
Abstract
Labelled compounds bearing hydrogen isotopes are keystones in diverse areas constituting a multi-billion dollar global market including drugs, diagnostics, biology, toxicology and smart materials. While hydrogen deuterium exchange (HDE) methods hold promise as relevant tools for the late-stage and one-step preparation of deuterium-labelled compounds, they often fall short in achieving sufficient isotopic purity combined either with a site-selectivity or with a full deuteration process, highlighting the need for further development and optimisation. This report pinpoints an approach to unlock the potential of HDE using the concept of iterative runs in continuous-flow technology (recirculation process). This closed-loop process grants access now to deuterated compounds with high isotopic purities, labelled at a precise site or perdeuterated on demand, in a fast, productive, and environmentally friendly way.
Similar content being viewed by others

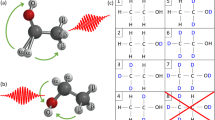
Introduction
The interest for the development of deuterium labelling methods has experienced a strong revival in the last few years1,2,3. This renewed focus on research and development has been driven by the increasing range of applications for deuterated compounds, from life and environmental sciences to the development of materials4. The functionalisation of organic compounds with deuterium is indeed essential to: (1) modify and improve the pharmacokinetic and/or toxicity profile of active pharmaceutical ingredients (API)5,6,7, (2) prepare stable isotope-labelled internal standards (SILS) for MS quantification4,8,9,10, (3) develop probes for imaging technics such as deuterium metabolic or Raman imaging11,12,13, or (4) increase performances and stability of fluorescent14,15 and electroluminescent emitters16,17,18,19,20,21 by limiting photobleaching processes or significantly increasing the lifetime of Organic Light Emitting Devices (OLEDs). In this active field of research, metal-catalysed hydrogen deuterium exchange (HDE) has been extensively explored and has recently provided a plethora of procedures for the late-stage deuteration of complex molecules using affordable liquid (D2O, deuterated organic solvents or acids) or gaseous (D2) isotopic sources1,2,3. Thanks to their numerous assets, they have become valuable alternatives to classical syntheses that rely on the availability of commercially deuterated reagents (so-called deuterated pool) or on the synthesis of deuterated precursors, and could be rather expensive, time-consuming and polluting. However, current HDE methodologies also display several limitations. They notably fall short in terms of isotopic enrichments when deuterated molecules applications require high isotopic purity (>95%) at a precise position, particularly for the preparation of heavy drugs and SILS. Indeed, classically, HDE procedures are performed in batch reactors where both the limited catalyst efficiency and the isotopic enrichment erosion of the deuterated source during the course of the reaction are the two main causes of the limited isotopic purity. Additionally, the high industrial demand for deuterated compounds in OLED manufacturing22 or for deuterated API production highlights the need for large scale deuteration processes in the most time-, cost-efficient and safest way. To circumvent the above-mentioned limitations and answer the needs, the implementation of flow chemistry HDE can be of great interest as flow strategies were shown promising in improving challenging reactions including C-H functionalization ones23. The ability of flow devices to enhance physical transfers thanks to large interfacial areas, is particularly relevant on systems involving ‘gas-liquid-solid’ mixtures by providing fast gas-liquid mixing and high contact surface between the substrate solution and the solid catalyst. In addition, the use of hazardous hydrogen gas in flow reactors is safer than in conventional batch ones especially when using high pressures and temperatures. Despite this potential, continuous-flow methods based on classical strategies such as deuterogenation24,25,26,27,28,29, deuterodehalogenation30 or decarboxylative deuteration31 reactions have been developed, but only two flow-based HDE approaches have been described so far (Fig. 1). Noël et al. published in 2015 a continuous-flow ortho-directed metal-catalysed H/D exchange using a supported iridium(I) complex loaded in a commercial packed-bed reactor (H-Cube®)32. Later Sajiki et al. successfully implemented a continuous-flow H/D exchange method for the direct perdeuteration of various aromatics using heterogeneous Pt catalysts33. Both studies showed that continuous-flow systems could benefit HDE reactions compared to their batch counterparts thanks to a fast liquid-gas mixing and an interesting reusability of the catalyst, leading to improved deuterium incorporations, an increased safety of processes and a possible large-scale industrial production. Nevertheless, the full potential of flow chemistry technology to overcome the current HDE limitations has not yet been fully exploited (Fig. 1). Flow reactors offer the great opportunity to work in a closed loop system and therefore to perform iterative runs of catalytic deuteration in a practical and safe manner. This recirculation strategy allied with a constant renewal of the isotopic source are keystones to unlock the HDE potential, granting access to complex regioselectively deuterated compounds with high isotopic purity in a productive fashion.

IE isotopic enrichment, CB carbon beads.
As a proof of concept, we report here an easy-to-implement continuous-flow HDE strategy, which allows a regioselective control of the isotope incorporation and maximal isotopic enrichments. The continuous-flow system developed uses commercially available ruthenium heterogeneous catalyst (Ru/C) and flow reactors, providing a wide range of labelled aza-compounds from simple alkylamines, formamides and purine building blocks to complex API with isotopic purities greater than 95% along with regioselective deuterium incorporations. In terms of productivity, almost all deuterated compounds were obtained after short reaction times (from 15 min to 4 h) in quantitative yields (on a 0.2 to 0.4 mmol scale) and without any purification step. Depending on the selected temperature of the reaction, the regioselectivity of the deuterium atoms incorporation can be finely tuned to afford, on demand, precise deuteration or perdeuteration. The usefulness of this process has notably been demonstrated by: (1) the one-step synthesis of a perdeuterated polyamine and a complex tetrapeptide fulfilling SILS requirements, (2) the precise deuteration of relevant API, (3) the deuterium labelling of formamides for which no HDE process had been described so far to the best of our knowledge and (4) the neat gram-scale synthesis of d4-azepane (40 mmol, 5.7 g) displaying a very high isotopic purity. During the process of revision, a paper describing a HDE method under flow conditions has been published by Minya et al.34. They demonstrated the superiority of continuous flow over batch deuteration processes. Using a heterogeneous catalyst, namely Raney Ni, they succeeded in labelling useful scaffolds as purine, imidazole and pyridine. However, contrary to our procedure, they used a single path protocol which do not allow them to gain access to site selective labelling and maximised isotopic enrichments, cornerstones in our strategy.
Results and discussion
Continuous-flow setup and procedure
Our continuous-flow setup (Fig. 2) was engineered on either an H-Cube Mini Plus® or an H-Cube Pro® apparatus (designated thereafter as HC) with a packed-bed-reactor filled with commercially available ruthenium particles on charcoal (Ru/C). The HC system displays several benefits for deuteration processes including an interesting design for gas-solid-liquid reactions using hydrogen gas, generated in situ from water electrolysis. Thus, in the same way, heavy water could be electrolysed to produce deuterium gas and used as a cheap isotopic source (D2 gas being 15 times more expensive in €/mol than D2O)2. Commercially available Ru/C was packed in a stainless steel cartridge (30 mm CatCart®) and heavy water was loaded in the cell reservoir for the in-line production of D2 gas. Iteration of runs (recirculation process) was made by simply connecting the inlet to the outlet of the flow line when the system was entirely filled with the substrate solution. In this manner, the system continuously pumped the THF solution of reactant through the reactor, allowing the H/D exchange of the whole mixture over time. Progress of the reaction was monitored by at-line ESI-MS analysis.

MS mass spectrum, HPLC high performance liquid chromatography.
The method described here was developed using commercially available H-Cube® apparatus to ensure: (1) the reliability of the process, thanks to standardised procedures notably for packing cartridges; (2) the safety, as high-pressure reactions can be performed (up to 100 bar) and no direct handling of D2 gas is necessary; (3) the scalability of the protocol, as deuterated molecules can be obtained from mg to g scale. It should be noted that the CatCart® cartridges used in H-Cube® systems mimic HPLC columns as they are stainless-steel tubes with filter systems at each end of the tube, allowing liquid to pass through the column but preventing catalyst from coming out. Therefore, using a home-made system equipped with a HPLC column filled with the catalyst, a D2 gas cylinder and HPLC pumps, can be considered in principle as equivalent to performing deuteration reactions at low pressures using an H-Cube® apparatus. It might be seen as an interesting alternative for laboratories not equipped with H-Cube systems®, which could be relatively expensive. Please note that this alternative pathway has not been used in our study.
Deuteration of model aza-building blocks
Amines are ubiquitous scaffolds in drugs, agrochemicals, metabolites, and materials. For instance, more than 40% of drug candidates contain an aliphatic amine that is almost exclusively a secondary (~35%) or a tertiary amine (~60%)35. In the past two decades, precision deuteration of alkylamines has proved to be a relevant functionalizing tool in drug design for heavy drugs5,6,7 and to enhance both fluorophores photochemical stability and quantum efficiency14,15. For the preparation of deuterated amines, conventional methods rely whether on N-alkylation reactions using the so-called deuterated pool36,37 or on the reductive deuteration of oxime38, imine39 or nitrile40 derivatives. These synthetic approaches can provide very high levels of isotopic purity but may require the preparation of costly precursors obtained through multi-step reactions, even for the preparation of small molecules. For instance, a synthetic procedure for dimethylamine-d6 was recently described involving four steps and the use of two deuterated electrophilic reagents41. Regarding HDE strategies, ruthenium and rhodium catalysts including Ru nanoparticles, were found to be convenient to perform C-H activation on amines42,43,44,45,46,47,48,49. These methods established direct H/D exchange procedures leading to good levels of isotopic enrichment on a wide range of substrates. However, high isotopic purity (>95%) and control of the regioselectivity (precise or per-deuteration) could not be achieved for most substrates, demonstrating that there is still an important gap to bridge.
In this context, our continuous-flow procedure was first applied to the deuteration of model aza-building blocks including linear amines and N-heterocycles. Deuterated dimethylamine 1 was obtained with 96% overall deuteration in 60 min at 100 °C and in quantitative yield (Fig. 3). Herein we describe a straightforward one-step synthesis of dimethylamine-d6 compared to the four-steps synthesis recently published in the literature41. Regarding the labelling of amines with longer alkyl chains, regioselectivity issues could be raised since metallic nanoparticles are prone to perform C-H activation on farther β positions45. In that regard, a quick optimisation study was performed to achieve the selective deuteration of diethylamine 2 using different temperatures (see S.I. page S97). It should be noted that the same cartridge was used for 15 catalytic runs without significant loss of activity. Moreover, these fast-to-implement experiments enabled the determination of suitable ranges of temperature for reaching either complete selectivity towards the α-position of the amine or perdeuteration (Fig. 4). Performed at 30 °C, the labelling of diethylamine afforded its labelled counterpart 2 with an excellent α-selectivity (99% of isotopic enrichment), highlighting the ability of this system to achieve precision deuteration. Alternatively, at 100 °C, perdeuterated diethylamine 2bis was obtained in 2 h. These two results illustrate the unique flexibility of our procedure that can provide, according to the need, site-controlled deuteration, multi-site deuteration or perdeuteration in quantitative yields.

Isolated yields (99% for all substrates) were determined after salification with HCl (to facilitate the work-up). The number of deuterium atoms indicated was determined using ESI-MS analysis. T temperature, P pressure.

The number of deuterium atoms indicated was determined using ESI-MS analysis. [XX] isotopic enrichment (NMR).
Similarly, a moderate temperature (20 °C) enabled the selective deuteration of dibutylamine 3 (Fig. 3). Molecule 4 bearing an acetal group was also effectively labelled at the α-position of the nitrogen atom without distinction between the primary and the secondary carbon. Regarding N-heterocycles, which are ubiquitous scaffolds in life sciences, 5, 6 and 7-membered cyclic amines were successfully labelled at the α-position of the nitrogen atom at 80 °C. Pyrrolidine 5 was found to be the most reactive heterocycle and was efficiently labelled in only 15 min. Chiral amine 6 was also effectively deuterated in a stereoretentive manner, in accordance with results previously obtained by our group with other nanoparticles45,46,47. Piperidine 7 was also labelled, even though the rate of deuteration was found to be lower compared to 5 and 7-membered cyclic amines. Additionally, fully labelled piperazine 8 was easily prepared in our conditions as well as azepane 9. Overall, most of the common aliphatic N-heterocycles were successfully labelled with an excellent regioselectivity for the α-position even at 80 °C. This could be explained by the cyclic nature of these compounds where farther C-H bonds might be less accessible and therefore less prompt to interact with the catalytic Ru surface than for linear alkyl chains. Our methodology was also successfully applied on tropine 10, containing a complex bicyclic system, which was regioselectively deuterated at the N-methyl group.
To highlight the benefits of our described flow chemistry approach over classical batch experiments, comparisons have been made using a selected set of substrates. Thus pyrrolidine, piperidine, tropine and desipramine were submitted to batch control conditions (see S.I page S89). Even if it is challenging to compare both procedures as the recirculation strategy and pressure from 20 to 100 bar cannot been easily applied in batch, similar conditions in terms of catalyst loading, temperature and number of deuterium equivalents were applied. Results are indisputable and in favour of our method as control of the selectivity and maximised isotopic enrichments are obtained solely in flow conditions.
Deuteration of biorelevant molecules including API
The scope of the reaction was then expanded to more complex structures and biomolecules to illustrate the potency of the developed continuous-flow HDE process (Fig. 5). Thus, API containing alkylamines and N-heterocycles scaffolds have been labelled. The methyl group of dextromethorphan 11 was fully labelled with an excellent regioselectivity and without any detectable degradation after 1 h of reaction (99% yield).

aP = 100 bar, 70% yield after salification with HCl; bP = 80 bar; cP = 80 bar, C = 0.0013 M. The number of deuterium atoms indicated was determined using ESI-MS analysis. T temperature, P pressure.
The antidepressant drug desipramine 12 was deuterated with a slightly lower selectivity as traces of exchange could be detected on the aromatic rings. Even so, almost 5 D atoms were incorporated at the α–position of the secondary amine unit. Zolmitriptan 13 was labelled efficiently with more than 10 D atoms incorporated using our procedure. It should be noted that the deuteration rate of indole’s position 2 was found superior to the one of the aliphatic amine. The aliphatic amine group as well as the indole position 8 were completely labelled with sufficient iterations, highlighting the usefulness of the developed recirculation strategy. Remarkably, the procedure could be applied for the perdeuteration of spermidine 14, which is an extremely challenging compound to prepare using a classical multi-step approach50. Seventy-nine mg of the pure perdeuterated polyamine was obtained in a few hours in a single step of synthesis without purification. In this case, only 70% of the polyamine was recovered despite the flushing of the cartridge with solvent for a long period. The cartridge was found to be inactive for further experiments, suggesting that the polyamine could have poisoned the catalyst by a strong coordination to the metal particles. Trans-4-hydroxy-L-proline 15, one of the major components of the collagen protein, can be regioselectively labelled with a maximised isotopic enrichment (99% on all labelled positions) on the pyrrolidine moiety without racemization. The method has been further applied to complex biologics such as peptides. Tuftsin 16, an immunoglobulin heavy-chain associated tetrapeptide with a stimulating activity of phagocytic cells51, has been labelled with an incorporation of four deuterium atoms, an overall yield of 95% and without degradation or racemization. In comparison with our previous batch labelling procedure (3.1 D incorporated using the non-commercially available RuNp@PVP catalyst)47, the flow recirculation strategy led to maximised isotopic enrichments for all labelled positions, effectively generating a valuable stable isotope-labelled internal standard.
Regarding the possibility of labelling N-heterocyclic Csp2-H bonds, several relevant scaffolds were tested. Nicotine 17 and the ibrutinib precursor 18 were efficiently labelled at the α–position of the nitrogen atom of their azine substructures. Purine derivatives adenosine 19 and inosine 20 were effectively labelled in 1 h and recovered in quantitative yields. Due to the poor solubility of these substrates both in water and organic solvents, concentrations were set lower than for the other amine derivatives and a heavy water/THF solution was used. The adenine scaffold of adefovir 21 and the caffeine moiety of doxofylline 22 were also labelled with very good levels of isotope incorporation.
Labelling of formamides
Subsequently, we sought to apply the potential of our continuous-flow concept to the development of underdeveloped HDE processes. In this context, we discovered that formamide substructures could be efficiently labelled using Ru nanocatalysis.
A series of formamide-containing substrates was successfully labelled at the formyl position using the developed flow procedure, paving the way for the HDE on formamides (Fig. 6). In this regard, N-formyldiisopropylamine 23, N-formylmorpholine 24 and N-formylpiperidine 25 were fully labelled (99% of isotopic enrichment) in only 20 min (approximatively 5 runs). Interestingly, the labelling of N-formylglycine ethyl ester 26 was much more challenging and led to a slightly lower deuterium incorporation even after 90 min (25 runs). While formamide substructures are present in a relatively small number of API, an easy access to regioselectively deuterated formamides can be seen as a valuable way to produce isotopically labelled formylation reagents. These are able to replace the commercially available DMF-d7 usually employed, with an improved isotope economy52,53.

The number of deuterium atoms indicated was determined using ESI-MS analysis.
Scale-up reactions: towards an efficient large-scale labelling
Eventually, owing to shorter reaction times compared to classical batch conditions and the flow setup ability to scale-up reactions easily, we considered the gram-scale reaction of a model substrate. In this context, 5.7 g of azepane-d4 (as an HCl salt) were obtained with a complete regioselectivity at the α-position of the nitrogen atom using neat conditions in only 6 h with 98% yield (Fig. 7).

a NMR spectra, b Mass spectrum of a mixture of the starting unlabelled compound and the labelled one. The number of deuterium atoms indicated was determined using ESI-MS analysis.
As shown by the NMR and MS spectra, our flow process afforded an isotopically labelled compound owning very high chemical and isotopic purities, useable as a SILS or a deuterated building block, without any purification step. This example highlights the potential of our strategy to produce large amounts of deuterated compounds in a productive (0.95 gram of compound per hour), safe (small volume of D2 at a given time) and environmentally friendly way (neat reaction). In comparison, gram scale deuteration performed in batch on another substrate (glycylglycine) using Ru/C led to a productivity of only 0.13 gram of compound per hour (19 g, 6 days, 76% yield)54 and gram-scale synthesis of perdeuterated phenol using a single-path flow strategy led to a productivity of 0.07 gram of compound per hour (1.69 g, 24 h, quantitative)33, highlighting the importance of the recirculation process.
In conclusion, a continuous–flow metal-catalysed HDE procedure was developed to overcome current conventional batch HDE weaknesses and in fine unlock its potential by transforming them in strengths. As proofs of concept, numerous ubiquitous aza-scaffolds to complex API and formamides substructures have been efficiently labelled using a commercially available catalyst without purification. This way, our environmentally friendly, safe, time-efficient, and flexible process combined very high isotopic purities with either on-demand precision deuteration or perdeuteration. Moreover, the neat gram-scale synthesis of d4-azepane owning very high isotopic and chemical purities highlighted the productivity of our recirculation strategy and its applicability in the industrial field. Application of this concept to the labelling of other useful moieties is currently ongoing in our laboratory.
Methods
Continuous-flow setup
Flow experiments were conducted using either a H-Cube Mini PlusTM or H-Cube ProTM apparatus (from ThalesNano®) filling the cell reservoir with heavy water. Iteration of runs was made by connecting the inlet to the outlet of the flow line when the system was entirely filled with the substrate solution. In this manner, the system continuously pumped the reactant in the reactor allowing the H/D exchange of the whole mixture over time. Progress of the reaction is monitored by at-line ESI-MS analysis at the outlet.
Gas flow rate was measured using a gas burette technique after equilibration of the system at 1 mL/min (THF), 20 bar and 20 °C (and 100 °C). An approximated gas flow rate of 30 mL/min was observed upon stabilisation of the system with D2 gas in accordance with data provided by ThalesNano® for the H-Cube Mini PlusTM and for the H-Cube ProTM apparatus in a 50% of deuterium gas production mode.
Volumes of the loop (3.9 mL for the H-Cube Mini PlusTM and 6 mL for the H-Cube ProTM) was determined via calculation from data provided by ThalesNano® and verified experimentally.
H/D exchange quantification
Deuterium incorporation was quantified by the decrease of 1H NMR integral intensities at the specified positions compared to the starting material. Integral intensities were calibrated against hydrogen signals that do not undergo H/D-exchange. Mass spectrometry quantification was performed by subtraction of the mean molecular masses of the product and substrate isotopologue clusters in order to eliminate the contribution of the natural isotope abundance to the total mass. When all positions are deuterated, the isotopic enrichment was evaluated by 1H NMR using mass spectrometry analysis.
General procedures for H/D exchanges
General procedure A: H-Cube Mini PlusTM apparatus
Once the system is equilibrated for a few minutes, a 4 mL solution of the corresponding substrate (1 equiv.) in distilled THF (C = 0.1 M) was passed through a 30 mm CatCart® cartridge (packed with a CatCart® packer apparatus) at 1 mL/min. Cartridges were filled with 120 mg of commercial Sigma-Aldrich Ru/C (5 wt%). Iterative runs were performed by continuously recirculating the solution after the first 3.9 mL of pure solvent (corresponding to the total volume of one loop) were discarded. After the correct amount of time (N × 220 s, N being the number of runs; 220 s = one loop at 1 mL/min), the product was collected and the loop was rinsed using distilled THF (5 mL). The final solution was evaporated under reduced pressure to give the desired product.
General procedure B: H-Cube ProTM apparatus
Once the system is equilibrated for a few minutes, a 6 mL solution of the corresponding substrate (1 equiv.) in distilled THF or deuterated water (C = 0.1 M) was passed through a 30 mm CatCart® cartridge (packed with a CatCart® packer apparatus) at 1 mL/min. Cartridges were filled with 120 mg of commercial Sigma-Aldrich Ru/C (5 wt%). D2 gas production was set to 50% (approximated gas flow rate of 30 mL/min). Iterative runs were performed by continuously recirculating the solution after the first 5.9 mL of pure solvent (corresponding to the total volume of one loop) were discarded. After the correct amount of time, the product was collected and the loop was rinsed using distilled THF or deuterated water (7 mL). The final solution was evaporated under reduced pressure to give the desired product.
General procedure C: batch experiments
A 100 mL Fisher-Porter glassware was charged with the corresponding substrate (1 equiv.) and Ru/C 5 wt%. Then distilled THF (C = 0.1 M) was added under argon. The solution was stirred under vacuum until bubbling then pressurised with D2 gas (3 bar) for 5 min. This operation was repeated twice. The reaction mixture was stirred at the corresponding temperature under pressure of D2 for the mentioned time. The final black solution was cooled down to room temperature and filtered through a syringe filter (nylon—0.45 µm) before being evaporated under reduced pressure to give the desired product.
Data availability
Materials, experimental setup and procedures and characterisation data are provided in the Supplementary Information. All data are available from the corresponding authors upon request. Correspondence and requests for materials should be addressed to S.F. and G.P.
References
Atzrodt, J., Derdau, V., Kerr, W. J. & Reid, M. C−H functionalisation for hydrogen isotope exchange. Angew. Chem. Int. Ed. 57, 3022–3047 (2018).
Kopf, S. et al. Recent developments for the deuterium and tritium labeling of organic molecules. Chem. Rev.122, 6634–6718 (2022).
Prakash, G., Paul, N., Oliver, G. A., Werz, D. B. & Maiti, D. C–H deuteration of organic compounds and potential drug candidates. Chem. Soc. Rev. 51, 3123–3163 (2022).
Atzrodt, J., Derdau, V., Kerr, W. J. & Reid, M. Deuterium‐ and tritium‐labelled compounds: applications in the life sciences. Angew. Chem. Int. Ed. 57, 1758–1784 (2018).
Di Martino, R. M. C., Maxwell, B. D. & Pirali, T. Deuterium in drug discovery: progress, opportunities and challenges. Nat. Rev. Drug Discov. 22, 562–584 (2023).
Gant, T. G. Using deuterium in drug discovery: leaving the label in the drug. J. Med. Chem. 57, 3595–3611 (2014).
Pirali, T., Serafini, M., Cargnin, S. & Genazzani, A. A. Applications of deuterium in medicinal chemistry. J. Med. Chem. 62, 5276–5297 (2019).
Jansen-van Vuuren, R. D., Jedlovčnik, L., Košmrlj, J., Massey, T. E. & Derdau, V. Deuterated drugs and biomarkers in the COVID-19 pandemic. ACS Omega 7, 41840–41858 (2022).
Derdau, V., Atzrodt, J., Zimmermann, J., Kroll, C. & Brückner, F. Hydrogen–deuterium exchange reactions of aromatic compounds and heterocycles by NaBD4‐activated rhodium, platinum and palladium catalysts. Chem. Eur. J. 15, 10397–10404 (2009).
Stokvis, E., Rosing, H. & Beijnen, J. H. Stable isotopically labeled internal standards in quantitative bioanalysis using liquid chromatography/mass spectrometry: necessity or not? Rapid Commun. Mass Spectrom. 19, 401–407 (2005).
De Feyter, H. M. & De Graaf, R. A. Deuterium metabolic imaging—back to the future. J. Magn. Reson. 326, 106932 (2021).
De Feyter, H. M. et al. Deuterium metabolic imaging (DMI) for MRI-based 3D mapping of metabolism in vivo. Sci. Adv. 4, eaat7314 (2018).
Moriyama, S. et al. Multiple deuteration of triphenylphosphine and live-cell Raman imaging of deuterium-incorporated Mito-Q. Chem. Commun. 59, 12100–12103 (2023).
Tacke, E. et al. Unprecedented perspectives on the application of CinNapht fluorophores provided by a “late-stage” functionalization strategy. Chem. Sci. 14, 6000–6010 (2023).
Grimm, J. B. et al. A general method to improve fluorophores using deuterated auxochromes. JACS Au 1, 690–696 (2021).
Ma, Z., Zhang, L., Cui, Z. & Ai, X. Improving the luminescence and stability of carbon-centered radicals by kinetic isotope effect. Molecules 28, 4805 (2023).
Wang, P. et al. Synthesis of all-deuterated tris(2-phenylpyridine)iridium for highly stable electrophosphorescence: the “deuterium effect. J. Mater. Chem. C 1, 4821 (2013).
Tsuji, H., Mitsui, C. & Nakamura, E. The hydrogen/deuterium isotope effect of the host material on the lifetime of organic light-emitting diodes. Chem. Commun. 50, 14870–14872 (2014).
Liu, X. et al. Isotope effect of host material on device stability of thermally activated delayed fluorescence organic light‐emitting diodes. Small Sci. 1, 2000057 (2021).
Jung, S. et al. Enhancing operational stability of OLEDs based on subatomic modified thermally activated delayed fluorescence compounds. Nat. Commun. 14, 6481 (2023).
Cheng, J.-F. et al. Positive isotope effect in thermally activated delayed fluorescence emitters based on deuterium-substituted donor units. Chem. Eng. J. 430, 132822 (2022).
Tullo, A. H. How LG and DuPont worked together to bring deuterated compounds to next-generation TVs. Chemical & Engineering News https://cen.acs.org/materials/electronic-materials/next-TV-contain-uncommon-isotopes/100/i9 (2022).
Govaerts, S., Nyuchev, A. & Noel, T. Pushing the boundaries of C–H bond functionalization chemistry using flow technology. J. Flow Chem. 10, 13–71 (2020).
Mándity, I. M., Martinek, T. A., Darvas, F. & Fülöp, F. A simple, efficient, and selective deuteration via a flow chemistry approach. Tetrahedron Lett. 50, 4372–4374 (2009).
Ötvös, S. B. et al. Continuous-flow synthesis of deuterium-labeled antidiabetic chalcones: studies towards the selective deuteration of the alkynone core. Molecules 21, 318 (2016).
Mészáros, R., Peng, B.-J., Ötvös, S. B., Yang, S.-C. & Fülöp, F. Continuous-flow hydrogenation and reductive deuteration of nitriles: a simple access to α,α-dideutero amines. ChemPlusChem 84, 1508–1511 (2019).
Ötvös, S. B., Mándity, I. M. & Fülöp, F. Highly selective deuteration of pharmaceutically relevant nitrogen-containing heterocycles: a flow chemistry approach. Mol. Divers. 15, 605–611 (2011).
Hsieh, C.-T. et al. Highly selective continuous-flow synthesis of potentially bioactive deuterated chalcone derivatives. ChemPlusChem 80, 859–864 (2015).
Chandrasekhar, S., Vijaykumar, B. V. D., Mahesh Chandra, B. Raji Reddy, Ch. & Naresh, P. Flow chemistry approach for partial deuteration of alkynes: synthesis of deuterated taxol side chain. Tetrahedron Lett. 52, 3865–3867 (2011).
Orsy, G., Fulop, F. & Mandity, I. M. Continuous-flow catalytic deuterodehalogenation carried out in propylene carbonate. Green Chem. 21, 956–961 (2019).
Mészáros, R. et al. Exploiting a silver–bismuth hybrid material as heterogeneous noble metal catalyst for decarboxylations and decarboxylative deuterations of carboxylic acids under batch and continuous flow conditions. Green Chem. 23, 4685–4696 (2021).
Habraken, E. R. M., Haspeslagh, P., Vliegen, M. & Noël, T. Iridium(I)-catalyzed ortho-directed hydrogen isotope exchange in continuous-flow reactors. J. Flow Chem. 5, 2–5 (2015).
Park, K., Ito, N., Yamada, T. & Sajiki, H. Efficient continuous-flow H–D exchange reaction of aromatic nuclei in D2O/2-PrOH mixed solvent in a catalyst cartridge packed with platinum on carbon beads. Bull. Chem. Soc. Jpn. 94, 600–605 (2021).
Minya, F. et al. Raney nickel-catalyzed deuterium labeling of nitrogen-containing heterocycles and pharmaceuticals under continuous flow conditions. Adv. Synth. Catal. (2025).
Roughley, S. D. & Jordan, A. M. The medicinal chemist’s toolbox: an analysis of reactions used in the pursuit of drug candidates. J. Med. Chem. 54, 3451–3479 (2011).
Wang, M., Zhao, Y., Zhao, Y. & Shi, Z. Bioinspired design of a robust d3-methylating agent. Sci. Adv. 6, eaba0946 (2020).
Shen, Z. et al. Trideuteromethylation enabled by a sulfoxonium metathesis reaction. Org. Lett. 21, 448–452 (2019).
Ning, L. et al. Synthesis of α-deuterated primary amines via reductive deuteration of oximes using D2O as a deuterium source. J. Org. Chem. 86, 2907–2916 (2021).
Fan, Y. et al. Metal-free electrochemically reductive deuteration of C═N bonds with D2O toward deuterated amines. Org. Lett. 25, 432–437 (2023).
Ding, Y., Luo, S., Weng, C. & An, J. Reductive deuteration of nitriles using D2O as a deuterium source. J. Org. Chem. 84, 15098–15105 (2019).
Liu, Z., Ren, X. & Wang, P. A practical synthesis of deuterated methylamine and dimethylamine. J. Chem. Res. 45, 265–268 (2021).
Lockley, W. J. S. & Hesk, D. Rhodium‐ and ruthenium‐catalysed hydrogen isotope exchange. J. Label. Compd. Radiopharm. 53, 704–715 (2010).
Chatterjee, B., Krishnakumar, V. & Gunanathan, C. Selective α-deuteration of amines and amino acids using D2O. Org. Lett. 18, 5892–5895 (2016).
Neubert, L. et al. Ruthenium-catalyzed selective α,β-deuteration of bioactive amines. J. Am. Chem. Soc. 134, 12239–12244 (2012).
Levernier, E. et al. Easy-to-implement hydrogen isotope exchange for the labeling of N-heterocycles, alkylkamines, benzylic scaffolds, and pharmaceuticals. JACS Au 2, 801–808 (2022).
Pieters, G. et al. Regioselective and stereospecific deuteration of bioactive Aza compounds by the use of ruthenium nanoparticles. Angew. Chem. Int. Ed. 53, 230–234 (2014).
Taglang, C. et al. Enantiospecific C-H activation using ruthenium nanocatalysts. Angew. Chem. Int. Ed. 54, 10474–10477 (2015).
Bhatia, S. et al. Stereoretentive H/D exchange via an electroactivated heterogeneous catalyst at sp3 C–H sites bearing amines or alcohols. Eur. J. Org. Chem. 2016, 4230–4235 (2016).
Lepron, M. et al. Nanocatalyzed hydrogen isotope exchange. Acc. Chem. Res. 54, 1465–1480 (2021).
Maruyoshi, K. et al. Synthesis and conformation of deuterated spermidine for investigating weak interaction with polyanionic biomolecules. Tetrahedron 60, 5163–5170 (2004).
Tzehoval, E. et al. Tuftsin (an Ig-associated tetrapeptide) triggers the immunogenic function of macrophages: implications for activation of programmed cells. Proc. Natl. Acad. Sci. USA 75, 3400–3404 (1978).
Wang, Z. & Wu, X.-F. Applications of DMF as a reagent in organic synthesis. In The Chemical Transformations of C1 Compounds. (eds Wu, X.-F., Han, B., Ding, K. & Liu, Z.) 1439–1474 (John Wiley & Sons, Ltd, 2022).
Heravi, M. M., Ghavidel, M. & Mohammadkhani, L. Beyond a solvent: triple roles of dimethylformamide in organic chemistry. RSC Adv. 8, 27832–27862 (2018).
Michelotti, A., Rodrigues, F. & Roche, M. Development and scale-up of stereoretentive α-deuteration of amines. Org. Process Res. Dev. 21, 1741–1744 (2017).
Acknowledgements
S.F. and G.P. disclose support for the research of this work from the European Union’s Horizon 2020 research and innovation program under the European Union’s Horizon 2020 research innovation program FET-OPEN No 862179 and Labex Charm3at (project FlowCatCasIX) and CEA. K.T. discloses support for the research of this work from the European Union’s Horizon 2020 research and innovation program under the European Union’s Horizon 2020 research innovation program FET-OPEN No 862179. M.L. discloses support for the research of this work from Labex Charm3at (project FlowCatCasIX). M.-C.S. discloses support for the research of this work from Labex Charm3at (project FlowCatCasIX). All authors thank T. D’Anfray, S. Lebrequier and D. -A. Buisson (DRF/JOLIOT/SCBM, CEA) for the excellent analytical support.
Author information
Authors and Affiliations
Contributions
S.F., M.-C.S. and G.P. conceived the idea and supervised the project. S.F., M.-C.S. and G.P. designed the experiments. K.T., M.L., S.F. and C.B. performed the experiments, synthesised and characterised the molecules, analysed the data and discussed the results. K.T., S.F. and G.P. prepared the manuscript with contributions from all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Troels Skrydstrup and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Tatoueix, K., Lepron, M., Barboux, C. et al. Unlocking the potential of hydrogen deuterium exchange via an iterative continuous-flow deuteration process. Nat Commun 16, 1314 (2025). https://doi.org/10.1038/s41467-025-56600-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-56600-8
