
Abstract
The aryl hydrocarbon receptor (AHR) possesses an extraordinary capacity to sense and respond to a wide range of small-molecule ligands, ranging from polycyclic aromatic hydrocarbons to endogenous compounds. Upon ligand binding, AHR translocates from the cytoplasm to nucleus, forming a transcriptionally active complex with aryl hydrocarbon receptor nuclear translocator (ARNT), for DNA binding and initiation of gene expression programs that include cellular detoxification pathways and immune responses. Here, we examine the molecular mechanisms governing AHR’s high-affinity binding and activation by a diverse group of ligands. Crystal structures of the AHR-ARNT-DNA complexes, bound with each of six established AHR ligands, including Tapinarof, 6-formylindolo[3,2-b]carbazole (FICZ), benzo[a]pyrene (BaP), β-naphthoflavone (BNF), Indigo and Indirubin, reveal an unconventional mode of subunit assembly with intimate association between the PAS-B domains of AHR and ARNT. AHR’s PAS-B domain utilizes eight conserved residues whose dynamic rearrangements account for the ability to bind to ligands through hydrophobic and π-π interactions. Our findings further reveal the structural underpinnings of a ligand-driven activation mechanism, whereby a segment of the AHR protein undergoes a structural transition from chaperone engagement to ARNT heterodimer stabilization, to generate the transcriptionally competent assembly. Our results provide key information for the future development of AHR-targeting drugs.
Similar content being viewed by others
Introduction
The aryl hydrocarbon receptor (AHR) was initially characterized as the intracellular receptor responsible for the actions of dioxin-like pollutants, playing a central role in their profoundly toxic effects1. AHR is abundant in the liver and barrier organs, including skin, lung, and gut2. This pattern of distribution positions AHR as the primary candidate for encountering and detoxifying an array of small-molecule ligands derived from environmental pollutants and microbial agents3,4. In addition, AHR binds and responds to dietary components, metabolic intermediates, and other endogenous molecules. AHR integrates all these signals into its transcriptional regulatory network, essential for detoxification and immune responses5. Dysregulation of AHR is associated with cancers, metabolic disorders, and inflammatory diseases6,7,8. The recent approval of Tapinarof (Benvitimod), a bacteria-derived AHR agonist treating plaque psoriasis, highlights numerous therapeutic opportunities associated with modulating AHR activity9,10,11,12.
AHR belongs to the basic helix-loop-helix-PER-ARNT-SIM (bHLH-PAS) family of transcription factors, characterized by a conserved bHLH DNA binding domain at its N-terminus, followed by tandem PAS domains (PAS-A and PAS-B), and an unstructured transactivation domain13,14,15. These domains collectively facilitate AHR’s heterodimerization with the aryl hydrocarbon receptor nuclear translocator (ARNT), crucial for forming a transcriptional complex binding to xenobiotic response elements (XREs)16,17,18. The PAS-B domain is the primary binding site for AHR’s repertoire of small-molecule ligands, yet the precise stereochemical basis for diverse ligand interactions has remained difficult to assess19,20.
In the absence of a ligand, AHR is cytoplasmic and complexed with two molecules of heat shock protein 90 (HSP90) and co-chaperones X-associated protein 2 (XAP2) and p2321,22. The bHLH and PAS-B domains of AHR are both involved in the interaction with HSP9023. Upon ligand binding to its PAS-B domain, the AHR in the cytosolic complex undergoes conformational changes, likely exposing the nuclear localization signal (NLS) near its bHLH domain to facilitate nuclear translocation24,25. In the nucleus, AHR dimerizes with ARNT to form the complex capable of recognizing XRE sequences associated with target genes26,27,28. Recent cryogenic-electron microscopy (cryo-EM) studies revealed interactions between the isolated PAS-B domain of AHR, HSP90, and co-chaperones, providing insights into AHR’s cytoplasmic complex29,30. However, visualization of the AHR bHLH and PAS-A domains was not attained in these studies, and the corresponding ligand-activated DNA-binding competent complex with ARNT has similarly remained structurally elusive. Consequently, comprehending the mechanisms underlying the bindings of promiscuous ligands and their capacity to induce dissociation of the chaperone complex to facilitate AHR’s association with ARNT for DNA binding has been challenging.
Here, we sought to elucidate the structural mechanisms underlying AHR’s binding and activation by a diverse group of ligands. We analyzed a series of structures of AHR-ARNT heterodimers bound to XRE, each engaged with a distinct small-molecule agonist. These ligands encompassed a spectrum of well-known compounds, including the first-in-class bacteria-derived psoriasis medication Tapinarof, the endogenous tryptophan derivative 6-formylindolo[3,2-b]carbazole (FICZ), the environmental pollutant benzo[a]pyrene (BaP), the synthetic flavone β-naphthoflavone (BNF), and the plant- or dietary-derived pigments Indigo and Indirubin. Our investigations further elucidated the mechanism by which agonists induce a uniform conformational rearrangement in AHR, promoting its state transition from chaperone-bound to ARNT-bound, thereby establishing the DNA-binding competence.
Results
Overall structural organization of multi-domain AHR-ARNT-DNA complex
To establish a robust structural framework for visualizing AHR-ARNT heterodimers, wherein ligand-binding and DNA-binding could be simultaneously analyzed at the highest resolution possible, we employed contiguous bHLH-PAS-A-PAS-B segments from both AHR and ARNT proteins for crystallization trials (Fig. 1a). Through numerous attempts using AHR and ARNT derived from many different species, while also attempting to incorporate a variety of loop-region truncations and DNA duplexes, we finally succeeded to obtain well-diffracting crystals using porcine AHR (pAHR) and human ARNT (hARNT) proteins in the presence of Tapinarof. This heterodimeric complex was co-crystallized with a 21-mer dsDNA segment harboring the 5’-TNGCGTG-3’ XRE sequence. The resolution of this crystal structure reached 3.0 Å (Supplementary Table 1), higher than previously reported crystal structures of DNA-bound bHLH-PAS proteins (3.6–4.7 Å)31,32,33. A single AHR-ARNT-DNA-Tapinarof complex resided in the asymmetric unit. The quaternary organization of this complex and the locations of DNA and ligand could be visualized clearly in the electron density maps (Fig. 1b and Supplementary Fig. 1a).

a Schematic representation showing the domain arrangements of AHR and ARNT. bHLH, basic helix-loop-helix; PAS, PER-ARNT-SIM; TAD, transactivation domain. b The 2Fo-Fc map (contour level = 1.0 σ, shown as gray mesh) of AHR-ARNT-DNA complex structure in two views with all domains labeled. AHR, ARNT, and DNA are colored in magenta, green, and orange, respectively. c Comparison between one representative projection of 2D class averages (left) and the crystal structure of AHR-ARNT-DNA (right). The spatial locations of DNA and bHLH, PAS-A and PAS-B domains of AHR-ARNT are labeled.
An intriguing feature of this structure is the distinct spatial arrangement of the two PAS-B domains of AHR and ARNT, positioned distinctively apart from their respective PAS-A domains and the DNA-bound bHLH domains (Fig. 1b and Supplementary Movie 1). Upon scrutinizing the AHR-ARNT-DNA complex structure using cryo-EM, we observed clear visualization of only the DNA-bound bHLH and PAS-A domains, while the two PAS-B domains appeared blurred (Fig. 1c and Supplementary Fig. 2a, b). This observation aligns with our crystallographic findings, where the two PAS-B domains are spatially distant from other AHR and ARNT segments but remain together as a PAS-B dimeric unit. The cryo-EM results suggest that our ability to visualize this double PAS-B unit clearly in the crystal structure was fortuitous, as its positioning became constrained by crystal packing forces, via direct interactions between this unit and two neighboring symmetric molecules (Fig. 1b and Supplementary Fig. 2c). These packing interactions do not interfere with the internal pockets or domain-domain interactions of the PAS-B domains of AHR and ARNT, but only constrain their overall location and dynamics to stabilize them in a distinct position within the crystal lattice to enable clear visualization of their features.
It is noteworthy that the sequence identity between porcine and human AHR proteins at their N-terminal half is 91%. Moreover, the AHR residues that directly interact with ARNT in the bHLH, PAS-A and PAS-B domains are highly conserved among these two species, with 66 out of 71 residues fully identical (Supplementary Fig. 1b). Therefore, we speculate that the heterodimeric structure of pAHR-hARNT would closely resemble the human AHR-ARNT structure, as well as those from other mammals given their conserved protein sequences.
We previously examined the crystal structures of other bHLH-PAS family members (Supplementary Fig. 3a), including the ARNT heterodimers with hypoxia-inducible factor (HIF)-1α, HIF-2α, HIF-3α, and three neuronal PAS proteins (NPAS1, NPAS3, NPAS4)31,32,33,34. These structures, like the current structure, all encompassed the bHLH, PAS-A, and PAS-B domains, and were categorized into two distinct interaction modes, represented by HIF-2α-ARNT and NPAS4-ARNT (Supplementary Fig. 3b). Interestingly, the orientation of the PAS-B domains of AHR-ARNT is markedly different from these two archetypes. For example, the PAS-B domains in HIF-2α and also in NPAS4 both strongly interact with their own PAS-A domains, and with ARNT’s PAS-A domain. In contrast, the AHR PAS-B domain does not have notable interactions with any of the PAS-A domains in its complex (Supplementary Fig. 3b). Therefore, the AHR-ARNT quaternary structure represents a distinct interaction mode in the bHLH-PAS family, in line with the feature of AHR as a ligand-dependent transcription factor distinguished from its fellow members (Supplementary Fig. 3a).
We then compared the rest of the complex with two previously reported AHR-ARNT structures encompassing only the PAS-A and DNA-bound bHLH domains35,36 (Supplementary Fig. 4a). By superimposition, the root-mean-square deviation (RMSD) values were calculated as 1.65 Å and 1.84 Å for Cα atoms, indicating a similar organization. The pAHR residues S36 and R40 interact with the CGC half site of XRE via hydrogen bonds, while hARNT residues H94, E98, and R102 contact the GTG half site (Supplementary Fig. 4b). This interaction pattern explains how AHR-ARNT heterodimer specifically recognizes the XRE sites, as opposed to the canonical E-boxes used by other bHLH-PAS family members.
The PAS-B unit relies on a conserved AHR segment
To understand how the two PAS-B domains form a stable unit in this complex, we began by comparing it to a recently published structure of PAS-B domains from drosophila AHR (dAHR) and mouse ARNT (mARNT)37. Our analysis revealed that an additional C-terminal loop from the pAHR PAS-B domain (hereafter denoted by the C-ter loop, residues 401–413) inserts directly between the PAS-B domains, providing critical stabilization to the PAS-B/PAS-B interface of our complex (Fig. 2a and Supplementary Fig. 5a). Intriguingly, the middle region of this C-ter loop (residues 407–409) forms a small β-sheet with the nearby β-strand (Bβ) of the ARNT PAS-B domain, contributing to the overall interface (Fig. 2a and Supplementary Fig. 5b). Furthermore, several residues in the C-ter loop participate in hydrogen bonds and/or hydrophobic interactions with ARNT (Fig. 2b), with varying contributions to energy decomposition (Supplementary Fig. 5c).

a Overall structure of the PAS-B domains of AHR-ARNT heterodimer. The C-ter loop of AHR PAS-B is colored in light pink. b The detailed interactions between the C-ter loop of AHR PAS-B and ARNT. The residues of AHR and ARNT are colored in magenta and green, respectively. These interactions were analyzed by the Ligplus+ program78. c Co-IP experiments showing the effect of C-ter loop deletion on the formation of AHR-ARNT heterodimers in cells. This experiment was performed three times with similar results. d, e XRE reporter assays (d) and qPCR assays (e) evaluating the effects of C-ter loop deletion on AHR transcriptional activity in HEK293 cells. f Enlarged view near the PAS-B dimer interface where H324 of AHR forms hydrogen bonds with A405 and G407 of C-ter loop. g, h XRE reporter assays (g) and qPCR assays (h) evaluating the effects of hAHR H326A mutation (corresponding to H324 in pAHR) on transactivation in HEK293 cells. For the panels d, e, g, h: error bars, mean ± s.d.; n = 3 (biological replicates); statistical significance, ** p < 0.01, *** p < 0.001, **** p < 0.0001 (p-values shown on the charts calculated using unpaired two-tailed t test versus the WT group).
Comparison of this PAS-B/PAS-B interface of AHR-ARNT with corresponding interfaces in other ARNT heterodimeric structures revealed that only the C-ter loop of the AHR PAS-B domain extends deeply into the heterodimer interface (Supplementary Fig. 5d). In addition, the protein sequence of this C-ter loop is highly conserved among AHR in vertebrates (Supplementary Fig. 6), but differs at corresponding positions in other bHLH-PAS proteins (Supplementary Fig. 3c). These findings indicate that this loop segment specifically and critically mediates interactions between AHR and ARNT PAS-B domains.
To explore the functional importance of the C-ter loop in PAS-B unit formation, we conducted co-immunoprecipitation (Co-IP) experiments in HEK293 cells to assess its influence on AHR-ARNT dimerization. Truncation of the C-ter loop (Δ401-415) showed no effect on heterodimerization (Fig. 2c), suggesting that the bHLH and PAS-A domains may exert a more dominant role in AHR-ARNT heterodimerization, allowing tolerance for the distant and independent states of their PAS-B domains (Fig. 1b).
Next, we evaluated the impact of the C-ter loop on AHR transcriptional activity using a dual-luciferase XRE reporter assay also in HEK293 cells. Truncation of the loop resulted in a clear decrease in AHR activity, both at basal levels and in the presence of agonists Tapinarof and FICZ (Fig. 2d). Moreover, to assess the potential interference of any endogenous AHR proteins in these cells on the assay results, we further utilized the human fibrosarcoma-derived HT1080 cell line alongside its AHR knockout (KO) variant38. The decreasing effects on AHR activity by the C-ter loop truncation exhibited a similar trend in the parental HT1080 and AHR KO cell lines (Supplementary Fig. 5e, f), with a relatively larger fold change in the KO cells, indicating that the endogenous AHR did not strongly disturb the detection of heterologous AHR activity. In addition, we observed a significant reduction in mRNA expression of the AHR downstream gene CYP1A1 upon truncation of the C-ter loop (Fig. 2e). These findings together suggest that although not essential for the overall AHR-ARNT dimerization stability, the C-ter loop contributes to AHR transcriptional activity.
Previous studies reported that the H320A mutation in mouse AHR (mAHR), located outside the PAS-B pocket (corresponding to H324A in porcine and H326A in human AHR), could reduce DNA-binding activity triggered by various agonists39. In our structure, we observed that residue H324 forms key hydrogen bonds with A405 and G407 on the C-ter loop of pAHR PAS-B domain (Fig. 2f), both of which are involved in interactions between AHR and ARNT (Fig. 2b and Supplementary Fig. 5c). Subsequent XRE reporter assays and qPCR experiments revealed that the H326A mutation in human AHR (hAHR) similarly reduced its transcriptional activity (Fig. 2g, h and Supplementary Fig. 5g, h), suggesting that this key histidine residue may contribute to stabilizing the C-ter loop, thereby influencing AHR transactivation.
Promiscuous ligand binding mediated by conserved residues
To unravel the mechanism behind AHR’s well-documented ligand promiscuity, we endeavored to obtain AHR-ARNT complex structures for various ligands, following a similar approach as employed for Tapinarof. Ultimately, we succeeded in elucidating the DNA-bound AHR-ARNT structures in complex with five additional ligands, originating from endogenous (FICZ), synthetic (BaP and BNF), or natural sources (Indigo and Indirubin) (Fig. 3a). These structures, with resolutions ranging from 2.6 Å to 3.1 Å (Supplementary Table 1), allowed for clear visualization of each ligand within the electron density maps in the PAS-B domain of AHR (Supplementary Fig. 1c–g). Comparative analysis of the overall structures of these six complexes revealed a remarkable similarity, with paired RMSD values for Cα atoms all < 0.7 Å (Fig. 3b).

a Chemical structure of AHR agonists, including Tapinarof, FICZ, BaP, BNF, Indigo, and Indirubin. b Superimposition of six AHR-ARNT-DNA complex structures respectively bound with Tapinarof, FICZ, BaP, BNF, Indigo, and Indirubin. c–h, Interactions between Tapinarof (c), FICZ (d), BaP (e), BNF (f), Indigo (g), and Indirubin (h) with surrounding AHR residues within the pocket, analyzed by the Ligplus+ program78. i A summarized diagram showing the residues of the AHR PAS-B domain directly interacting with each of the six ligands. Residues forming hydrophobic and van der Waals interactions are colored in green, and those forming hydrogen bonds are colored in magenta. j The spatial distribution of eight AHR residues involved in direct interactions with all six ligands are shown in two views.
Subsequently, we examined the direct interactions between these ligands and the AHR pocket residues to uncover potential clues explaining the ligand promiscuity. All six ligands bind into AHR through hydrophobic interactions, van der Waals interactions, and hydrogen bonds in a resembling yet not identical fashion (Fig. 3c–i and Supplementary Fig. 7a–f). Notably, compared to two recently published structures of AHR cytosolic complexes bound to Indirubin and BaP29,40, these two ligands demonstrate similar binding patterns within the AHR-ARNT complexes (Supplementary Fig. 7g, h), suggesting that engaging different protein partners might not substantially alter AHR’s binding to ligands.
According to the ligand-bound complex structures and the sequence alignment of pAHR and hAHR (Fig. 3b and Supplementary Fig. 1b), pocket residues directly outlining the cavity (calculated by CASTp algorithm41) are highly conserved among these two species, with 24 out of 26 residues fully identical. The two non-conserved pAHR residues are Y334 and A379 (corresponding to S336 and V381 in hAHR). Y334 forms hydrophobic interaction with FICZ only; whereas A379 interacts with all the ligands except Tapinarof, likely due to its relatively smaller compound size (Supplementary Fig. 7a–f). It is noteworthy that this A379 in pAHR also corresponds to the well-known A375V variant in mAHR, which has been shown to weaken binding to dioxin, accounting for the lower sensitivity to dioxin for the D-allele mice harboring this variation as compared with the normal B-allele mice42.
We observed that all six ligands bind at the same position within the AHR PAS-B pocket, with their ring structures well-superimposed (Fig. 3b). Among the twenty-one pAHR residues directly interacting with these ligands (Fig. 3i), eight of them (H289, F293, G319, C331, F349, L351, S363, and Q381) collectively define the outline of the binding pocket from directions perpendicular or parallel to the ring plane (Fig. 3j and Supplementary Movie 2). Importantly, these eight residues are highly conserved in vertebrate AHR proteins (Supplementary Fig. 6). Molecular dynamics (MD) simulations revealed relatively high binding energies for residues F293 and L351 in the six complex structures (Supplementary Fig. 8a–f). As depicted in Fig. 3j, H289, along with F293, and F349, along with L351, interact with the ligands via π-π and/or hydrophobic interactions from opposite sides of the PAS-B pocket. The coordinated and dynamic utilization of this network of conserved residues in the pocket underlies AHR’s remarkable ability to accommodate various chemical ligands.
To validate the involvement of these above four residues in ligand binding, we individually mutated them to alanine and conducted XRE reporter assays to assess the effects of mutations. These mutations reduced AHR transcriptional activity triggered by the six ligands (Supplementary Fig. 8g). In addition, these mutations also clearly decreased the activity induced by several other AHR agonists, including 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), 3-methylcholanthrene (3-MC), indole-3-lactic acid (ILA), and indolo[3,2-b]carbazole (ICZ) (Supplementary Fig. 8h). Similar ligand binding-decreasing effects were reported by other groups following point mutations of these key pocket residues39,43,44. Hence, we speculate that AHR agonists with polycyclic or planar chemical structures may bind into the PAS-B pocket in a similar manner, with these eight conserved residues accommodating the binding of promiscuous ligands through their precise spatial distribution (Supplementary Movie 2).
Ligand-specific binding and activation of AHR
Although all six agonists bind at the same location, surrounded by a group of conserved and interacting residues (Fig. 3i), subtle differences exist in their orientations. Tapinarof, FICZ, BaP, BNF, and Indigo share a similar distribution of rings in their chemical structures, defining a common major axis and unifying their binding mode (annotated as Mode 1, Fig. 4a and Supplementary Fig. 7i–m). In contrast, Indirubin exhibits a major axis shifted towards the Fα (annotated as Mode 2, Fig. 4a and Supplementary Fig. 7n), representing a notable deviation from the other five ligands.

a Superimposition of AHR PAS-B domain structures bound by FICZ (purple) or Indirubin (magenta). The binding modes of FICZ and Indirubin in the pocket are represented by arrows. b XRE reporter assays evaluating the effects of hAHR Y332A mutation (corresponding to Y330 in pAHR) on transactivation. c, d The apparent dissociation constants of Tapinarof, BaP, BNF, Indigo, and Indirubin to pAHR (c) and hAHR (d) as determined by the MST method from three technical replicates at each compound concentration. e, f XRE reporter assays evaluating the dose-responsive activation of AHR by six agonists in Hep3B (e) and HaCaT (f) cells. For Panels b,e,f: error bars, mean ± s.d.; n = 3 (biological replicates); statistical significance: ** p < 0.01, *** p < 0.001 (p-values shown on the charts calculated using unpaired two-tailed t test versus the WT group).
Two pAHR residues, Y330 and I347, exhibit different conformations to accommodate these two binding modes (Fig. 4a). For ligands sharing Mode 1, Y330 forms hydrogen bonds with L398 and L400, stabilizing the C-ter loop of AHR PAS-B domain. In the Indirubin-bound structure, the side chain of Y330 undergoes an inward movement, probably resulting from the altered side-chain orientation of nearby I347 induced by Indirubin binding in Mode 2. To test whether the conformational change of Y330 is related to AHR transcriptional activity, we mutated this tyrosine to alanine. XRE reporter assays showed that, indeed, this mutation reduced AHR activity in the absence or presence of ligands, including Tapinarof, FICZ, and Indirubin (Fig. 4b), suggesting that Y330 is critical for AHR’s transcriptional regulation. However, surprisingly, this alanine mutation exhibited a very similar impact on ligands in two different binding modes. To further investigate the possible role of Y330, we constructed more point mutations (arginine, glutamate, phenylalanine, and leucine) and tested them in the AHR KO HT1080 cell line (Supplementary Fig. 7o). Interestingly, among the three mutations (alanine, arginine, and glutamate) clearly decreasing the XRE reporter activities, the arginine mutation exhibited a relatively stronger impact on Tapinarof and FICZ (Mode 1) than on Indirubin (Mode 2). These above results collectively suggest that the Y330 residue is involved in AHR’s accommodation to the binding of diverse ligands, and the length and charge of the side chain at this position may influence the transcriptional activities of various ligands to differing degrees.
To quantitatively compare the binding affinities and cellular activities of the six AHR ligands, we utilized microscale thermophoresis (MST) and XRE reporter assays. For the pAHR protein complex, the dissociation constant (KD) values of Tapinarof, BaP, BNF, Indigo, and Indirubin were 292.0 nM, 444.7 nM, 1.6 μM, 1.3 μM, and 46.6 nM, respectively (Fig. 4c). For hAHR, the corresponding KD values of Tapinarof, BaP, BNF, Indigo, and Indirubin were 587.0 nM, 367.0 nM, 2.2 μM, 1.8 μM, and 294.9 nM, respectively (Fig. 4d). It is worth noting that these KD values should be viewed as relative indications of affinity, as we could not rule out the possibility that AHR proteins used in the assays were preoccupied by certain endogenous ligands from E. coli. We were unable to obtain a KD for FICZ due to its intrinsic fluorescence interfering with the measurement signals. Nevertheless, Indirubin demonstrated the strongest binding to AHR among these ligands, consistent with previous reports45,46.
Next, the dose-responsive activation of AHR by the six agonists was determined in Hep3B and HaCaT cells using XRE reporter assays (Fig. 4e, f). It is noteworthy that the calculated EC50 values were not found to be along the same orderings for the two cell lines. Tapinarof and Indirubin exhibited relatively high activities in both cells (Fig. 4e, f), roughly agreeing with their higher binding affinities (Fig. 4c, d). These findings underscore the complex regulatory mechanisms within the AHR pathway across different cell types, reflecting variations in ligand-binding affinities and subsequent cellular activities.
Dynamic AHR conformational shifts: from cytosol to nucleus
We then investigated the structural alterations occurring in AHR as it transitions between its chaperone-bound states (both ligand-free and ligand-bound) to its ligand-bound, chaperone-free heterodimeric complex. Comparisons among these three states initially revealed that the DE-loop, situated between Dα and Eα of the AHR PAS-B domain, undergoes a conformational change towards the pocket upon ligand binding29,30 (Fig. 5a). Furthermore, we observed distinct orientations of the C-ter loop of the AHR PAS-B domain among the three states (Fig. 5a), with its position closest to the AHR PAS-B in the context of the heterodimeric AHR-ARNT complex (Fig. 2a).

a Superimposition of the AHR PAS-B domain structures from heterodimeric pAHR in complex with Indirubin (light blue), chaperoned hAHR with Indirubin (marine blue), and chaperoned mAHR with no ligand (cyan) in two views. b Superimposition of the AHR PAS-B domain structures from heterodimeric pAHR with Indirubin (light blue) and chaperoned mAHR with no ligand (cyan). Residues D327/D323 in Fα, F349/F345 and V348/V344 in Gβ, and R396/R392 in Jα are shown as sticks. c XRE reporter assay evaluating the effects of hAHR R398E mutation (corresponding to R396 in pAHR) and hAHR D329K mutation (corresponding to D327 in pAHR) on transactivation (n = 3 biological replicates). d Representative confocal fluorescence microscopy images of cells expressing wild-type (WT) or R398E mutant of hAHR-GFP proteins in the presence of 50 nM Tapinarof (left) and calculated ratio of cytoplasmic: nuclear fluorescence ratio (right). Scale bar = 50 μm. This experiment was performed three times with similar results (n = 20 biological replicates). For the panels c, d: error bars, mean ± s.d.; statistical significance, **** p < 0.0001 (p-values shown on the charts calculated using unpaired two-tailed t test versus the WT group).
Upon closer examination, we found that when compared with the ligand-free cytosolic mAHR, ligand binding induces the outward movement of residues V348 and F349 on the Gβ strand of the pAHR PAS-B domain (Fig. 5b and Supplementary Movie 3). This movement facilitates the formation of a hydrogen bond between V348 and R396 on the Jα helix. In addition, R396 forms a salt bridge with D327 on the Fα helix, likely contributing to the stabilization of both the Jα helix and the C-ter loop, wherein the latter is critical for PAS-B interactions between AHR and ARNT (Fig. 5b). Similar residue interactions were observed in the ligand-bound hAHR in complex with chaperones, involving its corresponding residues (Supplementary Fig. 9a).
We then expanded our structural comparison to the dAHR, which is known to be constitutively active in a ligand-independent manner47. Intriguingly, in both the ligand-free and α-naphthoflavone (ANF)-bound states of dAHR, a similar interaction network is formed by conserved residues corresponding to those in agonist-bound pAHR or hAHR (Fig. 5b and Supplementary Fig. 9b), consistent with the constitutive activity of dAHR. Therefore, we hypothesize that this conserved network of residue interactions may mediate the allosteric regulatory effects of agonists on the conformation of the AHR C-ter loop following the Jα helix of the PAS-B domain (Supplementary Fig. 6), which could be a crucial step in AHR activation.
To test the above proposed mechanism, we mutated the fully conserved aspartate and arginine (the key interacting residues) in hAHR and measured the activities using XRE reporter and qPCR assays (Fig. 5c and Supplementary Fig. 9c). As expected, the D329K and R398E mutants exhibited clearly decreased transcriptional activities, implying that breaking the integrity of this interaction network compromises the activation of AHR. Given the complexity of AHR’s activation mechanism, especially the multiple steps from the cytosol to the nucleus that would rely on many precise intra- and inter-protein interactions, we also wondered if this network of residues would influence AHR translocation. We made a mutation of R398E on full-length hAHR fused with a GFP at the C-terminus and observed the cellular localization of AHR proteins under a confocal microscope (Fig. 5d). Compared with the wild-type AHR, this mutant showed more cytoplasmic distribution when cells were treated by Tapinarof. These above results suggest that the presence of proper conformational changes of this key arginine and likely the following C-ter loop is critical for AHR activation, including its nuclear translocation.
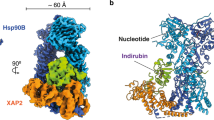
Finally, we aimed to understand how the C-ter loop of the AHR PAS-B domain influences inter-protein interactions through conformational changes. Previous studies have indicated that in the cytosolic AHR-HSP90-XAP2 complex29, this loop primarily interacts with XAP2 (Supplementary Fig. 9d). To visualize the potential movement of the AHR PAS-B C-ter loop during its transition into the AHR-ARNT heterodimer, we superimposed the PAS-B structures of AHR from both heterodimeric and chaperoned complexes (Fig. 6a). Interestingly, we observed that the spatial location of the ARNT PAS-B domain does not directly conflict with HSP90 or XAP2, suggesting the possible existence of a transitional state where ARNT can directly engage with AHR while it is still bound to HSP90 and XAP248. In this process, the C-ter loop of the AHR PAS-B domain may undergo a dramatic conformational change to insert into the dimer interface between the PAS-B domains of AHR and ARNT (Fig. 6a and Supplementary Movie 4), leading to reduced interactions between AHR and XAP2 to facilitate their dissociation. To investigate this further, we co-expressed AHR, ARNT, and XAP2 proteins in HEK293 cells and performed Co-IP to determine whether AHR can form stable complexes with ARNT and XAP2 simultaneously. The results from samples precipitated by ARNT revealed an absence of XAP2 in the ARNT-associated protein complexes regardless of Tapinarof treatment (Fig. 6b). Additional Co-IP results from samples precipitated by AHR showed that the presence of ARNT and/or Tapinarof could weaken the interaction between AHR and XAP2 (Supplementary Fig. 9e). These above findings suggest that upon AHR’s nuclear translocation triggered by ligand binding, ARNT binding may ultimately displace XAP2 from AHR.

a Structural superimposition of AHR PAS-B domains in the heterodimeric and cytosolic/chaperoned complexes. No steric hindrance apparently exists between the ARNT PAS-B domain and HSP90 or XAP2. b Co-IP experiments showing interactions between XAP2 and the AHR-ARNT heterodimer in the presence or absence of 1 μM Tapinarof. This experiment was performed three times with similar results. c Multi-step mechanism of AHR transformation. In the cytosol, ligand binding to AHR may weaken the interactions between the AHR PAS-B C-ter loop and XAP2. In the nucleus, ARNT binding to AHR (likely via their bHLH and PAS-A domains at the beginning) may result in the formation of a transient HSP90-XAP2-P23-AHR-ARNT complex. P23 and XAP2 may subsequently leave this complex, as the interactions between PAS-B domains of AHR and ARNT eventually lead to the complete formation of the heterodimer. Two HSP90 proteins are colored in blue; two P23 proteins are colored in yellow; XAP2, AHR, and ARNT are colored in brown, magenta, and green, respectively.
Discussion
The bHLH-PAS proteins represent a distinctive family of transcription factors with shared structural and functional characteristics (Supplementary Fig. 3a). These proteins play crucial roles in monitoring and responding to various environmental and physiological signals, such as day-night cycles (CLOCK), chemical pollutants (AHR), and fluctuations in oxygen levels (HIFs). Despite these commonalities, the regulatory mechanisms governing their response to these signals exhibit notable diversity. For instance, the HIF pathway primarily relies on oxygen-dependent hydroxylation of HIF-α subunits for regulation49. At the same time, small-molecule binding to the PAS-B domain of HIF-α subunits can trigger a distinct allosteric mechanism that modulates the transcriptional outputs of HIF-α-ARNT heterodimers in a bi-directional manner50,51. The PAS domains across this family are likely to act as ligand-sensing sites, as best evidenced by previously established AHR endogenous ligands such as FICZ, and a recently identified metabolite ligand for HIF-3α, oleoylethanolamide34.
Among the members of the bHLH-PAS family, AHR stands out due to its ligand-dependent activation, and its extensive repertoire of both endogenous and synthetic ligands. This wide range of ligands has allowed researchers to investigate the regulatory mechanisms governing ligand binding and transcriptional modulation within the family. However, for many years, studies on ligand actions have been primarily conducted at the cellular level, lacking the biochemical capacity to produce enough functional AHR proteins and/or complexes for high-affinity binding to ligands. Consequently, there has been a long-awaited need for biochemical advancements and detailed structural examinations to elucidate how diverse ligands physically bind to the AHR-ARNT complex and modulate the transcriptional function.
For AHR, agonist binding to the PAS-B domain initiates a process of transformation involving translocation from the cytosol to the nucleus, and dimerization with ARNT to allow direct binding to XRE sequences. The current study, relying on structural analyses, including both X-ray diffraction and cryo-EM, revealed a distinct dimerization pattern for AHR-ARNT compared to other members of the bHLH-PAS family. This pattern is characterized by a spatially isolated PAS-B dimeric unit (Fig. 1 and Supplementary Movie 1), explaining why certain ARNT point mutations previously shown to disrupt HIF-α-ARNT heterodimers could not similarly dissociate AHR-ARNT heterodimers32. It is noteworthy that AHR exhibits the least conserved protein sequence within the phylogenetic tree of mammalian bHLH-PAS family members (Supplementary Fig. 3d), consistent with its less stringent consistency for ARNT heterodimerization compared to other members such as HIF-α or NPAS1/3/4 proteins.
The detailed comparisons of six AHR-ARNT-DNA structures, each bound to a different chemical ligand, allowed us to identify a set of eight critical residues that together enable promiscuous ligand binding to AHR’s PAS-B pocket (Fig. 3 and Supplementary Movie 2). These residues are fully conserved across mammalian AHR proteins (Supplementary Fig. 6), with seven of them also conserved in vertebrates, except for pAHR S363, which corresponds to an alanine in chicken, frog, and fish. Collectively, these conserved residues create a formidable but adaptable binding space, wherein planar AHR ligands can be accommodated along two principal axes. Moreover, the special shape and chemical nature of different ligands, along with certain additional pocket residues not conserved across species, would allow diverse protein-ligand interactions (with possibly varied binding affinities), resulting in the ligand-specific or species-specific effects on AHR activation. It is also noteworthy that a large number of known ligands, especially those lacking of the hydrophobic and polycyclic properties, may not directly bind to AHR. They may rather function as “pro-ligands” through further conversion to “real” AHR ligands with much higher binding potencies, as exemplified by kynurenine and its trace derivatives52.
The studies conducted here further point to an allosteric mechanism that links ligand binding to the conformational state of the C-ter loop following AHR’s PAS-B domain. While vertebrate AHR activation relies on ligand binding, AHRs from invertebrates exhibit constitutive activity even in the absence of ligands. Intriguingly, a prior investigation employing chimeric AHR proteins from both mouse and drosophila demonstrated that the middle region of the AHR PAS-B domain governs ligand responsiveness in transactivation47 (Supplementary Fig. 9f). This region spans from Dα to Hβ, encompassing the DE-loop involved in ligand entry, as well as the Fα and Gβ participating in the conserved network of residue interactions (D327, V348, F349 and R396 in pAHR). Our studies found that this network may mediate allosteric effects, whereby ligand binding is transmitted to the Jα helix and the subsequent C-ter loop of the PAS-B domain, to configure the activation state of AHR (Fig. 5 and Supplementary Movie 3). Interestingly, this C-ter loop also locates within a previously identified “repressor” region of ~ 200 residues that could be “depressed” by the agonist-induced receptor transformation16. In addition, the C-ter loop undergoes significant conformational changes during AHR transformation and directly interacts with XAP2 or ARNT successively (Fig. 6a and Supplementary Movie 4). Therefore, in nucleus a transitional state may take place wherein ARNT binding to the AHR-HSP90-XAP2 complex is followed by displacement of XAP2, facilitated by the conformational changes of the C-ter loop (Fig. 6c). This specific loop segment of AHR, whose sequence is highly conserved in vertebrate AHRs (Supplementary Fig. 6) but not among bHLH-PAS proteins (Supplementary Fig. 3c), likely plays a key role in mediating the ligand-driven activation of AHR.
As our understanding of the structure-function relationships within the bHLH-PAS family continues to expand, it is increasingly likely to see further advances toward clinical candidates, echoing the development of drugs for the nuclear receptors16,53,54,55,56. With insights gained from detailed structural examinations and biochemical studies, one can also leverage chemical tools to further probe the functions and physiological pathways governed by these proteins. While transcription factors are often considered difficult to drug57, our understanding of ligand-binding and modulation of AHR, together with other bHLH-PAS family members, via agonists, antagonists, and allosteric modulators directed at their PAS domains, creates concrete strategies for modulating gene expression programs associated with physiological processes and disease pathways.
Methods
Chemicals
The compounds used in this study were all commercially available: Tapinarof (Bidepharmatech Ltd., BD01373851), 6-formylindolo[3,2-b]carbazole (FICZ, Sigma-Aldrich, SML1489), benzo[a]pyrene (BaP, Sigma-Aldrich, B1760), β-naphthoflavone (BNF, Sigma-Aldrich, N3633), Indigo (Sigma-Aldrich, 229296), Indirubin (Bidepharmatech Ltd., BD8483), 2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD, Dow Chemical Co. Ltd.), 3-Methylcholanthrene (3-MC, Sigma-Aldrich, 442388), Indole-3-lactic acid (ILA, Bidepharmatech Ltd., BD13033), Indolo[3,2-b]carbazole (ICZ, Bidepharmatech Ltd., BD182549).
Plasmid construction and site-directed mutagenesis
For protein overexpression in E. coli, the human ARNT (Uniprot P27540, residues 85–465, with the loop region 274-298 truncated) was cloned into the pMKH vector as previously described50. And this human ARNT was also cloned into the pMKH with a GFP-tag at its C-terminus. Meanwhile, the porcine AHR (Uniprot I3LF82, residues 26–414) and human AHR (Uniprot P35869, residues 28–414) were cloned into the pSJ2 vector with a C-terminal His-tag. For cell-based experiments, the full-length human AHR (residues 1–848) and its mutants were cloned into the pCMV-Tag4 vector, with a Myc-tag or GFP-tag at the C-terminus. Simultaneously, the full-length human ARNT (residues 1–789) was cloned into the pKH3 vector, with a Flag-tagged at the C-terminus. And the human XAP2 (Uniprot O00170, 1-330) were cloned into the pcDNA3.1 vector, with a HA-tag at the C-terminus. Site-directed mutagenesis was performed as previously34, and confirmed by DNA sequencing.
Protein expression and purification
To obtain heterodimeric AHR-ARNT proteins, the recombinant plasmids pSJ2-AHR and pMKH-ARNT were co-transformed into BL21-CodonPlus (DE3)-RIL competent cells (Agilent Technologies). The procedures of protein expression and purification followed a previously described protocol34. The protein complexes were expressed overnight at 16 °C in LB medium, and purified with a three-step chromatography protocol using Ni Bestarose FF (Bestchrom), SP Sepharose (Cytiva), and a Superdex 200 pg gel-filtration column (Cytiva). To obtain ligand-bound AHR-ARNT complexes, each of the small-molecule ligands were added into the medium up to 20 μM. To prepare AHR-ARNT-DNA complexes, a 21-mer double-strand DNA fragment (forward: 5’-CATCGGGCATCGCGTGACAAG-3’ and reverse: 5’-GCTTGTCACGCGATGCCCGAT-3’) was mixed with protein at a molar ratio of 1.2:1. The mixture was then purified using gel-filtration column in the running buffer containing 20 mM Tris (pH 8.0) and 150 mM NaCl. The pooled protein-DNA peak fractions were supplemented with 5 mM DTT. The heterodimeric proteins of AHR-ARNT-GFP used in the binding assay were prepared in a similar way as described above, with the replacement of pMKH-ARNT plasmid by pMKH-ARNT-GFP. No ligands were added into the medium during the expression of AHR-ARNT-GFP complexes.
Crystallization and X-ray data collection
The co-crystals of AHR-ARNT-DNA in complex with ligands were obtained by mixing equal volumes of protein (4 mg/ml) and the reservoir containing 200 mM potassium citrate tribasic monohydrate and 10% PEG3350 using the sitting-drop vapor diffusion method at 16 °C. 25% ethylene glycol was added into the reservoir solution to protect the crystals before flash frozen. Diffraction data were collected at 100 K using beamlines BL18U1, BL19U1, or BL02U1 at the Shanghai Synchrotron Radiation Facility (SSRF)58. The collected data were processed using either the HKL3000 program59 or XDS60.
Structure determination and refinement
The crystal structure of AHR-ARNT-DNA in complex with Tapinarof was determined by molecular replacement using Phaser61, employing the HIF-2α-ARNT-DNA structure (PDB: 4ZPK) as the initial search model31. Further manual model building and refinement were performed with Coot62 and Phenix.refine63. To determine the structures of other AHR-ARNT-DNA complexes with different ligands, molecular replacement was performed using the Tapinarof-bound complex structure as the initial model, followed by refinement using a similar approach. The statistics of diffraction data and final refinement are summarized in Supplementary Table 1. The Ramachandran statistics, calculated by Molprobity64, are 96.6%/0.17%, 97.6%/0.17%, 96.6%/0.17%, 96.8%/0.17%, 97.6%/0.17% and 97.5%/0.34% (favored/outliers) for the AHR-ARNT-DNA structures in complex with Tapinarof, BaP, FICZ, BNF, Indigo and Indirubin, respectively. All the structural figures were prepared using PyMOL (Schrödinger).
Dual-luciferase XRE reporter assay
Hep3B (Beijing Dingguo, CS0172), HaCaT (Procell, CL-0090), HT1080, and HT1080 AHR-KO (kindly gifted by Prof. Bo Chu of Shandong University) cells were cultured in 48-well plates using DMEM medium with 10% FBS at 37 °C in 5% CO2. When the cell density reached 70–80%, 0.2 μg pGL4.43 (Promega) and 1 ng pRL-CMV plasmids were transfected into cells using the jetPRIME reagent (Polyplus Transfection). To detect the effect of point mutation on AHR activity, HEK293 (Procell CL-0001) cells were also cultured in DMEM medium with 10% FBS in 48-well plates at 37 °C in 5% CO2. When the cell density reached 70–80%, 60 ng pCMV-Tag4-AHR-Myc or its mutants, 60 ng pGL4.43, and 0.5 ng pRL-CMV plasmids were transfected using the jetPRIME reagent. 4 h after transfection, the medium was refreshed either with or without different compounds. Another 24 h later, cells were lysed and analyzed using Dual Luciferase Reporter Gene Assay Kit (Beyotime). Final data were normalized by the relative ratio of firefly and Renilla luciferase activity.
Real-time quantitative PCR (qPCR)
HEK293 cells were cultured in DMEM medium with 10% FBS in 12-well plates at 37 °C in 5% CO2. When the cell density reached 70–80%, 0.4 μg pCMV-Tag4-AHR-Myc or its mutants, were transfected into cells using the jetPRIME reagent. After 4 h transfection, the medium was refreshed either with or without different compounds, and the cells were cultured for another 24 h. Then the cells were harvested, and RNA was isolated using RNAiso Plus kits (TaKaRa), followed by cDNA synthesis using PrimeScript RT reagent kits (TaKaRa). Real-time qPCR was performed on QuantStudio 3 (Thermo Fisher Scientific) using the SYBR Green Master Mix (Yeasen). The expression of CYP1A1 were normalized to the expression of β-actin (ACTB) in the same sample. PCR primers were as follows: ACTB: (F: 5′-GCACAGAGCCTCGCCTT-3′, R: 5′-GTTGTCGACGACGAGCG-3′); CYP1A1: (F: 5′-TCGGCCACGGAGTTTCTTC-3′, R: 5′-GGTCAGCATGTGCCCAATCA-3′).
Co-immunoprecipitation (Co-IP)
HEK293 cells were cultured in DMEM medium with 10% FBS in 6-well plates at 37 °C in 5% CO2. When the cell density reached 70–80%, 0.5 μg pCMV-Tag4-AHR-Myc or its mutants, and 0.5 μg pKH3-ARNT-Flag were transfected into cells using the jetPRIME reagent. After 4 h transfection, the medium was refreshed with 2 μM BNF, and the cells were cultured for another 24 h. Subsequently, the cells were harvested and immunoprecipitation was performed similarly to our previous work32. After the protein concentration measurement for each sample, 40 mg of supernatant was saved as input for western blot using an anti-Flag polyclonal antibody (Proteintech, 20543-1-AP), anti-Myc polyclonal antibody (Sangon Biotech, D155014), and Beta Actin Monoclonal Antibody (Proteintech, 66009-1-Ig). Immunoprecipitation was performed with the supernatant and 40 μl of anti-Flag affinity gel suspension (Beyotime, P2271) according to the manufacturer’s instructions, followed by western blot using the anti-Flag and anti-Myc antibodies. In order to investigate whether ARNT could form stable complexes with XAP2, 0.3 μg pCMV-Tag4-AHR-Myc, 0.3 μg pKH3-ARNT-Flag and 0.3 μg pcDNA3.1-XAP2-HA were transfected into HEK293 cells. The steps for immunoprecipitation were the same as described above. AHR, ARNT, and XAP2 proteins were detected by anti-Flag, anti-Myc, and anti-HA (Proteintech, 51064-2-AP) antibodies. The secondary antibodies were HRP-conjugated Goat Anti-Rabbit IgG (Sangon Biotech, D110058) and HRP-conjugated Goat Anti-Mouse IgG (Sangon Biotech, D110087). The primary antibodies and the secondary antibodies were used as 1:6000 dilutions.
Confocal microscopy
HEK293 cells were cultured in DMEM medium with 10% FBS in 12-well plates with coverslips at 37 °C in 5% CO2. When the cell density reached 70–80%, 0.4 μg pCMV-Tag4-AHR-GFP or its mutant pCMV-Tag4-AHR (R398E)-GFP, was transfected into cells using the jetPRIME reagent. After 12 h transfection, the medium was refreshed with 50 nM Tapinarof, and the cells were cultured for another 12 h. Primary cultures grown on coverslips were washed with PBS and then were fixed in 4% paraformaldehyde for 15 min. After fixing, coverslips were washed with PBS gently. Coverslips were then incubated in 0.1% Triton X-100 for 20 min, and then the coverslips were washed with PBS. After washing, coverslips were incubated with PBST containing 1% Hoechst (Solarbio, C0031). After incubating, coverslips were washed with PBST three times. The coverslips were placed upside down on the slides and observed using the confocal microscope (Zeiss LSM900) with a 63 × oil-immersion objective. The excitation/emission wavelengths for AHR-GFP and Hoechst were 488/509 nm and 353/465 nm, respectively. Fluorescence intensities were quantified using the ImageJ 1.53 software (National Institutes of Health).
Microscale thermophoresis (MST) binding assay
Compounds were diluted to various concentrations and mixed with AHR-ARNT-GFP (20 nM) protein complexes at room temperature (about 25 °C) in the assay buffer containing 20 mM Hepes (pH 7.5), 400 mM NaCl, 5 mM DTT, 0.05% Tween-20 and 1% DMSO. The mixed samples were loaded into Monolith standard-treated capillaries, and the thermophoresis was performed using a Monolith NT.115 instrument (NanoTemper Technologies). Binding was measured with 80% LED power and “medium” MST power. The data were analyzed using MO Control software (NanoTemper Technologies) to determine the KD values.
Molecular dynamics (MD) simulations
The initial structures used in the simulation were from the AHR-ARNT-DNA crystal structures in complex with Tapinarof, FICZ, BaP, BNF, Indigo, and Indirubin. We only employed the two PAS-B domains of AHR (residues 271–413) and ARNT (residues 360–464) when calculating the binding energy. For every system, the protonation state of neutral HIS residues was automatically determined by the pdb2gmx tool of GROMCAS65. Water molecules in the structure were removed. Possible missing atoms or residues in the structure are detected and automatically corrected (if present) by pdbfixer (https://github.com/openmm/pdbfixer). We used the amber-ff14SB force field66 for protein and the TIP3P model67 for water molecules. The force field parameters of small molecules came from gaff268, with an am1-bcc charge model69,70. The AmberTools71 was called to automatically generate force field parameters for small molecules. The simulation box was cubic, with a 10 Å boundary. 0.1 M NaCl buffer was added to ensure the system is electrically neutral. All simulations were performed using OpenMM72 (version 7.7). Each system was minimized for 1000 steps followed by a 500 ps equilibration run. Simulations were executed for 10 ns and conducted at 300 K and 1 atm using a Monte Carlo barostat. MD snapshots were saved every 25 ps after the systems were well equilibrated. The mass of the hydrogen atom and adjacent heavy atoms on the protein was redistributed so that the total mass of the chemical group did not change while the mass of the hydrogen atom increased to 4 atomic units to reduce the frequency of the hydrogen-related angular bending motion to increase the integration step size. The Langevin integrator in OpenMM was chosen with an integration step size of 2.5 fs. The MD simulation parameter files are available in Supplementary Data 1.
The binding free energies were calculated using the MM/GBSA method:
where \({\Delta E}_{{MM}}\) is the gas-phase interaction energy between protein and ligand, including the electrostatic and the van der Waals interaction energies; \({\Delta G}_{{GB}}\) is the electrostatic or polar contribution to the free energy of solvation, and the term \({\Delta G}_{{nonpolar}}\) is the non-polar or hydrophobic contribution to the solvation free energy; −TΔS is the change of conformational entropy upon ligand binding, which was not considered here since we evaluated the relative binding energies from different ligands and the contribution of entropy change to this was small.
The electrostatic solvation energy (ΔGGB) was calculated by using the GB models, more precisely, the GBn generalized Born model73. The value of the exterior dielectric constant was set to 80, and the solute dielectric constant was set to 2. The non-polar contribution was determined based on solvent-accessible surface area (SASA) with the LCPO method74,
We also assess the contributions from individual residues or energy terms by free energy decomposition analyses. For MM/GBSA calculations, molecular mechanics energies and the non-polar contribution to the solvation free energy were computed with gmx-MMPBSA75, which was used to convert Gromacs trajectories into AMBER format with ParmEd (https://github.com/ParmEd/ParmEd), and subsequently the mmpbsa.py76 module of AmberTools was used.
Sample preparation for cryo-EM and data collection
AHR-ARNT protein complexes were incubated with 21-mer double-strand DNA (forward: 5’-CGGGCATCGCGTGACAAGCCC-3’ and reverse: 5’-GGGCTTGTCACGCGATGCCCG-3’) at a molar ratio of 1.5:1. The mixture was then purified using gel-filtration column in the running buffer containing 20 mM Tris (pH 8.0) and 150 mM NaCl. Only the highest concentration fraction from the middle of the peak was used in further experiments. The AHR-ARNT-DNA complexes were diluted to a final concentration of 0.25 mg/mL, and 3 μL of this protein solution was applied to a newly glow discharged holey carbon film-coated gold grid (Au R1.2/1.3, 300 meshes, Quantifoil, Germany). The grids were blotted for 2.0 s at 100% humidity and room temperature, and then vitrified by plunge freezing into liquid ethane. The grids were stored in liquid nitrogen until data collection.
Cryo-EM data were collected on a Titan Krios G3i (Thermo Fisher Scientific) operating at 300 kV equipped with a Gatan K3 (Gatan). Each movie had an accumulated dose of 50 e-/Å2, fractionated into 32 frames in super-resolution mode with a pixel size of 0.669 Å. The defocus range was set between 1.0 and 2.0 μm. A total of 525 micrographs were collected.
Cryo-EM data processing
The data of 525 micrographs were processed in cryoSPARC77. Drift correction and dose-weighting were carried out using Patch Motion Corr. The contrast transfer function (CTF) of motion-corrected micrographs was estimated with Patch Gctf in cryoSPARC. A total of 402,293 particles from 516 micrographs were picked out for 2D classification. Finally, the selected 2D classification in Supplementary Fig. 2b was from 95,290 particles.
Statistical analysis
All statistical data were calculated using GraphPad Prism version 10.0. An unpaired two-tailed t test was used to compare the means of two groups.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data that support the findings of this study are available from the corresponding authors upon request. The refined structural models and corresponding structure-factor amplitudes for the AHR-ARNT-DNA complexes bound with Tapinarof, FICZ, BaP, BNF, Indigo, and Indirubin have been deposited in the PDB database under accession codes 8XS6, 8XS7, 8XS8, 8XS9, 8XSA, and 8XSB, respectively. The accession codes for the previously released structures HIF-2α-ARNT-DNA, NPAS4-ARNT-DNA, mAHR-hARNT-DNA, hAHR-mARNT-DNA, AHR-HSP90-XAP2-Indirubin, AHR-HSP90-XAP2-BaP, AHR-HSP90-XAP2 in ligand-free status, dAHR-ANF, and dAHR-dARNT in ligand-free status are 4ZPK, 7XI4, 5V0L, 5NJ8, 7ZUB, 8QMO, 8H77, 7VNH and 7VNI, respectively. Source data are provided in this paper as a Source Data file.
Code availability
The MD simulation files are available in Supplementary Data 1.
References
Poland, A., Glover, E. & Kende, A. S. Stereospecific, high affinity binding of 2,3,7,8-tetrachlorodibenzo-p-dioxin by hepatic cytosol. Evidence that the binding species is receptor for induction of aryl hydrocarbon hydroxylase. J. Biol. Chem. 251, 4936–4946 (1976).
Esser, C. & Rannug, A. The aryl hydrocarbon receptor in barrier organ physiology, immunology, and toxicology. Pharmacol. Rev. 67, 259–279 (2015).
Rothhammer, V. & Quintana, F. J. The aryl hydrocarbon receptor: an environmental sensor integrating immune responses in health and disease. Nat. Rev. Immunol. 19, 184–197 (2019).
Sladekova, L., Mani, S. & Dvorak, Z. Ligands and agonists of the aryl hydrocarbon receptor AhR: Facts and myths. Biochem. Pharmacol. 213, 115626 (2023).
Stockinger, B., Di Meglio, P., Gialitakis, M. & Duarte, J. H. The aryl hydrocarbon receptor: multitasking in the immune system. Annu. Rev. Immunol. 32, 403–432 (2014).
Murray, I. A., Patterson, A. D. & Perdew, G. H. Aryl hydrocarbon receptor ligands in cancer: friend and foe. Nat. Rev. Cancer 14, 801–814 (2014).
Quintana, F. J. & Sherr, D. H. Aryl hydrocarbon receptor control of adaptive immunity. Pharmacol. Rev. 65, 1148–1161 (2013).
Monteleone, I., MacDonald, T. T., Pallone, F. & Monteleone, G. The aryl hydrocarbon receptor in inflammatory bowel disease: linking the environment to disease pathogenesis. Curr. Opin. Gastroenterol. 28, 310–313 (2012).
Smith, S. H. et al. Tapinarof is a natural AhR agonist that resolves skin inflammation in mice and humans. J Invest. Dermatol. 137, 2110–2119 (2017).
Flegel, J. et al. The highly potent AhR agonist picoberin modulates Hh-dependent osteoblast differentiation. J. Med. Chem. 65, 16268–16289 (2022).
Ondrova, K. et al. Monoterpenoid aryl hydrocarbon receptor allosteric antagonists protect against ultraviolet skin damage in female mice. Nat. Commun. 14, 2728 (2023).
Wilkinson, I. V. L. et al. Chemical proteomics and phenotypic profiling identifies the aryl hydrocarbon receptor as a molecular target of the utrophin modulator ezutromid. Angew. Chem. Int. Ed. Engl. 59, 2420–2428 (2020).
Wu, D. & Rastinejad, F. Structural characterization of mammalian bHLH-PAS transcription factors. Curr. Opin. Struct. Biol. 43, 1–9 (2017).
Ema, M. et al. cDNA cloning and structure of mouse putative Ah receptor. Biochem. Biophys. Res. Commun. 184, 246–253 (1992).
Burbach, K. M., Poland, A. & Bradfield, C. A. Cloning of the Ah-receptor cDNA reveals a distinctive ligand-activated transcription factor. Proc. Natl. Acad. Sci. USA 89, 8185–8189 (1992).
Dolwick, K. M., Swanson, H. I. & Bradfield, C. A. In vitro analysis of Ah receptor domains involved in ligand-activated DNA recognition. Proc. Natl. Acad. Sci. USA 90, 8566–8570 (1993).
Lindebro, M. C., Poellinger, L. & Whitelaw, M. L. Protein-protein interaction via PAS domains: role of the PAS domain in positive and negative regulation of the bHLH/PAS dioxin receptor-Arnt transcription factor complex. EMBO J. 14, 3528–3539 (1995).
Sondermann, N. C. et al. Functions of the aryl hydrocarbon receptor (AHR) beyond the canonical AHR/ARNT signaling pathway. Biochem. Pharmacol. 208, 115371 (2023).
Coumailleau, P., Poellinger, L., Gustafsson, J. A. & Whitelaw, M. L. Definition of a minimal domain of the dioxin receptor that is associated with Hsp90 and maintains wild type ligand binding affinity and specificity. J. Biol. Chem. 270, 25291–25300 (1995).
Fukunaga, B. N., Probst, M. R., Reisz-Porszasz, S. & Hankinson, O. Identification of functional domains of the aryl hydrocarbon receptor. J. Biol. Chem. 270, 29270–29278 (1995).
Carlson, D. B. & Perdew, G. H. A dynamic role for the Ah receptor in cell signaling? Insights from a diverse group of Ah receptor interacting proteins. J. Biochem. Mol. Toxicol. 16, 317–325 (2002).
Kudo, I. et al. The regulation mechanisms of AhR by molecular chaperone complex. J. Biochem. 163, 223–232 (2018).
Perdew, G. H. & Bradfield, C. A. Mapping the 90 kDa heat shock protein binding region of the Ah receptor. Biochem. Mol. Biol. Int. 39, 589–593 (1996).
Bunger, M. K. et al. Abnormal liver development and resistance to 2,3,7,8-tetrachlorodibenzo-p-dioxin toxicity in mice carrying a mutation in the DNA-binding domain of the aryl hydrocarbon receptor. Toxicol. Sci. 106, 83–92 (2008).
Bunger, M. K. et al. Resistance to 2,3,7,8-tetrachlorodibenzo-p-dioxin toxicity and abnormal liver development in mice carrying a mutation in the nuclear localization sequence of the aryl hydrocarbon receptor. J. Biol. Chem. 278, 17767–17774 (2003).
Probst, M. R., Reisz-Porszasz, S., Agbunag, R. V., Ong, M. S. & Hankinson, O. Role of the aryl hydrocarbon receptor nuclear translocator protein in aryl hydrocarbon (dioxin) receptor action. Mol. Pharmacol. 44, 511–518 (1993).
Matsushita, N., Sogawa, K., Ema, M., Yoshida, A. & Fujii-Kuriyama, Y. A factor binding to the xenobiotic responsive element (XRE) of P-4501A1 gene consists of at least two helix-loop-helix proteins, Ah receptor and Arnt. J. Biol. Chem. 268, 21002–21006 (1993).
Ikuta, T., Eguchi, H., Tachibana, T., Yoneda, Y. & Kawajiri, K. Nuclear localization and export signals of the human aryl hydrocarbon receptor. J. Biol. Chem. 273, 2895–2904 (1998).
Gruszczyk, J. et al. Cryo-EM structure of the agonist-bound Hsp90-XAP2-AHR cytosolic complex. Nat. Commun. 13, 7010 (2022).
Wen, Z. et al. Cryo-EM structure of the cytosolic AhR complex. Structure 31, 295–308 (2023).
Wu, D., Potluri, N., Lu, J., Kim, Y. & Rastinejad, F. Structural integration in hypoxia-inducible factors. Nature 524, 303–308 (2015).
Wu, D., Su, X., Potluri, N., Kim, Y. & Rastinejad, F. NPAS1-ARNT and NPAS3-ARNT crystal structures implicate the bHLH-PAS family as multi-ligand binding transcription factors. Elife 5, https://doi.org/10.7554/eLife.18790 (2016).
Sun, X. et al. Structures of NPAS4-ARNT and NPAS4-ARNT2 heterodimers reveal new dimerization modalities in the bHLH-PAS transcription factor family. Proc. Natl. Acad. Sci. USA 119, e2208804119 (2022).
Diao, X. et al. Identification of oleoylethanolamide as an endogenous ligand for HIF-3alpha. Nat. Commun. 13, 2529 (2022).
Seok, S. H. et al. Structural hierarchy controlling dimerization and target DNA recognition in the AHR transcriptional complex. Proc. Natl. Acad. Sci. USA 114, 5431–5436 (2017).
Schulte, K. W., Green, E., Wilz, A., Platten, M. & Daumke, O. Structural Basis for Aryl Hydrocarbon Receptor-Mediated Gene Activation. Structure 25, 1025–1033 (2017).
Dai, S. et al. Structural insight into the ligand binding mechanism of aryl hydrocarbon receptor. Nat. Commun. 13, 6234 (2022).
Cui, W. et al. Gut microbial metabolite facilitates colorectal cancer development via ferroptosis inhibition. Nat. Cell Biol. 26, 124–137 (2024).
Soshilov, A. A. & Denison, M. S. Ligand promiscuity of aryl hydrocarbon receptor agonists and antagonists revealed by site-directed mutagenesis. Mol. Cell Biol. 34, 1707–1719 (2014).
Kwong, H. S. et al. Structural insights into the activation of human aryl hydrocarbon receptor by the environmental contaminant Benzo[a]pyrene and structurally related compounds. J. Mol. Biol. 436, 168411 (2023).
Tian, W., Chen, C., Lei, X., Zhao, J. & Liang, J. CASTp 3.0: computed atlas of surface topography of proteins. Nucleic Acids Res. 46, W363–W367 (2018).
Poland, A., Palen, D. & Glover, E. Analysis of the four alleles of the murine aryl hydrocarbon receptor. Mol. Pharmacol. 46, 915–921 (1994).
Pandini, A., Denison, M. S., Song, Y., Soshilov, A. A. & Bonati, L. Structural and functional characterization of the aryl hydrocarbon receptor ligand binding domain by homology modeling and mutational analysis. Biochemistry 46, 696–708 (2007).
Motto, I., Bordogna, A., Soshilov, A. A., Denison, M. S. & Bonati, L. New aryl hydrocarbon receptor homology model targeted to improve docking reliability. J. Chem. Inf. Model 51, 2868–2881 (2011).
Adachi, J. et al. Indirubin and indigo are potent aryl hydrocarbon receptor ligands present in human urine. J. Biol. Chem. 276, 31475–31478 (2001).
Faber, S. C., Soshilov, A. A., Giani Tagliabue, S., Bonati, L. & Denison, M. S. Comparative In Vitro and In Silico Analysis of the Selectivity of Indirubin as a Human Ah Receptor Agonist. Int. J. Mol. Sci. 19, https://doi.org/10.3390/ijms19092692 (2018).
Kudo, K., Takeuchi, T., Murakami, Y., Ebina, M. & Kikuchi, H. Characterization of the region of the aryl hydrocarbon receptor required for ligand dependency of transactivation using chimeric receptor between Drosophila and Mus musculus. Biochim. Biophys. Acta 1789, 477–486 (2009).
Soshilov, A. A., Motta, S., Bonati, L. & Denison, M. S. Transitional states in ligand-dependent transformation of the aryl hydrocarbon receptor into its DNA-binding form. Int. J. Mol. Sci. 21, https://doi.org/10.3390/ijms21072474 (2020).
Ivan, M. et al. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 292, 464–468 (2001).
Wu, D. et al. Bidirectional modulation of HIF-2 activity through chemical ligands. Nat. Chem. Biol. 15, 367–376 (2019).
Zhuang, J., Shang, Q., Rastinejad, F. & Wu, D. Decoding allosteric control in hypoxia-inducible factors. J. Mol. Biol. 436, 168352 (2024).
Seok, S. H. et al. Trace derivatives of kynurenine potently activate the aryl hydrocarbon receptor (AHR). J. Biol. Chem. 293, 1994–2005 (2018).
Khorasanizadeh, S. & Rastinejad, F. Visualizing the architectures and interactions of nuclear receptors. Endocrinology 157, 4212–4221 (2016).
Huang, P., Chandra, V. & Rastinejad, F. Structural overview of the nuclear receptor superfamily: insights into physiology and therapeutics. Annu. Rev. Physiol. 72, 247–272 (2010).
Rastinejad, F. Retinoic acid receptor structures: the journey from single domains to full-length complex. J. Mol. Endocrinol. 69, T25–T36 (2022).
Rastinejad, F. The protein architecture and allosteric landscape of HNF4alpha. Front. Endocrinol. 14, 1219092 (2023).
Zhuang, J. J., Liu, Q., Wu, D. L. & Tie, L. Current strategies and progress for targeting the “undruggable” transcription factors. Acta Pharmacol. Sin. 43, 2474–2481 (2022).
Sanchez-Weatherby, J. et al. VMXi: a fully automated, fully remote, high-flux in situ macromolecular crystallography beamline. J. Synchrotron Radiat. 26, 291–301 (2019).
Minor, W., Cymborowski, M., Otwinowski, Z. & Chruszcz, M. HKL-3000: the integration of data reduction and structure solution-from diffraction images to an initial model in minutes. Acta Crystallogr. D Biol. Crystallogr. 62, 859–866 (2006).
Kabsch, W. Xds. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 (2010).
McCoy, A. J. et al. Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 (2007).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010).
Chen, V. B. et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 (2010).
Mark James Abraham, T. M., Roland Schulz, Szil´ard P´all, Jeremy C. Smith, Berk Hess, Erik Lindahl. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1, 19–25 (2015).
Maier, J. A. et al. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 11, 3696–3713 (2015).
Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W. & Klein, M. L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 (1983).
Wang, J., Wolf, R. M., Caldwell, J. W., Kollman, P. A. & Case, D. A. Development and testing of a general amber force field. J. Comput. Chem. 25, 1157–1174 (2004).
Jakalian, A., Bush, B. L., Jack, D. B. & Bayly, C. I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: I. Method. J. Comput. Chem. 21, 132–146 (2000).
Jakalian, A., Jack, D. B. & Bayly, C. I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 23, 1623–1641 (2002).
Case, D. A. et al. AmberTools. J. Chem. Inf. Model. 63, 6183–6191 (2023).
Eastman, P. et al. OpenMM 7: Rapid development of high performance algorithms for molecular dynamics. PLoS Comput. Biol. 13, e1005659 (2017).
Mongan, J., Simmerling, C., McCammon, J. A., Case, D. A. & Onufriev, A. Generalized Born model with a simple, robust molecular volume correction. J. Chem. Theory Comput. 3, 156–169 (2007).
Weiser, J., Shenkin, P. S. & Still, W. C. Optimization of Gaussian surface calculations and extension to solvent-accessible surface areas. J. Comput. Chem. 20, 688–703 (1999).
Valdes-Tresanco, M. S., Valdes-Tresanco, M. E., Valiente, P. A. & Moreno, E. gmx_MMPBSA: A new tool to perform end-state free energy calculations with GROMACS. J. Chem. Theory Comput. 17, 6281–6291 (2021).
Miller, B. R. et al. MMPBSA.py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 8, 3314–3321 (2012).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Laskowski, R. A. & Swindells, M. B. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model 51, 2778–2786 (2011).
Acknowledgements
We thank Prof. Bo Chu from Cheeloo College of Medicine of Shandong University for providing the HT1080 and HT1080 AHR-KO cell lines, and Xiaoju Li, Zhifeng Li, Jing Zhu, Haiyan Yu, Xiaomin Zhao, Yuyu Guo and Sen Wang from Core Facilities for Life and Environmental Sciences of Shandong University for their assistance in experiments of X-ray diffraction, MST and laser scanning confocal microscopy imaging. We also thank the staff of BL19U1, BL18U1, and BL02U1 beamlines of Shanghai Synchrotron Radiation Facility for their assistance during diffraction data collection, as well as the Shuimu BioSciences for cryo-EM facility access and technical support during image acquisition. This work was supported by grants from the National Key R&D Program of China (2022YFA1304103), the National Natural Science Foundation of China (82373876, 22177063), Fundamental Research Funds of Shandong University (2023QNTD001), University Innovation Group Program of Jinan (202228080), Shandong Provincial Natural Science Foundation (ZR2021JQ30, ZR2022LSW019), Taishan Scholars Program of Shandong Province (tsqn201909004), and SKLMT Frontiers and Challenges Project (SKLMTFCP-2023-01) to D.W.; and grants from National Natural Science Foundation of China (32300089), China Postdoctoral Science Foundation (2023M732072), Shandong Provincial Natural Science Foundation (ZR2023QC183), and Qingdao Natural Science Foundation (23-2−1-29-zyyd-jch) to X.D.
Author information
Authors and Affiliations
Contributions
X.D. and Q.S. conducted experiments including clone construction, protein purification, crystallization, X-ray diffraction, structure refinement, binding assays, and cell-based experiments; M.G., M.Z., Y.L., and J.Z. participated in the cell-based experiments; Y.H. and X.C. participated in the protein purification and crystallization; X.S. participated in the clone construction; F.Z. performed the MD simulations; S.L., C.V., and F.R. provided critical experimental materials and revised the manuscript; D.W. conceived the study, supervised experiments, and wrote the paper with inputs from all authors.
Corresponding author
Ethics declarations
Competing interests
F.R. is a founder and consultant for Flare Therapeutics. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Chris Bradfield, Yun Zhu, and the other anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Diao, X., Shang, Q., Guo, M. et al. Structural basis for the ligand-dependent activation of heterodimeric AHR-ARNT complex. Nat Commun 16, 1282 (2025). https://doi.org/10.1038/s41467-025-56574-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-56574-7
This article is cited by
-
Computational discovery of novel aryl hydrocarbon receptor modulators for psoriasis therapy
Scientific Reports (2025)
