
Abstract
Anion exchange membrane seawater electrolysis is vital for future large-scale green hydrogen production, however enduring a huge challenge that lacks high-stable oxygen evolution reaction electrocatalysts. Herein, we report a robust OER electrocatalyst for AEMSE by integrating MXene (Ti3C2) with NiFe sulfides ((Ni,Fe)S2@Ti3C2). The strong interaction between (Ni,Fe)S2 and Ti3C2 induces electron distribution to trigger lattice oxygen mechanism, improving the intrinsic activity, and particularly prohibits the dissolution of Fe species during OER process via the Ti-O-Fe bonding effectively, achieving notable stability. Furthermore, the good retention of sulfates and the abundant groups of Ti3C2 provide effective Cl- resistance. Accordingly, (Ni,Fe)S2@Ti3C2 achieves high OER activity (1.598 V@2 A cm-2) and long-term durability (1000 h) in seawater system. Furthermore, AEMSE with industrial current density (0.5 A cm-2) and durability (500 h) is achieved by (Ni,Fe)S2@Ti3C2 anode and Raney Ni cathode with electrolysis efficiency of 70% and energy consumption of 48.4 kWh kg-1 H2.
Similar content being viewed by others
Introduction
Developing sustainable and clean energy is significant for alleviating energy shortages and carbon emissions caused by the utilization of fossil fuels1,2. Green hydrogen via water electrolysis using renewable electricity has attracted intensive interest3. Thereinto, anion exchange membrane water electrolysis (AEMWE) that could operate in low alkaline system emerges as a promising technology as it inherits the merits of both alkaline water electrolysis (AWE, cost-efficiency) and proton exchange membrane water electrolysis (PEMWE, compatible with intermittently renewable electricity), fulfilling the requirements for the sustainable development4,5. Beside the deliberations of the availability of electrolyzers and renewable electricity, water feed is a kernel for the scalable application of water electrolysis6. In terms of rare freshwater resources, direct electrolysis of earth-abundant seawater is extremely appealing as it not only alleviates the crisis of freshwater resources but also avoids the complex and cost purification steps7. Therefore, the development of AEM-based seawater electrolysis (AEMSE) is more meaningful and urgent. However, there is rare satisfactory progress in AEMSE to date. There are higher requirements for membrane and electrocatalysts in seawater electrolysis, especially for anodic oxygen evolution reaction (OER) due to the strong interference of Cl- ions, such as chloride evolution reaction (CER) and deterioration of electrodes. It is still a huge challenge to develop high-stable OER electrocatalysts, particularly for industrially current density and long-term durability, to ensure the efficient operation of AEMSE8.
Transition-metal materials (TMMs) have exhibited sublime performance for OER in alkaline seawater electrolysis9,10,11. For instance, layered double hydroxides (LDHs) are the most studied OER catalysts in alkaline seawater electrolysis, but suffering from low conductivity, infertile active sites, and weak Cl- corrosion resistance12,13,14,15. In this regard, several strategies have been reported to enhance and activity and Cl- corrosion resistance of LDHs for seawater electrolysis, including adding oxyanions in the electrolyte16, constructing oxide overlayer17, doping anions18, etc. Alternatively, recent studies have manifested that sulfides, phosphides, and nitrides own prominent conductivity, and unique electron configuration and particularly can alleviate Cl- corrosion to a certain extent due to in situ generated Cl--repelling species (e.g., sulfates, phosphates, and nitrates)19,20,21,22 on their surfaces based on electrostatic repulsion mechanism in seawater electrolysis23. Nevertheless, when these composites are utilized during high-potential and long-term electrolysis, the high-valence metal ions of (oxy)hydroxides generated from catalyst reconstruction are vulnerable to leach out24, along with the Cl- corrosion resistance effect decreases enormously as Cl--repelling species gradually dissolve. Thus, TMMs still face the common issue of activity and stability degradation induced by the dissolution of metals and or Cl- corrosion during seawater electrolysis. How to solve this dilemma is the key to the availability of TMMs in AEMSE. Recently, MXene, as a class of emergent 2D transition metal carbides or nitrides, serves as a brilliant catalyst support owing to its high electrical conductivity, abundant surface functional groups and notable stability25. In this context, employing a robust MXene matrix, particularly for Ti-based MXene (e.g., Ti3C2) with corrosion-resist and firm structure, may be a subtle strategy to enhance the corrosion resistance of TMMs for seawater electrolysis through the establishment of strong metal-support interaction. Besides, some reaction mechanisms of the catalysts could be modulated by introducing MXene via strong metal-support interaction26. Sun and co-workers utilized O-containing groups of MXene to effectively activate water molecules for promoting HER in seawater electrolysis27. Qiu et al. reported CoS2@C/Ti3C2Tx for HER in seawater electrolysis by construing a synergistic metal-support interface28. However, studies in this respect have not touched on the anode in seawater electrolysis.
Herein, we dexterously employ Ti3C2 to assist NiFe sulfides ((Ni,Fe)S2@Ti3C2), achieving large-current and high-stable OER performance in AEMSE. A series of experimental and theoretical results reveal that Ti3C2 traps the electrons from (Ni,Fe)S2 to increase the valences of Ni and Fe metals, which accelerates the catalyst reconfiguration to trigger lattice oxygen mechanism (LOM), improving the intrinsic activity. More importantly, the strong interaction between (Ni,Fe)S2 and Ti3C2 effectively prohibits the dissolution of Fe species (mainly active centers) during the OER process via the Ti-O-Fe bonding, achieving notable stability. Furthermore, the good retention of sulfates and the abundant negative charge functional groups of Ti3C2 provide dependable Cl- resistance. Accordingly, (Ni,Fe)S2@Ti3C2 achieves high OER activity (1.598 V at 2 A cm−2) and long-term durability (over 1000 h) in seawater. Furthermore, AEMSE with industrial current density and durability (500 h at 0.5 A cm−2) is achieved by (Ni,Fe)S2@Ti3C2 anode and Raney Ni cathode, manifesting the practicability of the catalyst.
Results
Material synthesis and structural characterization
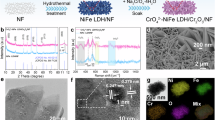
The synthetic procedure of (Ni,Fe)S2@Ti3C2 is illustrated in Fig. 1a. Briefly, the few-layer Ti3C2 nanosheets with an average thickness of ~ 5 nm were obtained by delaminating Ti3AlC2 MAX with the HCl + LiF solutions (Supplementary Figs. 1, 2). Subsequently, NiFe LDH was evenly grown on the Ti3C2 substrate (NiFe LDH@Ti3C2) via a reflux co-precipitation procedure in the presence of Ni and Fe sources, urea and N-Methylpyrrolidone (NMP) (Supplementary Figs. 3, 4). After controllable sulfurization of NiFe LDH@Ti3C2, yielding the (Ni,Fe)S2@Ti3C2 sample (Supplementary Fig. 5). The (Ni,Fe)S2, NiS2@Ti3C2 and FeS2@Ti3C2 samples were also prepared for comparison (Supplementary Figs. 6–8). Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images of (Ni,Fe)S2@Ti3C2 show the vertically interlaced nanosheet array architecture (Fig. 1b, c). Such vertically interlaced arrays could expose more active sites, increase the permeation of electrolytes, and provide rapid gas release. The high-resolution TEM (HRTEM) image (Fig. 1c insert) shows a clear lattice fringe of 0.278 nm, corresponding to the (200) plane of (Ni,Fe)S2. Energy dispersive X-ray spectroscopy (EDS) elemental mapping (Fig. 1d) demonstrates the homogeneous distribution of Ni, Fe, S, Ti, and C elements throughout the nanosheets. The molar ratio of Ni and Fe is 3:1, and the mass ratio of (Ni,Fe)S2 and Ti3C2 is 12:1 determined by inductively coupled plasma-emission spectroscopy (ICP- MS). The above results prove the successful synthesis of (Ni,Fe)S2 on Ti3C2 substrate. Besides, four probe electrical conductivity instrument and N2 adsorption analysis manifest that (Ni,Fe)S2@Ti3C2 owns higher conductivity and larger surface area than (Ni,Fe)S2 (Supplementary Figs. 9, 10), suggesting the enhanced conductivity and more active sites of (Ni,Fe)S2 by introducing Ti3C2, which are beneficial for rapid electron transfer and effective accessibility of active sites to gain large current performance.

a Schematic synthesis of (Ni,Fe)S2@Ti3C2. (NiFe LDH@Ti3C2: NiFe layered double hydroxides loading on the Ti3C2 surface. (Ni,Fe)S2@Ti3C2: NiFe sulfide loading on the Ti3C2 surface.) b SEM image and (c) TEM and HRTEM (insert) images of (Ni,Fe)S2@Ti3C2. d STEM image of (Ni,Fe)S2@Ti3C2 and corresponding EDS mappings of Ni, Fe, S, Ti, and C. e Ni 2p XPS spectra of (Ni,Fe)S2@Ti3C2 and (Ni,Fe)S2. f Ni K-edge XAFS spectra of (Ni,Fe)S2@Ti3C2, (Ni,Fe)S2, NiO, and Ni foil. g Fe 2p XPS spectra of (Ni,Fe)S2@Ti3C2 and (Ni,Fe)S2. h Fe K-edge XAFS spectra of (Ni,Fe)S2@Ti3C2, (Ni,Fe)S2, FeO, Fe2O3, and Fe foil. i Ti 2p XPS spectra of (Ni,Fe)S2 and Ti3C2. j ESR spectra of (Ni,Fe)S2@Ti3C2 and (Ni,Fe)S2.
The electronic states of (Ni,Fe)S2@Ti3C2 and the interaction between (Ni,Fe)S2 and Ti3C2 were investigated by X-ray photoelectron spectroscopy (XPS) and X-ray absorption spectroscopy (XAFS). The XPS survey spectrum confirms the co-existence of Fe, Ni, Ti, S, C, and O elements in (Ni,Fe)S2@Ti3C2 (Supplementary Fig. 11). The O element in (Ni,Fe)S2@Ti3C2 partly originates from the surface oxidation of sulfide29, and the other from the functional groups (-OH and -O) of Ti3C230. The rich functional groups on the surface of Ti3C2 offer Ti3C2 a strong Cl- repulsive capability. The Ni 2p spectra of (Ni,Fe)S2@Ti3C2 and (Ni,Fe)S2 show three doublet peaks for 2p3/2 and 2p1/2 cores, corresponding to Ni(II)-S bond, Ni(II)-O bond and satellite signals, respectively (Fig. 1e)31. The Ni peaks of (Ni,Fe)S2@Ti3C2 present a shift toward higher binding energy (0.7 eV) compared with those of (Ni,Fe)S2. Meanwhile, (Ni,Fe)S2@Ti3C2 owns a larger peak area of Ni(II)-O bond and a lower peak area of Ni(II)-S bond than (Ni,Fe)S2. These results indicate that Ti3C2 attracts electrons from Ni and makes the increase of valence state of Ni. The Ni K-edge XAFS spectra (Fig. 1f and Supplementary Fig. 12a) show that the Ni absorption edge for (Ni,Fe)S2@Ti3C2 shifts toward higher energy than that of (Ni,Fe)S2, further confirming the electron transfer from Ni to Ti3C232. Fe 2p region of (Ni,Fe)S2@Ti3C2 and (Ni,Fe)S2 shows six prominent peaks, assigning to Fe 2p3/2 and Fe 2p1/2 cores of Fe(II)-S, Fe(III)-O and satellite signals (Fig. 1g)31. Notably, two additional peaks at 714.3 and 727.4 eV indexed to Fe(III)-O-Ti bond are observed in (Ni,Fe)S2@Ti3C233, proving the strong electronic coupling between (Ni,Fe)S2 and Ti3C2. The existence of the bridge Fe(III)-O-Ti bond favors stabilizing the Fe ions. A shift toward higher binding energy (0.5 eV) of Fe in (Ni,Fe)S2@Ti3C2 relative to that of (Ni,Fe)S2 is observed, illustrating that Ti3C2 also strips electrons from Fe and endows the higher valence state of Fe. The Fe K-edge XAFS spectra (Fig. 1h and Supplementary Fig. 12b) manifest that the Fe absorption edge for (Ni,Fe)S2@Ti3C2 shifts to the higher energy than that of (Ni,Fe)S2, corroborating the electron transfer from Fe to Ti3C234. Ti 2p spectra (Fig. 1i) of (Ni,Fe)S2@Ti3C2 shows that five pairs of 2p3/2 and 2p1/2 doublets, corresponding to Ti-C, Ti(II)-O, Ti(III)-O, Ti(IV)-O and particularly Ti(IV)-O-M (Fe, Ni) bonds, respectively, further confirming the existence of Ti-O-M (M = Fe, Ni) bond in (Ni,Fe)S2@Ti3C230,33. Furthermore, the Ti peaks of (Ni,Fe)S2@Ti3C2 shift toward lower bonding energy in comparison with that of Ti3C2, further validating electron transfer from (Ni,Fe)S2 to Ti3C2, which is also substantiated by Mulliken charge analysis (Supplementary Fig. 13). Above observations reveal that Ti3C2 traps electrons from both Ni and Fe metals and induces the increase of metal valence states. The higher metal valence states endow the consolidation of the orbital hybridization between the metal 3 d and O 2p orbitals, leading to a strengthened M-O covalent bond that favors triggering the highly active LOM pathway for OER35. A further interaction between Ni and Fe in (Ni,Fe)S2@Ti3C2 is analyzed (Supplementary Figs. 14–16). There is an electron transfer from Fe to Ni in (Ni,Fe)S2@Ti3C2, resulting in a higher valence state of Fe that is more likely to be the active site for OER36. While the higher valent Fe easily dissolves during OER electrolysis, leading to the stability decay37. Fortunately, the strong Ti-O-M bond (especially for the Ti-O-Fe bond) between (Ni,Fe)S2 and Ti3C2 will help to stabilize the metal ions, enhancing the stability of the catalyst. In addition, electron spin resonance (ESR) manifests the presence of sulfur vacancies in both (Ni,Fe)S2@Ti3C2 and (Ni,Fe)S2, but lower content S-vacancies in (Ni,Fe)S2@Ti3C2 (Fig. 1j). (Ni,Fe)S2@Ti3C2 with S vacancies can facilitate the adsorption of H2O molecules and oxygen species for catalyst reconstruction to improve intrinsic activity38. Meanwhile, the less content of S vacancies ensures the stability of the structure39. Moreover, the lower S vacancies mean higher S content in (Ni,Fe)S2@Ti3C2, demonstrating that Ti3C2 has a role of fixing sulfur, which would increase Cl- resistance in seawater systems. Considering that wettability is the key parameter of OER catalysts, the contact angle (CA) test was carried out. The CA of (Ni,Fe)S2@Ti3C2 is nearly 0°, which is smaller than (Ni,Fe)S2 (82.3°) (Supplementary Fig. 17), confirming that demonstrating the enhanced hydrophilicity and good electrolyte permeability of (Ni,Fe)S2@Ti3C2, which facilitates the intimate contact with the electrolyte to accelerate mass transfer process. It should be noted that all the above structural characterizations on (Ni,Fe)S2@Ti3C2 were performed for the samples after optimization of the electrocatalyst properties.
Electrocatalytic OER performance
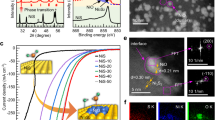
The electrocatalytic OER performance of (Ni,Fe)S2@Ti3C2 was first assessed in 1.0. M KOH. A series of control experiments were performed to select the optimal catalyst, including the molar ratios of Ni/Fe, mass ratios of (Ni,Fe)S2 and Ti3C2, and sulfurization conditions (Supplementary Figs. 18−23 and Table 1). The optimal (Ni,Fe)S2@Ti3C2 catalyst exerts the notable OER catalytic activity, requiring the low overpotentials of 266 (η0.5A), 285 (η1A) and 320 mV (η2A) to reach the industrial-level current density of 0.5, 1 and 2 A cm−2, respectively (Fig. 2a), which far exceeds (Ni,Fe)S2, RuO2, NiFe LDH@Ti3C2, NiS2@Ti3C2 and FeS2@Ti3C2 (Supplementary Fig. 24 and Tables 2 and 3). This activity surpasses the currently reported OER catalysts (Supplementary Fig. 25 and Table 4). (Ni,Fe)S2@Ti3C2 also exhibits the lower Tafel slope and charge-transfer resistance (26 mV dec−1 and 1.62 Ω) (Fig. 2b, c), as compared with (Ni,Fe)S2 (56 mV dec−1 and 7.50 Ω) and RuO2 (61 mV dec−1 and 8.86 Ω), indicating the fast catalytic reaction kinetics and charge-transfer capability of the (Ni,Fe)S2@Ti3C2. Moreover, (Ni,Fe)S2@Ti3C2 exhibits a higher electrochemically active surface area (ESCA) and the activity normalized by ECSA (Supplementary Figs. 26, 27), indicating that Ti3C2 contributes to the increased intrinsic activity. Furthermore, (Ni,Fe)S2@Ti3C2 shows notable long-term stability at 0.5 A cm−2 for 1000 h and even at 1 A cm−2 for 500 h without no obvious degradation of 62 and 158 μA·cm−2·h−1, respectively (Fig. 2d and Supplementary Fig. 28). In contrast, (Ni,Fe)S2 experiences a significant current decay about ~ 30% for only 80 h (Supplementary Fig. 29). This result manifests the important role of Ti3C2 in boosting the catalyst stability.

a Polarization curves with 85% iR compensation (The electrode area is 1 cm × 1 cm, and the resistance is 0.15 ± 0.01 Ω), (b) Tafel plots and (c) Nyquist plots without iR compensation of (Ni,Fe)S2@Ti3C2, (Ni,Fe)S2 and RuO2 in 1.0 M KOH. d I-t curve of (Ni,Fe)S2@Ti3C2 at 0.5 A·cm−2 in 1.0 M KOH. e Polarization curves with 85% iR compensation (The electrode area is 1 cm × 1 cm, and the resistance is 0.15 ± 0.01 Ω) and (f) Tafel plots of (Ni,Fe)S2@Ti3C2, (Ni,Fe)S2 and RuO2 in 1.0 M NaOH + 0.5 M NaCl. g Comparisons of overpotential, Tafel slope, and impedance value. h I-t curve of (Ni,Fe)S2@Ti3C2 at 0.5 A·cm−2 in 1.0 M NaOH + 0.5 M NaCl.
We then evaluated the OER activity of (Ni,Fe)S2@Ti3C2 in alkaline artificial seawater (1.0 M NaOH + 0.5 M NaCl). (Ni,Fe)S2@Ti3C2 still preserves splendid catalytic activity in alkaline seawater, requiring low overpotentials (η0.5A = 297, η1A = 332 and η2A = 368 mV) and Tafel slope (33 mV dec−1) (Fig. 2e, f). Furthermore, the activity of (Ni,Fe)S2@Ti3C2 also outperforms these counterparts (Fig. 2g, Supplementary Fig. 30 and Tables 5 and 6), and represents one of the notable currently reported OER catalysts in alkaline seawater (Supplementary Fig. 31 and Table 7). Furthermore, the outstanding stability of (Ni,Fe)S2@Ti3C2 was evaluated at 0.5 A cm−2 for 1000 h in alkaline seawater, exhibiting no apparent current fluctuation of 69 μA·cm−2·h−1 (Fig. 2h), while a more severe current decay over ~ 35% after 60 h for (Ni,Fe)S2 is observed (Supplementary Fig. 32). Together, the large current density and durable stability of (Ni,Fe)S2@Ti3C2 enable it suitability for practical seawater electrolysis. Furthermore, the combined SEM and XRD characterizations of the post-catalytic sample reveal that morphology of nanosheet experience no obvious change, while surface reconfiguration occurs to generate (oxy)hydroxides, which are active species for OER reported by literature40,41(Supplementary Fig. 33). In addition, the impedance of (Ni,Fe)S2@Ti3C2 did not exhibit obvious increase during the stability test, indicating that the surface reconstruction does not impact the conductivity of the material (Supplementary Fig. 34).
Origin of OER performance and Cl- resistance
We further investigate the origin of the enhanced OER activity, stability, and Cl- resistance effect on (Ni,Fe)S2@Ti3C2. In situ electrochemical impedance spectroscopy (in situ EIS) was conducted to analyze the surface reconstruction and electrochemical reaction kinetics. Bode plots reflect the oxidation reconstruction of catalyst at the middle-frequency region (101-102 Hz) and OER process at low-frequency region (10−1-100 Hz) (Fig. 3a, b)42. At the potential of 1.35 V, the phase angle of (Ni,Fe)S2@Ti3C2 lessens faster at the middle-frequency region, while a similar circumstance appears at the potential of 1.40 V for (Ni,Fe)S2, demonstrating that (Ni,Fe)S2@Ti3C2 has a faster oxidation reconstruction to generate MOOH species, which is also supported by the different pseudocapacitive charges (Supplementary Fig. 35). Since the higher metal valences induced by the electron-catching Ti3C2 endows the more easy oxidation reconstruction, which facilitates to start the active LOM pathway. Moreover, in the low-frequency region, (Ni,Fe)S2@Ti3C2 starts at 1.40 V for OER, while (Ni,Fe)S2 begins at 1.45 V, illustrating the fast reaction kinetics of (Ni,Fe)S2@Ti3C2. In addition, Nyquist plots show that (Ni,Fe)S2@Ti3C2 possesses a smaller charge transfer resistance than (Ni,Fe)S2 (Supplementary Fig. 36).

a, b Bode plots for (Ni,Fe)S2@Ti3C2 and (Ni,Fe)S2 in 1 M KOH. In situ XPS fitting results of (c) Ni 2p, (d) Fe 2p, and (e) O 1 s for (Ni,Fe)S2@Ti3C2. f The concentrations of Fe and Ni leaching from (Ni,Fe)S2@Ti3C2 and (Ni,Fe)S2 after electrocatalysis at 0.5 A·cm−2 in 1.0 M KOH for different reaction times. g Potentiodynamic polarization curves of (Ni,Fe)S2@Ti3C2 and (Ni,Fe)S2. h The ClO- concentration in the electrolyte after 24 h continuous operation for (Ni,Fe)S2@Ti3C2 and (Ni,Fe)S2.
In situ XPS spectra of (Ni,Fe)S2@Ti3C2 depict that the metal-S bond vanishes, and metal-OOH bond appears with the increase of the applied potential, indicating the surface reconfiguration of (Ni,Fe)S2@Ti3C2 to form NiFeOOH@Ti3C2 (Supplementary Fig. 37). It’s worth noting that from 1.32 V to 1.37 V, Ni-S bonds gradually decreases but do not disappear completely, illustrating that the oxidization of Ni species has started at 1.32 V. When increasing the potential to 1.42 V, Ni-S is completely vanished, which matches the Bode plots and CV curves. The same phenomenon can be observed in the S 2p spectra. From 1.32 V to 1.42 V, the peak of S22- gradually weaken. Specifically, Ni2+ is partly reconstructed to Ni3+, along with the content of NiOOH and the position of Ni 2p peak gradually rises with increase of the potential (Fig. 3c). In contrast, Fe2+ is entirely reconstructed to Fe3+, and the content of FeOOH and the position of Fe 2p peak elevates with increase of the potential (Fig. 3d). These results indicate catalyst reconstruction and the more rapid reconstruction rate of Fe than Ni. Besides, the sum of M-O and M-OH bonds hoists with the increase of potential (Fig. 3e), implying more NiFeOOH generation, which implies potentially following LOM43. Moreover, in situ XPS spectra of (Ni,Fe)S2@Ti3C2 are similar to those of NiFeOOH@Ti3C2 after the potential of 1.42 V, confirming that high-metal-valent NiFeOOH species induced by Ti3C2 are the actual active species for OER (Supplementary Fig. 38). To further unveil the origin for the enhanced activity and stability, the dissolved metal contents of catalysts after OER electrolysis at 0.5 A cm−2 were monitored (Fig. 3f). Ni dissolves slightly, but Fe dissolves obviously in (Ni,Fe)S2. Comparatively, after the introduction of Ti3C2, the dissolution of Fe is dramatically dampened. This can be attributed to the strong pulling effect of the Ti-O-Fe bond formed between Ti3C2 and (Ni,Fe)S2, which can effectively stabilize the Fe ions. Meanwhile, the leaching of Ni was further prohibited, manifesting that Ti3C2 contributes to stabilizing the overall structure of the catalyst. Furthermore, the negligible Ti content detected indicates the stability of Ti3C2 substrate (Supplementary Fig. 39 and Table 8). We further explore the role of Cl- resistance of (Ni,Fe)S2@Ti3C2 in alkaline seawater. The sulfates increase and steadily adsorb at the catalyst surface with an increase of potential (Supplementary Fig. 37d), which produces a positive role of electrostatic and spatial repulsion to Cl- in seawater electrolysis20. Furthermore, (Ni,Fe)S2@Ti3C2 presents a higher corrosion potential, a smaller corrosion current and a lower concentration of ClO- (1.28 V, 9.79*10−7 A and 2.10 × 10−7 mol L−1) than (Ni,Fe)S2 (1.12 V, 1.79*10−5 A, and 5.10 × 10−5 mol L−1) (Fig. 3g, h and Supplementary Fig 40)44. Moreover, the rotating ring-disk electrode tests show no occurrence of CER on (Ni,Fe)S2@Ti3C2 (Supplementary Fig. 41)45. These results demonstrate that the introduction of Ti3C2 substantially increases the Cl- corrosion resistance, which is due to the dual effects of Ti3C2 that owns good retention of sulfates and rich negative charge function groups. Besides, (Ni,Fe)S2@Ti3C2 presents a better Cl- resistance than NiFe LDH@Ti3C2, illustrating the sulfurization improves the catalytic performance to a great extent. (Supplementary Fig. 42). In addition, Cl- repulsion in catalysts surface with pH was studied, demonstrating the Cl- resistance is positively dependent on pH (Supplementary Fig. 43). To sum up, the introduction of Ti3C2 accelerates the reconstruction of (Ni,Fe)S2, which facilitates to trigger the LOM pathway and boosts the catalytic activity. Meanwhile, the bridging effect of the Ti-O-Fe bond enormously suppresses the leaching of Fe, thereby enhancing the stability. In addition, the good retention of sulfates and the abundant function groups of Ti3C2 substantially improve the Cl- resistance of (Ni,Fe)S2@Ti3C2.
Identification of OER mechanism and Cl- resistance
To accurately identify the actual mechanism of OER, a range of experiments and theoretical calculations were performed. The pH-dependent experiments (Fig. 4a and Supplementary Fig. 44) display that (Ni,Fe)S2@Ti3C2 has a stronger pH-dependent OER activity than (Ni,Fe)S2. Meanwhile, a larger proton reaction order (ρRHE = ∂log(j)/∂pH) of (Ni,Fe)S2@Ti3C2 (0.89) than (Ni,Fe)S2 (0.54) is observed, implying that (Ni,Fe)S2@Ti3C2 may undergo LOM while (Ni,Fe)S2 may experience AEM46. Furthermore, we use tetramethylammonium cation (TMA+) to detect O22- species produced from LOM based on strong electrostatic interaction47. (Ni,Fe)S2@Ti3C2 presents a larger reduction of OER activity in 1.0 M TMAOH compared to 1.0 M KOH (Fig. 4b and Supplementary Fig. 45), whereas there is a slight change for (Ni,Fe)S2. Besides, Raman spectroscopy shows (Fig. 4c) that the characteristic peaks of TMA+ at 750 and 947 cm−1 appear in (Ni,Fe)S2@Ti3C2, while no characteristic peaks are observed in (Ni,Fe)S2, verifying the existence of O22- species only in (Ni,Fe)S2@Ti3C2 during OER47. In situ Raman and in situ attenuated total reflectance-Fourier transform infrared (ATR-FTIR) spectra further showcase the presence of *O-O* bonds from O22- species in (Ni,Fe)S2@Ti3C2 not in (Ni,Fe)S2during OER process48,49 (Supplementary Fig. 46). These results validate that (Ni,Fe)S2@Ti3C2 experiences a LOM pathway and (Ni,Fe)S2 undertakes an AEM pathway.

a Polarization curves with 85% iR compensation (The scan rate is 5 mV s−1, and the resistance is 0.15 ± 0.01 Ω) measured in KOH electrolytes at different pH values (left) and j at 1.50 V vs. RHE plotted in log scale as a function of pH (right) of (Ni,Fe)S2@Ti3C2 and (Ni,Fe)S2. b Polarization curves in 1.0 M KOH and 1.0 M TMAOH (left) with 85% iR compensation (The scan rate is 5 mV s−1, and the resistance is 0.15 ± 0.01 Ω) and changes of overpotential at 0.5 A cm−2 (Δη0.5A) and Tafel slopes from KOH to TMAOH (right) of (Ni,Fe)S2@Ti3C2 and (Ni,Fe)S2. c Raman spectra of (Ni,Fe)S2@Ti3C2 and (Ni,Fe)S2, measured after running at 1.55 V vs. RHE for 1 h in 1.0 M KOH and 1.0 M TMAOH. d Projected density of states (EF: Fermi level, εO-2p: O2p band center) and (e) Schematic band diagrams of NiFeOOH@Ti3C2-xOx and NiFeOOH. f Calculated free energies of OER steps on NiFeOOH@Ti3C2-xOx and NiFeOOH. g Mechanism change from AEM to LOM after the introduction of Ti3C2. h The adsorption energy for *Cl of NiFeOOH@Ti3C2-xOx and NiFeOOH.
Density function theory (DFT) calculations were further performed to unveil the OER mechanism and the origin of Cl- repulsion. Owing to the surface reconstruction of the catalyst and substrate during OER, we selected NiFeOOH@Ti3C2-xOx and NiFeOOH as the models to study the OER mechanism (Supplementary Fig. 47 and Supplementary Data1)50. A charge transfer of 1.86 e- from NiFeOOH to Ti3C2-xOx, which is more than that between (Ni,Fe)S2 and Ti3C2 (0.64 e-). This confirms that the valence states of Ni and Fe metals are further elevated after the reconstruction process, which provides the possibility to trigger the LOM mechanism (Supplementary Fig. 48). The density of states (DOS) of the O 2p and metal 3 d orbitals were calculated to verify the lattice oxygen activity. The O 2p energy band center (εO-2p) of NiFeOOH@Ti3C2-xOx (− 2.934 eV) is much closer to Fermi energy level (EF) than that of NiFeOOH (− 3.500 eV) (Fig. 4d), suggesting the incorporation of Ti3C2-xOx promotes the release of lattice oxygen, which favors LOM process51. Based on molecular orbital theory, the strong d-d Coulomb interaction will promote the Mott-Hubbard splitting, producing a vacant upper Hubbard band (UHB) and an electron-filled lower Hubbard energy band (LHB). The energy difference (ΔU) between UHB and LHB is a critical descriptor to access the lattice oxygen activity, where a larger ΔU implies the experience of the LOM pathway52. Therefore, we compared ΔU values of NiFeOOH@Ti3C2-xOx and NiFeOOH (Fig. 4e and Supplementary Table 9). The introduction of Ti3C2-xOx reduces the LHB of metals and elevates the ΔU value, weakening the metal-oxygen bonding in NiFeOOH, which further confirms that NiFeOOH@Ti3C2-xOx is more inclined to the LOM mechanism.
The AEM and LOM pathways on NiFeOOH@Ti3C2-xOx and NiFeOOH and the corresponding Gibbs adsorption free energy diagrams were calculated and compared53,54. As shown, Ni sites exert a lower energy barrier than Fe sites for NiFeOOH@Ti3C2-xOx and NiFeOOH in AEM, whereas Fe sites present a lower energy barrier than Ni sites for them in LOM (Fig. 4f and Supplementary Figs. 49–52). In addition, both NiFeOOH@Ti3C2-xOx and NiFeOOH own the rate-determining step (RDS) of deprotonation in the AEM pathway. The RDS of the LOM pathway for NiFeOOH@Ti3C2-xOx is the deprotonation in the first step, but the oxygen vacancy refilling step for NiFeOOH55. Besides, the energy barrier of RDS for NiFeOOH in the AEM pathway is 0.54 eV, but 0.69 eV for LOM, suggesting NiFeOOH follows the AEM pathway. In contrast, NiFeOOH@Ti3C2-xOx needs to overcome the RDS with an energy barrier of 0.30 eV in the LOM pathway, which is lower than that in AEM (0.43 eV), suggesting NiFeOOH@Ti3C2-xOx follows the LOM pathway. Moreover, the energy barrier of RDS in the LOM pathway for NiFeOOH@Ti3C2-xOx is lower than that in the AEM pathway for NiFeOOH, indicating the rapid kinetics of NiFeOOH@Ti3C2-xOx (Supplementary Table 10). These observations demonstrate the change of mechanism from AEM to LOM and rapid kinetics by the introduction of Ti3C2-xOx (Fig. 4g). Furthermore, Cl- adsorption behavior on the NiFeOOH@Ti3C2-xOx and NiFeOOH models was evaluated. Strong Cl- absorption results in a poison of the active sites and destruction of catalysts56. NiFeOOH@Ti3C2-xOx presents a lower ∆ECl* (− 0.47 eV) than NiFeOOH (− 0.73 eV) (Fig. 4h and Supplementary Table 11), suggesting that NiFeOOH@Ti3C2-xOx owns weaker interaction with Cl-, which can inhibit CER and Cl- corrosion. Furthermore, the free energy of CER was calculated to support the viewpoint the ΔG values of the rate-determining step for CER for NiFeOOH@Ti3C2-xOx are distinctly larger than those of NiFeOOH, confirming that the introduction of Ti3C2 plays an important role in enhancing overall Cl- resistance of catalyst15 (Supplementary Fig. 53 and Table 10).
Electrolyzer performance
A two-electrode electrolyzer by using (Ni,Fe)S2@Ti3C2 anode and Raney Ni cathode was assembled to access the industrial performance at 70 ◦C (Fig. 5a). The pH monitoring of the electrolyte was shown in Supplementary Fig. 54. The influence of pore size of the gas diffusion layer (GDL), the composition of binder/catalyst, diffident anion exchange membranes on the performance of the electrolyzer were carried out (Supplementary Figs. 55–57). The optimal Raney Ni | |(Ni,Fe)S2@Ti3C2 exhibits notable industrial activities in both AEMWE and AEMSE, requiring merely 1.75 and 1.80 V to achieve 2 A, which are superior to Raney Ni | |RuO2 (1.97 and 2.06 V) (Fig. 5b and Supplementary Fig. 58). The performance can be further improved with the increase of temperature (Supplementary Fig. 59). Furthermore, Raney Ni | |(Ni,Fe)S2@Ti3C2 exerts low energy consumption of 4.19 and 4.31 kWh m−3 H2 and high electrocatalytic efficiencies of 72.00% and 70.00% for AEMWE and AEMSE, respectively. The activities and efficiencies surpass the currently reported electrolyzers (Supplementary Table 12). Besides, the gas-liquid-solid boundaries were conducted, demonstrating the rapid mass/electron transfer of Raney Ni | |(Ni,Fe)S2@Ti3C2 electrolyzer (Supplementary Fig. 60). To further investigate the origin of electrolyzer performance for Raney Ni | |(Ni,Fe)S2@Ti3C2, the ohmic overvoltage (ηohm), mass transport overvoltage (ηmt), and activation overpotential (ηact) were analyzed via performing electrochemical impedance spectroscopy and steady-state polarization without iR correction57 (Supplementary Fig. 61). Raney Ni | |(Ni,Fe)S2@Ti3C2 achieves the lower ηohm, ηmt and ηact than Raney Ni | |(Ni,Fe)S2 implying the multilevel optimization of anodic (Ni,Fe)S2@Ti3C2 catalyst layer. In addition, Raney Ni | |(Ni,Fe)S2@Ti3C2 exhibits outstanding long-term durability at 2 A for 500 h in AEMWE and AEMSE with negligible current degradation of 20 and 38 μA·cm−2·h−1, respectively, (Fig. 5c) whereas the large current degradation in short-time for Raney Ni | |RuO2 is observed. Moreover, the electrode and membrane of post-AEMWE and AEMSE testing maintain the same structure with those of before ones (Supplementary Fig. 62), and neither AEMWE nor AEMSE shows obviously decay in performance (Supplementary Fig. 63), confirming the notable stability of (Ni,Fe)S2@Ti3C2 under industrial condition. Furthermore, such AEMSE devices can be powered with a solar cell (Supplementary Fig. 64 and Supplementary Movie 1). The hydrogen purity and production rate are 99.997% and 13.5 mL min−1@0.5 A cm−2, respectively (Supplementary Fig. 65). Collectively, (Ni,Fe)S2@Ti3C2 can be a promising OER electrocatalyst for industrial application of AEMSE with renewable electricity input.

a Schematic diagram and reaction flow chart of the AEMSE device. b Polarization curves with 100% iR compensation of the Raney Ni | |(Ni,Fe)S2@Ti3C2 and Raney Ni | |RuO2 couples operating at 70 ◦C in 1.0 M KOH and 1.0 M NaOH + 0.5 M NaCl. (The scan rate is 5 mV s−1, and the resistance is 0.2 ± 0.045 Ω) (c) I-t curves of the Raney Ni | |(Ni,Fe)S2@Ti3C2 and Raney Ni | |RuO2 in 1.0 M KOH (upper) and in 1.0 M NaOH + 0.5 M NaCl (lower) at 70 ◦C.
Discussion
In summary, we report a robust Ti3C2-assisted NiFe sulfide electrocatalyst, achieving industrial-level OER activity, long-term durability, and corrosion Cl resistance in seawater. A series of characterizations and theory calculations verify that the strong interaction effect between (Ni,Fe)S2 and Ti3C2 regulates the electron distribution to trigger the active LOM mechanism, improves the durability by stabilizing metal ions, and enhances the resistance to Cl-. AEMSE with industrial-level density and durability is achieved by (Ni,Fe)S2@Ti3C2 anode and the Raney Ni cathode with a high electrolysis efficiency of 70% and low energy consumption of 48.4 kWh kg−1H2. This work provides an efficient approach to address the issue of deactivation of transition metal catalysts induced by the dissolution of active metals and or chloride corrosion in AEMSE.
Methods
Chemicals
Ti3AlC2 MAX was purchased from Laizhou Kai Kai Ceramic Materials Co., Ltd. Lithium fluoride (LiF), o-Toluidine, and Ruthenium dioxide (RuO2) was purchased from Shanghai Aladdin Chemical Reagent Co., Ltd. Commercial Raney Ni was purchased from Hebei Aegis Metal Materials Co., Ltd. Potassium hydroxide (KOH), hydrochloric acid (HCl), Sodium hydroxide (NaOH), and Iron(III) nitrate nonahydrate (Fe(NO3)3·9H2O) and solution was purchased from Tianjin Kermel Chemical Reagent Co., Ltd. 10% NaClO solution was purchased from Fuyu Chemical Reagent Co., Ltd. Nickel(II) nitrate hexahydrate (Ni(NO3)2·6H2O) was purchased from Tianjin Damao Chemical Reagent Co., Ltd. Sodium chloride (NaCl) and Urea (CH4N2O) was purchased from Tianjin Guangfu Chemical Reagent Co., Ltd. 1-Methyl-2-pyrrolidinone (NMP, analytical reagent) was purchased from Shanghai Rhawn Chemical Reagent Co., Ltd. commercial Titanium mesh (TM) was purchased from Kunshan Guangjiayuan Electronics Co. Ltd. All of the reagents were of analytical grade and directly used without further purification. Deionized (DI) water was used in the experiments.
Preparation of Ti3C2
Under magnetic stirring at 25 °C, 3 g of LiF were added into 80 mL of HCl solution (9 M) and allowed to mix for 20 min. Following this, 2 g of Ti3AlC2 MAX powder were slowly introduced into the solution and etched at 35 °C for 48 h with continuous stirring. Following that, the suspension underwent repeated washing with deionized water (DI water) through centrifugation at 342.31 × g for 2 min per cycle, continuing until the pH of the supernatant reached roughly 7. The final black sediment was redispersed in DI water, then subjected to ultrasonic treatment under an N2 flow for 1 h. After centrifugation at 342.31 × g for 1 h, the dark green Ti3C2 supernatant was collected with a concentration of 10 mg mL−158.
Preparation of (Ni,Fe)S2@Ti3C2
Ni(NO3)2·6H2O (3 mmol), Fe(NO3)3·9H2O (1 mmol) (Ni and Fe molar ratio was 3:1), urea (0.2 mmol) were dissolved in deionized water (20 mL) and stirred for 1 h to form a clear and uniform solution (named as Solution A). 10 mL of Ti3C2 colloidal solution (10 mg mL−1) was dispersed into 30 mL of NMP and stirred for 1 h to form a uniform solution (named Solution B). Then, Solution A was dropped into Solution B and the mixture was then subjected to reflux at 100 °C for 6 h in N2 flow. The products were harvested by several centrifugation-rinsing cycles with DI water followed by freeze-drying, named NiFe LDH@Ti3C2. Finally, 0.5 g sulfur powder and 0.05 g NiFe LDH@Ti3C2 precursor were put into the upstream and downstream of the quartz tube, respectively. The NiFe LDH@Ti3C2 precursor was heated at 400 °C with a ramp rate of 5 °C min−1 and maintained at 400 °C for 0.5 h, obtaining the final product (Ni,Fe)S2@Ti3C2. (The sample with the Ni:Fe molar ratio of 3:1, and the (Ni,Fe)S2:Ti3C2 mass ratio of 12:1 exhibits better catalytic activity, which is expressed as Ni0.75Fe0.25S2@0.06Ti3C2 in chemical formula.) To study the influence of active species and calcination temperature on the performance of the catalyst, control synthesis of (Ni,Fe)S2@Ti3C2 with different Ni and Fe molar ratios, different (Ni,Fe)S2 and Ti3C2 mass ratios and calcination temperatures were carried out. Specifically, Samples with different Ni and Fe molar ratios were adjusted to Ni: Fe = 2:2, 1:3, and 4:1. Samples with different (Ni,Fe)S2 and MXene mass ratios were prepared by adjusting the dosage of Ti3C2 solution to 5 mL and 20 mL (The mass ratios of (Ni,Fe)S2 and Ti3C2 are 24:1 and 6:1, respectively). Samples with different calcination temperatures were prepared at 300 °C and 500 °C.
Theoretical calculation
Density Functional Theory (DFT) simulations were conducted via employing the Cambridge Sequential Total Energy Package (CASTEP) module integrated within Material Studio (MS). The generalized gradient approximation (GGA) with a Perdew-Burke-Ernzerhof (PBE) functional was employed to describe the electronic exchange and correlation effects59. A (2 × 2 × 1) Monkhorst-Pack grid was utilized for Brillouin zone sampling, while a vacuum slab of 15 Å thickness was applied along the z-axis on each slab to prevent pseudo interactions between periodic images. The projector augmented-wave (PAW) method was employed with a planewave energy cutoff set at 500 eV. Geometry optimization was performed iteratively until convergence was achieved, with the total energy tolerance reaching 2 × 10−5 eV and the changes in atomic forces below 0.03 eV/Å60. Considering the lattice match, the heterojunction was modeled based on the (001) crystal plane of NiFeOOH and (001) crystal plane of Ti3C2 with O-termination. The plane fitness of NiFeOOH and Ti3C2 was tuned in order that the lattice mismatch was less than 5%.
OER calculation
In the case of the adsorbate evolution mechanism (AEM) approach, the steps follow the following reaction equation:
where “*” is for the adsorption sites61.
Regarding the lattice oxygen oxidation mechanism (LOM), the reaction steps are as follows:
where “Ol” denotes the lattice oxygen atoms48.
CER calculation
For the chlorine evolution reaction, the Volmer-Heyrovsky reaction was adopted15. The reaction formulas are as follows:
The Gibbs free energy differences (∆G) for each step of OER and CER were determined using the computational hydrogen electrode model in conjunction with the relevant equation as following:
where ∆E is the DFT energy difference; ∆S stands for entropy corrections based on vibrational analysis, and ∆EZPE means zero-point energy at 300 K62.
*Cl adsorption energy calculation:
The adsorption energy of *Cl defined as:
in which E(total) and E(surface) are the energies of all research systems with and without Cl adsorption, respectively. ECl2 is the energy of a Cl2 molecule.
Materials characterizations
The X-ray diffraction (XRD) was performed using a Rigaku Miniflex 600 diffractometer with Cu Kα radiation. The acceleration voltage is 40 kV with a scanning speed of 10o min−1. All test samples were pressed into the square groove of the quartz plate (50 mm*35 mm). The size of the groove is 20 mm*20 mm with a thickness of 0.5 mm. Scanning electron microscopy (SEM) tests were carried on a Hitachi S-4800 at an accelerating voltage of 5 kV. The samples were adhered to the conductive adhesive and tested after gold spraying on the surface. The transmission electron microscopy (TEM) and energy dispersive X-ray spectroscopy (EDS) elemental mapping characterization were carried out on a JEM-F200 electron microscope (JEOL, Japan) at an accelerating voltage of 200 kV. All test samples were uniformly dispersed in ethanol solution and then dripped onto the bilayered carbon coated on Lacey support film with a mesh size of 200 meshes for testing (AZD100/200, produced by Beijing Zhongjingkeyi Technology Co., Ltd.). X-ray photon-electron spectroscopy (XPS) tests were performed using a Thermo Fisher Scientific K-Alpha spectrophotometer with Al-K α radiation (1486.68 eV) operated at 15 kV with a filament current of 10 mA, and signals were accumulated over 5–10 cycles. High-resolution XPS spectra (HRXPS) were obtained through C1s spectrum (284.6 eV) correction. XAFS spectra were tested on the BL14W1 line station of the Shanghai Synchrotron Radiation Facility in transmission mode, in which the Si (111) double-crystal was chosen as the crystal monochromator for energy selection. After sweeping with 100 mg of catalyst at 150 oC for 4 h, the specific surface area (77 K) was calculated using the Brunauer-Emmett-Teller method on a Quantachrome autosorb iQ/ASiQwin. The UV-vis spectra were tested with a MADAPA UV-1800BPC spectrometer. Inductively Coupled Plasma Mass Spectrometry (ICP-MS) test was performed on Thermo DIONIEX ICS-5000 +. Raman spectra were conducted with Horiba Lab RAM Odyssey at 532 nm. Four-point resistance tester was conducted with ST2722. The electron spin resonance (ESR) test was determined by a Bruker EMX plus. The hydrogen purity is determined by hydrogen purity analyzer technology (NK-201A Portable hydrogen analyzer. In situ attenuated total reflectance-Fourier transform infrared (ATR-FTIR) spectra were conducted with Bruker INVENIO-S.
Electrochemical characterizations
For the OER measurements, a three-electrode system was employed, and the experiments were performed using a Gamry Interface 5000 electrochemical workstation. The data was automatically recorded by the workstation. A catalyst weighing 5 mg was dispersed in a mixture containing 30 μL of Nafion and 400 μL of a water-ethanol solution, where the volume ratio of water to ethanol was 1:163. The catalyst suspension was coated on a piece of commercial titanium mesh (TM, 1 × 1 cm−2) as a working electrode (loading amount is 5 mg cm−2). Before coating, the Ti mesh was pretreated with acetone, oxalic acid solution (15 % wt.), deionized water, and ethanol in sequence. A HgO/Hg (1.0 M KOH) electrode purchased from Tianjin Aidahengsheng Co., Ltd and a carbon rod purchased from Shanghai Ledonlab Co., Ltd were used as reference electrodes and counter electrodes, respectively. All of the potential results were processed relative to the reversible hydrogen electrode (RHE) through
The HgO/Hg electrode was calibrated under an H2 atmosphere, and a Pt wire was employed as the working electrode. In order to activate the catalyst, cyclic voltammetry (CV) tests were performed at a scan rate of 100 mV s−1. Polarization curves were obtained from linear sweep voltammetry (LSV) measurements. The voltage range is 1.1–1.7 V versus RHE, and the sweep rate is 5 mV s−1. 85% iR compensation was adopted in all the polarization curves, where R is the uncompensated resistance as determined at an open circuit voltage, which is 0.15 ± 0.01 Ω. The electrochemical OER for pure alkaline water and alkaline simulated seawater tests were conducted in 50 mL 1.0 M KOH and 1.0 M NaOH + 0.5 M NaCl solution, respectively. The 1.0 M KOH electrolyte is prepared by the following steps: (i). 56.11 g KOH solid was dissolved by 500 mL DI water in a beaker. (ii). After stirring 15 min, the KOH solids dissolved completely, and the solution was transferred into the 1 L volumetric flask. (iii). Bring to volume by DI water and sonicate for half an hour. For the 1.0 M NaOH + 0.5 M NaCl electrolyte, the solute is 40.00 g NaOH + 29.22 g NaCl. Other details are the same as the preparation with the 1.0 M KOH electrolyte. All the electrolytes were prepared one hour before the test stored at room temperature, and protected from light. The pH of the electrolyte is 13.95 ± 0.01. Electrochemical impedance spectroscopy (EIS) measurements were conducted over a frequency range from 10 kHz to 0.01 Hz, with an amplitude of 10 mV. The electrochemically surface area (ECSA) of the catalysts was estimated according to the following equal (14):
where the double-layer capacitance (Cdl) was derived from the scan-rate-dependent cyclic voltammograms (CVs) measured in the non-Faradaic potential region of 1.0–1.2 V versus RHE, using 1 M KOH as the electrolyte at scan rates of 20, 40, 60, 80, 100, 120, 140, 160, 180, and 200 mV s−164. The specific capacitance (CS) of 0.04 mF cm−2 was used. The long-term stability was obtained by chronoamperometry tests were performed without iR compensation.
For the electrolyzer, the performance is determined by measuring the LSV curves within a range of 1.2–2.5 V at a scan rate of 5 mV s−1 with 100% iR compensation, where R is 0.2 ± 0.045 Ω. The electrolytes for AEMWE and AEMSE were 2 L 1.0 M KOH and 1.0 M NaOH + 0.5 M NaCl solution, respectively. For the stability test, the electrolyzer continuously operated with a peristaltic pump flow rate of 40 mL min‒1 to circulate the electrolyte. EIS measurements are conducted at a current density of 0.5 A cm−2 within a frequency range of 10 kHz to 0.01 Hz, with an amplitude of 10 mV.
Electrolyzer characterizations
For the anode, the prepared catalysts were sprayed on the 2 cm × 2 cm Ti mesh (The porosity is 50%, the PPI is 80 mesh per inch and the thickness is 0.32 mm) to make the gas diffusion electrodes (GDE) with the loading of 5 mg cm−2 catalyst by using UAC4000L-advanced (Cheersonic, China). Before coating, the Ti mesh was pretreated with acetone, oxalic acid solution (15 % wt.), deionized water, and ethanol in sequence. The specific sprayed process as follows: the ink was prepared with 20 mg of catalyst, 180 µL Nafion (5%) solution as a binder, and 6 mL of 1:1 aqueous ethanol solution. After sonication for 1 h, the ink was injected into the liquid reservoir of the machine. Spraying length and width were 20 mm. The spraying height was 50 mm. The working speed was 9000 mm/mir. The fast-forward speed was 10000 mm/mir. The liquid supply flow rate was 0.5 mL/min. The ultrasonic module and the heating module worked throughout the process. The heating temperature is 70 oC.
For the cathode, the commercial Raney Ni mesh (The porosity is 40%, the PPI is 60 mesh per inch and the thickness is 0.52 mm) was used as the GDE. The PPI of the Raney Ni mesh is 60 pores per inch, and the thickness is 0.52 mm. Before use, the Raney Ni mesh was activated with an alkaline solution (1 M KOH) for 12 h. Prior to assembly, the GDEs were pressurized at 40 MPa for 2 min to make them dense and reduce their thickness to prevent liquid leakage.
The anion exchange membrane (Fumasep FAA-3-PK-130 with a thickness of 130 μm) was pre-treated in 1 M KOH for over 24 h and subsequently rinsed with distilled water to exchange the membrane ions into hydroxide form. Furthermore, the pretreated membrane should be stored under humid and CO2-free conditions to avoid CO2 contamination. The pretreated membrane is cut into 2.2 cm × 2.2 cm and sandwiched between the cathode and anode GDEs. Then the MEA was loaded into a fixture with other components (rubber gaskets and titanium flow) and clamped with a torque of 4 N·m.
Data availability
The data generated in this study are provided within this paper the Supplementary Information. Source data are provided in this paper.
References
Logan, B. E. et al. Conversion of wastes into bioelectricity and chemicals by using microbial electrochemical technologies. Science 337, 686–690 (2012).
Jin, Y. et al. Energy production and water savings from floating solar photovoltaics on global reservoirs. Nat. Sustain. 6, 865–874 (2023).
Liang, Q. et al. Efficient osmosis-powered production of green hydrogen. Nat. Sustain. 7, 628–639 (2024).
Xu, Q. et al. Anion exchange membrane water electrolyzer: Electrode design, lab-scaled testing system and performance evaluation. EnergyChem 4, 100087 (2022).
Liu, R.-T. et al. Recent advances in proton exchange membrane water electrolysis. Chem. Soc. Rev. 52, 5652–5683 (2023).
Khan, M. A. et al. Seawater electrolysis for hydrogen production: a solution looking for a problem? Energy Environ. Sci. 14, 4831–4839 (2021).
Dresp, S. et al. Direct electrolytic splitting of seawater: Opportunities and challenges. ACS Energy Lett. 4, 933–942 (2019).
Song, L. et al. Boosting hydrogen adsorption via manipulating the d-band center of ferroferric oxide for anionexchange membrane-based seawater electrolysis. ACS Catal. 14, 6981–6991 (2024).
Zhou, Q. et al. Innovative strategies in design of transition metal-based catalysts for large-current-density alkaline water/seawater electrolysis. Mat. Today Phys. 26, 100727 (2022).
Kawashima, K. et al. A review of transition metal boride, carbide, pnictide, and chalcogenide water oxidation electrocatalysts. Chem. Rev. 123, 12795–13208 (2023).
Zhang, Y. et al. Amorphous/Crystalline heterostructure transition-metal-based catalysts for high-performance water splitting. Coord. Chem. Rev. 475, 214916 (2023).
Lv, L. et al. 2D Layered double hydroxides for oxygen evolution reaction: From fundamental design to application. Adv. Energy Mater. 9, 1803358 (2019).
Tan, L. et al. Partial sulfidation strategy to NiFe‐LDH@FeNi2S4 heterostructure enable high‐performance water/seawater oxidation. Adv. Funct. Mater. 32, 2200951 (2022).
Kuang, Y. et al. Solar-driven, highly sustained splitting of seawater into hydrogen and oxygen fuels. Proc. Natl. Acad. Sci. USA 116, 6624–6629 (2019).
Fan, R. et al. Ultrastable electrocatalytic seawater splitting at ampere-level current density. Nat. Sustain. 7, 158–167 (2024).
Ye, L. et al. Unraveling the crucial contribution of additive chromate to efficient and stable alkaline seawater oxidation on Ni-based layered double hydroxides. J. Colloid Interf. Sci. 665, 240–251 (2024).
Wang, Z. et al. MnOx Film-coated NiFe-LDH nanosheets on Ni foam as selective oxygen evolution electrocatalysts for alkaline seawater oxidation. Inorg. Chem. 61, 15256–15265 (2022).
Wang, Z. et al. Boron modification promoting electrochemical surface reconstruction of NiFe-LDH for efficient and stable freshwater/seawater oxidation catalysis. J. Colloid Interf. Sci. 668, 607–617 (2024).
Liu, W. et al. Self-protecting CoFeAl-layered double hydroxides enable stable and efficient brine oxidation at 2 A cm−2. Nat. Commun. 15, 4712 (2024).
Ren, J. et al. Rational synthesis of core‐shell‐structured Nickel Sulfide‐based nanostructures for efficient seawater electrolysis. Small 19, 2300194 (2023).
Shen, X. et al. Construction of heterojunction‐rich metal Nitrides Porous nanosheets electrocatalyst for alkaline water/seawater splitting at large current density. Small 20, 2310535 (2024).
Xia, Y. et al. Manipulating electronic structure of nickel phosphide via asymmetric coordination interaction for anion-exchange membrane based seawater electrolysis. Appl. Catal. B Environ. Energy 351, 123995 (2024).
Yu, M. et al. Anionic formulation of electrolyte additive towards stable electrocatalytic oxygen evolution in seawater splitting. J. Energy Chem. 72, 361–369 (2022).
Cao, J. et al. Improved electrocatalytic activity and stability by single iridium atoms on iron‐based layered double hydroxides for oxygen evolution. Angew. Chem. Int. Ed. 62, e202310973 (2023).
Wang, Y. et al. Challenges and opportunities in utilizing MXenes of carbides and nitrides as electrocatalysts. Adv. Energy Mater. 11, 2002967 (2021).
Zhao, X. et al. Dual-atom Co/Ni electrocatalyst anchored at the surface-modified Ti3C2Tx MXene enables efficient hydrogen and oxygen evolution reactions. ACS Nano 18, 4256–4268 (2024).
Sun, F. et al. Energy-saving hydrogen production by chlorine-free hybrid seawater splitting coupling hydrazine degradation. Nat. Commun. 12, 4182 (2021).
Zhang, L. et al. Energy‐saving hydrogen production by seawater electrolysis coupling sulfion degradation. Adv. Mater. 34, 2109321 (2022).
Yang, Y. et al. Hierarchical nanoassembly of MoS2/Co9S8/Ni3S2/Ni as a highly efficient electrocatalyst for overall water splitting in a wide pH range. J. Am. Chem. Soc. 141, 10417–10430 (2019).
Natu, V. et al. A critical analysis of the X-ray photoelectron spectra of Ti3C2Tz MXenes. Matter 4, 1224–1251 (2021).
Liu, C. et al. (NiFe)S2 nanoparticles grown on graphene as an efficient electrocatalyst for oxygen evolution reaction. Electrochim. Acta 286, 195–204 (2018).
Zhang, Y. et al. Coupling donor doping and anion vacancy in Ni3Se4 battery-type cathode for large-capacity and high-rate charge storage. Energy Storage Mater. 67, 103284 (2024).
Li, J. et al. Direct hydro-alcohol thermal synthesis of special core–shell structured Fe-doped titania microspheres with extended visible light response and enhanced photoactivity. Appl. Catal. B Environ. Energy 85, 162–170 (2009).
Liu, Y. et al. Tuning the spin state of the iron center by bridge‐bonded Fe‐O‐Ti ligands for enhanced oxygen reduction. Angew. Chem. Int. Ed. 61, e202117617 (2022).
Zhang, N. et al. Lattice oxygen activation enabled by high-valence metal sites for enhanced water oxidation. Nat. Commun. 11, 4066 (2020).
Zhang, J. et al. Oxygen-vacancy-mediated electron localization at the nickel sites in nickel/iron layered double hydroxide towards efficient oxygen evolution reaction. J. Mater. Chem. A 12, 2044–2052 (2024).
Kuai, C. et al. Phase segregation reversibility in mixed-metal hydroxide water oxidation catalysts. Nat. Catal. 3, 743–753 (2020).
He, L. et al. S-vacancy-rich NiFe-S nanosheets based on a fully electrochemical strategy for large-scale and quasi-industrial OER catalysts. Appl. Catal. B Environ. Energy 345, 123686 (2024).
Ji, Q. et al. The role of oxygen vacancies of ABO3 perovskite oxides in the oxygen reduction reaction. Energy Environ. Sci. 13, 1408–1428 (2020).
Zhong, H. et al. Fundamental understanding of structural reconstruction behaviors in oxygen evolution reaction electrocatalysts. Adv. Energy Mater. 13, 2301391 (2023).
Wang, C. et al. Identification of the origin for reconstructed active sites on oxyhydroxide for oxygen evolution reaction. Adv. Mater. 35, 2209307 (2023).
Chen, W. et al. Unveiling the electrooxidation of urea: Intramolecular coupling of the N−N bond. Angew. Chem. Int. Ed. 60, 7297–7307 (2021).
Wang, Y. et al. Unraveling oxygen vacancy site mechanism of Rh-doped RuO2 catalyst for long-lasting acidic water oxidation. Nat. Commun. 14, 1412 (2023).
Feng, S. et al. Self-assembled heterojunction CoSe2@CoO catalysts for efficient seawater electrolysis. Electrochim. Acta 463, 142870 (2023).
Vos, J. G. et al. Measurement of competition between oxygen evolution and chlorine evolution using rotating ring-disk electrode voltammetry. J. Electroanal. Chem. 819, 260–268 (2018).
Tomar, A. K. et al. Enabling lattice oxygen participation in a triple perovskite oxide electrocatalyst for the oxygen evolution reaction. ACS Energy Lett. 8, 565–573 (2023).
Huang, Z.-F. et al. Tuning of lattice oxygen reactivity and scaling relation to construct better oxygen evolution electrocatalyst. Nat. Commun. 12, 3992 (2021).
Wang, F. et al. Activating lattice oxygen in high-entropy LDH for robust and durable water oxidation. Nat. Commun. 14, 6019 (2023).
Luo, X. et al. Fe-S dually modulated adsorbate evolution and lattice oxygen compatible mechanism for water oxidation. Nat. Commun. 15, 8293 (2024).
Niu, S. et al. Mitigating the reconstruction of metal sulfides for ultrastable oxygen evolution at high current density. CCS Chem. 6, 137–148 (2024).
Shi, Z. et al. Confined Ir single sites with triggered lattice oxygen redox: Toward boosted and sustained water oxidation catalysis. Joule 5, 2164–2176 (2021).
Zhang, N. et al. Lattice oxygen redox chemistry in solid-state electrocatalysts for water oxidation. Energy Environ. Sci. 14, 4647–4671 (2021).
Xue, Z. et al. Revealing Ni-based layered double hydroxides as high-efficiency electrocatalysts for the oxygen evolution reaction: a DFT study. J. Mater. Chem. A 7, 23091–23097 (2019).
Li, Y. et al. Introducing phosphorus into spinel nickel ferrite to enhance lattice oxygen participation towards water oxidation electrocatalysis. Appl. Catal. B Environ. Energy 355, 124116 (2024).
Huang, Z.-F. et al. Chemical and structural origin of lattice oxygen oxidation in Co-Zn oxyhydroxide oxygen evolution electrocatalysts. Nat. Energy 4, 329–338 (2019).
Dionigi, F. et al. Operating conditions, and nickel-iron hydroxide catalyst materials for selective seawater electrolysis. ChemSusChem 9, 962–972 (2016).
Xie, Z. et al. Honeycomb‐structured IrOx foam platelets as the building block of anode catalyst layer in PEM water electrolyzer. Angew. Chem. Int. Ed. 64, e202415032 (2024).
Guo, D. et al. A dual-active Co-CoO heterojunction coupled with Ti3C2-MXene for highly-performance overall water splitting. Nano Res. 15, 238–247 (2022).
Yang, G. et al. Unraveling the mechanism for paired electrocatalysis of organics with water as a feedstock. Nat. Commun. 13, 3125 (2022).
Wang, S. et al. Tuning electronic structure of Ni3S2 with tungsten doping for high-performance electrooxidation of 5-hydroxymethylfurfural. Sci. China Chem. 66, 3636–3644 (2023).
Zhong, X. et al. Constructed interfacial oxygen‐bridge chemical bonding in core‐shell transition metal phosphides/carbon hybrid boosting oxygen evolution reaction. ChemSusChem 14, 2188–2197 (2021).
Zhang, W. et al. Multi-interfacial engineering of an interlinked Ni2P-MoP heterojunction to modulate the electronic structure for efficient overall water splitting. J. Mater. Chem. A 11, 15033–15043 (2023).
Dong, Y. et al. Lower‐temperature synthesis of nitrogen‐rich molybdenum nitride/nickel (Cobalt) heterojunctional assembly for the effective water electrolysis. Adv. Funct. Mater. 35, 2412979 (2024).
Wu, H. et al. Atomically engineered interfaces inducing bridging oxygen-mediated deprotonation for enhanced oxygen evolution in acidic conditions. Nat. Commun. 15, 10315 (2024).
Acknowledgements
This research was supported by the National Key R&D Program of China (2023YFA1507204, H.F.,), the National Natural Science Foundation of China (No. U20A20250, H.F., 22322104, H.Y., 22171074, H.Y.), and the Basic Research Fund of Heilongjiang University in Heilongjiang Province (No. 2021-KYYWF-0031, H.Y.).
Author information
Authors and Affiliations
Contributions
H.Y. and H.F. conceived the idea. J.W. performed the experiments. Y.L. contributed to the DFT calculations studies and set up the AEMSE devices. G.Y. did the analysis of energy consumption. Y.D. performed the XPS analysis and helped with electrochemistry experiments. C.T. performed the SEM and TEM characterizations. H.Y. and Y.J. designed and revised the structure and logic of the manuscript. J.W., H.Y., and H.F. wrote the manuscript with input from all co-authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, J., Liu, Y., Yang, G. et al. MXene-Assisted NiFe sulfides for high-performance anion exchange membrane seawater electrolysis. Nat Commun 16, 1319 (2025). https://doi.org/10.1038/s41467-025-56639-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-56639-7
This article is cited by
-
Quantum-Size FeS2 with Delocalized Electronic Regions Enable High-Performance Sodium-Ion Batteries Across Wide Temperatures
Nano-Micro Letters (2026)
-
Rational design of nitrate-intercalated NiFe LDH with dual chloride-blocking mechanisms for stable alkaline seawater oxidation
Science China Chemistry (2026)
-
Dual Chloride Confinement in Noble Metal‐Doped NiV LDH Catalysts Enables Stable Industrial-Level Seawater Electrolysis
Nano-Micro Letters (2026)
-
TiNbC MXene cathode for high-performance aluminum-ion batteries
Communications Materials (2025)
-
Advances in modifying porous carbon fibers for boosting electrocatalytic reactions
Ionics (2025)
