
Abstract
Metal halide perovskites are attractive for optoelectronic applications, but the existing solution-based synthetic methods rely on hazardous solvents and lack reproducibility. To overcome these challenges, solvent-free mechanochemical synthesis of perovskites can be conducted, although traditional opaque ball milling equipment hinder real-time monitoring of the reaction progress. Herein, mechanochemistry was performed with time-lapsed in situ (TLIS) measurements to elucidate the optical properties during the synthesis. The TLIS spectrometer enabled real-time observation of the impact of varying the compositions on the absorption properties of formamidinium/methylammonium lead iodide (FAxMA1-xPbI3, x = 0 to 1) and facilitated optimization of the synthesis duration. Moreover, we can accelerate the evaluation and directly observe the effects of different synthetic strategies for enhancing the photoluminescence quantum yield and stability of the perovskites, providing near-infrared (NIR) emitting tin-based perovskites FASnIzBr3-z (where z = 0 to 3) with core-shell structures. Intriguingly, halide ion migration was deduced in lead-free double perovskites Cs2NayAg1-yBiCl6 (y = 0 to 1.0) at room temperature during aging after mechanochemical activation. Our work provides insights into the mechanochemistry of compounds with low thermodynamic barriers.
Similar content being viewed by others


Introduction
Since Miyasaka’s seminal report on metal halide perovskites as light harvesters in dye-sensitized solar cells1, perovskites have re-emerged with numerous optoelectronic and energy applications2,3. Consequently, there is strong interest in affordable, green, and scalable synthetic methods for perovskites2,4, including the use of essentially solvent-free mechanochemistry5,6. Owing to the compositional versatility of perovskites, an infinite number of materials can, in principle, be readily synthesized with little to no waste, simply by stoichiometric mechanochemical mixing of the constituent salts7. The mechanochemical synthesis of metal halide perovskites represents a revitalized approach in materials science (e.g., for hard materials such as metal chalcogenides8, carbides9, or borides10) that has led to improved environmental sustainability11, scalability12, and superior material properties13, and has driven research and industrial applications in optoelectronic materials14. However, research on the direct kinetic or mechanistic insights during mechanochemical synthesis remains sparse, which can limit its future deployment.
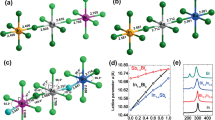
Although mechanochemistry has been practiced in metallurgy and mineral processing for centuries15, the modern form of mechanochemistry, as exemplified by automated ball milling for solvent-free solid-state syntheses, has been revitalized and named by the International Union for Pure and Applied Chemistry (IUPAC) in 2019 as one of the ten chemical innovations that will change the world16. Nonetheless, despite growing interest in mechanochemistry, time-lapsed in situ (TLIS) methods to probe mechanochemical processes and provide real-time kinetic insights remain in their infancy, owing to the traditional use of sealed, opaque steel reactor jars that are not amenable to monitoring (Fig. 1a).

a Difficulties associated with measurements during in situ mechanochemistry. b Previously reported characterization tools for in situ observations during mechanochemistry. c TLIS monitoring of functional optical properties during mechanochemistry via UV–visible diffuse reflectance and photoluminescence spectroscopy.
In 2013, Friščić et al. pioneered the application of synchrotron powder X-ray diffraction (pXRD) for the in situ kinetic study on the mechanochemistry of metal organic frameworks aided by organic liquids or ionic compounds using transparent poly(methyl)methacrylate (PMMA) jars17,18. The latest research has sought to enhance the quality of the X-ray diffraction results through the use of thinner or smaller PMMA jars, with a time resolution of 5 s, and improved data processing19. However, several major challenges remain for these existing in situ techniques (Fig. 1b), such as the need for synchrotron radiation to obtain suitable diffraction data, which may achieve successful results for crystalline but not amorphous samples20. Subsequently, in situ Raman spectroscopy20, thermal measurements21,22, and combinations of these methods23,24,25,26 were developed to monitor representative classes of the mechanochemistry of small molecules and materials systems27,28,29,30,31,32. Curić and Juribašić et al. observed that the fluorescence signals at certain wavelengths were strongly correlated with the structure of intermediates or products formed during their in situ Raman spectroscopic study of a C-H bond activation mechanism under ball milling conditions33. Friščić et al. later integrated Raman spectroscopy with fluorescence emission spectroscopy, highlighting the ability to monitor the transformations between crystalline and amorphous phases during ball milling34. Nonetheless, the customized PMMA jars are easily damaged during ball milling35, cannot be reused many times, and have interfering Raman signals that limit their general usage29, whereas the newer sapphire jars are more robust but are significantly costlier36. Most critically, previous in situ techniques mainly provide characterization and kinetic information rather than a time-resolved description of the functional attributes of the material formed.
Here, we present TLIS mechanochemical measurements for UV–visible reflectance and photoluminescence (PL) measurements with millisecond time-resolution (Fig. 1c), correlated with post-synthetic characterization techniques that provide quantitative short range information and dynamics of the chemical environment37,38,39,40,41,42,43,44,45. The experiments are conducted in glass or quartz jars that can be repeatedly used and are more affordable than sapphire. Furthermore, the reflectance and PL data facilitates not only TLIS kinetic information about the sample composition, but we are also able to ascertain their functional optical properties concurrently. We illustrate the capabilities of our TLIS spectrometer in three representative technological applications for three classes of perovskites. (i) We demonstrate how this technique was used to obtain in situ data to optimize the synthetic duration and light harvesting by formamidinium (FA)/methylammonium (MA) lead iodide (FAxMA1−xPbI3, x = 0 to 1), where we observed the formation and synchronous phase transition of the metastable α-FAPbI3. (ii) Moreover, we showcase how we can adopt in situ measurements during mechanochemistry to rapidly identify strategies to maximize the photoluminescence quantum yield (PLQY) of near-infrared (NIR) emitting tin-based perovskites FASnIzBr3−z (where z = 0 to 3)46,47,48, which can accelerate their translation for optoelectronic applications. (iii) Most intriguingly, during the course of screening for broadband luminescent perovskites with high PLQYs, we observed a mechanochemically activated, post-synthetic ion migration after the cessation of ball milling in double perovskites such as Cs2Na0.9Ag0.1BiCl6, which is distinct from traditional slow chemical transformations that is typically accompanied by changes in the crystal structure49,50, giving us insights into solid-state ion migration phenomena in the absence of light and electric fields.
Results and discussion
Time-lapsed in situ optimization and kinetic analysis during the mechanochemical synthesis of light harvesting lead halide perovskites
FAPbI3 is one of the most extensively studied organic–inorganic hybrid perovskite systems because of its market potential in photovoltaics. However, the photovoltaically active black α-FAPbI3 can spontaneously transition to an inactive yellow δ-FAPbI3 at room temperature51, which imposes limitations on applying FAPbI3 in photovoltaics. Strategies to enhance the structural stability of α-FAPbI3 include introducing MA cations at the A-site, which strengthens Coulombic attractions and enhances the interactions between FA and I, eventually leading to improved stability of FAxMA1−xPbI3 compared to α-FAPbI352,53,54. Nonetheless, traditional synthetic methods fall short in enabling TLIS observation of the functional properties within materials, which can provide valuable insights into the structural dynamics under different synthetic strategies. Accordingly, we customized a spectrometer for TLIS UV–visible reflectance and PL spectroscopy during mechanochemistry. The spectrometer setup consists of an illumination arm that provides the light source to a sample loaded in a quartz jar, and a spectrometer arm that receives the return signal reflected or emitted from the sample (Fig. 1c). The light focused on and either reflected or emitted from the sample are collimated, and then collected and directed to the spectrometer. The optical signals during ball milling can be collected separately in both diffuse reflectance and PL measurement modes, which allows us to obtain the sample’s absorption and PL spectra, respectively. This spectrometer offers TLIS optical measurements with millisecond time resolution, allowing us to probe the dynamic processes that occur during mechanochemistry.
We mechanosynthesized FAxMA1−xPbI3 (x = 0, 0.2, 0.4, 0.6, 0.8, 1.0) and observed each sample for 90-120 min using our TLIS spectrometer. The precursors showed an absorption edge of ~540 nm at the beginning, which red-shifted over tens of minutes and is consistent with the formation of the perovskite phases (Fig. 2a–c). For FAPbI3, the yellow PbI2 turned orange and eventually brown (Supplementary Movie 1), with the absorption edge red-shifting to reach a maximum of 825 nm after 48 min, consistent with the formation of the black cubic phase α-FAPbI3 (Fig. 2a). However, at around 53 min, the absorption intensity gradually decreased and the sample’s band edge blue-shifted, indicating a phase transition from the black α-FAPbI3 to the yellow trigonal phase δ-FAPbI3. To further investigate the origins of these spectral changes, we performed pXRD characterization on a sample that had undergone ball milling for 48 min only and another that had been ball milled for 90 min and then stored for 20 h under N2 (Supplementary Fig. 1). Clearly, the sample contained a mixture of α-FAPbI3, δ-FAPbI3, and PbI2 after 48 min of ball milling, but only the more thermodynamically stable δ-FAPbI3 remained after 20 h.

TLIS spectra for a x = 1, b x = 0.8, and c x = 0, and the respective pXRD results for d x = 1, e x = 0.8, and f x = 0.
To probe the effects of introducing MA cations at the A-site, we collected TLIS spectra during the mechanochemistry of FAxMA1−xPbI3 with decreasing values of x from 0.8 to 0 (Fig. 2b, c and Supplementary Fig. 2). Unlike FAPbI3 that showed continuously evolving absorption spectra during the 90-min ball milling, the TLIS spectrum of each remaining sample reached a steady-state after different durations, remaining constant thereafter, suggesting that these latter samples did not undergo additional reactions during the mechanochemistry. For example, the absorption intensity and band edge of FA0.8MA0.2PbI3 reached their maxima at approximately 59 min and remained stable (Fig. 2b), with the corresponding times at 73 min for FA0.6MA0.4PbI3 (Supplementary Fig. 2a), 60 min for FA0.4MA0.6PbI3 (Supplementary Fig. 2b), 78 min for FA0.2MA0.8PbI3 (Supplementary Fig. 2c), and 80 min for MAPbI3 (Fig. 2c).
Upon introducing MA into FAPbI3, the diffraction peaks of α-FA0.8MA0.2PbI3 at 13.9° and 28.1° were clearly observed (Fig. 2e), alongside a reduction in most δ-FAPbI3 diffraction peaks. Notably, the PbI2 peak at 12.6° observed in FAPbI3 (Fig. 2d) was absent, indicating that MA facilitated the cubic phase crystal formation. A comparative pXRD analysis of the samples (x = 1, 0.8, 0.6, 0.4, 0.2 and 0) confirmed the cubic and tetragonal phases present and revealed that increasing amounts of MA led to shifts of the (001) facet’s diffraction peaks to higher angles due to the smaller ionic radius of MA (Supplementary Fig. 3)55. With the TLIS data, we are able to conduct a kinetic analysis (Supplementary Figs. 4 and 5), similar to a previous study on the in situ mechanochemistry of metal-organic frameworks. Overall, our observations concur with prior reports that MA can substitute for FA within the A-sites in FAxMA1−xPbI3 synthesized by two-step sequential solution deposition and lead to lattice contraction without lattice distortion; moreover, approximately 20% MA stabilizes α-FAPbI3 and suppresses the transition to the undesired δ-FAPbI353,56.
Mechanochemically activated, post-synthetic solid-state ion migration in lead-free double perovskites
Another functionality of the TLIS spectrometer is the ability to measure PL intensities during mechanochemistry. Thus, we conducted TLIS PL measurements during the mechanochemistry of the broadband emissive, lead-free, solid solution Cs2NayAg1−yBiCl6 (y = 0 to 1.0) double perovskites, with supporting post-synthesis pXRD (Supplementary Fig. 6), 133Cs and 23Na magic angle spinning (MAS) nuclear magnetic resonance (NMR) data, and PLQY being acquired. The pXRD data is indicative of the longer-range periodicity but insensitive to short range structural alteration and disorder, while the corresponding MAS NMR data will exhibit sensitivity to short range structural behavior and ion dynamics. As shown in Supplementary Fig. 6a, the pXRD patterns of the as-synthesized Cs2NayAg1−yBiCl6 remained similar upon alloying, matching the Fm\(\bar{3}\)m space group of Cs2NaBiCl6, Cs2AgInaBi1–aCl6 (a = 0–1), and Cs2NabAg1−bInCl6 (b = 0.2–1)44,57. Owing to the shorter bond length of Ag–Cl (2.708 Å) compared to Na–Cl (2.736 Å), increasing B-site substitution by Ag is expected to induce a lattice contraction identified by a shift of the (220) facet peaks toward higher 2θ (Supplementary Fig. 6b)44,58.
The corresponding MAS NMR data for this system indicate that the B-site substitutions are accompanied by largely monotonically changing 133Cs (Fig. 3a) and 23Na (Fig. 3b) isotropic chemical shifts. The 133Cs MAS NMR data (Supplementary Fig. 7) shows that each composition exhibits a single narrow 133Cs resonance assigned to A-site positions that becomes more deshielded (from δ ~ 67 to 71 ppm) upon increasing Ag substitution (see Fig. 3a). In contrast, the 23Na speciation exhibits greater complexity (Supplementary Fig. 7) with broad (2.5 ppm, full width at half maximum FWHM ~ 700 Hz) and narrow (7.3 ppm, FWHM ~ 140 Hz) resonances readily observed. Although the 23Na chemical shifts and linewidths of the 7.3 ppm resonance are largely invariant to Ag substitution (Fig. 3b), quantitative estimates of their intensities relative to the broad Cs2NayAg1−yBiCl6 peak at 2.5 ppm demonstrate that this species grows with Ag substitution up to ~0.6 equivalents (y = 0.4, Fig. 3c), after which it noticeably disappears. However, taking into account the different amounts of Ag doping on the Na site across the series, Fig. 3d shows that the intensity of this resonance relative to the whole composition of the sample remains largely unchanged until the y = 0.2 composition, where a marked drop off is evident. We assign this species to [NaCl6]5− domains that display short-range order, but of insufficient size and long-range order to be detected by pXRD, which may have arisen from incomplete reaction during ball milling that created inhomogeneous nanoparticles with pseudo core–shell structures. As shown in Supplementary Fig. 8, we performed transmission electron microscopy (TEM) and high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) on Cs2Na0.9Ag0.1BiCl6 and Cs2Na0.1Ag0.9BiCl6 as representative examples. The results clearly revealed the distribution of Na and Ag elements within single particles. When y = 0.9 (Supplementary Fig. 8c), the Na signal near the center was stronger compared to Ag, indicating that [NaCl6]5− formed the core in the pseudo core–shell structure. Similarly, when y = 0.1 (Supplementary Fig. 8d), the results were the opposite: [AgCl6]5− formed the core of the structure.

a 133Cs and b 23Na isotropic NMR shifts of Cs2NayAg1−yBiCl6 (y = 0–1.0, Supplementary Fig. 7). c Measured relative intensities of the two 23Na NMR lineshapes for the spectra presented in Supplementary Fig. 7. d Calculated intensities of the [NaCl6]5− NMR resonance relative to the whole sample composition, taking into account the Ag substitution at the Cs2NayAg1−yBiCl6 B-site. e Plotted 23Na center-of-gravity shifts (δcg) the spectra (Supplementary Fig. 9) measured at various magnetic fields. f Both 133Cs T1 components (T1α and T1β) extracted from biexponential fitting of the 133Cs T1 data (Supplementary Fig. 10), measured at magnetic fields of 11.7 T (gray) and 14.1 T (red) for the corresponding systems. g 23Na T1 values acquired at a magnetic field of 9.4 T and extracted by simulation of the build-ups (Supplementary Fig. 11). Data points that branch represent 23Na T1 values that became two-component. h Experimental PLQYs of Cs2NayAg1−yBiCl6 measured after storage under N2 for 14 days. The error bars for isotropic chemical shifts and integrated intensities in parts a–g were determined based on typical uncertainties associated with NMR lineshapes of similar linewidths and signal-to-noise ratios. The error estimates for T1 values incorporate the propagated errors from the integrated intensities of slices within the T1 dataset, as well as the fitting uncertainties from the exponential build-up analysis of the T1 data points.
The characteristic narrow 23Na linewidth indicates that they are unaffected by a loss of translational symmetry and ionic mobility, which would be more prevalent near nanoparticle surface and subsurface layers39,40, and is supported by the XPS data (vide infra). In contrast, the broader 2.5 ppm 23Na resonance is consistent with greater disorder and chemical shift dispersion, presumably associated with Cl volatility, defect formation, and migration. This resonance represents Na speciation within the double perovskite since a shift to higher shielding (from 3.3 to 1.7 ppm) is observed upon increasing Ag substitution (Fig. 3b). Indeed, the analysis of the 2.5 and 7.3 ppm shifts with varying external magnetic field strengths (B0) shows that the center-of-gravity chemical shifts (δcg) of these Gaussian resonances are invariant to B0 (Fig. 3e and Supplementary Fig. 9), suggesting that there is no second-order quadrupolar contribution to their linewidths. This is consistent with the cubic point symmetry in both cases, thus confirming that chemical shift dispersion and disorder dominate the resonance linewidths of both species.
The 133Cs T1 values from biexponential fitting of the saturation-recovery data (Fig. 3f) reveal that each system is described by a shorter T1 relaxation time component (50 – 100 s) that is predominantly invariant with Ag/Na composition, and a longer T1 component (120 – 700 s) that is dependent on the extent of Ag substitution (Supplementary Fig. 10). The 23Na T1 data (Fig. 3g and Supplementary Fig. 11) exhibits a similar trend in a less pronounced fashion and without the multicomponent behavior. Notably, the compositional dependence of the PLQY data (Fig. 3h) correlates with the slow component of the 133Cs T1 (Fig. 3f), indicating that the PLQY is associated with surface passivation, as explained in detail in the Supplementary Information.
In addition to the above study that focused on aged (2–3 weeks) samples stored under an inert atmosphere prior to post-synthesis characterization, we performed in-depth measurements on Cs2Na0.9Ag0.1BiCl6, which exhibited the optimum PLQY characteristics. We prepared Cs2Na0.9Ag0.1BiCl6 by 6 h of ball milling and observed negligible PL intensity by TLIS (sample #1, Fig. 4a). After it was ball milled for a further 3 h and stored under N2 for 18 h (27 h total), negligible PL intensity was detected still (Fig. 4a). Intriguingly, after storage for a further 45 h under N2 (72 h total) without ball milling, we observed an intense signal at 680 nm by TLIS PL measurements, marking a dramatic 106-fold increment in the PL intensity after this post-mechanochemical aging process (sample #2, Fig. 4a). The PLQY values of samples #1 and #2 under 355 nm excitation light were 0.74% and 17.3%, respectively.

a TLIS PL spectra, b pXRD data, HRTEM images of c sample #1 and d sample #2, and high-resolution XPS spectra of e Bi 4f, f Ag 3d, g Cl 2p, and h Na 1s binding energy regions. Sample #1 is Cs2Na0.9Ag0.1BiCl6 after 6 h of ball milling, while sample #2 is the same Cs2Na0.9Ag0.1BiCl6 after a total of 9 h of ball milling and 63 h of storage under N2.
Ex situ pXRD analyses of samples #1 and #2 show that the periodicity of these systems over 20–40 unit cell length scales are ostensibly indistinguishable and remain unaltered from the original Fm\(\bar{3}\)m space group (Fig. 4b). These observations are supported by the high-resolution transmission electron microscopy (HRTEM) data (Fig. 4c, d) that exhibit observable lattice fringes for the (220) facet of the cubic phase in both samples. The Rietveld refinement results (Supplementary Fig. 12) suggested the formation of Cs2Na0.9Ag0.1BiCl6 that crystallized in a cubic Fm\(\bar{3}\)m space group. The Rietveld refined position coordinates and lattice parameters are shown in Supplementary Table 1. The fitting results indicated that the crystallite sizes increased from 112 nm (#1) to 402 nm (#2). This phenomenon was also verified by the HRTEM, which showed that sample #1 exhibited particle agglomeration, whereas sample #2 showed larger particles (Supplementary Fig. 13a, b), confirming growth of the nanocrystalline domains post-mechanosynthesis. After statistically evaluating the sizes of 100 particles from each sample, we determined that sample #2 has an average particle size of 185 nm, which is substantially larger than the average particle size of 17.1 nm found in sample #1 (Supplementary Fig. 13c, d).
Furthermore, we determined the valence states and elemental distribution of samples #1 and #2 using X-ray photoelectron spectroscopy (XPS). Supplementary Fig. 14a shows the XPS survey spectra of both samples, confirming the presence of Cs, Na, Ag, Bi, and Cl signals from the Cs2Na0.9Ag0.1BiCl6 sample. The Cs 3d5/2 and 3d3/2 binding energies are located at 728.2 and 742.1 eV, respectively, for sample #1, whereas they decrease to 726.9 and 740.9 eV, respectively, for sample #2 (Supplementary Fig. 14b). Conversely, the binding energies of the Bi (Fig. 4e) and Ag (Fig. 4f) signals increased slightly, suggesting that the metal-Cl bonds in the [BiCl6]3– and [AgCl6]5– double perovskite sublattices strengthened after storage. Likewise, the Cl 2p3/2 and 2p1/2 binding energies at 201.0 and 202.5 eV in sample #1, respectively, are typically associated with Cl ions near surfaces, whereas the corresponding binding energies at 201.5 eV and 203.0 eV in sample #2 (Fig. 4g) can be attributed to bulk Cl ions59, which is consistent with greater ordering of the Cl chemical environment. The most notable changes in the XPS data relate to an increase of 1.1 eV in the Na 1 s binding energy from 1074.3 eV in sample #1 to 1075.4 eV in sample #2 (Fig. 4h), which is strongly indicative of a more electron-deficient environment surrounding each Na position after 72 h of ball milling and aging. This can only be achieved if Cl vacancies around each Na position have become quenched and populated through ion migration, thus reducing the bulk disorder.
We corroborated this hypothesis by 23Na and 133Cs MAS NMR spectroscopy (Fig. 5a). The 23Na spectrum highlights a prominent resonance at 7.3 ppm in sample #1, which is assigned to [NaCl6]5– nanoclusters in the bulk (see above) and is present through incomplete formation of the double perovskite framework. The small sizes and dispersed locations of these nanoclusters render them undetectable by pXRD and XPS. However, additional ball milling and aging up to a total duration of 72 h eliminated this species and induced a more ordered framework in conjunction with larger particle domains and optoelectronic performance, consistent with the TLIS and HRTEM data. This contrasts with the corresponding 133Cs MAS NMR data, which suggests that both the surface and bulk Cs speciation (i.e., the [AgCl6]5– sublattice) remains essentially unperturbed throughout the additional mechanochemistry and aging periods. In particular, the two-component 133Cs T1 data (insets, Fig. 5a) that characterize both the particle core and surface environments appear largely preserved, with the longer T1 components evidencing the retention of more covalent Ag passivation of the surface regions.

(a) 133Cs (left) and 23Na (right) single pulse MAS NMR data of Cs2Na0.9Ag0.1BiCl6 measured directly after ball milling; 133Cs T1 build-up curves fitted biexponentially are shown in the inset. The associated NMR parameters are presented in Supplementary Table 2. (b) Structure of Cs2Na0.9Ag0.1BiCl6 with the Na and Ag ion migration pathways indicated by paths 1 and 2 in red and gray arrows, respectively. The BI vacancy site is outlined by the octahedron with dashed orange lines. (c) Structure of Cs2Na0.9Ag0.1BiCl6 with the Cl ion migration pathways from the Na-Cl and Ag-Cl bonds to the X-site vacancy in the Na-Cl octahedron indicated by paths 3 and 4 in red and gray arrows, respectively. The Cl vacancy site is outlined by orange circles. (d) Energy profiles of the Na (red) and Ag (black) ion migration path 1 and 2, respectively. (e) Energy profiles of the Cl migration paths 3 and 4.
First-principles density functional theory (DFT) calculations were undertaken on the Cs2Na0.9Ag0.1BiCl6 system to understand the origins of the solid-state ion migration after mechanochemical activation. We modeled the BI and X site vacancies of Cs2Na0.9Ag0.1BiCl6 by removing one Na atom from a BI site or one Cl atom from a X site, respectively, using a 2 × 1 × 1 supercell. The climbing-image nudged elastic band (NEB) method was used to estimate the activation barriers for the migration of the Na or Ag cations at the nearest BI sites to the created BI site vacancy through paths 1 and 2, respectively (Fig. 5b), and for the migration of Cl ions from either a neighboring Na-Cl or Ag-Cl unit to the created X site vacancy through paths 3 and 4, respectively (Fig. 5c)60,61,62. The calculated energy barriers for the BI cation migrations are significantly higher at 29.7 kcal mol−1 for Na ions through path 1 and 24.7 kcal mol−1 for Ag ions through path 2, respectively (Fig. 5d). The energy barriers for the Cl ion migration to fill the X site vacancy are substantially lower compared to BI ion migration, suggesting that Cl ions are likely the predominant migrating species. Notably, the Cl ion migration pathways differ in their energy barriers when originating from the Na-Cl or the Ag-Cl octahedra. The energy barrier of path 4 (11.1 kcal mol−1) is 3.3 kcal mol−1 lower than that of path 3 (14.4 kcal mol−1, Fig. 5e). This suggests that the filling of the X-site vacancies in the Na-Cl octahedron is more favorable when the migrating Cl ion is in closer proximity to Ag instead of Na. This is consistent with our findings from the XPS studies that the Na-Cl interactions in the [NaCl6]5− octahedra of the perovskite structure strengthened after storage and ion migration. Thus, we propose that the increased nanocrystal domain sizes after mechanochemical activation arose predominantly from vacancy-assisted Cl ion migration at the X sites.
To further probe the influence of the Ag dopants on the Cl ion migration, similar DFT calculations were performed on Cs2NaBiCl6. The calculated energy barrier for the equivalent vacancy-assisted Cl ion migration in Cs2NaBiCl6 is 11.0 kcal mol−1 (Supplementary Fig. 15a, b), which is very similar to that of path 4 for Cs2Na0.9Ag0.1BiCl6. This suggests that the 10% Ag ion doping is not the main origin of the Cl ion migration. To experimentally verify that the mechanochemically activated ion migration is not limited to Cs2Na0.9AgBi0.1Cl6, we conducted TLIS spectroscopy on Cs2Na0.95Ag0.05BiCl6 and observed a dramatic 75 times increase in PL intensity at ~680 nm, albeit requiring a longer total duration of 195 h likely because of its intrinsically lower maximum PLQY (Supplementary Fig. 16).
In situ optimization of NIR-emissive tin perovskites with core-shell structures
NIR-emissive materials have been attractive for biomedical optoelectronic applications in the therapeutic window from 650-1350 nm, which is the wavelength range of highest penetration depth in tissues. Consequently, we used our TLIS PL spectrometer to discover and maximize the PLQY of lead-free formamidinium tin iodide (FASnI3) derivatives, which is known to have a direct bandgap of 1.41 eV63. Moreover, we can tune the bandgap and reduce the likelihood of tin vacancy defects in FASnI3 by introducing Br64. Accordingly, we mechanosynthesized FASnIzBr3-z (where z = 3.0, 2.7, 2.6, 2.5, 2.4, 2.0, 0) with concurrent TLIS PL spectroscopy.
For FASnI3, we observed a weak PL signal at 870 nm that reached a maximum after 12 min, but the PL intensities gradually decayed slightly after prolonged ball milling (Fig. 6a). With increasing Br content, the PL peak position of FASnIzBr3-z gradually blue-shifted. (Supplementary Fig. 18). During our screening of the Br loadings for the highest PL intensities, we observed the highest intensity for FASnI2.5Br0.5 that was five times as high as that of FASnI3, which was reached after 7 min of milling, with the PL peak at 827 nm (Fig. 6b). Although the Br substantially increased the NIR PL intensities, FASnI2.5Br0.5 also appeared to be unstable and the PL signals decayed owing to the instability of SnII65. Further increase in the Br contents led to weaker and no NIR PL for FASnI2Br and FASnBr3, respectively (Supplementary Fig. 18d, e). The pXRD data across the entire compositional range retained the cubic Pm\(\bar{3}\)m phase at both extremes, with a shift to higher angles from z = 3.0 to 0, in agreement with the decrease in lattice spacings because of the smaller Br ionic radius relative to I (Fig. 6c).

TLIS PL spectra of FASnIzBr3−z for a z = 3 and b z = 2. c pXRD patterns of FASnIzBr3-z (where z = 3.0, 2.5, 2.0, 0). Electronic band structure and partial density of states of FASnIzBr3−z for d z = 3.0 and e z = 2.5.
To further investigate the origin of the enhanced PL intensity of FASnI2.5Br0.5 compared to FASnI3, we employed DFT calculations to study the influence of the halide (Br/I) compositions on the electronic structures of the FASnIzBr3−z (details in the Supplementary Information). The DFT-computed band structure and partial densities of state of FASnIzBr3−z for z = 3.0 (FASnI3) and 2.5 (FASnI2.5Br0.5) (Fig. 6d, e) and those of z = 2.0 and 0 (Supplementary Fig. 19) suggest that the additional electronic states (brown, Br) near the valence band edge increases the transition probability from the valence band maximum to the conduction band minimum and thus enhance the PL intensity, which could account for the enhanced PL of FASnI2.5Br0.5 compared to FASnI3.
To improve the PLQYs and overcome the decay of the NIR-emissive FASnI2.5Br0.5, we decided to explore the in situ formation of core–shell structures, which has been established to reduce non-radiative recombination, fine-tune electronic properties, and enhance PL efficiency and stability66,67. Accordingly, we mechanosynthesized FASnI2.5Br0.5 by 15 min of ball milling with TLIS PL monitoring, transferred the jar into a N2 glove box, added different equivalents (relative to FASnI2.5Br0.5) of 1:1 FACl and SnCl2, and then continued the ball milling with TLIS monitoring. With 80 mol% of FACl/SnCl2 added, the NIR PL intensities increased 1.77 times over 90 min of ball milling (Fig. 7a), exceeding other proportions of added FACl/SnCl2 (40 mol%: 1.05 times; 60 mol%: 1.19 times; 100%: 1.32 times, Supplementary Fig. 20). The pXRD data (Fig. 7b) confirmed the formation of FASnCl3 as an independent phase mixed with FASnI2.5Br0.5. From the scanning transmission electron microscopy (STEM) coupled with energy dispersive X-ray spectroscopy (EDS) measurements (Supplementary Fig. 21), Sn is uniformly distributed across the entire micron-sized particle, suggesting that the PL enhancement did not arise because of a passivating shell containing only FACl. Interestingly, the core region predominantly contained I, while Cl was sparsely distributed across the particle. An overlay of the I and Cl EDS data confirmed that Cl was more prominent at the boundaries of the particle (Fig. 7c), which validated our approach in enhancing and stabilizing the NIR PL properties by mechanochemistry of FASnI2.5Br0.5@FASnCl3 core-shell systems. The XPS data for FASnI2.5Br0.5@FASnCl3 verified the presence of Sn2+ and Cl on the surface (Fig. 7d, e).

a TLIS PL spectra, b pXRD data, c STEM images and the STEM energy dispersive EDS maps of FASnI2.5Br0.5@FASnCl3 after 1:1 FACl and SnCl2 was added under N2 to the mechanosynthesized FASnI2.5Br0.5. The dark-field image of a particle and the I (red), Cl (blue), and co-localized I and Cl EDS maps are shown, while the Sn data can be found in Supplementary Fig. 21. High-resolution XPS spectra of d the Sn 3d binding energies of FASnI3, FASnI2.5Br0.5, and FASnI2.5Br0.5@FASnCl3, and e the Cl 2p binding energies of FASnI2.5Br0.5@FASnCl3.
Through TLIS measurements in conjunction with mechanochemistry, we were able to observe the solid-state synthesis progress and evolution of the functional optical properties of metal halide perovskites. The TLIS spectral data from increasing amounts of MA+ while mechanosynthesizing α-FAPbI3 indicated the synchronous formation and decay of the metastable α-FAPbI3 phase and the stabilizing effects of the MA+ cations. We employed TLIS in the PL mode to acquire insights about the mechanochemical activation of Cs2Na0.9Ag0.1BiCl6 and subsequent post-synthetic halide ion migration that led to growth of the crystalline domains. We also exploited the TLIS PL mode to mechanosynthesize a NIR-emissive FASnI2.5Br0.5, which was structurally stabilized by introducing chloride ions to form a core–shell structure. Thus, our study underscores the potential of TLIS optical spectroscopy combined with mechanochemistry to accelerate the discovery and optimization of functional materials for optoelectronic applications. Furthermore, the TLIS optical spectrometer developed here can be readily extended to solution-based synthesis protocols.
Methods
Chemicals
The following chemicals were purchased and used as received. Formamidine hydrochloride (FACl): Sigma-Aldrich, 97%; lead(II) iodide (PbI2): Sigma-Aldrich, 99%; bismuth(III) chloride (BiCl3): Tokyo Chemical Industry, > 97%; cesium chloride (CsCl): Sigma-Aldrich, Reagent Plus, 99.9%; cesium bromide (CsBr): Aldrich, 99.9% (trace metal basis); silver chloride (AgCl): Sigma-Aldrich, 99%.
General procedure for the synthesis of alkylammonium halides
Formamidinium bromide (FABr), and formamidinium iodide (FAI): In a typical reaction, 28 mL (0.25 mol) of 48% HX (X = Br, I) solution was gradually added dropwise into 60 mL (0.17 mol) of methylamine or formamidine. The solution was stirred for 16 h, after which it was concentrated using a rotary evaporator at 65 °C. Subsequently, the residue was washed at least three times with 100 mL of diethyl ether. Finally, a colorless powder was collected after drying under high vacuum for 24 h and the product was characterized by nuclear magnetic resonance (NMR) spectroscopy.
FABr: 1H NMR (400 MHz, DMSO-d6) δ = 7.88 (s, 1 H), 8.08 (br s, 3 H) ppm. (29.64 g, 95%).
FAI: 1H NMR (400 MHz, DMSO-d6) δ = 1.00 (s, 2 H), 3.20 (s, 2 H) ppm. (40.78 g, 95%).
TLIS experiments
The precursors of each metal halide perovskite were weighed and loaded into a 12 mL quartz jar (Chuangkai Quartz Products Co., Ltd.) at room temperature in a glovebox filled with N2. Then, a milling ball (customized and purchased from Shanghai Ningci Industrial Co., Ltd.) consisting of 8 mm diameter stainless steel coated with a 3 mm thick layer of polytetrafluoroethylene (PTFE) shell (total ~15 mm diameter) was placed in the jar. The jar was sealed and taken out from the glovebox, after which it was placed within a Retsch MM400 mixer mill. Subsequently, the TLIS spectrometer, developed and commercialized in collaboration with LightHaus Photonics Pte Ltd., was mounted on the mixer mill as part of the cover. Following optical calibration, the TLIS experiment was initiated with the quartz jars oscillated at a frequency of 20 Hz for different time durations. The TLIS spectrometer demonstrated stable operation for at least 24 h. After the TLIS experiments, the final products were collected in a fumehood and transferred back to the glovebox immediately. The detailed amounts and durations of each reaction are shown in Supplementary Table 5. In this study, we used a total of 16 quartz jars, each of which could be used at least 50 times within 12 months without accidental damage.
Diffuse reflectance measurement mode
A reference measurement is first performed before the sample measurement. The reference measurement consists of measuring the spectrum of the light that has been diffusely reflected from a reference jar containing a PTFE film with a well-characterized broadband reflectance mounted against the inner walls of the sample jar. The experiment is performed with a broadband light source connected to the Illumination Arm, which is then aimed and focused on the reference jar, after which the measurement is recorded. After the reference measurement, the lamp of the Illumination Arm is kept on to maintain the stability of the light source. Subsequently, the reference jar is replaced with the sample jar, which is calibrated with the light source by adjusting the height of the Spectrometer Arm to saturate the signal. Once the focal plane is established, TLIS of the mechanochemical experiments can begin. The measurement duration time is typically set for a cycle of 90 min, with updates occurring every minute. Supplementary Movie 1 shows the synchronous operation of the ball mill and the software interface in real-time during the TLIS spectroscopy of the FAPbI3 mechanochemistry. The reflectance spectrum from 340 to 1000 nm is determined by comparing the ratio of the sample to the reference data using a graphical user interface (GUI), which is then converted to an absorption spectrum by the Kubelka–Munk function (Eq. 1) for display.
where, F and R are the absorption and remission fraction of sample, respectively.
Photoluminescence measurement mode
For the PL measurements, the light source is switched from the reflection mode to a laser (370 nm) with the other steps remaining the same as the ones for the diffuse reflectance measurements. The integration time can be set in a range from 1 to 1000 ms. For this study, the integration time was set at 500 ms for both double halide perovskites and Sn-based perovskites, while it was set at 50 ms for Pb-based perovskites. The PL spectrum was recorded in the range from 340 nm to 1000 nm. Using CsPbBr3 as an example, Supplementary Movie 2 shows the synchronous operation of the ball mill and the software interface in real-time during a TLIS measurement in PL mode for a 30 min mechanochemical synthesis.
Characterization
pXRD
The pXRD studies were performed on a Bruker D8 X-ray diffractometer with a Cu-Kα radiation (λ = 1.5406 Å) source set to 40 kV and 40 mA. A Bragg–Brentano geometry was used in the pXRD experiments, which were conducted in the θ–2θ scan mode with a scan rate of 4° per min. The data were analyzed using the “Match!” software.
TEM
TEM samples were prepared by dispersing 2.0 mg of the perovskite powder in 4 mL of toluene. The mixture was sonicated for 20 min, after which 20 μL of the suspension was drop-cast onto a standard Cu grid coated with carbon film. The Cu grid was dried under vacuum for 3 h. TEM, HRTEM, HAADF-STEM, and EDS mapping images were taken with a JEM-2100F microscope with a ZrO/W(100) Schottky electron source at an acceleration voltage of 200 kV, equipped with a 2 K × 2 K Gatan Ultrascan 1000XP charge-coupled device camera, Gatan Digiscan and STEM detectors, and an EDS detector.
Solid-state MAS NMR spectroscopy
All reported 133Cs solid-state MAS NMR data were measured at external B0 field strengths of 11.7 and 14.1 T using Bruker Avance III (Larmor frequency υ0(133Cs) = 65.60 MHz) and Bruker Avance Neo (Larmor frequency υ0(133Cs) = 78.62 MHz) spectrometers, respectively. Each 133Cs solid-state MAS NMR experiment was undertaken using a Bruker 4 mm HX probe spinning at MAS frequencies of 12 kHz. The 133Cs pulse calibration was achieved using solid CsCl where a non-selective (“solution”) π/2 pulse length of 8 μs was determined, corresponding to a selective (“solid”) pulse length of 8 μs for the I = 7/2 133Cs nucleus. Single pulse/direct excitation experiments were employed for the acquisition of the 133Cs MAS NMR data using selective π/4 pulses of 1 μs duration, a recycle delay of 2000 s, and the acquisition of 16 transients per spectrum. Accompanying 133Cs T1 measurements were performed using the saturation-recovery pulse sequence that employed a saturation “train” of 300 π/2 pulses of 2 μs, followed by variable τ delays ranging from 0.01 to 1600 s. Each T1 build-up curve was plotted by measuring the integrated area under each 133Cs resonance for each τ delay using the Bruker Dynamics Center package. All 133Cs MAS NMR data were indirectly referenced against the IUPAC recommended reference of 0.10 M CsNO3 in D2O (δiso = 0.0 ppm) via a secondary solid reference of CsCl (δiso = 223.2 ppm)68,69.
The 23Na MAS NMR data were acquired at external B0 field strengths of 2.3, 9.4, and 11.7 T using Bruker Avance III HD spectrometers for the two lower field measurements (Larmor frequencies υ0(23Na) = 26.49 and 105.50 MHz, respectively), and a Bruker Avance III spectrometer for the higher field measurement (Larmor frequency υ0(23Na) = 132.29 MHz). For low field 2.3 T measurements, a Bruker HFX 2.5 mm probe was used, which enabled MAS frequencies of 20 kHz, while the higher field 9.4 and 11.7 T measurements used Bruker 3.2 mm HX probes, which enabled MAS frequencies of 20 kHz. At all fields, non-selective (“solution”) π/2 pulse durations of 4 μs were calibrated using a 0.10 M solution of NaCl in D2O, corresponding to a selective (“solid”) π/2 pulse of 2 μs for the I = 3/2 23Na nucleus. Single pulse/direct detection 23Na MAS NMR data using π/4 pulses of 1 μs and a recycle delay of 20 s ensured full T1 relaxation of the 23Na spin system and quantitative results. In similar fashion to the 133Cs MAS NMR study, 23Na T1 measurements were performed at 9.4 T using a saturation-recovery pulse sequence consisting of a saturation train of 100 π/2 pulses of 2 μs duration followed by variable τ delays ranging from 0.001 to 100 s, while each T1 build-up curve was processed by measuring the integrated area using the Bruker Dynamics Center package. All 23Na MAS NMR data were directly referenced against the IUPAC recommended reference of 0.10 M NaCl in D2O (δiso = 0.0 ppm)68,69.
For both the 133Cs and 23Na MAS NMR measurements, all samples were loaded into rotors in a glovebox under a high-purity N2 atmosphere. All MAS NMR data were acquired using the Bruker Topspin program. The processed data were simulated and plotted using a combination of DMFit70, Adobe Illustrator, and Origin software packages.
XPS
XPS were performed using a Phoibos 100 spectrometer and a monochromatic Mg-Kα (hv = 1254.6 eV) X-ray radiation source (SPECS, Germany).
PLQY measurement
PLQY measurement of each powder sample was recorded by an Otsuka Quantum Efficiency Measurement System QE2000. Firstly, a reference measurement is conducted where the excitation light is reflected off a diffuse reflectance standard (BaSO4), ensuring that the excitation wavelength is the same for the sample as well. For the excitation of different types of perovskites, the selected excitation wavelengths are 355, 370, and 450 nm for double perovskites, Pb-based perovskites, and Sn-based perovskites, respectively. The reference holder is then switched to the sample, where both the reflected excitation light and the emission from the sample are recorded from 300 to 950 nm. The PLQY is calculated by taking the number of photons emitted by the sample divided by the number of photons absorbed by the sample.
Rietveld refinement for the pXRD data
The Rietveld refinement fitting of the experimental pXRD data was performed using the Topas software (version 6, Bruker AXS, Germany)71.
Computational methods
The DFT calculations were performed using the Quantum ESPRESSO package72 within the Atomic Simulation Environment (ASE)73 with the Perdew–Burke–Ernzerhof (PBE) functional and the generalized gradient approximation (GGA)74. The wavefunctions were represented with plane-wave basis sets and the core-valence interactions were described with scalar relativistic ultrasoft pseudopotentials75. The D3 correction method was used to account for the van der Waals interactions76. For all calculations, the plane-wave and density cutoffs were 500 eV and 5000 eV, respectively. The coordinates of the optimized structures have been included in the Supplementary Data 1 file.
Several compositions of FASnIzBr3−z were modeled, each using a 1 × 1 × 1 supercell. Every geometry optimization was performed with a quasi-Newton algorithm as implemented in ASE. The convergence criteria for geometry optimization were chosen such that the maximum forces acting on each relaxed atom were less than 0.02 eV/Å and the energy change was less than 10−7 eV. The Brillouin zone was sampled with a 6 × 6 × 6 and an 18 × 18 × 18 k-point mesh for geometry optimizations and electronic structure calculations, respectively. Band structures were calculated along the high-symmetry points using the Brillouin zone path outlined by the AFLOW software framework77.
Cs2Ag0.1Na0.9BiCl6 was modeled using a 2 × 1 × 1 supercell (80 atoms in total) with a structural formula Cs2Ag0.125Na0.875BiCl6, which is not far from the experimental composition. The BI site vacancy was modeled by removing one Na atom, and the X site vacancy was modeled by removing one Cl atom from the Na–Cl octahedral. Each geometry optimization was performed with a quasi-Newton algorithm as implemented in ASE and the convergence criterion was chosen such that the maximum forces acting on each relaxed atom were less than 0.05 eV/Å and the energy change was less than 10−5 eV, respectively. Transition states were determined using the climbing-image NEB approach60,61,62. Nine images were interpolated between the initial and final states linearly and the convergence criteria for the forces were set to 0.05 eV/Å. The optimized structures were visualized using the VESTA software78.
Data availability
The authors declare that the data supporting the findings of this study are available within the paper and its Supplementary Information files. The data in Figs. 2–7 generated in this study are provided in the Source Data file. The Supplementary Information data used in this study are available in the NTU data repository with the identifier https://doi.org/10.21979/N9/W0M8KL. Should any raw data files be needed in another format, they are available from the corresponding authors upon request. Source data are provided with this paper.
References
Kojima, A., Teshima, K., Shirai, Y. & Miyasaka, T. Organometal halide perovskites as visible-light sensitizers for photovoltaic cells. J. Am. Chem. Soc. 131, 6050–6051 (2009).
Li, Z. et al. Scalable fabrication of perovskite solar cells. Nat. Rev. Mater. 3, 18017 (2018).
Liu, X.-K. et al. Metal halide perovskites for light-emitting diodes. Nat. Mater. 20, 10–21 (2021).
García de Arquer, F. P., Armin, A., Meredith, P. & Sargent, E. H. Solution-processed semiconductors for next-generation photodetectors. Nat. Rev. Mater. 2, 16100 (2017).
Leupold, N. & Panzer, F. Recent advances and perspectives on powder‐based halide perovskite film processing. Adv. Funct. Mater. 31, 2007350 (2021).
Karmakar, A. et al. Mechanochemical synthesis of methylammonium lead mixed–halide perovskites: unraveling the solid-solution behavior using solid-state NMR. Chem. Mater. 30, 2309–2321 (2018).
Hong, Z. et al. Completely solvent-free protocols to access phase-pure, metastable metal halide perovskites and functional photodetectors from the precursor salts. iScience 16, 312–325 (2019).
Zhang, Z., Zhao, X. & Li, J. SnSe/carbon nanocomposite synthesized by high energy ball milling as an anode material for sodium-ion and lithium-ion batteries. Electrochim. Acta 176, 1296–1301 (2015).
Ljubek, G., Kralj, M. & Kraljić Roković, M. Fluorine-free mechanochemical synthesis of MXene. Mater. Sci. Technol. 39, 1645–1649 (2023).
Gaykwad, B., Thakur, A., Buch, A. & Jasuja, K. Large-scale synthesis of electrochemically active titanium diboride-based nanosheets by high-energy ball milling. J. Phys. Chem. C 127, 15887–15900 (2023).
Alić, J., Schlegel, M.-C., Emmerling, F. & Stolar, T. Meeting the UN sustainable development goals with mechanochemistry. Angew. Chem. Int. Ed. 63, e202414745 (2024).
Bi, D. et al. Multifunctional molecular modulators for perovskite solar cells with over 20% efficiency and high operational stability. Nat. Commun. 9, 4482 (2018).
Prochowicz, D., Saski, M., Yadav, P., Grätzel, M. & Lewiński, J. Mechanoperovskites for photovoltaic applications: preparation, characterization, and device fabrication. Acc. Chem. Res. 52, 3233–3243 (2019).
Palazon, F., El Ajjouri, Y. & Bolink, H. J. Making by grinding: mechanochemistry boosts the development of halide perovskites and other multinary metal halides. Adv. Energy Mater. 10, 1902499 (2020).
Takacs, L. & Takacs, L. The historical development of mechanochemistry. Chem. Soc. Rev. 42, 7649–7659 (2013).
Gomollón-Bel, F. Ten chemical innovations that will change our world: IUPAC identifies emerging technologies in Chemistry with potential to make our planet more sustainable. Chem. Int. 41, 12–17 (2019).
Friscic, T. et al. Real-time and in situ monitoring of mechanochemical milling reactions. Nat. Chem. 5, 66–73 (2013).
Halasz, I. et al. In situ and real-time monitoring of mechanochemical milling reactions using synchrotron X-ray diffraction. Nat. Protoc. 8, 1718–1729 (2013).
Lampronti, G. I. et al. Changing the game of time resolved X-ray diffraction on the mechanochemistry playground by downsizing. Nat. Commun. 12, 6134 (2021).
Gracin, D., Strukil, V., Friscic, T., Halasz, I. & Uzarevic, K. Laboratory real-time and in situ monitoring of mechanochemical milling reactions by Raman spectroscopy. Angew. Chem. Int. Ed. 53, 6193–6197 (2014).
Kulla, H. et al. Warming up for mechanosynthesis—temperature development in ball mills during synthesis. Chem. Commun. 53, 1664–1667 (2017).
Uzarevic, K. et al. Enthalpy vs. friction: heat flow modelling of unexpected temperature profiles in mechanochemistry of metal-organic frameworks. Chem. Sci. 9, 2525–2532 (2018).
Haferkamp, S., Fischer, F., Kraus, W. & Emmerling, F. Mechanochemical Knoevenagel condensation investigated in situ. Beilstein J. Org. Chem. 13, 2010–2014 (2017).
Kulla, H. et al. In Situ Investigations of Mechanochemical One-Pot Syntheses. Angew. Chem. Int. Ed. 57, 5930–5933 (2018).
Lukin, S. et al. Tandem in situ monitoring for quantitative assessment of mechanochemical reactions involving structurally unknown phases. Chem. Eur. J. 23, 13941–13949 (2017).
Uzarevic, K., Halasz, I. & Friscic, T. Real-time and in situ monitoring of mechanochemical reactions: a new playground for all chemists. J. Phys. Chem. Lett. 6, 4129–4140 (2015).
Michalchuk, A. A. L. et al. Challenges of mechanochemistry: is in situ real-time quantitative phase analysis always reliable? A case study of organic salt formation. Adv. Sci. 4, 1700132 (2017).
Halasz, I. et al. Real-time in situ powder X-ray diffraction monitoring of mechanochemical synthesis of pharmaceutical cocrystals. Angew. Chem. Int. Ed. 52, 11538–11541 (2013).
Julien, P. A., Malvestiti, I. & Friscic, T. The effect of milling frequency on a mechanochemical organic reaction monitored by in situ Raman spectroscopy. Beilstein J. Org. Chem. 13, 2160–2168 (2017).
Petersen, H. et al. In situ synchrotron X-ray diffraction studies of the mechanochemical synthesis of ZnS from its elements. Chem. Eur. J. 27, 12558–12565 (2021).
Emmerling, S. T. et al. In situ monitoring of mechanochemical covalent organic framework formation reveals templating effect of liquid additive. Chem 7, 1639–1652 (2021).
Germann, L. S. et al. Monitoring polymer-assisted mechanochemical cocrystallisation through in situ X-ray powder diffraction. Chem. Commun. 56, 8743–8746 (2020).
Bjelopetrović, A. et al. Mechanism of mechanochemical C−H bond activation in an azobenzene substrate by PdII catalysts. Chem. Eur. J. 24, 10672–10682 (2018).
Julien, P. A. et al. Illuminating milling mechanochemistry by tandem real-time fluorescence emission and Raman spectroscopy monitoring. Chem. Sci. 14, 12121–12132 (2023).
Rathmann, T. et al. In situ synchrotron x-ray diffraction studies monitoring mechanochemical reactions of hard materials: challenges and limitations. Rev. Sci. Instrum. 92, 114102 (2021).
Julien, P. A. et al. In situ monitoring of mechanochemical synthesis of calcium urea phosphate fertilizer cocrystal reveals highly effective water-based autocatalysis. Chem. Sci. 11, 2350–2355 (2020).
Xie, Y.-M. et al. Direct observation of cation-exchange in liquid-to-solid phase transformation in FA1−xMAxPbI3 based perovskite solar cells. J. Mater. Chem. A 6, 9081–9088 (2018).
Vashishtha, P. et al. Cesium copper iodide tailored nanoplates and nanorods for blue, yellow, and white emission. Chem. Mater. 31, 9003–9011 (2019).
Vashishtha, P. et al. Investigating the structure–function relationship in triple cation perovskite nanocrystals for light-emitting diode applications. J. Mater. Chem. C 8, 11805–11821 (2020).
Vashishtha, P. et al. Performance enhanced light-emitting diodes fabricated from nanocrystalline CsPbBr3 with in situ Zn2+ addition. ACS Appl. Electron. Mater. 2, 4002–4011 (2020).
Sun, Y. et al. Lewis base-mediated perovskite crystallization as revealed by in situ, real-time optical absorption spectroscopy. J. Phys. Chem. Lett. 12, 5357–5362 (2021).
Mrkyvkova, N. et al. Combined in situ photoluminescence and X-ray scattering reveals defect formation in lead-halide perovskite films. J. Phys. Chem. Lett. 12, 10156–10162 (2021).
Schötz, K. & Panzer, F. Using in situ optical spectroscopy to elucidate film formation of metal halide perovskites. J. Phys. Chem. A 125, 2209–2225 (2021).
Vashishtha, P. et al. Elucidation of the structural and optical properties of metal cation (Na+, K+, and Bi3+) incorporated Cs2AgInCl6 double perovskite nanocrystals. J. Mater. Chem. A 10, 3562–3578 (2022).
Dintakurti, S. S. H. et al. A powder XRD, solid state NMR and calorimetric study of the phase evolution in mechanochemically synthesized dual cation (Csx(CH3NH3)1−x)PbX3 lead halide perovskite systems. Phys. Chem. Chem. Phys. 24, 18004–18021 (2022).
Li, X. et al. Lead-free halide perovskites for light emission: recent advances and perspectives. Adv. Sci. 8, 2003334 (2021).
Liang, H. et al. High color purity lead-free perovskite light-emitting diodes via Sn stabilization. Adv. Sci. 7, 1903213 (2020).
Yuan, F. et al. Color-pure red light-emitting diodes based on two-dimensional lead-free perovskites. Sci. Adv. 6, eabb0253 (2020).
Huskić, I., Lennox, C. B., Friščić, T. & Huskić, I. Accelerated ageing reactions: towards simpler, solvent-free, low energy chemistry. Green Chem. 22, 5881–5901 (2020).
Budny‐Godlewski, K. et al. Uncovering factors controlling reactivity of metal‐TEMPO reaction systems in the solid state and solution. Chem. Eur. J. 30, e202401968 (2024).
Heo, J. H., Song, D. H. & Im, S. H. Planar CH3NH3PbBr3 hybrid solar cells with 10.4% power conversion efficiency, fabricated by controlled crystallization in the spin‐coating process. Adv. Mater. 26, 8179–8183 (2014).
Prochowicz, D. et al. Mechanosynthesis of pure phase mixed-cation MAxFA1−xPbI3 hybrid perovskites: photovoltaic performance and electrochemical properties. Sustainable Energy Fuels 1, 689–693 (2017).
Binek, A., Hanusch, F. C., Docampo, P. & Bein, T. Stabilization of the trigonal high-temperature phase of formamidinium lead iodide. J. Phys. Chem. Lett. 6, 1249–1253 (2015).
Kim, M. et al. Methylammonium chloride induces intermediate phase stabilization for efficient perovskite solar cells. Joule 3, 2179–2192 (2019).
Prochowicz, D. et al. Mechanosynthesis of the hybrid perovskite CH3NH3PbI3: characterization and the corresponding solar cell efficiency. J. Mater. Chem. A 3, 20772–20777 (2015).
Pellet, N. et al. Mixed‐organic‐cation Perovskite photovoltaics for enhanced solar‐light harvesting. Angew. Chem. Int. Ed. 53, 3151–3157 (2014).
Yao, M. M. et al. Improving lead‐free double perovskite Cs2NaBiCl6 nanocrystal optical properties via ion doping. Adv. Opt. Mater. 8, 1901919 (2020).
Pei, Y. et al. Boosting Near-Infrared Luminescence of Lanthanide in Cs2AgBiCl6 Double Perovskites via Breakdown of the Local Site Symmetry. Angew. Chem. Int. Ed. 61, e202205276 (2022).
Jeevaraj, M. et al. Structural, optical and charge density investigations on lead free Mn2+ doped Cs2NaBiCl6 double perovskite microcrystals. Mater. Today Commun. 33, 104715 (2022).
Torres, J. A. G., Jennings, P. C., Hansen, M. H., Boes, J. R. & Bligaard, T. Low-scaling algorithm for nudged elastic band calculations using a surrogate machine learning model. Phys. Rev. Lett. 122, 156001 (2019).
Koistinen, O.-P., Dagbjartsdóttir, F. B., Ásgeirsson, V., Vehtari, A. & Jónsson, H. Nudged elastic band calculations accelerated with Gaussian process regression. J. Chem. Phys. 147, 152720 (2017).
del Río, E. G., Mortensen, J. J. & Jacobsen, K. W. Local Bayesian optimizer for atomic structures. Phys. Rev. B. 100, 104103 (2019).
Wu, T. et al. Heterogeneous FASnI3 absorber with enhanced electric field for high-performance lead-free perovskite solar cells. Nano-Micro Lett. 14, 99 (2022).
Lee, S. J. et al. Reducing carrier density in formamidinium tin perovskites and its beneficial effects on stability and efficiency of perovskite solar cells. ACS Energy Lett. 3, 46–53 (2017).
López‐Fernández, I. et al. Lead‐free halide perovskite materials and optoelectronic devices: progress and prospective. Adv. Funct. Mater. 34, 2307896 (2023).
Chen, G. et al. Core/shell NaGdF4: Nd3+/NaGdF4 nanocrystals with efficient near-infrared to near-infrared downconversion photoluminescence for bioimaging applications. ACS Nano 6, 2969–2977 (2012).
Haque, A., Zechel, F., Vretenár, V., Roy, M. & Sýkora, M. Type‐II CdSe/ZnO core/shell nanorods: nanoheterostructures with a tunable dual emission in visible and near‐infrared spectral ranges. Adv. Funct. Mater. 34, 2305296 (2024).
Harris, R. K., Becker, E. D., Cabral De Menezes, S. M., Goodfellow, R. & Granger, P. NMR nomenclature. Nuclear spin properties and conventions for chemical shifts(IUPAC Recommendations 2001). Pure Appl. Chem. 73, 1795–1818 (2001).
Harris, R. K. et al. Further conventions for NMR shielding and chemical shifts (IUPAC Recommendations 2008). Pure Appl. Chem. 80, 59–84 (2008).
Massiot, D. et al. Modelling one‐ and two‐dimensional solid‐state NMR spectra. Magn. Reson. Chem. 40, 70–76 (2002).
Wang, X., Shi, J., Wang, X. & Li, Y. Structural, strong magnetic exchange, and room temperature multiferroic properties of single-phase 0.75BiFeO3–BaFe1/2Nb1/2O3 bulk ceramics. Ceram. Int. 46, 18707–18715 (2020).
Giannozzi, P. et al. QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials. J. Phys. 21, 395502 (2009).
Larsen, A. H. et al. The atomic simulation environment—a Python library for working with atoms. J. Phys. 29, 273002 (2017).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865 (1996).
Garrity, K. F., Bennett, J. W., Rabe, K. M. & Vanderbilt, D. Pseudopotentials for high-throughput DFT calculations. Comput. Mater. Sci. 81, 446–452 (2014).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Setyawan, W. & Curtarolo, S. High-throughput electronic band structure calculations: challenges and tools. Comput. Mater. Sci. 49, 299–312 (2010).
Momma, K. & Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 44, 1272–1276 (2011).
Acknowledgements
This work is supported by the Ministry of Education (Singapore) under its AcRF Tier 2 grant (MOE-T2EP10123-0007) and an AcRF Tier 1 grant RG 9/22. H.S.S. is also grateful that this project is partly supported by the National Research Foundation (NRF) Singapore under the grant NRF-CRP27−2021-0001. H.S.S. and Y.-F.L. acknowledge that this project was partly supported by A*STAR under the AME IRG grant no. A2083c0050 and an IAF-PP grant no. A19E9a0103. T.C.S. and V.K.R. acknowledge the support from the Ministry of Education (Singapore) under its AcRF Tier 2 grant (MOE-T2EP50123-0001) and the NRF Singapore under its Competitive Research Program (NRF-CRP25-2020-0004). F.G. would like to thank Monash University for start-up funds. J.V.H. acknowledges financial support for the solid-state NMR instrumentation at Warwick used in this research, which was funded by EPSRC (grants EP/M028186/1 and EP/K024418/1) and the University of Warwick, and the Birmingham Science City AM1 and AM2 projects that were supported by Advantage West Midlands (AWM) and the European Regional Development Fund (ERDF). We would like to acknowledge the Facility for Analysis, Characterization, Testing and Simulation, Nanyang Technological University, Singapore, for use of their electron microscopy/X-ray facilities. We would like to acknowledge the Nanyang Technological University High Performance Computing Center for the use of their resources. X. Zhou is supported by a Discovery Early Career Researcher Award (ARC DECRA DE190100144) from the Australian Research Council.
Author information
Authors and Affiliations
Contributions
F.G., Y.-F.L., and H.S.S. conceived and designed the project. H.S.S. supervised the project. Y.X. and X.H. performed the TLIS mechanochemical measurements of perovskites, structural characterizations, and data analyses. D.G.B. and J.V.H. performed the measurement and analysis of the solid-state MAS NMR data. W.X.C. and X.Zhou conducted the DFT calculations. Z.L., Z.Y.T., C.L., and Y.H.T. assisted in the TLIS mechanochemical measurements on the perovskites. L.X., Y.X. and H.S. performed the PLQY measurements on the perovskites. X.Zhao and M.S. completed the XPS measurements. V.K.R. and T.C.S. carried out the TEM measurements. X.W. assisted in the Rietveld refinement of the pXRD data. S.C.L. and P.B.P. performed the installation and calibration of the TLIS system. Y.X., D.G.B., W.X.C., F.G., X.Zhou, Y.-F.L., J.V.H., and H.S.S. analyzed the results and prepared the manuscript. Y.X., D.G.B., and W.X.C. were involved in revising the figures. All the authors reviewed and contributed to this paper.
Corresponding authors
Ethics declarations
Competing interests
H.S.S., Y.X., X.H., P.B.P., and S.C.L. are inventors of a Singapore provisional patent application number 10202401942U covering the TLIS mechanochemical spectrometer filed by NTUitive. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Matej Baláž, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xiao, Y., Bradley, D.G., Chan, W.X. et al. Optical time-lapsed in situ mechanochemical studies on metal halide perovskite systems. Nat Commun 16, 1362 (2025). https://doi.org/10.1038/s41467-025-56571-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-56571-w
