al. Nitrogen reduction to ammonia at high efficiency and rates based on a phosphonium proton shuttle. Science 372, 1187–1191 (2021).
Lim, J., Fernández, C. A., Lee, S. W. & Hatzell, M. C. Ammonia and Nitric acid demands for fertilizer use in 2050. ACS Energy Lett. 6, 3676–3685 (2021).
Ye, D. & Tsang, S. C. E. Prospects and challenges of green ammonia synthesis. Nat. Synth. 2, 612–623 (2023).
Guo, J. & Chen, P. Ammonia history in the making. Nat. Catal. 4, 734–735 (2021).
Ye, T. N. et al. Vacancy-enabled N2 activation for ammonia synthesis on an Ni-loaded catalyst. Nature 583, 391–395 (2020).
Fu, X. et al. Continuous-flow electrosynthesis of ammonia by nitrogen reduction and hydrogen oxidation. Science 379, 707–712 (2023).
Lv, C. et al. Selective electrocatalytic synthesis of urea with nitrate and carbon dioxide. Nat. Sustain. 4, 868–876 (2021).
Zou, H., Rong, W., Wei, S., Ji, Y. & Duan, L. Regulating kinetics and thermodynamics of electrochemical nitrogen reduction with metal single-atom catalysts in a pressurized electrolyser. Proc. Natl. Acad. Sci. USA 117, 29462–29468 (2020).
He, W. et al. Splicing the active phases of copper/cobalt-based catalysts achieves high-rate tandem electroreduction of nitrate to ammonia. Nat. Commun. 13, 1129 (2022).
Hao, R. et al. Pollution to solution: A universal electrocatalyst for reduction of all NOx-based species to NH3. Chem Catal. 2, 622–638 (2022).
Li, K. et al. Enhancement of lithium-mediated ammonia synthesis by addition of oxygen. Science 374, 1593–159 (2021).
Zhang, S. et al. Atomically dispersed bimetallic Fe–Co electrocatalysts for green production of ammonia. Nat. Sustain. 6, 169–179 (2022).
Fu, X. B., Zhang, J. H. & Kang, Y. J. Recent advances and challenges of electrochemical ammonia synthesis. Chem Catal. 2, 2590–2613 (2022).
Rosca, V., Duca, M., de Groot, M. T. & Koper, M. T. M. Nitrogen cycle electrocatalysis. Chem. Rev. 109, 2209–2244, (2009).
Centi, G. & Perathoner, S. in Handbook of Climate Change Mitigation and Adaptation, pp. 1759-1802 (Springer International Publishing, 2022).
Sun, J. et al. A hybrid plasma electrocatalytic process for sustainable ammonia production. Energy Environ. Sci. 14, 865–872 (2021).
John, J., MacFarlane, D. R. & Simonov, A. N. The why and how of NOx electroreduction to ammonia. Nat. Catal. 6, 1125–1130 (2023).
Zhang, L. et al. High-performance electrochemical NO reduction into NH3 by MoS2 nanosheet. Angew. Chem. Int. Ed. 60, 25263–25268 (2021).
Li, Y. et al. Electrocatalytic reduction of low-concentration Nitric Oxide into Ammonia over Ru nanosheets. ACS Energy Lett. 7, 1187–1194 (2022).
Long, J. et al. Direct electrochemical Ammonia synthesis from Nitric Oxide. Angew. Chem. Int. Ed. 59, 9711–9718 (2020).
Wang, D. et al. Hexagonal cobalt nanosheets for high-performance electrocatalytic NO reduction to NH3. J. Am. Chem. Soc. 145, 6899–6904 (2023).
Zhang, H. et al. Isolated electron-rich Ruthenium atoms in intermetallic compounds for boosting electrochemical Nitric Oxide reduction to ammonia. Angew. Chem. Int. Ed. 62, e202213351 (2022).
Chen, K., Zhang, G., Li, X., Zhao, X. & Chu, K. Electrochemical NO reduction to NH3 on Cu single atom catalyst. Nano Res. 16, 5857–5863 (2023).
Bai, C. et al. Hollow Cu2O@CoMn2O4 nanoreactors for electrochemical NO reduction to NH3: Elucidating the void‐confinement effects on intermediates. Adv. Funct. Mater. 32, 2205569 (2022).
Wan, H., Bagger, A. & Rossmeisl, J. Electrochemical Nitric Oxide reduction on metal surfaces. Angew. Chem. Int. Ed. 60, 21966–21972 (2021).
Li, H., Long, J., Jing, H. & Xiao, J. Steering from electrochemical denitrification to ammonia synthesis. Nat. Commun. 14, 112 (2023).
Deng, Y., Handoko, A. D., Du, Y., Xi, S. & Yeo, B. S. In situ Raman spectroscopy of copper and copper oxide surfaces during electrochemical oxygen evolution reaction: Identification of CuIII oxides as catalytically active species. ACS Catal. 6, 2473–2481 (2016).
Dorakhan, R. et al. A silver–copper oxide catalyst for acetate electrosynthesis from carbon monoxide. Nat. Synth. 2, 448–457 (2023).
Wang, Y., Zhou, W., Jia, R., Yu, Y. & Zhang, B. Unveiling the activity origin of a copper-based electrocatalyst for selective Nitrate reduction to Ammonia. Angew. Chem. Int. Ed. 59, 5350–5354 (2020).
Katayama, Y., Okanishi, T., Muroyama, H., Matsui, T. & Eguchi, K. Enhanced supply of hydroxyl species in CeO2-modified Platinum catalyst studied by in situ ATR-FTIR spectroscopy. ACS Catal. 6, 2026–2034 (2016).
Katsounaros, I., Figueiredo, M. C., Chen, X., Calle-Vallejo, F. & Koper, M. T. M. Structure- andcoverage-sensitive mechanism of NO reduction on Platinum electrodes. ACS Catal. 7, 4660–4667 (2017).
Ataka, K.-i, Yotsuyanagi, T. & Osawa, M. Potential-dependent reorientation of water molecules at an electrode/electrolyte interface studied by surface-enhanced infrared absorption spectroscopy. J. Phys. Chem. 100, 10664–10672 (1996).
Yao, Y., Zhu, S., Wang, H., Li, H. & Shao, M. A spectroscopic study on the Nitrogen electrochemical reduction reaction on gold and Platinum surfaces. J. Am. Chem. Soc. 140, 1496–1501 (2018).
Hou, J., Chang, X., Li, J., Xu, B. & Lu, Q. Correlating CO coverage and CO electroreduction on Cu via high-pressure in situ spectroscopic and reactivity investigations. J. Am. Chem. Soc. 144, 22202–22211 (2022).
Zhang, G. et al. Tandem electrocatalytic Nitrate reduction to Ammonia on MBenes. Angew. Chem. Int. Ed. 62, e202300054 (2023).
Chen, T. et al. Actinide-uranium single-atom catalysis for electrochemical nitrogen fixation. Sci. Bull. 67, 2001–2012 (2022).
Wang, Y. et al. Highly selective NH3 synthesis from N2 on electron-rich Bi0 in a pressurized electrolyzer. Proc. Natl. Acad. Sci. USA 120, e2305604120 (2023).
Yang, W. et al. Boosting C–C coupling to multicarbon products via high-pressure CO electroreduction. J. Energy Chem. 85, 102–107 (2023).
Kresse, G. & Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B Condens Matter 47, 558–561 (1993).
Kresse, G. & Furthmuller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Kresse, G. & Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Blochl, P. E. Projector augmented-wave method. Phys. Rev. B Condens Matter 50, 17953–17979 (1994).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Grimme, S., Ehrlich, S. & Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 32, 1456–1465 (2011).
Acknowledgements
Financial support was provided by the National Key R&D Program of China (No. 2022YFA1504500 to D.D.), the National Natural Science Foundation of China (Nos. 21988101, 22225204 to D.D., 22272174 to X.Cui and 22272170 to L.Y.), the CAS Project for Young Scientists in Basic Research (No. YSBR-028 to X.Cui), and the Fundamental Research Funds for the Central Universities (No. 20720220008 to D.D.). We thank the BL14W1 beamline of the Shanghai Synchrotron Radiation Facility.
Author information
Authors and Affiliations
Contributions
D.D. conceived and designed the project. W.Y. performed the experiments and the manuscript preparation. H.L. conducted the DFT calculations. X.Cui supervised the experiments and manuscript revision. L.Y. supervised the DFT calculations. X.Chang, Y.Z., Y.Cai, Y.L., Y.Cui, and B.X. assisted in the materials characterization and experiments measurement. W.Y., H.L., L.Y., X.Cui, and D.D. wrote the paper. All the authors discussed and revised the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Yang, W., Liu, H., Chang, X. et al. Electrosynthesis of NH3 from NO with ampere-level current density in a pressurized electrolyzer. Nat Commun 16, 1257 (2025). https://doi.org/10.1038/s41467-025-56548-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-56548-9
