
Abstract
Designing an electrocatalyst that simultaneously satisfies high catalytic activity and surface stability is essential for realizing high-performance lithium-sulfur (Li||S) batteries. Here, we propose an advanced electrocatalyst by constructing a thin protective catalytic layer (PCL) on the surface of metal nanoparticle catalysts. This few atomic layer thicknesses of the PCL composed of pyridinic N embedded graphitic carbon allows electrons to transfer from a metal nanoparticle to pyridinic N, resulting in an optimized p-orbital level of pyridinic N of PCL favorable for highly active conversion reaction of lithium sulfide. Further, PCL suppresses the direct contact of sulfur species with metal electrocatalysts. This surface protection effect inhibits the phase change of metal electrocatalysts to metal sulfide impurities, which maintains a highly active Li||S electrocatalysis for long-term cycling. Consequently, A h-level Li||S pouch cell with >500 W h kg−1 (specific energy based on current collector, anode, separator, electrolyte, and cathode), Coulombic efficiency (>95%), and stable life of 20 cycles was successfully realized.
Similar content being viewed by others
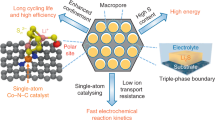

Introduction
Despite significant progress in the field of lithium-ion batteries (LIBs) in the past 30 years1, the current specific energy (WG) of LIBs (300–350 W h kg-1) remains far from 400 W h kg−1 2. As a next-generation battery system, lithium-sulfur (Li||S) batteries have attracted attention because of their great potential to achieve higher WG than LIBs, and ~14 years have been extensively invested in academia to realize a competitive WG and cycle life3,4,5. So far, despite intensive efforts, WG of Li|| batteries is not realistically at a level which can compete with state-of-the-art Li metal anode-based LIBs owing to their fundamentally lower cell operating potential than LIBs6,7. To attain Li||S batteries with WG > 400 W h kg−1 at the pouch cell level, an extremely low electrolyte-to-sulfur (E/S) and negative-to-positive (N/P) ratio of 2.0 is required to minimize the excess weight of the cell component8,9,10. However, intrinsic issues of cathode electrochemistry (i.e., shuttle effect and sluggish redox kinetics) are exacerbated in these harsh conditions of E/S and N/P ratios11,12, and therefore, approaching a low electrolyte-to-capacity (E/C) ratio and a stable cycle life have been restricted9. Recently, the development of metal-based electrocatalysts has been at the frontline of Li||S battery technology because of their merits in enhancing the chemical/electrochemical behaviors of active sulfur species13,14. Numerous precedent studies have made significant improvements of WG and cycle life by designing metal-based electrocatalysts through a multifaceted approach6,15,16. However, the actual WG of the A h-level pouch cell remains less than 400 W h kg−1 (Supplementary Fig. 1a). Although a few studies realized an A h-level pouch cell with WG > 400 W h kg-1, however, the lifespan was limited to less than 20 cycles (Supplementary Fig. 1b). Thus, in-depth studies of designing advanced electrocatalysts are required for realizing A h-level Li||S pouch cell with a satisfactory WG and improved cycle life.
Catalytic activity that determines the overall redox kinetics of electrically/ionically insulating species (i.e., Li2S2 and Li2S) is a crucial factor for developing highly efficient electrocatalysts17,18. Considerable effort has been invested toward achieving the high catalytic activity of electrocatalysts by modulating the chemical adsorption strength of lithium polysulfide (LiPS) on an electrocatalyst surface19,20,21. However, it is intrinsically limited to attain discharge capacity that is sufficiently high for realizing the satisfactory WG of the pouch cell by only considering the chemical adsorption behaviors of active materials22. This is because i) chemical adsorption, ii) surface diffusion, and iii) electrochemical nucleation occur sequentially in the conversion reaction of sulfur species on the catalyst surface. Therefore, all three parameters need to be considered carefully for achieving a high capacity.
More importantly, the stable maintenance of initial catalytic activity can be a key point to improve the cycle stability of the Li||S pouch cell22. In other battery research fields of developing electrocatalyst (e.g., metal-air and seawater batteries), a variety of techniques to inhibit the side reaction on electrocatalyst surface and improve the long-term surface stability of electrocatalyst has been intensively studied23. In Li||S battery field, however, most research has focused on increasing initial electrocatalytic activity of electrocatalysts, and their surface stability during the long-term cycling has been overlooked so far19. Only a few pioneering researchers have reported the possibility of phase change on the surface of metal-based catalysts at the beginning of cycling (i.e., the formation of metal sulfide species caused by strong chemical bonding between metal and sulfur species)20,24. This phase change on the surface of electrocatalyst causes changes in catalytic activity, which implies that the catalytic effect cannot be sustained during long-term cycling even if electrocatalysts with high intrinsic catalytic activity are designed. In this regard, the design of advanced electrocatalysts satisfying both high catalytic activity and high surface stability is necessary for simultaneously pushing the WG and cycle life of Li||S pouch cell to the next stage.
In this paper, we propose a concept to design electrocatalysts with high catalytic activity and surface stability by constructing a thin protective catalytic layer (PCL) on the surface of metal-based electrocatalyst (Fig. 1). The encapsulation of metal nanoparticle catalysts by PCL with a few atomic layers inhibits metal nanoparticles to directly contact chemically active LiPS. This thin protection of metal nanoparticles by PCL suppresses an undesirable phase change, which leads to the high surface stability of the electrocatalyst. Thus, the catalytic effect is well-maintained even for the long-term cycling of the pouch cell. Second, the thin PCL contains chemically stable catalytic pyridinic N site which has important mechanistic role to improve the discharge capacity with facile lithium sulfide redox reaction on its surface under lean electrolyte conditions25. Unlike the conventional role of metal nanoparticle catalysts in which the catalytic reaction occurs directly on their surface19, metal nanoparticles encapsulated by PCL act as an effective modulator for controlling the p-band center of the pyridinic N site in PCL by the electron transfer phenomenon. Therefore, moderate control of the p-band center of pyridinic N in PCL by controlling electron transfer properties with different types of metal nanoparticles enables optimizing all three steps relevant to electrocatalysis (i.e., chemical adsorption, surface diffusion, and electrochemical nucleation); this results in high catalytic activity and the realization of high-energy Li||S pouch cell.

The thin PCL on the surface of metal nanoparticles is expected to i) protect metal nanoparticles from directly contacting sulfur species, which mitigates the undesirable phase change during long-term cycling and ii) facilitate electron transfer between metal nanoparticles and the catalytic sites of PCL, which modulates the electronic structure of the catalytic site of PCL, that maximizes the catalytic activity for powering the Li||S electrochemistry. The yellow, green, cyan, and gray sphere indicate sulfur, lithium, nitrogen, and carbon atoms, respectively. The specific energy of pouch cell was calculated by considering the mass of current collector, anode, cathode, electrolyte, and separator.
Results and discussion
Construction of thin PCL on metal electrocatalysts
To construct multifunctional PCL with a few atomic layers on the surface of metal-based electrocatalysts (M-PCL, M: Fe, Co, and Ni), a simple synthetic process for forming a dopamine complex with a metal chloride precursor followed by polymerization on carbon nanotube (CNT) was performed (see Method) (Supplementary Fig. 2). The homogeneous distribution of metal nanoparticles in each M-PCL is clearly shown in the scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images (Fig. 2a–c and Supplementary Fig. 3). High-resolution TEM (HRTEM) images indicate that the size of metal nanoparticles is ~10 nm in all M-PCL (Fig. 2d–f). Metal nanoparticles are encapsulated completely by graphitic carbon with a few atomic layers (Fig. 2d–f, Supplementary Figs. 4 and 5). Meanwhile, X-ray diffraction (XRD) patterns of M-PCL indicate that body-centered cubic (bcc) crystalline phase of Fe metal and face-centered cubic (fcc) crystalline phases of Co and Ni metals are well-developed without any impure crystalline phases (Fig. 2g) with ~10 wt% contents (Supplementary Fig. 6). X-ray photoelectron spectroscopy (XPS) results demonstrate that graphitic carbon-based PCL contains N heteroatoms on its surface (Supplementary Fig. 7). An X-ray absorption spectroscopy (XAS) analysis was performed to characterize the local atomic structures and electronic states of each M-PCL. The Co K-edge X-ray absorption near-edge structure (XANES) spectrum of Co-PCL exhibits the profiles of pre-edge and white lines, which are almost the same as those of the Co metallic foil; however, they are different from those of cobalt monoxide (CoO) (Fig. 2h). Moreover, Fourier-transformed extended X-ray absorption fine structure (FT-EXAFS) spectrum of Co-PCL is almost the same with the Co metallic foil, which exhibits Co-Co metallic bonds at ~2.1 Å without any Co-N/O bonds observed at ~1.4 Å (Fig. 2i)26. Similar to Co-PCL, the XANES spectra of Fe-PCL and Ni-PCL show that the profiles of pre-edge and white lines are almost the same with those of Fe and Ni metallic foils, respectively (Supplementary Fig. 8a and b). In the FT-EXAFS spectra of Fe-PCL and Ni-PCL, the metallic Fe-Fe and Ni-Ni bonds at ~2.1 Å are observed without Fe-N/O and Ni-N/O bonds at ~1.4 Å14, which is similar to the Fe and Ni metallic foils (Supplementary Fig. 8c and d). This implies the formation of metal nanoparticles encapsulated by the PCL of the N-doped graphitic carbon, without any other catalytic sites (e.g., Metal-N-C or metal nitride).

Low-magnification TEM images of (a) Fe-PCL, (b) Co-PCL, and (c) Ni-PCL. High-magnification TEM images of (d) Fe-PCL, (e) Co-PCL, and (f) Ni-PCL. Thin PCL was derived from the carbonization of polydopamine and the subsequent graphitization on the metal surface. g XRD patterns of M-PCL. The XRD patterns of bcc structured Fe, fcc structured Co, and fcc structured Ni metals are well-matched with JCPDS No. 06-0696, 15-0806, and 04-0850, respectively. h Co K-edge XANES and (i) FT-EXAFS spectra of Co-PCL, reference metallic Co foil, and cobalt monoxide (CoO).
In situ and ex-situ analyses of Li||S electrochemistry
To investigate the Li||S electrochemistry in each M-PCL embedded cathode, in situ XRD analysis was performed (Fig. 3a–c). A crystalline peak at ~27o corresponding to Li2S (111) appears with an increase in the depth of discharge (DOD) because of the nucleation and growth of insoluble Li2S in the cathode. In Fe-PCL, this Li2S (111) peak starts to be observed from the DOD of ~75% (Fig. 3a). The Li2S (111) peak appears even at the DOD of ~50% in the Co-PCL cathode. Moreover, Co-PCL exhibits a more drastic increase in the Li2S peak intensity than Fe-PCL, and therefore, the final Li2S peak intensity at a DOD of 100% in Co-PCL is higher than that of Fe-PCL (Fig. 3d and e). This implies that Co-PCL is more favorable for the utilization of sulfur by the deposition of larger amount of Li2S than the Fe-PCL during the discharge process. However, the Li2S peak intensity of Ni-PCL cathode is considerably lower than that of Co-PCL and Fe-PCL, which indicates the poor utilization of sulfur for the deposition of Li2S (Fig. 3c and f). A chronoamperometry technique was used to further understand the origin of a different behaviors for the nucleation and growth of lithium sulfide (Fig. 3g). Pristine CNT showed lower Tm values (i.e., time at which maximum current (Im) appears) and steeper slopes than M-PCL, indicating that lithium sulfide nucleation and growth reaction stopped quickly because of the poor transfer of electrons and Li+ ions. Co-PCL showed relatively high Tm values and smooth slopes, which implies that lithium sulfide nucleation and growth reaction occurs continuously with a facile transfer of electrons and Li+ ions. The correlation between I/Im and T/Tm was fitted to four classical models of electrochemical deposition for characterizing the lithium sulfide growth behaviors; Bewick, Fleischmann, and Thirsk models for 2D instantaneous (2DI) and 2D progressive (2DP) nucleation and growth27 and Scharifker and Hills models for 3D instantaneous (3DI) and 3D progressive (3DP) nucleation and growth28 are used to understand the deposition behaviors of lithium sulfide in each cathode (Fig. 3h). The I/Im profiles of pristine CNT and Ni-PCL cathodes are well-matched with 2DI model, whereas the I/Im profiles of Fe-PCL and Co-PCL cathodes are well-matched with the 3DI and 3DP models, respectively. These different behaviors of lithium sulfide nucleation and growth in M-PCL surface were visually characterized by ex situ SEM analysis (Fig. 3i). After the initial discharge at 83.8 mA g-1, the SEM images of pristine CNT and Ni-PCL show the 2D film of lithium sulfide passivating the cathode surface, which is consistent with the chronoamperometry results. The 3D particle of lithium sulfide is observed in the ex situ SEM images of Fe-PCL and Co-PCL without the severe passivation of the cathode surface with a 2D film of lithium sulfide. Consequently, Co-PCL exhibits the high utilization of sulfur for lithium sulfide deposition reaction because of the 3D morphological growth of lithium sulfide, whereas CNT and Ni-PCL are not suitable for the high utilization of sulfur because of the 2D morphological growth of lithium sulfide29,30,31.

In situ XRD results of (a) Fe-PCL, (b) Co-PCL, and (c) Ni-PCL. At the beginning, a peak intensity corresponding to the sulfur decreases in all M-PCL, which indicates the complete reduction of sulfur to soluble LiPS species. The magnified in situ XRD patterns of Li2S (111) crystalline peak for (d) Fe-PCL, (e) Co-PCL, and (f) Ni-PCL. For the in situ XRD analysis, each coin cell was galvanostatically tested at 167.5 mA g−1 with cut-off voltage of 1.7–3.0 V. The in situ XRD analysis was performed for initial discharge under 30 oC. g Chronoamperometry analysis results of pristine CNT and M-PCL. After the pre-discharge process at 2.2 V to completely remove the soluble LiPS species, the current corresponding to nucleation and growth of lithium sulfide was experimentally measured at 2.05 V. h The I/Im vs T/Tm curves of pristine CNT and M-PCL. The I/Im vs T/Tm curves were reproduced from the results of chronoamperometry analysis. i Ex situ SEM images of pristine CNT and M-PCL cathodes after the initial discharge process at 83.8 mA g−1 to 1.7 V with 30 oC. For the in situ XRD and ex situ SEM analysis, the areal S loading and E/S ratio were fixed to 2.0 mg cm-2 and 10 μL mg-1, respectively.
Origin of different lithium sulfide conversion behaviors
Density functional theory (DFT) calculations were performed to elucidate the relationship between the surface properties of each M-PCL and three sequential behaviors relevant to electrocatalysis of active sulfur species (i.e., adsorption, diffusion, and nucleation) (Supplementary Fig. 9). Although XPS results indicated that two different N sites (i.e., graphitic and pyridinic N) were developed on the surface of M-PCL, it is believed that the pyridinic N site dominantly acts as an adsorption and catalytic site for Li||S electrochemistry, because of its considerably stronger adsorption strength with active sulfur species (−1.53 eV) than that with graphitic N (−0.44 eV) (Supplementary Fig. 10)32,33.
First, the chemical adsorption energy (ΔEads) of nonpolar S8 on the surface of M-PCL models was almost same (Supplementary Fig. 11). Soluble polar LiPS (i.e., Li2S6 and Li2S4) exhibit the difference in ΔEads in each M-PCL model. Ni-PCL shows the lowest ΔEads of LiPS (i.e., the strongest adsorption strength), whereas Fe-PCL shows the high ΔEads of LiPS (i.e., the weakest adsorption strength) (Fig. 4a, b, and Supplementary Fig. 12). Static adsorption test results with the Li2S6 solution confirm that the Li2S6 adsorption strength is the weakest in Fe-PCL and the highest in Ni-PCL (Supplementary Fig. 12b). This difference in the LiPS adsorption strength in M-PCL is attributed to the different chemical bonding strength between the pyridinic N site of M-PCL and the Li atoms of LiPS. The ΔEads of LiPS species is considerably different in M-PCL, despite the same surface (i.e., PCL consisting of the pyridinic N doped graphitic carbon) being exposed on all M-PCL models. Therefore, the detailed electronic structures of pyridinic N catalytic sites in M-PCL were investigated. The Bader charge distribution results show the different electron-donating properties of each metal in Fe-PCL, Co-PCL, and Ni-PCL (Fig. 4c). The Fe metal donates the largest amounts of electrons to the pyridinic N catalytic site of PCL, which results in the lowest p-band center, as characterized by the density of states (DOS) calculation (Fig. 4d). The Ni metal donates the least amounts of electrons to the pyridinic N catalytic site of PCL, which results in the highest p-band center. Co-PCL shows a moderate p-band center with a moderate electron donating phenomenon from the Co metal to the pyridinic N catalytic site of PCL (Supplementary Fig. 13).

a Molecular configuration of Li2S4 adsorption on the surface of Ni-PCL, Co-PCL, and Fe-PCL. b Calculated adsorption energies of Li2S4 on Ni-PCL, Co-PCL, and Fe-PCL. c Bader charge distribution results of Ni-PCL, Co-PCL, and Fe-PCL. The negative and positive values indicate the donation and withdrawal of electrons, respectively. d DOS diagrams of Ni-PCL, Co-PCL, and Fe-PCL. e Energy barrier for the surface diffusion of Li2S4 on the surfaces of Ni-PCL, Co-PCL, and Fe-PCL. f Relative energy diagrams for the sulfur conversion reaction on each M-PCL surface, calculated at U = 1.5 V (vs. Li/Li+). The yellow, green, dark gray, cyan, brown, blue, and light gray spheres indicate sulfur, lithium, carbon, nitrogen, iron, cobalt, and nickel atoms, respectively.
Second, the surface diffusion behaviors of LiPS species were studied theoretically (Supplementary Figs. 14 and 15). The theoretically calculated energy barriers for the surface diffusion of LiPS in Fe-PCL, Co-PCL, and Ni-PCL are 0.37, 0.52, and 0.58 eV, respectively (Fig. 4e), which implies that the Fe-PCL surface is the most suitable for the facile surface diffusion of LiPS species attributed to the weakest adsorption strength of LiPS. The surface diffusion behaviors of LiPS on the surface of each M-PCL were further characterized by electrochemical analysis. Though limited to estimating the diffusivity of LiPS electrochemically, Li+ ion diffusivity is a reasonable indicator for comparing the diffusivity of LiPS on each M-PCL surface because the mechanism of LiPS adsorption on the M-PCL surface is based on the chemical interaction between the Li+ ion of LiPS and the pyridinic N site of PCL34. To evaluate the Li+ ion diffusivity through the Randles-Sevcik equation, current vs scan rate1/2 curves of each M-PCL were collected using cyclic voltammetry analysis (Supplementary Fig. 16 and Supplementary Table 1). The Fe-PCL exhibits the highest slope (i.e., the highest diffusivity of Li+ ion and LiPS), whereas Ni-PCL shows the lowest slope (i.e., the lowest diffusivity of Li+ ion and LiPS). The slope of Co-PCL is higher than that of Ni-PCL; however, it is lower than that of Fe-PCL, which is consistent with the DFT calculation results.
Third, relative energy diagrams for the sulfur conversion reactions of discharging process in M-PCL were explored (Fig. 4f, Supplementary Figs. 17–23 and Supplementary Table 2, see Computational details for model structure). Figure 4f represents a specific portion of the overall reaction thermodynamics, particularly Section B of Supplementary Fig. 17, which highlights the decomposition of S8 into Li2S2. In Ni-PCL, the ΔG for potential-limiting step (PLS) (ΔGPLS) is 0.04 eV at U = 1.5 V (vs. Li/Li+), which is the lowest among the three M-PCL models, and it indicates the fast conversion of Li2S2 to Li2S. The Fe-PCL exhibits the highest ΔGPLS value of 0.50 eV at U = 1.5 V (vs. Li/Li+), which indicate the slow conversion of Li2S2 to Li2S. This activity trend comes from their different LiPS adsorption strength (Supplementary Fig. 24). During the Li||S battery discharging, the number of LiPSs adsorbates on the surface is increased by combining S atoms from *S8 decomposition and lithium ions from the electrolyte. Therefore, stronger LiPSs adsorption could facilitate the nucleation of Li2S. Since the transition metal core enhances LiPS adsorption in the order of Ni > Co > Fe, it exhibits better Li2S nucleation in the same order. To experimentally investigate this different Li2S nucleation behaviors on each M-PCL electrocatalyst, activation energy (Eact) for the lithium sulfide deposition was quantified by Arrhenius equation13,35,36. Voltammetry (LSV) curves of M-PCL electrocatalysts were collected at the different temperatures (Supplementary Fig. 25a–c). The current of cathodic peak which was observed at 1.9–2.0 V (i.e., corresponding to the deposition of solid-phase lithium sulfide from the soluble LiPS species) was evaluated from these LSV curves (Supplementary Fig. 25d) and Eact value of each M-PCL electrocatalyst was calculated from the slope of the Arrhenius plot (Supplementary Fig. 25e and f). The calculated Eact values of Fe-PCL and Co-PCL were 19.2 and 12.6 kJ mol−1, while Ni-PCL exhibited the lowest Eact values (6.2 kJ mol−1) among three electrocatalysts. Electrochemical impedance spectroscopy (EIS) analysis was further performed with different temperature to thoroughly investigate the Li2S deposition reaction on M-PCL electrocatalysts (Supplementary Fig. 25g–i). The charge transfer resistance (Rct) was estimated from each Nyquist plot (Supplementary Fig. 25j) and Arrhenius plot was evaluated considering the inversely proportional relationship between current and impedance (Supplementary Fig. 25k). The slope (i.e., indicative of activation barrier for nucleation of Li2S) of Ni-PCL was lower than that of Fe-PCL and Co-PCL, in good agreement with the LSV and DFT calculation results, which corroborates that nucleation of Li2S is facile in the order of Ni > Co > Fe.
Studies on Li||S coin cell performance
To evaluate coin-cell performance, M-PCL embedded cathodes with an areal S loading of 5.0 mg cm−2 were fabricated and tested at 167.5 mA g−1 with an E/S ratio of 4.5 µL mg-1 (Supplementary Table 3). An initial discharge capacity of 1016 mA h g−1 was observed in pristine CNT without M-PCL electrocatalysts (Supplementary Fig. 26 and Supplementary Table 4). As cycle number increased, discharge capacity and Coulombic efficiency (CE) gradually decreased. After 200 cycles, capacity retention ratio and CE were 53.3% and 97.6%, respectively. The cathode with Ni-PCL exhibited an initial discharge capacity of 1029 mA h g-1 and a capacity retention ratio of 61.7 and 55.6% for 200 and 300 cycles, respectively (Fig. 5a). Fe-PCL and Co-PCL showed improved coin-cell performances compared with that of the CNT and N-PCL. The initial discharge capacities of Fe-PCL and Co-PCL were 1053 and 1147 mA h g−1, respectively (Fig. 5a and b). Co-PCL exhibited a discharge capacity of 999 and 856 mA h g−1 with a capacity retention ratio of 87.1 and 74.6% after 200 and 300 cycles, respectively. The discharge capacity of Fe-PCL was 782 and 727 mA h g−1 with a capacity retention ratio of 74.3 and 69.0% for 200 and 300 cycles. The polarization potential at the 1st cycle of Co-PCL was 136 mV, whereas that of Fe-PCL was 217 mV, respectively (Fig. 5b and Supplementary Fig. 27). Although Fe-PCL and Co-PCL showed a similar 3D morphological growth of lithium sulfide, more facile deposition of lithium sulfide from the soluble LiPS species of Co-PCL than Fe-PCL led to lower polarization, higher reversible capacity, and a more stable cycle life of Co-PCL than that of Fe-PCL. The polarization potential of Ni-PCL and pristine CNT were 253 and 300 mV, respectively, higher than those of Co-PCL and Fe-PCL (Supplementary Table 4 and Supplementary Fig. 27), which was attributed to the 2D morphological growth of lithium sulfide. The capacity ratio of high (QH) and low voltage plateaus (QL) was calculated from the initial voltage profiles of each M-PCL (Supplementary Fig. 28). Even though QH values are almost similar in all M-PCL cathodes (Fig. 5b and Supplementary Table 5), Co-PCL showed a considerably lower QH/QL ratio than those of Fe-PCL and Ni-PCL, implying the highly enhanced kinetics for the conversion reaction from Li2S4 to the solid lithium sulfide products on the Co-PCL surface. To further demonstrate this electron donating effect of metal particles to modulate Li||S cell performance, M-PCL electrocatalysts with thick PCL (~7 nm thickness) were prepared by increasing the concentration of the dopamine solution (Supplementary Fig. 29). The galvanostatic coin-cell test results indicate that the initial discharge capacity and polarization potential are almost the same in all M-PCL cathodes with thick PCL (Supplementary Fig. 30). This similarity is attributed to the thick PCL impeding electron donating behaviors from metal nanoparticles to the pyridinic N catalytic site of PCL, which limit the modulation of the p-band center. Therefore, a thin thickness (a few atomic layers) of PCL is crucial for the facile electron transfer between metal nanoparticles and the pyridinic N catalytic site of PCL, which optimizes the catalytic activity of the Li||S electrochemistry.

a Cycle performances of M-PCL for 450 cycles at 167.5 mA g−1. b Initial voltage profiles of M-PCL at 167.5 mA g−1. c Galvanostatic test of Co-PCL embedded cathode at 83.8 mA g−1. d Cycle performance of the Co nanoparticle catalyst embedded cathode without constructing PCL at 167.5 mA g−1. Despite the initial discharge capacity similar to that of Co-PCL, the capacity retention ratio of the bare Co catalyst was only 70.2% for ~200 cycles, considerably lower than that of Co-PCL. e Magnified capacity vs cycle number plot of Co catalyst without PCL and Co-PCL. f Ex situ XRD patterns of Co-PCL and Co without PCL after 5 cycles (charged state). For ex situ XRD, cell was cycled at 167.5 mA g−1 at 30 oC. (CoS2 peaks: JCPDS No. 89–1492). g Calculation of the formation energy for CoS2 solid products through the chemical reaction of Co metals with Li2S8 species. h Calculated Gibbs free energies of the conversion reaction from liquid phase Li2S8 to solid phase S8 on the surface of CoS2 and Co-PCL. The blue, yellow, and green spheres indicate cobalt, sulfur, and lithium atoms, respectively.
These series of Li||S coin cell tests and DFT calculation with experimental results corroborate that cell performance (i.e., discharge capacity, overpotential, and cycle stability) of Ni-PCL which showed the strongest adsorption, the lowest diffusivity of LiPS, and the most facile nucleation of lithium sulfide on its surface is the worst among M-PCL electrocatalysts owing to the nature of 2D growth of lithium sulfide which deteriorates the reversible sulfur utilization ratio. The Fe-PCL which showed the weakest adsorption and the highest diffusivity of LiPS exhibited Li||S coin cell performance better than that of Ni-PCL owing to its nature of 3D growth of lithium sulfide, but worse than that of Co-PCL owing to poor nucleation activity of lithium sulfide. Meanwhile, Co-PCL which showed the moderate adsorption, diffusivity of LiPS, and nucleation activity of lithium sulfide exhibited the high Li||S coin cell performance among the M-PCL electrocatalysts. Even though we don’t want to mislead the researchers in this field by describing the relationship between these three parameters (i.e., adsorption, diffusion, and nucleation of active species) of electrocatalysts with lithium sulfide growth model without any clear evidence supported by advanced analysis technology (e.g., operando observation of LiPS dynamic behaviors on electrocatalyst surface), our findings in this work suggest that the design of electrocatalysts moderately balancing the adsorption-diffusion-nucleation behaviors of active sulfur species potentially enables the high reversible utilization of sulfur (Supplementary Fig. 31).
Further, even at specific current of 1675 and 3350 mA g−1, Co-PCL achieved the high rate-capability under harsh cell operating conditions of areal S loading (≥ 5 mg cm−2) and low E/S ratio (≤ 5 µL mg−1) (Supplementary Fig. 32 and Supplementary Fig. 33). A Co-PCL cathode with an areal S loading of 6.0 mg cm−2 and E/S ratio of 4.5 µL mg−1 exhibited an initial areal discharge capacity of ~6.0 mA h cm−2, which is considerably higher than that of conventional LIBs (4.0 mA h cm-2), without capacity decay for ~100 cycles (Fig. 5c and Supplementary Table 6). The CE of Co-PCL was well-maintained and > 99.5% after 100 cycles (Supplementary Fig. 34). Further, the Co-PCL cathode with an areal S loading of 7.2 mg cm−2 and E/S ratio of 4.0 µL mg−1 showed the initial areal capacity of ~8.0 mA h cm−2, by achieving a low E/C ratio of 3.6 µL [mA h]−1 (Fig. 5c and Supplementary Table 7). In the Co-PCL cathode with S loading of 8.5 mg cm−2 and E/S ratio of 3.0 µL mg−1, the initial areal capacity of ~9.3 mA h cm−2 was exhibited with a low E/C ratio of 2.8 µL [mA h]−1 (Fig. 5c and Supplementary Table 8). As areal S loading increased and E/S ratio decreased, CE tended to slightly decrease but well-maintained with > 98.0% for 100 cycles even under this harsh cell operating condition (Supplementary Fig. 34). Even after 100 cycles, the areal capacity of ~7.2 mA h cm−2 was retained with a capacity retention ratio of 78.2%. The E/S ratio of 3.0 µL mg−1, E/C ratio of 2.8 µL [mA h]−1, areal S loading of 8.5 mg cm−2, and areal capacity of 9.3 mA h cm−2 achieved in this work are comparable with Li||S coin-cell performance of recent reports (Supplementary Fig. 35 and Supplementary Table 9).
Most importantly, a CNT and Co nanoparticle composite without the pyridinic N doped graphitic carbon-based PCL was prepared to characterize the effect of constructing a thin PCL on metal-based electrocatalysts (Supplementary Fig. 36). Compared to the Co-PCL catalyst, the bare Co catalyst without PCL showed a poor stability of discharge capacity and CE (Fig. 5d and Supplementary Fig. 37). Especially, bare Co catalysts showed drastic capacity fading after 5 cycles, whereas this capacity decay was not observed in the case of Co-PCL (Fig. 5e). On the other hand, the ex situ SEM image of a cycled pristine Co catalysts embedded cathode (disassembled after 5th discharge process) indicates that lithium sulfide was deposited in a mixture of 2D film-like and 3D morphology but 2D film-like deposition was dominant, which severely passivates the conductive CNT framework (Supplementary Fig. 38a). In the case of PCL without any Co (i.e., PCL construction on the surface of CNT without any Co metal nanoparticles), ex situ SEM image obviously showed the 2D film-like growth of lithium sulfide (Supplementary Fig. 38b). Further, chronoamperometry analysis results indicate that I/Im profile of PCL without Co was well-matched with 2DI model, whereas that of Co without PCL was matched with 2DI model to some extent, but there is also a mix of 3DP model as observed in the ex situ SEM result (Supplementary Fig. 39). Meanwhile, the ex situ SEM image of Co-PCL after the 5th discharge process indicates that the 3D particle-like growth of lithium sulfide is still well-maintained (Supplementary Fig. 40). This high surface stability of the catalytic site in Co-PCL was further demonstrated by ex situ XRD and XPS analyses. After 5 cycles at 167.5 mA g−1, the XRD pattern of Co-PCL showed only crystalline peaks corresponding to S and (111) plane of fcc structured Co metals, indicating that the initial crystalline phase of Co nanoparticles was well-maintained without an undesirable phase change (Fig. 5f). The ex situ XPS analysis of Co-PCL after 5 cycles also demonstrated that the pyridinic N catalytic site of Co-PCL was maintained in a stable manner without any surface phase evolution, indicating the high surface stability of the pyridinic N catalytic site during cycling (Supplementary Fig. 41). However, in the case of PCL-absent Co nanoparticles, crystalline peaks corresponding to CoS2 (200) and (210) planes were observed, and the Co (111) peak disappeared after 5 cycles. This implies that Co nanoparticles partially converted to CoS2 during cycling because of the direct contact of chemically active sulfur species on the surface of Co metals (Fig. 5f). The additional DFT calculations suggest that Co metals without the PCL exhibited considerably stronger adsorption strengths (i.e., ΔEads of −10.18 and −13.46 eV for Li2S4 and Li2S6, respectively) than those of Co-PCL (i.e., ΔEads of −1.38 and −1.66 eV for Li2S4 and Li2S6, respectively), implying the feasibility of an undesirable phase change on the catalyst surface (Supplementary Fig. 42 and Fig. 4b). The formation energy of CoS2 was theoretically calculated as -1.77 eV (Fig. 5g), which indicates that the formation of CoS2 through the chemical reaction of Co metals with a soluble LiPS species is thermodynamically favorable. Meanwhile, CoS2 has been widely studied as an electrocatalyst for Li||S battery, so Li||S electrochemistry activity on the surface of CoS2 was also characterized. The CoS2-CNT (i.e., composite of CoS2 nanoparticles with CNT) was prepared by post-sulfurization process (see Method) (Supplementary Fig. 43). The Arrhenius plots of as-prepared CoS2-CNT and PCL without Co nanoparticle (as a control sample) were evaluated from the Rct value of each Nyquist plot with different temperature (Supplementary Fig. 44). Considering that the slope of Arrhenius plot is inversely proportional to the electrocatalytic activity of electrocatalyst, the slope of CoS2-CNT which is lower than that of PCL without Co indicates that electrocatalytic activity of CoS2-CNT is higher than that of pure N-doped carbon surface (without any optimization of p-band center of N functionalities). However, the slope of CoS2-CNT is higher than that of Co-PCL, implying that N-doped carbon surface of PCL whose p-band center was optimally controlled by electron transfer with Co nanoparticle is more favorable than CoS2 surface for facile polysulfide conversion reaction. This analysis result is in good agreement with cycle stability of Co without PCL, which exhibited the drastic capacity decay after the phase change of Co to CoS2 at 5th cycle. To elucidate the origin of lower electrocatalytic activity of CoS2-CNT than Co-PCL, DFT calculation was performed. The CoS2 surface exhibits much stronger LiPS adsorption compared to Co-PCL (Supplementary Fig. 24), making the discharging process highly efficient (Supplementary Fig. 45). However, this strong adsorption property of CoS2 surface becomes disadvantageous during the charging process. In the charging process, contrary to the discharging process, small LiPS such as *Li2S and *LiS combine while releasing lithium ions, reducing the number of LiPS adsorbates on the surface. As a result, CoS2 requires significantly more energy than Co-PCL throughout the entire charging process (Supplementary Figs. 46–48 and Supplementary Table 10). The CoS2 surface covered with LiPS is challenging to be recovered, leading to a loss of electrocatalytic activity in the Li||S battery. It appears that the degree of the charging difficulty due to the strong LiPS adsorption property of CoS2 outweighs the ease of discharging.
To correlate surface stability of electrocatalyst with the change of electrocatalytic activity during cell cycling, ex situ EIS analysis was performed at 15th and 50th cycle for Co-PCL and Co without PCL (Supplementary Fig. 49a–d). In the case of Co-PCL, semicircle size of Nyquist plot after 15 and 50 cycles slightly decreased compared to that of 1st cycle (Supplementary Fig. 25h) because LiPS species dissolved in electrolyte during cycling can improve the kinetics of sulfur conversion reaction to some extent37,38. Meanwhile, it is noteworthy that the slope of Arrhenius plot in Co without PCL after 15 cycles is much higher than that of Co-PCL after 15 cycles, indicating higher activation barrier for Li||S electrochemistry in Co without PCL than Co-PCL after 15 cycles (Supplementary Fig. 49e). It is attributed to the irreversible phase change of Co to CoS2 (owing to the absence of PCL) whose electrocatalytic activity is lower than Co-PCL, based on aforementioned our findings from the ex situ XRD (Fig. 5f), EIS analysis results (Supplementary Fig. 44), and DFT calculation results. Even after 50 cycles, the slope of Arrhenius plot in Co-PCL was well-maintained implying the high retention of electrocatalytic activity during cell cycling. On the other hand, the Co without PCL exhibited drastic increase of slope in Arrhenius plot after 50 cycles indicating the deterioration of electrocatalytic activity as cycle number increases. Consequently, a series of ex situ XRD analysis, Arrhenius plot with EIS analysis, and DFT calculations confirm that the construction of PCL on the surface of metal-based catalysts is a key point for retaining an initial catalytic activity and achieving the long-term cycle stability of Li||S batteries by inhibiting the undesirable phase change (i.e., formation of metal sulfide phase), which decreases the electrocatalytic activity (Supplementary Fig. 50).
Realization of high-energy Li||S pouch cell
To realize multi-stacked Li||S pouch cell at practical level, scalability of Co-PCL is one of the most important factors for facile double-side coated electrodes with high areal loading level. To evaluate scalability of synthetic route of Co-PCL, we optimized the 60 times scale-up synthetic process to prepare tens of gram scale (~24 g) of Co-PCL at a batch (Supplementary Fig. 51). The characterization results of TEM, TGA, and XRD corroborate the property consistency of Co-PCL, similar as before scale-up. Then, we fabricated a multi-stacked Li||S pouch cell with a Co-PCL catalyst embedded cathode to investigate the WG and cycle stability at the pouch cell level. To study the pouch cell performance at the practical level, a 30 µm thin Li metal foil was used as an anode and the total E/S and N/P ratios were set to 3.0 µL mg−1 and 1.9, respectively, with 5 layers of cathode and 6 layers of anode (Fig. 6a). In the case of Li||S pouch cell with Co-PCL catalysts in cathode, initial discharge capacity was 1219.7 mA h g−1 (corresponding to the E/C ratio of 2.5 μL [mA h]−1) and initial CE was 99.9% at 167.5 mA g−1. The WG of this pouch cell was 412 W h kg−1 without considering the weight of pouching case and tab and 330 W h kg−1 with considering all the weight of pouch cell. (Fig. 6b and Supplementary Fig. 52). Discharge capacity decreased to 998.7 mA h g−1 which corresponds to the capacity retention ratio of ~82% and CE was 93.9% after 50 cycles (Fig. 6c). On the other hand, in the case of Li||S pouch cell assembled without any Co-PCL electrocatalysts in cathode, discharge capacity was <700 mA h g−1 after 50 cycles, indicating the poor cycle stability. To further improve the WG of the Li||S pouch cell, we decreased the E/S and N/P ratios to 2.4 µL mg−1 and 1.5 (only 50% Li excess), respectively (Fig. 6d and Supplementary Fig. 53). The total capacity was fixed to ~1.0 A h-level with 7 stacks of electrode parts and the areal S loading in the cathode was set to 3.4 mg cm−2. The as-fabricated ~1 A h-level Li||S pouch cell with Co-PCL electrocatalysts exhibited an initial discharge capacity of 1228.9 mA h g−1 at 83.8 mA g−1 (Fig. 6e and f). The calculated WG of pouch cell was ~507 W h kg−1 (without considering the weight of pouching case and tab) and ~400 W h kg−1 (with considering all the weight of pouch cell). After 20 cycles, a discharge capacity of 1027.9 mA h g−1 was observed with a capacity retention ratio of 83.6%. The ~1 A h-level Li||S pouch cell without Co-PCL catalysts showed a drastic decrease in the discharge capacity and CEs only after 7 cycles. After 20 cycles, the discharge capacity and CE were only <200 mA h g−1 and ~50%, respectively (Supplementary Fig. 54). The Li||S pouch cell performance achieved in this work by designing the advanced Co-PCL electrocatalysts is one of the finest among the state-of-the-art Li||S pouch cell performances that have been recently reported (Fig. 6g and Supplementary Table 11).

a Summary of detailed cell parameters for fabricating a Li||S pouch cell with a WG of ~412 W h kg-1. b Cycle performance of the Li||S pouch cell with a WG of ~412 W h kg-1 at 167.5 mA g−1 and (inset) an actual photo of the as-fabricated Li||S pouch cell. The filled and empty circles indicate the capacity and CE, respectively. c Voltage profiles of the Li||S pouch cell after 50 cycles. d Summary of the detailed cell parameters for the fabrication of the Li||S pouch cell with a WG of ~507 W h kg−1. e Cycle performance of the Li||S pouch cell with a WG of ~507 W h kg−1. The 1st cycle was performed under 83.8 mA g−1. From the 2nd cycle, the pouch cell was performed under 335.0 mA g−1. f Voltage profiles of Li||S pouch cell with a WG of ~507 W h kg−1 after 20 cycles. g Comparison of performances of the state-of-the-art Li||S pouch cells. All WG of Li||S pouch cell was re-calculated based on the total weight of pouch cell including the mass of pouching case and tab (see Supplementary Table 10 for details).
We proposed that the construction of pyridinic N doped graphitic carbon-based multifunctional PCL on the surface of metal-based electrocatalysts can open a promising avenue for simultaneously enhancing the catalytic activity and surface stability to sufficiently high values to develop a high-energy and long-cycle Li||S pouch cell. First, PCL with a thickness of a few atomic layers on a metal catalyst surface allows electrons to transfer from the metal catalyst to PCL, modulating the p-band center of the pyridinic N catalytic site in PCL. The use of such metal particles leads to balancing the adsorption-diffusion-conversion behaviors of sulfur species favorable for the high catalytic activity via the facile 3D growth of lithium sulfide. Second, the PCL inhibits undesirable phase change on metal catalyst surface by protecting the metal catalyst from the direct contact with active sulfur species. This enables it to maintain the catalytic activity at high level continuously, even for long-term cycling. Thus, a high-energy and long-cycle A h-level Li||S pouch was successfully realized. Meanwhile, i) versatility of whether as-suggested methodology of constructing the PCL can be extended to other types of metal species or other carbonaceous support materials and ii) large scalable production of hundreds of kg for fabricating practical multi-stacked pouch cell should be additionally investigated. Further, beyond the rational design of advanced cathode materials, continuous efforts to develop the functional electrolyte, separator, and Li metal anode39,40,41 are also crucial for Li||S battery to jump to the next level. We believe that our insights into the rational design of advanced electrocatalysts reported in this work with additional efforts to address the potential limitations for versatility and scalability of this work can pave the way for commercializing Li||S batteries in the near future. Moreover, our findings in this work to protect the surface of metal-based electrocatalysts by constructing thin PCL can be an effective strategy to suppress the surface deterioration phenomena of metal-based electrocatalysts in the multiple battery fields of Li-air, Zn-air, and Zn-I2 batteries.
Methods
Materials
Iron chloride hexahydrate (FeCl3·6H2O, 99%), cobalt chloride hexahydrate (CoCl2·6H2O, 99%), and nickel chloride hexahydrate (NiCl2·6H2O, 99%), dopamine hydrochloride (98%), ethanol (70%), sulfur (99.98%), and Tris(hydroxymethyl) aminomethane (Tris base, ACS reagent) were purchased from Sigma-Aldrich. Multiwalled carbon nanotube (CNT) was obtained from Carbon Nano-material Technology Co., LTD. All chemicals were used as received without further purification.
Synthesis of M-PCL
For the preparation of Co-PCL, Ni-PCL, and Fe-PCL, certain amount of metal chloride hexahydrate precursors; CoCl2·6H2O (1.004 g), NiCl2·6H2O (2.508 g), and FeCl3·6H2O (0.568 g) were homogeneously dissolved in deionized (D.I.) water (500 mL). Then, dopamine hydrochloride (0.4 g, 0.8 g, and 2.0 g for as-prepared FeCl3·6H2O, CoCl2·6H2O, and NiCl2·6H2O solution, respectively) was additionally added in metal chloride hexahydrate solution to form the metal-dopamine complex. After stirring for 30 min at 25 ± 3 °C, CNT (1.2 g) was added into the solution and vigorously stirred for 6 h at 25 ± 3 °C. After CNT was dispersed in solution, certain amounts of Tris were added into the solution to adjust the pH of 8.5 and keep stirring for 24 h. The suspension was filtered and washed with D.I. water and ethanol several times. The collected solid products were dried overnight at 60 oC, then heat-treated at 800 oC for 2 h under inert Argon atmosphere with the heating rate of 5 oC min−1. The pristine Co nanoparticle catalyst without PCL on its surface was prepared by same synthetic route with Co-PCL, but without using dopamine hydrochloride in solution. The PCL without Co nanoparticle catalyst was prepared by same synthetic route with Co-PCL, but without the Co precursors. The CoS2-CNT was prepared by post-sulfurization method. As-prepared Co without PCL was mixed with sulfur in a weight ratio of 1:3 and followed by heat treatment at 350 oC for 2 h under inert atmosphere with the heating rate of 5 oC min−1. For all of the heat-treatment, the temperature was naturally cooled down.
Material characterization
A transmission electron microscopy (TEM; G2 F30 S-Twin, Tencai) and a scanning electron microscopy (SEM; S-4800 field emission, Hitachi) were used to observe the morphology, size, and surface layer of the as-synthesized metal nanoparticle composites with CNT. X-ray diffraction (XRD) analysis was performed using a RIGAKU D/MAX-2500V X-ray diffractometer (Cu Kα radiation, λ = 1.541 Å). X-ray photoelectron spectroscopy (XPS) spectra were collected by VG Scientific Escalab 250 (Al Kα). High annular dark-field scanning TEM (STEM) and high-resolution TEM (HR-TEM; Titan cubed G2 60–300) analyses were conducted for the in-depth characterization of N-doped graphitic carbon-based PCL with a few atomic layers. X-ray absorption spectroscopy (XAS) analysis was performed at the 10 C XAFS beamline at Pohang Accelerator Laboratory (PAL, Korea) to investigate the local electronic structures of PCL. Thermogravimetric analysis (TGA) was performed under O2 atmosphere with scan rate of 10 oC min−1.
Computational details
Density functional theory (DFT) calculations were performed using the Vienna ab initio Simulation Package (VASP)42,43,44. The pseudo-potential, projector augmented-wave (PAW), and Perdew–Burke–Ernzerhof (PBE) exchange–correlation functional45,46 were used with spin-polarization. A plane-wave cutoff energy was controlled as 400 eV for the optimization of geometry and 520 eV for electronic structure calculation. The convergence criterion of total energy was controlled as 10−4 eV for geometric optimization, and the atoms were relaxed until the force acting on each atom was less than 0.01 eV Å−1. DFT-D3 method47 was adopted for van der Waals correction. Brillouin-zone was sampled with 18 × 18 × 18, 15 ×15 × 15, 15 × 15 × 15, 7 × 7 × 7, 7 × 6 × 1, and 3 × 3 × 1 Monkhorst-Pack grid48 for bcc-Fe bulk, fcc-Co bulk, fcc-Ni bulk, CoS2 bulk, M-PCL slab, and CoS2 slab models, respectively. The surface models were represented by Fe (110), Co (111), Ni (111), and CoS2 (101) facets according to the XRD data. The optimized lattice parameters agree with the experimental results (Supplementary Table 12). One layer of graphene with one pyridinic nitrogen site representing PCL was added on the metal supercells in a direction that minimized the lattice misfit (Supplementary Figs. 18–20 and 55). The lattice misfit (f) is defined by the equation:
where lupper and lbottom represent the side lengths of the upper and bottom layers (graphene and metal, respectively, in this study). CoS2 (101) surface model was composed of two layers (Supplementary Fig. 47). To avoid the virtual interaction between periodic cells, thick vacuum layer corresponding to ~15 Å was constructed between each slab model. The method for the correction of solvation and the reaction mechanism of Li||S electrochemistry during discharge/charge process is described in Supplementary Notes 1 and 2, respectively. To estimate Gibbs free energies, DFT energy (E) with entropy (S) energy was corrected at room temperature (T = 298.15 K), and zero-point energy (ZPE).
The vibrational entropy of adsorbed or isolated molecules was obtained via the finite difference method. The electrode potential U is reflected by equating the energy of Li to that of Li++e- and adjusting the electron potential by the term of -eU. The binding energies of lithium polysulfides (LiPSs) were obtained from the energies of the pristine surface (E*), the LiPS-adsorbed surface (E*LiPS) and the isolated LiPS molecule (ELiPS).
The band centers were obtained via the following equation,
where E is the energy referred to the Fermi level and ρ is density of state (DOS). For the Li2S4 diffusion barrier calculation, the climbing image nudged elastic band (CINEB) method was used to find the transition paths and corresponding energy barriers. Force-based quick-min optimizers with a force criterion of 0.01 eV were used in each CI-NEB calculation and the path was divided into seven images. Each structure file of Co-PCL, Fe-PCL, and Ni-PCL with and without LiPS molecules is provided in Supplementary Data 1, 2, and 3, respectively.
Fabrication of Li||S coin cell and characterization
A composite of each M-PCL (30 wt%) with sulfur (70 wt%) were prepared by heating the mixture at 155 °C for 8 h. To prepare the working electrodes, a slurry was fabricated by mixing the sulfur composite with each M-PCL catalyst materials and polymeric binder (LA132, Shandong Gelon Lib Co., Ltd.) in a weight ratio of 9:1 using D.I. water as a solvent. The prepared slurry was coated on carbon-coated Al foil by Doctor blade, and completely dried at 60 °C for 8 h. Then, as-prepared electrode was cut into a coin shape (14 mm) by hand operated punching tool (WC-H125, Wellcos Ltd.). A porous polypropylene-based membrane (Celgard 2400, Wellcos Ltd.) and circular stainless-steel plate with the thickness of 1.0 mm and the diameter of 14 mm were utilized as a separator and spacer disk, respectively. Lithium metal (Honjo Co., Ltd., > 99.9%) with the thickness of 200 μm was used as an anode without any surface-treatment. Battery performance was characterized by 2032 coin-type cell. The 1.0 M bis(trifluoromethane) sulfonamide lithium salt (LiTFSI) in an organic solvent of dimethoxymethane and 1,3-dioxolane (DME/DOL, 1:1 volume ratio, PANAX E-TEC Co., Korea) was used as an electrolyte with 2.0 wt.% of lithium nitrate (LiNO3, 99.99% metal basis, Sigma-Aldrich). For the coin cell test to investigate the cell performance, areal S loading and E/S ratio were fixed to 5.0 mg cm−2 and E/S ratio of 4.5 µL mg-1. For the ex situ SEM, XPS, XRD, and chronoamperometry analyses, cathode with areal S loading of 2.0 mg cm−2 and E/S ratio of 10 µL mg−1 was used. In particular, cell was disassembled in Ar-filled glove box, and the cycled cathode was washed with pure DME and DOL 3 times to completely remove residual salts for ex situ analyses. In the case of ex situ XPS and SEM analyses, cathode was placed into sealing container in Ar-filled glove box and then transferred to the vacuum chamber of analysis equipment to avoid any exposure to air. Meanwhile, in the case of ex situ XRD analyses, cathode was additionally taped and sealed by Kapton tape to prevent air exposure during analysis. Areal sulfur loading in cathode and E/S ratio were controlled as 6.0–8.5 mg cm−2 and 4.5 µL mg−1, respectively, to study the cell performance at more practical level. Cut-off voltage range was 1.7–2.8 V (vs. Li/Li+) and specific current was controlled from 167.5 to 3350 mA g−1. The specific capacity was calculated considering the weight of active material in the electrode. Linear sweep voltammetry (LSV) analysis was performed in the potential range of 1.7–3.0 V (vs. Li/Li+) and scan rate was set as 0.1 mV s−1. To collect in situ XRD pattern for the 1st cycle, 1.5 mm hole sealed by polyethylene film on the 2032 coin-type cell was used as beam window and to avoid the evaporation of electrolyte during in situ analysis. 2D mode XRD patterns were collected for satisfactory peak intensity. The areal S loading and E/S ratio for in situ XRD analysis were fixed to 2.0 mg cm−2 and 10 µL mg−1. Each electrode was galvanostatically discharged at 167.5 mA g−1 to clearly characterize the nucleation and growth of crystalline lithium sulfide species. All the in situ XRD data were collected at Korea Advanced Institute of Science and Technology Analysis Center for Research Advancement (KARA). Electrochemical impedance spectroscopy (EIS) analysis was performed using Wonatech ZIVE MP2 by applying sine wave with potential amplitude of 10 mV from 0.01 to 105 Hz frequency. The EIS analysis was performed in potentiostatic mode with the 10 points per decade of frequency. EIS analysis was carried out in an environmental chamber by controlling the temperature from 30 °C to 60 °C to estimate the relationship between charge transfer resistance and temperature. To perform the EIS analysis for the cycled Li||S batteries, each cell was rested at open-circuit state for 30 min to reach quasi open circuit voltage before the analysis. Each Nyquist plot was fitted by constructing the equivalent circuits to estimate the Rct values (Supplementary Fig. 56) and R-squared (R2) value was controlled as > 0.98 (Supplementary Table 13). All the Li||S coin cell performance in Fig. 5 was double-checked with different three sets (two sets in Supplementary Fig. 57 and one set in Fig. 5) to demonstrate the result consistency. All electrochemical measurement was carried out in an environmental chamber with constant temperature of 30 °C. Anode and cathode correspond to negative and positive electrode, respectively.
Fabrication of Li||S pouch cell and characterization
A cathode composite was composed of Co-PCL electrocatalysts (5 wt%), conductive carbon agent (22.5 wt%), sulfur (67.5 wt%), and binder (5 wt%) for the fabrication of Li||S pouch cell. It is noteworthy that only 5 wt% Co-PCL electrocatalysts was used to prepare the cathode for Li||S pouch cell, which indicates that the total amount of Co nanoparticle in cathode is only 0.5 wt%. Considering that the cost of Co has been a big issue in the cathode (e.g., NCM and NCA) of commercial LIBs, the use of significantly small amount of Co in sulfur-based cathode for fabricating the Li||S pouch cell is economically favorable in practical perspective. The Li metal with thickness of 30 µm was used as an anode. The assembly of pouch cell was performed in dry room (dry air environment) with dew point of 30–40 oCdp and injection of electrolyte and sealing the pouching case were performed by vacuum-involved instrument. For the fabrication of ~0.5 A h level Li||S pouch cell with WG of ~400 W h kg−1, areal S loading, E/S ratio, and N/P ratio were fixed to 2.7 mg cm−2, 3.0 µL mg−1, and 1.9. For the fabrication of ~1 A h level Li||S pouch cell with WG of ~500 W h kg−1, areal S loading, E/S ratio, and N/P ratio were fixed to 3.4 mg cm−2, 2.4 µL mg−1, and 1.5 (only 50% excess Li metal). The temperature for testing as-fabricated Li||S pouch cell was controlled as 30 oC. Specific capacity was calculated considering the total amount of sulfur in cathode, whereas specific energy (WG) was estimated considering the weight of all pouch cell components by below equation:
where Mj indicates the weight (g) of Li||S pouch cell components including Cu current collector, Li metal, separator, electrolyte, sulfur, binder, carbon/catalysts composite, and Al current collector, while Q and V indicate the total capacity (mA h) and average operating voltage (V), respectively. Note that specific energy values presented in this work are calculated on the basis of pouch cell configuration, rather than anode and cathode material only. All the Li||S pouch cell performance in Fig. 6 was double-checked with different three sets (two sets in Supplementary Fig. 58 and one set in Fig. 6) to demonstrate the result consistency.
Data availability
The data generated in this study are provided in the Supplementary Information, Supplementary Data files, and Source Data file. Source data are provided with this paper. All data are available from the corresponding authors upon request. Source data are provided with this paper.
References
Cao, R., Xu, W., Lv, D., Xiao, J. & Zhang, J. G. Anodes for rechargeable lithium‐sulfur batteries. Adv. Energy Mater. 5, 1402273 (2015).
Zhao, M., Li, B.-Q., Zhang, X.-Q., Huang, J.-Q. & Zhang, Q. A perspective toward practical lithium–sulfur batteries. ACS Cent. Sci. 6, 1095–1104 (2020).
Cao, D. et al. Threshold potentials for fast kinetics during mediated redox catalysis of insulators in Li–O2 and Li–S batteries. Nat. Catal. 5, 193–201 (2022).
Lim, W. G. et al. Approaching ultrastable high‐rate Li–S batteries through hierarchically porous titanium nitride synthesized by multiscale phase separation. Adv. Mater. 31, 1806547 (2019).
Li, Z., Guan, B. Y., Zhang, J. & Lou, X. W. D. A compact nanoconfined sulfur cathode for high-performance lithium-sulfur batteries. Joule 1, 576–587 (2017).
Lim, W. G., Kim, S., Jo, C. & Lee, J. A comprehensive review of materials with catalytic effects in Li–S batteries: enhanced redox kinetics. Angew. Chem. 131, 18920–18931 (2019).
Li, Q., Yang, Y., Yu, X. & Li, H. A 700 W h kg−1 rechargeable pouch type lithium battery. Chin. Phys. Lett. 40, 048201 (2023).
Dörfler, S. et al. Challenges and key parameters of lithium-sulfur batteries on pouch cell level. Joule 4, 539–554 (2020).
Chen, Z.-X. et al. Failure analysis of high-energy-density lithium‒sulfur pouch cells. Energy Storage Mater. 53, 315–321 (2022).
Kim, S. et al. Polymer interface-dependent morphological transition toward two-dimensional porous inorganic nanocoins as an ultrathin multifunctional layer for stable lithium–sulfur batteries. J. Am. Chem. Soc. 143, 15644–15652 (2021).
Shin, W. et al. Fluorinated co-solvent promises Li-S batteries under lean-electrolyte conditions. Mater. Today 40, 63–71 (2020).
Li, Y. et al. Two birds with one stone: interfacial engineering of multifunctional Janus separator for lithium–sulfur batteries. Adv. Mater. 34, 2107638 (2022).
Hua, W. et al. Optimizing the p charge of S in p-block metal sulfides for sulfur reduction electrocatalysis. Nat. Catal. 6, 174–184 (2023).
Lim, W.-G. et al. Synergistic effect of molecular-type electrocatalysts with ultrahigh pore volume carbon microspheres for lithium–sulfur batteries. ACS Nano 12, 6013–6022 (2018).
Kim, S. et al. Simultaneous suppression of shuttle effect and lithium dendrite growth by lightweight bifunctional separator for Li–S batteries. ACS Appl. Energy Mater. 3, 2643–2652 (2020).
Zhong, Y. et al. Surface chemistry in cobalt phosphide-stabilized lithium–sulfur batteries. J. Am. Chem. Soc. 140, 1455–1459 (2018).
Yang, Y. et al. Electrocatalysis in lithium sulfur batteries under lean electrolyte conditions. Angew. Chem. 130, 15775–15778 (2018).
Li, X.-Y. et al. Regulating lithium salt to inhibit surface gelation on an electrocatalyst for high-energy-density lithium–sulfur batteries. J. Am. Chem. Soc. 144, 14638–14646 (2022).
Xu, Q. et al. Co nanoparticles anchored on the Co-Nx active centers grafted nitrogen-doped graphene with enhanced performance for lithium-sulfur battery. J. Alloys Compd. 890, 161552 (2022).
Zhao, M. et al. Electrochemical phase evolution of metal‐based pre‐catalysts for high‐rate polysulfide conversion. Angew. Chem. 132, 9096–9102 (2020).
Ye, C. et al. A Mo5N6 electrocatalyst for efficient Na2S electrodeposition in room-temperature sodium-sulfur batteries. Nat. Commun. 12, 7195 (2021).
Li, H. et al. Unraveling the catalyst‐solvent interactions in lean‐electrolyte sulfur reduction electrocatalysis for Li−S batteries. Angew. Chem. Int. Ed. 61, e202213863 (2022).
Cheng, F. & Chen, J. Metal–air batteries: from oxygen reduction electrochemistry to cathode catalysts. Chem. Soc. Rev. 41, 2172–2192 (2012).
Zhao, M. et al. Activating inert metallic compounds for high‐rate lithium–sulfur batteries through in situ etching of extrinsic metal. Angew. Chem. Int. Ed. 58, 3779–3783 (2019).
Doerfler, S. et al. On the mechanistic role of nitrogen-doped carbon cathodes in lithium-sulfur batteries with low electrolyte weight portion. Nano Energy 54, 116–128 (2018).
Sa, Y. J. et al. Heterogeneous Co–N/C electrocatalysts with controlled cobalt site densities for the hydrogen evolution reaction: structure–activity correlations and kinetic insights. ACS Catal. 9, 83–97 (2018).
Bewick, A., Fleischmann, M. & Thirsk, H. Kinetics of the electrocrystallization of thin films of calomel. Trans. Faraday Soc. 58, 2200–2216 (1962).
Scharifker, B. & Hills, G. Theoretical and experimental studies of multiple nucleation. Electrochim. Acta 28, 879–889 (1983).
Zhao, M. et al. Dictating high‐capacity lithium–sulfur batteries through redox‐mediated lithium sulfide growth. Small Methods 4, 1900344 (2020).
Li, Z., Zhou, Y., Wang, Y. & Lu, Y. C. Solvent‐mediated Li2S electrodeposition: a critical manipulator in lithium–sulfur batteries. Adv. Energy Mater. 9, 1802207 (2019).
Yang, J.-L. et al. Rich heterointerfaces enabling rapid polysulfides conversion and regulated Li2S deposition for high-performance lithium–sulfur batteries. ACS Nano 15, 11491–11500 (2021).
Xu, J., Su, D., Zhang, W., Bao, W. & Wang, G. A nitrogen–sulfur co-doped porous graphene matrix as a sulfur immobilizer for high performance lithium–sulfur batteries. J. Mater. Chem. A 4, 17381–17393 (2016).
Yuan, H. et al. Facilitation of sulfur evolution reaction by pyridinic nitrogen doped carbon nanoflakes for highly-stable lithium-sulfur batteries. Energy Storage Mater. 10, 1–9 (2018).
Tao, X. et al. Balancing surface adsorption and diffusion of lithium-polysulfides on nonconductive oxides for lithium–sulfur battery design. Nat. Commun. 7, 1–9 (2016).
Shen, Z. et al. Cation-doped ZnS catalysts for polysulfide conversion in lithium–sulfur batteries. Nat. Catal. 5, 555–563 (2022).
Peng, L. et al. A fundamental look at electrocatalytic sulfur reduction reaction. Nat. Catal. 3, 762–770 (2020).
Yang, Y. et al. High-capacity micrometer-sized Li2S particles as cathode materials for advanced rechargeable lithium-ion batteries. J. Am. Chem. Soc. 134, 15387–15394 (2012).
Vizintin, A. et al. The mechanism of Li2S activation in lithium-sulfur batteries: can we avoid the polysulfide formation? J. Power Sources 344, 208–217 (2017).
Zhang, S. S. Problem, status, and possible solutions for lithium metal anode of rechargeable batteries. ACS Appl. Energy Mater. 1, 910–920 (2018).
Zhang, S. S., Fan, X. & Wang, C. Preventing lithium dendrite-related electrical shorting in rechargeable batteries by coating separator with a Li-killing additive. J. Mater. Chem. 6, 10755–10760 (2018).
Cao, X. et al. Stability of solid electrolyte interphases and calendar life of lithium metal batteries. Energy Environ. Sci. 16, 1548–1559 (2023).
Kresse, G. & Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 47, 558 (1993).
Sholl, D. S. & Steckel, J. A. Density functional theory: a practical introduction. (John Wiley & Sons, 2022).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169 (1996).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953 (1994).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758 (1999).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Monkhorst, H. J. & Pack, J. D. Special points for Brillouin-zone integrations. Phys. Rev. B 13, 5188 (1976).
Acknowledgements
The authors acknowledge financial support from LG Energy Solution, Ltd., and the R&D Program for Forest Science Technology (Project No.2023499B10-2325-AA02; J. Lee) provided by Korea Forest Service (Korea Forestry Promotion Institute). This research was also supported by National Research Foundation of Korea (NRF) grant funded by the Korea Government (RS-2024-00335715; J. Lee) and Nano-Material Technology Development Program through the NRF funded by Ministry of Science and ICT (RS-2023-00235596; J. Lee).
Author information
Authors and Affiliations
Contributions
S. Kim, W.-G. Lim, H. Jung, K. Sohn, J. W. Han, and J. Lee conceived the research concept. S. Kim, W.-G. Lim, C.-Y Park, and J. Lee designed the experiments and analyzed the results. H. Jung and J. W. Han performed DFT calculations and analysis. Y. C. Jeong, S. B. Yang, C. H. Lee, and K. Sohn affiliated with LG Energy Solution contributed to the scientific discussion on the analyses results and the fabrication of Li||S pouch cell. D. Wang discussed the results and reviewed the manuscript. S. Kim, W.-G. Lim, H. Jung, J. W. Han, and J. Lee wrote the manuscript. J. W. Han and J. Lee supervised the project. S. Kim, W.-G. Lim, and H. Jung contributed equally to this work. All authors commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kim, S., Lim, WG., Jung, H. et al. Protective catalytic layer powering activity and stability of electrocatalyst for high-energy lithium-sulfur pouch cell. Nat Commun 16, 1649 (2025). https://doi.org/10.1038/s41467-025-56606-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-56606-2
This article is cited by
-
MOF-Derived CoOx–CeO2−y/C Heterojunction Synergistically Promotes the Deposition and Decomposition of Li2S in Lithium–Sulfur Batteries
Transactions of Tianjin University (2025)
