
Abstract
Single-cell ATAC-seq technology advances our understanding of single-cell heterogeneity in gene regulation by enabling exploration of epigenetic landscapes and regulatory elements. However, low sequencing depth per cell leads to data sparsity and high dimensionality, limiting the characterization of gene regulatory elements. Here, we develop scAGDE, a single-cell chromatin accessibility model-based deep graph representation learning method that simultaneously learns representation and clustering through explicit modeling of data generation. Our evaluations demonstrated that scAGDE outperforms existing methods in cell segregation, key marker identification, and visualization across diverse datasets while mitigating dropout events and unveiling hidden chromatin-accessible regions. We find that scAGDE preferentially identifies enhancer-like regions and elucidates complex regulatory landscapes, pinpointing putative enhancers regulating the constitutive expression of CTLA4 and the transcriptional dynamics of CD8A in immune cells. When applied to human brain tissue, scAGDE successfully annotated cis-regulatory element-specified cell types and revealed functional diversity and regulatory mechanisms of glutamatergic neurons.
Similar content being viewed by others


Introduction
Gene expression is regulated by millions of active DNA regulatory elements exposed in accessible chromatin regions of the genome1. The technique of Single-cell Assay for Transposase-Accessible Chromatin using sequencing (scATAC-seq) enables the exploration of the chromatin accessibility landscape via the hyperactive Tn5 transposase2. Consequently, scATAC-seq has provided notable insights into cellular heterogeneity by profiling chromatin accessibility beyond transcriptome measurements, shedding light on epigenetic regulatory mechanisms at the single-cell level3. However, during whole-genome profiling, the capture rate of open chromatin sites is relatively low, leading to high dimensionality and sparsity in scATAC-seq data, which poses a significant analytical challenge. This sparsity obscures important genomic information and complicates downstream analyses, including peak calling and motif discovery. Furthermore, the inherent limitations in DNA copy numbers exacerbate the binary nature of the data, making it difficult to distinguish signal from noise. These issues collectively create substantial challenges for computational analysis, and the rapidly expanding scATAC-seq datasets4 highlight the urgent need for better and more robust analytical tools.
Accurate identification of cell types from single cell resolution ATAC-seq data is fundamental in understanding gene regulation and epigenetic heterogeneity within complex tissues. To date, most scATAC-seq studies have directly applied clustering algorithms that were developed for single-cell RNA sequencing (scRNA-seq) data to identify cell types5,6,7. However, their performance may vary when directly applied to scATAC-seq data, due to distinct differences in data sparsity and distribution characteristics. In recent years, computational methods have been aimed at transforming data into a low-dimensional space while preserving the relevant structure of the data; for example, scABC8, cisTopic9, SnapATAC10, chromVAR11, and Cicero12 all compute a low-dimensional embedding for each individual cell. However, the performance of these methods is surpassed by subsequent deep learning-based models, including SCALE13, SAILER14, and PeakVI15, which have demonstrated their ability to perform nonlinear transformations and create low-dimensional embeddings by automatically, effectively extracting key information from the scATAC-seq data. Although these models are continually complicating the variational autoencoder (VAE) architecture to improve the ability to model scATAC-seq data, they have always been based on an autoencoder approach that inevitably ignores complex cell relationships of the global cell population.
Beyond cell type identification, scATAC-seq research aims to reveal gene regulatory networks and epigenetic heterogeneity by providing deep insights into single-cell chromatin accessibility data through precise peak selection and imputation. However, most existing work ignores this or directly employs methods designed for gene selection in scRNA-seq data analysis, such as Scanpy6. To better accommodate scATAC-seq data, EpiScanpy16 was introduced as a toolkit for the analysis of single cell epigenomic data, which performs peak selection based on fixed predefined statistical indicators that do not vary depending on the dataset. Another critical task is imputation, which aims to eliminate noise signals and recover true chromatin accessibility landscapes, thus enhancing downstream analyses, including the identification of regulatory elements and transcription factor motifs. Currently, SCALE is one of the few methods specifically designed to address the unique imputation challenges in scATAC-seq data13. However, SCALE and similar methods may not fully capture the intricate cell-cell interactions and topological structure present in chromatin accessibility landscapes. Emerging graph neural networks (GNNs) have demonstrated a natural ability to capture structural information from graphs by propagating neighbor information17. These networks typically integrate cell graphs with deep autoencoders to uncover compact latent representations, thereby exploring both rich content and structural information. Compared to traditional autoencoders used in scATAC-seq analysis18,19, the incorporation of cell graphs and GNNs enables the learning of low-dimensional representations of graph topology, which enhances the modeling of cell-cell relationships, which are driven by complex mechanisms such as underlying cellular interactions or communication in the context of their microenvironments, offering a more comprehensive understanding of the structural and functional context of the data.
To this end, we propose scAGDE, a single-cell chromatin accessibility model-based deep graph embedded learning method designed to efficiently process sparse single-cell ATAC-seq data. scAGDE distinguishes itself from previous tools in four key aspects. First, scAGDE reconstructs both the chromatin accessibility profiles and the neighboring graph from the same low-dimensional cell representation. This design allows scAGDE to retain the chromatin accessibility profiles of the analyzed cells and their neighboring cells, as well as the cell-cell interaction relationships between them. In contrast, other methods do not construct graphs and merely analyze chromatin accessibility profiles, lacking the ability to learn relationships between neighboring cells. Second, scAGDE uses a chromatin accessibility-based autoencoder to measure the importance of the peaks and select the key peaks, enabling scAGDE to focus on highlighting the chromatin accessibility profiles of each analyzed cell or the most critical peak regions. Third, scAGDE employs a dual-decoder component that can reconstruct the cell topology and estimate the distribution of data. This ensures that scAGDE accurately models scATAC-seq data while retaining the relationship of each cell with its neighboring cell profiles in representation. Lastly, scAGDE defines a dual cluster optimization objective to guide the preservation of the information about cell heterogeneity in its representation. This explicit cluster optimization task is overlooked by other methods and prompts our model to focus on distinguishing cell heterogeneity. Experimental tests have demonstrated that scAGDE outperforms existing scATAC-seq analysis methods across multiple synthetic datasets derived from ATAC-seq data of bone marrow and several real-world datasets characterized by different degrees of sparsity, sequencing platforms, and species diversity. Moreover, scAGDE effectively supports dimensionality reduction, visualization, and dropout event correction. Specifically, through the imputation of a mouse forebrain dataset, scAGDE identified potential accessible peaks that contain insightful regulatory elements, including crucial transcription factor binding motifs. Extended analysis in a human brain dataset revealed that scAGDE could successfully annotate cis-regulatory element (CRE)-specified cell types while uncovering functional diversity within glutamatergic neurons.
Results
Methodology overview of scAGDE
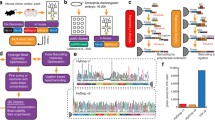
The primary objective of scAGDE is to revolutionize the analysis of high-dimensional sparse scATAC-seq data by learning low-dimensional topological embedding representations. The summary of scAGDE is shown in Fig. 1a, which mainly consists of processing scATAC-seq data using a chromatin accessibility-based autoencoder and the graph embedding learning procedure. The proposed chromatin accessibility-based autoencoder is used to comprehensively learn the latent representation of the raw data matrix. This step involves selecting significant peaks as features for cell characterization and constructing a cell graph based on this information. In the next step, scAGDE integrates a Graph Convolutional Network (GCN) as an encoder, which extracts crucial information and considers cell-cell relationships in the cell graph, and then applies a Bernoulli-based decoder to model the probability of chromatin opening events. Additionally, scAGDE infers clustering assignments using a cluster layer as soft labels from latent embedding representations by optimizing two loss functions and incorporates a self-supervised learning mechanism to refine the representation learning process. The scAGDE produces the final topological embeddings, providing efficient and accurate representations of scATAC-seq data.

a A summary graphical illustration of the scAGDE workflow. scAGDE takes the binary cell-by-peak matrix as input first into a chromatin accessibility-based autoencoder and then performs the graph embedding learning. b The chromatin accessibility-based autoencoder maps data into a latent space, where each individual cell connects its nearest cell as neighbours to construct a cell graph. The variation in the encoder’s weights can be translated into the importance score of peaks for the peak selection procedure. c The well-prepared cell graph and filtered data are simultaneously handled by a two-layer GCN encoder (i) and mapped into the latent space (ii). On the one hand, the latent embedding serves as input to the dual decoders (iii), which include a graph decoder module to reconstruct cell graph from embedding, and a Bernoulli-based decoder module to estimate the probability of a peak being accessible, which are estimates of the true chromatin landscape in each cell. On the other hand, the dual clustering optimizations are introduced into scAGDE (iv), where a network of cluster layer, which is initialized by K-means results on the embedding, infers soft clustering labels. The target distribution and one-hot pseudo label are sequentially calculated and used for label prediction loss and distribution alignment loss. d scAGDE facilitates critical downstream applications of clustering, visualization, imputation, enrichment analysis and discovery of regulators.
Specifically, the chromatin accessibility-based autoencoder takes a binary preprocessed scATAC-seq data matrix as input to learn the preliminary compressed representations, while retaining essential chromatin accessibility information and minimizing noise in an unsupervised manner. The encoder module is expected to identify key accessible patterns within the accessibility landscapes, and thus is applied to extract crucial information for cell topology inference and informative peak selection, remarkably enhancing subsequent analyses. In particular, we define a cell neighborhood graph to structure the cell topological information, where cells constitute the nodes in the graph, and the edges between cells indicate the neighborhood relationships. To construct a cell graph, we search for a certain number of nearest cells as neighbors for each cell in the low-dimensional space obtained from the encoder outputs (Fig. 1b). Additionally, to balance the benefits of graph learning with resource overhead in scAGDE, we employ a weight distribution-based strategy on the encoder module for peak selection.
To extract essential chromatin accessibility information while simultaneously capturing cell-cell topology from both the cell graph and the filtered count matrix, scAGDE employs a GCN encoder module (Fig. 1c). Guided by the cell graph, this encoder module encodes each cell’s filtered accessibility profiles, while aggregating features from its neighbors. This approach significantly advances previous autoencoder-based methods, which often overlook the inter-cell relationships. Eventually, the GCN encoder module outputs the embeddings for cells in the low-dimensional latent space, where each cell is characterized by restricted features. Subsequently, the embeddings are decoded via dual decoder modules to reconstruct topological and accessibility information. First, a graph decoder module recovers a cell graph by similarity measurement on cell embeddings, minimizing differences between the corrupted and input cell graph, thus preserving the inter-cellular topology as accurately as possible. Second, scAGDE designs a Bernoulli-based decoder to represent the global probabilistic structure of the dataset by simulating the probability of chromatin site opening events. This choice of Bernoulli distribution aligns with the binary nature of chromatin site accessibility.
To self-optimize the clustering task, scAGDE incorporates the sequential clustering optimization (Fig. 1c). The first optimization uses KL divergence to align clustering assignments with an auxiliary target distribution, both derived from the cell embeddings. Here, the assignments act as soft labels that guide the iterative refinement of the clustering assignments. Clustering centers are initialized through pretraining and K-means to mitigate potential deviations in clustering distribution due to unreliable assignments. The second optimization uses cross-entropy loss to align soft labels with pseudo labels, which are determined by high-confidence assignments within the soft labels. By optimizing a comprehensive set of losses, scAGDE can jointly evaluate clustering label assignments and feature learning of the topological structure by training the entire graph autoencoder framework. This comprehensive approach allows scAGDE to accurately balance clustering and feature learning, ensuring reliable performance in scATAC-seq data analysis.
We comprehensively demonstrated the effectiveness of scAGDE on multiple synthetic datasets considering five sequencing depths, five noise levels, and seven dropout rates, as well as on several single cell experimental datasets differing by sequencing platform and other data characteristics. Comparative analysis with other scATAC-seq and scRNA-seq methodologies elucidates scAGDE’s superior performance on scATAC-seq data. In addition, scAGDE demonstrated exceptional ability in dimensionality reduction, visualization, dropout correction, and cell-type-specific enhancer discovery (Fig. 1d). It is notable that scAGDE’s imputation capacity has uncovered a wide spectrum of chromatin accessibility, enhancing the identification of critical regulatory elements associated with cellular identity. Furthermore, scAGDE effectively characterized single-cell heterogeneity and revealed functional diversity within a human brain dataset.
scAGDE achieves state-of-the-art performance on simulated scATAC-seq scenarios
We evaluated the clustering performance of scAGDE on simulated single cell ATAC-seq datasets, having various dataset characteristics. To achieve this, we sampled synthetic datasets from extensive bulk Bone Marrow ATAC-seq data according to reference20. Simulation datasets were varied in terms of read depth, noise level, and dropout rate to simulate different biological scenarios and generated with six annotated cell populations, including hematopoietic stem cells (HSC), common myeloid progenitor cells (CMP), erythroid cells (Ery), natural killer cells (NK), and CD4 and CD8 cells. To evaluate the consistency of generated clustering results and the true labels, we employed several metrics: the normalized mutual information (NMI), adjusted rand index (ARI) and F1 score. For the evaluation metrics, a score closer to 1 indicates superior clustering performance. On this basis, we conducted a comparative analysis of scAGDE to eleven other scATAC-seq data clustering methods, including ArchR21, SnapATAC222, SCALE13, SIMBA23, cisTopic9, Signac24, PeakVI15, BAVARIA18, SnapATAC10, SAILER14, and scABC8. This comparison aimed to comprehensively evaluate clustering performance in various conditions and explore the potential of scAGDE to analyze scATAC-seq data.
We first assessed the performance of the clustering methods on single-cell ATAC-seq datasets with adjusted read depth, represented by the number of fragments per cell (Fig. 2a). We systematically varied the fragment count, starting from a low depth represented by 250 or 500 fragments, a moderate depth of 1500 or 2500 fragments, and a high depth of 5000 fragments. The NMI values of the clustering performance are depicted in Fig. 2a and additional results for ARI and F1 values, as well as visualizations of the clustering assignments are provided in Supplementary Figs. 1a and 2. From the results we see several observations that firstly, the performance of all the computational methods decreased as the read depth decreased, suggesting that read depth has a critical effect on clustering accuracy. Secondly, we observe that the deep learning methods were more susceptible to read depth. At low read depths (250 or 500 fragments), SCALE struggled to differentiate between Ery cells and other cell populations, while PeakVI, SAILER, and BAVARIA failed to differentiate entirely. In contrast, ArchR, SnapATAC2, cisTopic, Signac and SnapATAC had ARI values above 0.5 at 500 fragments, successfully distinguishing both Ery and HSC cells. ArchR improved on Signac using an iterative LSI algorithm and consistently outperformed Signac. SnapATAC2 exhibited comparable performance to scAGDE yet did not obtain an ARI value near 1.0 until 2500 fragments. SIMBA worked well but not for low sequencing depths. Of note, scAGDE consistently surpassed the other methods across all read depths, with metric values closer to 1.00. Specifically, scAGDE demonstrated robustness to variation in read depth, providing accurate clustering even at low fragment counts. Remarkably, when the number of fragments per cell was increased to 1500, scAGDE approached a clustering accuracy of almost 1.00, a level of performance that other methods only reached at high fragments coverage. This suggests that scAGDE can effectively mine information contained in chromatin accessibility profiles, thereby revealing cellular heterogeneity via accurate and stable clustering results.

a Comparison of ARI values obtained for each method across five simulated datasets of varying sequencing depth. A value approach 1 indicates optimal clustering. The sequencing depth is represented by fragment numbers per cell, which covers a low depth of 250 to 500, moderate depth of 1500 to 2500 and high depth of 5000. b Table displaying NMI average results between scAGDE and compared methods under five noise levels. c UMAP visualization annotated by the true cell-type labels (first column, True-label) and obtained clustering labels from each method (subsequent columns). The first to last rows, respectively show clustering on simulated datasets with low (10%) to high (40%) noise levels. d Table displaying NMI values for each method across seven simulation datasets with varying dropout rates (top, 10–70%). The violin plots (bottom) aligned with the top horizontal axis displaying the increasing sparsity distribution of corresponding datasets (n = 1200 cells in each group; center black dot, median; box limits, upper and lower quartiles; whiskers, the 95% confidence intervals; violin plot edges represent minima and maxima). Source data are provided as a Source Data file.
Next, we evaluated the robustness of the computational methods under various levels of added noise. We increased the proportion of fragments sampled from the random distribution from zero to 0.4 in steps of 0.1. From Fig. 2b and Supplementary Fig. 1b, scAGDE demonstrated superior performance, maintaining stability across different noise levels. It is observed that NMI values of all the methods decreased with an increase in noise and in a noise-free environment, scAGDE achieved perfect cell assignment. ArchR, SnapATAC2, SIMBA, cisTopic and SnapATAC also performed well, reaching NMI values close to 1.0. Even with increased noise level, the performance of scAGDE remained robust, with an NMI value close to 1.0 and only a slight decrease even at the highest noise level, in contrast to the seven compared methods that had NMI values below 0.8. A couple of up-to-date methods such as ArchR, SnapATAC2 and SIMBA also showed decent robustness to noise, but it was slightly less resilient to dropout events compared to SnapATAC2 and scAGDE. Additionally, we visualized the raw data by projecting it onto a 2D space. We then annotated the visualization with the clustering results derived from the computational methods evaluated (Fig. 2c and Supplementary Fig. 3). From the results, we find that the introduction of noise obscured cellular heterogeneity, causing closely related cell types (such as HSC and CMP, CD4 and CD8) to become less distinguishable, as evidenced by the overlapping of these cell populations in the visualization plots. Nonetheless, scAGDE successfully resolved even the challenging cell populations and had a minimum misclassification rate, demonstrating its remarkable adaptability to noise.
Finally, we examined the influence of dropout events on the performance of our scAGDE model. Like scRNA-seq, scATAC-seq is also plagued by “dropout” issues, resulting in a sparse and high-dimensional count matrix that complicates downstream analysis, as noted in reference25. To address this, we simulated seven datasets with increasing dropout rates, defined as the proportion of peaks omitted from the read counts relative to all accessible peaks in each cell. The experimental results are summarized in Fig. 2d and Supplementary Fig. 1c of the clustering performance. The visualization of the overwhelming overlap of cell populations is listed in Supplementary Fig. 4. From the figures, we can observe that the performances of scABC, SAILER and BAVARIA were significantly affected by the increased dropout rates and there is a large number of methods had degraded performance sharply at dropout rates of 40% or higher, such as scABC, SAILER, SIMBA and ArchR. Most impressively, scAGDE proved to be resilient against increasing dropout frequencies as scAGDE maintained an NMI value close to 1.0 up to the 60% dropout rate, and remained above 0.85 even as dropout rates increased further. SnapATAC2 performed comparably to scAGDE, but degraded more severely at the highest dropout rate.
In summary, we can conclude that these simulation experiments highlight the superiority and adaptability of scAGDE with varying sequencing depth, diverse noise levels, and increasing rates of dropout events, demonstrating the potential of scAGDE for effective application to real-world scATAC-seq datasets under a variety of experimental conditions.
scAGDE provides promising performance than other scATAC-seq clustering methods across multiple real-world scATAC-seq datasets
We extended our experiments to real-world single-cell ATAC-seq datasets to investigate the clustering capabilities of scAGDE under real experimental conditions, compared to the other 11 computational methods. Specifically, we collected nine datasets of typical size and two datasets with considerably larger scales generated in different sequencing platforms. These datasets presented cell counts spanning from 300 to nearly 200,000 and peak numbers ranging from 7000 to 70,000, reflecting various degrees of sparsity, as detailed in Supplementary Note 20 and Supplementary Figs. 41 and 42. According to the prior benchmarking of single-cell genomics26, we assess the bio-conservation quality of the cell embeddings through a suite of metrics, including the ARI, NMI, F1, cell-type average silhouette width (ASW), with larger values of these metrics indicating better performance. To derive an overview of the comparison, we first for each method, averaged the value of each metric over all datasets. Then we normalized each metric using min-max scaling to aggregate these individual metrics into a unified score. In addition, an overall score was computed by taking the mean of all metrics, with a score of 1 indicating optimal performance. Fig. 3a, b represent the bio-conservation metrics and the ARI values of each method across all datasets, respectively. Our results reveal that scAGDE outperformed and maintained a large lead with other methods. Specifically, scAGDE reached the highest ARI on eight out of the eleven datasets and comparably high performance as SCALE and SnapATAC2 on the GM12878vsHEK, GM12878vsHL and InSilico datasets. The superiority of scAGDE on each dataset can be further seen in Supplementary Figs. 5 and 6. After scAGDE, SnapATAC2, ArchR and SCALE had the next highest overall scores, but were significantly less effective than scAGDE on datasets with more than 20,000 cells, such as the DropBlood, Lung and Liver datasets. Methods like BAVARIA, PeakVI, and SAILER generally had lower performance than SCALE, especially on sparser datasets. For instance, on the Leukemia and Forebrain datasets, their ARI values were significantly lower than those of scAGDE. Additionally, PeakVI failed completely on the two notably sparse datasets (GM12878vsHEK and GM12878vsHL). Given that ArchR also features its own unique peak selection, we compared it with scAGDE’s peak selection. When exchanging peak selection with each other, both performance degraded, but scAGDE still performed better (Supplementary Note 3 and Supplementary Fig. 12). The comparison of all algorithms using scAGDE’s peak selection strategy, alongside a cross-comparison with ArchR’s peak selection strategy, showed that scAGDE’s advantage lies in its model design itself, rather than peak selection alone (Supplementary Note 4 and Supplementary Fig. 13).

a The normalized scores (0–1 range) of six metrics used to evaluate each method’s bio-conservation across all datasets, with score of 1 indicating optimal performance. The overall score is the average of all metric values. b Each method’s ARI scores on all datasets. c UMAP visualization annotated by true cell type labels and obtained clustering labels on the Splenocyte dataset. The two dotted ellipses in each box indicate four T cell types (large dotted circle) and dendritic cells and macrophages (smaller dotted circle). d The agreement between the clustering labels and true cell type labels. Matrices with a clear diagonal indicate better performance. Source data are provided as a Source Data file.
Despite the density of the Splenocyte dataset, all methods except scAGDE and SIMBA produced suboptimal results on this dataset, with ARI values of 0.6 at the most. scAGDE, however, achieved an ARI of 0.84, illustrating its adeptness in clustering complex datasets. The visual clustering representation in Fig. 3c shows that scAGDE and SIMBA successfully segregated the four T cell types into distinct clusters and the accurate alignment of known B cell labels. The “reference” plot displays the ground truth and compared to this, scAGDE was able to differentiate two rare cell populations, Dendritic and Macrophage cells, which other methods such as SnapATAC2, SnapATAC, SCALE, SAILER and PeakVI failed to do accurately, often confusing the three B cell types into multiple overlapping groups. This indicates that scAGDE surpassed the other methods in distinguishing cell subpopulations across various datasets (Supplementary Figs. 7–10). In addition, scAGDE effectively separated three subpopulations of excitatory neuron cells (EX1, EX2, and EX3) with minimal overlap, as illustrated in Fig. 3d. To explore the reasons for SnapATAC’s poor performance on the Splenocyte dataset, we conducted exhaustive algorithmic component analysis and ablation experiments (Supplementary Note 5 and Supplementary Fig. 14).
We also compared the runtime and memory usage of scAGDE on the datasets to the other methods (Supplementary Fig. 11). It is noteworthy that scAGDE divides the data into multiple batches for learning when handling datasets with more than 50,000 cells. As depicted in Supplementary Fig. 11a, the runtime for most methods correlated with the overall data scale, which is determined by the number of cells and peaks. SnapATAC, ArchR and Signac emerged as the three fastest methods across all datasets, particularly at lower scales. In contrast, the performance of scAGDE is in line with the other deep learning methods, balancing speed and accuracy. Although scAGDE is inherently slower than the non-deep learning methods, its runtime remains in a competitive range. We also evaluated the runtime of deep learning methods on one core of a 2.20 GHz Intel Xeon Gold 5220 CPU, which is an alternative solution when a GPU is not available (Supplementary Fig. 11c). The results reveal that scAGDE takes as long as SCALE when clustering datasets with <15K cells and requires comparable runtime with PeakVI on large-scale datasets such as Lung and Liver. For the memory usage, we found scAGDE maintains reasonable memory requirements (Supplementary Fig. 11b), and there is not much difference between scAGDE and the deep learning methods when clustering large-scale datasets although scAGDE needs to process the entire cell graph due to its graph learning framework. In general, scAGDE’s architecture proved advantageous by optimizing the balance of speed and accuracy. This is probably attributable to the fewer model parameters (Supplementary Fig. 11d) and its peak importance filtering operation, a notable feature of scAGDE, simplified processing by focusing computational resources on the most informative parts of the data, thereby reducing unnecessary computation. Therefore, we can conclude that the balance between speed and analytical depth affirms scAGDE’s suitability for expansive single-cell ATAC-seq data analysis. To further demonstrate scAGDE’s scalability, we applied it to the human fetal atlas dataset from Domcke et al.27, which contains approximately 800,000 single cells from 59 human fetal samples, spanning 15 organs and ranging from 89 to 125 days in estimated postconceptual age. The results and analysis on this dataset are detailed in Supplementary Note 6 and Supplementary Fig. 15. Moreover, we investigated whether the methods developed for the scRNA-seq data could be directly applied to the scATAC-seq data analysis and the results demonstrated the necessity for specialized and tailored approaches to scATAC-seq data (Supplementary Note 7 and Supplementary Figs. 16–18).
scAGDE performs efficient dimensionality reduction correction and significantly improves visualization in scATAC-seq data
Dimensionality reduction and visualization in high-throughput single-cell ATAC-seq data are crucial for identifying potential cell subpopulations and investigating functional differences. To demonstrate the effectiveness of scAGDE, we compared it with widely-used dimensionality reduction tools: PCA28, t-SNE29, and UMAP30 and ArchR’s iterative LSI which is a specialized algorithm for sparse scATAC-seq data. We chose these tools due to their prevalent use and distinct methodological approaches to data analysis. We employed the ASW, the Calinski-Harabasz index (CHI), and the Davies-Bouldin index (DBI), calculated using ground truth cell-type labels and the resulting low-dimensional data matrices. The metrics used, namely ASW, CHI, and DBI, are essential in evaluating the quality of clustering. The ASW ranges from −1.0 to 1.0, where higher values indicate a better definition of clusters. Both the CHI and the DBI are positive metrics, with higher CHI values signifying more distinct inter-cluster separation and lower DBI values indicating tighter intra-cluster aggregation. These metrics collectively provide a comprehensive assessment of clustering performance across various computational methodologies.
Our results showed that scAGDE consistently achieved the highest average ASW and CHI values and the lowest DBI value, indicating its superior performance compared to PCA, t-SNE, UMAP, and LSI (Fig. 4b). As depicted in Fig. 4a, scAGDE effectively segregated cells into distinct groups, demonstrating a high degree of consistency with known cell types in the visualization space. In particular, on the GM12878vsHEK dataset, scAGDE and LSI divided cells from different cell lines into two compact and separated groups while the other methods dramatically expanded the two cell populations, with some of the cells even closer together in the results of PCA. On the Blood2K dataset which profiled ~2000 hematopoietic cells, scAGDE demonstrated its effectiveness by providing a clear and distinct separation between various cell types. Notably, it accurately depicted the complex dynamics of hematopoiesis, which was evidenced by the adjacent but distinct distribution patterns of HSCs, multipotent progenitor cells (MPPs), and CMPs, as well as the discernible separation between CMPs and lymphoid-primed multipotent progenitor cells (LMPPs)31. Although LSI showed a similar continuous distribution, unlike scAGDE, it could not separate MPP and HSC cells, and GMP and LMPP cells. The visualization and ASW results across all other datasets are presented in Supplementary Figs. 19 and 20.

a Visualization of scAGDE and four widely-used dimensionality reduction tools (PCA, t-SNE, UMAP, and LSI) on the GM12878vsHEK and Blood2K datasets. The ASW values are displayed at the top-left edge of the boxes, which ranges from −1.0 to 1.0 with a value approaching 1.0 indicating better performance. b Performance comparison by ASW, CHI and DBI across 11 datasets as boxplots (n = 11 datasets in each group; center line, median; box limits, upper and lower quartiles; whiskers, 1.5 × interquartile range). c, d The UMAP results of scAGDE and two single-cell ATAC-seq analysis packages (Signac and EpiScanpy) labeled with true cell-type labels (c), and the evaluation using benchmark metrics of clustering (ARI, NMI, F1, and V-Measure) and visualization (ASW, CHI, and DBI) (d). The CHI values shown are in logarithmic scale for better visualization. Source data are provided as a Source Data file.
Moreover, we conducted an extensive comparative analysis of scAGDE with two leading single-cell chromatin data analysis tools, Signac24 and EpiScanpy16, focusing on dimensionality reduction and visualization aspects. Among them, Signac employs latent semantic indexing (LSI) for dimensionality reduction, while EpiScanpy relies on PCA transformation. To effectively compare and visualize the clustering performance, we used UMAP plots labeled with both the clustering results and the real labels of the cells to provide a clear and intuitive visual representation of the performance of the delineated cellular data. To further demonstrate effectiveness, we conducted an in-depth analysis of scAGDE on three datasets: Leukemia, Forebrain, and InSilico. We calculated a series of visualization-related metrics (ASW, CHI and DBI) and clustering-related metrics (ARI, NMI, F1, and V-Measure) to thoroughly assess scAGDE’s performance. These metrics not only reflect the quality of clustering but also reveal the effectiveness of data visualization post-dimensionality reduction. As illustrated in Fig. 4c, d, scAGDE demonstrated exceptional performance in dimensionality reduction and clustering in scATAC-seq data analysis, particularly in differentiating various cell types.
Specifically, in the Leukemia dataset, scAGDE effectively isolated six distinct cell groups. Notably, the Mono cells and LMPP cells were well-separated, aligning with their vastly different characteristics and roles in the progression of Leukemia, as corroborated by existing studies32. In contrast, the six clusters identified by EpiScanpy16 were closely placed without clear demarcations, resulting in a mixture of multiple cell types within most clusters. Signac, on the other hand, grouped the six cell types into four clusters, suggesting a different clustering approach. On the Forebrain dataset, scAGDE and Signac showed comparable visualization capabilities. However, scAGDE was particularly proficient in distinguishing and clearly separating the three subpopulations of excitatory neuron cells (EX1, EX2, and EX3). EpiScanpy, meanwhile, struggled to differentiate these excitatory neuron subpopulations and ended up classifying oligodendrocyte cells (OC) into two distinct groups. Further, Supplementary Figs. 21 and 22 show that scAGDE consistently outperformed both Signac and EpiScanpy across the nine datasets. This evidence confirms the superior effectiveness of scAGDE in achieving accurate dimensionality reduction correction and visualization, highlighting its proficiency in ATAC-seq data analysis.
Evaluation of an ablation study and hyperparameter selection
We conducted comprehensive experiments to evaluate the effectiveness of individual components of scAGDE for scATAC-seq data analysis, including the cell graph generation based on the chromatin accessibility-based autoencoder, the employment of a Bernoulli-based decoder and the integration of GNNs. In addition, we evaluated the effect of the different hyperparameters involved in scAGDE on clustering performance, providing insights for future enhancement.
First, we evaluated scAGDE’s cell graph construction against analogous methods in scATAC-seq data analysis. In our study, scAGDE typically employs a chromatin accessibility-based autoencoder to build the cell graph. To demonstrate the effectiveness of scAGDE, we respectively substituted this approach with several alternatives: the PCA transformation followed by the KNN method (KNN), the Shared Nearest-Neighbor graph employed by Seurat and a graph method based on positive pointwise mutual information matrix (PPMI) recently developed in the scMGCA framework33 for scRNA-seq analysis. Fig. 5a indicates scAGDE’s superior clustering performance with a mean NMI value of 0.83, surpassing the other methods whose performance may be limited by imprecise topological relationships between cells (Supplementary Fig. 16). In particular, the PPMI method, while effective for scRNA-seq, did not yield reliable clustering results on the four sparsest scATAC-seq datasets including the Forebrain, GM12878vsHEK, GM12878vsHL, and leukemia, implying the limited applicability of cell graph strategies from scRNA-seq in scATAC-seq data. For a fair comparison, we explored a range of the number of components in the PCA and found that 50 components had the highest average ARI value of 0.70, which was still lower than 0.81 for scAGDE (Supplementary Note 9 and Supplementary Fig. 23).

a Table displaying the impact on clustering performance of several components in scAGDE, including the graph construction method, distribution of the decoder and fundamental model architecture. b UMAP visualization of embeddings at three progressive stages, with each cell type label and annotated by ASW value. c The heatmap of Pearson correlation coefficients on the average embeddings of distinct cell types. A value approaching 1 implies a higher correlation between two cell populations. Source data are provided as a Source Data file.
Next, we explored different distribution models for scATAC-seq count data by modifying the decoder to suit various distribution assumptions, including zero-inflated negative binomial (ZINB) distribution, multinomial distribution (Multinomial), negative multinomial distribution (Neg-Multinomial). In brief, the decoder network was utilized to estimate the various parameters of the distributions and get trained with the corresponding modified loss functions and we detailed this part in Supplementary Note 10. The results indicated that within scAGDE, the Bernoulli-based decoder outperformed the other distribution models, followed closely by the multinomial distribution, which exhibited significant differences compared to scAGDE on only four datasets, and the ZINB and negative multinomial distributions, which involve multiple parameters to estimate, demonstrated similar average performance, with no clear leader emerging across all datasets (Fig. 5a and Supplementary Fig. 24). Actually, we developed scAGDE considering the concise parameter settings, which makes it less susceptible to outliers and noise in sparse scATAC-seq data, and the ablation analysis provides further evidence.
Then, we explored the impact of incorporating graph learning in scAGDE. We experimented by substituting the original graph neural network structures (scAGDE-GNN) with linear layers, which are standard in autoencoders, to create scAGDE-AE as detailed in Supplementary Note 11. Besides, we also included an experiment using original GNN structures with identity matrix as cell graph input (scAGDE-Identity), to verify the impact of the cell graph information. As depicted in Fig. 5a and Supplementary Fig. 25, discarding the graph learning mechanism or utilizing incorrect cell graph input significantly reduced the clustering accuracy, which highlights the crucial role of intercellular topological information in scATAC-seq data analysis. To further explore this improvement, we extracted the low-dimensional embeddings obtained at the initial, mid, and near the end stages of the training process, from scAGDE-GNN and scAGDE-AE, respectively. The UMAP visualizations of embeddings revealed a progressively clearer distinction among cell populations, characterized by wider inter-group distances and narrower intra-group distances, with scAGDE-GNN demonstrating a better separation, as reflected in higher ASW scores (Fig. 5b). Notably, scAGDE-GNN was able to differentiate cell types such as IN1, OC, IN2, MG, and excitatory neuron cells earlier in the training process compared to scAGDE-AE, which exhibited a more sequential arrangement with significant overlaps. As the model was trained, the excitatory neurons remained highly similar yet increasingly distinguishable from each other, as demonstrated in the correlation coefficient heatmap in Fig. 5c.
Finally, we explored the impact of various hyperparameters of the scAGDE framework. A primary focus was the number of neighboring nodes, denoted as K, in the cell graph construction. Optimal performance was achieved at K = 15, as demonstrated in Supplementary Fig. 26. Notably, increasing the number of neighboring nodes beyond this threshold led to a decline in overall performance, highlighting the importance of this parameter in the graph construction process. Then, we benchmarked the architecture of scAGDE in terms of the number of consecutive GCN layers and the count of neuron nodes in the hidden layers, and the results evaluated using NMI and ARI are shown in Supplementary Fig. 27. The most effective configuration was determined to be a two-layer GCN encoder with a size of 128-10, providing adequate capacity for compressing information into latent embeddings while avoiding over-complication of the hidden space. Additionally, we explored various weight coefficients for each loss function as outlined in Eq. 29 and identified the optimal settings for scAGDE (Supplementary Fig. 28). By default, we selected the top 10,000 peaks based on the peak importance score as our analysis showed that the performance plateaued at this point (Supplementary Fig. 29).
scAGDE enhances discovery of cell type-specific enhancer regions to infer regulatory mechanisms of key functions
One of the key abilities of scAGDE is its capacity to evaluate and filter peaks based on our proposed peak importance scores, thereby mitigating the curse of dimensionality in downstream analysis. These scores, reflecting the contribution of each peak to the learning of its low-dimensional representation, are derived from the weights of the encoder network, which learns from chromatin accessibility profiles. Thus, they indicate the peak selection preferences of scAGDE. In this study, we detailed the characteristics of the peaks selected by scAGDE and compared these to the peaks identified using EpiScanpy16, which served as our baseline method.
First, we selected 10,000 peaks from the preprocessed 10×Blood dataset using scAGDE and EpiScanpy, respectively, to determine the accessibility of human immune cells and analyze their accessibility-related statistics. Specifically, after identifying the peaks from scAGDE or EpiScanpy, we generated pseudo-bulk data by merging the accessibility profiles of each peak across different cell types, resulting in a summarized accessibility profile for each peak within nine distinct cell types. Based on this pseudo-bulk data, we calculated the median accessibility and the range (the difference between the maximum and minimum accessibility values across cell types) for each peak, and presented these in the top density plot of Fig. 6a. Additionally, we measured the Shannon entropy of each peak and the percentage of cells where each peak is accessible, which are visualized in the bottom density plot of Fig. 6a. In particular, the peaks identified by scAGDE exhibited lower accessibility and a larger range of accessibility variability, whereas the peaks from EpiScanpy showed higher accessibility and a smaller range of variability. In addition, the variance in accessibility within each cell type in the pseudo-bulk data generated using scAGDE peaks was greater than that of EpiScanpy peaks (Supplementary Fig. 30a). Additionally, we observed an increase in the number of peaks with lower Shannon entropy and a smaller percentage of accessibility, alongside a decrease in peaks with high Shannon entropy, indicating that scAGDE tended to select cell type-specific accessible peaks. To further illustrate this, we performed differential accessibility region analysis and identified 6162 cell type-specific accessible peaks out of the 10,000 peaks, which is significantly more than the 462 identified by EpiScanpy (both with a false discovery rate <0.01). Furthermore, scAGDE demonstrated a higher accessibility difference with larger values of the log-fold-change (Supplementary Fig. 30b, c).

a Density plots showing various statistics for the peaks selected by scAGDE and EpiScanpy. In the top density plot, the y-axis represents the median accessibility of these peaks across the nine cell types and the x-axis shows the log-transformed accessibility range. In the bottom density plot, the y-axis represents the Shannon entropy based on the pseudo-bulk data for each peak and the x-axis indicates the percentage of cells in which each peak is accessible. b Peak annotation results using ChIPseeker on peaks from scAGDE and EpiScanpy. c Genome track around CD37, CTLA4 and MS4A1. The peak regions discovered by scAGDE and EpiScanpy are separately displayed below the aggregated signals. d The heatmap of peak importance scores during the entire training process. e The numbers of enhancer peaks in discovered regions of each method (n = 40 peaks in EpiScanpy and n = 78 peaks in scAGDE; Fisher’s two-sided test). f Triangle heatmap shows chromatin interaction from the Hi-C experiment data of peripheral blood CD8+ T cells isolated from the peripheral blood of two human donors102. g Genome track around loci of CD8A with regions of discovered peaks and the super-enhancer, and the results of de novo motif analysis on the highlighted peak regions are displayed. The one-sided hypergeometric test was used and p-values were not corrected for multiple testing. Source data are provided as a Source Data file.
To further dissect the identity of these candidate peaks, we annotated them with peak type using ChIPseeker34 based on their proximity to the nearest transcription start sites (TSS). Fig. 6b shows that peaks from scAGDE were primarily annotated as promoters near TSSs (19.9%), intronic regions (31.0%), and distal intergenic regions (32.1%). In contrast, peaks from EpiScanpy were almost exclusively composed of promoters near TSSs (83.8%). Based on the distance to the TSSs, 77% of the scAGDE peaks can be defined as distal peaks (i.e., greater than 2000 bp), while 83% of the peaks from EpiScanpy are promoters (i.e., less than 200 bp) (Supplementary Fig. 30d). In detail, peaks selected by scAGDE comprised promoters that are useful for cell-type annotation, which were also identified by EpiScanpy, as well as promoters not in EpiScanpy. Additionally, scAGDE included distal and intergenic peaks that were not identified by EpiScanpy (Fig. 6c and Supplementary Fig. 32). For instance, scAGDE uniquely identified the CTLA4 gene-associated promoter and distal peaks, which are predominantly accessible in specific T cell groups. Therefore, scAGDE exhibited a higher preference for peaks in intronic and distal intergenic regions.
To further illustrate these preferences, we uniformly sampled 500 moments from the entire training process of our autoencoder model, and computed the importance scores of all peaks at each moment, as presented in Fig. 6d. To characterize the distinct preference patterns, we organized the 39,565 peak importance profiles into 6 modules by K-means clustering based on the elbow method35. As a result, the identified 6 modules have similar internal distribution and are distinct from each other. Specifically, at the initial stage, these modules exhibited a nearly uniform distribution and as the training progressed, the first two modules (M1 and M2) received increasing attention, whereas other modules, particularly M6, garnered less focus. The variation in peak importance scores is greatest at the 20% stage and gradually stabilizes as the model converges. As expected, M1 and M2 were primarily composed of peaks from intronic and distal intergenic regions, and nearly half of M6 was composed of promoter peaks. At this point, the preference analysis, together with prior annotation results, shows that scAGDE tends to select peaks from intronic and distal intergenic regions. Therefore, we can conclude that scAGDE identified cell type-specific accessible peaks which were predominantly located in intronic and distal intergenic regions, and this discovery of the strong association between specific accessible peaks and their identities is highly consistent with a previous study in which a single-cell ATAC-seq analysis of immune cell populations in the mouse spleen reported that the marker peaks were found primarily in these regions36. Additionally, Chung et al. demonstrated that distal regions are more cell-type specific accessible and informative than promoter-proximal regions37, further supporting the significance of our discovery. Besides, enhancer-associated regions are well known to exhibit cell-type-dependent accessibility38 and have proven to be more effective in distinguishing cell types than promoters or transcriptomic data39. These findings decipher the underlying association between peak attention bias and their unique accessibility patterns in our deep learning models, providing a marked perspective on model interpretability.
To validate the potential functions of the peaks we identified, we compared them to two publicly available enhancer databases: VISTA40 and EnDB41. The VISTA enhancer browser provides catalogs of mammalian enhancers validated in transgenic mice, and EnDB documents over 700 validated enhancers along with related information. We retrieved enhancers corresponding to the human genome from both databases for comparison and discovered that the peaks identified by scAGDE matched more known enhancers, demonstrating a significance of p = 4.28 × 10−4. Notably, among the scAGDE peaks that did not match acknowledged enhancers, we observed patterns of chromatin accessibility reminiscent of enhancers around key genes. For instance, CTLA4, an immune checkpoint responsible for inhibiting T cell activation and function42, is constitutively expressed in regulatory T cells (Treg)43. Within scAGDE peaks proximal to CTLA4, we observed that both conventional T cells (CD4 Helper T cells, Memory CD4/CD8+ T cells, and Naive CD4/CD8+ T cells) and Treg cells demonstrated high chromatin accessibility at the promoter site. Memory CD8+ T cells, in particular, showed even greater accessibility at the promoter site compared to Treg cells (Fig. 6c). In contrast, Treg cells displayed a unique chromatin accessibility peak at approximately 20kb upstream of the CTLA4 promoter, which was uniquely found by scAGDE and we found it overlaps with a super-enhancer (SE_18993) listed in dbSUPER (Supplementary Fig. 31). The overlap with the known super-enhancer suggests that this accessible region may play a key role in regulating the constitutive expression of CTLA4 in Treg cells, potentially highlighting a cell type-specific regulatory mechanism. A similar regulatory pattern may be present near the CD8A gene locus (Fig. 6g). The CD8+ T cells exhibited high levels of chromatin accessibility at both the promoter and 3’UTR regions of the CD8A gene, which encodes the molecular marker of CD8+ T cells44. We found that the super-enhancer (SE_12469) from dbSUPER overlapped with two peaks identified by scAGDE near the 3’UTR and confirmed the chromatin interactions between these peaks and the promoter of CD8A (Fig. 6f), supporting our hypothesis that these regions are candidate enhancers. The chromatin interactions can be further validated with additional biologically relevant Hi-C datasets, with consistent results across multiple tissues or cell lines (Supplementary Fig. 33). Interestingly, studies have shown that CD8A is expressed in a subset of NK cells45. In our findings, NK cells exhibited similar chromatin accessibility at promoter sites as other immune cell populations, yet displayed increased accessibility in our identified candidate enhancer region. This high accessibility emphasizes the essential role of this enhancer in regulating the transcriptional activity of CD8A, suggesting its substantial impact on gene expression. In summary, scAGDE directs us to regions with potentially functional and unique regulatory programs, offering notable insights into the regulatory mechanisms of key genes.
scAGDE can recover the potential open sites in chromatin open sites and infers cell type-specific cis-regulatory elements
The limitations of the diploid genome structure and the technical constraints inherent in scATAC-seq experiments pose similar challenges as scRNA-seq, leading to the phenomenon of “dropout” where signals from the actual open chromatin sites may be absent46. To address this issue, we developed a Bernoulli-based distribution decoder aimed at recovering these dropout events and reducing noise signals present in the original data, thereby enhancing downstream analysis. We first evaluated the effectiveness of scAGDE in peak recovery by comparing it with three state-of-the-art methods specifically tailored to scATAC-seq data analysis, namely SCALE13, PeakVI15, and SAILER14.
We computed meta-cells for each cell-type group by aggregating individual profiles within the same group, which represented the ground truth representation of chromatin opening landscapes without any dropouts. Subsequently, we computed Pearson correlation coefficients between imputed cells and their corresponding meta-cells for each cell type, with values approaching 1 indicating superior peak recovery performance. Our analysis encompassed nine datasets, and we present the distribution of correlation coefficients in Fig. 7a. The correlation analysis of raw data at the cell level demonstrated significant dispersion, with the lowest mean value recorded at 0.39 (indicated by the dotted line). This dispersion is likely attributed to the presence of accessibility variation, independent of cellular heterogeneity arising from dropout events and noise in the data. After the application of scAGDE’s recovery method, the correlation coefficients exhibited a notably narrower distribution, with a peak approaching 1.0 and a highly concentrated pattern, maintaining an average value of 0.88. This performance was followed by SCALE’s correlation coefficient of 0.85 and PeakVI’s coefficient of 0.78. However, it’s disappointing that SAILER failed to improve the consistency of meta-cell accessibility, and even demonstrated larger variability.

a The cell-wise correlations with meta-cells in original data and post-imputation data for each method across all datasets. b The average performance of K-means clustering on all datasets, both before and after imputation by each method. c The UMAP visualizations of imputed data, where the ASW values are displayed at the top-left edge inside the boxes. d The heatmaps of cell-by-peak matrix that contains only the top 15 differential accessibility regions of each cell type from raw, imputed and binary imputed data. Rows represent DARs and columns represent cells, which are both arranged according to eight different cell types. Tiny spacing was plotted between the different cell types therefore the x and y axis were both divided into eight parts. e The recovered cell type-specific peaks by scAGDE and enrichment analysis of known transcription factor motifs and de novo motifs in the peaks. The one-sided hypergeometric test was used for both TF motifs enrichment and de novo motifs enrichment and p-values were not corrected for multiple testing. f The significant results of chromVAR deviation score profile of identified motifs of each cell type. g Enhanced TF deviation scores of NEUROG2, Dlx2 and MEIS2, compared with raw data, displayed on the T-SNE embedding, which was calculated based on TF deviation scores. Source data are provided as a Source Data file.
Subsequently, we applied conventional clustering techniques, specifically PCA transformation followed by the K-means algorithm, to all imputed datasets for evaluation. As expected, scAGDE demonstrated superior performance compared to the other methodologies, achieving the highest average values across all evaluated metrics, as illustrated in Fig. 7b. Conversely, no significant enhancement in clustering outcome was observed following imputation by either PeakVI or SAILER.
Further analysis was conducted on the imputed Forebrain dataset to investigate the observed performance discrepancies. As presented in Fig. 7c, the 2D UMAP visualization demonstrates the ability of scAGDE to effectively delineate different cell populations, closely agreeing with the true labels, especially in the case of excitatory neuronal cells. Additionally, the ASW score for each method, as indicated in Fig. 7c, shows that scAGDE achieved significantly higher scores relative to other methods. Although SCALE and PeakVI preserved the continuous distribution characteristic of the raw data, they tended to blur the boundaries between different cell populations, which elevated the risk of misidentifying cell types. In SAILER, although there was no significant overlap between cell groups, the positioning of these groups was remarkably close, with an almost uniform internal distribution. This arrangement within the UMAP embedding space introduced additional variability at the cellular level, which accounts for the reduced intra-cluster consistency and the suboptimal clustering performance illustrated in Fig. 7a, b.
Beyond this, we found that scAGDE could recover cell type-specific accessibility signals through imputation, thereby enhancing the identification of genomic elements. Specifically, we identified differential accessibility regions (DARs) within the raw, imputed, and binary imputed Forebrain dataset with the Signac toolkit24. As a result, we uncovered 5263 significant DARs by applying the Signac toolkit24 with the default parameters, marking a significant increase from the 626 DARs identified using identical parameter settings prior to imputation. This increase in the number of DARs highlights the impact of imputation on enhancing the detection of differential accessibility regions, particularly in cell types where low coverage may have obscured relevant peaks. For each cell type, we ranked their DARs according to the log2 fold-change values and selected the top 15 DARs, which were used for subsequent analyses. Then, the cell-by-peak matrices from the original, imputed, and binary imputed datasets, containing only these top DARs, were visualized in heatmaps as depicted in Fig. 7d, where the rows represent the DARs and the columns represent the cells, which are both arranged according to cell type. Clear separations are drawn between different cell type groups, reflecting distinct accessibility patterns across the cell populations. The alignment of rows and columns according to cell types ensures that DARs with strong cell type specificity appear along the main diagonal, showing high accessibility in their respective cell types. The heatmaps further revealed that the imputed and binary imputed data exhibited distinct clustering patterns, sharply contrasting with the original data. Specifically, imputation led to a more pronounced clustering of cell types, suggesting that missing or incomplete accessibility information in the original data may have masked these biological patterns. To further understand the biological significance behind this increase in cell type-specific peaks post-imputation, we proceeded with enrichment analysis. The analysis revealed that the enhanced cell type-specific peaks after imputation are significantly associated with cell identity, rather than being arbitrary occurrences (Fig. 7e).
On this basis, we explored the biological significance by characterizing the relevant genes within these recovered regions. Utilizing rGREAT47, we meticulously examined the genes associated with the recovered regions, concentrating on a ±15 kb range from the TSS. This analysis revealed a significant emergence of cell type-specific marker genes. For instance, Aldh1l1 and Slc1a3, markers for astrocyte cells (cluster AC)48, along with Neuorod6, Stab2 and other genes significant for excitatory cells (clusters EX1-3)49, were identified within the differential accessibility regions (DARs). Similarly, Gad1 and Gad2, associated with inhibitory cells (clusters IN1 and IN2)50, and the marker gene Mobp for oligodendrocyte cells (cluster OC)51, were also discovered. These findings indicate that scAGDE is proficient in enriching the signals of chromatin accessible regions tied to key genes that define cellular identity, which may be the fundamental reason for the significant improvement in clustering results observed with imputed data.
In addition, to further investigate the additional discovered cell type-specific cis-regulatory elements, we performed motif enrichment analysis, seeking to identify known motifs via Signac24 and de novo motifs through HOMER52. For the enrichment of known motifs, after filtering out non-significant findings based on ‘p-value’ and ‘fold.enrichment’, we discovered 427 transcription factor binding motifs that displayed cell type-specific enrichment patterns (Fig. 7e). Of note, these included binding motifs of key transcription factors (TFs) crucial for defining cell identity. For instance, motifs for NFIX and NFIB were enriched in regions associated with astrocytes, which have been shown to significantly express these factors53, which are linked to the specification, differentiation, and migration of astrocytes in conjunction with other NFI family members54,55,56. Excitatory neuron regions displayed enrichment for MEF2C and bHLH family members57 motifs, while Dlx2 motifs were enriched in inhibitory neuron regions58. Additionally, we identified ETS-factor PU.1 in microglial cell regions59 and SOX10 in oligodendrocyte cell regions60. In the de novo motif identification, we found that the TFs correlating best with the top enriched motifs were either directly mentioned as key TFs in the specific cell types or belonged to the same motif family, such as the NFI transcription factor NFIA in the AC cluster and the MEF2 transcription factor MEF2A in the EX1 cluster.
The deviation score takes the accessibility matrix as input to calculate accessibility variations for TFs, and scAGDE can restore or erase the accessibility of certain elements harboring TF motifs, thus affecting the chromVAR deviation score. Finally, we evaluated the chromVAR deviation score for each TF motif on the imputed data11. scAGDE can restore or erase the accessibility of certain elements harboring TF motifs, thus affecting the chromVAR deviation score. As expected, we found that the imputation offered more highly significant enriched TF motifs (from 141 to 702) and enhanced the cell type-specific TF activity schema, especially for the EX2 cells within excitatory neurons and the OC cells. (Fig. 7f and Supplementary Fig. 35a). Furthermore, the significantly enhanced specificity improved the t-SNE visualization derived from the TF deviation scores, facilitating clearer distinction between different cell types as shown in Supplementary Fig. 35b. From this figure, we observe that by filling missing values, scAGDE strengthened the patterns of variability for these crucial TF motifs across clusters (Fig. 7g and Supplementary Fig. 35c). Remarkably, the deviation score distribution between IN1 and IN2 cells showed substantial differentiation post-imputation for the MEIS2 motif that plays a critical role in the development of medium spiny neurons61 (Supplementary Fig. 35d). This observation lends further support to the hypothesis proposed in the literature that cluster IN1 comprises medium spiny neurons62. To provide a comprehensive evaluation, we also applied scAGDE to a single-cell multi-omic dataset, demonstrating that scAGDE achieves superior imputation performance (Supplementary Fig. 34). In summary, these findings indicate that scAGDE can effectively uncover potential chromatin accessible regions during the modeling of scATAC-seq data distribution, thereby enhancing the identification of insightful chromatin regulatory elements.
scAGDE reveals subtle functional diversity and regulatory mechanisms in human brain neurons
To investigate the application of scAGDE to single-cell datasets with significant epigenetic variation, we applied the scAGDE algorithm to a brain dataset comprising 14,906 nuclei from single-cell chromatin accessibility assays of adult human frontal cortex tissue63, a region known for its complex and diverse epigenetic landscape. To address data sparsity, the dataset was first preprocessed and subsequently clustered using scAGDE. As depicted in Fig. 8a, we identified eight cell groups, most of which were individually aggregated into clusters, with clusters 2 and 3, and clusters 5 and 6 being adjacent to each other. To further characterize these clusters, we next identified the signature genes of each cluster using the ArchR “gene expression score” matrix21. In the eight clusters, we identified 8126 signature genes, many of which displayed unique patterns of expression and aligned with markers found in the PanglaoDB64 and CellMarker65 databases (Fig. 8b and Supplementary Fig. 36a). On this basis, we assigned specific cell types to each cluster, including oligodendrocyte cells (OG), blood-brain barrier endothelial cells (BBB), microglial cells (MG), GABAergic neurons (GABA+), astrocytes (AC), oligodendrocyte precursor cells (OPC), and two groups of glutamatergic neurons (vGlut+ 1 and vGlut+ 2) (Fig. 8c). Furthermore, our Gene Ontology (GO) analysis of the top 100 marker genes uncovered the biological processes integral to cellular identity, validating the reliability of the clustering and cell type annotation. For instance, oligodendrocytes myelinate and support the axons of the central nervous system (CNS), with their damage leading to demyelination66. MG are characterized by their rapid response to even minor pathological changes in the CNS67. Additionally, ‘Gliogenesis’ describes the development of OPC into mature oligodendrocytes. Therefore, these cell clusters are distinguished by their unique access to the promoter loci of marker genes (Fig. 8d).

a The UMAP visualization of brain cell clusters. b Heatmap of gene score matrix obtained by ArchR across all clusters. Selected GO terms and correlated genes are shown on the right. c Cluster connectivity changes between initial cluster, refined cluster and scRNA clusters shown as Sankey plots. d Genome track around marker genes from the aggregated scATAC data in all clusters. e Heatmap of 48,578 differentially-accessible CREs in the eight clusters. f Known and de novo motif enrichment results of CREs restricted to all clusters. The top de novo motifs, their best matches and p-values are displayed. The one-sided hypergeometric test was used and p-values were not corrected for multiple testing. g Heatmap of differentially-accessible CREs between two glutamatergic neuron clusters (left) and top-ranked TFs enriched in the two sets of CREs. The top 16 TFs are listed. A one-sided hypergeometric test was performed, and the p-values were adjusted using the Bonferroni correction. Source data are provided as a Source Data file.
Next, we investigated the cis-regulatory elements (CREs) in our eight cell clusters, identifying 48,578 differential chromatin accessibility peaks. As illustrated in Fig. 8e, the largest number of cell type-specific CREs are found in glutamatergic neurons, with comparable amounts of CREs across the four glial cell types. To further dissect sequence features across these cell-type-specific regulatory elements, we measured the enrichment of motifs with HOMER and ArchR in the CREs. This revealed a total of 125 enriched motifs displaying cell type-specific distribution (Fig. 8f); for instance, the SOX TF family are crucial regulators in various developmental processes in oligodendrocytes68; The microglial-specific CREs show enrichment for motifs associated with pro-inflammatory TFs, such as STAT2 and IRF1, and the lineage-specific master TF, PU.169. Additionally, ASCL1 is highly enriched in OPC, as evidenced by its active expression in subventricular zone progenitors and OPCs during myelination and remyelination processes70. Interestingly, the two groups of glutamatergic neurons exhibited similar motif enrichment patterns, yet they displayed notable differences that warrant further discussion. Notably, the basic Helix-loop-Helix family members such as NEUROG1, NEUROG2, and NEUROG3, known regulators of cell proliferation and neuronal specification71 were identified in both neuron groups. However, compared to the vGlut+ 2 cells, the vGlut+ 1 cells had an increased number of accessible peaks, with 4957 peaks gained and 1300 peaks lost (Fig. 8g). For the motif enrichment across the gained peaks, we found TFs could be broadly categorized into major families, including FOS and JUN, MEF2, POU and FOX. The FOS and JUN family members can form heterodimers that constitute the transcription factor activator protein-1 (AP-1), regulating the expression of multiple genes in response to external stress stimuli72. The MEF2 family members regulate synaptic plasticity, affecting learning and memory formation73. In the lost accessible peaks, motifs from significant TF families such as BHLH and OLIG were identified. Taken together, these findings suggest that there may be even more subtle heterogeneities within the glutamatergic neuron cell groups.
To further explore the subtle heterogeneities, we employed a re-clustering strategy by adjusting the resolution settings of the Leiden algorithm implemented in Seurat5, guided by changes in the ASW value. The ASW metric measures both the intra- and inter-class distances, thereby when the number of clusters is larger than the number of major cell types, the clustering resolution increases yet at the cost of a decrease in the ASW values. Here we need to make a trade-off between the cluster numbers and ASW values. As the resolution increased from 0 to 1.0, the ASW decreased more gradually at 0.4, with an ASW value of 0.34 (Supplementary Fig. 36b). This setting refined the cluster assignments as shown in Fig. 8g, where we see GABA+ cells are now divided into two clusters (C1 and C2) and glutamatergic neurons into three clusters (C3–C5). Differential analysis of peak accessibility and gene scores revealed prominent patterns in the three subgroups of glutamatergic neurons (Fig. 9b and Supplementary Fig. 36c).

a The GO (biological process) enrichment results with correlated genes among clusters C3–C5. The biological processes and negative log10p values are displayed in the histogram and are linked by lines to correlated genes, and the corresponding gene score data are described in the heatmap. A two-sided Wilcoxon test was performed and p-values were not corrected for multiple testing. b Heatmap of differentially accessible CREs between cluster C3–C5 (left) and selected top de novo enriched TFs and negative log10 p values. The one-sided hypergeometric test was used and p-values were not corrected for multiple testing. c, d Genome tracks around the RGS4 (c), and the PCDH8 loci (d). Loops shown below these tracks indicate the peak-to-gene linkages and co-accessibility linkages among peaks. The scRNA expression levels are shown in the violin plot for each cluster on the right side. Blue vertical bars spanning each panel highlight CREs linked to gene locus or being co-accessible with gene promoters. Source data are provided as a Source Data file.
To reveal the functional differences between clusters C3–C5, we applied GO enrichment analysis to uncover the biological processes. The results showed that genes associated with cluster-specific CREs of the three clusters were involved in several biological processes (Fig. 9a). Genes from cluster C3 were involved in processes related to synaptic assembly, such as regulation of synapse assembly (GO:0051963), regulation of synapse structure and activity (GO:0051960), and positive regulation of synapse assembly (GO:0051965). Additional processes such as the regulation of synaptic vesicle priming and recycling (GO:1901890 and GO:1901888) are essential for the release and reuse of neurotransmitters74,75. Cluster C4 was associated with synaptic plasticity (GO:0048167), synaptic transmission (GO:0051966, GO:0050804, and GO:0099177), and sensory perception, notably olfaction (GO:005090, GO:0050911, and GO:0007608). Cluster C5 showed enrichment in synapse regulation-related processes, including synapse organization (GO:0050808) and the regulation of synapse structure or activity (GO:0050803). Expression of genes linked to these processes is specific to the clusters. Furthermore, cell label prediction using single-cell RNA-seq data from Ma et al.76 validated our annotation of cell types (Fig. 8c), e.g., clusters C3 and C5 were identified as intratelencephalic neurons from layers 3–5 and 2–3 of the brain, respectively, and cluster C4 was identified predominantly as layer 2–3 intracerebral neurons.
In addition, we applied motif enrichment analysis on the cell type-specific CREs to reveal the correlated TFs for each sub-cluster. For instance, CREs restricted to cluster C3 revealed binding sites of Smad3, which is involved in TGFß-Smad3 signaling pathway that regulates synaptogenesis77, and NEUROD1 and Mef2c which are critical for the development and function of the central nervous system78,79. From CREs restricted to cluster C5, we found MEF2A, which plays a key role in activity-dependent gene expression during synaptic remodeling80, and CREB5 of CREB family that is essential for synaptic plasticity81. CREs restricted to cluster C5 were also enriched in PAX6, a transcription factor regulating the expression of many synaptic proteins82.
To reveal transcriptional regulatory differences showing functional heterogeneity, we performed peak co-accessibility analysis and detected peak-to-gene linkages using integrated scRNA data for the three clusters. We identified 3390 peak-to-gene links (Supplementary Fig. 36d). Based on these links, we pinpointed differences in accessibility at non-promoter regions of functionally correlated genes, potentially representing candidate enhancers. For example, within the region upstream of the RGS4 promoter, cluster C3 exhibited higher accessibility and higher expression of gene RGS4 in the scRNA-seq predicted cell population (Fig. 9c). RGS4 is involved in amine transport and regulates G protein-coupled receptor signaling pathways83 and function of glutamate receptors84. This suggests that cluster C3 fine controls the transmission of excitatory signals. Additionally, we found higher accessibility in cluster C5 at three distal regions of the PCDH8 gene, which is consistent with observed high PCDH8 scRNA expression levels (Fig. 9d). PCDH8 encodes a cell adhesion molecule that is associated with the regulation of synapse structure and activity, indicating that cluster C5 may help support the maintenance of neural networks.
In summary, our scAGDE algorithm successfully annotated various neuron types with unique regulatory landscapes previously uncharacterized in human frontal cortex tissue. Indeed, scAGDE accurately localized specific markers of each neuron type and thereby provided notable insights into their functional roles and interactions in the brain.
Discussion
Single cell ATAC-seq is an advanced technique for genome-wide profiling of chromatin accessibility at the individual cell level, enabling the study of epigenetic heterogeneity and further characterization of regulatory mechanisms beyond the transcriptional level. However, computational analysis of scATAC-seq data remains challenging due to its limited sensitivity, inherent binarization, and high dimensionality. Here, we propose scAGDE, a multi-step model-based deep graph embedded learning framework to efficiently analyze the extensive and complex scATAC-seq data. We first use a chromatin accessibility-based autoencoder to learn representations from a preprocessed count matrix, then extract peak importance scores from the model parameters of the trained encoder network to filter out less significant peaks. Following this, we utilize a Bernoulli-based graph autoencoder to simultaneously process the data and the cell graph matrix, effectively capturing critical cell-cell topology. To accurately model the distribution of the scATAC-seq data, we introduce a Bernoulli-based decoder network capable of estimating chromatin openness and integrate a combined self-supervised clustering optimization objective into the training guidance, along with other loss functions within scAGDE, to improve overall model performance. Notably, scAGDE incorporates automated cluster estimation to reduce reliance on prior knowledge and allows to bypass clustering components based on task requirements, enhancing flexibility and preserving robust embedding quality (Supplementary Note 16 and Supplementary Figs. 37 and 38).
To investigate the clustering superiority of scAGDE, we performed a comprehensive benchmarking analysis on synthetic and real-world single-cell datasets. In this analysis, scAGDE outperformed eleven other comparison methods in all simulated and real-world datasets, achieving excellent results, even in conditions of poor data quality, confirming the robustness of scAGDE. We found that scAGDE effectively reduced dimensionality and improved data visualization, outperforming five other widely-used methods, evaluated by several metrics, including the ASW value. Notably, scAGDE’s visualization ability could distinguish cell subpopulations and preserve the associations of cell identity in low-dimensional space. Furthermore, our exploration of the direct application of scATAC-seq data for scRNA-seq analysis methods revealed the potential applicability of GNN-based models, although performance was partly hindered by suboptimal cell graph design specific to scRNA-seq data.
Our study revealed that scAGDE exhibited a bias towards chromatin accessible regions with variable regulatory programs in specific cell types during the training process, particularly showing a preference for intronic and distal intergenic regions, in contrast to the promoter-dominated peaks identified by EpiScanpy. Further enhancer enrichment analysis provides evidence of the regulatory significance of scAGDE-identified peaks. Notably, we observed specific chromatin accessibility patterns around key genes such as CTLA4 and CD8A, suggesting the accessible chromatin regions function as candidate enhancers. This offers a deeper understanding of transcriptional regulatory mechanisms in immune cells and highlights the utility of scAGDE in revealing complex regulatory landscapes.
Using a mouse forebrain dataset, we demonstrated that scAGDE not only significantly reduced variation in chromatin accessibility across different cell populations but also enhanced clustering performance and the quality of visualization. Notable increases in the accessibility of gene regions associated with cellular identity were detected. In addition, we found that scAGDE improved the identification of chromatin regulatory elements after recovery of cell type-specific signals, coupled with enhanced enrichment of transcription factor motifs within these chromatin regulatory elements (CREs).
Moreover, scAGDE successfully elucidated epigenetic heterogeneity and regulatory mechanisms in the human brain frontal cortex. When clustering a human brain dataset, scAGDE distinguished several neurons and glial cell types through associations between accessible chromatin regions and marker genes. These annotations were validated by cell-type labels derived from integrated scRNA-seq data. In particular, within glutamatergic neurons, we identified three distinct subgroups, each enriched with subtly varied biological processes, highlighting the further complexity of these cells and the increased diversity of glutamatergic neuronal function.
Additionally, it is worth noting that although scAGDE is not exclusively designed for batch-effect correction, it effectively addresses batch effects while preserving biological heterogeneity using a two-step Harmony-based post-processing strategy (Supplementary Note 17 and Supplementary Fig. 39). This approach enables scAGDE to construct a cross-batch cell graph, facilitating robust integration across different cell batches. Evaluation across multi-batch datasets showed that scAGDE achieves competitive integration performance and effective batch mixing, comparable to dedicated batch-correction tools. Looking forward, scAGDE could be further enhanced by incorporating batch covariates directly into its neural network architecture to directly learn batch-corrected embeddings. This is a promising future direction to strengthen the versatility of scAGDE’s versatility in multi-batch data integration.
As with other deep learning-based approaches, scAGDE also has certain limitations that could affect its performance in specific contexts. One limitation of scAGDE is scalability when dealing with notably large datasets. While scAGDE is designed to efficiently handle typical single-cell ATAC-seq datasets, when the number of cells or peaks increases substantially (e.g., to millions of cells), memory consumption and runtime may rise significantly, leading to potential performance bottlenecks. In such cases, subsampling strategies or distributed computing methods may be necessary to mitigate this issue. Another limitation arises in dealing with notably sparse datasets. Although scAGDE is specifically designed to handle the sparsity commonly found in single-cell ATAC-seq data, leveraging its autoencoder and graph embedding components to extract meaningful patterns from moderately to highly sparse datasets, challenges may still occur when accessibility signals are detected in only a few peaks across most cell types. In these scenarios, the lack of sufficient signal may make it difficult for the model to differentiate between cell types or identify cell type-specific regulatory elements. While scAGDE is robust for sparse data, low signal-to-noise ratios may hinder its ability to extract meaningful patterns, leading to reduced clustering accuracy or embedding performance. This is not a limitation unique to scAGDE; many other algorithms face similar challenges when dealing with notably sparse data. Additionally, as the sparsity increases, managing batch effects becomes more difficult. While scAGDE incorporates the Harmony algorithm for batch effect correction, highly sparse data may require further enhancements to effectively address batch effects. The last limitation is the parameter sensitivity. Like other deep learning-based methods, the clustering results and overall performance of scAGDE can be influenced by the selection of hyperparameters, such as the weighting factors for different loss terms in the objective function. If these parameters are not optimally tuned for the specific dataset, it may result in suboptimal performance.
In summary, scAGDE has demonstrated strong performance across various tasks, including clustering, dimensionality reduction, and imputation, providing notable insights into cellular diversity and epigenetic regulation. In the future, we plan to extend the application of scAGDE to additional omics data types, such as single-cell RNA-seq and proteomics data. Given that scAGDE enables topological information integration, it is particularly suitable for handling the spatial neighborhood relationships inherent in spatial transcriptomics, epigenomics, or even multi-omics data. In addition, inspired by approaches such as scBasset85, CellSpace86, and SANGO87, we have explored a variant of scAGDE, named scAGDE-M, to integrate DNA sequence information with chromatin accessibility to enhance its adaptability in multi-omics integration tasks (Supplementary Note 18 and Supplementary Fig. 40). We are confident that scAGDE has significant potential to enhance genomic research by revealing complex insights across diverse datasets.
Methods
Data preprocessing
In our study, we took the scATAC-seq count matrix \({{{\mathcal{X}}}}\in {R}^{N\times M}\) as input, where N and M denote the number of cells and peaks, respectively. The first step is to filter out peaks that are not expressed in cells, reducing the impact of high dropout rates in scATAC-seq data following the scABC model8. Indeed, we retained the peaks present in a minimum of 10 cells with ≥2 reads for the InSilico, GM12878vsHEK and GM12878vsHL datasets; 5 cells with ≥2 reads for the Leukemia dataset; at least 50 cells with ≥2 reads for the Forebrain dataset and at least 50 with ≥1 reads for the 10XBlood, Blood2K and DropBlood dataset. In addition, we kept all the peaks in the Splenocyte dataset. Note that these parameters were determined on the basis of the original sparsity of each dataset.
Then, to facilitate network training and optimization, we excluded rare peaks, occurring in less than δ% of cells with more than two reads, or nearly universal, present in at least (100-δ)% of cells with one or more reads according to reference7. Subsequently, we converted the count matrix obtained into binary data by applying a thresholding method where values greater than zero were set to one and all others were set to zero. Finally, we produced the final matrix Xpre ∈ RN×K to ensure the stability of the analysis, where K denotes the number of filtered peaks. A final binarization was applied to each dataset prior to analysis using our scAGDE method, with no further processing for simulated datasets.
Chromatin accessibility-based autoencoder
We first adopted a chromatin accessibility-based autoencoder to filter out insignificant peaks for potential representation of the scATAC-seq data. The analysis begins with a binarized input matrix M ∈ [0, 1]N×K defined in the range [0, 1] for N cells and K peaks, representing the cell-by-peak matrix of the scATAC-seq data. The chromatin accessibility profile of a single cell is defined as m and modeled as a Bernoulli distribution. We assign mi,j a value of 1 if the sequencing fragments are detected at the j-th peak and 0 otherwise. The chromatin accessibility-based autoencoder begins by estimating the latent representation zi of each observed mi, using the principles of variational Bayes88. The chromatin accessibility-based autoencoder utilized in our study is structured into two main components: the encoder and the decoder. The encoder is responsible for transforming the high-dimensional input data into a lower-dimensional latent space, thereby capturing the essential features of the data. This transformation is mathematically represented by:
where ϕ denotes a Rectified Linear Unit activation function, which introduces non-linearity into the encoding process, enhancing the model’s ability to learn complex patterns. The variables μ (mean) and σ (standard deviation) represent the parameters of the Gaussian distribution N(μ, σ2). By employing the re-parameterization trick, we can perform efficient backpropagation and stochastic gradient descent during training. Specifically, the sampling process from the Gaussian distribution is reparameterized as follows:
where z is the latent representation; ϵ is a random variable sampled from the standard normal distribution N(0, 1). This step ensures that the model remains differentiable and allows the loss gradient to propagate back through the stochastic nodes, thus facilitating effective learning of the representation of latent space.
The decoder, on the other hand, aims to reconstruct the input data from the compressed latent representation, attempting to minimize the loss between the original input and the reconstructed output. The mathematical form of the decoder function is:
where \(\,{\mbox{sigmoid}}\,\left(\cdot \right)=1/1(1+{e}^{-\cdot })\) denotes the sigmoid activation function. The primary objective of model training is to minimize the negative lower bound of the marginal log-likelihood probability of the observed data, which is achieved through a combination of reconstruction loss and Kullback-Leibler (KL) divergence loss between the Gaussian distribution N(μ, σ2) and the standard normal distribution, which are formulated as follows:
where \({{{\mathcal{L}}}}\) represents the loss function of the chromatin accessibility-based autoencoder and β is a weighting coefficient for the KL divergence term, often used to balance the influence of the KL term relative to the reconstruction error. This coefficient was set at 0.5 in our study according to the desired emphasis on the quality of the representation over the disentanglement of embeddings, aligning with the central principle of beta-VAE89.
Autoencoder-based peak selection in chromatin accessibility data
We implemented a model weight-based peak selection strategy to identify crucial peaks from the original count matrix, leveraging a previously well-trained Chromatin Accessibility-based Autoencoder. The encoder network directly processes the accessibility patterns observed across each peak in all cells, capturing this information within the encoder weight matrix \({{{{\bf{W}}}}}_{e}\in {{{{\bf{R}}}}}^{d\times {d}_{h}}\), as demonstrated in Eq. 1. Consequently, each peak is connected to hidden nodes dh with connection weights denoted as wi, which signify the importance of each peak in the model training process. To quantify the importance si for j-th peak, we calculate:
Here, si measures the variance of the connection weights from the peaks to the hidden nodes, serving as a metric to identify peaks of higher importance. By default, we retain the top 10,000 peaks based on their calculated importance scores. If fewer than 10,000 peaks meet this criterion, all peaks are retained. Then, we can obtain the accessibility profiles X after the autoencoder-based peak selection model.
Cell graph
After refining the feature matrix through our feature selection process, we obtained a data matrix in which each feature (peak) is considered critical for preserving cell heterogeneity. Then, we constructed a cell graph to explore the topological relationships between cells, which was based on the embedded features learned from the previously chromatin accessibility-based autoencoder. By focusing on representation learning, we aimed to improve the accuracy of defining cell-wise relationships.
Indeed, we used the K-nearest neighbors (KNN) algorithm, implemented in the scikit learn package, to build the graph where each node represents an individual cell, and the edges represent relationships between cells. For each cell, its neighbors were identified as the closest k cells, including the cell itself, based on the Euclidean distance between their embedded features. The number k was predefined to control the scale of interaction between the cells. By defining the adjacency of the cell graph, where a connection exists if another cell is within the nearest k neighbors, we effectively mapped out the domain of each cell and its immediate network. The neighborhood \({{{{\mathcal{N}}}}}_{k}(i)\) includes the nearest k cells with the i-th cell as the center. Eventually, we constructed the cell graph A as follows:
In this manner, we identified the domain of each cell and its neighbors through a local search method in a low-dimensional space, an efficient method proven to model biological networks90.
Bernoulli-based graph convolutional autoencoder
To leverage both the accessibility and cell-wise topological connections within the cell graph, we proposed a graph autoencoder that incorporates a GCN and Bernoulli distribution. Specifically, the GCN encoder processes the cell graph and chromatin accessibility profiles simultaneously to learn the highly compressed, low-dimensional embeddings. Subsequently, we employ two decoders: one to reconstruct the cell graph matrix, thus ensuring the accurate preservation of inter-cellular topology; and another to reconstruct the count matrix, thereby capturing the global probabilistic structure.
Indeed, our graph autoencoder operates within the framework of the variational graph autoencoder91 for its proficiency in managing complex graph structures and enhancing dimensionality reduction. The GCN encoder processes the input X, representing the refined feature data, to estimate the distributional parameters of the latent variables Z, which can be formulated as below:
where \(\bar{{{{\bf{A}}}}}={{{{\bf{D}}}}}^{-\frac{1}{2}}{{{\bf{A}}}}{{{{\bf{D}}}}}^{-\frac{1}{2}}\) represents the adjacency matrix of the cell graph and \({{{\bf{D}}}}=diag\left\{({{{\bf{I}}}}+{{{\bf{A}}}}){{{{\bf{1}}}}}_{N}\right\}\) denotes the degree matrix; We, Wμ, and Wσ are the model parameters of the corresponding layers. These parameters are critical for defining the Gaussian distribution from which the latent variable Z is sampled, leveraging the re-parameterization trick for efficient gradient descent:
Here, ϵ denotes a random variable sampled from a standard normal distribution, ensuring that the model training is differentiable and that the stochastic gradient descent can effectively optimize the variational lower bound; z is one column of the latent variable Z, and μ and σ are elements in certain column of Zμ and Zσ, respectively. Following this, we employ an inner product decoder to reconstruct the cell graph matrix from the low-dimensional embeddings. The sigmoid activation function is chosen to map the inner product to probabilities between 0 and 1, which effectively transforms the latent embeddings back into a graph structure. This can be expressed mathematically as:
where ZTZ represents the inner product of the latent representation matrix Z, and the sigmoid function maps these products to the [0, 1] range, indicating the probability of edge presence between nodes.
On this basis, we define our loss function in line with auto-encoding variational Bayes, which includes the reconstruction loss and the Kullback-Leibler (KL) divergence loss between the Gaussian distribution N(μ, σ2). These are formulated as follows:
After that, a Bernoulli-based decoder is employed to reconstruct the count matrix, assuming each element follows a multiple Bernoulli distribution. Specifically, for a given cell xi, the state of accessibility in j-th peak is either open or closed, taking the value 0 or 1. Here, xij is modeled as an independent Bernoulli random variable, with the probability of being open (bij) at j-th peak. Therefore, the likelihood of the observed accessibility pattern for cell xi across all peaks is expressed as:
To learn the probabilities bij, we employ a neural network decoder with a sigmoid activation function at its output, which can be defined as follows:
where fd represents a single layer fully connected neural network which can be formulated as: fd(Z) = sigmoid(ZWd), and B is a matrix where each element bi,j represents the probability of accessibility at the j-th peak for the i-th cell. We can then directly use floating probability values as imputed data, reflecting the degree of chromatin accessibility in each peak for every cell, thereby avoiding the complexity and randomness associated with the sampling operation. For training the model and optimizing the parameters B, the objective is to minimize the negative log-likelihood of observation data, serving as our another loss function \({{{{\mathcal{L}}}}}_{m}\):
This loss function effectively aggregates the individual log probabilities across all cells and peaks, thereby guiding the neural network to better identify the state of each peak in terms of accessibility.
Self-supervised clustering optimization
We implemented a dual clustering optimization objective that integrates the clustering task directly into the training guidance of our graph autoencoder. Initially, we define the cluster centroids of latent embeddings as cj, j = 1, ⋯ , N. The soft clustering assignment, which is crucial to our loss function, measures the similarity between each embedded point zi and centroid cj using Student’s t-distribution as the kernel. This similarity is defined as:
where the embedding zi ∈ Z corresponds to cell xi ∈ X and the initial cluster centroids cj are determined by k-means clustering after pre-training. Additionally, an auxiliary target distribution pij can be formulated as follows:
where fj = ∑iqij represents the soft cluster frequencies. The objective of soft clustering optimization is to minimize the KL divergence between the soft assignment matrix Q and the target distribution P, defined as follows:
Furthermore, we introduce a self-supervised learning strategy utilizing high-confidence predictions. Initially, we derive the cluster prediction yi as a pseudo-label from the clustering assignment qi using \(\arg \max\) function, followed by a one-hot transformation:
where ϕOH denotes the one-hot encoding transformation. The hard self-supervised loss function is then defined as:
where ϕCE denotes the cross-entropy loss function and \({q}_{ij}^{{\prime} }\) serves as a weighting factor that filters out low-confidence predictions from qij by applying a threshold r, which is adjusted from 0.60 to 0.80 to reflect improvements in cluster assignment accuracy during the training process. The combined clustering loss is defined as:
Training of scAGDE
The chromatin accessibility-based autoencoder is first trained using the loss function presented in Eq. (8), which balances the impact of the KL divergence term relative to the reconstruction error through the weighting coefficient β. In our training, we set β to 0.5 to place more emphasis on representation quality than on disentanglement, as suggested in beta-VAE89. Next, we initiated training of our graph autoencoder by pretraining it without incorporating the clustering loss function \({{{{\mathcal{L}}}}}_{c}\). During this pretraining phase, we determine the initial cluster centroids using the k-means algorithm on the learned embedding space, setting the stage for further training. In the subsequent phase, scAGDE optimizes a composite loss function defined as follows:
where \({{{{\mathcal{L}}}}}_{recon}\) represents the cell graph reconstruction loss, ensuring the faithful recreation of the original graph structure; \({{{{\mathcal{L}}}}}_{kl}\) regularizes the distribution of the latent variables, encouraging them to follow a predefined standard distribution; \({{{{\mathcal{L}}}}}_{m}\) corresponds to the Bernoulli-based decoder loss, focusing on minimizing discrepancies in the reconstructed count matrix; and \({{{{\mathcal{L}}}}}_{c}\) refers to the self-supervised clustering loss. The coefficients γ1, γ2, γ3, and γ4 are weight factors assigned to each loss term, controlling the relative importance of the different training objectives. To optimize this composite loss function, we use the Adam optimizer92.
Implementation details and comparisons with baseline methods
scAGDE was implemented in Python, taking advantage of the PyTorch framework that is publicly available at https://pypi.org/project/scAGDE/. The framework included both a chromatin accessibility-based autoencoder and a Bernoulli-based graph autoencoder, each featuring a two-layer encoder network (128-10) and a single-layer decoder network. For the coefficients of the loss function, we assigned values of 10, 5, 1, 1 to γ1, γ2, γ3, γ4, respectively. In this study, scAGDE was benchmarked against several state-of-the-art scATAC-seq data analysis methods including seven non-deep learning approaches and four deep learning-based models, as detailed below:
-
Single cell accessibility based clustering (scABC, https://github.com/timydaley/scABC)8. scABC employs a weighted K-medoids clustering approach to partition cells and refines the clustering based on landmarks for each cluster, ensuring more accurate clustering assignments.
-
An unsupervised Bayesian framework based on topic modeling (cisTopic, http://github.com/aertslab/cistopic)9. cisTopic utilizes latent Dirichlet allocation (LDA) with a collapsed Gibbs sampler to iteratively classify regions into regulatory topics and cluster cells based on their regulatory topic contributions.
-
Single nucleus analysis pipeline for ATAC-seq (SnapATAC, https://github.com/r3fang/SnapATAC)10. SnapATAC applies dimensionality reduction on a Jaccard similarity matrix that measures the overlap in accessibility between cells and accounts for sequencing depth variations with regression-based normalization.
-
A comprehensive toolkit for single-cell chromatin data (Signac, https://github.com/timoast/signac/)24. The Signac toolkit employs Latent Semantic Index (LSI), leveraging the frequency-inverse document frequency transformation and principal component analysis to reduce the dimensionality of the binarized accessibility matrix.
-
An integrative and scalable software package for scATAC-seq data (ArchR, https://github.com/GreenleafLab/ArchR)21. The ArchR package implements an iterative LSI for dimensional reduction and call clusters in this reduced dimension sub-space.
-
A fast, scalable and versatile tool for scATAC-seq data (SnapATAC2, https://github.com/kaizhang/SnapATAC2. SnapATAC2 is a pairwise-similarity based method and obtains cell embeddings through Laplacian eigenmaps. SnapATAC2 is an enhancement of SnapATAC.
-
A graph embedding method that jointly embeds cells and features (SIMBA, https://github.com/sgoldenlab/simba). SIMBA explicitly learns low-dimensional representations of single cells and their defining features, such as chromatin accessible regions, and DNA sequences into a common latent space.
-
Single-cell ATAC-seq analysis via latent feature extraction (SCALE, https://github.com/jsxlei/SCALE)13. SCALE integrates a probabilistic Gaussian Mixture Model into the VAE to learn latent embeddings.
-
Scalable and accurate invariant representation learning (SAILER, https://github.com/uci-cbcl/SAILER)14. SAILER adopts the VAE model with additional constraints to ensure that the learned representations are independent of confounding factors.
-
A deep generative model for scATAC-seq data analysis (PeakVI, https://github.com/YosefLab/scvi-tools/)15. PeakVI employs a VAE with an additional neural network to estimate technical factors that impact the probability of observing an accessible region.
-
A batch-adversarial variational auto-encoder approach (BAVARIA, https://github.com/BIMSBbioinfo/bavaria)18. BAVARIA uses a VAE and incorporates dimensionality reduction and batch correction through an adversarial learning strategy.
Additionally, we compared five scRNA-seq analysis methods, including both graph-based and nongraph-based, detailed below:
-
R toolkit for single-cell genomics (Seurat, https://github.com/satijalab/seurat)5. Seurat is an R package specifically designed for analyzing and exploring single-cell RNA-seq data.
-
Single-cell variational inference approach (scVI, https://github.com/YosefLab/scVI)93. scVI utilizes stochastic optimization and deep neural networks to analyze single-cell RNA-seq data, effectively aggregating information across similar cells and genes to model the distribution underlying observed gene expression.
-
Single-cell model-based deep learning method (scDeepCluster, https://github.com/ttgump/scDeepCluster)94. scDeepCluster employs a ZINB model-based autoencoder to simultaneously learn feature representations and perform clustering.
-
A single-cell model-based deep graph embedding clustering method (scTAG, https://github.com/Philyzh8/scTAG95. scTAG integrates a ZINB-based model with a topology adaptive graph convolutional autoencoder to learn low-dimensional latent representations.
-
A graph convolutional autoencoder framework (scMGCA, https://github.com/Philyzh8/scMGCA)33. scMGCA leverages a graph-embedding autoencoder and introduces a notable method for constructing cell graphs.
The detailed implementation, including function specifications and parameter settings, is provided in Supplementary Note 19.
Benchmarking metrics for clustering and visualization
We employed multiple metrics to evaluate the performance of the clustering algorithms, including NMI, ARI, F1 score, V-Measure and Homogeneity score. The NMI is a metric that normalizes the mutual information (MI) score to a range between 0 and 1 and can be defined as:
where I() is the mutual information metric and H() is the entropy metric. The ARI score is the corrected for chance of the rand index (RI) score and can be defined as:
where nij represents the entries in the contingency table formed by the true partition and predicted partition. The F1 score combines the precision and recall scores of a prediction and is calculated as the harmonic mean of the precision and recall scores, as shown below:
V-Measure is an external entropy-based clustering evaluation metric, which can be defined as:
where \(\frac{{{{\rm{H}}}}\left(C| K\right)}{{{{\rm{H}}}}(C)}\) represents the conditional entropy of cell type division given the cluster division condition; h is the homogeneity measure and c is the completeness measure. Eventually, the V-Measure value v is the harmonic mean of h and c measures, and the value is larger when two divisions are closely separated. The Homogeneity score is used to evaluate the clustering algorithm’s ability to assign cells of a single cell type to a single cluster. The score, denoted as h, ranges from 0 to 1, with a higher value indicating better homogeneity. It’s defined as:
where \({{{\rm{H}}}}({Y}_{true}| {Y}_{pred})\) is the probability of assigning true cells to a set of predicted cells, and \({{{\rm{H}}}}({Y}_{true})\) are the labels of the cells. These five metrics all range from 0 to 1, and a higher score denotes a better-quality performance.
Then, to evaluate the quality of dimensionality reduction and visualization, we used the ASW, the CHI, and the DBI. The ASW is calculated using the average intra-cluster distance a and the average nearest-cluster distance b across all samples and the ASW can be defined as:
The CHI measures the compactness and separation of clustering results by calculating the ratio of the between-cluster separation SSB to the within-cluster dispersion SSW, normalized by their number of degrees of freedom:
where n is the number of samples and k is the corresponding number of clusters. The DBI measures the average maximum similarity of each cluster. Its calculation involves both the inter-cluster distances denoted and the intra-cluster separation degree, denoted as Si, as shown below:
where \({\parallel {w}_{i}-{w}_{j}\parallel }_{2}\) calculates the Euclidean distance between i-th cluster and j-th cluster, and \(\overline{{S}_{i}}\) represents the average distance of all samples of i-th cluster to the cluster centroid. These metrics range from 0 to 1, where a higher score generally indicates a better clustering or visualization quality, except for DBI where a lower value indicates better clustering.
Peaks annotation with ChIPseeker
We conducted an analysis of the peak categories identified by scAGDE in scATAC-seq data, specifically examining their proximity to the nearest transcription start site (TSS) regions. To achieve this, we utilized the ‘annotatePeak’ function from ChIPseeker, an R package specifically developed for ChIP peak annotation34. Considering that our samples were derived from human tissues, we set the parameters ‘TxDb=TxDb.Hsapiens.UCSC.hg19.knownGene’ and ‘annoDb=org.Hs.eg.db’ to ensure alignment with the human genome annotation.
Identifying differentially accessible peaks
To carry out differential peaks analysis, we followed Stuart et al.24 and performed a Wilcoxon rank sum test implemented in Seurat package, using the ‘FindAllMarkers’ function. We set the parameters ‘min.pct=0.15’ and ‘logfc.threshold=0.15’ to keep significantly differentially accessible peaks. When using ArchR package, we applied the ‘getMarkerFeatures’ function with default parameter setting to identify marker peaks.
Identifying peak-associated genes using rGREAT
In our study, to examine the biological relevance between uncovered peaks and cell identity, we used the R package of rGREAT47 to identify peak-associated genes. We applied the ‘great’ function with parameters set to ‘basal_upstream = 15,000’ and ‘basal_downstream = 15,000’. The source of TSS was set to ‘txdb:mm10’ and the gene sets collections were obtained from MSigDB96 by using the ‘msigdbr’ function with parameters set to ‘species = Mus musculus’.
Known and de novo motifs enrichment analysis
To identify underlying regulators within chromatin accessible regions, we conducted both de novo and known motif enrichment analyses on the datasets. For the de novo motifs discovery, we employed the HOMER tool52 with parameters set to ‘-size 200 -mask’, which helps to identify marked regulatory elements that may be associated with the regulation of gene expression. For the enrichment of known motifs, we applied the ‘FindMotifs’ function of Signac24 and the ‘peakAnnoEnrichment’ function from ArchR21 across the data. This comprehensive analysis allowed us to contrast the presence and potential functional impact of both newly discovered and established motifs within the chromatin accessible regions of our scATAC-seq data.
Transcription factor deviation scores
We measured transcription factor activity using the chromVAR algorithm, integrated into the Signac package24 in R. Initially, we utilized the ‘addMotifs’ function to incorporate motif information into the Seurat object, sourcing motif position frequency matrices from the JASPAR database via ‘getMatrixSet’. Subsequently, we executed the ‘RunChromVAR’ function on the raw, imputed, and binary imputed count data, setting the genome parameter to ‘BSgenome.Mmusculus.UCSC.mm10’.
GO enrichment analysis
We performed GO enrichment analysis on these datasets using the R package ClusterProfiler97. Initially, we selected the top 100 genes ranked by the lowest false discovery rate (FDR) from differentially expressed genes based on each cluster. These genes were identified from the ‘GeneScoreMatrix’ data calculated by ArchR21. We employed the ‘enrichGO’ function for this analysis, setting the parameters to ‘OrgDb = org.Hs.eg.db, p-valueCutoff = 0.05, q-valueCutoff = 0.05, with all other parameters at their default values. For enrichment analysis of three groups of glutamatergic neurons, we selected differentially expressed genes with FDR ≤ 0.05 and Log2FC ≥ 0.75. The parameters for this subsequent analysis are consistent with those described previously.
Integrative analysis of human brain scATAC-seq and scRNA datasets
To further evaluate our cell type identifications and detect regulatory differences between three glutamatergic neuron clusters, we integrated the scRNA-seq data of the primate dorsolateral prefrontal cortex76. To perform integrative analysis, we first manually filtered scRNA-seq data from human samples. Second, we added the cell embeddings from scAGDE to the ArchR project. Next, we used the function ‘addGeneIntegrationMatrix’ to perform the final integration, with the parameter ‘reducedDims’ to the slot name of scAGDE embeddings. This operation employs the function ‘FindTransferAnchors’ in Seurat to integrate datasets. Following the integration, we identified peak-to-gene linkages by leveraging the integrated scRNA-seq data to detect correlations between chromatin accessibility and the expression of genes of interest. Specifically, we utilized the ‘addPeak2GeneLinks’ function, similarly setting the ‘reducedDims’ parameter to the slot name of scAGDE embeddings to ensure consistency in data handling.
Annotating cell types to cell clusters
In our study, we annotated cell types in the scATAC-seq data by evaluating the cell-type enrichment score that measures the inferred activity of marker genes for each cell type. We first sourced the top 50 marker genes for a range of cell types from two marker gene databases: PanglaoDB64 and CellMarker65. We then extracted gene activity data from the ‘GeneScoreMatrix’ by ArchR tool, and calculated the average activities across clusters with reads per million mapped reads (RPM) normalization. For each defined cell type, we computed the raw enrichment scores as the mean RPM values across all the marker genes within each cluster. Subsequently, a double z-score transformation was applied on the raw enrichment scores, first across cell types and then across cell clusters, to derive the final enrichment scores. The cell type with the highest enrichment score was selected as the definitive annotation for each cell cluster.
Statistics and reproducibility
The detailed statistical tests were explained in each figure legend. Sample data were obtained from public repositories. Sample size was not predetermined and is the maximum number of samples available for each datasets. No data were excluded from the analyses. No experimental groups were assigned. Our study does not involve group allocation that requires blinding. To reproduce the results, please find the Source Data file we provided.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
We generated a total of 17 simulated datasets from bulk ATAC-seq data of bone marrow98, which contains six FACS-sorted cell populations. Following a previously published benchmarking framework for scATAC-seq tools20, we set the parameter n, which determines fragment counts within a single cell, at 250, 500, 1500, 2500, and 5000, respectively, thus obtaining five datasets of varying sequencing depth. We set the parameter q, which controls the proportion of cell-specific reads at 0, 0.1, 0.2, 0.3, 0.4, thus obtaining five datasets of differing noise levels. Lastly, we randomly dropped valid reads at rates ranging from 10% to 70%, generating seven datasets with a varying degree of dropout. The simulated data generated in this study have been deposited both in the Zenodo database at https://doi.org/10.5281/zenodo.11609252 and in the Figshare databse at https://doi.org/10.6084/m9.figshare.28076054.v199. Additionally, we collected 11 publicly available scATAC-seq datasets with given cell type labels for benchmarking to validate the effectiveness of scAGDE. These datasets, generated from different platforms and including human and mouse samples, vary in sparsity and scalability. Four datasets annotated through computational approaches included ‘Forebrain’ under accession code GSE10003362 [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE100033], ‘Splenocyte’ under accession code E-MTAB-671436 [https://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-6714/], ‘GM12878vsHEK’ and ‘GM12878vsHL’ under accession code GSE6536025 [http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE65360], ‘Lung’ and ‘Liver’ under accession code GSE14968327 [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE149683]. Three datasets containing FACS-sorted cell populations were ‘Blood2K’ under accession code GSE9677231 [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE96772], ‘10XBlood’ under accession code GSE1297852 [http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE129785], and ‘DropBlood’ under accession code GSE123581100 [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE123581]. The remaining two datasets were ‘Leukemia’ under accession code GSE7431039 [http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE74310], which mixes cells from a healthy donor with leukemia cells from two acute myeloid leukemia patients, and ‘InSilico’ under accession code GSE653608 [http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE65360] combining six individual scATAC-seq data from distinct cell lines. Details of these datasets and statistics plots are provided in Supplementary Note 20 and Supplementary Figs. 41 and 42. The human fetal atlas dataset from Domcke et al.27, can be obtained from the public resource at https://descartes.brotmanbaty.org/bbi/human-chromatin-during-development/. The human brain dataset is available under accession code GSE18446263 [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE184462], comes from a single-cell atlas of scATAC-seq of the human genome. The reference single-cell RNA-seq dataset from brain tissue used in our study is available under accession code GSE20733476 [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE207334] and we utilizes data from human samples. All processed datasets for benchmarking analysis and the human brain dataset have been deposited both in the Zenodo database at https://doi.org/10.5281/zenodo.11609252 and in the Figshare databse at https://doi.org/10.6084/m9.figshare.28076054.v199. All other relevant data supporting the key findings of this study are available within the article and its Supplementary Information files or from the corresponding author upon request. Source data are provided with this paper.
Code availability
The source code of MIT-licensed scAGDE software is freely available at GitHub https://github.com/Hgy1014/scAGDE101. scAGDE is also released as a Python package at: https://pypi.org/project/scAGDE/. The online documentation website can be accessed at https://scagde-tutorial.readthedocs.io/en/latest/index.html to learn how to use scAGDE and reproduce the study with the provided tutorials.
References
Kundaje, A. et al. Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330 (2015).
Satpathy, A. T. et al. Massively parallel single-cell chromatin landscapes of human immune cell development and intratumoral t cell exhaustion. Nat. Biotechnol. 37, 925–936 (2019).
Cusanovich, D. A. et al. Multiplex single-cell profiling of chromatin accessibility by combinatorial cellular indexing. Science 348, 910–914 (2015).
Yan, F., Powell, D. R., Curtis, D. J. & Wong, N. C. From reads to insight: a hitchhiker’s guide to atac-seq data analysis. Genome Biol. 21, 1–16 (2020).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587 (2021).
Wolf, F. A., Angerer, P. & Theis, F. J. Scanpy: large-scale single-cell gene expression data analysis. Genome Biol. 19, 1–5 (2018).
Kiselev, VladimirYu et al. Sc3: consensus clustering of single-cell rna-seq data. Nat. Methods 14, 483–486 (2017).
Zamanighomi, M. et al. Unsupervised clustering and epigenetic classification of single cells. Nat. Commun. 9, 2410 (2018).
González-Blas, CarmenBravo et al. cistopic: cis-regulatory topic modeling on single-cell atac-seq data. Nat. Methods 16, 397–400 (2019).
Fang, R. et al. Comprehensive analysis of single cell atac-seq data with snapatac. Nat. Commun. 12, 1337 (2021).
Schep, A. N., Wu, B., Buenrostro, J. D. & Greenleaf, W. J. chromvar: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat. Methods 14, 975–978 (2017).
Pliner, H. A. et al. Cicero predicts cis-regulatory dna interactions from single-cell chromatin accessibility data. Mol. Cell 71, 858–871 (2018).
Xiong, L. et al. Scale method for single-cell atac-seq analysis via latent feature extraction. Nat. Commun. 10, 4576 (2019).
Cao, Y. et al. Sailer: scalable and accurate invariant representation learning for single-cell atac-seq processing and integration. Bioinformatics 37, i317–i326 (2021).
Ashuach, T., Reidenbach, D. A., Gayoso, A. & Yosef, N. Peakvi: a deep generative model for single-cell chromatin accessibility analysis. Cell Rep. Methods 2, 100182 (2022).
Danese, A. et al. Episcanpy: integrated single-cell epigenomic analysis. Nat. Commun. 12, 5228 (2021).
Zeng, Y., Zhou, X., Rao, J., Lu, Y. & Yang, Y. Accurately clustering single-cell rna-seq data by capturing structural relations between cells through graph convolutional network. In 2020 IEEE International Conference on Bioinformatics and Biomedicine (BIBM) (eds Park, T. et al.) 519–522 (IEEE, 2020).
Kopp, W., Akalin, A. & Ohler, U. Simultaneous dimensionality reduction and integration for single-cell atac-seq data using deep learning. Nat. Mach. Intell. 4, 162–168 (2022).
Xiong, L. et al. Online single-cell data integration through projecting heterogeneous datasets into a common cell-embedding space. Nat. Commun. 13, 6118 (2022).
Chen, H. et al. Assessment of computational methods for the analysis of single-cell atac-seq data. Genome Biol. 20, 1–25 (2019).
Granja, J. M. et al. Archr is a scalable software package for integrative single-cell chromatin accessibility analysis. Nat. Genet. 53, 403–411 (2021).
Zhang, K., Zemke, N. R., Armand, E. J. & Ren, B. A fast, scalable and versatile tool for analysis of single-cell omics data. Nat. Methods 21, 217–227 (2024).
Chen, H., Ryu, J., Vinyard, M. E., Lerer, A. & Pinello, L. Simba: single-cell embedding along with features. Nat. Methods 21, 1003–1013 (2024).
Stuart, T., Srivastava, A., Madad, S., Lareau, C. & Satija, R. Single-cell chromatin state analysis with signac. Nat. Methods https://doi.org/10.1038/s41592-021-01282-5 (2021).
Buenrostro, J. D. et al. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 523, 486–490 (2015).
Luecken, M. D. et al. Benchmarking atlas-level data integration in single-cell genomics. Nat. Methods 19, 41–50 (2022).
Domcke, S. et al. A human cell atlas of fetal chromatin accessibility. Science 370, eaba7612 (2020).
Wold, S., Esbensen, K. & Geladi, P. Principal component analysis. Chemom. Intell. Lab. Syst. 2, 37–52 (1987).
Van der Maaten, L. & Hinton, G. Visualizing data using t-sne. J. Mach. Learn. Res. 9, 2579−2605 (2008).
McInnes, L., Healy, J. & Melville, J. Umap: uniform manifold approximation and projection for dimension reduction. Preprint at arXiv preprint arXiv:1802.03426 (2018).
Buenrostro, J. D. et al. Integrated single-cell analysis maps the continuous regulatory landscape of human hematopoietic differentiation. Cell 173, 1535–1548 (2018).
Goardon, N. et al. Coexistence of lmpp-like and gmp-like leukemia stem cells in acute myeloid leukemia. Cancer Cell 19, 138–152 (2011).
Yu, Z. et al. Topological identification and interpretation for single-cell gene regulation elucidation across multiple platforms using scmgca. Nat. Commun. 14, 400 (2023).
Wang, Q. et al. Exploring epigenomic datasets by chipseeker. Curr. Protoc. 2, e585 (2022).
Thorndike, R. L. Who belongs in the family? Psychometrika 18, 267–276 (1953).
Chen, X., Miragaia, R. J., Natarajan, KedarNath & Teichmann, S. A. A rapid and robust method for single cell chromatin accessibility profiling. Nat. Commun. 9, 5345 (2018).
Chung, Chi-Yeh et al. Single-cell chromatin analysis of mammary gland development reveals cell-state transcriptional regulators and lineage relationships. Cell Rep. 29, 495–510 (2019).
Vierstra, J. et al. Mouse regulatory dna landscapes reveal global principles of cis-regulatory evolution. Science 346, 1007–1012 (2014).
Corces, M. R. et al. Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat. Genet. 48, 1193–1203 (2016).
Frazer, K. A., Pachter, L., Poliakov, A., Rubin, E. M. & Dubchak, I. Vista: computational tools for comparative genomics. Nucleic Acids Res. 32, W273–W279 (2004).
Bai, X. et al. Endb: a manually curated database of experimentally supported enhancers for human and mouse. Nucleic Acids Res. 48, D51–D57 (2020).
Chikuma, S. Ctla-4, an essential immune-checkpoint for t-cell activation. In Emerging Concepts Targeting Immune Checkpoints in Cancer and Autoimmunity (ed. Yoshimura, A.) 99–126 (Springer International Publishing, 2017).
Syn, N. L., Teng, MicheleW. L., Mok, TonyS. K. & Soo, R. A. De-novo and acquired resistance to immune checkpoint targeting. Lancet Oncol. 18, e731–e741 (2017).
Gao, G. F. & Jakobsen, B. K. Molecular interactions of coreceptor cd8 and mhc class i: the molecular basis for functional coordination with the t-cell receptor. Immunol. Today 21, 630–636 (2000).
Addison, E. G. et al. Ligation of cd8α on human natural killer cells prevents activation-induced apoptosis and enhances cytolytic activity. Immunology 116, 354–361 (2005).
Hicks, S. C., Townes, F. W., Teng, M. & Irizarry, R. A. Missing data and technical variability in single-cell rna-sequencing experiments. Biostatistics 19, 562–578 (2018).
Gu, Z. & Hübschmann, D. rgreat: an r/bioconductor package for functional enrichment on genomic regions. Bioinformatics 39, btac745 (2023).
Rousseau, A. et al. Expression of oligodendroglial and astrocytic lineage markers in diffuse gliomas: use of ykl-40, apoe, ascl1, and nkx2-2. J. Neuropathol. Exp. Neurol. 65, 1149–1156 (2006).
Zeisel, A. et al. Cell types in the mouse cortex and hippocampus revealed by single-cell rna-seq. Science 347, 1138–1142 (2015).
La Manno, G. et al. Molecular diversity of midbrain development in mouse, human, and stem cells. Cell 167, 566–580 (2016).
Pernet, V., Joly, S., Christ, F., Dimou, L. & Schwab, M. E. Nogo-a and myelin-associated glycoprotein differently regulate oligodendrocyte maturation and myelin formation. J. Neurosci. 28, 7435–7444 (2008).
Heinz, S. et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and b cell identities. Mol. Cell 38, 576–589 (2010).
Fraser, J. et al. Cell-type-specific expression of nfix in the developing and adult cerebellum. Brain Struct. Funct. 222, 2251–2270 (2017).
Shu, T., Butz, K. G., Plachez, C., Gronostajski, R. M. & Richards, L. J. Abnormal development of forebrain midline glia and commissural projections in nfia knock-out mice. J. Neurosci. 23, 203–212 (2003).
Deneen, B. et al. The transcription factor nfia controls the onset of gliogenesis in the developing spinal cord. Neuron 52, 953–968 (2006).
Campbell, C. E. et al. The transcription factor nfixis essential for normal brain development. BMC Dev. Biol. 8, 1–18 (2008).
Barbosa, A. C. et al. Mef2c, a transcription factor that facilitates learning and memory by negative regulation of synapse numbers and function. Proc. Natl. Acad. Sci. USA 105, 9391–9396 (2008).
Nord, A. S., Pattabiraman, K., Visel, A. & Rubenstein, JohnL. R. Genomic perspectives of transcriptional regulation in forebrain development. Neuron 85, 27–47 (2015).
Kierdorf, K. et al. Microglia emerge from erythromyeloid precursors via pu. 1-and irf8-dependent pathways. Nat. Neurosci. 16, 273–280 (2013).
Glasgow, S. M. et al. Mutual antagonism between sox10 and nfia regulates diversification of glial lineages and glioma subtypes. Nat. Neurosci. 17, 1322–1329 (2014).
Yuan, F. et al. Efficient generation of region-specific forebrain neurons from human pluripotent stem cells under highly defined condition. Sci. Rep. 5, 18550 (2015).
Preissl, S. et al. Single-nucleus analysis of accessible chromatin in developing mouse forebrain reveals cell-type-specific transcriptional regulation. Nat. Neurosci. 21, 432–439 (2018).
Zhang, K. et al. A single-cell atlas of chromatin accessibility in the human genome. Cell 184, 5985–6001 (2021).
Franzén, O., Gan, Li-Ming & Björkegren, JohanL. M. Panglaodb: a web server for exploration of mouse and human single-cell rna sequencing data. Database 2019, baz046 (2019).
Hu, C. et al. Cellmarker 2.0: an updated database of manually curated cell markers in human/mouse and web tools based on scrna-seq data. Nucleic Acids Res. 51, D870–D876 (2023).
Kuhn, S., Gritti, L., Crooks, D. & Dombrowski, Y. Oligodendrocytes in development, myelin generation and beyond. Cells 8, 1424 (2019).
Kreutzberg, G. W. Microglia: a sensor for pathological events in the cns. Trends Neurosci. 19, 312–318 (1996).
Wittstatt, J., Reiprich, S. & Küspert, M. Crazy little thing called sox—new insights in oligodendroglial sox protein function. Int. J. Mol. Sci. 20, 2713 (2019).
Das, A. et al. Transcriptome sequencing reveals that lps-triggered transcriptional responses in established microglia bv2 cell lines are poorly representative of primary microglia. J. Neuroinflammation 13, 1–18 (2016).
Nakatani, H. et al. Ascl1/mash1 promotes brain oligodendrogenesis during myelination and remyelination. J. Neurosci. 33, 9752–9768 (2013).
Jiang, F. et al. Simultaneous profiling of spatial gene expression and chromatin accessibility during mouse brain development. Nat. Methods 20, 1048–1057 (2023).
Raivich, G. & Behrens, A. Role of the ap-1 transcription factor c-jun in developing, adult and injured brain. Prog. Neurobiol. 78, 347–363 (2006).
Linseman, D. A. et al. Inactivation of the myocyte enhancer factor-2 repressor histone deacetylase-5 by endogenous ca2//calmodulin-dependent kinase ii promotes depolarization-mediated cerebellar granule neuron survival. J. Biol. Chem. 278, 41472–41481 (2003).
Südhof, T. C. Neurotransmitter release: the last millisecond in the life of a synaptic vesicle. Neuron 80, 675–690 (2013).
Rizo, J. & Xu, J. The synaptic vesicle release machinery. Annu. Rev. Biophys. 44, 339–367 (2015).
Ma, S. et al. Molecular and cellular evolution of the primate dorsolateral prefrontal cortex. Science 377, eabo7257 (2022).
Yu, Chuan-Yong et al. Neuronal and astroglial tgfβ-smad3 signaling pathways differentially regulate dendrite growth and synaptogenesis. Neuromolecular Med. 16, 457–472 (2014).
Tutukova, S., Tarabykin, V. & Hernandez-Miranda, L. R. The role of neurod genes in brain development, function, and disease. Front. Mol. Neurosci. 14, 662774 (2021).
Harrington, A. J. et al. Mef2c regulates cortical inhibitory and excitatory synapses and behaviors relevant to neurodevelopmental disorders. elife 5, e20059 (2016).
Assali, A., Harrington, A. J. & Cowan, C. W. Emerging roles for mef2 in brain development and mental disorders. Curr. Opin. Neurobiol. 59, 49–58 (2019).
Lonze, B. E. & Ginty, D. D. Function and regulation of creb family transcription factors in the nervous system. Neuron 35, 605–623 (2002).
Tomas-Roca, L. et al. Developmental disruption and restoration of brain synaptome architecture in the murine pax6 neurodevelopmental disease model. Nat. Commun. 13, 6836 (2022).
Gold, S. J., Ni, Y. G., Dohlman, H. G. & Nestler, E. J. Regulators of g-protein signaling (rgs) proteins: region-specific expression of nine subtypes in rat brain. J. Neurosci. 17, 8024–8037 (1997).
De Vries, L., Zheng, B., Fischer, T., Elenko, E. & Farquhar, M. G. The regulator of g protein signaling family. Annu. Rev. Pharmacol. Toxicol. 40, 235–271 (2000).
Yuan, H. & Kelley, D. R. scbasset: sequence-based modeling of single-cell atac-seq using convolutional neural networks. Nat. Methods 19, 1088–1096 (2022).
Tayyebi, Z., Pine, A. R. & Leslie, C. S. Scalable and unbiased sequence-informed embedding of single-cell atac-seq data with cellspace. Nat. Methods 21, 1–9 (2024).
Zeng, Y. et al. Deciphering cell types by integrating scatac-seq data with genome sequences. Nat. Comput. Sci. 4, 1–14 (2024).
Kingma, D. P. & Welling, M. Auto-encoding variational bayes. In International Conference on Learning Representations (ICLR, 2014).
Higgins, I. et al. beta-vae: learning basic visual concepts with a constrained variational framework. In International Conference on Learning Representations, Poster (ICLR, 2017).
Goldenberg, A., Mostafavi, S., Quon, G., Boutros, P. C. & Morris, Q. D. Unsupervised detection of genes of influence in lung cancer using biological networks. Bioinformatics 27, 3166–3172 (2011).
Kipf, T. N. & Welling, M. Variational graph auto-encoders. Preprint at arXiv preprint arXiv:1611.07308 (2016).
Kingma, D. P. & Ba, J. Adam: a method for stochastic optimization. In International Conference for Learning Representations (ICLR, 2015).
Lopez, R., Regier, J., Cole, M. B., Jordan, M. I. & Yosef, N. Deep generative modeling for single-cell transcriptomics. Nat. Methods 15, 1053–1058 (2018).
Tian, T., Wan, J., Song, Q. & Wei, Z. Clustering single-cell rna-seq data with a model-based deep learning approach. Nat. Mach. Intell. 1, 191–198 (2019).
Yu, Z. et al. Zinb-based graph embedding autoencoder for single-cell rna-seq interpretations. Proc. AAAI Conf. Artif. Intell. 36, 4671–4679 (2022).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005).
Wu, T. et al. clusterprofiler 4.0: a universal enrichment tool for interpreting omics data. Innovation 2, 100141 (2021).
Ulirsch, J. C. et al. Interrogation of human hematopoiesis at single-cell and single-variant resolution. Nat. Genet. 51, 683–693 (2019).
Hao, G. & Li, X. Topological identification and interpretation for single-cell epigenetic regulation elucidation in multi-tasks using scAGDE. Hgy1014/scAGDE. https://doi.org/10.6084/m9.figshare.28076054.v1 (2024b).
Lareau, C. A. et al. Droplet-based combinatorial indexing for massive-scale single-cell chromatin accessibility. Nat. Biotechnol. 37, 916–924 (2019).
Hao, G. Topological identification and interpretation for single-cell epigenetic regulation elucidation in multi-tasks using scAGDE. Hgy1014/scAGDE. https://doi.org/10.5281/zenodo.14512100 (2024).
Bediaga, N. G. et al. Multi-level remodelling of chromatin underlying activation of human t cells. Sci. Rep. 11, 528 (2021).
Acknowledgements
The work was supported by the China National Natural Science Foundation under Grant No. 62472195 (X.L.), No. 62076109 (X.L.), No. 623B2041 (Z.Y.) and No. 62401131 (Y.Y.). The work described in this article was also substantially supported by the Jilin Province Outstanding Young Scientist Program (Grant NO. 20230508098RC) (X.L.) and the ‘Fundamental Research Funds for Central Universities’ (X.L.).
Author information
Authors and Affiliations
Contributions
X.L. conceived and supervised the project. G.H. developed and implemented the algorithms under the guidance of X.L. and G.H. wrote the manuscript. G.H. conducted the experiments. X.L., G.H., K.-C.W., X.C., and G.W. did the biological interpretation. G.H., Y.F., and Z.Y. completed the figures and manuscript. G.H., X.L., K.-C.W., Y.S., F.W., and Y.Y. revised the manuscript. H.Z. provided advice in method development. All authors approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Peizhuo Wang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hao, G., Fan, Y., Yu, Z. et al. Topological identification and interpretation for single-cell epigenetic regulation elucidation in multi-tasks using scAGDE. Nat Commun 16, 1691 (2025). https://doi.org/10.1038/s41467-025-57027-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-57027-x
