
Abstract
Nitrogen bioavailability, governed by fixation and loss processes, is crucial for oceanic productivity and global biogeochemical cycles. The key nitrogen loss organisms—denitrifiers and anaerobic ammonium-oxidizing (anammox) bacteria—remain poorly understood in deep-sea cold seeps. This study combined geochemical measurements, 15N stable isotope tracer analysis, metagenomics, metatranscriptomics, and three-dimensional protein structural simulations to explore cold-seeps nitrogen loss processes. Geochemical evidence from 359 sediment samples shows significantly higher nitrogen loss rates in cold seeps compared to typical deep-sea sediments, with nitrogen loss flux from surface sediments estimated at 4.96–7.63 Tg N yr-1 (1.65–2.54% of global marine sediment). Examination of 147 million non-redundant genes indicates a high prevalence of nitrogen loss genes, including nitrous-oxide reductase (NosZ; 6.88 genes per million reads, GPM), nitric oxide dismutase (Nod; 1.29 GPM), and hydrazine synthase (HzsA; 3.35 GPM) in surface sediments. Analysis of 3,164 metagenome-assembled genomes expands the nitrous-oxide reducers by three phyla, nitric oxide-dismutating organisms by one phylum and two orders, and anammox bacteria by ten phyla going beyond Planctomycetota. These microbes exhibit structural adaptations and complex gene cluster enabling survival in cold seeps. Cold seeps likely are previously underestimated nitrogen loss hotspots, potentially contributing notably to the global nitrogen cycle.
Similar content being viewed by others
Introduction
Cold seeps are specialized marine environments primarily located along continental slopes and subduction zones, where subsurface fluids rich in hydrogen sulfide and hydrocarbons, such as methane, seep through the seabed1,2,3. These environments support complex ecosystems centered on the anaerobic oxidation of methane (AOM), a process involving methane-consuming archaea and sulfate-reducing bacteria. All life in these deep-sea oases depends on bioavailable nitrogen to support growth, which is a critical factor limiting biological productivity. Thus, understanding the processes that balance the nitrogen budget in cold seeps is essential4,5. Diazotrophs, organisms that convert dinitrogen gas (N2) into bioavailable ammonium (NH4+) through biological nitrogen fixation, are widespread in these environments, supported by diverse energy sources from either cultivated or uncultivated lineages6. Concurrently, nitrogen loss microbes convert bioavailable nitrogen back into N2 to maintain a balanced nitrogen cycle. However, studies on nitrogen loss processes in deep-sea cold seeps and the responsible microbial communities are relatively limited7,8,9. Understanding these processes is crucial for comprehending how cold seeps contribute to the broader nitrogen cycle in marine systems.
In marine sediments, two primary microbial processes for nitrogen loss are denitrification and anaerobic ammonium oxidation (anammox)10,11. The relative contributions of denitrification and anammox to nitrogen loss vary across different marine sediments. Most studies have focused on areas within 1000 meters of water depth, where denitrification typically accounts for over 80% of nitrogen loss10,12,13. Meanwhile, in deeper sea sediments (> 1000 meters water depths), anammox emerges as a crucial process, which contributes up to 50% to the N2 production rates measured in sediments of the Cascadia Basin14, Peru margin15, and East China Sea16. These observations indicate that both denitrification and anammox may play a role in nitrogen loss at cold seeps. However, the exact contributions of these processes in balancing the nitrogen budget at cold seep sites remain unclear.
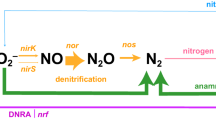
Denitrification occurs in two forms: classical and oxygenic. Classical denitrification reduces NO3– to NO2– and then to nitric oxide (NO), which is subsequently converted to nitrous oxide (N2O). This N2O is further reduced to N2 by the enzyme nitrous-oxide reductase (N2OR)17. N2OR, encoded by nosZ clade I and II, is the only enzyme that biologically converts N2O to N218. NosZ clade I is primarily found in some members of Alpha-, Beta-, and Gammaproteobacteria that possess the complete denitrification pathway. In contrast, NosZ clade II has been found in diverse bacterial groups that lack nitrite reductase genes (nirS or nirK), including Gemmatimonadetes, Verrucomicrobia, Gammaproteobacteria, Campylobacterota, Myxococcota, Planctomycetota and Chloroflexi19,20,21,22. While NosZ clade I is well-documented, recent studies suggest that NosZ clade II may play a more important role in reducing N2O in certain ecosystems than previously assumed19,21,23,24. Oxygenic denitrification simplifies this process by converting NO into N2 and molecular oxygen (O2) through the action of nitric oxide dismutase (Nod), bypassing the production of N2O25,26. The nod genes were first identified in the genomes of Candidatus Methylomirabilis oxyfera-like bacteria (also known as NC10 or nitrite-dependent methane-oxidizing bacteria)26,27,28, which use Nod to generate N2 and O2. The generated O2 is utilized to catalyze methane oxidation, thereby coupling oxygenic denitrification with aerobic methanotrophic pathways in anoxic environments27,29,30. These organisms have been detected in globally distributed cold seeps and other seafloor sediments through metagenomic and amplicon sequencing of pmoA or 16S rRNA genes31,32,33. Other bacteria like the Gammaproteobacterium strain HdN1, and species from the genera Sediminibacterium and Algoriphagus (within the phylum Bacteroidota)34, which also possess nod genes, may produce oxygen via dismutation, suggesting the potential involvement of microbes in oxygen production at cold seeps.
Compared to denitrification, anammox combines NO2– with NH4+ to form N2 using carbon dioxide (CO2) or carbonate as the carbon source, providing another nitrogen removal pathway with lower organic carbon requirements10,35. This process includes the reduction of NO2– to NO, which then reacts with NH4+ to create hydrazine (N2H4), a highly reactive and toxic compound with a low redox potential which then is oxidized to N2. The hydrazine-forming reaction, facilitated by hydrazine synthase (Hzs) is essential for the anammox pathway due to its unique biochemical properties. Known anammox bacteria are all affiliated with the Planctomycetota phylum, specifically within five families in the Brocadiales order: Candidatus Scalinduaceae, Candidatus Brocadiaceae, Candidatus Anammoxibacteraceae, Candidatus Bathyanammoxibiaceae, and Candidatus Subterrananammoxibiaceae36,37,38. Although these lineages are widely distributed across marine ecosystems and found in sediments from diverse marine environments, pure cultures have not yet been obtained39,40,41. Further studies using amplicon sequencing of 16S rRNA, hydrazine dehydrogenase (hzo), and hzsB genes have discovered diverse anammox bacteria in deep-sea cold seep sediments of the Okhotsk Sea and the South China Sea8,9. These findings suggest that the deep-sea cold seep environment might harbor previously undescribed anammox bacteria outside the Planctomycetota phylum.
In this study, we aim to investigate the contributions of denitrifying and anammox microbial communities in nitrogen loss at cold seep habitats, along with their diversity. We first provide geochemical evidence for nitrogen losses in cold seeps, based on data from 324 sediment samples collected from three sites—Lingshui, Haima, and Site F (Fig. 1 and Supplementary Fig. 1). This evidence is further supported by measurements of denitrification and anammox activities, conducted through slurry incubation experiments with 15N-labeled tracers. Subsequently, we explore the genes associated with nitrogen losses (nosZ, nod, and hzsA) and the diversity of microbes linked to these processes. This is achieved through sequence- and structure-based analyses using a detailed gene and genome catalog compiled from 165 metagenomes from 16 cold seep sites. Our findings reveal that cold seeps are overlooked areas for nitrogen loss in marine sediments under high pressure and low temperatures. Nitrogen loss in this habitat is mediated by diverse microbial populations, including newly identified phyla of anammox bacteria, contributing to this process.

a–d Depth concentration profiles of nutrients (SO42–, NH4+, NO3– and NO2–) in porewater (0 − 26 cmbsf, n = 231) collected from Lingshui, Haima and Site F cold seeps. e, f Depth profiles of N2 and their δ15N values in the headspace gas of Site F-10 and Site F-14 cold seep sediments. g–i Depth profiles of TOC, TN, and TOC: TN in cold seep and non-seep sediments (0 − 36 cmbsf, n = 209) collected from Lingshui, Haima, Site F, Shenhu, and Qiongdongnan. j–m Depth concentration profiles of dissolved metals (Cu2+, Zn2+, Ca2+, and Fe2+) in porewater (0 − 26 cmbsf, n = 231) collected from Lingshui, Haima and Site F cold seeps. Black, green, orange, dark blue, and red points represent the concentration of geochemical parameters in Lingshui, Haima, Site F, Shenhu, and Qiongdongnan, respectively. Source data are provided as a Source Data file.
Results and Discussion
Geochemical evidence for nitrogen loss in cold seeps
Our geochemical measurements provide evidence for nitrogen loss in cold seep sediments. A total of 324 sediment samples from 37 cores with depths of 0–36 cm below the seafloor (cmbsf) were collected from three different cold seeps—Lingshui, Haima, and Site F (Supplementary Fig. 1). These sites represent different stages of cold seep activity with varying methane seeping intensity42. Correspondingly, sulfate (SO42–) concentrations in the porewater showed coherent downcore variations, either decreasing rapidly or remaining relatively stable depending on methane fluxes (Fig. 1a and Supplementary Data 1). The predominant form of dissolved inorganic nitrogen was NH4+ (Fig. 1b), with the highest concentrations found at Site F (8.6–867.2 μM, average 266.0 μM) and lower concentrations at Lingshui and Haima (averaged at 61.5 and 15.0 μM, respectively). Porewater NO3– and NO2– concentrations (averaged at 1.6 and 0.6 μM, respectively) were generally lower than NH4+ across all sites (Fig. 1c-d). Following this, we detected the signature of active N2 production, as evident by the downcore increasing concentrations of N2 (1.89–6.12% of the headspace gas) in two cores, with δ15N values in the range of − 3.08–6.11‰ (Fig. 1e, f). The depleted values of δ15N (− 3.09–0.10‰) indicated intense denitrification and anammox activities at Site F-1443,44. Together, our extensive geochemical measurements indicate the occurrence of nitrogen loss in cold seep sediments.
Total organic carbon (TOC), total nitrogen (TN), and TOC:TN ratio varied considerably between sites, with both TOC and TN generally decreasing with sediment depth (Fig. 1g–i). TOC levels at the Lingshui-6/-10/-11/-12, Haima-7/-8/-9, and Site F−14/-15 sites were much higher than other sites, suggesting that these sites could provide more electron donors for heterotrophic denitrification and leading to potentially higher nitrogen loss rates (Supplementary Fig. 2). Enzymes involved in microbial nitrogen loss processes require metal cofactors45,46,47, which were abundant in all studied sediments. In Lingshui, copper (Cu2+) reached the highest concentration, averaging 26.6 nM, while zinc (Zn2+) peaked in Haima, averaging 100.4 nM (Fig. 1j, k). Calcium (Ca2+) displayed a decreasing trend in Site F, while iron (Fe2+) was highest in Haima, averaging 11.9 μM (Fig. 1l, m).
Slurry experiments indicate substantial nitrogen loss rates in cold seep sediments
We examined potential nitrogen loss rates and the contribution of anammox to N2 production by conducting slurry experiments with nitrogen isotope tracing on sediment samples up to 36 cm deep from Lingshui, Haima, Site F, Shenhu, and Qiongdongnan (Supplementary Fig. 1) at 4 °C. We detected both denitrification and anammox, with rates significantly higher in cold seep regions (n = 60) compared to non-seep regions (n = 34; P < 0.0001; Fig. 2b and Supplementary Data 2). In cold seeps, average potential denitrification rates were measured at 2.30 ± 4.03 nmol cm−3 h−1, and anammox rates were 0.14 ± 0.21 nmol cm−3 h−1. In contrast, non-seep regions had denitrification and anammox rates of only 0.1 ± 0.1 nmol cm−3 h−1 and 0.04 ± 0.05 nmol cm−3 h−1, respectively. It should be noted that the lesser importance of anammox in the cold seep sediments is mainly attributed to higher organic carbon contents (Fig. 1g), which favor denitrification over anammox11,48.

a Potential N2 production rates (denitrification, n = 681; anammox, n = 648) and the percentage of N2 production attributed to anammox (ra, %; n = 698) across various environments, measured via slurry incubations with nitrogen isotope tracing. DEN and ANA represent denitrification and anammox rates respectively. Environmental differences were computed using two-sided Kruskal-Wallis rank-sum tests; non-seeps vs. cold seeps were compared using a two-sided student’s t test (**: P < 0.01). Data are presented as mean ± SD. b Potential N2 production rates and ra in non-seep (n = 34) and cold seep (n = 60) measured in this study. Differences were computed using a two-sided student’s t test (***: P < 0.001; ****: P < 0.0001). Boxplot: center line, median; box limits, upper and lower quartiles; whiskers, 1.5 × interquartile range; points, outliers. c Relative abundance (GPM) of nosZ, nod, hzsA, nifH, reductive dsrA, and oxidative mcrA genes in sediment metagenomes (n = 163). Inserted plots highlight genes: all nosZ genes (dark blue), nosZ Clade I genes (orange), nosZ Clade II genes (green), nod genes (dark red), hzsA genes (dark purple), nifH genes (brown), reductive dsrA genes (pink), oxidative mcrA genes (dark yellow). Differences were computed using Kruskal-Wallis rank-sum tests. Data are presented as mean ± SD. d Expression (TPM) of nosZ, nod, hzsA, and nifH genes in sediment metatranscriptomes (n = 33). Inserted plots show the transcriptional abundance of nitrogen loss genes, with differences computed using Kruskal-Wallis rank-sum tests. Data are presented as mean ± SD. e Relationships between gene abundance (GPM) of nosZ, nod, hzsA, and depths (cmbsf) in cold seeps. Average abundance per sample is shown, with regression lines and R values. Source data are provided as a Source Data file.
Although the anammox contributions were lower than those measured previously in deep-sea sediments (Fig. 2a), they were comparable to values measured in sediments from continental slope15,41 and deep-sea14 environments. Denitrification is the primary nitrogen loss process in these environments, with anammox contributing to roughly 26.14 ± 28.93% of the total nitrogen loss in cold seeps (Supplementary Fig. 2). The rich organic carbon and tight correlation between denitrification rates and TOC contents in cold seeps (P < 0.01; Supplementary Fig. 3a) suggest that heterotrophic denitrification is a likely metabolic pathway7. However, deep-sea cold seeps are chemosynthetic ecosystems where seepage fluids rich in hydrogen sulfide, methane, and other hydrocarbons provide energy for diverse microbial communities49. These chemosynthetic communities may also play an important role in the denitrification process. For instance, chemosynthetic Campylobacterota are abundant in cold seep sediments (Fig. 3a), which are likely responsible for sulfide oxidation coupled with NO3– reduction50. In addition, Beggiatoaceae, a chemolithoautotrophic sulfur-oxidizing bacterium51, is one of the most prevalent NosZ-encoding organisms in cold seep sediments (Supplementary Data 3). A tight correlation between anammox and denitrification rates was observed in cold seeps and non-seep (P < 0.01; Supplementary Fig. 3c and d), aligning with the fact that denitrification from nitrate to nitrite provides the necessary NO2– for anammox in marine sediments52,53. Despite low-temperature conditions, several cold seep sites—specifically Haima-6, Haima-7, Haima-8, Lingshui-10, and Lingshui-11—exhibited exceptionally high rates of nitrogen loss (up to 17.65 ± 1.24 nmol cm−3 h−1; Supplementary Fig. 2 and Supplementary Data 2) possibly related to abundant carbon sources (1.01–2.37%; Supplementary Data 1). These rates are comparable to those in estuarine and coastal environments of higher temperatures, as well as other known cold seep sites such as those in the Gulf of Mexico7, but are considerably higher than typical deep-sea sediments (Fig. 2a and Supplementary Data 3). However, it should be noted that these comparisons are approximations, as variations in organic matter concentrations and composition across regions introduce uncertainties that limit the reliability of direct comparisons.

a Structure trees created with Foldtree and affiliated taxonomic phylum of NosZ in MAGs. nosZ Clade I (NosZG1, NosZG2) is shown in orange, while nosZ Clade II (NosZG3, NosZG4, NosZG5, NosZG6, NosZG8) is shown in green. NosZG8, belonging to nosZ Clade II, was only found through structural analysis. The scale bar indicates the mean number of substitutions per site. Surrounding the tree, the affiliated taxonomic phylum and AlphaFold-predicted 3D structures of representative NosZ are displayed. b Comparison of nos clusters from genomes harboring Clade I or Clade II nosZ genes. The nos clusters of Clade II harbor the atypical nosZ and encode predicted iron-sulfur-binding proteins (labeled “4Fe-4S” or “2Fe-2S”) and c-type cytochromes (cy-c). Accessory genes (nosD, nosF, nosL, and nosY) are generally conserved across nos clusters with both typical (Clade I) and atypical nosZ. Non-colored genes in the operons have no orthologs in any other known nos cluster. nosR and nosX are associated exclusively with typical nos clusters. Details for the taxonomy and annotations of nosZ-containing contig are provided in Supplementary Data 9, 12, and 13.
These findings suggest that certain denitrifying or anammox microbes have adapted or evolved to thrive in deep-sea cold seeps with high methane and sulfide concentrations. Although our experiments did not replicate the high hydrostatic pressure found in natural settings, research has shown that N2 production rates measured in the laboratory closely matched those observed in situ using a benthic lander in hadal sediments of the Atacama Trench11. We extrapolated the potential nitrogen loss rates measured in our study across the globally estimated seepage area using methods from a previous study54. Assuming a sediment mean bulk dry density of about 1.3 g cm−3 55 and an estimated global cold seep area of 2.05–3.15 × 105 km256,57, we calculated a global nitrogen loss flux from denitrification and anammox in the top 5 cm of cold seeps sediments to be around 4.96–7.63 Tg N per year. This flux represents roughly 1.65–2.54% of the global marine sediment nitrogen loss (estimated at 300 Tg N per year)58, underscoring that, despite covering only about 0.057–0.087% of the global marine area, cold seeps are potential nitrogen loss hotspots in deep-sea sediments. It is important to note that the measured rates are potential rates, meaning that they could be higher than actual in situ rates by a variable and unknown degree. Our use of slurry incubations provided a controlled method for assessing nitrogen loss potential but did not fully replicate natural conditions and, therefore, might not capture exact in situ rates. In addition, the extent of global cold seep areas remains imprecise, as cold seep sizes can vary significantly, adding uncertainty to our estimate. Nonetheless, this calculation provides a first-order quantification that highlights the potential importance of cold seeps in global sedimentary nitrogen loss. Future studies with in situ techniques and advanced modeling approaches will help refine these estimates and better capture the true nitrogen loss rates in these unique environments.
Near-surface sediments are hotspots of nitrogen loss gene diversity
To investigate the diversity of nitrogen loss genes in cold seeps, we further focus on their evolution, taxonomy, and potential horizontal transfer via mobile genetic elements (MGEs). Using a gene catalog of 147 million non-redundant genes from cold seeps59, we delved into the diversity of genes linked to nitrogen loss, focusing on nitrous-oxide reductase (NosZ), nitric oxide dismutase (Nod), and hydrazine synthase (HzsA) (Supplementary Fig. 4). We identified 530 NosZ sequences containing cupredoxin-related protein active domains (Supplementary Fig. 5). These sequences are categorized into two groups based on their signal peptides: clade I (n = 164) utilizes the twin-arginine translocation (Tat) pathway60, while clade II (n = 366) has an additional c-type heme domain at the C-terminus, associated with the secretory (Sec) pathway20,24. Furthermore, we discovered a sequence-divergent branch called NosZG7 (n = 27), which, despite sequence differences, showed structural congruence with canonical NosZ (Supplementary Fig. 5b). In addition, we identified 151 putative Nod sequences, all bearing conserved catalytic site residues similar to those of the putative nitric oxide dismutase34 from the NO-dismutating bacterium Methylomirabilis oxyfera (Supplementary Fig. 6). The sequence-divergent branches Cluster1 (n = 28) and Cluster5 (n = 8) were only found through phylogenetic and structural analysis, respectively. We also identified 644 HzsA sequences with hydrazine synthase alpha subunit domains and the pentacoordinated c-type heme47. The HzsA sequences were classified into six clades, with five branches identified through structural analysis as divergent from those of known anammox bacteria (Supplementary Fig. 7). Among the genes of the nosZ clade II, the most abundant contig belongs to Gammaproteobacteria, while that of nosZ clade I genes is affiliated with Alphaproteobacteria (Supplementary Data 4). Furthermore, the predominant contigs of nod and hzsA genes are assigned to Scalindua (Planctomycetota) and Bacteroidales (Bacteroidota), respectively (Supplementary Data 4). Notably, while no hzsA and nod protein sequences were detected in MGEs, we found 31 nosZ sequences in MGEs, with clade I (n = 24; Supplementary Fig. 8) being more prevalent than clade II (n = 7). This observation suggests that horizontal gene transfer via MGEs may play a significant role in the diversification of nosZ-bearing denitrifying microorganisms in deep-sea cold seeps, supporting the hypothesis that MGEs are crucial for genetic exchange and adaptation in these environments61.
The abundance, expression, and depth distribution of nitrogen loss genes in cold seep sediments were explored, revealing their role in nitrogen cycling dynamics and their relationship with environmental drivers. The average abundances of nosZ, nod, and hzsA genes were 4.84, 0.90, and 2.78 genes per million reads (GPM; 0–6855 cmbsf; Fig. 2c, Supplementary Fig. 9a and Supplementary Data 5), respectively. These abundances are one-quarter that of the reductive dsrA gene (averaging 21.32 GPM), indicative of sulfate reduction, and only one-tenth of the oxidative mcrA gene (averaging 46.15 GPM), which is indicative of methane oxidation. Similarly, they are one-tenth the abundance of the nifH gene (averaging 55.45 GPM), associated with nitrogen fixation. However, our measurements found that sediment nitrogen fixation rates were significantly lower than nitrogen loss rates in cold seeps (P < 0.001; Supplementary Fig. 10 and Supplementary Data 2). This apparent discrepancy can be explained by the energy-intensive nature of nitrogen fixation, which requires substantial energy and reducing equivalents62, thereby limiting its activity despite higher gene abundance. In addition, alternative nitrogen sources, like ammonium from subsurface seepage56, and efficient recycling processes such as ammonification43, sustain nitrogen availability, offsetting losses and maintaining productivity in cold seep ecosystems. The expression levels of nitrogen loss genes indicate that microbial nitrogen loss processes are active, particularly at the surface of cold seep sediments, with average values of 2.38 transcripts per million reads (TPM) for nosZ, 0.20 TPM for nod, and 0.48 TPM for hzsA (Fig. 2d, Supplementary Fig. 9b and Supplementary Data 6). Specifically, nosZ clade II genes were more abundant than clade I genes (P < 0.0001; Fig. 2c, Supplementary Fig. 9a and Supplementary Data 5). The expression level of nosZ clade II genes (averaging 3.71 TPM) was also higher than that of nosZ clade I genes (averaging 1.13 TPM; P < 0.0001; Supplementary Fig. 9b), indicating that nosZ clade II may play a more important role in N2O consumption in cold seeps19,23,24. The distributions of nosZ, nod, and hzsA genes varied with sediment depth (0–300 cmbsf), showing generally negative trends with increasing depth, indicating diminished nitrogen loss activity deeper in the sediments (Fig. 2e). However, within the shallow surface layers (up to 40 cmbsf), the abundance of nosZ, nod, and hzsA genes positively correlated with depth (P < 0.05; Supplementary Fig. 9c and Supplementary Data 8). In addition, cold seep nitrogen loss gene abundance exhibited statistically significant differences across surface and middle/deep depths, decreasing with sediment depth, with lower abundances in deeper sediments. Specifically, gene abundances ranged from 1.29 to 6.88 GPM at the surface (0–50 cmbsf), 0.08 to 1.41 GPM in the middle depth (50–500 cmbsf), and 0.20 to 0.98 GPM in deep sediments (> 500 cmbsf) (P < 0.0001; Fig. 2c, e and Supplementary Data 5). To identify environmental drivers in the shallow surface layers (up to 40 cmbsf), we correlated the relative abundance of nitrogen loss genes with environmental factors (Supplementary Fig. 9c). Overall, NH4+ and NO3– were the strongest correlates of nosZ, nod, and hzsA genes in the surface layer (P < 0.05). The correlation points towards a dominant contribution of denitrifiers and anammox bacteria, which is linked to NH4+ and NO3– conversion.
NosZ and Nod proteins from seep denitrifiers exhibit considerable structural diversity
To explore the diversity, distribution, and structural features of nosZ genes in cold seep environments, we then focus on their phylogenetic variation, transcriptional activity, and metabolic diversity across different microbial phyla. From a cold seep genome catalog of 3164 metagenome-assembled genomes (MAGs), we identified 142 nosZ sequences, which were categorized into nosZ clade I (n = 52) and nosZ clade II (n = 90) based on phylogenetic and structural analyses (Supplementary Fig. 11 and Supplementary Data 9). The relative abundance of the MAGs that comprised nosZ genes ranged from 0.0001 to 0.0263% in the 165 sediment samples of cold seeps (Supplementary Data 9). Structurally, the N-terminal of cold seep nosZ clades I and II N2ORs configurations differ to align with their physiological functions (Fig. 3a), adopting signal peptides63 for either the Tat pathway in clade I or the Sec pathway in clade II (Supplementary Figs. 12, 13 and Supplementary Data 15). In addition, some nosZ clade II variants possess a C-terminal α-helix, which surrounds the periphery of CuA and CuZ sites of adjacent monomers in the structure (Fig. 3a and Supplementary Fig. 13b), potentially enhancing the stability of their active sites. Details of protein folding are provided in the Supplementary Note 1.
The distribution of the 142 nosZ genes across MAGs spans one archaeal and 18 bacterial phyla, reflecting the wide phylogenetic breadth of potential nitrous-oxide reducers in these environments (Supplementary Fig. 11a and Supplementary Data 9). The most common phyla containing nosZ genes are Pseudomonadota (n = 56) and Bacteroidota (n = 36), with fewer occurrences in Campylobacterota, Myxococcota, Chloroflexota, Desulfobacterota, Gemmatimonadota and other phyla (Fig. 3a). NosZ clade I genes are present in Alphaproteobacteria, Gammaproteobacteria, and a few other phyla, while clade II genes are more widespread, found in 15 bacterial groups and the archaeal phylum Thermoplasmatota (Supplementary Fig. 11 and Supplementary Data 9). Among the MAGs containing nosZ clade II genes, the most abundant MAG belongs to Desulfobacterota (Desulfobacterales, 0.0263%), whereas the most abundant MAG with nosZ clade I genes is affiliated with Pseudomonadota (Photobacterium frigidiphilum, 0.0137%). Desulfobacterales and Photobacterium frigidiphilum function as chemoautotrophs64 and heterotrophs65,66, respectively, highlighting the metabolic diversity of denitrifying microbes in cold seeps. This reveals three additional phyla (Desulfobacterota, Krumholzibacteriota, and Zixibacteria) capable of reducing N2O, considerably expanding the known genetic diversity of potential N2O reducers in deep-sea cold seep sediments. Most clade II MAGs contain multiple nosZ genes, with six MAGs containing two copies and one containing three copies, contrasting with the single nosZ gene typically found in clade I MAGs from cold seeps (Fig. 3b, Supplementary Fig. 14 and Supplementary Data 13). Notably, we identified a MAG, RS_10_sbin_88, from Gammaproteobacteria that carries one copy of both nosZ clade I and II genes. The nosZ clade I genes of Pseudomonadota were transcribed at high levels, up to 150.71 TPM, while nosZ clade II genes were transcribed at moderate to high levels in several phyla, up to 41.92–200.31 TPM, especially in Campylobacterota and Pseudomonadota (Supplementary Data 10, 11).
In addition, the diversity, distribution, and structure of nod genes involved in nitric oxide dismutation in cold seeps were explored. From 3164 MAGs, we identified five nod genes characterized by conserved enzymatic active center residues, distinguishing them from the related nitric oxide reductase (nor) by the substitution of “Thr” with “Ile”, “His” with “Asp”, and “Glu” with “Gln”34 (Fig. 4). The relative abundance of MAGs comprising nod genes ranged from 0.0004 to 0.0133% in the 165 samples of cold seeps (Supplementary Data 16). Structurally, the nitric oxide dismutase from cold seeps consistently exhibited conserved α-helix domains—four enzymes had 14 α-helices, and one had 13, reflecting notable structural conservation (Supplementary Fig. 15a–e and Supplementary Data 17). These nod genes were found in five MAGs across two bacterial phyla, indicating a broader presence of oxygenic denitrifiers than previously understood (Fig. 4a, b and Supplementary Data 16)25,27,34. Specifically, three of these MAGs are attributed to the Planctomycetota phylum (two from UBA1135 and one from Scalinduaceae), and two to the Bacteroidota phylum (as Maribacter_A sp023141835 and Cecembia rubra from the orders Flavobacteriales and Cytophagales, respectively). This expanded diversity suggests more widespread bacterial capabilities for nitric oxide dismutation at cold seeps than previously appreciated through metagenomic or environmental studies67. The gene cluster for nitric oxide dismutase in UBA1135 is conserved across two genomes, featuring a helix-turn-helix domain and electron transport-associated proteins, including ferredoxin oxidoreductase and a 4Fe-4S dicluster domain (Supplementary Data 18). In Cecembia rubra, the nod gene cluster includes not only the standard nos cluster (nosZ, nosD, nosF, nosY, and nosL) but also elements that regulate nitric oxide signaling and electron transport. However, in the other two MAGs from Scalinduaceae and Maribacter_A sp023141835, the genes downstream of nod are categorized as having domains of unknown function. In addition, most of these putative nod-containing MAGs possess genes for other nitrogen metabolic processes (Supplementary Data 16) such as genes for hydroxylamine dehydrogenase (hao), nitric oxide reduction (nor), nitrous oxide reduction (nosZ), nitrate reduction (nap/nir), nitrite reduction to nitric oxide (nir) and nitrite reduction to ammonia (nrf/nir).

a Maximum-likelihood phylogenetic tree of nod genes. b Structure tree of Nod created with Foldtree. Each Nod is labeled in dark blue and dark green in the phylogenetic tree and structure tree, respectively. The scale bar indicates the mean number of substitutions per site. c Alignment of Nod and reference sequences. It shows the same diagnostic substitutions as the putative nod gene of the known NO-dismutating microbe Methylomirabilis oxyfera. Details for Nod MAGs are provided in Supplementary Data 16.
Genetic potential for anammox is found in multiple phyla beyond Planctomycetota
To explore the diversity, distribution, and structural characteristics of hzsA genes involved in anammox processes in cold seep sediments, further analyses of the 3,164 MAGs identified 265 hzsA genes, the diagnostic gene of bacteria capable of anammox (Fig. 5a and Supplementary Data 19). The relative abundances of these hzsA-contain MAGs ranged from 0.0002% to 0.0201% in the 165 samples from cold seeps (Supplementary Data 19). These hzsA gene sequences, characterized by a motif binding to a pentacoordinated c-type heme in the hydrazine synthase alpha subunit47, were distributed across 94 bacterial MAGs spanning 10 bacterial phyla (Supplementary Fig. 16 and Supplementary Data 19). This distribution extends beyond the previously recognized anammox bacteria, which contain traditional HzsA found in the Brocadiales order of the Planctomycetota phylum36,37,38,68, to include previously undescribed HzsA in species outside of Brocadiales. These hzsA-containing MAGs are members of Planctomycetota (n = 55), Bacteroidota (n = 16), Acidobacteriota (n = 10), Verrucomicrobiota (n = 6), Sumerlaeota (n = 1), JABMQX01 (n = 1), Calditrichota (n = 1), Desulfobacterota (n = 1), Fibrobacterota (n = 1), Gemmatimonadota (n = 1) and Zixibacteria (n = 1) (Supplementary Fig. 16 and Supplementary Data 19). The hzsA genes of Acidobacteriota, Bacteroidota, Calditrichota, Planctomycetota, and Sumerlaeota were transcribed at moderate to high levels, up to 56.75–414.23 TPM, whereas fewer transcripts from Verrucomicrobiota were detected (Supplementary Data 20).

a Structure tree created with Foldtree and affiliated taxonomic phylum of HzsA in MAGs. Each cold seep HzsA is labeled in blue in the structure tree. The scale bar indicates the mean number of substitutions per site. Surrounding the tree, the affiliated taxonomic phylum and AlphaFold-predicted 3D structures of representative HzsA are displayed. b Reference structure of HZS in 5C2V belonging to Candidatus Kuenenia stuttgartiensis (Planctomycetota) and c FR_S1_sbin_24 belonging to Verrucomicrobiales (Verrucomicrobiota). In the HZS complex structure: α-subunits are colored green and yellow, β-subunits are colored blue and pink, and γ-subunits are colored gray and magenta. Details for the taxonomy of hzsA-containing contig are provided in Supplementary Data 19.
Among the 94 MAGs, 27 contain 42 hzd genes, which encode hydrazine dehydrogenase for oxidizing N2H4 to N2 (Supplementary Data 22). Notably, only two belong to the Ca. Scalinduaceae family within the Planctomycetota phylum, and both do not contain the complete hzsBC genes (Fig. 6 and Supplementary Data 23). However, 37 MAGs do contain these genes (Supplementary Data 23, 24), forming a multienzyme complex HZS-αβγ69. Of these, five MAGs comprise multiple hzsABC clusters and are associated with Phycisphaerae (n = 3), Verrucomicrobiae (n = 1), and Bacteroidia (n = 1). These findings considerably broaden the recognized phylogenetic diversity and environmental presence of potential anammox bacteria.

a Conservation of key genes encoding hydrazine metabolism in anammox bacteria genomes. b Comparison of the metabolic potential of anammox bacteria. Filled circles represent the presence of genes encoding the metabolic process, gray circles denote partial presence, and open circles indicate absence. Details for hzsA-containing annotations are provided in Supplementary Data 21–24.
We carefully examined the five MAGs with multiple hzsABC clusters and two Scalinduaceae MAGs, confirming they were not contaminated during binning (Supplementary Data 25). We found that the MAGs not only shared a similar gene cluster arrangement (Fig. 6a and Supplementary Data 23) but also structural similarities with known anammox bacteria (Fig. 5a). In addition to the essential genetic machinery for anammox metabolism, including hydrazine synthase (hzs) for converting NO and NH4+ to N2H4, and hydrazine dehydrogenase (hzd) for oxidizing N2H4 to N2, the MAGs also possess additional nitrogen metabolic processes such as genes for nitrate reduction (nap/nir), nitrite reduction to nitric oxide (nir) and nitrite reduction to ammonia (nrf/nir)70 (Fig. 6b and Supplementary Data 26). Notably, we identified a MAG affiliating with Verrucomicrobiae, FR_S1_sbin_24, which contained at least two copies of the hzsA, hzsB, and hzsC genes in a single contig (Fig. 6a and Supplementary Data 23), interspersed with several unknown proteins and upstream genes related to c-type cytochromes and Ca-activated chloride channels, which were annotated as belonging to the Verrucomicrobiae against NCBI’s non-redundant database. This MAG also encodes both hydroxylamine oxidoreductase (Hao) for nitrite reduction71 and NosZ (Fig. 6b), suggesting a versatile metabolic capability.
Both traditional and previously undescribed HzsA proteins encoded in the seven MAGs are structured into three distinct domains (Fig. 5a, Supplementary Figs. 17a, b, and 18a, b and Supplementary Note 2)47. However, traditional HzsA proteins lack an N-terminal transmembrane α-helix (Supplementary Fig. 17c, d), as do the previously undescribed HzsA proteins described here (Supplementary Fig. 18c, d). Only the previously undescribed HzsA proteins include N-terminal signal peptides indicative of the Sec secretion pathway (Supplementary Fig. 18e, f). In addition, these proteins exhibit significant variation in their C-terminal structures, particularly the previously undescribed HzsA proteins, which include a C-terminal α-helix that may stabilize the binding at the heme αI active center (Fig. 5a and Supplementary Figs. 17a, b, 18a, b). These proteins assemble into one or two heterotrimer complexes as predicted by AlphaFold Multimer (Fig. 5c and Supplementary Fig. 19). In each complex, one of the β and γ subunits of the previously undescribed HzsA are fused into a single polypeptide chain, consistent with earlier studies72. The previously undescribed HzsA proteins retain essential domains for heme binding and electron transfer, indicating they are functionally capable of catalyzing anammox reactions. Structural adaptations, such as N-terminal signal peptides and C-terminal α-helices, may optimize these proteins for the unique conditions of cold seeps, showcasing the evolutionary adaptability of anammox bacteria in nitrogen cycling.
Implications of nitrogen loss in cold seeps
In the past, gene annotations primarily relied on sequence similarity, which often overlooked genes that are distantly related yet functionally similar. However, over long evolutionary time scales, multiple substitutions at the same site can cause uncertainty in sequence alignment. Structures evolve at a slower rate than the underlying sequence mutations and are more conserved, emphasizing the importance of protein structural analysis73. By employing protein structural similarities and phylogenetic analysis, we have discovered that the NosZ clade II is notably more diverse and abundant in deep-sea cold seeps than previously assumed. This diversity may have significant implications for the role of NosZ clade II in N2O consumption in cold seeps. Members of the Planctomycetota phylum, as well as the orders Flavobacteriales and Cytophagales, might be notable contributors to nitrogen loss through nitric oxide dismutation. These organisms might also act as oxygen producers which could be linked to the presence of aerobic methane and sulfide oxidizers within apparently anoxic layers of cold seep ecosystems74. Crucially, our findings also indicate that anammox bacteria are found in multiple phyla (e.g., Bacteroidota, Acidobacteriota, and Verrucomicrobiota) beyond Planctomycetota and that these overlooked lineages actively express anammox genes. Our findings highlight the importance of nitrogen loss through denitrification and anammox processes at cold seeps. Overall, this study provides evidence supporting the presence of numerous previously undescribed, cold-adapted microbial lineages involved in denitrification (including oxygenic denitrification) and anammox processes. It establishes cold seeps as considerable nitrogen loss hotspots in the deep sea and as important contributors to the global nitrogen cycle, broadening the recognized roles of cold seeps beyond serving as oases of diversity, productivity, and methane removal.
Methods
Sampling and geochemical measurements
A total of 324 samples from 37 push cores were collected by a remotely operated vehicle (ROV) at Lingshui, Haima, and Site F cold seeps in the South China Sea between 2020 and 2024 (Supplementary Fig. 1). Upon retrieval on deck, sediment cores were immediately placed in a helium-filled glove bag. Porewater was extracted at 2 cm depth intervals using Rhizon samplers with 0.2 μm pore size (Rhizosphere, Netherlands). Collected porewater samples for use for metal analysis were acidified to pH ~2 with HNO3 (Optima grade, Thermo Scientific, USA), and stored at 4 °C before analysis. For nitrogen gas measurement in sediments, 3 mL of sediments at 2 cm intervals was transferred using a 5 mL cut-off syringe into a 22 mL serum vial. The vial was immediately crimp-sealed and allowed to equilibrate to enable the dissolved gases to partition into the headspace at 4 °C. Samples of porewater and sediment for analyzing nutrients, sulfate, and calcium were stored at − 20 °C until analysis.
The concentrations of N2 in the headspace gas, along with its δ15N values were measured using gas chromatography with thermal conductivity detection (Agilent, USA) and a continuous-flow isotope-ratio mass spectrometer (SerCon, UK). The N2 concentration is expressed as the percentage of N2 in the total headspace gas volume of the 22 mL serum vial. The concentrations of ammonium (NH4+), nitrite (NO2–), and nitrate (NO3–) in porewater were determined using a Quattro continuous flow analyzer (SEAL Analytical AA3, Germany). Sulfate (SO42–) and calcium (Ca2+) concentrations were determined using a Dionex Ion Chromatograph (Thermo Scientific Dionex, USA). Dissolved metals (Fe2+, Cu2+, and Zn2+) in porewater were measured using ICP-MS (Thermo Scientific, USA). Sediment total organic carbon (TOC) and total nitrogen (TN) were measured using a Vario Micro Cube elemental analyzer (Elementar, Germany) after the sediments were treated with 1 M HCl to remove carbonates.
Determination of nitrogen loss rates
Nitrogen loss rates were determined using 60 samples from 12 push cores (Lingshui-10/-11/-12, Haima-3/-4/-5/-6/-7/-8/-9, and Site F-15/-16) collected by an ROV during 2023 and 2024 from the cold seeps of Lingshui, Haima, and Site F, with water depths ranging from 1171 to 1758 meters. In addition, 34 samples from 8 push cores (Shenhu-1/-2/-3/-4/-5, Qiongdong-1, Lingshui-13, and Site F-17) were collected from the non-seep area in Shenhu, Lingshui, Site F, and Qiongdongnan, at depths of 489 to 1761 meters in the South China Sea (Supplementary Fig. 1). Potential nitrogen loss rates were measured using N isotope-tracing techniques as follows75,76.
Potential denitrification and anammox rates were measured using the N-isotope tracing technique based on previous studies with some modifications48,75,77. Briefly, slurries were prepared using collected sediments and artificial seawater matching in situ salinity, at a sediment/water volume ratio of 1:7. The mixture was purged with helium for 30 min and stirred vigorously to ensure homogeneity. Gas-tight borosilicate vials (n = 21; Exetainers, Labco, UK) were then filled with slurries under a helium atmosphere. Subsequently, the vials were pre-incubated at near in situ temperature (4 °C) for 48 h to remove residual NOx– (NO3– + NO2–) and dissolved oxygen. After the pre-incubation, three vials were used to measure the ambient NOx−. The residual slurries (n = 18) were divided into three groups, each amended with different nitrogen compounds in helium-purged stock solutions: (1) 15NH4+ (99.12%), (2) 15NH4+ + 14NO3–, and (3) 15NO3– (99.21%). The final concentration of 15N compounds in each vial was 75 μM. At 0 and 8 h incubation times, triplicate vials were sacrificed with 200 μL 50% ZnCl2 solution as initial and final samples, respectively. The 15N labeled N2 were measured using a membrane inlet mass spectrometer (MIMS, HPR-20, Hiden Analytical, UK) with a detection limit of 0.1 μM. Denitrification and anammox rates were estimated from the accumulation of 29N2 and 30N2 during the slurry incubation75,78. The respective contributions of denitrification and anammox to 29N2 production were quantified using Eq. (1).
where P29 (nmol cm−3 h−1) denotes the total 29N2 production rates, D29 (nmol cm−3 h−1) and A29 (nmol cm−3 h−1) denote the production rates of 29N2 from denitrification and anammox, respectively. Here, D29 was obtained by Eq. (2), assuming random paring of 14N and 15N from 14NO3− or 15NO3−79,80.
where P30 (nmol cm−3 h−1) denotes the total 30N2 production rates, FN (%) denotes the fraction of 15N in NO3−, which was obtained from the added 15NO3− and the measured residual ambient NOx−, ranging from 97.8% to 99.7%. The potential rates of denitrification and anammox were quantified by Eqs. (3) and (4).
where Dt and A29 (nmol cm−3 h−1) denote the denitrification and anammox rates, respectively. By convention, the percent of N2 production accounted for anammox is abbreviated as ra (%).
Metagenomic data processing
The metagenomic datasets from 165 samples were collected from 16 cold seep sites worldwide, including oil and gas seeps, methane seeps, gas hydrates, asphalt volcanoes, and mud volcanoes (Supplementary Fig. 1). Non-redundant gene and genome catalogs were constructed in our previous study59. Briefly, metagenomic sequence data were quality-controlled and assembled into contigs. Protein-coding sequences were then predicted and clustered to create a non-redundant gene catalog consisting of 147,289,169 representative clusters. Salmon (v1.10.2)81 was used to calculate gene abundance in each metagenome, which was then normalized to genes per million reads (GPM)82. Functional annotations were performed using eggNOG-mapper (v2.1.9) with default parameters83,84. MMseqs2 taxonomy (v13.45111; parameter: --tax-lineage 1)85 was used to assign taxonomic labels to each representative amino acid sequence with reference to GTDB R207 database86.
Contigs longer than 1000 bp were selected for subsequent binning, and the produced metagenome-assembled genomes (MAGs) underwent dereplication at 95% average nucleotide identity59. Then, the MAGs were checked by GUNC (v1.0.5; default parameters)87 to remove genomes potentially containing chimerism, retaining only those classified as “pass.GUNC”. A total of 3164 representative MAGs (completeness > 50% and contamination < 10%) were obtained. The relative abundance of each MAG was calculated using CoverM (v0.6.1; https://github.com/wwood/CoverM; parameters: -m relative_abundance --trim-min 0.10 --trim-max 0.90 --min-read-percent-identity 0.95 --min-read-aligned-percent 0.75). The taxonomy of each MAG was assigned using GTDB-Tk v2.1.1 with reference to GTDB R207 database86.
Identification of nitrous oxide reductase gene (nosZ)
For nosZ gene database search (see workflow in Supplementary Fig. 4a), we first queried protein sequences from the non-redundant gene catalog and 3164 MAGs against NCycDB88 using DIAMOND (version 2.0.14)89 in blastp mode (-k 1 -e 0.0001 -p 5). Subsequently, the nosZ reference sequences (n = 403) from the Greening lab metabolic marker gene databases90 were used to search for potential nosZ sequences in the non-redundant gene catalog and 3164 MAGs with DIAMOND blastp (version 2.0.14; --id 50)89. In addition, hidden Markov models (HMMs) of nosZ clade I (TAT-dependent nitrous-oxide reductase) and nosZ clade II (Sec-dependent nitrous-oxide reductase) were obtained from NCBI’s Protein Family Models using accession “TIGR04244.1”91 and “TIGR04246.1”92, respectively. These HMMs were used to screen proteins from the non-redundant gene catalog and 3164 MAGs with hmmsearch in HMMER v3.3.2 using the parameter -E 1e-5. All nosZ genes identified with the above three methods were merged, and any sequences shorter than 400 amino acids were excluded.
The phylogenetic trees of the amino acid sequences of filtered genes were constructed to validate the phylogenetic clades of nosZ against reference sequences. Sequences were aligned using MUSCLE (v3.8.1551)93 and trimmed with TrimAL (v1.4.1)94 with default settings. Maximum-likelihood trees were constructed with IQ-TREE (v2.2.0.3)95 with the “-m MFP -B 1000” options. The produced tree was visualized and beautified using the Interactive Tree of Life (iTOL; v6)96. Meanwhile, ESMFold97 was applied to predict the structure of each filtered gene. The 154 reference protein structures of NosZ were downloaded from AlphaFoldDB98 and Protein Data Bank (PDB)99. A structural tree of NosZ was constructed using Foldtree (https://github.com/DessimozLab/fold_tree) based on a local structural alphabet100 and visualized using iTOL (v6)96. The structure of each gene in both the phylogenetic and structural trees was predicted using AlphaFold2101 and aligned against PDB using Foldseek (v8.ef4e960)102 with parameters “--tmscore-threshold 0.5 -e 0.001”. Cupredoxin-related protein active domains of nitrous-oxide reductase were investigated against the ECOD (Evolutionary Classification Of protein Domains) database103 with a TM-score > 0.5 using the Foldseek easy-search module102.
Identification of nitric oxide dismutase gene (nod)
For the nod gene database search (see the workflow in Supplementary Fig. 4b), we initially downloaded reference protein sequences (n = 1036) in NCBI’s databases to build an HMM model (available at https://doi.org/10.6084/m9.figshare.25650927). Then, nod genes in the non-redundant gene catalog and 3,164 MAGs were extracted using the above HMM model with hmmsearch in HMMER v3.3.2 using “-E 1e-5”. All potential nod genes shorter than 200 amino acids were excluded from further analysis. Following the methodology used for nosZ genes, these nod genes underwent verification through phylogenetic and structural trees. The reference protein structures for 44 Nod proteins were predicted using AlphaFold2101. The structures of the filtered nod genes were then predicted using AlphaFold2 and aligned with the reference protein structures using Foldseek (v8.ef4e960)102. In addition, diagnostic amino acid residues in the active center of the enzyme34 were identified in all proteins with a TM-score > 0.5 using MAFFT (EMBL-EBI)104 and visualized with Jalview105.
Identification of hydrazine synthase and hydrazine dehydrogenase
To identify hzsA genes (see the workflow in Supplementary Fig. 4c), protein sequences in the non-redundant gene catalog and 3164 MAGs were firstly searched against NCycDB88, with the program DIAMOND blastp (version 2.0.14)89. Then, reference hzsA sequences (n = 14) from the Greening lab metabolic marker gene databases90 were used to search for potential hzsA sequences in the non-redundant gene catalog and 3164 MAGs with DIAMOND blastp (version 2.0.14; --id 50)89. In addition, hzsA genes of non-redundant gene catalog and 3164 MAGs were extracted using HMMER v3.3.2 with the HMM profile “PF13486” from the InterPro database47. All identified hzsA genes were merged, and sequences shorter than 400 amino acids were filtered out. These genes were then verified by constructing phylogenetic and structural trees. The structure of each gene was predicted using AlphaFold2101 and aligned with PDB99 using Foldseek (v8.ef4e960)102. In addition, the active domain of the hydrazine synthase alpha subunit was searched in protein structures with a TM-score > 0.5 using Foldseek’s easy-search module102.
To ensure the accuracy of identifying MAGs with hzsA genes, MAGpurify (v2.1.2) was used to detect contamination in MAGs through a combination of features and algorithms, including phylo-markers, clade-markers, tetra-freq, GC-content, and known-contam106. In addition, hzsB and hzsC sequences in MAGs were extracted with HMMER v3.3.2 using the “nitro.cycle.sub.hmm” model from the Metascan metabolic HMM database107. To further refine our search, the protein complexes of the previously undescribed clade hydrazine synthase (HzsABC) were predicted using AlphaFold (v2.0; model_preset = multimer)101. All structures were visualized and exported as images using PyMOL (http://www.pymol.org)108. In addition, the hzd gene associated with hydrazine dehydrogenase was identified in MAGs containing hzsA genes. To begin, we searched for reference protein sequences (n = 49) in NCBI’s databases to construct the HMM model (available at https://doi.org/10.6084/m9.figshare.25650927). Subsequently, hzd genes were extracted using hmmsearch in HMMER, applying the “-E 1e-5” parameter. The structures were then predicted using AlphaFold2 and aligned with reference proteins in the PDB databases using Foldseek (v8.ef4e960) with settings “--tmscore-threshold 0.5 -e 0.001”.
MAG annotations and topological structure predictions
The MAGs were annotated using DRAM (v1.3.5)82 and Prokka (v1.14.6)109 with the default settings against KEGG, Pfam, MEROPS and dbCAN databases. Gene context was visualized using Chiplot (https://www.chiplot.online/), with the files produced by DRAM and Prokka as the input. DeepTMHMM (v1.0.24)110 and SignalP (v6.0)111 were employed to predict transmembrane topology and signal peptides of NosZ, Nod and HzsA proteins, respectively.
Identification of nifH, dsrA, mcrA and mobile genetic elements
To identify nifH genes associated with nitrogen fixation, protein sequences in the non-redundant gene catalog was first searched against NCycDB88, using the program DIAMOND (version 2.0.14)89 as mentioned above. Then, we utilized nifH reference sequences (n = 1271) from the Greening lab metabolic marker gene databases90 to search for potential nifH sequences in the non-redundant gene catalog using DIAMOND blastp (version 2.0.14; --id 50)89. Subsequently, nifH genes were extracted using the NCBI’s Protein Family Models with HMM accession “TIGR01287.1” by HMMER v3.3.2. The genes were then verified through phylogenetic analysis as described in our previous study6. Conserved motifs (CXXR)6 were analyzed using MAFFT(EMBL-EBI)104 and visualized with Jalview105.
For the identification of dsrA and mcrA genes associated with sulfate reduction and anaerobic methane oxidation respectively, we employed eggNOG-mapper (v2.1.9; default parameters) for initial detection83,84. The reductive dsrA genes were further detected using DiSco (v1.0.0)112, while oxidative mcrA genes were confirmed via phylogenetic analysis as described in our previous study59. Classification of contigs belonging to mobile genetic elements (plasmids, proviruses, and viruses) was performed using Genomad v.1.5.0 with default parameters113.
Transcriptional activities of nitrogen loss genes
A total of 33 samples from various cold seeps—Haima, Qiongdongnan Basin, Shenhu area, and Jiaolong—were analyzed for metatranscriptomes, as outlined in our previous study114. Briefly, raw reads were quality filtered (parameters: --skip-bmtagger) using the Read_QC module within the metaWRAP (v1.3.2) pipeline115. SortMeRNA (v2.1)116 was employed to remove ribosomal RNA from quality-controlled reads using default settings. The abundances of transcripts for nosZ, nod, and hzsA genes were quantified by mapping filtered reads to a non-redundant gene catalog and genes of MAGs using Salmon (v.1.9.0; parameters: -validateMappings -meta)81. The results were expressed in transcripts per million reads (TPM)82.
Identification of nitrogen loss genes in surface sediments
A total of 34 samples from the Lingshui cold seeps in surface sediments, varying from 0 to 40 cmbsf, were analyzed in detail, as outlined in our previous study114. Briefly, the abundances of nosZ, nod and hzsA genes were quantified by mapping reads filtered using the Read_QC module within the metaWRAP to the non-redundant gene catalog and genes of 3164 MAGs using Salmon (v.1.9.0; parameters: -validateMappings -meta)81. The results were expressed in GPM.
Statistical analyses
Statistical analyses were performed using R v4.2.3. The normality of the data was assessed using Shapiro–Wilk tests prior to further analyses. The Kruskal-Wallis rank-sum test was used to compare the abundances of nitrogen loss genes across different depths, and their rates across different types of environments. Student’s t tests were utilized to examine differences in the relative abundance of nosZ, nod, and hzsA genes at varying depths, as well as nitrogen loss rates across non-seep and cold seep environments. Pairwise comparisons of environmental factors were conducted using Pearson’s correlation coefficients. In addition, relationships between the relative abundance of nosZ, nod, and hzsA genes and environmental factors was analyzed using Mantel tests.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The non-redundant gene catalog and the metagenome-assembled genomes (MAGs) catalog can be accessed at figshare (https://doi.org/10.6084/m9.figshare.22568107) and the NCBI database under BioProject PRJNA950938. Details for MAGs containing nosZ, nod, and hzsA genes, along with the protein structures and phylogenetic trees for NosZ, Nod, and HzsA, as well as all the HMM models developed in this study, are available at https://doi.org/10.6084/m9.figshare.25650927. The raw sequencing reads generated in surface sediments of the Lingshui cold seeps have been deposited in NCBI under BioProject ID PRJNA1169195. All additional data supporting the findings of this study are provided within the article and its Supplementary Information Files. Source data are provided in this paper.
Code availability
The present study did not generate codes, and the mentioned tools used for the data analysis were applied with default parameters unless specified otherwise.
References
Vigneron, A. et al. Contrasting pathways for anaerobic methane oxidation in Gulf of Mexico cold seep sediments. mSystems 4, e00091–00018 (2019).
Dong, X. et al. Thermogenic hydrocarbon biodegradation by diverse depth-stratified microbial populations at a Scotian Basin cold seep. Nat. Commun. 11, 5825 (2020).
Niu, M., Fan, X., Zhuang, G., Liang, Q. & Wang, F. Methane-metabolizing microbial communities in sediments of the Haima cold seep area, northwest slope of the South China Sea. FEMS Microbiol. Ecol. 93, fix101 (2017).
Tyrrell, T. The relative influences of nitrogen and phosphorus on oceanic primary production. Nature 400, 525–531 (1999).
Zehr, J. P. & Capone, D. G. Changing perspectives in marine nitrogen fixation. Science 368, eaay9514 (2020).
Dong, X. et al. Phylogenetically and catabolically diverse diazotrophs reside in deep-sea cold seep sediments. Nat. Commun. 13, 4885 (2022).
Bowles, M. & Joye, S. High rates of denitrification and nitrate removal in cold seep sediments. ISME J. 5, 565–567 (2011).
Shao, S. et al. Deep-sea methane seep sediments in the Okhotsk Sea sustain diverse and abundant anammox bacteria. FEMS Microbiol. Ecol. 87, 503–516 (2014).
Wu, J. et al. Unexpectedly high diversity of anammox bacteria detected in deep-sea surface sediments of the South China Sea. FEMS Microbiol. Ecol. 95, https://doi.org/10.1093/femsec/fiz013 (2019).
Devol, A. H. Denitrification, anammox, and N2 production in marine sediments. Annu. Rev. Marine Sci. 7, 403–423 (2015).
Thamdrup, B. et al. Anammox bacteria drive fixed nitrogen loss in hadal trench sediments. Proc. Natl. Acad. Sci. 118, https://doi.org/10.1073/pnas.2104529118 (2021).
Sun, L. et al. Biotic and abiotic controls on dinitrogen production in coastal sediments. Global Biogeochem. Cycles 35, e2021GB007069 (2021).
Wu, S. et al. Nitrogen cycling in China marginal seas: Progress and challenges. Marine Chem. 265-266, 104421 (2024).
Engström, P., Penton, C. R. & Devola, A. H. Anaerobic ammonium oxidation in deep‐sea sediments off the Washington margin. Limnol. Oceanogr. 54, 1643–1652 (2009).
Rich, J. J., Arevalo, P., Chang, B. X., Devol, A. H. & Ward, B. B. Anaerobic ammonium oxidation (anammox) and denitrification in Peru margin sediments. J. Marine Syst. 207, 103122 (2020).
Na, T. et al. N2 production through denitrification and anammox across the continental margin (shelf-slope-rise) of the Ulleung Basin, East Sea. Limnol. Oceanogr. 63, S410–S424 (2017).
Zumft, W. G. & Kroneck, P. M. Respiratory transformation of nitrous oxide (N2O) to dinitrogen by Bacteria and Archaea. Adv. Microb. Physiol. 52, 107–227 (2006).
Zhang, L., Wüst, A., Prasser, B., Müller, C. & Einsle, O. Functional assembly of nitrous oxide reductase provides insights into copper site maturation. Proc. Natl. Acad. Sci. USA 116, 12822–12827 (2019).
Xu, X. et al. NosZ clade II rather than clade I determine in situ N2O emissions with different fertilizer types under simulated climate change and its legacy. Soil Biol. Biochem. 150, 107974 (2020).
Sanford, R. A. et al. Unexpected nondenitrifier nitrous oxide reductase gene diversity and abundance in soils. Proc. Natl. Acad. Sci. USA 109, 19709–19714 (2012).
Lin, Y., Hu, H. W., Deng, M., Yang, P. & Ye, G. Microorganisms carrying nosZ I and nosZ II share similar ecological niches in a subtropical coastal wetland. Sci. Total Environ. 870, 162008 (2023).
Intrator, N., Jayakumar, A. & Ward, B. B. Aquatic nitrous oxide reductase gene (nosZ) phylogeny and environmental distribution. Front. Microbiol. 15, 1407573 (2024).
Jones, C. M. et al. Recently identified microbial guild mediates soil N2O sink capacity. Nat. Clim. Change 4, 801–805 (2014).
Hallin, S., Philippot, L., Loffler, F. E., Sanford, R. A. & Jones, C. M. Genomics and ecology of novel N2O-reducing microorganisms. Trends Microbiol. 26, 43–55 (2018).
Zhu, B. et al. Nitric oxide dismutase (nod) genes as a functional marker for the diversity and phylogeny of methane-driven oxygenic denitrifiers. Front. Microbiol. 10, 1577 (2019).
Ettwig, K. F. et al. Nitrite-driven anaerobic methane oxidation by oxygenic bacteria. Nature 464, 543–548 (2010).
Yao, X. et al. Methane-dependent complete denitrification by a single Methylomirabilis bacterium. Nat. Microbiol. 9, 464–476 (2024).
Zhu, B. et al. A novel Methylomirabilota methanotroph potentially couples methane oxidation to iodate reduction. mLife 1, 323–328 (2022).
Schmitz, E. V., Just, C. L., Schilling, K., Streeter, M. & Mattes, T. E. Reconnaissance of oxygenic denitrifiers in agriculturally impacted soils. mSphere 8, e00571-00522 (2023).
Zhu, B. et al. Long-read amplicon sequencing of nitric oxide dismutase (nod) genes reveal giverse oxygenic denitrifiers in agricultural soils and lake sediments. Microb. Ecol. 80, 243–247 (2020).
Jing, H., Wang, R., Jiang, Q., Zhang, Y. & Peng, X. Anaerobic methane oxidation coupled to denitrification is an important potential methane sink in deep-sea cold seeps. Sci. Total Environ. 748, 142459 (2020).
Jiang, Q., Jing, H., Liu, H. & Du, M. Biogeographic distributions of microbial communities associated with anaerobic methane oxidation in the surface sediments of deep-sea cold seeps in the South China Sea. Front. Microbiol. 13, 1060206 (2022).
Ruff, S. E. et al. A global comparison of surface and subsurface microbiomes reveals large-scale biodiversity gradients, and a marine-terrestrial divide. Sci. Adv. 10, eadq0645 (2024).
Ruff, S. E. et al. Hydrogen and dark oxygen drive microbial productivity in diverse groundwater ecosystems. Nat. Commun. 14, 3194 (2023).
Kartal, B., Kuenen, J. V. & Van Loosdrecht, M. Sewage treatment with anammox. Science 328, 702–703 (2010).
Suarez, C. et al. Metagenomic evidence of a novel family of anammox bacteria in a subsea environment. Environ. Microbiol. 24, 2348–2360 (2022).
Zhao, R., Biddle, J. F. & Jørgensen, S. L. Introducing Candidatus Bathyanammoxibiaceae, a family of bacteria with the anammox potential present in both marine and terrestrial environments. ISME Commun. 2, 42 (2022).
Zhao, R., Le Moine Bauer, S. & Babbin, A. R. Candidatus Subterrananammoxibiaceae,” a new anammox bacterial family in globally distributed marine and terrestrial subsurfaces. Appl. Environ. Microbiol. 89, e00800–e00823 (2023).
Yang, Y. et al. Activities and metabolic versatility of distinct anammox bacteria in a full-scale wastewater treatment system. Water Res. 206, 117763 (2021).
Cao, W., Guan, Q., Li, Y., Wang, M. & Liu, B. The contribution of denitrification and anaerobic ammonium oxidation to N2 production in mangrove sediments in Southeast China. J. Soils Sediments 17, 1767–1776 (2017).
Na, T. et al. N2 production through denitrification and anammox across the continental margin (shelf–slope–rise) of the Ulleung Basin, East Sea. Limnol. Oceanogr. 63, S410–S424 (2018).
Feng, D. South China Sea Seeps. (Springer Nature Singapore, Singapore; 2023).
Kuypers, M. M. M., Marchant, H. K. & Kartal, B. The microbial nitrogen-cycling network. Nat. Rev. Microbiol. 16, 263–276 (2018).
Tiirola, M. A., Rissanen, A. J., Sarpakunnas, M., Arvola, L. & Nykänen, H. Stable isotope profiles of nitrogen gas indicate denitrification in oxygen‐stratified humic lakes. Rapid Communi. Mass Spectrom. 25, 1497–1502 (2011).
Lu, Y. et al. The content of trace element iron is a key factor for competition between anaerobic ammonium oxidation and methane-dependent denitrification processes. Chemosphere 198, 370–376 (2018).
Müller, C. et al. Molecular interplay of an assembly machinery for nitrous oxide reductase. Nature 608, 626–631 (2022).
Dietl, A. et al. The inner workings of the hydrazine synthase multiprotein complex. Nature 527, 394–397 (2015).
Engström, P., Dalsgaard, T., Hulth, S. & Aller, R. C. Anaerobic ammonium oxidation by nitrite (anammox): implications for N2 production in coastal marine sediments. Geochim. Cosmochim. Acta 69, 2057–2065 (2005).
Levin, L. A. Oceanography and Marine Biology. (CRC Press, 2005).
Sun, Q. et al. Nitrogen and sulfur cycling driven by Campylobacterota in the sediment-water interface of deep-sea cold seep: a case in the South China Sea. mBio 14, e00117–e00123 (2023).
Desai, M. S., Assig, K. & Dattagupta, S. Nitrogen fixation in distinct microbial niches within a chemoautotrophy-driven cave ecosystem. ISME J. 7, 2411–2423 (2013).
McTigue, N., Gardner, W., Dunton, K. & Hardison, A. Biotic and abiotic controls on co-occurring nitrogen cycling processes in shallow Arctic shelf sediments. Nat. Commun. 7, 13145 (2016).
Zhao, R. et al. Geochemical transition zone powering microbial growth in subsurface sediments. Proc. Natl. Acad. Sci. USA 117, 32617–32626 (2020).
Lin, X. et al. Nitrogen losses in sediments of the East China Sea: spatiotemporal variations, controlling factors, and environmental implications. J. Geophys. Res. Biogeosci. 122, 2699–2715 (2017).
Lin, H. et al. Mercury methylation by metabolically versatile and cosmopolitan marine bacteria. ISME J. 15, 1810–1825 (2021).
Boetius, A. & Wenzhöfer, F. Seafloor oxygen consumption fuelled by methane from cold seeps. Nat. Geosci. 6, 725–734 (2013).
Li, J. et al. Deep sea cold seeps are a sink for mercury and source for methylmercury. Commun. Earth Environ. 5, 324 (2024).
Codispoti, L. A. et al. The oceanic fixed nitrogen and nitrous oxide budgets: Moving targets as we enter the anthropocene? Sci. Mar. 65, 85–105 (2001).
Han, Y. et al. A comprehensive genomic catalog from global cold seeps. Sci. Data 10, 596 (2023).
Graf, D. R., Jones, C. M. & Hallin, S. Intergenomic comparisons highlight modularity of the denitrification pathway and underpin the importance of community structure for N2O emissions. PLoS ONE 9, e114118 (2014).
Zhao, J. et al. Novel viral communities potentially assisting in carbon, nitrogen, and sulfur metabolism in the upper slope sediments of Mariana Trench. mSystems 7, e01358–01321 (2022).
Dixon, R. & Kahn, D. Genetic regulation of biological nitrogen fixation. Nat. Rev. Microbiol. 2, 621–631 (2004).
Bagos, P. G., Nikolaou, E. P., Liakopoulos, T. D. & Tsirigos, K. D. Combined prediction of Tat and Sec signal peptides with hidden Markov models. Bioinformatics 26, 2811–2817 (2010).
Chen, X. et al. Phylogenetically and metabolically diverse autotrophs in the world’s deepest blue hole. ISME Commun. 3, 117 (2023).
Ortega‐Retuerta, E. et al. Dissolved organic matter released by two marine heterotrophic bacterial strains and its bioavailability for natural prokaryotic communities. Environ. Microbiol. 23, 1363–1378 (2021).
Liu, Y. et al. Photobacterium sp. NNA4, an efficient hydroxylamine-transforming heterotrophic nitrifier/aerobic denitrifier. J. Biosci. Bioeng. 128, 64–71 (2019).
Ruff, S. E. et al. Widespread occurrence of dissolved oxygen anomalies, aerobic microbes, and oxygen-producing metabolic pathways in apparently anoxic environments. FEMS Microbiol. Ecol. 100, https://doi.org/10.1093/femsec/fiae132 (2024).
Lodha, T., Narvekar, S. & Karodi, P. Classification of uncultivated anammox bacteria and Candidatus Uabimicrobium into new classes and provisional nomenclature as Candidatus Brocadiia classis nov. and Candidatus Uabimicrobiia classis nov. of the phylum Planctomycetes and novel family Candidatus Scalinduaceae fam. nov to accommodate the genus Candidatus Scalindua. Syst. Appl. Microbiol. 44, 126272 (2021).
Strous, M. et al. Deciphering the evolution and metabolism of an anammox bacterium from a community genome. Nature 440, 790–794 (2006).
Kartal, B. et al. How to make a living from anaerobic ammonium oxidation. FEMS Microbiol. Ecol. 37, 428–461 (2013).
Ferousi, C. et al. Characterization of a nitrite-reducing octaheme hydroxylamine oxidoreductase that lacks the tyrosine cross-link. J. Biol. Chem. 296, https://doi.org/10.1016/j.jbc.2021.100476 (2021).
van de Vossenberg, J. et al. The metagenome of the marine anammox bacterium ‘Candidatus Scalindua profunda’illustrates the versatility of this globally important nitrogen cycle bacterium. Environ. Microbiol. 15, 1275–1289 (2013).
Illergård, K., Ardell, D. H. & Elofsson, A. Structure is three to ten times more conserved than sequence-a study of structural response in protein cores. Proteins Struct. Funct. Bioinf. 77, 499–508 (2009).
Ruff, S. E. et al. Global dispersion and local diversification of the methane seep microbiome. Proc. Natl. Acad. Sci. USA 112, 4015–4020 (2015).
Thamdrup, B. & Dalsgaard, T. Production of N2 through anaerobic ammonium oxidation coupled to nitrate reduction in marine sediments. Appl. Environ. Microbiol. 68, 1312–1318 (2002).
Song, G. et al. Response of benthic nitrogen cycling to estuarine hypoxia. Limnol. Oceanogr. 66, 652–666 (2021).
Brin, L. D., Giblin, A. E. & Rich, J. J. Environmental controls of anammox and denitrification in southern New England estuarine and shelf sediments. Limnol. Oceanogr. 59, 851–860 (2014).
Trimmer, M., Nicholls, J. C. & Deflandre, B. Anaerobic ammonium oxidation measured in sediments along the Thames estuary, United Kingdom. Appl. Environ. Microbiol. 69, 6447–6454 (2003).
Nielsen, L. P. Denitrification in sediment determined from nitrogen isotope pairing. FEMS Microbiol. Lett. 86, 357–362 (1992).
Risgaard-Petersen, N., Nielsen, L. P., Rysgaard, S., Dalsgaard, T. & Meyer, R. L. Application of the isotope pairing technique in sediments where anammox and denitrification coexist. Limnol. Oceanogr. Methods 1, 63–73 (2003).
Patro, R., Duggal, G., Love, M. I., Irizarry, R. A. & Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419 (2017).
Shaffer, M. et al. DRAM for distilling microbial metabolism to automate the curation of microbiome function. Nucleic Acids Res. 48, 8883–8900 (2020).
Cantalapiedra, C. P., Hernández-Plaza, A., Letunic, I., Bork, P. & Huerta-Cepas, J. eggNOG-mapper v2: functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol. Biol. Evol. 38, 5825–5829 (2021).
Huerta-Cepas, J. et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 47, D309–D314 (2019).
Mirdita, M., Steinegger, M., Breitwieser, F., Söding, J. & Levy Karin, E. Fast and sensitive taxonomic assignment to metagenomic contigs. Bioinformatics 37, 3029–3031 (2021).
Parks, D. H. et al. GTDB: an ongoing census of bacterial and archaeal diversity through a phylogenetically consistent, rank normalized and complete genome-based taxonomy. Nucleic Acids Res. 50, D785–D794 (2022).
Orakov, A. et al. GUNC: detection of chimerism and contamination in prokaryotic genomes. Genome Biol. 22, 1–19 (2021).
Tu, Q. et al. NCycDB: a curated integrative database for fast and accurate metagenomic profiling of nitrogen cycling genes. Bioinformatics 35, 1040–1048 (2019).
Buchfink, B., Reuter, K. & Drost, H. G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 18, 366–368 (2021).
Leung, P. M. & Greening, C. Greening lab metabolic marker gene databases. (2020).
Heikkilä, M. P., Honisch, U., Wunsch, P. & Zumft, W. G. Role of the Tat transport system in nitrous oxide reductase translocation and cytochrome cd 1 biosynthesis in Pseudomonas stutzeri. J. Bacteriol. 183, 1663–1671 (2001).
Liu, X. et al. The nos gene cluster from gram-positive bacterium Geobacillus thermodenitrificans NG80-2 and functional characterization of the recombinant NosZ. FEMS Microbiol.y Lett. 289, 46–52 (2008).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Capella-Gutiérrez, S., Silla-Martínez, J. M. & Gabaldón, T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973 (2009).
Nguyen, L.-T., Schmidt, H. A., Von Haeseler, A. & Minh, B. Q. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274 (2015).
Letunic, I. & Bork, P. Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296 (2021).
Lin, Z. et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 379, 1123–1130 (2023).
Varadi, M. et al. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 50, D439–D444 (2022).
Berman, H. M. et al. The protein data bank. Nucleic Acids Res. 28, 235–242 (2000).
Moi, D. et al. Structural phylogenetics unravels the evolutionary diversification of communication systems in gram-positive bacteria and their viruses. Preprint at https://doi.org/10.1101/2023.09.19.558401 (2023).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
van Kempen, M. et al. Fast and accurate protein structure search with Foldseek. Nat. Biotechnol. 42, 243–246(2023).
Cheng, H. et al. ECOD: an evolutionary classification of protein domains. PLoS Comput. Biol. 10, e1003926 (2014).
Madeira, F. et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 47, W636–W641 (2019).
Waterhouse, A. M., Procter, J. B., Martin, D. M., Clamp, M. & Barton, G. J. Jalview Version 2–a multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191 (2009).
Nayfach, S., Shi, Z. J., Seshadri, R., Pollard, K. S. & Kyrpides, N. C. New insights from uncultivated genomes of the global human gut microbiome. Nature 568, 505–510 (2019).
Cremers, G., Luecker, S. & op den Camp, H. Metascan metabolic HMM database and auxilliary files (Version 1). Zenodo. https://doi.org/10.5281/zenodo.6365663 (2022).
DeLano, W. L. Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr. 40, 82–92 (2002).
Seemann, T. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069 (2014).
Hallgren, J. et al. DeepTMHMM predicts alpha and beta transmembrane proteins using deep neural networks. Preprint at https://doi.org/10.1101/2022.04.08.487609 (2022).
Teufel, F. et al. SignalP 6.0 predicts all five types of signal peptides using protein language models. Nat. Biotechnol. 40, 1023–1025 (2022).
Neukirchen, S. & Sousa, F. L. DiSCo: a sequence-based type-specific predictor of Dsr-dependent dissimilatory sulphur metabolism in microbial data. Microb. Genom. 7, https://doi.org/10.1099/mgen.0.000603 (2021).
Camargo, A. P. et al. Identification of mobile genetic elements with geNomad. Nat. Biotechnol. 42, 1303–1312 (2023).
Dong, X. et al. A vast repertoire of secondary metabolites potentially influences community dynamics and biogeochemical processes in cold seeps. Sci. Adv. 10, eadl2281 (2024).
Uritskiy, G. V., DiRuggiero, J. & Taylor, J. MetaWRAP—a flexible pipeline for genome-resolved metagenomic data analysis. Microbiome 6, 1–13 (2018).
Kopylova, E., Noé, L. & Touzet, H. SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 28, 3211–3217 (2012).
Acknowledgements
The work was supported by National Science Foundation of China (No. 92351304 to X.D., No. 42376115 to X.D., No. 42371104 to X.L. and No. 42030407 to L.C.), Natural Science Foundation Project of Xiamen City (No. 3502Z202373076 to X.D.), Natural Science Foundation of Fujian Province (No. 2023J06042 to X.D.), Scientific Research Foundation of Third Institute of Oceanography, MNR (No. 2022025 and No. 2023022 to X.D.) and the Fundamental Research Funds for the Central Universities (202262007 to X.L.). SER was supported by the Simons Foundation (824763) and funds from the Human Frontier Science Program (RGEC34/2023). We thank Chengpeng Li and Xinyue Liu for assistance in determining geochemical parameters, Weichao Wu for providing sediment samples, and Chris Greening for helpful discussions. We also express our gratitude to the captains, crews, and pilots of the R/V KEXUE, as well as the ROV Faxian operation team for their support in collecting the samples.
Author information
Authors and Affiliations
Contributions
X.D. and Q.J. designed this study. Q.J. and Z.Z. performed the omics analysis. L.C., J.P., and M.W. analyzed environmental factors. X.L. analyzed nitrogen cycling rates. Y.H. contributed to discussions and methodology. S.L., R.Z., X.Z., S.E.R., and B.Z. participated in discussions and data interpretations. L.C., M.W., and J.L. collected cold seep sediment samples. Q.J., X.L., L.C., and X.D. wrote the paper, with input from other authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Maxim Rubin-Blum, and the other anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Jiang, Q., Cao, L., Han, Y. et al. Cold seeps are potential hotspots of deep-sea nitrogen loss driven by microorganisms across 21 phyla. Nat Commun 16, 1646 (2025). https://doi.org/10.1038/s41467-025-56774-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-56774-1
