
Abstract
Perovskite/organic tandem solar cells (PO-TSCs) exploit the advantages of cost-effective fabrication, orthogonal solvent processing for perovskite and organic absorber layers, and compatibility with low-temperature, high-throughput deposition techniques. However, their performance remains hampered by energy losses of subcells and interconnecting layers (ICLs). Here, an energy loss management strategy for PO-TSCs is proposed, focusing on the simultaneous regulation of defect states in perovskite front subcells and the reduction of optical and electrical losses in the ICL. The synergistic effect of hydrogen bonding and coordination interactions between the pyridinium bromide perbromide and perovskite layer effectively mitigates ion migration, thereby minimizing energy losses. Meanwhile, the optimized V2O5-based ICL structure not only demonstrates excellent transmissivity for near-infrared photons but also allows for barrier-free extraction of charge carriers. Such structure can provide a low-loss interface, facilitating light management within the bulk heterojunction, which effectively balances the current between the front and rear subcells. Taken together, the resulting PO-TSCs deliver a power conversion efficiency of 25.1% with a high open-circuit voltage of 2.10 V.
Similar content being viewed by others
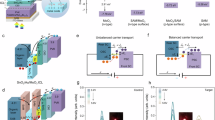


Introduction
The pursuit of superior power conversion efficiency (PCE) has spurred the development of tandem solar cells (TSCs), which hold the potential to surpass the Shockley–Queisser (S–Q) limit inherent in single-junction solar cells1,2. Perovskite/organic tandem solar cells (PO-TSCs) typically consist of three components, including a wide-bandgap (WBG) front perovskite subcell that absorbs high-energy photons, a narrow-bandgap rear organic subcell that absorbs transmitted low-energy photons, and an optically transparent interconnecting layer (ICL) that establishes electrical contact between the two subcells. Monolithic PO-TSCs have garnered significant interest due to their unique advantages, including low-cost fabrication, the use of orthogonal solvents for the perovskite and organic absorbers, and compatibility with low-temperature, high-throughput manufacturing processes like roll-to-roll and blade coating3,4. However, the practical implementation of PO-TSCs has been hindered by significant issues, particularly related to energy losses in the WBG perovskite layers and optoelectronic losses in the ICL5,6.
Despite the considerable efforts exerted to optimize the performance of PO-TSCs, achieving efficiencies exceeding 25% remains a significant challenge. Energy losses in WBG perovskites are a critical barrier to achieving high-efficiency PO-TSCs. WBG perovskite layers, essential for capturing high-energy photons, are susceptible to phase segregation and defect formation, leading to non-radiative recombination7,8,9. This recombination results in significant energy losses, thereby reducing the overall efficiency of the PO-TSCs. To mitigate energy losses, various strategies have been conducted, primarily targeting the reduction of non-radiative recombination losses, crystallographic inhomogeneity, or strain within the bulk perovskite phase10,11. Enhancing the bulk properties of a perovskite subcell often involves controlling the crystallization process or adjusting the perovskite composition. Methodologies such as introducing crystallization agents, designing charge transporting layers with respectable stability and energy level alignment, using non-stoichiometric solutions, tuning the anions fraction on the X-site and cations fraction on the A-site, or incorporating hybrid 2D–3D perovskite heterojunctions at the top surface of 3D perovskites structures have all proven effective in reducing energy losses12,13,14,15.
To electrically connect the front and rear subcells in series within PO-TSCs, an ICL is necessary, where a recombination layer joins the electron transporting layer to the hole transporting layer. An effective ICL must enable efficient charge transfer between subcells, ensure optical transparency, and provide mechanical and chemical protection for the bottom subcell. Nevertheless, the designs of ICLs often suffer from optical and electrical losses, leading to unbalanced short-circuit current density (JSC) between the front and bottom subcells. Early work typically used ultrathin metal layers (thermally evaporated Ag or Au) to render the ohmic contact16,17. However, even with an ultrathin metal layer with an equivalent thickness in the range of 0.5–1 nm, significant optical losses can still occur. The transparent metal oxides, such as InOx and SnOx, are also a great option for recombination layers18. Nonetheless, the ICLs constructed with InOx and SnOx as recombination layers still has large photoelectric losses. It still remains a significant challenge to construct interlayer structures with low photoelectric loss to meet the demands of burgeoning PO-TSCs.
Here, we fabricated high-efficiency PO-TSCs by minimizing the optical and electric energy losses in subcells and ICL layer. Pyridinium bromide perbromide (PBP) was introduced to the WBG single-junction perovskite solar cells (PerSCs). By leveraging the synergistic enhancement effects of hydrogen bonding and coordination between PBP and perovskites, halide ion migration was effectively restricted, and defect density was reduced. The resulting 1.79 eV WBG single-junction PerSCs exhibited a reduced energy loss of 0.46 eV, achieving a maximum PCE of 19.08% with an open-circuit voltage (VOC) of 1.33 V. Furthermore, a high-performance ICL structure was developed. The V2O5-based ICL demonstrated excellent near-infrared (NIR) transmittance, good conductivity, and appropriate energy level alignment, minimizing charge accumulation in the ICL layer and balancing the current between the front and bottom subcells. These combined effects enabled the PO-TSCs to achieve a VOC of 2.10 V, a fill factor (FF) of 81.1%, a JSC of 14.68 mA cm−2, and a high PCE of 25.1%.
Results
Reducing energy loss in WBG perovskite subcells
PBP consists of an anionic part where the central bromine atom is bonded to two adjacent bromine atoms and a cationic part from pyridinium salts (Fig. 1a, b). It can occupy a halide site or equivalently fill a halide vacancy, coordinating with the perovskite. The pure solution of PBP, the mixed solution of PBP and PbI2, and the mixed solution of PBP and CsI exhibited the color of orange, red-brown, and brown, respectively. These noticeable color differences indicate strong interactions between PBP and the perovskite precursors. Proton nuclear magnetic resonance (1H NMR) spectroscopy was employed to investigate the existing interaction. For clarity in analysis, we labeled the hydrogen atoms at different positions of the PBP as Ha, Hb, and Hc, respectively. As depicted in Fig. 1c, compared to the pure PBP solution, all proton signals shift upfield in the PBP and PbI2 mixed solution. However, it remains unclear whether this shift is due to the formation of N−H···I hydrogen bonds, coordination between N−H and Pb2+ or a combination of both effects. To elucidate this, we compared the 1H NMR differences between the PBP and CsI mixed solution and the PBP solution. Only the peaks of Ha and Hb shift upfield, while the peak of Hc remains almost unchanged. Since PBP and CsI interact mainly through hydrogen bonding, the upfield shift of Ha and Hb is attributed to the formation of N−H···I hydrogen bonds19,20. Therefore, the upfield shift of Hc in the mixed solution of PBP and PbI2 is solely due to the deshielding effect from the coordination of N−H with Pb2+ 21. Moreover, combining the effects of hydrogen bonding and coordination, the shift of Ha and Hb peaks in the mixed solution of PBP and PbI2 is significantly greater than that in the mixed solution of PBP and CsI. These results reveal the existence of N−H···I hydrogen bonds and coordination between PBP and perovskite precursors, and this strategy may reduce the trap states in the bulk perovskite22,23,24.

a Molecular structure of PBP. b Photographs of the PBP, PBP + PbI2 and PBP + CsI solutions. c 1H NMR spectra of PBP, PBP + PbI2 and PBP + CsI solutions. d, e High-resolution XPS spectra of (d) Pb 4f and (e) I 3d for the pristine and PBP-treated perovskite films. f, g PL mapping of (f) pristine and (g) PBP-treated perovskite films. h, i Top-surface SEM images of the (h) pristine and (i) PBP-treated perovskite films. j, k AFM images of (j) the pristine and (k) PBP-treated perovskite films.
Further confirmation of the interactions between PBP and perovskites was obtained through X-ray photoelectron spectroscopy (XPS). The Pb 4 f XPS spectra (Fig. 1d) show that the Pb 4f5/2 and 4f7/2 peaks of the perovskite surface shift from 143.04 and 138.22 eV (untreated) to 142.85 and 138.00 eV (PBP-treated), suggesting alterations in the electron cloud density surrounding Pb. This change could be attributed to electron sharing between the −NH− group in PBP and Pb2+ in the perovskite25. Similarly, as shown in the I 3d XPS spectrum in Fig. 1e, we find that the I 3d3/2 and 3d5/2 peaks of the untreated perovskite surface are located at 630.44 and 618.97 eV, respectively. After PBP optimization, these peaks shift to 630.09 and 618.63 eV, indicating the formation of N−H···I hydrogen bonds between PBP and I− in the perovskite26. In contrast, the Cs 3d XPS spectra (Supplementary Fig. 1) exhibit no notable shift, indicating the unchanged chemical environment around Cs, which is consistent with results obtained from the 1H NMR analysis. The possible interactions between PBP and perovskites are schematically depicted in Supplementary Fig. 2.
As for the uniformity and defect distribution details of the untreated and PBP-treated perovskite films, photoluminescence (PL) mapping displays the corresponding information (Fig. 1f, g). It can be seen that the PL peak distribution in the untreated perovskite film is more dispersed with poor distribution uniformity, indicating the presence of numerous defects or non-uniform region. In contrast, the PL peak distribution in the PBP-treated perovskite film is more concentrated and uniform, reflecting the improved film quality with higher uniformity with fewer defects27. The PL spectra of the untreated film exhibited redshifted peaks, suggesting that halide phase segregation occurs upon illumination (Supplementary Fig. 3). This phenomenon is attributed to the tendency of photogenerated carriers to relax into lower-energy states within the I-rich phase. In contrast, the perovskite modified with PBP displayed negligible shifts in the PL peaks, indicating improved photostability of the perovskite phase due to the suppression of ion migration.
The influence of PBP on perovskite morphology was verified through scanning electron microscopy (SEM) and atomic force microscopy (AFM) measurements. As shown in the SEM images (Fig. 1h, i), it is evident that while both untreated and PBP-treated perovskite films exhibit distinct perovskite grains and grain boundaries, the grain size of untreated perovskite films is uneven, with sizes primarily ranging from 350 to 500 nm, showing a broad distribution. In contrast, the grain size of the PBP-treated perovskite is concentrated within a narrower range of 450 to 500 nm, indicating a more uniform internal structure of the film. This helps reduce grain boundary defects caused by size differences, facilitates smoother carrier transport between grains and reduces recombination losses at the grain boundaries. Fig. 1j, k illustrates the AFM images of untreated and PBP-treated perovskite films, where the perovskite film after PBP treatment possessed slightly larger grain domain sizes, leading to the surface roughness increase from 13 to 18 nm, which is conducive to forming better physical contact and reduce defects. In addition, the (1 0 0) and (1 1 0) orientation peak of the target perovskite film is stronger than that of the control one, indicating that the PBP-modified perovskite film exhibits stronger crystallinity. Furthermore, the PBP-modified film demonstrated a decrease in the full-width at half-maximum values, with reductions from 0.32° to 0.28° for the (1 0 0) peak and from 0.18° to 0.15° for the (1 1 0) peak, confirming that PBP modification can enlarge grain size (Supplementary Fig. 4).
To exclude the defect evolution of WBG perovskite, PL spectra under continuous illumination were performed (Fig. 2a, b). During the initial 10 min of illumination, the PL intensity of the pristine perovskite film exhibits a rapid decline, indicating an increase in recombination centers on the perovskite surface. Following this period, the PL intensity continues to decrease, albeit at a slower rate, implying a gradual accumulation of defect states within the perovskite film. In contrast, the perovskite film with PBP treatment shows no significant reduction in PL intensity throughout the entire duration of illumination. The PL intensity of the perovskite film after PBP optimization decreased to 98% of its original value, while that of the untreated perovskite film decreased to 72%, which proves that PBP can effectively suppress the reduction of PL intensity.

a, b PL spectra of (a) pristine and (b) PBP-treated perovskite films under continuous illumination. c The trap state density in the perovskites. d J–V curves of the PerSCs with and without PBP modification. e The corresponding EQE spectra with integrated JSC. f Comparison of energy losses of the PerSCs with and without PBP modification.
The significant reduced trap density of the PBP-treated perovskite devices indicates that the introduction of PBP can effectively improve the crystalline quality and mitigate defects in the perovskite films (Fig. 2c). The carrier mobility (µ)was calculated by using the Mott-Gurney formula:
where JD is the current density, and V is the applied voltage. For the electron-only devices (Supplementary Fig. 5), the electron mobility increased from 1.17 × 10−3 cm2 V−1 s−1 for untreated perovskite devices to 1.70 × 10−3 cm2 V−1 s−1 for PBP-treated perovskite devices, an enhancement of 0.53 × 10−3 cm2 V−1 s−1. For the hole-only devices, the hole mobility of the PBP-treated perovskite film is 1.65 × 10−3 cm2 V−1 s−1, much higher than that of the untreated film (9.14 × 10−4 cm2 V−1 s−1), indicating faster carrier transport in PBP-treated devices. Additionally, the hole-to-electron mobility ratio in untreated and PBP-treated devices is 0.78 and 0.97, respectively, suggesting more balanced carrier transport in PBP-treated perovskites. This balance minimizes recombination and asymmetric transport losses in the devices.
Time-resolved photoluminescence (TRPL) spectroscopy was used to analyze the carrier dynamics and recombination kinetics in perovskite films. As depicted in Supplementary Fig. 6, a double exponential decay model was used to fit the TRPL decay curves, obtaining carrier lifetime parameters. Detailed fitting parameters are listed in Supplementary Table 1. For the untreated perovskite film, the fast recombination process τ1 is 50.0 ns, while for the PBP-treated perovskite film, τ1 increases to 102.3 ns, suggesting that the shallow traps are reduced and carriers can exist for a longer duration without undergoing fast recombination in the PBP-treated film. Additionally, compared to the untreated film, which has a slow recombination process τ2 of 204.3 ns, the PBP-treated film exhibits a significantly higher τ2 of 563.0 ns, indicating a reduction in deep defects and more efficient carrier transport. Furthermore, the average decay lifetime of the untreated perovskite devices is 187.3 ns, increasing to 550.2 ns for the PBP-treated devices, which implies a reduced defect density and suppressed non-radiative recombination28. This indicates that the introduction of PBP into perovskite films effectively improves charge separation efficiency and minimizes charge recombination losses.
Sequentially, electrochemical impedance spectroscopy (EIS) was performed to study the impact of PBP treatment on carrier transport and recombination mechanisms in PerSCs. Supplementary Fig. 7 shows the Nyquist plots obtained from the EIS measurements of untreated and PBP-treated PerSCs under dark conditions and zero bias. The Nyquist plots include a semicircle in the high-frequency region and an arc in the mid-frequency region. The semicircle in the high-frequency region reflects charge transfer resistance (Rct), while the arc in the mid-frequency region reflects recombination resistance (Rrec). The fitting parameters are listed in Supplementary Table 2. With the reduction in Rct from 9.6 × 104 Ω in untreated perovskite solar cells to 2.52 × 103 Ω cm2 after PBP treatment, the interfacial charge transport efficiency is improved, enabling carriers to traverse the interface more quickly and effectively. The increase in Rrec from 3.04 × 104 Ω cm2 in untreated PerSCs to 4.48 × 104 Ω cm2 in PBP-treated PerSCs indicates a more efficient charge separation process, resulting in reduced charge recombination29. The charge transfer and recombination behaviors of the carriers of PerSCs were further evaluated by EIS under illumination (Supplementary Fig. 8). The fitted impedance parameters are summarized in Supplementary Table 3. Compared to the control device, the optimized PerSCs exhibited a smaller Rct, indicating a more efficient charge transfer process. Additionally, the optimized PerSCs demonstrated a larger Rrec, which hindered carrier recombination within the device, thereby facilitating more effective carrier extraction. We also offer a light dependence of the EIS of PerSCs through our proposed equivalent circuit (Supplementary Fig. 9). When the light intensity increases, the charge transfer resistance decreases, indicating that the charge transfer at the interface becomes more efficient, which is beneficial to charge transport. Supplementary Fig. 10 displays the current density-voltage (J–V) characteristics of PerSCs tested under dark conditions. The dark current density of PBP-treated PerSCs is significantly lower than that of untreated PerSCs, suggesting reduced leakage current paths and fewer recombination centers30. Subsequently, to investigate the carrier dynamics in devices, we measured the J–V characteristics under varying light intensities from 1 to 100 mW/cm2. The relationship between light intensity and VOC can be used to assess the recombination mechanisms in PerSCs. Theoretically, the slope of the curve corresponds to the ideality factor (n), which can be calculated using the following formula31:
where KB is the Boltzmann constant, T is the absolute temperature, I is the light intensity. When n approaches 1, it indicates that bimolecular radiative recombination is the dominant recombination mechanism. When n approaches 2, it implies that Shockley-Read-Hall recombination and non-radiative recombination caused by trap states are dominant. As shown in Supplementary Fig. 11a, the value of n decreases from 1.74 in untreated PerSCs to 1.61 in PBP-treated PerSCs, reflecting a significant suppression of charge recombination and a reduction in defect state density. Next, we investigated the trend of JSC with varying light intensities to further verify the carrier recombination mechanisms. Generally, the relationship between JSC and light intensity can be described by the following formula: JSC ∝ \({P}_{{light}}^{{{\rm{\alpha }}}}\), where α is an exponent related to the recombination mechanism. An α value less than 1 indicates the presence of defect-assisted non-radiative recombination. As illustrated in Supplementary Fig. 11b, the α values for untreated and PBP-treated devices are 0.97 and 0.99, respectively, indicating the lower bimolecular recombination after PBP treatment.
On the basis of the above-mentioned results, we fabricated inverted WBG perovskite devices to study the impact of PBP treatment on device performance. The optimal concentration of PBP was found to be 0.5 mg/mL (Supplementary Fig. 12 and Supplementary Table 4). Fig. 2d describes the J–V curves of the best-performing untreated and PBP-treated PerSCs. The corresponding optimal performance parameters are given in Supplementary Table 5. The untreated PerSC achieves a VOC of 1.29 V, a JSC of 17.59 mA cm−2, an FF of 77.6%, and a PCE of 17.73%. In comparison, the optimized PBP-treated PerSC exhibits a higher VOC of 1.33 V, a JSC of 17.72 mA cm−2, and an improved FF of 81.2%, yielding a high PCE of 19.08%. The external quantum efficiency (EQE) spectra of the PerSCs are shown in Fig. 2e. The integrated current density for the untreated PerSC is 16.73 mA cm−2, while that for the PBP-treated PerSC is 17.34 mA cm−2, which are consistent with the JSC values obtained from the J–V curve. The bandgap of the target device is determined to be 1.79 eV by differentiating its EQE spectrum, which exhibited an inflection point at 691 nm (Supplementary Fig. 13). Fig. 2f makes a clear comparison of energy loss. Energy losses are 0.5 and 0.46 eV for untreated and PBP-treated PerSCs, respectively. Supplementary Fig. 14 presents the statistical photovoltaic parameters of 15 individual PerSCs from different batches. The significant improvement in the photovoltaic parameters of PBP-treated PerSCs can be attributed to the passivation effect of PBP on perovskite defects, which results in more efficient charge extraction and reduced recombination losses. These results demonstrate the reliability of our method in improving the performance of PerSCs. Supplementary Fig. 15 shows the operational stability of the PerSCs. The PBP-treated devices exhibit significantly enhanced stability, retaining 80% of their initial efficiency after 160 h of continuous LED illumination. Additionally, we also achieved improved thermal stability for the PBP-treated PerSC, which retained 90% of its initial PCE after heating at 65 °C for 600 h in the N2 atmosphere (Supplementary Fig. 16).
Design of ICL with high transmittance and conductivity
In PO-TSCs, the ICLs control charge collection and recombination from subcells and are critical in determining the performance of PO-TSCs. The design of high-quality ICLs aims to avoid or minimize reverse junction formation, parasitic light absorption, and junction resistance. Here, we designed a V2O5-based ICL structure with low optical/electrical loss and compared it with the traditional MoO3-based ICLs. To investigate the differences in optical and electrical properties of the V2O5-based and MoO3-based ICLs, we conducted a series of characterizations, including transmittance, conductivity, and hole mobility. First, we compared the optical properties of the MoO3-based and V2O5-based ICLs. As displayed in Fig. 3a and Supplementary Fig. 17, within the wavelength range of 550 to 1100 nm, the transmittance of the MoO3-based ICLs is significantly lower than that of V2O5-based ICLs. The low optical loss of the V2O5-based ICLs in the NIR region indicates that the front junction perovskite subcell and the ICL have lower utilization efficiency for the NIR part of the solar spectrum, favoring the transmission of photons in the NIR region. This contributes to the enhancement of current in the organic bottom cells, which promotes current matching between the front and rear subcells in the PO-TSCs.

a Transmittance spectra of the perovskite subcells with different ICLs. b Dark J−V characteristics of ICL devices with a structure of ITO/C60/PEI/ITO/V2O5 or MoO3/Ag. c J–V characteristics of the hole-only devices with a structure of ITO/PEDOT:PSS/V2O5 or MoO3/Ag. d, e Energetic line-up of (d) MoO3-based and (e) V2O5-based ICL. f, g Distributions of photon absorption in the (f) MoO3-based and (g) V2O5-based OSCs. h J–V curves of the MoO3-based and V2O5-based OSCs. i EQE spectra and integrated JSC for the corresponding OSCs. ΔEQE is the difference between the EQE value of the MoO3-based and V2O5-based OSCs.
Regarding conductivity (σ), both MoO3-based and V2O5-based ICLs show a linear relationship between current density and voltage, indicating the formation of good ohmic contacts (Fig. 3b and Supplementary Fig. 18)32. The σ of the ICLs was calculated using the formula33: σ = Jd/V, where d represents the thickness of the ICL, J represents the current density, and V represents the voltage. The V2O5-based ICLs show a higher σ value of 2.23 × 10−3 mS cm−1 compared with MoO3-based ICLs (1.84 × 10−3 mS cm−1). Obviously, the hole mobility of V2O5 is also superior to that of MoO3 (Fig. 3c). These results indicate that V2O5 can effectively facilitate rapid and efficient charge transport, which enhances hole selectivity34,35.
The ultraviolet photoelectron spectroscopy (UPS) measurement of V2O5 shows the work function (WF) and the valence band maximum of 5.10 eV and 7.87 eV (Supplementary Fig. 19). Then the conduction band minimum of the V2O5 film is determined to be 5.52 eV based on its bandgap of 2.35 eV (Supplementary Fig. 20). Supplementary Fig. 21 illustrates the energy level diagram. The detailed energy diagrams following Fermi level alignment are depicted in Fig. 3d, e. A significant band bending occurs at the C60/MoO3 interface due to the considerable work function mismatch. This bending is expected to induce a large Schottky barrier at the interface, which hampers efficient charge recombination within the ICL.
We selected the combination of PBDB-T-2F (PM6), BTP-eC9 and PC71BM as the photoactive layer for the organic subcells36,37. The detailed chemical structures of the donor and acceptor materials are illustrated in Supplementary Fig. 22. UV–Vis absorption spectroscopy was then used to compare the absorbance and transmittance of MoO3 and V2O5 thin films. As seen in Supplementary Fig. 23, within the absorption range of 600 to 1100 nm, the absorption intensity of the MoO3 films is higher than that of V2O5 films. The higher absorption intensity of MoO3 films in the NIR region reduces the effectiveness of the organic subcell in capturing low-energy photons, leading to current mismatch in the PO-TSCs. Correspondingly, the transmittance of the MoO3 films is lower than that of V2O5 films. We then established an optical transfer matrix model to analyze the light field distribution within the absorber layers. The device structure used for the simulation is ITO/MoO3 or V2O5/PM6:BTP-eC9/TPMA/Ag. As presented in Fig. 3f, g, the MoO3-based and V2O5-based OSCs exhibit similar simulated JSC within the 300–600 nm range owing to the comparable absorption capacity of both for high-energy photons. However, the V2O5-based OSCs improve the utilization of low-energy photons in the NIR range of 600 to 1000 nm, leading to a higher simulated JSC, which is beneficial for the current balancing between the front and rear subcells in PO-TSCs.
Transient photovoltage (TPV) and transient photocurrent (TPC) experiments were performed to gain insight into the charge recombination and extraction kinetics process38. As for TPC decay lifetimes (Supplementary Fig. 24), V2O5-based OSCs exhibit a shorter extraction time of 1.91 μs compared to 2.13 μs for MoO3-based OSCs, suggesting enhanced charge transport capacity. Photogenerated carrier lifetimes obtained from TPV measurements are 5.26 and 8.27 μs for MoO3-based and V2O5-based OSCs, respectively, indicating charge recombination has been suppressed in the V2O5-based OSCs.
Utilizing AFM, we studied the surface morphology of the films. The AFM images (Supplementary Fig. 25) reveal stripe-like structures for both MoO3 and V2O5 films, likely caused by anisotropic growth during thermal evaporation39. Additionally, the MoO3 films show a calculated root mean square roughness (RMS) of 0.50 nm, while V2O5 films exhibit an RMS of 0.39 nm. The extremely low roughness and smooth surface morphology of V2O5 films facilitate the formation of well-ordered molecular arrangements and crystallization in the active layer. Subsequently, absorber layers were fabricated on ITO/V2O5 and ITO/MoO3 films to validate the above conclusions. The smoother surface with a roughness of 2.25 nm is achieved for the V2O5-based absorber layer, while a rougher surface with a roughness of 3.53 nm is observed for the MoO3-based absorber layer. According to our previous work, the interface plays an important role in controlling the vertical phase distribution40. To studied the vertical phase distribution of the active layer, we carried out the contact angle tests on water and diiodomethane, and the results are presented in Supplementary Fig. 26. Through the contact angles, we could obtain the surface tensions (γ) for donor and acceptor materials. The γ was calculated using the Owens-Wendt-Rabel-Kaelble method. The relevant calculation results are listed in Supplementary Table 6. Compared with the MoO3-based blend, the γ of the V2O5-based active layer (39.5 mN m−1) is much closer to that of the acceptor on V2O5 films (41.3 mN m−1), suggesting that the acceptor phase tends to enrich on the surface of the absorber layer. Such favorable vertical phase distribution is conducive to efficient charge transport in the conventional device structure.
Based on the above analysis, we investigate the impact of V2O5 or MoO3 on the device performance. The corresponding J–V curves are exhibited in Supplementary Fig. 27, and the photovoltaic parameters are summarized in Supplementary Table 7. Fig. 3h displays the J–V curves of the best-performing MoO3-based and V2O5-based OSCs. The MoO3-based OSCs show a PCE of 17.34% with a VOC of 0.844 V, a JSC of 26.75 mA cm−2, and an FF of 76.8%. In comparison, the V2O5-based OSC exhibits a VOC of 0.859 V, a JSC of 27.29 mA cm−2, an FF of 78.4%, and a PCE of 18.39%. Supplementary Fig. 28 presents the statistical photovoltaic parameters of 15 individual OSCs from different batches, showing significant enhancement in V2O5-based OSCs compared to MoO3-based OSCs. Moreover, we observed an enhanced EQE in V2O5-based OSCs from 300 to 850 nm (Fig. 3i). The integrated current density derived from the EQE curves increased from 25.67 mA cm−2 for the MoO3-based device to 26.31 mA cm−2 for the V2O5-based device.
Photovoltaic performance of Monolithic PO-TSCs
Encouraged by the reduced energy loss of subcells and minimized optical/electrical loss of ICL, we fabricated PO-TSCs with the architecture of ITO/NiOx/4PADCB/perovskite/EDADI/C60/PEI/ITO/V2O5 or MoO3/PM6:BTP-eC9:PC71BM/TPMA/Ag (Fig. 4a). The device structure is schematically depicted in Supplementary Fig. 29. We compared the performance of PO-TSCs prepared with different thicknesses of ICLs. The J–V curves of the PO-TSCs are depicted in Supplementary Fig. 30. Supplementary Table 8 summarizes the corresponding photovoltaics parameters. We found that about 15 nm is the optimal thickness for device performance (Supplementary Fig. 31). The control PO-TSCs yield a PCE of 23.20%, with a VOC of 2.07 V, a JSC of 14.41 mA cm−2 and an FF of 77.7%. In contrast, the optimized PO-TSCs exhibited an FF of 81.1%, a JSC of 14.68 mA/cm², a VOC of 2.10 V, and a PCE of 25.1% (Fig. 4b). As summarized in Supplementary Tables 9 and 10, our champion cells showed high performance among reported solar cells. As shown in Fig. 4c, the optimized PO-TSCs show good current matching, where the JSC values of 14.11 and 13.98 mA cm−2 are integrated from the EQE spectra of the perovskite and organic subcells, respectively. For the control PO-TSCs, the integrated current densities are 13.97 and 13.56 mA cm−2, respectively. Additionally, the steady-state power output of the optimized PO-TSCs was 25.02% at a bias of 1.77 V (Fig. 4d). The statistical photovoltaic parameters of 30 individual PO-TSCs from different batches confirmed the advantages of the PO-TSCs with V2O5-based ICL (Fig. 4e and Supplementary Fig. 32). The significant enhancement in the performance of PO-TSCs can be attributed to the reduced energy loss of perovskite subcells and the low electrical and optical loss of V2O5-based ICL. Fig. 4f, g depicts the light field distribution simulated by the optical transfer matrix model. The simulated JSC values of the PO-TSCs with V2O5-based ICL are higher than those with MoO3-based ICL, which could be due to the higher light field intensity in the 650 to 850 nm wavelength range for the PO-TSCs with V2O5-based ICL.

a Cross-sectional SEM image of the PO-TSCs. b J–V curves of the best PO-TSCs with MoO3-based and V2O5-based ICLs. c EQE spectra of the perovskite and organic subcell for the corresponding PO-TSCs. d The steady-state power output of the PO-TSCs with V2O5-based ICL. e Statistics of PCE for 30 PO-TSCs with various ICLs over different batches. f, g Distributions of photon absorption in the PO-TSCs with (f) MoO3-based and (g) V2O5-based ICLs. h Thermal stability of the PO-TSCs with MoO3-based and V2O5-based ICLs stored in N2 atmosphere. i Operational stability of the PO-TSCs with MoO3-based and V2O5-based ICLs under LED illumination.
Subsequently, we evaluated the stability of the PO-TSCs. The thermal stability of the unencapsulated PO-TSCs in a nitrogen atmosphere was verified. After 200 h of storage in a nitrogen atmosphere at 65 °C fixed temperature (Fig. 4h), the PCE of the optimized PO-TSCs decreased by 9%, while the PCE of the control PO-TSCs decreased by 31%. As depicted in Fig. 4i and Supplementary Fig. 33, we also assessed the operational stability of the PO-TSCs. The control PO-TSCs exhibited obvious performance degradation (the PCE dropped to 80% of the initial value after 250 h), whereas the optimized PO-TSCs maintained 90% of the initial PCE after storing for 450 h. The fact that the operational stability of optimized PO-TSC significantly enhanced clearly demonstrates that the halide phase segregation in perovskite subcell has been effectively suppressed. In addition, replacing MoO3 with V2O5, which is a metal oxide demonstrating greater morphological robustness, leads to a dramatic improvement in operational stability of optimized PO-TSC. As summarized in Supplementary Table 11, our optimized cells showed good stability among reported solar cells.
Discussion
In summary, efficient PO-TSCs with a high PCE of 25.1% has been obtained via minimizing the optical and electrical energy losses simultanously. By passivating the defects in WBG perovskites, we have significantly reduced the Eloss, boosting the VOC of perovskite subcells. We also demonstrated a designed ICL structure with low optical and electrical loss. Compared with the MoO3-based ICL, the V2O5-based ICL exhibits higher transmittance in the NIR region. In addition, the V2O5-based ICL has better electrical conductivity, higher carrier mobility, and more compatible energy level alignment, which helps improve charge transport and extraction, reduce charge recombination, and minimize charge accumulation at the ICL, thereby balancing the current between the perovskite and organic subcell. These results emphasize the importance of simultaneously minimizing energy losses in subcells along with optical and electrical losses in ICLs, which is crucial for constructing high-performance PO-TSCs.
Methods
Materials
All of the materials were used without further purification. Caesium iodide (CsI), formamidinium iodide (FAI), methylammonium iodide (MAI), lead bromide (PbBr2), EDADI, 4PADCB, fullerene (C60), were purchased from Xi’an Yuri Solar Co., Ltd. Lead iodide (PbI2), NiOx, and BCP were purchased from Advanced Election Technology. PM6, BTP-eC9 and PC71BM were purchased from Solarmer Materials (Beijing). PEI was purchased from Beijing Innochem Science & Technology Co., Ltd. MoO3, V2O5, and Ag were purchased from ZhongNuo Advanced Material (Beijing) Technology Co., Ltd. TPMA were obtained from the previous literature41. Solvents were purchased from Acros Organics. PBP was purchased from Alfa.
Perovskite subcell fabrication
The ITO glass substrates were sequentially cleaned by sonication with detergent, deionized water and ethanol for 15 min, respectively. The cleaned ITO glass substrates were then transferred into an oven at 100 °C for 10 min and treated with ultraviolet ozone for 20 min before use. The NiOx (10 mg ml−1) dispersed in deionized water was deposited on the ITO glass substrates by spin-coating at 2000 rpm for 45 s and then annealed at 100 °C for 15 min in air. Then, the samples were transferred into a nitrogen glovebox for deposition of the 4PADCB and perovskite layers. 4PADCB (0.5 mg ml−1) dissolved in isopropanol (IPA) was deposited on the NiOx substrate by spin-coating at 4000 rpm for 40 s, followed by annealing at 100 °C on a hot plate for 10 min. The precursor solution (FAMACsPb(I0.6Br0.4)3) was prepared by mixing 0.1 M CsI, 0.8 M FAI, 0.1 M MAI, 0.6 M PbBr2, and 0.9 M PbI2 in DMF/DMSO (4:1) with 1 M solution. PBP with different concentrations were incorporated into the precursors for the target sample. The precursor solution was spin-coated onto the NiOx/4PADCB layer in the first step at 1000 rpm for 10 s and the second step at 4000 rpm for 40 s. During the spin-coating process, 150 μL CB antisolvent was dropped at 25 s of the second step before the end of the spin-coating program. Then, the films were annealed at 100 °C for 10 min. After cooling to room temperature, 50 µL EDADI solution (0.5 mg ml–1) in IPA was dripped on the perovskite surface and spin-coated at 5000 r.p.m. for 30 s followed by annealing at 100 °C for 10 min. Finally, all the samples were transferred into a high-vacuum chamber to evaporate 25 nm C60, 6 nm BCP and 100 nm Ag electrode.
Organic subcell fabrication
MoO3 and V2O5 were thermally evaporated as hole transporting layers on the ITO glass substrates. The PM6:BTP-eC9:PC71BM was dripped on the MoO3 or V2O5 at 4000 r.p.m. for 30 s, followed by annealing at 80 °C for 10 min. After cooling, 50 µL TPMA solution (0.5 mg ml–1) in methanol was spin-coated onto the active layer at 4000 r.p.m. for 30 s. Finally, all the samples were transferred into a high-vacuum chamber to evaporate Ag electrode (100 nm).
PO-TSC fabrication
After the thermal evaporation of C60 for wide-bandgap subcells, 50 µL PEI solution (1 mg ml–1) in IPA was spin-coated onto the C60 layer at 5000 r.p.m. for 30 s. A radio frequency sputtering technique (Eminex, ED500) was applied to deposit ITO recombination layers. The ITO target, made of 90% In2O3 and 10% SnO2, was used. Pure argon was used as a process gas. The work pressure was kept at 2 × 10–3 torr, and the sputtering power was fixed at 100 W. Subsequently, MoO3 or V2O5 was thermally evaporated on top of the ITO, to form an ICL structure for the TSCs. Next, the PM6:BTP-eC9:PC71BM active layer was spin-coated on MoO3 or V2O5. TPMA was spin-coated on the organic active layer at 4000 r.p.m. for 30 s. Finally, all the samples were transferred into a high-vacuum chamber to evaporate 100 nm Ag electrode.
Characterization of the solar cells
J−V characteristics of the devices were measured with a Keithley B2901A source meter under the simulated AM 1.5 G illumination (100 mW·cm−2). The devices were measured both in reserve and forward scan with a step of 0.02 V. Light intensity was calibrated by a standard silicon cell (SRC-2020, EnliTech). The EQE spectra were performed using the Enlitech QE-R system. For TSCs, when measuring the EQE values of the perovskite subcells, a bias illumination with an 800 nm optical filter was applied to saturate the corresponding organic subcells. Similarly, when measuring the EQE values of the organic subcells, a bias illumination with a 500 nm optical filter was applied to saturate the corresponding perovskite subcells. No bias voltage was applied during the EQE measurements of either subcell. The dark I–V characteristics curves were recorded using a Keithley B2901A source meter. The steady-state power output measurements were recorded by tracing the current density at a bias (voltage at maximum power point).
Characterization of the films
Absorption and transmittance spectra were measured using a Shimadzu UV–Vis-NIR 3600 spectrometer. PL and TRPL were measured by FLS980 Photoluminescence Spectrophotometer (Edinburgh Instruments Ltd.). Krüss DSA30 optical contact angle meter. XPS measurements were performed on a THERMO VG ESCALAB 250. UPS measurements were performed on an AXIS ULTRA DLD, Kratos (by using an He I source) under high vacuum (10−8 Torr). The top-view and cross-sectional morphology were analyzed by a SEM (SU8600, HITACHI). The AFM characterizations were conducted by Bruker DMFASTSCAN2-SYS. Ellipsometry was carried out by an ellipsometer (ES01, ELLITOP SCIENTIFIC CO., LTD.). The EIS was measured by the CHI660E electrochemical workstation.
Other characterizations
1H NMR spectra were measured on Bruker AVANCE ΙΙΙ (400 MHz) spectrometers.
Characterization of the device stability
Operational stability tests were performed on the encapsulated solar cells. The tests were conducted using a white-light LED array under a continuous simulation intensity of 100 mW cm−2 (Suzhou D&R instruments, PVLT-G8001M-32B). For thermal stability measurements, the devices without encapsulation were kept at a fixed temperature in the dark except during the J–V measurement, and the J–V curves were recorded at specific time intervals.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All data supporting the results of this study are provided in this article and its supplementary information. Any other information can be requested from the corresponding author. Source data are provided with this paper.
References
Shockley, W. & Queisser, H. J. Detailed balance limit of efficiency of p‐n junction solar cells. J. Appl. Phys. 32, 510–519 (1961).
Green, M. A. Limiting photovoltaic efficiency under new ASTM International G173-based reference spectra. Prog. Photovoltaics 20, 954–959 (2012).
Guo, X. et al. Stabilizing efficient wide-bandgap perovskite in perovskite-organic tandem solar cells. Joule 8, 2554–2569 (2024).
Wu, S., Liu, M. & Jen, A. K. Y. Prospects and challenges for perovskite-organic tandem solar cells. Joule 7, 484–502 (2023).
Yang, H. et al. Regulating charge carrier recombination in the interconnecting layer to boost the efficiency and stability of monolithic perovskite/organic tandem solar cells. Adv. Mater. 35, 2208604 (2023).
Chen, X. et al. Efficient and reproducible monolithic perovskite/organic tandem solar cells with low-loss interconnecting layers. Joule 4, 1594–1606 (2020).
Yang, C.-Q. et al. Atomically deciphering the phase segregation in mixed halide perovskite. Adv. Funct. Mater. 34, 2400569 (2024).
Shi, R. et al. Structural disorder in higher-temperature phases increases charge carrier lifetimes in metal halide perovskites. J. Am. Chem. Soc. 144, 19137–19149 (2022).
Kerner, R. A., Xu, Z., Larson, B. W. & Rand, B. P. The role of halide oxidation in perovskite halide phase separation. Joule 5, 2273–2295 (2021).
Tian, J. et al. Dual interfacial design for efficient CsPbI2Br perovskite solar cells with improved photostability. Adv. Mater. 31, 1901152 (2019).
Shen, X. et al. Chloride-based additive engineering for efficient and stable wide-bandgap perovskite solar cells. Adv. Mater. 35, 2211742 (2023).
Xu, J. et al. Triple-halide wide–band gap perovskites with suppressed phase segregation for efficient tandems. Science 367, 1097–1104 (2020).
Wang, G. et al. Molecular engineering of hole-selective layer for high band gap perovskites for highly efficient and stable perovskite-silicon tandem solar cells. Joule 7, 2583–2594 (2023).
Wang, Z. et al. Suppressed phase segregation for triple-junction perovskite solar cells. Nature 618, 74–79 (2023).
Ye, Q. et al. Cesium lead inorganic solar cell with efficiency beyond 18% via reduced charge recombination. Adv. Mater. 31, 1905143 (2019).
Xie, Y.-M. et al. Understanding the role of interconnecting layer on determining monolithic perovskite/organic tandem device carrier recombination properties. J. Energy Chem. 71, 12–19 (2022).
Li, Z. et al. Hybrid perovskite-organic flexible tandem solar cell enabling highly efficient electrocatalysis overall water splitting. Adv. Energy. Mater. 10, 2000361 (2020).
Brinkmann, K. O. et al. Perovskite–organic tandem solar cells with indium oxide interconnect. Nature 604, 280–286 (2022).
Chao, L. et al. Direct and stable α-phase formation via ionic liquid solvation for formamidinium-based perovskite solar cells. Joule 6, 2203–2217 (2022).
Hui, W. et al. Stabilizing black-phase formamidinium perovskite formation at room temperature and high humidity. Science 371, 1359–1364 (2021).
Li, M. et al. Stabilizing Perovskite precursor by synergy of functional groups for NiO-based inverted solar cells with 23.5 % efficiency. Angew. Chem. Int. Ed. 61, e202206914 (2022).
Long, R., Liu, J. & Prezhdo, O. V. Unravelling the effects of grain boundary and chemical doping on electron–hole recombination in CH3NH3PbI3 perovskite by time-domain atomistic simulation. J. Am. Chem. Soc. 138, 3884–3890 (2016).
Agiorgousis, M. L., Sun, Y.-Y., Zeng, H. & Zhang, S. Strong covalency-induced recombination centers in perovskite solar cell material CH3NH3PbI3. J. Am. Chem. Soc. 136, 14570–14575 (2014).
Yun, S., Zhou, X., Even, J. & Hagfeldt, A. Theoretical treatment of CH3NH3PbI3 perovskite solar cells. Angew. Chem. Int. Ed. 56, 15806–15817 (2017).
Li, R. et al. Customizing aniline-derived molecular structures to attain beyond 22% efficient inorganic perovskite solar cells. Angew. Chem. Int. Ed. 63, e202410600 (2024).
Wu, Z. et al. The tricyclic alkaloid catalyzed crystallization of α-FAPbI3 for high performance antisolvent-free perovskite solar cells. Energ. Environ. Sci. 17, 4670–4680 (2024).
Bai, Y. et al. Initializing film homogeneity to retard phase segregation for stable perovskite solar cells. Science 378, 747–754 (2022).
Ma, Z. et al. Crosslinkable and chelatable organic ligand enables interfaces and grains collaborative passivation for efficient and stable perovskite solar cells. Small 18, 2201820 (2022).
Meng, Y. et al. Pre-buried ETL with bottom-up strategy toward flexible perovskite solar cells with efficiency over 23%. Adv. Funct. Mater. 33, 2214788 (2023).
Xie, L. et al. A deformable additive on defects passivation and phase segregation inhibition enables the efficiency of inverted perovskite solar cells over 24%. Adv. Mater. 35, 2302752 (2023).
Yuan, X. et al. Synergistic crystallization modulation and defects passivation via additive engineering stabilize perovskite films for efficient solar cells. Adv. Funct. Mater. 33, 2215096 (2023).
Xie, G. et al. Phase segregation and voltage loss mitigated highly efficient perovskite–organic tandem solar cells with a simple ambipolar SnO interconnecting layer. Adv. Funct. Mater. 33, 2308794 (2023).
Zhuang, J. et al. Rubidium fluoride modified SnO2 for planar n-i-p perovskite solar cells. Adv. Funct. Mater. 31, 2010385 (2021).
Liu, M. et al. Localized oxidation embellishing strategy enables high-performance perovskite solar cells. Angew. Chem. Int. Ed. 63, e202318621 (2024).
Huang, K. et al. Universal strategy with structural and chemical crosslinking interface for efficient and stable perovskite solar cells. Adv. Energy. Mater. 14, 2304073 (2024).
Yu, R. et al. Thermodynamic phase transition of three-dimensional solid additives guiding molecular assembly for efficient organic solar cells. Angew. Chem. Int. Ed. 62, e202308367 (2023).
Ma, Z. et al. Transparent recombination electrode with dual-functional transport and protective layer for efficient and stable monolithic perovskite/organic tandem solar cells. Adv. Mater. 35, 2307502 (2023).
Yu, H. et al. Effects of halogenation of small-molecule and polymeric acceptors for efficient organic solar cells. Adv. Funct. Mater. 33, 2300712 (2023).
Xia, X. et al. Uncovering the out-of-plane nanomorphology of organic photovoltaic bulk heterojunction by GTSAXS. Nat. Commun. 12, 6226 (2021).
Yu, R. et al. Self-assembly metal chelate as ultraviolet filterable interface layer for efficient organic solar cells. Adv. Energy. Mater. 12, 2201306 (2022).
Gao, H. et al. Self-aggregated light-trapping nanodots for highly efficient organic solar cells. Small 18, 2205128 (2022).
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (52373169 and 22379011).
Author information
Authors and Affiliations
Contributions
Z.T. and Z.H. proposed the idea. Z.H. and R.Y. designed the experiments. Z.H., Y.D., R.W., and Y.Z. fabricated and characterized the solar cells. Z.H. conducted data analysis. The first draft of manuscript was prepared by Z.H. and R.Y. revised and finalized the manuscript. Z.T. directed and supervised the project. All authors participated in the discussion and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Yiqiang Zhang, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
He, Z., Yu, R., Dong, Y. et al. Minimized optical/electrical energy loss for 25.1% Monolithic perovskite/organic tandem solar cells. Nat Commun 16, 1773 (2025). https://doi.org/10.1038/s41467-025-57093-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-57093-1
