
Abstract
The ever-evolving SARS-CoV-2 variants necessitate the development of additional oral antivirals. This study presents the systematic design of quinoline-containing SARS-CoV-2 papain-like protease (PLpro) inhibitors as potential oral antiviral drug candidates. By leveraging the recently discovered Val70Ub binding site in PLpro, we designed a series of quinoline analogs demonstrating potent PLpro inhibition and antiviral activity. Notably, the X-ray crystal structures of 6 lead compounds reveal that the 2-aryl substitution can occupy either the Val70Ub site as expected or the BL2 groove in a flipped orientation. The in vivo lead Jun13296 exhibits favorable pharmacokinetic properties and potent inhibition against SARS-CoV-2 variants and nirmatrelvir-resistant mutants. In a mouse model of SARS-CoV-2 infection, oral treatment with Jun13296 significantly improves survival, reduces body weight loss and lung viral titers, and prevents lung tissue damage. These results underscore the potential of quinoline PLpro inhibitors as promising oral SARS-CoV-2 antiviral candidates, instilling hope for the future of SARS-CoV-2 treatment.
Similar content being viewed by others

Introduction
The COVID-19 pandemic has accelerated antiviral and vaccine development1. Drug repurposing was the primary strategy early in the pandemic, but genuinely new strategies have also emerged2,3. One of the ultimate goals is to develop broad-spectrum oral antivirals that can be used to prevent and treat pathogens with pandemic potential4. Priorities have been given to conserved viral targets, including polymerases and proteases. The SARS-CoV-2 RNA-dependent RNA polymerase (RdRp) inhibitors remdesivir and molnupiravir are known to have broad-spectrum antiviral activities against viruses in different families. For example, remdesivir, developed initially against the hepatitis C virus, has well-documented antiviral efficacy against filoviruses (e.g., Ebola, Marburg), coronaviruses (e.g., MERS-CoV, SARS-CoV, and SARS-CoV-2), Pneumoviridae (e.g., respiratory syncytial virus), and paramyxoviruses (e.g., Nipah, Hendra, measles, and mumps)5,6. Molnupiravir is effective against influenza viruses and coronaviruses7. Similarly, the SARS-CoV-2 main protease (Mpro) inhibitor nirmatrelvir has broad-spectrum antiviral activity against SARS-CoV, MERS-CoV, SARS-CoV-2, and common human coronaviruses HCoV-OC43, 229E, NL63, and HKU18,9. Additional antivirals with alternative mechanisms of action are needed to combat viruses for which no antivirals are available or for which drug resistance issues with current antivirals are associated.
Structurally disparate compounds have been developed as SARS-CoV-2 Mpro inhibitors through high-throughput screening (HTS), fragment-based and structure-based design1,10. Paxlovid, a combination of the Mpro inhibitor nirmatrelvir and metabolic enhancer ritonavir, is approved by the FDA9. Ensitrelvir is a noncovalent Mpro inhibitor approved in Japan and Singapore11. Several additional Mpro inhibitors are at different stages of clinical trials, including Pfizer’s second-generation inhibitor PF-07817883 (Ibuzatrelvir)12 and Merck’s MK-784513. Mutations have emerged in Mpro among circulating strains14, including the predominant mutation P132H15. Fortunately, the P132H mutant remains sensitive to nirmatrelvir15, and drug-resistant variants have yet to become prevalent in the clinic. Nevertheless, nirmatrelvir-resistant mutants have been identified from in vitro viral passage and enzymatic assay experiments16,17,18,19. The Mpro L50F/E166A/L167F triple mutant is of particular concern as the corresponding recombinant virus showed a high level of drug resistance while maintaining similar fitness of replication in cell culture and animals19,20. The Nsp5-L50F/E166V mutant was found in an immunocompromised human patient who underwent prolonged Paxlovid treatment21.
The SARS-CoV-2 papain-like protease (PLpro) is the second cysteine protease expressed by SARS-CoV-2 in addition to Mpro during viral replication. PLpro is part of the non-structural protein 3 (nsp3) and cleaves viral polyproteins at the nsp1/2, nsp2/3, and nsp3/4 junctions22. PLpro also has deubiquitinase and deISGylase activities and cleaves ubiquitin and ISG15 conjugates from host proteins, a mechanism to antagonize host immune responses23,24. Since the COVID-19 pandemic, the interest in developing PLpro inhibitors has resurged, and several PLpro inhibitors have been identified from HTS, drug repurposing, and virtual screening25. PLpro is a cysteine protease with shallow S1 and S2 substrate binding sites (corresponds to Gly-Gly in substrates)22, which accounts for the challenges in designing covalent and noncovalent PLpro inhibitors26. The noncovalent naphthalene compound GRL0617 and its analogs are the major PLpro inhibitors with a validated mechanism of action25,27. Our recent study revealed a first-in-class rationally designed noncovalent inhibitor Jun12682 showing in vivo antiviral efficacy in a mouse model of SARS-CoV-2 infection28. Jun12682 binds to the same site as GRL0617 in the blocking loop 2 (BL2) region. In addition, Jun12682 extends to a previously unexplored binding site, Val70Ub, which accounts for its high potency.
In this work, we design a series of quinoline analogs to similarly target the Val70Ub site. The X-ray crystal structures of PLpro with six lead compounds reveal interesting findings: while five compounds, Jun13317, Jun13306, Jun13307, Jun13308, and Jun13296 have their 2-aryl substituent occupying the Val70Ub site as expected, Jun12665 adopts a flipped orientation with the 2-pyrazolyl substitution fitting in the BL2 groove site. The in vivo lead Jun13296 displays more potent enzymatic inhibition and antiviral activity than the previously reported biarylphenyl inhibitor Jun12682. In a SARS-CoV-2 infection mouse model, oral treatment of Jun13296 displays improved in vivo antiviral efficacy than Jun12682. Collectively, Jun13296 represents a promising PLpro antiviral drug candidate for further development.
Results
Design of quinoline SARS-CoV-2 PLpro inhibitors
We recently designed a covalent inhibitor, Jun11313, showing potent enzymatic inhibition with an IC50 of 0.12 µM28. The X-ray crystal structure of PLpro-Jun11313 (PDB: 8UVM) uncovered a previously unexplored drug binding site, Val70Ub, which accommodates the Val70 and Leu71 side chains from ubiquitin as well as the thienyl substituent in Jun11313 (Fig. 1a). Superposition of the X-ray crystal structures of SARS-CoV-2 PLpro with Jun11313 (green) (PDB: 8UVM) and GRL0617 (yellow) (PDB: 7JRN) (Fig. 1b) suggests that the benzene ring in Jun11313 can be expanded to naphthalene to engage additional hydrophobic interactions with residues in the BL2 loop region (Fig. 1c). For the convenience of installing various aromatic substitutions to target the Val70Ub binding site, we replaced the naphthalene ring with quinoline in our inhibitor design (Fig. 1c). In addition, a terminal positively charged amino group is added at the meta-position of the benzoic acid through a linker to bridge the electrostatic interaction with Glu167. Previous studies have shown that this design strategy improves the binding affinity of PLpro inhibitors28,29. The position of the linker attachment and the types of linkers chosen for the design were informed by previous structure-activity relationship results and the molecular docking results of the virtual library of designed compounds28,29,30.

a Superposition of the X-ray crystal structures of SARS-CoV-2 PLpro with Jun11313 (green) (PDB: 8UVM) and ubiquitin (orange) (PDB: 6XAA). b Superposition of the X-ray crystal structures of SARS-CoV-2 PLpro with Jun11313 (green) (PDB: 8UVM) and GRL0617 (yellow) (PDB: 7JRN). c Design of the quinoline PLpro inhibitors based on Jun11313 and GRL0617. Source data of (c) are provided as a Source Data file.
Guided by the design hypothesis illustrated in Fig. 1c, we initiated a stepwise lead optimization on the quinoline PLpro inhibitors (Fig. 2, Supplementary Fig. 1). We first fixed the amine substituent and explored 2-aryl substitutions in the quinoline (Supplementary Fig. 1a), followed by varying the amine substituents (Supplementary Fig. 1b, c). A virtual library sharing the common scaffold, as shown in Fig. 1c, was created and docked in PLpro using Schrödinger Glide’s extra precision. Prioritized hits were then synthesized and tested in the Förster resonance energy transfer (FRET) enzymatic assay using the FRET substrate containing the viral polyprotein nsp2/3 cleavage sequence and cytotoxicity assay in Vero E6 cells. Promising leads were tested in the PLpro FlipGFP assay, a cell-based PLpro-specific protease assay that correlates well with antiviral activity31,32. Next, the leads were tested in the SARS-CoV-2 antiviral assay in the Caco-2-ACE2-TMPRSS2 cell line and profiled for mouse liver microsomal stability (T1/2). Representative potent lead compounds are shown in Fig. 2. The biarylphenyl PLpro inhibitor Jun12682 was included as a positive control (Fig. 2a)28. Thienyl, furanyl, thiazole, pyrazole, and pyridine substituents were all tolerated at the 2-aryl position (Supplementary Fig. 1a), suggesting the Val70Ub site can accommodate a diverse set of aromatic substituents through hydrophobic interactions. This result is consistent with the previous structure-activity relationship (SAR) result of the biarylphenyl series of inhibitors28. Compounds with pyrazole substituents, including Jun12315, Jun1285, Jun12226, and Jun1275, had potent enzymatic inhibition (IC50 = 0.19–0.25 µM) and low cytotoxicity (CC50 = 63.1–146.3 µM). Jun12665 had potent enzymatic inhibition (IC50 = 0.14 µM, Ki = 32.1 nM), but displayed weak cellular activity in the FlipGFP assay (EC50 = 32 µM), possibly due to poor membrane permeability. Next, we examined different amine substitutions (Supplementary Fig. 1b, c). Nine lead compounds had potent enzymatic inhibition, including the pyrrolidine analogs Jun13306 (IC50 = 0.18 µM, Ki = 25.8 nM) and Jun13307 (IC50 = 0.16 µM, Ki = 25.4 nM), the azetidine analogs Jun1393 (IC50 = 0.28 µM, Ki = 45.0 nM) and Jun13338 (IC50 = 0.17 µM, Ki = 39.6 nM), the piperazine analogs Jun13126 (IC50 = 0.27 µM, Ki = 64.8 nM), Jun13317 (IC50 = 0.20 µM, Ki = 47.4 nM), and Jun13308 (IC50 = 0.14 µM, Ki = 19.3 nM), the 3,8-diazabicyclo[3.2.1]octane analog Jun13296 (IC50 = 0.13 µM, Ki = 8.8 nM), and the octahydropyrrolo[3,4-c]pyrrole analog Jun13556 (IC50 = 0.19 µM, Ki = 63.7 nM) (Fig. 2). The positive control Jun12682 had an antiviral EC50 of 1.0 µM. Seven compounds showed comparable or enhanced antiviral potency, including Jun13306 (EC50 = 0.9 µM), Jun13307 (EC50 = 0.6 µM), Jun13126 (EC50 = 1.0 µM), Jun13308 (EC50 = 0.5 µM), Jun13296 (EC50 = 0.1 µM), Jun1393 (EC50 = 0.4 µM), and Jun13556 (EC50 = 0.7 µM). Among the list of analogs, Jun13296 is the most active lead compound with an antiviral EC50 value of 0.1 µM, a 10-fold improvement compared to Jun12682 (EC50 = 1.0 µM). The potent antiviral activity of Jun13296 correlates with its high enzymatic inhibitory activity (IC50 = 0.13 µM, Ki = 8.8 nM) and cellular PLpro inhibitory activity in the Flip-GFP assay (EC50 = 1.5 µM). Selective compounds were profiled for mouse liver microsomal stability to prioritize lead candidates for the in vivo PK and antiviral efficacy studies. Jun12665, Jun13126, Jun13308, and Jun13296 showed the highest microsomal stability (T1/2 > 100 min).

a Chemical structure and in vitro activities of Jun12682, Jun12665, and Jun13338. b Chemical structures and in vitro activities of quinoline analogs with diverse amine substituents. IC50, half maximal inhibitory concentration in the FRET enzymatic assay; Ki, inhibitory constant in the FRET enzymatic assay; CC50: half maximal toxicity concentration in Vero cells, CC50 values are mean ± S.D. of three technical repeats; EC50, half maximal effective concentration in the FlipGFP and antiviral assays, EC50 values are mean ± standard deviation of three technical repeats; T1/2, half-life in mouse liver microsomal stability assay. Source data are provided as a source data file.
In vivo pharmacokinetic (PK) properties of quinoline SARS-CoV-2 PLpro inhibitors
Eight compounds were selected for in vivo oral snapshot PK studies in mice based on their antiviral activity and mouse liver microsomal stability (Fig. 2, Supplementary Fig. 1). Compounds were formulated in 0.5% methylcellulose and 2% Tween 80 in water and dosed in 3 male C57BL/6J mice through oral gavage. Blood samples were collected from the tail vein at 0.5, 1, 3, and 5 h to quantify plasma drug concentrations by LC-MS/MS. Jun13306, Jun13317, Jun13338, and Jun13126 had fast absorption and reached the maximum plasma concentrations at the first time point of 0.5 h (Fig. 3a, b). In contrast, Jun13308 and Jun13296 absorbed relatively slower and reached the maximum plasma concentrations at 3 h. Jun13296 displayed the highest drug concentrations at 3 h (3530 ng/mL) and 5 h (3040 ng/mL) and was thus selected for a 24 h in vivo PK study to determine the oral bioavailability. Interestingly, both the intravenous (i.v.) and per os (p.o., oral) administrations of Jun13296 showed a biphasic drug exposure pattern (Fig. 3c). Following p.o. administration at 50 mg/kg, Jun13296 reached the peak plasma concentration (Cmax) of 6,957 ng/mL at 2 h, followed by a decline to 664 ng/mL at 4 h, and an increase to 2413 ng/mL at 8 h. Similarly, for the i.v. administration at 10 mg/kg, the drug concentrations decreased from 3553 ng/mL at 0.25 h to 1527 ng/mL at 2 h, followed by an increase to 5637 ng/mL at 4 h. The half-lives (T1/2) were 2.4 h and 3.4 h for i.v. and p.o. administrations (Fig. 3d). The oral bioavailability of Jun13296 was 32.8%. The plasma concentration of Jun13296 was maintained above the antiviral EC90 values for over 8 h with oral dosing at 50 mg/kg (Fig. 3c).

a Plasma drug concentration of Jun13306, Jun13307, Jun13308, and Jun13317 in C57BL/6J mice (6 to 8 weeks old) following p.o. administration of 50 mg/kg of compounds in 0.5% methylcellulose and 2% Tween 80 in water (n = 3 per group). b Plasma drug concentrations of Jun13338, Jun1393, Jun13126, and Jun13296 in C57BL/6J mice (6 to 8 weeks old) following p.o. administration of 50 mg/kg of compounds in 0.5% methylcellulose and 2% Tween 80 in water (n = 3 per group). c Plasma drug concentrations of Jun13296 in C57BL/6J mice (6 to 8 weeks old) following p.o. administration of 50 mg/kg and i.v. injection of 10 mg/kg (n = 3 per group) of compound. The error bars are mean ± s.d. d In vivo pharmacokinetic parameters of Jun13296 in C57BL/6J mice. e In vitro pharmacokinetic parameters of Jun13296. T1/2, half-life; Tmax, time for maximal concentration; Cmax, maximum plasma concentration; AUC0-t, area under the curve from time zero to time t; AUCt-∞, area under the curve from time t to infinity; CL, clearance; MRT, mean residence time; Vss, volume of distribution; F, oral bioavailability. Source data are provided as a source data file.
Jun13296 was selected for further in vitro PK profiling. Jun13296 is stable in mouse and human liver microsomal stability assays with T1/2 of 111.2 and 73.6 min, respectively. Jun13296 did not inhibit five representative CYP isoforms 1A2, 2C9, 2C19, 2D6, and 3A-M (IC50 > 50 µM) (Fig. 3e), indicating it might not have the drug-drug interaction issue associated with the Mpro inhibitor nirmatrelvir. The mouse plasma protein binding for Jun13296 is 94.1%. The high plasma protein binding ratio of Jun13296 might account for its in vivo biphasic drug exposure. Jun13296 has moderate permeability in Egg- Parallel Artificial Membrane Permeability Assay (PAMPA) and blood-brain barrier specific (BBB)-PAMPA assays with Pe values of 11.0 and 10.9 nm/s. Overall, Jun13296 has favorable in vitro and in vivo PK properties that warranted advancing to the in vivo antiviral efficacy study in mice.
Mechanism of action of Jun13296
Jun13296 displayed dose-dependent stabilization of SARS-CoV-2 PLpro in the differential scanning fluorimetry assay (Fig. 4a), with a larger stabilization (and hence higher binding affinity) than Jun12682, consistent with the enzymatic FRET protease assay results (Fig. 1). To characterize whether Jun13296 inhibits the deubiquitinase and deISGylase activities of SARS-CoV-2 PLpro, we tested Jun13296 in the PLpro enzymatic assays using ubiquitin-AMC (7-amino-4-methylcoumarin) and ISG15-AMC substrates, respectively. Jun13296 inhibited the deubiquitinase and deISGylase activities of PLpro with Ki values of 5.5 nM and 5.4 nM (Fig. 4b, c), similar to its activity in inhibiting the hydrolysis of the viral polyprotein nsp2/3 substrate (Ki = 8.8 nM) (Fig. 2). To profile the selectivity of Jun13296, we tested it against a panel of host proteases, including human deubiquitinases USP2, USP7, USP8, USP14, USP15, USP30, UCH-L1, cathepsin B, cathepsin K, calpain-1, trypsin, and caspase 329,33. Jun13296 did not inhibit these host proteases at up to 30 µM (Fig. 4d), suggesting Jun13296 is specific for the viral PLpro. Other PLpro inhibitors, including GRL0617 analogs and PF-07957472, similarly showed a high selectivity over host proteases as reported33,34.

a Differential scanning fluorimetry assay of Jun13296 in stabilizing SARS-CoV-2 PLpro. Jun12682 was included as a positive control for comparison. Data from Jun12682 is the mean of two repeats, and data from Jun13296 is the mean ± standard deviation of three technical repeats. b Ki plot of Jun13296 in inhibiting SARS-CoV-2 PLpro hydrolysis of ISG15-AMC. c Ki plot of Jun13296 in inhibiting SARS-CoV-2 PLpro hydrolysis of Ub-AMC. d Counter screening of Jun13296 against host proteases USP2, USP7, USP8, USP14, USP15, USP30, UCH-L1, cathepsin B, cathepsin K, calpain-1, trypsin, and caspase 3. Data in (d) are presented as mean ± standard deviation of two technical repeats. Source data are provided as a source data file.
X-ray crystal structures of SARS-CoV-2 PLpro with quinoline inhibitors
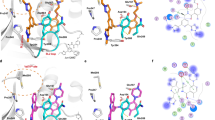
The inhibitor-bound X-ray co-crystal structures of SARS-CoV-2 wild-type PLpro were determined for six noncovalent quinoline series compounds: Jun12665, Jun13317, Jun13306, Jun13307, Jun13308, and Jun13296, with resolutions ranging from 2.30 Å to 2.95 Å (Supplementary Table 1). The electron density maps clearly delineate the protein molecule, coordinated zinc, and inhibitors (Supplementary Fig. 2), confirming their orientations and interactions with the surrounding environment (Fig. 5a–f). All inhibitors consistently bind at the same site, interacting with the BL2 loop, the Glu167 side chain, and the recently identified Val70Ub site within the extended peptide binding region of PLpro, distal from the active site triad (Cys111, His272, and Asp286) (Fig. 5a–f and Supplementary Fig. 2a, b). This binding follows a conserved interaction pattern involving Asp164, Glu167, Pro247, Pro248, Tyr264, Tyr268, and Gln269 of wild-type PLpro. All inhibitors’ 2-aryl substituted quinoline aromatic rings, except Jun12665, orient toward the Val70Ub site, consistent with our design hypothesis (Fig. 1b). The quinoline moiety of the inhibitors is stabilized by hydrophobic interactions and π-stacking interactions with Tyr268, Tyr264, Pro248, and Pro247, as well as CH–π and S–π interactions with Met208, as previously reported28. Jun12665, with its amide pyrazole substitution, occupies the previously reported BL2 groove site and forms hydrophobic interactions with Pro247, Pro248, Tyr264, and Tyr268 (Fig. 5a). The terminal dimethylamide carbonyl oxygen forms an additional hydrogen bond with the main-chain amide nitrogen of Tyr268 (Fig. 5a, Supplementary Fig. 2a), which might account for the flipped orientation of Jun12665 compared with the other inhibitors (Fig. 5g). The central amide linker regions of all six inhibitors are stabilized through hydrogen bonds with the carboxylate side chain of Asp164 and the main-chain amide nitrogen of Tyr268. Various substituents at the positions of previously published dimethylamino moieties engage in electrostatic interactions with the Glu167 carboxylate across all structures, showing slight variations in bonding distances and the number of Glu167 oxygen atoms involved in the interactions (Fig. 5). Jun13306 and Jun13307 containing the R and S pyrrolidine substitutions both form similar electrostatic interactions with the Glu167 carboxylate (Fig. 5b, c, h, Supplementary Fig. 2g), corroborating their equivalent enzymatic inhibitory activities (Ki = 25.8 and 25.4 nM) (Fig. 2).

Binding of quinoline inhibitors to SARS-CoV-2 PLpro. a–f Interactions of Jun12665 (orange), Jun13306 (yellow), Jun13307 (green), Jun13317 (light blue), Jun13308 (magenta), and Jun13296 (bright orange) with PLpro are conserved (residues within 5 Å of the inhibitor are shown as light brown sticks). Hydrogen bonds are indicated by black dashed lines, van der Waals contacts by red dashed lines, and π–π interactions by light green dashed lines. g The superposition of Jun12665 onto Jun13296 represents the flipped orientation of terminal amide carbonyl groups toward the BL2 groove. h Superposition of Jun13306 (yellow) and Jun13307 (green) containing the R and S pyrrolidine substituents both form similar electrostatic interactions with the Glu167 carboxylate.
The superimposed X-ray crystal structure of PLpro/Jun13296 with host deubiquitinases, USP2, USP7, and USP14, showed that the Val70Ub and BL2 groove binding sites are absent in these host deubiquitinases (Supplementary Fig. 3), which accounts for the high selectivity of Jun13296.
In vitro and in vivo antiviral activities of Jun13296
Given the distinct mechanism of action of PLpro inhibitors from Mpro inhibitor nirmatrelvir and the sequence conservation of PLpro, Jun13296 is expected to have broad-spectrum antiviral activity against nirmatrelvir-resistant mutants and SARS-CoV-2 variants. For this, we tested Jun13296 against the nirmatrelvir-resistant SARS-CoV-2 viruses rNsp5-L50F/E166V and rNsp5-L50F/E166A/L167F, and the recently circulating SARS-CoV-2 omicron variants XBB.1.16 and JN.1 in plaque assay (Fig. 6, Supplementary Fig. 4). Nsp5-L50F/E166V mutant was identified from an immunocompromised human patient with prolonged Paxlovid treatment21. Nsp5-L50F/E166A/L167F mutant was selected under drug selection pressure in the cell culture viral passage experiment18,19. The Mpro inhibitor nirmatrelvir and the previously reported PLpro inhibitor Jun12682 were included as controls. All three inhibitors showed potent inhibition against SARS-CoV-2 WA1 and the omicron variants XBB1.16 and JN.1 (Fig. 6a–c). The rNsp5-L50F/E166V and rNsp5-L50F/E166A/L167F viruses had significant resistance against nirmatrelvir with EC50 values of >1 µM, and 0.63 µM, respectively (Fig. 6a). In contrast, Jun13296 displayed consistent antiviral potency against the rNsp5-L50F/E166V and rNsp5-L50F/E166A/L167F mutants with EC50 values of 0.47 and 0.22 µM, respectively (Fig. 6c), and was more potent than Jun12682 (Fig. 6b). Collectively, these results show that PLpro inhibitors have consistent antiviral activity against SARS-CoV-2 variants and nirmatrelvir-resistant strains.

The antiviral activity of nirmatrelvir, Jun12682, and Jun13296 was tested in the plaque assay using Vero-ACE2-TMPRSS2 (Vero-AT) cells. a Plaque assay EC50 plots of nirmatrelvir. b Plaque assay EC50 plots for Jun12682. c Plaque assay EC50 plots of Jun13296. EC50 values are mean ± standard deviation of two technical repeats. Source data are provided as a source data file.
To evaluate the in vivo antiviral efficacy of Jun13296, we utilized a lethal SARS-CoV-2 mouse model that has been widely accepted for assessing SARS-CoV-2 therapeutics and vaccine candidates35,36,37,38. We previously reported the antiviral efficacy of Jun12682 using this model28. Hence, Jun12682 was included as a control in this study. To this end, young female BALB/c mice were intranasally inoculated with 6,000 PFU of SARS2-N501YMA30 and orally administered PLpro inhibitors or vehicle one hour after virus inoculation, followed by twice daily dosing with a 10- and 14-h interval within 24 h for 3 consecutive days (Fig. 7a). A subgroup of 5 mice from each treatment group were necropsied at 2- and 4-day post-infection (DPI) to collect lung tissues for viral load and histopathological assessment. We first evaluated the antiviral efficacy of a 125 mg/kg dose. The weight loss plot (Fig. 7b) illustrates that vehicle-treated mice experienced rapid body weight loss exceeding 20% by 4 DPI. In contrast, both Jun12682- and Jun13296-treated mice exhibited reduced weight loss and had maximal weight loss at 4 DPI, following body weight regain. Specifically, Jun13296-treated mice had an average of 11% maximal weight loss, significantly less than the maximal weight loss of Jun12682-treated mice (17%, p < 0.001) and the vehicle group (21%, p < 0.0001). Survival analyses demonstrated that inhibitor-treated groups had statistically higher survival rates compared to the vehicle group (0% survival rate): Jun13296 (90%, p < 0.0001), and Jun12682 (75%, p < 0.0001) (Fig. 7c). We previously demonstrated a 125 mg/kg dosage of Jun12682, administered with an 8-hour and 16-hour interval between doses within 24 h, provided only 20% protection28. Our current results indicate that administrating doses with 10-h and 14-h intervals offers improved protection, likely due to maintaining effective plasma concentrations as suggested by the in vivo PK data28. Viral load analyses of mouse lungs revealed that the vehicle-treated mice had robust infections in the lungs at 2 DPI with a mean titer of log10 8.87 ± 0.079 PFU/mL. In contrast, the Jun12682- and Jun13296-treated mice had statistically lower lung viral titers, log10 8.25 ± 0.064 PFU/mL (p = 0.0005), and log10 8.11 ± 0.137 PFU/mL (p < 0.0001), respectively (Fig. 7d). The titer reductions were more evident at 4 DPI with a half-log lower titer (log10 5.94 ± 0.115 PFU/mL, p = 0.0004) for the Jun12682-treated mice and over a log titer reduction (log10 5.38 ± 0.059 PFU/mL, p < 0.0001) for the Jun13296-treated mice compared to the mean titer (log10 6.57 ± 0.115 PFU/mL) of the vehicle group (Fig. 7d). The 4 DPI titer of the Jun13296 group is significantly lower than that of the Jun12682 group (p = 0.0014). These results corroborate the weight loss and survival data (Fig. 7b, c).

a Experimental design for twice-a-day (BID) treatment for 3 days. b Mouse body weight loss and c survival rate of the mice receiving a 125 mg/kg BID_3 treatment. Data in (b, c) are pooled results of four independent experiments and are shown as mean ± standard error of the mean (SEM) (n = 14, 20, 19 mice for the Vehicle, Jun12682, and Jun13296 group, respectively). d Viral titers in lungs (n = 5 per group). Quantitative PCR analysis of viral nucleocapsid gene (e) and cellular cytokines (f) in lungs (n = 5 per group). g Haematoxylin and eosin (H&E) staining of lungs collected 4 DPI (n = 5 per group). Lungs exhibited airway edema (asterisks), hyaline membranes (HM, arrowheads), perivascular cuffing (arrows), and interstitial thickness (number sign). Scale bars, 100 mm (top) and 50 mm (bottom). h Quantification of the pathological lesions (g). i Immunostaining of lungs collected 4 DPI (n = 5 per group) for SARS-CoV-2 nucleocapsid protein (brown color staining). Scale bars, 100 µm (top) and 50 µm (bottom). j Summary scores of nucleocapsid immunostaining of lungs. k Mouse body weight loss and l survival rate of the mice receiving a 75 mg/kg BID_3 treatment. Data are pooled results of two independent experiments (n = 10 per group) and are shown as mean ± SEM. m Viral titers in lungs (n = 5 per group). n Quantitative PCR analysis of cellular cytokines in lungs collected 2 DPI (n = 5 per group). Data in (c, l) are shown as mean ± SEM, and the p values were determined using a log-rank (Mantel-Cox) test. Data in (d, m) are shown as mean ± SEM and analyzed with a two-way ANOVA Tukey’s multiple comparison test. Data in (e, f, n) are shown as mean ± SEM and analyzed with a one-way ANOVA multiple comparison test for each tested gene. Data in (h, j) are mean ± SEM and analyzed with the Krushal-Wallis multiple comparisons test for each category. Source data are provided as a source data file. a was created with Biorender.com. Li, K. (2025) https://BioRender.com/q16l908.
Quantitative PCR analysis of the RNA samples extracted from 2 DPI mice lungs showed that the 125 mg/kg dosage of either PLpro inhibitor significantly reduced the expression of the viral nucleocapsid (N) gene (Fig. 7e) and multiple inflammatory cytokines, including IL-1β, CXCL10, IL-6, and IFN-γ (Fig. 7f). The relative mRNA levels of these cytokines in the lungs of Jun13296-treated mice are significantly lower than those of Jun12682-treated mice. Histopathological analysis of mouse lungs collected at 4 DPI reveals that the vehicle-treated, SARS2-N501YMA30-infected mice exhibited multifocal pulmonary lesions, including lymphocytic perivascular cuffing, pulmonary edema, hyaline membrane formation, and interstitial thickening and inflammation (Fig. 7g). In contrast, the mice that received 125 mg/kg BID of PLpro inhibitors exhibited overall reduced lung histopathological manifestations with statistically significant improvement in the Jun13296-treatment group (Fig. 7h). Immunohistochemical (IHC) analysis of mouse lungs using a monoclonal antibody to detect SARS-CoV-2 N protein demonstrated strong and expansive antigen staining in lungs from vehicle-treated, infected mice, whereas inhibitor-treated considerably decreased viral antigen staining levels with a few sporadic positive cells (Fig. 7i). Lungs of Jun13296-treated mice had significantly less antigen staining than the lungs of the vehicle group (Fig. 7j), consistent with the lung viral titer results (Fig. 7d). The reduced viral replication in the lung and the expression of inflammatory cytokines (Fig. 7d–f) corroborate with the reduced lung inflammation and N protein staining at 4 DPI (Fig. 7g–j).
We further evaluated a lower dosage of 75 mg/kg. During this treatment, despite the mice administered with either inhibitor experiencing more maximal weight loss than the mice receiving the 125 mg/kg dosage (Fig. 7k), the Jun13296 treatment rendered 90% protection, significantly higher than vehicle treatment (p < 0.0001) (Fig. 7l). Jun12682 treatment instead provided only 40% protection, higher than the vehicle group (p = 0.0044) but significantly lower than the Jun13296 treatment (p = 0.0182) (Fig. 7l). Viral load analysis of mouse lungs revealed statistically insignificant titer reductions at 2 and 4 DPI (Fig. 7m). Quantitative PCR analysis of the RNA samples extracted from 2 DPI mice lungs showed that the 75 mg/kg dosage of either inhibitor significantly reduced the expression of IL-6 and IFN-γ but not IL-1β and CXCL10 (Fig. 7n). These results suggest that a 75 mg/kg dosage of Jun13296 or Jun12682 still provides evident protection without markedly lowering viral loads in the lung. IL-6 is a biomarker associated with adverse clinical outcomes in SARS-CoV-2 infection, and blockade of IL-6 signaling has been shown to improve outcomes in critically ill COVID-19 patients39,40,41,42,43,44. A high level of IFN-γ was observed in severe COVID-19 cases and was associated with lung fibrosis45,46,47,48. Significantly reduced expression of IL-6 and IFN-γ in the inhibitor-treated groups may contribute to improved mouse survival (Fig. 7k, l).
Discussion
Compared to the successful development of SARS-CoV-2 Mpro inhibitors, PLpro has proven to be a challenging antiviral drug target25. Designing PLpro inhibitors with in vivo antiviral efficacy has only become a reality very recently28,34. With the potential threat of drug-resistant mutants against Mpro and RdRp inhibitors, there is a dire need for additional antivirals with alternative mechanisms of action. Given its sequence conservation and multi-faceted roles in viral replication, PLpro has been enlisted as a high-profile antiviral drug target25. We recently reported the design of a biarylphenyl inhibitor Jun12682 showing in vivo antiviral efficacy in a SARS-CoV-2 infection mouse model28. The high antiviral potency of Jun12682 stems from its dual targeting mode by binding to the Val70Ub site and the previously reported BL2 groove28. Given the high triage rate in translational development, additional PLpro inhibitors with novel scaffolds are needed as backups. In this study, we designed quinoline-containing PLpro inhibitors to similarly target the Val70Ub site and the BL2 groove. Jun13296 was nominated as an in vivo lead candidate through iterative cycles of structure-activity and structure-property relationship studies.
Literature mining has identified a similar compound recently reported by Pfizer, known as PF-0795747234 (Supplementary Fig. 5). This compound closely resembles our quinoline series compounds, with a few notable differences. Specifically, the hydrophobic cyclopropyl substituent in PF-07957472 at the methyl group position in Jun13296 forms CH-π interactions with Tyr264 and engages with polar residues within the pocket, such as Thr301 and Asp164. The reported structure in this study, along with other solved structures28, shows the consistent orientation of N-methyl pyrazole-quinoline aromatic rings (Supplementary Fig. 5b) (with one exception, Jun12665, Supplementary Fig. 5e) towards the Val70Ub site, which is opposite to the PF-07957472 orientation (towards the BL2 groove).
The superposition of the PLpro structure complexed to Jun13296 with the corresponding PF-07957472 structure (Supplementary Fig. 5d) shows that the main difference between them is the orientation of N-methyl pyrazole substituent. Meanwhile, the superposition of the PLpro structure complexed to Jun12665 with the PF-07957472 model (Supplementary Fig. 5e) shows the same orientation of the substituents towards the BL2 groove, with Jun12665 exhibiting the aforementioned additional hydrogen bond with the main-chain amide of Tyr268.
Jun13296 displayed more potent antiviral activity than the previously reported in vivo lead Jun12682 in inhibiting SARS-CoV-2 variants and nirmatrelvir-resistant strains in cell culture-based assays. In vivo PK profiling showed that Jun13296 has an oral bioavailability of 32.8% and the plasma concentration of Jun13296 was maintained above the antiviral EC90 values for over 8 h with oral dosing at 50 mg/kg (Fig. 3c). In vivo assessment with a SARS-CoV-2 mouse model reveals that a 125 mg/kg BID-3 dosage of Jun13296 provides improved mouse protection compared to Jun12682 as demonstrated by the reduced body weight loss and mortality (Fig. 7a, b), lower lung viral load (Fig. 7d, e, i, j), and alleviated lung inflammation (Fig. 7f–h). In the experiment with a lower dosage of 75 mg/kg, Jun13296 still rendered 90% survival protection and outperformed Jun12682. Intriguingly, no significant reduction in the lung viral titers was detected. Through measuring the transcriptional levels of several pro-inflammatory cytokines, we found that the relative mRNA levels of IL-6 and IFN-γ, two key cytokines associated with disease severity, were significantly reduced in the lungs of inhibitor-treated mice. We postulate that in the scenario of unaffected viral replication, the surviving mice benefited from the reduced expression of some proinflammatory cytokines (e.g., IL-6 and IFN-γ), potentially through inhibiting the deubiquitinase and deISGylase activities of PLpro (Fig. 4b, c). These activities mediate the deconjugation of ubiquitin or ISG15 from substrates and have been associated with modulating host immune responses through reversing the ubiquitinating and ISGylating modifications of host immune-related molecules49,50,51. In addition, free ISG15 can be secreted and play immunomodulatory functions as an extracellular cytokine52,53,54. PLpro has been linked to the increasing release of ISG15 during SARS-CoV-2 infection of human macrophages55. Despite strong evidence supporting the immunomodulatory role of PLpro, the role of its deubiquitinase and deISGylase activities, particularly during in vivo infection, has been challenging to study, due to the integrated nature of these activities with PLpro polyprotein processing activity56,57,58,59. Reduced cytokine expression in treated mice could result from decreased viral infection or inhibition of the deubiquitinase and deISGylase activities of PLpro or a combination of both. Further investigation is needed to confirm the benefit of inhibiting deubiquitinase and deISGylase activities of PLpro.
Methods
Ethical statement
All procedures with live SARS-CoV-2 were performed in certified biosafety level 3 (BSL3) facilities at Oklahoma State University using biosafety protocols approved by the Institutional Biosafety Committee (IBC), which comprises scientists, biosafety and compliance experts, and members of the local community. All mouse experiments involving SARS-CoV-2 were performed within a certified animal biosafety level 3 (ABSL3) facility at Oklahoma State University. All animal studies were reviewed and approved by the Oklahoma State University Animal Care and Use Committee and met stipulations of the Guide for the Care and Use of Laboratory Animals. All research personnel received rigorous biosafety, biosecurity, and BSL3 training before participating in experiments. Personal protective equipment, including scrubs, disposable overalls, shoe covers, double-layered gloves, and powered air-purifying respirators, were used. Biosecurity measures are built in the environment through building and security systems and are reinforced through required training programs, standing meetings, and emergency exercises. The researchers involved in working with live viruses received the SARS-CoV-2 vaccines before the study was started. Finally, all researchers were medically cleared by the Oklahoma State University Occupational Health Program.
Compound synthesis and characterization
Synthesis procedures are shown in Supplementary Fig. 6-8. Compound characterization is included in the Supplementary Methods section.
Cell lines and virus
A Vero-E6 cell line, a gift from Dr. Susan Baker (Loyola University Chicago), was grown in Dulbecco’s modified Eagle medium (DMEM) (Corning, 10013CM) containing 10% heat-inactivated fetal bovine serum (FBS) (Gibco, 10-438-026), 1% Pen/Strep (30-001-CI), nonessential amino acid (NEAA) (Cytiva HyClone, SH30238.01). A Vero-E6 line expressing human angiotensin-converting enzyme 2 (hACE2) and human transmembrane protease, serine 2 (hTMPRSS2) (Vero-AT), obtained through NIH-BEI Resources (NR-54970) was grown in DMEM containing 10% FBS, 1% Pen/Strep, 1× NEAA, and 10 μg/mL puromycin (InVivogen, ant-pr-1) to maintain the expression of hTMPRSS2 and hACE2. A Caco-2 line expressing hACE2 and hTMPRSS2 (Caco2-AT)60, a gift from Dr. Mohsan Saeed (Boston University), was propagated in DMEM containing 10% FBS, 1% Pen/Strep, 1× NEAA, 1 μg/mL puromycin, and 1 μg/mL blasticidin (InVivogen, ant-bl-05).
The mouse-adapted SARS2-N501YMA30 virus, a gift from Dr. Stanley Perlman (University of Iowa), was propagated once with Vero-AT cells and titrated with Vero-E6 cells. The following SARS-CoV-2 strains/isolates were obtained through BEI Resources, NIAID, NIH: Washington strain 1 (WA1) (NR-52281), recombinant SARS-CoV-2 expressing nano-luciferase reporter (icSARS-CoV-2-nLuc) (NR-54003), Omicron JN.1 variant (NR-59693), and Omicron XBB1.16 variant (NR-59442). These viruses were propagated once with Vero-AT cells to obtain large viral stocks and titrated with Vero-AT cells. A recombinant wild-type SARS-CoV-2 WA1 strain (rSARS-CoV-2) and nirmatrelvir-resistant mutants rL50F/E166V and rL50F/E166A/L167F were generated previously14. These viruses were full-genome sequenced using the ARTIC method61.
Protein expression, purification and crystallization
The PLpro bacterial expression plasmid, obtained from BEI Resources, NIAID, NIH (Vector pMCSG53 containing the SARS-related coronavirus 2, Wuhan-Hu-1 papain-like protease gene, NR-52897), was used to produce SARS-CoV-2 PLpro following a previously described procedure with minor modifications28. The protein construct was expressed in E. coli BL21 (DE3) cultured in LB medium supplemented with ampicillin (100 µg/ml). Protein overexpression was induced by adding 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) at an OD600 of 0.8. After 18 hours of growth at 16 °C with slow agitation, cells were pelleted by centrifugation at 7000×g, washed with PBS, and stored at -80 °C until purification.
The cell pellets were resuspended in 12.5 ml of lysis buffer (500 mM NaCl, 5% (v/v) glycerol, 50 mM HEPES pH 8.0, 20 mM imidazole, 1 mM TCEP, 1 µM ZnCl2) per liter of culture and sonicated at 120 W for 12 minutes (5 s ON, 20 s OFF). The lysate was clarified by centrifugation at 37,000×g for 90 minutes at 4 °C. Ni-NTA purification was performed according to the manufacturer’s recommendations (Qiagen, Valencia, CA, USA) using the lysis buffer. Bound PLpro was eluted with 20 ml of lysis buffer supplemented with 500 mM imidazole pH 7.5, followed by treatment with tobacco etch virus (TEV) protease at a 1:25 protease ratio at 4 °C overnight in dialysis buffer, and a reverse Ni-NTA purification.
After confirming His-tag cleavage by SDS-PAGE, the dialyzed protein solution was passed over a 10-mL Ni-NTA gravity column to remove His-tagged impurities. The column flowthrough was collected, analyzed by SDS-PAGE, and concentrated using a 10 kDa molecular weight cutoff Amicon Ultra-15 ultrafiltration membrane. Size exclusion chromatography was performed on a Superdex 200 Increase 10/300 GL column equilibrated with 150 mM NaCl, 50 mM Tris-HCl pH 7.5, 1 μM ZnCl2, and 1 mM TCEP. Peak fractions were pooled, and the buffer was exchanged for a final buffer with a similar composition, replacing the reducing agent TCEP with either 10 mM DTT or 10 mM BME as needed. The final PLpro concentration was 20 mg/ml.
A similar approach was utilized for the crystallization of PLpro and six inhibitor complexes, as described previously28. Pure PLpro protein at 20 mg/ml in 50 mM Tris-HCl pH 7.5, 150 mM NaCl, and 1 mM TCEP/10 mM DTT/10 mM BME was mixed with inhibitors at a molar ratio of 1:10 and incubated for 1 hour at 4 °C. The PLpro-inhibitor complexes were clarified by high-speed centrifugation at 4 °C for 15 minutes. The sitting drop vapor diffusion method was employed using an Oryx8 robot (Douglas Instruments Ltd) in a 96-well Intelli-Plate (Art Robbins Instruments). Co-crystallizations were attempted with protein-to-well solution ratios of 1:1, 2:1, and 1:2 at 4 °C, using a focused screen based on the SARS-CoV-2 PLpro PDB deposited conditions. This screen specifically included a grid prepared with 200 mM zinc acetate, 0.1 M Bis-Tris pH 5.5-6.6, and 6-20% PEG 8000. Pyramidal-shaped crystals ( ~ 200 μm) grew overnight, belonging to space group I4122, with one copy of the complex in the asymmetric unit. Crystals selected for data collection were transferred into 1-2 μl of a cryoprotective solution consisting of crystallization buffer (without inhibitor) supplemented to a final concentration of 20% glycerol/ethylene glycol. After a few seconds, the crystals were looped up and flash-cooled in liquid nitrogen. All crystal manipulations were conducted at 4 °C.
Data collection, structure determination, and refinement
Diffraction data were collected at 100 K for the PLpro complex with Jun12665 on beamline 12-2 at the Stanford Synchrotron Radiation Lightsource (Menlo Park, CA) at a wavelength of 0.97 Å, while data for the PLpro complexes with Jun13317, Jun13306, Jun13307, Jun13308, and Jun13296 were collected on beamline 17-ID-1 (AMX) at NSLS-II at Brookhaven National Laboratory (Upton, NY) at a wavelength of 0.92 Å, using Pilatus 6 M and Eiger 9 M detectors, respectively. The datasets were collected remotely, with crystal rastering performed prior to data collection to locate the best diffraction positions. Diffraction data for the complexes were collected with 200° of data per crystal and a 0.1° oscillation per image, except for the PLpro-Jun12665 complex, for which 360° was collected using a 0.2° oscillation.
For all the datasets, autoprocessed files from XDS were subsequently scaled using Aimless62 or autoPROC from Global Phasing63. Initial phases for each inhibitor structure were obtained by molecular replacement using PDB 7JIT as the starting model in Phaser64. The inhibitor electron density was examined, and the inhibitor molecules were fitted using Coot65. To improve the inhibitor and protein models, iterative refinement was performed by manual model building in Coot alternating with refinement in Phenix66. Solvent molecules were added using 2mFo-Fc and Fo-Fc maps as guides. Statistics for diffraction data processing and the converged refinement statistics for all six datasets, along with R, Rfree, and other validation measures, are presented in Table S1. The inhibitor restraints were generated using the Grade online server67. PLpro-inhibitor complex structures were analyzed, and figures showing the protein-inhibitor co-crystal structures were created with PyMOL (The PyMOL Molecular Graphics System, Version 1.2, Schrödinger, LLC).
Differential Scanning Fluorimetry (DSF)
The drug-binding assay was performed in the Thermo Fisher SuantStudio 5 Real-time PCR system. 4 µM of SARS-CoV-2 PLpro dissolved in the reaction buffer (HEPES 50 mM, DTT 5 mM, Trion X-100 0.01%, pH 7.2) was mixed with different concentrations of compounds. After 30 min of 30 °C incubation, 1x final concentration of SYPRO orange (Invitrogen, #S6650) was added to the mixture. The fluorescence was monitored with temperature rising from 25 °C to 95 °C at the increment of 0.05 °C/s. The melting temperature (Tm) of each sample was obtained by the mid-log of the transition phase from the native to denatured protein using the Boltzmann model in Protein Thermal Shift Software v1.3.
Enzymatic assays
The enzymatic assay of SARS-CoV-2 PLpro digesting Ub-AMC (UBPBIO, #M3010) was performed by mixing 50 nM of SARS-CoV-2 PLpro with various concentrations of compounds in the reaction buffer (HEPES 50 mM, DTT 5 mM, Triton X-100 0.01%, pH 7.2). After the incubation of 10 min at 30 °C, Ub-AMC was added to the reaction to a final concentration of 2.5 µM. The fluorescence was monitored every 71 s with the excitation of 360/40 nm and emission of 460/40 nm for 3 h at 30 °C, using a Cytation 5 plate reader. The Ki was determined by plotting the first 1 h of initial velocity against the compound concentrations using the Morrison equation for tight binding (Y = V0*(1 − ((((Et + X + (Ki*(1 + (S/Km )))) − (((Et + X +(Ki*(1 + (S/Km )))) 2̂) − 4*Et*X)0.5̂))/(2*Et ))), X = inhibitor concentration; Y = enzyme velocity; Et = enzyme concentration; V0 = enzyme velocity in the absence of inhibitor). The enzymatic assay of SARS-CoV-2 PLpro digesting ISG15-AMC (R&D, #UL-553) was performed similarly with the changes in protein and substrate concentrations. The final concentration of SARS-CoV-2 PLpro was 2 nM and of ISG15-AMC was 0.5 µM in the ISG15 assays. All reported data were the average of 2 replicates with standard error with a 95% confidence interval calculated as SE = (upper limit – lower limit)/3.92.
The counter-screening assays with USP14 (ProSci, 91-171) and USP7(Sino Biological, #11681-H20B) were performed in the USP reaction buffer (HEPES 50 mM, pH 7.2, BSA 0.1 mg/mL, Triton X-100 0.01%, DTT 5 mM, Glycerol 5%). Compounds were incubated with 40 nM of USP7 and 800 nM of USP14 respectively for 10 min at 30 °C. 5 µM of Ub-AMC was added to initiate the reaction. The fluorescence was monitored, and initial velocities of each group were taken to compare with the DMSO-treated group. Values reported were performed in 2 replicates and error bars represent standard deviation.
Cell-Based FlipGFP-PLpro assay
The FlipGFP-PLpro assay was performed following the published procedures28,31,32. Briefly, the reporter plasmid pcDNA3-TEV-flipGFP-T2A-mCherry was obtained from Addgene (catalog no.124429). SARS-CoV-2 PLpro cleavage site LRGG ↓ APTK was introduced into pcDNA3-FlipGFP-T2AmCherry via overlapping PCRs to generate a fragment with SacI and HindIII sites at the ends. SARS-CoV-2 PLpro expression plasmid pcDNA3.1 SARS2 PLpro was ordered from Genscript (Piscataway, NJ) with codon optimization. For transfection, HEK293T cells were seeded in 96-well Greiner plate (catalog no. 655090) and grown overnight to achieve 70–80% confluency. A total of 9 μL of Opti-MEM, 0.1 μL of 500 ng/μL pcDNA3-flipGFP-T2A-mCherry plasmid, 0.1 μL of 500 ng/μL protease expression plasmid pcDNA3.1, and 0.3 μL of transIT-293 (Mirus) were added to each well of a 96-well plate. Three hours after transfection in a cell culture incubator (humidified, 5% CO2/95% air, 37 °C), 1 μL of testing compound was added to each well at 100-fold dilution. Images were acquired 48 h after transfection with a Celigo Image Cytometer (Nexcelom) and were analyzed with Gen5 3.10 software (Biotek). SARS-CoV-2 PLpro protease activity was calculated by the ratio of GFP signal sum intensity over the mCherry signal sum intensity. The FlipGFP-PLpro assay IC50 value was calculated by plotting the GFP/mCherry signal over the applied compound concentration with a four-parameter dose-response function in Prism 8. The mCherry signal alone was utilized to determine the compound cytotoxicity.
Enzymatic counter screenings against host proteases
The enzymatic assay of human ubiquitinase in digesting Ub-AMC substrate (UBPBIO, M3010) was performed in a 384-well plate. The ubiquitinases were dissolved in the reaction buffer (50 mM HEPES, 0.1 mg/mL BSA, 0.01% Triton X-100, 5 mM DTT, and 5% glycerol in 50 µL) to the final concentrations: 1 nM USP2 (R&D systems, E504), 20 nM USP7 (Sino Biological, 11681-H20B), 20 nM USP8 (Boston Biochem. E-520-025), 500 nM USP14 (ProSci, 91-171), 1 nM USP15 (R&D systems, E594), 20 nM USP30 (R&D systems, E582), and 1 nM UCH-L1 (R&D systems, 6007-CY). Then, 1 µL of Jun13296 with various concentrations or DMSO control was added to the PLpro protein solution. The PLpro protein with Jun13296 was mixed by shaking and incubating at 30 °C for 30 min. The enzymatic reaction was initiated by adding 1 µL Ub-AMC substrate (UBPBIO, M3010) to a final concentration of 2 µM. The fluorescent signal was monitored at excitation of 360 nm and emission of 460 nm. The first 500 s of the initial velocity of each reaction was calculated by GraphPad Prism 8.0 and was compared with the DMSO-treated group to determine the inhibition of Jun13296.
The enzymatic assays of Cathepsin B and K were performed as previously described68. 100 µl of cathepsin B or K was dissolved in the reaction buffer (20 mM sodium acetate, 1 mM EDTA, 5 mM DTT). The final concentration of the enzymes were 1.5 nM of cathepsin B and 100 nM of cathepsin K. The enzyme solutions were incubated at 30 °C for 30 minfor activation, followed by the addition and incubation of Jun13296. The reaction was initiated by the addition of the substrate Z-FR-AMC to a final concentration of 5 µM.
Calpain 1 (Sigma, C6108) were dissolved in the reaction buffer (50 mM HEPES, 50 mM NaCl, 10 mM DTT, and 5 mM CaCl2) in 100 µL. The final concentration of calpain 1 was 0.01 µg/mL. Then, 1 µL of Jun13296 in various concentrations or DMSO control was added to the PLpro protein solution. The PLpro protein with Jun13296 was mixed by shaking and incubating at 30 °C for 30 min. The enzymatic reaction was initiated by the addition of 1 µL of 4 mM N-succinyl-Leu-Leu-Val-Tyr-7-amino-4-methylcoumarin (Sigma, S6510) substrate.
Caspase-3 (BioVision, 1083) was dissolved in the reaction buffer (20 mM sodium acetate, 1 mM EDTA, 5 mM DTT) in 100 µL. The final concentration of Caspase-3 was 0.01 unit/µL. Then, 1 µL of Jun13296 with various concentrations or DMSO control was added to the Caspase-3 solution. The Caspase-3 with Jun13296 was mixed by shaking and incubating at 30 °C for 30 min. The enzymatic reaction was initiated by the addition of 1 µL of 2 mM Ac-DEAD-AMC substrate.
Trypsin (Sigma, T6763) was dissolved in the reaction buffer (50 mM HEPES, 50 mM NaCl, 10 mM DTT) in 100 µL. The final concentration of trypsin was 20 µg/mL. Then, 1 µL of Jun13296 with various concentrations or DMSO control was added to the trypsin solution. The trypsin with Jun13296 was mixed by shaking and incubating at 30 °C for 30 min. The enzymatic reaction was initiated by the addition of 1 µL of 2 mM Bz-R-AMC (Bachem, 4002540.0050) substrate. The fluorescent signal was monitored at excitation of 360 nm and emission of 460 nm. The first 500 seconds of the initial velocity of each reaction was calculated by GraphPad Prism 8.0 and was compared with the DMSO-treated group to determine the inhibition of Jun13296.
Antiviral assays with live SARS-CoV-2
A SARS-CoV-2 reporter virus assay and an antiviral plaque assay were employed for antiviral effect evaluation. The reporter virus assay was performed as described previously30. Briefly, Caco2-AT cells at a density of 1.5 × 104 cells/well were batch inoculated with icSARS-CoV-2-nLuc at a MOI of 0.2 and then added to 96-well plates with 50 μL per well of DMEM containing 4% FBS and 2 μM CP-100356. To prepare compound solutions, the test compounds and positive control (Jun12682) were 3-fold serially diluted in DMEM, starting at 30 μM final concentration for the testing compounds. After dilution, 50 μL diluted compound solution was transferred and mixed (1:1 volume ratio) with the cell-virus mixture pre-seeded in 96-well plates. Cells were incubated for 24 h at 37 °C with 5% CO2 and then subjected to a Nano-Glo Luciferase assay (Promega, G9243) on a Promega GloMax Discover microplate reader (Promega, GM3000) following the manufacturer’s instructions. The raw data were analyzed with Prism 10 using the non-linear regression curve fit method.
The antiviral plaque assay was performed similarly as we described previously28. Briefly, Vero-AT cells (3.0 × 105 cells/well) were seeded in 12-well plates a day before viral infection. The PLpro inhibitors dissolved in DMSO were serially diluted in DMEM with 3-fold dilutions between test concentrations, starting at 10 μM final concentration for PLpro inhibitors or 1 μM final concentration for nirmatrelvir. Cells in 12-well plates were infected with approximately 20 plaque-forming units (PFU) per well for 1 h. After incubation, the inoculum was removed and 1 mL 1X DMEM-1.2% Avicel (FMC polymers) mixture containing serially-diluted compound and 2 μM CP-100356 was added to each well. After 48 h of incubation at 37 °C, the DMEM-Avicel mixture was removed and the cells were stained using 0.1% crystal violet solution. Plates were photographed and measured for the area of cells affected by infection using ImageJ.
Mouse experiments
Nine to twelve-week-old female BALB/c mice (Strain #: 000651) were procured from the Jackson Laboratory and acclimated for a week before viral inoculation in standard vivarium conditions of dark/light cycle, ambient temperature, and humidity. The animal sex was not considered in the study design as both sexes of mice are susceptible to SARS2-N501YMA30 infection. Mice were randomly assigned to different groups, with numbers per group sufficient to obtain statistical significance. For virus inoculation, mice were briefly anesthetized with isoflurane and inoculated intranasally (i.n.) with 6000 PFU of SARS2-N501YMA30 in a total volume of 50 μL DMEM. PLpro inhibitors were dissolved in 0.5% methylcellulose solution containing 2% Tween-80 and administered via oral gavage using 20 G/30 mm plastic feeding tubes (Instech, FTP203050). Oral treatment with PLpro inhibitors or vehicle started one-hour post-virus inoculation, following a twice-daily, 10 h–14 h interval administration for 3 days. Mouse body weight and health were monitored daily. Mice losing 20% of body weight were humanely euthanized (predefined endpoint). The weight loss and mortality results were analyzed with Prism 10. A group of 5 mice for each treatment group were euthanized at 2- and 4-day post-infection (DPI) for necropsy. The left lungs were collected in pre-filled bead tubes (Fisher Scientific, 15-340-153) filled with 1 mL DMEM for viral load determination. For histopathological analyses, the remaining portion of the lungs was infused with 1 mL zinc-buffered formalin solution (Fisher Scientific, STLBFZ1), followed by removal and fixation with 30 mL zinc-buffered formalin solution for at least two days before removal from ABSL3 per an approved IBC protocol.
Lung viral titer determination by plaque assay
The left lung tissues were homogenized in pre-filled bead tubes using an automated homogenizer (Fisherbrand™, 15-340-164), followed by centrifugation (500 × g for 5 min). The clarified tissue homogenate supernatants were aliquoted and stored at −80 °C or subjected to a standard plaque assay with Vero-AT cells. The supernatants were serially diluted in DMEM and inoculated onto Vero-AT cells in six-well plates and maintained at 37 °C in 5% CO2 for 1 h with gentle rocking every 15 min. After removing the inoculum, plates were overlaid with 1.2% agarose (Fisherbrand, BP160-500)-1X DMEM mix containing 2% FBS. After 2 days, overlays were removed, and plaques were visualized by staining with 0.1% crystal violet. Viral titers were quantified as PFU per mL tissue homogenate.
Lung histology and immunohistochemistry (IHC)
Fixed tissues were trimmed and processed using a Sakura Tissue-Tek VIP® 6 AI tissue processor (Sakura Finetek USA, Inc. 1750 214th St, Torrence CA, 90501) on a delayed short cycle program and embedded in paraffin (Leica Surgipath Paraplast Infiltration and Embedding Medium; Leica Biosystems). Paraffin blocks were cut into 4 μm-thick sections and mounted on VistaVision HistoBond adhesive glass slides from VWR (Radnor, PA). Hematoxylin and eosin (H&E) staining was performed following standard operating procedures with the Sakura Finetek DRS601 (Sakura Finetek USA, Inc., 1750 West 214th Street, Torrance, CA 90501). For IHC, slides were rehydrated with water, following HIER (Heat Induced Epitope Retrieval) performed at 95 °C for 20 min in Citrate Unmasking solution (H-3300, Vector Laboratories, Newark, CA). SARS-CoV-2 Nucleocapsid antibody [HL448] (Genetex, GTX635686) was diluted 1:5000 in TBS-Tween 20 buffer with 10% normal goat serum and slides were incubated for 1 h at room temperature (RT). Slides were washed in TBS-Tween 20 buffer and then quenched of endogenous peroxidase using 0.3% H2O2 for 10 min. Slides were washed and detection was carried out using VECTASTAIN® Elite® ABC-HRP Kit, Peroxidase (Rabbit IgG) (Vector Laboratories, PK-6101) per the manufacturer’s instructions. Hematoxylin diluted 1:10 was used as a counterstain. Stained tissue sections were evaluated by a board-certified veterinary pathologist for three parameters: presence of edema or hyaline membranes, perivascular lymphoid inflammation, and interstitial inflammation. Edema or hyaline membranes were evaluated using a distribution-based ordinal scoring on a scale of 0 to 5 with 0, none; 1, <5%; 2, 6–25%; 3, 26–50%; 4, 51–75%; and 5, >75% of tissue affected. Perivascular lymphoid inflammation and interstitial pneumonia were evaluated using a severity-based ordinal scoring system on a scale of zero to four: 0 (absent), 1 (minimal), 2 (mild), 3 (moderate), and 4 (severe). An accumulative score is calculated by summing the ordinal scores of the three parameters. IHC was scored using a distribution-based ordinal scoring of 0 to 5 with 0, none; 1, <5%; 2, 6–25%; 3, 26–50%; 4, 51–75%; and 5, >75% of tissue affected.
RNA extraction and Real-time PCR quantification
Total RNA was extracted from the lung homogenate supernatants using TRIzol (Invitrogen, 15596018). A total of 1000 ng RNA was used for cDNA synthesis using the RT² HT First Strand Kit (QIAGEN, 330411), which contains a component to eliminate genomic DNA contamination. Quantitative PCR was performed with specific primers (Table 1) using PowerUp SYBR Green Master mix (Fisher, A25918) on QuantStudio 6 Pro (ThermoFisher, A43160). Cycle threshold values were normalized to 18S rRNA levels by using the 2-ΔCt method.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Crystal structures generated during the current study are available in the Protein Data Bank (PDB) under accession codes 9DO3 (PLpro bound to Jun13317), 9DNV (PLpro bound to Jun13308), 9DO1 (PLpro bound to Jun13307), 9DOI (PLpro bound to Jun13306). 9DNU (PLpro bound to Jun13296) and 9DO5 (PLpro bound to Jun12665). The mouse-adapted SARS2-N501YMA30 virus was obtained from Dr. Stanley Perlman at the University of Iowa and the availability of this virus is restricted under a materials transfer agreement (MTA) with the University of Iowa. The request for this virus should be directed to Dr. Stanley Perlman. The recombinant SARS-CoV-2 nsp5 mutant viruses are available upon request by contacting Xufang Deng through an MTA with Oklahoma State University. Requests will be processed within 8 weeks, and if approved, arrangements will be made for materials transfer. The following SARS-CoV-2 strains/isolates were obtained through and available at BEI Resources (https://www.beiresources.org/), NIAID, NIH: Washington strain 1 (WA1) (cat# NR-52281), recombinant SARS-CoV-2 expressing nano-luciferase reporter (icSARS-CoV-2-nLuc) (cat# NR-54003), Omicron JN.1 variant (cat# NR-59693), and Omicron XBB1.16 variant (cat# NR-59442). These viruses may be further distributed to other entities for any legitimate purpose required to rapidly prevent, detect, prepare for, and respond to, the spread or transmission of SARS-CoV-2, as described in an Emergency Use Simple Letter Agreement (EUSLA) Article 1. An MTA is needed for further distribution and must be reported to BEI (contact@beiresources.org). Source data are provided with this paper as Source Data file. Source data are provided with this paper.
References
Li, G., Hilgenfeld, R., Whitley, R. & De Clercq, E. Therapeutic strategies for COVID-19: progress and lessons learned. Nat. Rev. Drug Discov. 22, 449–475 (2023).
Shoichet, B. K. & Craik, C. S. Preparing for the next pandemic. Science 382, 649–650 (2023).
Boby, M. L. et al. Open science discovery of potent noncovalent SARS-CoV-2 main protease inhibitors. Science 382, eabo7201 (2023).
Karim, M., Lo, C. W. & Einav, S. Preparing for the next viral threat with broad-spectrum antivirals. J. Clin. Investig. 133, e170236 (2023).
Radoshitzky, S. R. et al. Expanded profiling of Remdesivir as a broad-spectrum antiviral and low potential for interaction with other medications in vitro. Sci. Rep. 13, 3131 (2023).
Malin, J. J., Suárez, I., Priesner, V., Fätkenheuer, G. & Rybniker, J. Remdesivir against COVID-19 and Other Viral Diseases. Clin. Microbiol. Rev. 34, e00162–20 (2020).
Kabinger, F. et al. Mechanism of molnupiravir-induced SARS-CoV-2 mutagenesis. Nat. Struct. Mol. Biol. 28, 740–746 (2021).
Joyce, R. P., Hu, V. W. & Wang, J. The history, mechanism, and perspectives of nirmatrelvir (PF-07321332): an orally bioavailable main protease inhibitor used in combination with ritonavir to reduce COVID-19-related hospitalizations. Med. Chem. Res. 31, 1637–1646 (2022).
Owen, D. R. et al. An oral SARS-CoV-2 M(pro) inhibitor clinical candidate for the treatment of COVID-19. Science 374, 1586–1593 (2021).
Tan, B., Joyce, R., Tan, H., Hu, Y. & Wang, J. SARS-CoV-2 main protease drug design, assay development, and drug resistance studies. Acc. Chem. Res. 56, 157–168 (2023).
Unoh, Y. et al. Discovery of S-217622, a Noncovalent Oral SARS-CoV-2 3CL protease inhibitor clinical candidate for treating COVID-19. J. Med. Chem. 65, 6499–6512 (2022).
Allerton, C. M. N. et al. A second-generation oral SARS-CoV-2 main protease inhibitor clinical candidate for the treatment of COVID-19. J. Med. Chem. 67, 13550–13571 (2024).
Shurtleff, V. W. et al. Invention of MK-7845, a SARS-CoV-2 3CL protease inhibitor employing a novel difluorinated glutamine mimic. J. Med. Chem. 67, 3935–3958 (2024).
Hu, Y. et al. Naturally occurring mutations of SARS-CoV-2 main protease confer drug resistance to nirmatrelvir. ACS Cent. Sci. 9, 1658–1669 (2023).
Sacco, M. D. et al. The P132H mutation in the main protease of Omicron SARS-CoV-2 decreases thermal stability without compromising catalysis or small-molecule drug inhibition. Cell Res. 32, 498–500 (2022).
Iketani, S. et al. Multiple pathways for SARS-CoV-2 resistance to nirmatrelvir. Nature 613, 558–564 (2022).
Zhou, Y. et al. Nirmatrelvir-resistant SARS-CoV-2 variants with high fitness in an infectious cell culture system. Sci. Adv. 8, eadd7197 (2022).
Jochmans, D. et al. The Substitutions L50F, E166A, and L167F in SARS-CoV-2 3CLpro Are Selected by a protease inhibitor in vitro and confer resistance to Nirmatrelvir. mBio, 14, e0281522 (2023).
Kiso, M. et al. In vitro and in vivo characterization of SARS-CoV-2 strains resistant to nirmatrelvir. Nat. Commun. 14, 3952 (2023).
Abdelnabi, R. et al. Nirmatrelvir-resistant SARS-CoV-2 is efficiently transmitted in female Syrian hamsters and retains partial susceptibility to treatment. Nat. Commun. 14, 2124 (2023).
Zuckerman, N. S., Bucris, E., Keidar-Friedman, D., Amsalem, M. & Brosh-Nissimov, T. Nirmatrelvir resistance - de novo E166V/L50V mutations in an immunocompromised patient treated with prolonged nirmatrelvir/ritonavir monotherapy leading to clinical and virological treatment failure - a case report. Clin. Infect. Dis. 78, 352–355 (2023).
Rut, W. et al. Activity profiling and crystal structures of inhibitor-bound SARS-CoV-2 papain-like protease: A framework for anti-COVID-19 drug design. Sci. Adv. 6, eabd4596 (2020).
Shin, D. et al. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature 587, 657–662 (2020).
Wydorski, P. M. et al. Dual domain recognition determines SARS-CoV-2 PLpro selectivity for human ISG15 and K48-linked di-ubiquitin. Nat. Commun. 14, 2366 (2023).
Tan, H., Hu, Y., Jadhav, P., Tan, B. & Wang, J. Progress and challenges in targeting the SARS-CoV-2 Papain-like protease. J. Med. Chem. 65, 7561–7580 (2022).
Sanders, B. C. et al. Potent and selective covalent inhibition of the papain-like protease from SARS-CoV-2. Nat. Commun. 14, 1733 (2023).
Ghosh, A. K., Mishevich, J. L., Mesecar, A. & Mitsuya, H. Recent drug development and medicinal chemistry approaches for the treatment of SARS-CoV-2 infection and COVID-19. ChemMedChem 17, e202200440 (2022).
Tan, B. et al. Design of a SARS-CoV-2 papain-like protease inhibitor with antiviral efficacy in a mouse model. Science 383, 1434–1440 (2024).
Shen, Z. et al. Design of SARS-CoV-2 PLpro inhibitors for COVID-19 antiviral therapy leveraging binding cooperativity. J. Med. Chem. 65, 2940–2955 (2022).
Jadhav, P. et al. Structure-based design of SARS-CoV-2 papain-like protease inhibitors. Eur. J. Med. Chem. 264, 116011 (2024).
Tan, H., Hu, Y. & Wang, J. FlipGFP protease assay for evaluating in vitro inhibitory activity against SARS-CoV-2 M(pro) and PL(pro). STAR Protoc. 4, 102323 (2023).
Ma, C. et al. Discovery of SARS-CoV-2 papain-like protease inhibitors through a combination of high-throughput screening and a FlipGFP-based reporter assay. ACS Cent. Sci. 7, 1245–1260 (2021).
Klemm, T. et al. Mechanism and inhibition of the papain-like protease, PLpro, of SARS-CoV-2. EMBO J. 39, e106275 (2020).
Garnsey, M. R. et al. Discovery of SARS-CoV-2 papain-like protease (PL(pro)) inhibitors with efficacy in a murine infection model. Sci. Adv. 10, eado4288 (2024).
Zhang, L. et al. Viral anti-inflammatory serpin reduces immuno-coagulopathic pathology in SARS-CoV-2 mouse models of infection. EMBO Mol. Med. 15, e17376 (2023).
Wang, G. et al. Universal subunit vaccine protects against multiple SARS-CoV-2 variants and SARS-CoV. NPJ Vaccines 9, 133 (2024).
Fumagalli, V. et al. Antibody-independent protection against heterologous SARS-CoV-2 challenge conferred by prior infection or vaccination. Nat. Immunol. 25, 633–643 (2024).
Fumagalli, V. et al. Nirmatrelvir treatment of SARS-CoV-2-infected mice blunts antiviral adaptive immune responses. EMBO Mol. Med. 15, e17580 (2023).
Moore, J. B. & June, C. H. Cytokine release syndrome in severe COVID-19. Science 368, 473–474 (2020).
Coomes, E. A. & Haghbayan, H. Interleukin-6 in Covid-19: A systematic review and meta-analysis. Rev. Med. Virol. 30, 1–9 (2020).
Gordon, A. C. et al. Interleukin-6 receptor antagonists in critically Ill patients with Covid-19. N. Engl. J. Med. 384, 1491–1502 (2021).
Jones, S. A. & Hunter, C. A. Is IL-6 a key cytokine target for therapy in COVID-19? Nat. Rev. Immunol. 21, 337–339 (2021).
Shankar-Hari, M. et al. Association between administration of IL-6 antagonists and mortality among patients hospitalized for COVID-19: a meta-analysis. JAMA 326, 499–518 (2021).
Chen, L. Y. C., Hoiland, R. L., Stukas, S., Wellington, C. L. & Sekhon, M. S. Confronting the controversy: interleukin-6 and the COVID-19 cytokine storm syndrome. Eur. Respir. J. 56, 2003006 (2020).
Todorović-Raković, N. & Whitfield, J. R. Between immunomodulation and immunotolerance: the role of IFNγ in SARS-CoV-2 disease. Cytokine 146, 155637 (2021).
Karki, R. et al. Synergism of TNF-α and IFN-γ triggers inflammatory cell death, tissue damage, and mortality in SARS-CoV-2 infection and cytokine shock syndromes. Cell 184, 149–168.e117 (2021).
Krishna, B. A. et al. Spontaneous, persistent, T cell-dependent IFN-γ release in patients who progress to Long Covid. Sci. Adv. 10, eadi9379 (2024).
Li, C. et al. Comparative single-cell analysis reveals IFN-γ as a driver of respiratory sequelae after acute COVID-19. Sci. Transl. Med. 16, eadn0136 (2024).
Mielech, A. M., Chen, Y., Mesecar, A. D. & Baker, S. C. Nidovirus papain-like proteases: multifunctional enzymes with protease, deubiquitinating and deISGylating activities. Virus Res. 194, 184–190 (2014).
Báez-Santos, Y. M., John, S. E. S. & Mesecar, A. D. The SARS-coronavirus papain-like protease: structure, function and inhibition by designed antiviral compounds. Antiviral Res. 115, 21–38 (2015).
Xiong, Y. et al. The substrate selectivity of papain-like proteases from human-infecting coronaviruses correlates with innate immune suppression. Sci. Signal. 16, eade1985 (2023).
Swaim, C. D., Scott, A. F., Canadeo, L. A. & Huibregtse, J. M. Extracellular ISG15 signals cytokine secretion through the LFA-1 integrin receptor. Mol. Cell 68, 581–590.e585 (2017).
Perng, Y. C. & Lenschow, D. J. ISG15 in antiviral immunity and beyond. Nat. Rev. Microbiol. 16, 423–439 (2018).
Sarkar, L., Liu, G. & Gack, M. U. ISG15: its roles in SARS-CoV-2 and other viral infections. Trends Microbiol. 31, 1262–1275 (2023).
Munnur, D. et al. Altered ISGylation drives aberrant macrophage-dependent immune responses during SARS-CoV-2 infection. Nat. Immunol. 22, 1416–1427 (2021).
Clasman, J. R., Everett, R. K., Srinivasan, K. & Mesecar, A. D. Decoupling deISGylating and deubiquitinating activities of the MERS virus papain-like protease. Antiviral Res. 174, 104661 (2020).
Deng, X. et al. Structure-guided mutagenesis alters deubiquitinating activity and attenuates pathogenesis of a murine coronavirus. J. Virol. 94, e01734-19 (2020).
Myeni, S. K. et al. Engineering potent live attenuated coronavirus vaccines by targeted inactivation of the immune evasive viral deubiquitinase. Nat. Commun. 14, 1141 (2023).
van Huizen, M. et al. Deubiquitinating activity of SARS-CoV-2 papain-like protease does not influence virus replication or innate immune responses in vivo. PLoS Pathog. 20, e1012100 (2024).
Chen, D. Y. et al. Cell culture systems for isolation of SARS-CoV-2 clinical isolates and generation of recombinant virus. iScience 26, 106634 (2023).
Deng, X. et al. Breakthrough infections with multiple lineages of SARS-CoV-2 variants reveals continued risk of severe disease in immunosuppressed patients. Viruses 13, 1743 (2021).
Evans, P. R. & Murshudov, G. N. How good are my data and what is the resolution? Acta. Crystallogr. D Biol. Crystallogr. 69, 1204–1214 (2013).
Vonrhein, C. et al. Data processing and analysis with the autoPROC toolbox. Acta. Crystallogr. D Biol. Crystallogr. 67, 293–302 (2011).
McCoy, A. J. et al. Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 (2007).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta. Crystallogr. D Biol. Crystallogr. 60, 2126–2132 (2004).
Afonine, P. V. et al. Towards automated crystallographic structure refinement with phenix.refine. Acta. Crystallogr. D Biol. Crystallogr. 68, 352–367 (2012).
Smart, O. S. et al. Grade2 version 1.6.0. Cambridge, United Kingdom: Global Phasing Ltd. (2021).
Ma, C. et al. Discovery of Di- and Trihaloacetamides as Covalent SARS-CoV-2 main protease inhibitors with high target specificity. J. Am. Chem. Soc. 143, 20697–20709 (2021).
Acknowledgements
We thank Shannon Cowan and Caden Miller at the Immunopathology Core of the Oklahoma Center for Respiratory and Infectious Diseases (OCRID) for their technical assistance. J.W. is funded by National Institutes of Health grants R01AI158775 and Rutgers HealthAdvance funding. Research reported in this publication was supported by the National Heart, Lung, And Blood Institute of the National Institutes of Health under Award Number U01HL150852. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. E.A. is funded by National Institutes of Health grant U19AI171110; X.D. is funded by National Institutes of Health grant R01AI158775. Research reported in this publication was made possible in part by the services of the Immunopathology Core, part of the OCRID which is supported by the NIH COBRE grant under award number P30GM149368.
Author information
Authors and Affiliations
Contributions
J.W., X.D., E.A., and F.X.R. conceived and supervised the research and designed the experiments; J.W., P.J., and B.T. designed the inhibitors; P.J. and B.T. performed chemical syntheses, separation, purification, and structural characterizations; A.A. and F.X.R. performed gene expression, protein purification, crystallization, and diffraction data collection; A.A., and F.X.R., and E.A. determined and analyzed the crystal structures; B.T., H.T., and K.L. performed enzymatic inhibition assays, DSF assays, and cellular cytotoxicity assays; X.L. performed in vitro cellular antiviral assays and in vivo mouse model studies; A.F. performed the histopathology and immunohistochemistry (IHC) assessment; X.C. performed the mouse tissue analysis and assisted in the in vivo antiviral studies; J.W., P.J., B.T., X.L., A.A., A.F., X.C., F.X.R., X.D., and E.A. analyzed and discussed the data with the assistance of H.T., and K.L.; and J.W., A.A., F.X.R., E.A., and X.D. wrote the manuscript with the assistance of B.T., X.L., P.J., H.T., K.L., and A.F.
Corresponding authors
Ethics declarations
Competing interests
Rutgers, the State University of New Jersey, has applied for a patent US2024382494A1 covering the PLpro inhibitors reported in this study and related compounds, which has been published and is pending. J.W. is listed as an inventor. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Chamandi Dampalla, and the other, anonymousreviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Jadhav, P., Liang, X., Ansari, A. et al. Design of quinoline SARS-CoV-2 papain-like protease inhibitors as oral antiviral drug candidates. Nat Commun 16, 1604 (2025). https://doi.org/10.1038/s41467-025-56902-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-56902-x
This article is cited by
-
YL1004 is a SARS-CoV-2 papain-like protease inhibitor with immunomodulatory and antiviral activity in mice
Nature Communications (2026)
-
Chasing allosteric inhibition of the SARS-CoV-2 PLpro via molecular dynamics simulations with flooding fragments (MDFFr)
Journal of Computer-Aided Molecular Design (2026)
-
A Comprehensive Review on Broad Spectrum of Medicinal Applications of Schiff Base Metal Complexes Derived from Quinoline Scaffolds
Journal of Inorganic and Organometallic Polymers and Materials (2025)
