
Abstract
Environmental surveillance of antibiotic resistance genes (ARGs) is critical for understanding and mitigating the spread of antimicrobial resistance. Current short-read-based ARG profiling methods are limited in their ability to provide detailed host information, which is indispensable for tracking the transmission and assessing the risk of ARGs. Here, we present Argo, a novel approach that leverages long-read overlapping to rapidly identify and quantify ARGs in complex environmental metagenomes at the species level. Argo significantly enhances the resolution of ARG detection by assigning taxonomic labels collectively to clusters of reads, rather than to individual reads. By benchmarking the performance in host identification using simulation, we confirm the advantage of long-read overlapping over existing metagenomic profiling strategies in terms of accuracy. Using sequenced mock communities with varying quality scores and read lengths, along with a global fecal dataset comprising 329 human and non-human primate samples, we demonstrate Argo’s capability to deliver comprehensive and species-resolved ARG profiles in real settings.
Similar content being viewed by others

Introduction
Over the past few decades, antimicrobial resistance (AMR), specifically bacterial AMR, has emerged as one of the greatest global concerns because of the growth of infections by drug-resistant pathogens and the lack of novel drug discoveries1. In 2021 alone, AMR was reported to be directly responsible for an estimated 1.14 million deaths worldwide, and this number is forecast to rise to 1.91 million by 2050 if no concerted and collaborative global action is taken2. This high AMR-associated death toll imposes a substantial burden on human public health and highlights the urgency of surveillance to monitor the emergence and spread of AMR, both spatially and temporally3.
Traditionally, AMR surveillance has predominantly focused on pathogenic isolates of antibiotic-resistant bacteria (ARB) cultured from clinical samples4. While these methods provide comprehensive information about resistance phenotypes through antimicrobial susceptibility testing (AST), they are time-consuming, costly, and often overlook non-pathogenic species, which are equally capable of developing antibiotic resistance genes (ARGs) under certain selective pressures and transmitting them via horizontal gene transfer (HGT)5. Cultivation-independent methods, specifically metagenomics, directly extract genetic material from all microorganisms as a whole, thus circumventing the issue of culture bias and are increasingly being applied for AMR surveillance6. A typical workflow of this kind involves quantifying ARGs by mapping sequenced reads to a known ARG reference database, such as CARD (Comprehensive Antibiotic Resistance Database)7, NDARO (National Database of Antibiotic Resistant Organisms)8, or SARG (Structured Antibiotic Resistance Gene database)9, and subsequently normalizing the estimated number of ARG copies with respect to a constant to account for varied sequencing depths10. With this approach, surveillance efforts have been expanded to include a range of environmental compartments in addition to clinical isolates, such as oceans11, soil12, cities13, wastewater14, and feces15.
Despite these advances, current surveillance methods utilizing second-generation short reads are known to be limited in linking ARGs to their specific microbial hosts6. To overcome this, short reads are often assembled into contiguous sequences (contigs) to improve the maximally achievable taxonomic resolution, at the cost of reduced sensitivity in ARG detection15. However, assigning species-level taxonomy to ARG-containing contigs assembled from short reads may still not be trivial, given that they tend to be fragmented due to the high frequency of repetitive regions surrounding ARGs, especially in complex environmental metagenomes16. Third-generation long-read sequencing, with its ability to generate reads tens of thousands of bases in length, holds potential for addressing this issue17. Due to their length advantage, long reads can span not only ARGs at full-length but also include their contextual information, thereby markedly increasing the likelihood of correct taxonomic classification18. Recently, the use of long-read sequencing for AMR surveillance has become prevalent. Dai et al. adopted the Antimicrobial Resistance Mapping Application (ARMA) workflow developed by Oxford Nanopore Technologies (ONT), which first identifies ARGs by aligning long reads to CARD and then performs taxonomic classification of these ARG-carrying reads with Centrifuge19,20, to track the hosts and fates of ARGs during wastewater treatment processes. Similarly, Yang et al. employed Kraken2 to assign GTDB taxonomy to ARG-carrying reads and incorporated these assignments into a framework for quantitative microbial risk assessment of beach water21,22. Although these approaches show great promise, their performance—particularly in using taxonomic classifiers for ARG-carrying reads—has rarely been justified. Furthermore, as ARGs are prone to horizontal gene transfer, they may appear in multiple genetic locations (e.g., plasmids or chromosomes) across different species, making it even more challenging to classify ARG-containing reads accurately on a per-read basis16.
Here, we introduce Argo, a novel ARG profiler designed to enhance the accuracy of host-tracking in long-read metagenomics. Unlike existing methods such as Kraken2 and Centrifuge, which assign taxonomic labels to each individual read, Argo operates on read clusters—identified through graph clustering of read overlaps. Taxonomic labels of reads are determined on a per-cluster basis with base-level alignment to GTDB and refined via greedy set covering. Using simulated data, we demonstrate this read-overlapping approach substantially reduces the number of misclassifications in host identification compared to traditional taxonomic assignment strategies, while maintaining high sensitivity and speed by avoiding the computationally intensive assembly step.
We further validate Argo’s high accuracy in profiling ARGs using four real metagenomic mock samples, each exhibiting unique read characteristics. By leveraging a dataset comprising 329 human and non-human primate fecal samples, we illustrate Argo’s effectiveness in surveilling ARGs within complex environments. Our analysis reveals that human-specific co-diversification of the gut microbiota significantly increases ARG abundance, and this increase is primarily driven by non-pathogenic commensal lineages rather than pathogenic ones. Moreover, using Escherichia coli (E. coli) as a global indicator, we observe distinct geographical patterns in its ARG types and potential horizontal ARG transfers between it and other non-pathogenic species in the gut.
Results
Overview of Argo and its database
Argo is a read-based ARG profiler that takes long reads as input and generates ARG profiles for each detected species. Given a reference ARG database, Argo first identifies reads carrying at least one ARG using DIAMOND’s frameshift-aware DNA-to-protein alignment (Fig. 1a, “Methods”)23. This step serves as a preliminary filter that substantially reduces the number of reads requiring further processing. ARGs, along with their detailed coordinates on reads, are recorded for subsequent computation. Since long reads exhibit highly diverse quality scores, which consequently affect the accuracy of alignments, Argo adaptively sets an identity cutoff by default to ensure profiles are comparable across samples generated by different sequencing platforms. This identity cutoff is initially estimated based on the per-base sequence divergence derived from the overlaps of the first 10,000 reads, obtained using minimap2’s approximate mapping24, and is later recalculated once the overlaps of ARG-containing reads become available (Supplementary Fig. S1, “Methods”).

a Screening of ARG reads. Given input long reads, those that carry at least one ARG are extracted using DIAMOND’s frameshift-aware DNA-to-protein alignment with an adaptive identity cutoff. b Collection of host information. ARG-containing reads are mapped to an ARG-tailored reference taxonomy database (GTDB) to obtain candidate species sets and alignment scores using minimap2’s base-level alignment. c Assignment of taxonomic labels. ARG-containing reads are overlapped with each other to form an overlap graph using minimap2’s approximate mapping (without base-level details). The graph (each vertex represents a read, and each edge the identity between reads) is segmented into components (read clusters) using the MCL algorithm. Taxonomic labels of reads within each cluster are determined by solving a weighted set cover problem with a greedy approximation. The final output of Argo is a tab-delimited table listing ARG abundances by species. a–c Semi-transparent gray dots stand for ARGs. Colors indicate different species.
Here, the reference ARG database, which we refer to as SARG+, is a manually curated compendium of protein sequences collected from CARD, NDARO, and SARG (“Methods”). We do not use any pre-existing databases since they are either not comprehensive enough or not designed primarily for read-based environmental surveillance at species-level resolution. These databases typically contain only a single or just a few representative sequences per ARG. For example, eptA is a phosphoethanolamine transferase present in both Escherichia coli and Salmonella enterica25, yet only the sequence of Escherichia coli is included in CARD. This may lead to an underestimation of ARG abundance if any identity-based filter is applied, given that the protein sequences of eptA in Salmonella enterica and Escherichia coli share only 82.4% identity. By contrast, SARG+ augments these databases by including all RefSeq protein sequences annotated through the same evidence (BlastRules or Hidden Markov Models provided by the NCBI Prokaryotic Genome Annotation Pipeline, PGAP) as those for experimentally validated ARGs26, thereby covering not only the eptA gene of Escherichia coli but also the ones from a variety of other species, including Salmonella enterica. This database expansion allows Argo to employ more stringent thresholds while maintaining high sensitivity in ARG identification. Furthermore, we discard all regulators (e.g., activators and repressors of ARGs), housekeeping genes, and ARGs that arise from point mutations, since the presence of these genes is not necessarily indicative of direct antibiotic resistance15. We also group highly similar ARGs—such as blaOXA-1 and blaOXA-1042, which differ by only a single amino acid (R101G, 99.6% identical)—to avoid potential ambiguities. After deduplication via clustering, SARG+ currently encompasses 104,529 protein sequences organized in a consistent hierarchy of type (class/family), subtype (subclass/gene), and sequence (“Methods”).
ARG-containing reads then undergo two major steps for taxonomic classification. First, these reads are mapped to a reference taxonomy database using minimap2’s base-level alignment to generate a list of candidate species labels for each read. These labels are subsequently aggregated into candidate species sets, where each set contains at least one read, and each read belongs to at least one set (Fig. 1b, “Methods”). The reference taxonomy database is derived by extracting ARG-containing genomic regions, each with at most 10,000 bp, from 596,663 assemblies (113,104 species, excluding 196 assemblies deprecated by NCBI) of GTDB release 09-RS220 (Supplementary Fig. S2, “Methods”). We use GTDB by default since it is more comprehensive, better quality controlled, and has fewer issues with confused taxonomic annotations compared to NCBI RefSeq27. Note that the database is constructed with the entire collection of GTDB rather than just the species representatives. This redundancy is important, given that a single representative genome may not cover all possible ARGs of a species, especially those acquired through HGT in complex environmental samples28. To account for ARGs carried by plasmids, ARG-containing reads are marked as “plasmid-borne” if they additionally map to a decontaminated subset of RefSeq plasmid29, which currently includes 39,598 sequences (“Methods”). We omit ARGs carried by phages since phage-associated ARGs are highly rare, and even if detected with permissive thresholds, these genes are unlikely to be bona fide ARGs that confer antibiotic resistance30,31.
Second, reads containing ARGs are overlapped with each other to build an overlap graph. This graph—a large sparse matrix representing the pairwise identities between reads—is then segmented into components (i.e., read clusters) using the Markov Cluster (MCL) algorithm (Fig. 1c, Supplementary Table S1, “Methods”)32. Reads originating from the same genomic region tend to have higher overlap identity and are therefore more likely to be clustered together. Ideally, each read cluster should represent a single ARG from a specific species. However, some clusters may span multiple closely located ARGs or ARGs shared across species. In these cases, taxonomic assignments are determined by solving a weighted set cover problem using a greedy algorithm, with species sets defined previously and weights given by alignment scores (Fig. 1c, “Methods”). Within each cluster, reads with the same taxonomic assignments are deemed plasmid if over 50% of them are labeled as “plasmid-borne”. The motivation behind using read clusters is that some ARGs can be lengthy, making individual fragmented reads (e.g., 1000 bp) insufficient to cover entire genes, thus leading to inaccurate per-read taxonomic classification. With read-overlapping, Argo properly handles multi-mapped, ambiguous reads by reassigning them to the most likely lineages based on local information collected from their neighbors.
For a given ARG, its abundance within a specific species is calculated by first summing up the coverage of all its associated reference sequences, and then normalizing this sum with respect to the estimated genome copies of the species, as determined by Melon33. This results in ARG abundance estimates expressed as “ARG copies per genome” (cpg), which is equivalent to “ARG copies per cell” (cpc) if each cell is assumed to contain a single genome10. The final output of Argo is an ARG abundance table, with different ARGs as rows and species as columns (Fig. 1c).
Benchmarking read-overlapping with other taxonomic assignment strategies
To benchmark the host identification performance of read-overlapping in relation to several existing methods, we simulated two samples with distinct read characteristics—one with higher mean quality score q but shorter mean read length l (HQ, q = 19.04, l = 5028), and one with lower accuracy but longer reads (LQ, q = 13.19, l = 10,365)—using the metagenomic mode of NanoSim34. Each sample comprised 1,000,000 reads from 25 widespread primary or opportunistic pathogens with even taxonomic abundance (Supplementary Table S2, “Methods”). We specifically extracted reads carrying ARGs and used them to assess whether various taxonomic assignment strategies could correctly identify their taxonomy. For evaluation, we focused on the percentage of misclassified reads. The types of misclassification include (1) true-positive (the misclassified species is within the 25 species), (2) false-positive (the misclassified species is not among the 25 species), and (3) unclassified (the read is either not utilized by the classifier or its classification does not reach species-level taxonomy).
As the performance of taxonomic classifiers hinges on their underlying databases35, we rebuilt the databases for Kraken222, Centrifuger36, MetaMaps37, MEGAN-LR38, and minimap224 exclusively using RefSeq complete genomes as of May 10, 2024, to ensure fairness (“Methods”). Regarding these methods, minimap2 refers to the native base-level alignment of ONT reads (preset ‘map-ont’). minimap2+BH retains a single best-hit (BH) for each read based on the alignment score and adopts its taxonomy. minimap2+EM reassigns the taxonomic labels of multi-mapped reads using the expectation-maximization (EM) algorithm. minimap2+RO is the taxonomic assignment strategy of Argo, where taxonomic labels are determined through read-overlapping (RO) and subsequent set covering. MEGAN-LR aggregates the alignments of minimap2 using a lowest common ancestor (LCA) algorithm called interval-union LCA. MetaMaps employs minimizer-based approximate mapping and EM for post-correction. Centrifuger and Kraken2 are both alignment-free methods. Centrifuger is a successor of Centrifuge20, designed to scale with large and growing databases. Kraken2 represents the most widely used taxonomic classifier in the field.
As shown in Fig. 2a–b, minimap2+RO clearly outperformed all other methods, achieving overall misclassification rates of 0.19% and 0.09% for HQ and LQ, respectively. Kraken2 performed poorly, with its misclassification rates varying strongly across species and reaching up to 16.63% for Mycobacterium tuberculosis in HQ. Unlike Kraken2, Centrifuger displayed reasonable performance, maintaining below 1.32% overall misclassification rates for both samples, despite also being alignment-free. MetaMaps was the only tool exhibiting higher accuracy for HQ (0.79% misclassified) than for LQ (1.64% misclassified), suggesting that its approximate mapping algorithm may be more sensitive to read accuracy than to read length and/or sequencing depth. MEGAN-LR also employs minimap2, but its LCA algorithm resulted in a large proportion of reads being unclassified at the species level, especially for Bacillus cereus (up to 46.09% unclassified and 46.78% misclassified). minimap2+BH demonstrated the second-lowest overall misclassification rates, with 0.55% for HQ and 0.27% for LQ, highlighting the advantages of base-level alignment over approximate and alignment-free methods. minimap2+EM did not perform as well as minimap2+BH for certain species. Although the additional EM step greatly reduced the number of false-positive misclassifications (HQ: 0.39% to 0.01%, LQ: 0.25% to 0.05%), it also inadvertently skewed the relative abundance estimates by introducing true-positive misclassifications (HQ: 0.16% to 0.79%, LQ: 0.02% to 0.43%) due to the presence of highly similar species, leading to overall misclassification rates above 0.47% for both HQ and LQ.

a Species-specific misclassification rate in percentage (mis. %). The 25 species represent the top 25 primary or opportunistic pathogens given by NCBI. All species have at least one ARG. b Overall misclassification rate in percentage. Colors indicate three different types of classification, including true-positive, false-positive, and unclassified. c Sequence cover (reference ARG sequence) in percentage. The lower and upper hinges of the boxplot correspond to the first quartile (Q1) and third quartile (Q3), respectively, while the center line represents the median. The whiskers extend to the smallest and largest values within Q1 − 1.5 × IQR (lower whisker) and Q3 + 1.5 × IQR (upper whisker), where IQR (interquartile range) is the difference between Q3 and Q1. Data outside this range are considered outliers and are displayed individually. The number of high-scoring segment pairs (HSPs) within length groups for HQ (LQ): 0–1k, n = 10,600 (19,935); 1–2k, n = 24,250 (43,566); 2–3k, n = 1177 (2103); >3k, n = 5725 (9521). Colors denote read types: raw reads (individual cover) and clustered reads (collective cover). Dashed gray lines represent 90% subject-cover cutoffs. d Cluster size distribution. Clusters with a single read were omitted. Sample statistics, including no. clusters (clu.), mean coverage (cov.), and mean size of clusters, are shown in tables. Dashed gray lines represent samples’ mean coverages. a–d Samples with high quality (HQ, q = 19.04, l = 5028) and low quality (LQ, q = 13.19, l = 10,365).
Notably, some species were considerably harder to classify. For instance, while all methods accurately identified Vibrio parahaemolyticus, most failed to classify Enterobacter cloacae without errors (Fig. 2a). This difficulty in classifying Enterobacter cloacae likely originated from the presence of a plasmid sequence (NZ_OW968330.1) in its genome that is shared by a wide range of species, including Klebsiella pneumoniae, Escherichia coli, Citrobacter freundii, and Enterobacter hormaechei. Resolving this ambiguity solely with metagenomics is unlikely to be feasible.
To better understand the roles of read-overlapping in facilitating host identification, we analyzed the alignment patterns and taxonomic compositions of reads within the derived clusters. For each cluster, reference ARG sequences—regardless of their lengths—could be adequately covered by reads collectively (Fig. 2c). This property aided in the elimination of spurious ARGs by applying a subject-cover cutoff (typically 90%), without compromising fragmented long reads that could not sufficiently cover reference sequences on their own. Additionally, the majority of read clusters containing at least two reads tended to match the sample’s mean coverage in size (Fig. 2d). This trend suggests a preference for clusters with unique ARG-species combinations, which is expected given that reads from the same genomic region generally exhibit lower sequence divergence and are more likely to be grouped together. Furthermore, 99.48% and 97.73% of these clusters achieved perfect species-level purity for HQ and LQ, respectively, confirming that read clusters successfully segregated reads into species-specific bins, thereby simplifying taxonomic classification.
Comparison between read-based, assembly-based and binning-based methods for ARG profiling
Assembly can enhance the effective length and accuracy of sequences by combining overlapping reads to construct longer contigs, and is a must when studying ARGs in the context of mobility39. However, it is well-established that assembly can lead to reduced sensitivity in ARG detection due to the requirement of a minimum coverage (average sequencing depth), typically 3×15. On the other hand, assembled contigs can be binned to form metagenome-assembled genomes (MAGs) for more consolidated taxonomic classification, but this may result in further information loss as contigs not selected for binning will be discarded. To compare read-based, assembly-based, and binning-based ARG profiling methods, we downsampled the two simulated samples (HQ and LQ) to achieve a range of coverages at 1×, 2×, 4×, 8×, 16×, and 32×. Reads were assembled into contigs using metaFlye40. Contigs were binned into MAGs using SemiBin241. Taxonomic labels for contigs and MAGs were obtained with Kraken222 and GTDB-Tk42, respectively (“Methods”).
Overall, the performance of all profiling methods improved with higher coverage, as evidenced by increased F1-scores and correlation coefficients, as well as decreased L1/L2 distances (Fig. 3a–d). Binning-based profiling (SemiBin2 + GTDB-Tk) consistently underperformed assembly-based profiling (metaFlye + Kraken2), which was expected given that the genomes of the 25 species were all present in Kraken2’s database, making the advantages of genome-centric taxonomic classification less apparent. Additionally, since the assemblies could be highly fragmented, with N50 values ranging from 15,689 bp (HQ, 1×) to 3,972,440 bp (LQ, 32×) and numbers of contigs from 2089 (HQ, 4×) to 72 (LQ, 32×), a large proportion of contigs were not properly binned due to their short lengths, resulting in considerably lower recall and, consequently, inaccurate ARG abundance estimates. We also note that binning these contigs might be particularly difficult because modern binning algorithms typically leverage differential abundance, in addition to sequence composition, to distinguish species, yet here, all species were simulated with equal taxonomic abundance (Supplementary Fig. S3).

a Expected and estimated ARG abundances (cpg). F1-scores at the type level (typ.) and subtype level (sub.) are summarized in tables. ARGs with unclassified taxonomy were not used in F1-score calculations. Dashed gray lines indicate 1:1 ratio lines. b Pearson correlation coefficient measuring the linear relationship between estimated and expected ARG abundances. c, d L1 (c) or L2 (d) distances measuring the deviation between estimated and expected ARG abundances. a–d Colors represent samples: HQ (q = 19.04, l = 5028) and LQ (q = 13.19, l = 10,365). Shapes of points and line types denote ARG profiling methods: read-based (Argo), assembly-based (metaFlye + Kraken2), and binning-based (SemiBin2 + GTDB-Tk).
Read-based profiling (Argo) featured much lower detection thresholds compared to assembly- and binning-based profiling, as reflected by its relatively high F1-scores (types: 0.811–0.935, subtypes: 0.692–0.892) at 1–2× (Fig. 3a). In contrast, assembly- and binning-based profiling hardly reached F1-scores above 0.5 at such low coverage, primarily due to their low recall. For coverage at 4× and above, read-based profiling maintained F1-scores exceeding 0.9 for both ARG types and subtypes, whereas assembly-based profiling required at least 8× and binning-based profiling more than 32× to achieve similar scores. This disparity likely stems from the inherent challenges in assembling and binning ARGs, which are frequently embedded in repetitive regions across diverse genomic contexts16. Moreover, although read-based profiling is known to be prone to false-positive predictions due to spurious alignments39, Argo’s precision remained high even at coverage down to 1× (types: 0.977–0.992, subtypes: 0.982–0.988). This reinforced the robustness of using read clusters for ARG profiling.
Regarding correlation coefficients and L1/L2 distances, read-based profiling proved less advantageous than assembly- and binning-based profiling owing to its susceptibility to intra-genomic sequencing depth variation (Fig. 3b–d). This variation (approximately Poisson assuming uniform read distribution) prevents read-based profiling from achieving perfect correlations and zero distances by introducing noise to estimates of both ARG copies (numerator of cpg) and genome copies (denominator of cpg). In real metagenomics where species can actively grow, this issue is expected to be further exacerbated due to the uneven sequencing depth caused by bidirectional DNA replication from the origin to the terminus43. Assembly- and binning-based methods, by contrast, are less affected by this variation and can directly yield ARG abundances in terms of cpg (assuming repeats can be resolved) without normalization. Here, while read-based profiling generally demonstrated the best performance at low coverage (1–2×), assembly-based profiling tended to overtake at 4–8× for both correlation coefficients and L1/L2 distances. Nevertheless, the differences between these two at high coverage (16–32×) were minimal, as indicated by their similar type-level correlation coefficients (assembly-based: 0.959–0.999, read-based: 0.986–0.993) and L2 distances (assembly-based: 20.833–2.646, read-based: 12.332–9.011). Subtypes exhibited slightly different performance patterns compared to types, but the trend of improvement with increased coverage held true.
Finally, we observed in this simulation experiment that more accurate reads did not necessarily guarantee improved performance for either assembly- or binning-based profiling (Fig. 3a–d). Both methods appeared to be more sensitive to read length than to read accuracy, especially at higher coverage, as suggested by their peak F1-scores (HQ: 0.959, LQ: 0.997). Conversely, while read-based profiling did show some improvement with increased read accuracy (HQ: 0.993, LQ: 0.978), this effect was likely marginal.
Performance evaluation using sequenced mock communities
We next investigated whether Argo works with real metagenomic samples. We applied Argo to four sequenced mock communities possessing different mean read lengths l (4771–10,365) and mean quality scores q (10.77–38.77), including Zymo D6300 (8 bacteria and 2 yeasts, ONT), Zymo D6331 (15 bacteria and 2 yeasts, ONT), ATCC MSA2006 (12 bacteria, ONT), and ATCC MSA1003 (20 bacteria, PacBio). Expected ARG abundances were obtained by aligning the reference genomes of these mocks (provided by the respective suppliers) to SARG+ and counting ARG occurrences, while estimated ARG abundances were directly output by Argo (Supplementary Table S3).
Argo performed reasonably well at the type level for all mock communities except D6331, with Pearson correlation coefficients of 0.672 for D6331, but 0.976, 0.981, and 0.990 for MSA2006, MSA1003, and D6300, respectively (Fig. 4, Supplementary Fig. S4). D6331 had a staggered distribution of species and a relatively low total number of genomes. Consequently, most species in this mock were of low coverage (less than 1×). ARGs of these rare species could hardly be detected, leading to a substantially low type-level recall of only 0.652. However, this issue is likely to improve with increased sequencing depth. For example, MSA1003, despite also having a staggered distribution, achieved a recall of 0.930 for ARG types. To verify whether increasing sequencing volume enhances recall, we deeply sequenced D6331 using a single PromethION flow cell (R10.4.1) with the latest chemistry (Kit 14, Q20+), yielding a total of 23.43 million reads (base-called with Dorado v7.2.13) and 128.4 Gbp. As expected, recall improved to 0.761, 0.826, and 0.913 at sequencing volumes of 4 Gbp, 64 Gbp, and 128 Gbp, respectively (Supplementary Fig. S5).

Expected and estimated ARG abundances (cpg). Sample statistics, including the total number of genomes (no. gen.), mean quality scores (q), and mean read lengths (l), are shown in the upper left tables. Precision (pr.) and recall (re.) for ARG types (typ.) and subtypes (sub.) are displayed in the lower right tables. Dashed gray lines indicate 1:1 ratio lines. Species names were manually adjusted to reflect updates in taxonomy and resolve inconsistencies between NCBI and GTDB. Schaalia odontolytica was renamed to Pauljensenia odontolytica (MSA1003), Lactobacillus fermentum to Limosilactobacillus fermentum (D6300 and D6331), Bacillus subtilis to Bacillus spizizenii (D6300), Fusobacterium nucleatum to Fusobacterium animalis (D6331), and Clostridium perfringens to Sarcina perfringens (D6331). MLS: macrolide-lincosamide-streptogramin.
Additionally, Argo’s precision remained above 0.9 for all these mocks at both the type and subtype levels, irrespective of the identity cutoff chosen for each sample, which ranged from 67.47 (median sequence divergence 0.0901, MSA2006) to 89.58 (median sequence divergence 0.0017, MSA1003). However, we did observe some increase in precision with respect to q. This improvement is reasonable given that the primary goal of using adaptive cutoffs is to minimize the deviation between estimated and expected ARG abundances. With a default cutoff of 90 − 2.5 × median sequence divergence in percentage, we aim to ensure that even reads of the lowest quality can be retained. As a result, it is inevitable to have false positives when reads exhibit a wide range of qualities, as indicated by an interdecile range (IDR) in sequence divergence of 0.046 for MSA2006 (q = 10.77) and 0.007 for MSA1003 (q = 38.77). On the other hand, completely eliminating false positives can also be challenging due to the presence of cross-species chimeric reads, as Argo assumes each read has a single taxonomy.
It is worth noting that Argo properly discerned all plasmid-associated ARGs, including ant(4’)-I, qacG, and tet(L) for D6300; sul2, erm(C), mupA, tet(K), and blaZ for MSA1003; aph(2”)-I, erm(B), and qacH for MSA2006, although the assigned taxonomic labels might not be fully correct due to the presence of shared plasmids across different species. Furthermore, some plasmid-borne ARGs, such as aph(2”)-I (4.256 cpg), erm(B) (2.524 cpg), and qacH (2.586 cpg) of Enterococcus faecalis (MSA2006), exhibited higher abundances compared to others. This confirms the multicopy nature of plasmids, which serves as a catalyst for the evolution of ARGs44.
Application to human and non-human primate fecal samples
Non-human primates are close evolutionary relatives of humans, sharing genetics, physiology, behavior, and social structures akin to those of humans45. These similarities make them excellent models for studying gut microbial communities and their roles in human health and disease46. Furthermore, the minimal exposure of these primates to antibiotics renders them ideal baselines for comparisons with their human counterparts. To examine the distribution patterns of ARGs in human and non-human (NH) primates, we downloaded fecal long-read metagenomic samples of 317 healthy humans from five countries spanning three continents: China (CN, n = 170)47,48, South Korea (KR, n = 5)49,50, Singapore (SG, n = 109)51, Germany (DE, n = 11)52, and the United States (US, n = 22)53,54, along with 12 NH primates (chimpanzees and bonobos) residing in the wild of equatorial Africa46. Using Argo, we generated ARG profiles for these samples (Supplementary Fig. S6).
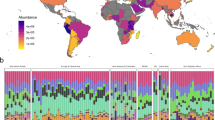
Overall, the estimated total ARG abundances in terms of cpg differed significantly between these datasets (Kruskal–Wallis, χ2 = 98.4, adjusted p < 2e−16), showing a decreasing trend from CN to NH (Fig. 5a). This trend, excluding NH, roughly mirrored the Human Development Index (HDI) rankings, corroborating a global-scale study observing an inverse proportionality between ARG abundances and HDI values (i.e., higher development resulting in lower ARG abundances) using sewage55. NH had distinctly lower cpg values than all countries, with adjusted p-values (Mann–Whitney) below 0.05 for all pairwise comparisons. Moreover, this difference was more pronounced for abundant and functional gut genera such as Blautia and Faecalibacterium than for Escherichia and Streptococcus, suggesting that the co-diversification of humans and their gut microbes may primarily contribute to increased ARGs in commensal rather than pathogenic lineages. Nonetheless, noting that NH primates are much less studied than humans, we cannot rule out the possibility that they harbor unique ARGs currently not covered by RefSeq and consequently not represented in SARG+. As such, the observed large differences may be exaggerated.

a Estimated ARG abundance (cpg). Pairwise comparisons (Mann–Whitney) between humans and NH primates are shown at the top. Overall group comparisons (Kruskal–Wallis) are shown at the bottom. The number of asterisks indicates the significance levels of p-values returned by these tests (after Bonferroni correction): *p < 0.05, **p < 0.01, ***p < 0.001. The lower and upper hinges of the boxplot correspond to the first quartile (Q1) and third quartile (Q3), respectively, while the center line represents the median. The whiskers extend to the smallest and largest values within Q1 − 1.5 × IQR (lower whisker) and Q3 + 1.5 × IQR (upper whisker), where IQR (interquartile range) is the difference between Q3 and Q1. Data outside this range are considered outliers and are displayed individually. b, c Relationship between ARG abundance and the mean genome size of prokaryotes (b), or the Shannon diversity index (c). Black lines depict regression lines, while gray ribbons represent 95% confidence intervals. Model fits of GAMs (generalized additive models), including R2 and p-values, are shown as text. d, e PCoA (principal coordinate analysis) of the estimated total ARG abundances with respect to genera (d), or ARG types (e). The five most abundant genera or ARG types and their projections are shown by arrows. R2 and p-values of PERMANOVA (permutational multivariate analysis of variance) are shown as text. f Abundances of different ARG types for E. coli across countries and NH primates. Colors indicate the number of unique subtypes within ARG types. e, f MLS: macrolide-lincosamide-streptogramin. g Abundances of sul and mcr for E. coli across countries and NH primates. h Network analysis with sul and mcr. Edge widths represent Spearman’s rank correlation coefficients. Coefficients below 0.1 are not displayed. Pathogens include Enterobacter kobei, Enterobacter roggenkampii, Klebsiella pneumoniae, and Klebsiella quasipneumoniae.
We then attempted to identify potential causes of this difference between humans and NH primates. Exploratory analysis revealed that total ARG abundances within the gut were positively correlated with the mean genome size of prokaryotes (generalized additive model, R2 = 0.187, F = 8.1, p < 2e−16) and negatively correlated with the Shannon diversity index (R2 = 0.593, F = 51.5, p < 2e−16) (Fig. 5b–c). NH possessed a relatively low average genome size but markedly high diversity. The former implies NH primates harbor more specialists than generalists in terms of gut microbiota56, as larger genome sizes are typically associated with a broader range of functional traits, possibly including antibiotic resistance57. The latter reflects the loss of ancestral microbial taxa and core microbial biodiversity in humans58,59, a phenomenon that likely contributes to the development of antibiotic resistance, as species diversity is known to function as an ecological barrier to ARGs60.
To further test whether the difference is primarily associated with the carriers of ARGs or ARGs themselves, we decomposed the estimated total ARG abundances with respect to genera and ARG types using principal coordinate analysis. Interestingly, ARG types (permutational multivariate analysis of variance, R2 = 0.373, F = 38.5, p = 1e−4) explained much more variance compared to genera (R2 = 0.169, F = 13.2, p = 1e−4) (Fig. 5d–e). This suggests that the widespread use of antibiotics in modern medicine and agriculture, which developed in the post-industrial era, may lead to selective pressures within genera, favoring antibiotic-resistant species despite their similar functional roles compared to other species of the same genus. This evolutionary process in the gut microbiota over the long term may also provide evidence that human-associated species tend to have larger genome sizes, as discussed above. Furthermore, ARG types that contain a high proportion of antibiotic inactivators (which are known for their high mobility) were found to be enriched in humans61. Examples included aminoglycoside (humans: 0.224 cpg, NH primates: 0.007 cpg) and beta-lactam (humans: 0.412 cpg, NH primates: 0.005 cpg).
In addition to the difference in overall ARG abundances, it is also of interest to investigate whether the same species carry identical ARGs in both humans and NH primates. To verify this, we used E. coli as an indicator and computed its type-level ARG abundances across different datasets. Strikingly, we observed strong geographical variations among countries, as well as clear distinctions between humans and NH primates (Fig. 5f). CN exhibited the most diverse ARG types, with all but streptothricin detected. Some ARG types were highly country-specific. For example, rifamycin, quinolone, fosfomycin, and bleomycin were only identified in CN and SG, whereas streptothricin was exclusive to KR and US. The number of subtypes within each ARG type also varied across countries. For instance, aminoglycoside in CN had the largest number of subtypes detected (n = 12), whereas US the smallest (n = 1). Although this pattern could be influenced by the uneven sequencing efforts among countries, it implies CN may harbor more variants of ARGs within the samples collected. Notably, NH covered the fewest ARG types. ARGs against most man-made antibiotics, including quinolone, phenicol, and sulfonamide, were absent from NH.
Using two common acquired ARGs of E. coli, sul and mcr, as examples, we observed a widespread distribution of sul2 across countries, with the highest abundances (0.705 cpg) in CN and the lowest (0.063 cpg) in KR (Fig. 5g). In contrast, mcr-6 was detected only in SG but not in CN, despite CN having the largest sample size. All sul and mcr were absent from NH primates. We then conducted a simple network analysis using sul and mcr. Interestingly, we observed positive correlations between pathogens (including E. coli) and non-pathogens, indicating the potential for HGT between these species in the gut (Fig. 5h). This finding aligns with existing evidence that commensals can serve as reservoirs for ARG dissemination, underscoring the importance of studying the gut microbiota as a whole62.
Discussion
Argo provides a fast and robust framework for quantifying ARGs and tracking their hosts in complex metagenomes. By combining the merits of both read-based and assembly-based profiling methods through long-read overlapping (the first step of overlap-layout-consensus assembly), Argo enables accurate ARG abundance estimation and host identification without the computational overhead of full genome assembly. This integration not only reduces the chance of false-positive ARG predictions due to spurious alignments arising from local sequence homology, but also aids in correct species labeling with aggregated evidence collected from local read clusters. Using simulated, synthetic, and real samples, we demonstrate Argo’s superior performance in various settings.
Argo uses base-level alignment to determine the hosts of ARG-containing reads. Though accurate, base-level alignment is known to be computationally intensive compared to alignment-free methods. To resolve this, we implement Argo following a two-stage taxonomic classification scheme. First, since most reads do not carry ARGs, filtering these ARG-free reads out effectively bypasses unnecessary alignments. Second, considering that the median read length from current sequencing platforms (e.g., ONT or PacBio) for metagenomic samples is frequently below 10,000 bp, using reference sequences of this maximum length allows an over 100-fold compression of GTDB (from more than 1.5 Tbp to less than 15 Gbp). This combination overcomes the computational bottleneck associated with base-level alignment, making detailed ARG profiling feasible on a standard laptop computer.
Here, we specifically opt for the full collection of GTDB due to its comprehensiveness, despite its inherent redundancy. This redundancy is crucial since many ARGs, especially the acquired ones, are not covered by species-level representative genomes. Within species, types and copies of ARGs can vary substantially, and these variations typically lead to different AMR phenotypes. Additionally, Argo’s framework is not exclusive to ARGs in metagenomics. For other relevant genes, such as the β-subunit of RNA polymerase rpoB63, the origin of replication oriC64, or other universal single-copy marker genes65, the framework does apply. The only requirement is to construct a customized database for taxonomic classification.
In our simulation experiment, we compared Argo with two commonly used ARG profiling approaches: assembly-based (metaFlye + Kraken2) and binning-based (SemiBin2 + GTDB-Tk). Argo demonstrated consistently good performance across a range of coverages, from low (1×) to high (32×). However, assembly-based and binning-based methods may offer advantages in real-world settings where species can exhibit differential abundance and remain uncharacterized. These methods may be better suited for de novo identification of ARGs’ genomic contexts and hosts, particularly in new, understudied environments. In contrast, Argo is inherently reference-based and may face classification issues with unseen species or novel plasmid syntenies. Nevertheless, the simple structure of Argo’s reference databases (plain sequences) allows for straightforward expansion by incorporating user-specific reference sequences (derived from MAGs), in addition to those from GTDB and RefSeq. This flexibility enhances Argo’s utility, supporting effective ARG profiling even in complex metagenomes with many unknowns.
Argo is a read-based profiler, which implies challenges in distinguishing single nucleotide polymorphisms (SNPs) from sequencing errors. As a consequence, ARGs originating from point mutations are consistently excluded from its database. Likewise, highly similar ARGs are clustered to avoid potential ambiguities. For instance, blaOXA-1 and blaOXA-1042, which differ by only one amino acid, are grouped into a single ARG subtype, blaOXA. This clustering lowers the resolution of ARGs and could be a drawback for applications requiring open reading frame (ORF)-based annotation or detailed sequence typing.
Another potential limitation of Argo lies in its detection threshold. By default, Argo considers ARGs of species with less than one genome copy (1×) as “unclassified”. This masking is reasonable, given that insufficient coverage can not only lead to unstable normalization but also hinder accurate taxonomic classification due to inadequate read overlaps and less effective read clustering. Such a high detection threshold makes Argo less suitable for profiling ARGs of extremely rare species, similar to other tools for metagenomic analysis. Nevertheless, with ongoing advancements in long-read sequencing technologies, including both increased sequencing depth and decreased sequencing cost, this limitation is expected to diminish in the near future.
In conclusion, as the integration of long-read sequencing into environmental surveillance of ARGs continues to grow, we believe that Argo will help standardize ARG quantification and enhance our ability to trace their origins and dissemination pathways, ultimately contributing to tackling the global health threat posed by AMR.
Methods
Database construction
SARG+
SARG+ was constructed by combining protein reference sequences from CARD (ver. 3.2.9), NDARO (ver. 2024-05-02.2), and SARG (ver. 3.2.1). Sequences were manually curated to ensure they follow a consistent hierarchy: type (class/family), subtype (subclass/gene), and sequence. Sequences that potentially belong to more than one subtype were labeled with an asterisk. For example, cml* represents sequences of either cmlA or cmlB. Regulators (e.g., activators and repressors of ARGs), housekeeping genes, and ARGs that arise from point mutations were removed from the list. This resulted in a reference database containing 39 types, 1039 subtypes, and 7041 sequences. The database was subsequently augmented by including all RefSeq protein sequences annotated with the same evidence, specifically BlastRules or Hidden Markov Models from the NCBI Prokaryotic Genome Annotation Pipeline (PGAP), as those for experimentally validated ARGs26, leading to an extension database containing 476,546 sequences. After deduplication via clustering with MMseqs2 v15.6f45266 (reference: ‘easy-cluster -s 7.5 -c 0.995 --min-seq-id 0.995 --cov-mode 0 --cluster-reassign’, extension: ‘easy-cluster -s 7.5 -c 0.95 --min-seq-id 0.95 --cov-mode 0 --cluster-reassign’), the final database contained 104,529 sequences, constituting 39 types and 1053 subtypes, with 81 of these subtypes classified as Risk Rank I.67.
ARG-tailored GTDB
We downloaded 596,663 assemblies (GTDB R09-RS220, including 107,235 bacterial and 5869 archaeal species) from NCBI genomes (https://ftp.ncbi.nlm.nih.gov/genomes/). At the time of download, 196 assemblies had been deprecated by NCBI and were therefore excluded. Assemblies were aligned to SARG+ using the frameshift alignment mode of DIAMOND v2.1.823 with an e-value cutoff of 10−15, an identity cutoff of 90%, and a subject-cover cutoff of 90% (‘blastx --evalue 1e-15 --id 90 --subject-cover 90 --range-culling --frameshift 15 --range-cover 25 --max-target-seqs 25 --max-hsps 0’). The resulting high-scoring segment pairs (HSPs) were filtered using in-house scripts. Specifically, for each query sequence (contig), we sorted its HSPs by e-value in ascending order, and then iteratively added HSPs to a collection if and only if they exhibited less than 25% pairwise query range overlaps (computed as percentages of the shorter ranges involved) with any HSPs already in the collection. It is important to note that we allowed multiple copies of a specific reference sequence to be detected by setting ‘--max-hsp 0’. For each of the filtered HSPs, a sequence of length 10,000 bp (left and right 5000 bp flanking regions of the HSP’s center) was extracted using SeqKit v2.8.268 (‘subseq’), wherever possible, yielding a total of 6,990,421 ARG-containing sequences. To further reduce redundancy, these ARG-containing sequences were clustered by ARG subtypes and species at an identity cutoff of 0.9995 and a cover cutoff of 0.9995 with MMseqs2 (‘easy-cluster -s 7.5 -c 0.9995 --min-seq-id 0.9995 --cov-mode 1 --cluster-reassign’), resulting in a reference taxonomy database comprising 1,394,827 unique sequences.
Plasmid database
47,163 complete genome or chromosome-level assemblies were retrieved from NCBI RefSeq as of June 30, 2024. Sequences were considered candidate plasmid sequences if their headers contained keywords “plasmid” or “megaplasmid”. To reduce the number of falsely labeled sequences, we used geNomad v1.8.069 to reclassify these candidate sequences (‘end-to-end --disable-find-proviruses’). Sequences classified as chromosomes were directly discarded, while those as viruses were retained only if they carried at least one plasmid hallmark and their plasmid scores were at least twice their chromosome scores. 10,000 bp ARG-containing sequences were obtained from the filtered plasmid sequences using the same method as above (see section “ARG-tailored GTDB” for more details), with the exception that sequences of circular topology were concatenated before extraction, as many plasmids were much shorter than 10,000 bp. We reran geNomad on these extracted sequences using permissive cutoffs (‘--min-virus-marker-enrichment -100 --min-plasmid-marker-enrichment -100 --max-uscg 0 --min-score 0 --disable-find-proviruses’) since we observed that some sequences were in fact chimeras of plasmids and chromosomes and could not be properly discerned when considered as a whole. 75,994 sequences with plasmid scores greater than two times their chromosome scores were kept for further analysis. After clustering with MMseqs2 (‘easy-cluster -s 7.5 -c 0.9995 --min-seq-id 0.9995 --cov-mode 1 --cluster-reassign’), 39,598 plasmid sequences were obtained.
Argo implementation
Extraction of ARG-containing reads
Argo identifies ARG-containing reads again using DIAMOND’s frameshift alignment mode and SARG+, albeit without direct identity and subject-cover cutoffs, as reads may be short and vary in accuracy (‘blastx --evalue 1e-15 --range-culling --frameshift 15 --range-cover 25 --max-target-seqs 25 --max-hsps 0’). These reads are subsequently overlapped using the approximate mapping of minimap2 v2.2824 (‘-x ava-ont’). By default, Argo employs an adaptive identity cutoff inferred from the ‘dv’ tag (approximate per-base sequence divergence) of overlaps. Assuming reads are indexed by \(i,{i}^{\, {\prime} }\in \{1,\ldots,n\}\), \({{{\rm{dv}}}}_{i,{i}^{\, {\prime} }}\) represents the sequence divergence between reads i and \({i}^{\, {\prime} }\) as returned by minimap2, where \({{{\rm{dv}}}}_{i,{i}^{\, {\prime} }}=1\) if reads i and \({i}^{\, {\prime} }\) do not overlap. We define \(\,{{\rm{dv}}}\,=\left[{{{\rm{dv}}}}_{i,{i}^{\, {\prime} }}| {{{\rm{dv}}}}_{i,{i}^{\, {\prime} }}\ne 1\right]\) and set the identity cutoff as \(100\times \left(0.9-\left(2.5 \times {{\rm{median}}}\left({{\rm{dv}}}\right)\right)\right)\). Overlaps for which \({{{\rm{dv}}}}_{i,{i}^{\, {\prime} }} \, > \,\max \left(2.5\times{{\rm{median}}}\left({{\rm{dv}}}\right),0.05\right)\) are discarded, since they may arise from alignments across similar species. The filtered overlaps are used later for graph clustering (“Graph clustering”).
Collection of host information
Reads containing at least one ARG are mapped to the ARG-tailored GTDB for taxonomic classification using the base-level alignment of minimap2 (‘-cx map-ont -f 0 -N 2147483647 -p 0.9’). Alignments are sorted by the alignment score (AS) in descending order. For each read-species combination, only the top alignment is recorded. Assuming species are indexed by \(j,{j}^{{\prime} }\in \{1,\ldots,m\}\), an alignment between read i and species j is considered valid, i.e., ai,j = 1, if its alignment score ASi,j meets the criterion of \(\max ({{{\rm{AS}}}}_{i,j} \div 0.995,{{{\rm{AS}}}}_{i,j}+50)\, > \,{\max }_{{j}^{{\prime} }}{{{\rm{AS}}}}_{i,{j}^{{\prime} }}\), and ai,j = 0 otherwise. With this setup, we define species sets sj as sj = {i∣ai,j = 1} ⊆ {1, …, n}, which are used later for set covering (“Set covering”).
Graph clustering
The overlap graph is clustered using the MCL algorithm, which exploits random walks to simulate flow within the graph and identifies clusters as regions of high flow density32. Briefly, we construct an n × n matrix \({{{\bf{X}}}}={({x}_{i,{i}^{\, {\prime} }})}_{1\le i,{i}^{\, {\prime} }\le n}\) using the overlaps obtained above, where \({x}_{i,{i}^{\, {\prime} }}=1-{{{\rm{dv}}}}_{i,{i}^{\, {\prime} }}\) if \(i\ne {i}^{\, {\prime} }\), and \({x}_{i,{i}^{\, {\prime} }}=1\) otherwise. X is then converted into a stochastic matrix Z, which represents the transition probabilities for random walks on the graph, through column-wise L1 normalization:
Z undergoes two operations, expansion and inflation, until convergence:
-
1.
Expansion—raise Z to the power of e (default e = 2), which simulates taking e steps in a random walk:
$${{{\bf{Z}}}}={{{{\bf{Z}}}}}^{e}.$$(2) -
2.
Inflation—raise each element of Z to the power of r (default r = 2) and renormalize:
$${z}_{i,{i}^{\, {\prime} }}=\frac{{({z}_{i,{i}^{\, {\prime} }})}^{r}}{{\sum }_{i}{({z}_{i,{i}^{\, {\prime} }})}^{r}}.$$(3)
At the end of each iteration, values close to zero, i.e., \({z}_{i,{i}^{\, {\prime} }}\, < \,\frac{1}{n}\), are pruned for speed. After convergence, clusters are extracted from the final matrix by identifying non-zero blocks. These blocks, where matrix entries remain unpruned, indicate high probabilities of reads staying connected within the same cluster during random walks.
Set covering
With species sets and read clusters defined, we assign taxonomic labels to reads on a per-cluster basis by solving a weighted set cover problem using a greedy heuristic. Specifically, given a universe of reads U ⊆ {1, …, n}, a collection of species sets S = {s1, …, sm}, and a weight function w(sj) = − ∑iASi,j, we iterate:
-
1.
Initialize \(C\leftarrow {{\emptyset}}\) and T ← U.
-
2.
While \(T \, \ne \, {{\emptyset}}\) and there exists sj ∈ S⧹C such that \({s}_{j}\cap T \, \ne \, {{\emptyset}}\):
-
a.
Select sj from S⧹C with the minimal weight, i.e., \({s}_{j}=\arg {\min }_{{s}_{{j}^{{\prime} }}\in S\setminus C}-{\sum }_{i\in {s}_{{j}^{{\prime} }}\cap T}{{{\rm{AS}}}}_{i,{j}^{{\prime} }}\).
-
b.
Update C ← C ∪ {sj} and T ← T⧹sj.
-
a.
C represents possible species of a cluster. If \(\left\vert C\right\vert\, > \,1\), a post-refinement is conducted by reassessing the alignment scores of species ASi,j, where j ∈ C. This gives the final taxonomic assignments of reads. Within each cluster, reads with the same taxonomic assignments are considered plasmid if over 50% of them map to the plasmid database. We also remove HSPs if their reference sequences are covered by less than 90% by all reads jointly in that cluster. After this, redundant HSPs are filtered on a per-read basis using the same scripts as before (see section “ARG-tailored GTDB” for more details), yielding a final list of ARGs carried by reads.
Simulation experiment
Taxonomic classifiers and their databases
To ensure fair comparisons, we rebuilt the databases for all taxonomic classifiers using RefSeq complete genomes (collected on May 10, 2024), which contained 588 archaeal and 40,461 bacterial assemblies. Kraken2’s database (v2.1.322) was built with ‘kraken2-build’ (note that we modified script ‘rsync_from_ncbi.pl’ to force it to download only complete genome assemblies). Centrifuger’s database (v1.0.136) was built with ‘centrifuger-download’ and ‘centrifuger-build’. MetaMaps’s database (v0.137) was built with script ‘buildDB.pl’. minimap2’s database (v2.2824) was constructed using ‘-x map-ont -d’ (the database of minimap2 was shared by minimap2+BH, minimap2+EM, minimap2+RO, and MEGAN-LR v6.25.938). All tools were run with default settings, except for MetaMaps, where we set ‘--maxmemory 20’ to reduce its peak memory usage.
Simulation
We generated two error models, high-quality (HQ) and low-quality (LQ), using script ‘read_analysis’ (‘metagenome’) from NanoSim v3.1.034. These error models were specifically trained on Zymo D6331 for HQ and Zymo D6300 for LQ, respectively (see section “Performance evaluation using sequenced mock communities” for more details). Reference genomes of the top 25 pathogens were sourced from NCBI and all present in RefSeq complete genomes as of May 10, 2024. Using the two error models and script ‘simulator’ (‘metagenome’) from NanoSim, we simulated samples HQ and LQ, each containing the 25 species at even taxonomic abundance and comprising 1,000,000 reads.
Assembly
HQ and LQ were downsampled to achieve expected coverages at 1×, 2×, 4×, 8×, 16×, and 32×. Reads from each downsampled set were assembled into contigs using metaFlye v2.9.340 (‘--nano-raw --meta’). These assembled contigs were subsequently binned into MAGs using Semibin2 v2.1.041 (‘single_easy_bin --sequencing-type=long_read’). Taxonomic labels for contigs and MAGs were predicted with Kraken2 v2.1.322 and GTDB-Tk v2.4.042 (‘classify_wf’), respectively. Since GTDB and NCBI can have inconsistencies in taxonomy, we manually converted GTDB taxonomy into NCBI taxonomy for the 25 species to facilitate comparison.
Expected and estimated ARG abundances
Expected ARG abundances were computed by (1) mapping the reference genomes of the 25 species to SARG+ using DIAMOND (‘blastx --evalue 1e-15 --id 90 --subject-cover 90 --range-culling --frameshift 15 --range-cover 25 --max-target-seqs 25 --max-hsps 0’), (2) filtering HSPs with previously described scripts (see section “ARG-tailored GTDB” for more details), and (3) counting ARG occurrences. Estimated ARG abundances were provided by Argo v0.1.0, which normalizes ARG copies relative to genome copies, resulting in ARG abundance estimates expressed as “ARG copies per genome” (cpg). We set ‘--plasmid -z 0’ to ensure plasmids and species with low coverage (less than 1×) were taxonomically classified.
Evaluation metrics
Misclassification rate, defined as the percentage of reads where the assigned taxonomic labels differ from the ground truths, was employed for comparing taxonomic assignment strategies. Three types of misclassification were considered: (1) true-positive (within-species misclassification, where the misclassified species is within the 25 species), (2) false-positive (out-of-species misclassification, where the misclassified species is not among the 25 species), and (3) unclassified (the read is either not utilized by the classifier or its classification fails to reach species-level taxonomy).
For evaluation of ARG profiling methods, we used precision, recall, F1-score, L1/L2 distances, and Pearson correlation coefficient. These metrics were assessed at either the type or subtype level of ARG abundances. Precision is defined as the ratio of true positives to the sum of true positives and false positives, whereas recall is defined as the ratio of true positives to the sum of true positives and false negatives. F1-score is the harmonic mean of precision and recall. Note that in this context, the definition of positive differs from the one mentioned above. Here, a true positive refers to the correct identification of both a species and its specific ARG type/subtype, while a true negative refers to the correct exclusion of both a species and its specific ARG type/subtype. L1/L2 distances and Pearson correlation coefficient are measures that describe the deviations or linear relationships between expected and estimated ARG profiles. Their definitions are provided elsewhere35.
Metagenome experiment
Sample quality control and preprocessing
All samples were quality-controlled using Porechop v0.2.470 (‘--discard_middle’) and nanoq v0.10.071 (‘--min-qual 10 --min-len 1000’). For mock communities, we mapped reads back to their reference genomes using minimap2 (‘-ax map-ont --secondary=no’). Reads that mapped to yeasts or remained unmapped were discarded with customized scripts. Sequencing depth at all genomic locations were determined for each species using samtools v1.1872 (‘sort’ and ‘depth -J -a’). To avoid the influence of multi-copy plasmids, we considered the coverage (average sequencing depth) of each species’ longest contig as its expected genome copy. The total number of genomes of a sample was obtained by summing up the expected genome copies of all its species. Plasmid sequences within the reference genomes of these mock communities were identified using geNomad (‘end-to-end --disable-find-proviruses’). ARG profiles were computed and evaluated using the same methods as before (see sections “Expected and estimated ARG abundances” and “Evaluation metrics” for more details).
Genome copies, relative abundances, average genome sizes, and Shannon diversity indices
Species-level, genus-level, and total genome copies of the fecal samples were estimated using Melon v0.2.033, with Kraken2’s PlusPF database (ver. 2024-06-05) for pre-filtering non-prokaryotic reads. Species- and genus-level relative abundances were computed by normalizing their respective genome copies to the total number of genome copies (sum-scaled to one). Mean genome sizes of prokaryotes were calculated by dividing the total number of bases (excluding contributions from humans and other non-prokaryotes) by the total number of genome copies. Shannon diversity indices were computed using in-house scripts, with species-level relative abundances as input73.
Statistical analyzes
All statistical analyzes were carried out in R v4.3.174. Principal coordinate analysis (PCoA) and permutational multivariate analysis of variance (PERMANOVA) were performed using functions ‘wcmdscale’ and ‘adonis2’ from package ‘vegan’ v2.6-475, with Bray–Curtis dissimilarities of genus-level relative abundances or type-level ARG abundances as input and 9999 permutations. Mann–Whitney and Kruskal–Wallis tests were conducted using functions ‘wilcox.test’ and ‘kruskal.test’, respectively. Generalized additive models (GAMs) were fitted using ‘gam’ from package ‘mgcv’76 with penalized cubic regression splines (‘cs’) as the smoothing basis. ARG abundances were log10-transformed to reduce skewness and ensure normality where necessary. All figures were generated with ‘ggplot2’ v3.4.377. Results were deemed statistically significant if p < 0.05. No multiple testing correction was applied unless otherwise stated.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Deep ONT Q20+ sequencing of Zymo D6331 is uploaded to NCBI Sequence Read Achieve (SRA) under BioProject ID PRJNA1181840. Other sequenced mock communities, including Zymo D6331 (ONT), ATCC MSA1003 (PacBio), and ATCC MSA2006 (ONT) are available under BioProject IDs PRJNA1028177, PRJNA546278, and PRJNA508395, respectively. Zymo D6300 (ONT) can be downloaded from https://lomanlab.github.io/mockcommunity/r10.html. Human and non-human primate fecal samples can be found under BioProject IDs PRJNA820119 (CN), PRJNA763692 (CN), PRJDB8879 (KR), PRJNA798244 (KR), PRJEB49168 (SG), PRJNA929328 (DE), PRJNA940499 (US), PRJNA508395 (US), and PRJNA842693 (NH). Reference genomes of the top 25 pathogens and the two simulated samples (HQ and LQ) are deposited at Zenodo (https://doi.org/10.5281/zenodo.13283127).
Code availability
Argo is available under the MIT License on GitHub (https://github.com/xinehc/argo) and can be installed via Bioconda (https://anaconda.org/bioconda/argo). SARG+ is available at https://github.com/xinehc/sarg-curation. Scripts (Jupyter notebooks) for reproducing the source data of Figs. 2–5 can be found at https://github.com/xinehc/argo-evaluation. Detailed instructions on building and extending the reference taxonomy & plasmid databases are available at https://github.com/xinehc/argo-supplementary. Argo v0.1.0’s source code is deposited at https://doi.org/10.5281/zenodo.1483730178.
References
Darby, E. M. et al. Molecular mechanisms of antibiotic resistance revisited. Nat. Rev. Microbiol. 21, 280–295 (2023).
Naghavi, M. et al. Global burden of bacterial antimicrobial resistance 1990–2021: a systematic analysis with forecasts to 2050. Lancet 404, 1199–1226 (2024).
Mao, X. et al. Standardization in global environmental antibiotic resistance genes (args) surveillance. Crit. Reviews Environ. Sci. Technol. 54, 1633–1650 (2024).
Burnham, C.-A. D., Leeds, J., Nordmann, P., O’Grady, J. & Patel, J. Diagnosing antimicrobial resistance. Nat. Rev. Microbiol. 15, 697–703 (2017).
Larsson, D. & Flach, C.-F. Antibiotic resistance in the environment. Nat. Rev. Microbiol. 20, 257–269 (2022).
Djordjevic, S. P. et al. Genomic surveillance for antimicrobial resistance—a one health perspective. Nat. Rev. Genet. 25, 142–157 (2024).
Alcock, B. P. et al. Card 2023: expanded curation, support for machine learning, and resistome prediction at the comprehensive antibiotic resistance database. Nucleic acids Res. 51, D690–D699 (2023).
Feldgarden, M. et al. Curation of the amrfinderplus databases: applications, functionality and impact. Microb. Genomics 8, 000832 (2022).
Yin, X. et al. Args-oap v3. 0: Antibiotic-resistance gene database curation and analysis pipeline optimization. Engineering 27, 234–241 (2023).
Yin, X. et al. Toward a universal unit for quantification of antibiotic resistance genes in environmental samples. Environ. Sci. Technol. 57, 9713–9721 (2023).
Cuadrat, R. R., Sorokina, M., Andrade, B. G., Goris, T. & Davila, A. M. Global ocean resistome revealed: exploring antibiotic resistance gene abundance and distribution in tara oceans samples. Gigascience 9, giaa046 (2020).
Zheng, D. et al. Global biogeography and projection of soil antibiotic resistance genes. Sci. Adv. 8, eabq8015 (2022).
Danko, D. et al. A global metagenomic map of urban microbiomes and antimicrobial resistance. Cell 184, 3376–3393 (2021).
Prieto Riquelme, M. V. et al. Demonstrating a comprehensive wastewater-based surveillance approach that differentiates globally sourced resistomes. Environ. Sci. Technol. 56, 14982–14993 (2022).
Lee, K. et al. Population-level impacts of antibiotic usage on the human gut microbiome. Nat. Commun. 14, 1191 (2023).
Abramova, A., Karkman, A. & Bengtsson-Palme, J. Metagenomic assemblies tend to break around antibiotic resistance genes. BMC genomics 25, 959 (2024).
Agustinho, D. P. et al. Unveiling microbial diversity: harnessing long-read sequencing technology. Nat. Methods 21, 954–966 (2024).
Portik, D. M., Brown, C. T. & Pierce-Ward, N. T. Evaluation of taxonomic classification and profiling methods for long-read shotgun metagenomic sequencing datasets. BMC Bioinforma. 23, 541 (2022).
Dai, D. et al. Long-read metagenomic sequencing reveals shifts in associations of antibiotic resistance genes with mobile genetic elements from sewage to activated sludge. Microbiome 10, 20 (2022).
Kim, D., Song, L., Breitwieser, F. P. & Salzberg, S. L. Centrifuge: rapid and sensitive classification of metagenomic sequences. Genome Res. 26, 1721–1729 (2016).
Yang, Y. et al. Qmra of beach water by nanopore sequencing-based viability-metagenomics absolute quantification. Water Res. 235, 119858 (2023).
Wood, D. E., Lu, J. & Langmead, B. Improved metagenomic analysis with kraken 2. Genome Biol. 20, 1–13 (2019).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using diamond. Nat. methods 12, 59–60 (2015).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Needham, B. D. & Trent, M. S. Fortifying the barrier: the impact of lipid a remodelling on bacterial pathogenesis. Nat. Rev. Microbiol. 11, 467–481 (2013).
Li, W. et al. Refseq: expanding the prokaryotic genome annotation pipeline reach with protein family model curation. Nucleic acids Res. 49, D1020–D1028 (2021).
Parks, D. H. et al. Gtdb: an ongoing census of bacterial and archaeal diversity through a phylogenetically consistent, rank normalized and complete genome-based taxonomy. Nucleic acids Res. 50, D785–D794 (2022).
Wang, X. et al. Inter-plasmid transfer of antibiotic resistance genes accelerates antibiotic resistance in bacterial pathogens. ISME J. 18, wrad032 (2024).
O’Leary, N. A. et al. Reference sequence (refseq) database at ncbi: current status, taxonomic expansion, and functional annotation. Nucleic acids Res. 44, D733–D745 (2016).
Enault, F. et al. Phages rarely encode antibiotic resistance genes: a cautionary tale for virome analyses. ISME J. 11, 237–247 (2017).
Nayfach, S. et al. Metagenomic compendium of 189,680 dna viruses from the human gut microbiome. Nat. Microbiol. 6, 960–970 (2021).
Enright, A. J., Van Dongen, S. & Ouzounis, C. A. An efficient algorithm for large-scale detection of protein families. Nucleic acids Res. 30, 1575–1584 (2002).
Chen, X. et al. Melon: metagenomic long-read-based taxonomic identification and quantification using marker genes. Genome Biol. 25, 226 (2024).
Yang, C. et al. Characterization and simulation of metagenomic nanopore sequencing data with meta-nanosim. GigaScience 12, giad013 (2023).
Sun, Z. et al. Challenges in benchmarking metagenomic profilers. Nat. methods 18, 618–626 (2021).
Song, L. & Langmead, B. Centrifuger: lossless compression of microbial genomes for efficient and accurate metagenomic sequence classification. Genome Biol. 25, 106 (2024).
Dilthey, A. T., Jain, C., Koren, S. & Phillippy, A. M. Strain-level metagenomic assignment and compositional estimation for long reads with metamaps. Nat. Commun. 10, 3066 (2019).
Huson, D. H. et al. Megan-lr: new algorithms allow accurate binning and easy interactive exploration of metagenomic long reads and contigs. Biol. direct 13, 1–17 (2018).
Gupta, C. L., Tiwari, R. K. & Cytryn, E. Platforms for elucidating antibiotic resistance in single genomes and complex metagenomes. Environ. Int. 138, 105667 (2020).
Kolmogorov, M. et al. metaflye: scalable long-read metagenome assembly using repeat graphs. Nat. methods 17, 1103–1110 (2020).
Pan, S., Zhao, X.-M. & Coelho, L. P. Semibin2: self-supervised contrastive learning leads to better mags for short-and long-read sequencing. Bioinformatics 39, i21–i29 (2023).
Chaumeil, P.-A., Mussig, A. J., Hugenholtz, P. & Parks, D. H. Gtdb-tk: a toolkit to classify genomes with the genome taxonomy database. Bioinformatics 36, 1925–1927 (2020).
Brown, C. T., Olm, M. R., Thomas, B. C. & Banfield, J. F. Measurement of bacterial replication rates in microbial communities. Nat. Biotechnol. 34, 1256–1263 (2016).
San Millan, A., Escudero, J. A., Gifford, D. R., Mazel, D. & MacLean, R. C. Multicopy plasmids potentiate the evolution of antibiotic resistance in bacteria. Nat. Ecol. evolution 1, 0010 (2016).
Manara, S. et al. Microbial genomes from non-human primate gut metagenomes expand the primate-associated bacterial tree of life with over 1000 novel species. Genome Biol. 20, 1–16 (2019).
Sanders, J. G. et al. Widespread extinctions of co-diversified primate gut bacterial symbionts from humans. Nat. Microbiol. 8, 1039–1050 (2023).
Chen, L. et al. Short-and long-read metagenomics expand individualized structural variations in gut microbiomes. Nat. Commun. 13, 3175 (2022).
Jin, H. et al. A high-quality genome compendium of the human gut microbiome of inner mongolians. Nat. Microbiol. 8, 150–161 (2023).
Kim, C. Y., Ma, J. & Lee, I. Hifi metagenomic sequencing enables assembly of accurate and complete genomes from human gut microbiota. Nat. Commun. 13, 6367 (2022).
Song, E.-J. et al. Early response of the gut microbiome and serum metabolites to cheonggukjang intake in healthy korean subjects. J. Funct. Foods 101, 105420 (2023).
Gounot, J.-S. et al. Genome-centric analysis of short and long read metagenomes reveals uncharacterized microbiome diversity in southeast asians. Nat. Commun. 13, 6044 (2022).
Spohr, P. et al. Insights into gut microbiomes in stem cell transplantation by comprehensive shotgun long-read sequencing. Sci. Rep. 14, 4068 (2024).
Maghini, D. G. et al. Quantifying bias introduced by sample collection in relative and absolute microbiome measurements. Nat. Biotechnol. 42, 328–338 (2024).
Moss, E. L., Maghini, D. G. & Bhatt, A. S. Complete, closed bacterial genomes from microbiomes using nanopore sequencing. Nat. Biotechnol. 38, 701–707 (2020).
Hendriksen, R. S. et al. Global monitoring of antimicrobial resistance based on metagenomics analyses of urban sewage. Nat. Commun. 10, 1124 (2019).
Nayfach, S. & Pollard, K. S. Average genome size estimation improves comparative metagenomics and sheds light on the functional ecology of the human microbiome. Genome Biol. 16, 1–18 (2015).
Wang, C. et al. Bacterial genome size and gene functional diversity negatively correlate with taxonomic diversity along a ph gradient. Nat. Commun. 14, 7437 (2023).
Sonnenburg, E. D. & Sonnenburg, J. L. The ancestral and industrialized gut microbiota and implications for human health. Nat. Rev. Microbiol. 17, 383–390 (2019).
Bello, M. G. D., Knight, R., Gilbert, J. A. & Blaser, M. J. Preserving microbial diversity. Science 362, 33–34 (2018).
Klümper, U. et al. Environmental microbiome diversity and stability is a barrier to antimicrobial resistance gene accumulation. Commun. Biol. 7, 706 (2024).
Nielsen, T. K., Browne, P. D. & Hansen, L. H. Antibiotic resistance genes are differentially mobilized according to resistance mechanism. GigaScience 11, giac072 (2022).
Sommer, M. O., Dantas, G. & Church, G. M. Functional characterization of the antibiotic resistance reservoir in the human microflora. science 325, 1128–1131 (2009).
Ogier, J.-C., Pagès, S., Galan, M., Barret, M. & Gaudriault, S. rpob, a promising marker for analyzing the diversity of bacterial communities by amplicon sequencing. BMC Microbiol. 19, 1–16 (2019).
Dong, M.-J., Luo, H. & Gao, F. Doric 12.0: an updated database of replication origins in both complete and draft prokaryotic genomes. Nucleic Acids Res. 51, D117–D120 (2023).
Wang, S., Ventolero, M., Hu, H. & Li, X. A revisit to universal single-copy genes in bacterial genomes. Sci. Rep. 12, 14550 (2022).
Steinegger, M. & Söding, J. Mmseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 35, 1026–1028 (2017).
Zhang, A.-N. et al. An omics-based framework for assessing the health risk of antimicrobial resistance genes. Nat. Commun. 12, 4765 (2021).
Shen, W., Le, S., Li, Y. & Hu, F. Seqkit: a cross-platform and ultrafast toolkit for fasta/q file manipulation. PloS one 11, e0163962 (2016).
Camargo, A. P. et al. Identification of mobile genetic elements with genomad. Nat. Biotechnol. 42, 1303–1312 (2024).
Wick, R. R., Judd, L. M., Gorrie, C. L. & Holt, K. E. Completing bacterial genome assemblies with multiplex minion sequencing. Microb. genomics 3, e000132 (2017).
Steinig, E. & Coin, L. Nanoq: ultra-fast quality control for nanopore reads. J. Open Source Softw. 7, 2991 (2022).
Li, H. et al. The sequence alignment/map format and samtools. Bioinformatics 25, 2078–2079 (2009).
Magurran, A. E. et al. Ecological Diversity and Its Measurement (Princeton University Press, 1988).
R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria https://www.R-project.org/ (2024).
Oksanen, J. et al. Package ‘vegan’. Community ecology package, version 2 (2019).
Wood, S. N. et al. Generalized additive models: an introduction with R (Chapman and hall/CRC, 2017).
Wickham, H.ggplot2: Elegant Graphics for Data Analysis https://ggplot2.tidyverse.org (Springer-Verlag New York, 2016).
Chen, X., Yin, X., Xu, X. & Zhang, T. Argo v0.1.0 https://doi.org/10.5281/zenodo.14837301 (2025).
Acknowledgements
We thank Xianghui Shi for assistance in PromethION sequencing of Zymo D6331. This study was financially supported by the Theme-based Research Scheme (T21-705/20-N) of Hong Kong. The computations were performed using research computing facilities offered by Information Technology Services, the University of Hong Kong.
Author information
Authors and Affiliations
Contributions
X.C. and T.Z. conceived the study. X.C. developed the tool and analyzed the data. X.C. and T.Z. interpreted the results. X.C. wrote the original draft of the manuscript. X.C., X.Y., X.X., and T.Z. contributed to the revision of the manuscript. All authors approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Daniel Paiva Agustinho and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chen, X., Yin, X., Xu, X. et al. Species-resolved profiling of antibiotic resistance genes in complex metagenomes through long-read overlapping with Argo. Nat Commun 16, 1744 (2025). https://doi.org/10.1038/s41467-025-57088-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-57088-y
This article is cited by
-
Assessing antimicrobial resistance connectivity across One Health sectors
Nature Water (2025)
