
Abstract
Closely related genes typically display common essential functions but also functional diversification, ensuring retention of both genes throughout evolution. The histone lysine acetyltransferases KAT6A (MOZ) and KAT6B (QKF/MORF), sharing identical protein domain structure, are mutually exclusive catalytic subunits of a multiprotein complex. Mutations in either KAT6A or KAT6B result in congenital intellectual disability disorders in human patients. In mice, loss of function of either gene results in distinct, severe phenotypic consequences. Here we show that, surprisingly, 4-fold overexpression of Kat6b rescues all previously described developmental defects in Kat6a mutant mice, including rescuing the absence of hematopoietic stem cells. Kat6b restores acetylation at histone H3 lysines 9 and 23 and reverses critical gene expression anomalies in Kat6a mutant mice. Our data suggest that the target gene specificity of KAT6A can be substituted by the related paralogue KAT6B, despite differences in amino acid sequence, if KAT6B is expressed at sufficiently high levels.
Similar content being viewed by others


Introduction
In eukaryotic cells, transcription is influenced by the nucleosomal barriers imposed by histone proteins. Post-translational modifications of chromatin, for example histone acetylation, regulate chromatin structure and influence the nucleosome landscape such as to promote or suppress the expression of target loci1. Lysine acetylation is catalysed by acetyltransferase enzymes and generally associated with increased gene expression and the regulation of fundamental cellular functions2,3 Three families of histone acetyltransferases, with well-described acetylation domains4,5, are currently defined based on structural and sequence conservation: the MYST family6, the GNAT family7 and the p300/CBP family8.
Interestingly, histone lysine acetyltransferases typically occur as pairs of highly similar proteins. MYST family members KAT6A (MOZ) and KAT6B (QKF/MORF) share identical domain structure and high sequence similarity across all functional domains6. Likewise, both the GNAT family proteins, KAT2A (GCN5) and KAT2B (PCAF)9 and the p300/CBP family proteins KAT3A (CBP) and KAT3B (P300)8,10,11 share identical domain structure and high sequence similarity across all functional domains. Sequence and structural similarities between family members likely resulted from ancestral gene duplication events and subsequent neofunctionalisation12. Indeed, in vivo studies have demonstrated that these proteins have acquired independent functions13,14; however, the extent to which one protein within a pair of closely related chromatin regulatory proteins can replace the other, has not been assessed.
KAT6A and KAT6B are mutually exclusive catalytic subunits of a shared multi-protein complex including chromatin adaptor proteins of the BRPF family, primarily BRPF1, ING family, ING5 and ING4, and MEAF15,16. Both proteins have been shown to acetylate H3K9 and H3K23 in a range of cell and tissue systems17,18,19,20,21,22,23. The KAT6A and KAT6B genes are oncogenes. KAT6A (MOZ, monocytic leukaemia zinc finger gene) was originally identified in an aggressive form of acute myeloid leukaemia resulting from a translocation fusing it to KAT3A24. A number of other recurrent translocations of the KAT6A locus have since been identified in leukaemia with a variety of translocation partners25. Interestingly, loss of just one allele of KAT6A greatly enhances survival and disease latency in a mouse model of MYC driven lymphoma26. Similarly, KAT6B translocations have been identified in leukaemia and leiomyomata27,28. KAT6A and KAT6B mRNAs are commonly upregulated across a range of different cancer types29. This association with cancer has fuelled the development of drugs targeting both KAT6A and KAT6B proteins30,31, which have shown promise in clinical trials32.
In human patients, de novo mutations in KAT6A or KAT6B result in congenital disorders defined by cognitive impairment, developmental delay and craniofacial dysmorphogenesis33,34,35,36. Heterozygous mutations in the KAT6A gene result in Arboleda-Tham syndrome (ARTHS)33,34, while mutations in the KAT6B gene result in the Say-Barber-Biesecker-Young Simpson variant of Ohdo syndrome (SBBYSS)35 or Genitopatellar syndrome (GPS)36.
Deletion of the Kat6a or Kat6b gene in mice has identified their unique functions during development. Most Kat6bgt/gt mice, deficient in 90% of Kat6b mRNA, die at birth on a 129 Sv inbred genetic background, while the 20% surviving Kat6bgt/gt mice show a failure to thrive, short stature and a squared skull37,38. Kat6bgt/gt mice show abnormalities of the brain, including a reduced number of GAD67-positive interneurons and reduced numbers of layer V pyramidal neurons37, as well as fewer neural stem cells (NSCs) in the adult subventricular zone. Kat6bgt/gt NSCs show reduced proliferation and self-renewal and a reduced capacity to differentiate into neurons39. Mice homozgous for a null allele of Kat6b show premature ossification and increased bone density in the skull and long bones40. Kat6b+/− show learning and memory defects that can be ameliorated by increasing histone acetylation41. These findings suggest that KAT6B function is primarily required for normal brain, craniofacial and skeletal development. Consistently, Kat6b is most strongly expressed in the primordia of these tissues during embryonic development37.
Loss of KAT6A, either through deletion of the carboxy terminus or deletion of exons 5 to 917,18, results in lethality at embryonic day (E) 14-18, depending on the genetic background18, an absence of transplantable haematopoietic stem cells (HSCs)42,43, an extensive anterior homeotic transformation17, cleft palate18,44 and cardiac and large vessel defects18,45. The roles of KAT6A within the foetal haematopoietic system, have been shown to depend upon its histone acetyltransferase function46,47. These tissues are not affected in mice lacking KAT6B, with the exception of a reduction, but not absence of hematopoietic stem cells in Kat6b−/− embryos23.
The recent development of drugs targeting both KAT6A and KAT6B has highlighted the need to better understand the cellular functions of these chromatin regulators. While it is clear from single knockout studies that one factor within the pair cannot compensate for the other at endogenous levels, it remains to be determined if there are truly unique, non-redundant functions of these proteins. To assess this, we overexpressed Kat6b mRNA 4-fold above endogenous levels in mice lacking Kat6a. We show here that Kat6b overexpression can restore all anomalies previously described in Kat6a mutant mice, including the 100% penetrant lethality, gene expression anomalies and histone acetylation deficits at histone lysine targets perturbed by loss of KAT6A. These data indicate that KAT6B can completely replace loss of essential KAT6A functions, if expressed at sufficiently high levels, despite KAT6B not normally regulating critical processes dependent on KAT6A.
Results
Structure and expression of KAT6A and KAT6B
KAT6A and KAT6B have identical protein domain structures and high sequence similarity in all functional domains (Fig. 1a). During development, Kat6a mRNA is expressed at higher levels than Kat6b mRNA (Fig. 1b). Heterozygous mutations in the human KAT6A gene result in ARTHS33,34, while mutations in the KAT6B gene result in the Say-Barber-Biesecker-Young-Simpson variant of Ohdo syndrome (SBBYSS)35 or genitopatellar syndrome (GTPTS)36. Despite being clinically distinct, there is notable overlap in the clinical presentation across these disorders (Supplementary Fig. 1b; Supplementary Data 1).

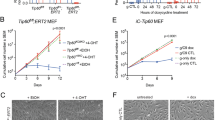
a Schematic of KAT6A and KAT6B mRNA with exon structure and protein domains/regions encoded are shown in colour. White boxes indicate an uncharacterised region. The percentage amino acid sequence similarity between protein domains is indicated. Non-coding exons are indicated in grey. b Kat6a and Kat6b mRNA levels from RNA-seq experiments analysing E8.5 embryos75, E9.5 embryos (this work), E10.5 embryos54, E10.5 pharyngeal arches44, E10.5 heart75, E12.5 MEFs69, E14.5 LSK cells76. c Western immunoblot detecting histone H3 acetylated on lysine 23 (H3K23ac) and pan-H3 as a loading control in acid extracted histones from E14.5 mouse embryonic fibroblasts (MEFs). Each lane represents histones from MEFs isolated from an individual E14.5 foetus. 250 ng protein loaded per lane. d Quantification of the western immunoblot in (a). Each circle represents one lane of the immunoblot in (a). e Cumulative growth curve of MEFs isolated from Kat6a+/+Kat6b+/+, Kat6a–/–Kat6b+/+, Kat6a+/+Tg(Kat6b) and Kat6a–/–Tg(Kat6b) E14.5 foetuses, passages 0–10. f Representative images of MEFs from Kat6a+/+Kat6b+/+, Kat6a–/–Kat6b+/+, Kat6a+/+Tg(Kat6b) and Kat6a–/–Tg(Kat6b) E14.5 foetuses at passage 3. Scale bar = 50 μm. g–j Western immunoblot detecting H3K9ac (e) and H3K23ac (g) with pan-H3 as a loading on acid extracted histones from E9.5 embryos. Each lane represents histones from an individual E9.5 embryo. 500 ng (g) or 250 ng () (i) protein loaded per lane. Quantification of (g) in (h) and of (i) in (j). k Representative images of E9.5 Kat6b+/+Kat6b+/+, Kat6a–/–Kat6b+/+, Kat6a–/–Tg(Kat6b) and Kat6a+/+Tg(Kat6b) embryos. Scale bar = 1 mm. N = 3–8 embryos or foetuses per genotype (a), MEF cultures derived from 3 foetuses per genotype (c, d, e, f). 3 embryos per genotype (g, h, i, j, k). Circles represent individual embryos or foetuses (b, d, h, j). Data are presented as mean ± s.e.m. and were analysed using a one-way ANOVA with Dunnett post hoc correction (b, g, i) or two-way ANOVA with Sidak post-hoc correction (c). Drawings created in BioRender, Bergamasco, M. (2025) https://BioRender.com/m13f247.
To test if transgenic overexpression of the Kat6b gene was able to rescue the phenotypic anomalies resulting from loss of the Kat6a gene, we generated transgenic mice overexpressing the Kat6b gene from pBACe3.6 clone RP23-360F23. This clone contained all coding exons as well as 21 kb 5 prime and 42 kb 3 prime of the expressed sequence (Supplementary Fig. 1a, b). Seven copies of the pBACe3.6 inserted into the mouse genome, resulting in the Tg(Kat6b) allele, which caused a 4-fold increase in Kat6b mRNA levels (Supplementary Fig. 1c). Mice were maintained on an FVB x BALB/c hybrid background as Kat6b overexpressing mice were not viable on inbred backgrounds.
To assess the potential of KAT6B to compensate for KAT6A, we crossed Tg(Kat6b) heterozygous mice to mice lacking exons 5–9 of Kat6a (Kat6a+/–)17,18. Embryonic day (E) 9.5 Tg(Kat6b) embryos and E14.5 and E18.5, Tg(Kat6b) foetuses were present at expected Mendelian ratios (N = 15, p = 0.2; N = 21, p = 0.6; N = 18, p = 0.6; at E9.5, E14.5 and E18.5, respectively; binomial probability p values). Kat6a–/– foetuses were underrepresented at E18.5 (N = 16, p = 0.8; N = 6, p = 0.1; N = 4, p = 0.02; at E9.5, E14.5 and E18.5, respectively). In contrast, Kat6a–/–Tg(Kat6b) foetuses that lacked Kat6a, but overexpressed Kat6b, were obtained at the expected Mendelian ratio (N = 9, p = 1.0; N = 8, p = 1.0; N = 10, p = 1.0; at E9.5, E14.5 and E18.5, respectively).
Kat6b overexpression restored histone acetylation and cell proliferation in Kat6a –/– Tg(Kat6b) fibroblasts and histone acetylation in Kat6a –/– Tg(Kat6b) embryos
To determine whether KAT6B was capable of compensating for the loss of KAT6A at the biochemical level, histone acetylation levels at previously identified KAT6A lysine targets, H3K9 and H3K23, as well as H3K14, were assessed chromatin-wide by Western blotting in primary mouse embryonic fibroblasts (MEFs) and in whole embryonic day 9.5 (E9.5) embryos.
Kat6a–/– MEFs had a 40% reduction in acetylation at H3K23 (H3K23ac), relative to wild type controls (p = 0.03; Fig. 1c, d). Overexpression of Kat6b in Kat6a–/–Tg(Kat6b) MEFs returned H3K23ac levels to wild-type levels (Fig. 1c, d). H3K23ac was similar in Kat6b+/+Tg(Kat6b) and wild-type Kat6a+/+Kat6b+/+ control MEFs (Fig. 1c, d). Chromatin-wide H3K9 and H3K14 acetylation levels were not affected by KAT6A or KAT6B status in this cell type (Supplementary Fig. 2a–d).
Consistent with previous reports30,47,48, Kat6a–/–Kat6b+/+ MEFs underwent cell cycle arrest after only 3 passages in culture (p = 0.00007; Fig. 1e, f). Overexpression of Kat6b restored cell proliferation in Kat6a–/–Tg(Kat6b) MEFs compared to Kat6a–/–Kat6b+/+ cells to levels similar to wild-type Kat6a+/+Kat6b+/+ control MEFs (Fig. 1e, f).
In E9.5 Kat6a–/–Kat6b+/+ embryos, acetylation levels at H3K9 (p = 0.046; Fig. 1g, h) and H3K23 (p = 0.02; Fig. 1i, j) were 32% and 43% reduced, respectively, compared to Kat6a+/+Kat6b+/+ control embryos. Overexpression of Kat6b returned the histone acetylation levels to normal in Kat6a–/–Tg(Kat6b) embryos compared to Kat6a+/+Kat6b+/+ control embryos (Fig. 1c, d, g–j). Interestingly, H3K9 and H3K23 were elevated by 41% and 39%, respectively, in Kat6a+/+Tg(Kat6b) samples compared to controls (p = 0.01–0.03); (Fig. 1g–j). H3K14ac was not affected by KAT6A or KAT6B status in E9.5 embryos (Supplementary Fig. 2e, f). Representative images of E9.5 embryos are shown in (Fig. 1k).
Kat6b overexpression reversed most gene expression changes present in Kat6a –/– E9.5 embryos
Loss of KAT6A has previously been shown to reduce expression of gene families encoding embryonic patterning transcription factors, most notably Hox genes17, Tbx genes18 and Dlx genes44. To determine how mRNA levels affected by loss of KAT6A were altered when KAT6B was overexpressed, we performed RNA-sequencing of Kat6a–/–Kat6b+/+, Kat6a+/+Kat6b+/+, Kat6a–/–Tg(Kat6b) and Kat6a+/+Tg(Kat6b) E9.5 embryos (Supplementary Data 2–5). E9.5 was chosen as a timepoint when major patterning genes previously shown to be perturbed by loss of KAT6A are highly expressed.
Samples clustered within genotype and segregated between genotypes (Fig. 2a). Interestingly, Kat6a–/–Tg(Kat6b) RNA profiles were more closely related to wild-type control profiles in dimension 1, as assessed by multidimensional scaling, compared to Kat6a–/–Kat6b+/+ profiles, but segregated from the other genotypes in dimension 2, indicating that, despite more closely resembling wild-type controls than Kat6a–/–Kat6b+/+samples, Kat6a–/–Tg(Kat6b) samples still maintained distinct RNA expression profiles.

a–i RNA-sequencing data of Kat6a+/+Kat6b+/+, Kat6a–/–Kat6b+/+, Kat6a+/+Tg(Kat6b) and Kat6a–/–Tg(Kat6b) E9.5 embryos. N = 4 embryos per genotype. Data were analysed as described in the ‘methods’ section. A false discovery (FDR) < 0.05 was used as the cut off for significance. a Multidimensional scaling plot of the leading gene expression differences between samples in pair-wise comparisons showing Kat6a+/+Kat6b+/+, Kat6a–/–Kat6b+/+, Kat6a+/+Tg(Kat6b) and Kat6a–/–Tg(Kat6b) E9.5 embryo RNA samples. b M (log ratio) and A (mean average) plot showing Kat6a–/–Kat6b+/+ vs. Kat6a+/+Kat6b+/+ and Kat6a–/–Tg(Kat6b) vs. Kat6a+/+Kat6b+/+ embryos. The total numbers of upregulated and downregulated genes at FDR < 0.05 are indicated in each comparison. Upregulated genes are represented in red, downregulated in blue and unchanged genes in grey. c Heatmap showing genes differentially expressed in the contrast of Kat6a–/–Kat6b+/+ vs. Kat6a+/+Kat6b+/+ embryos but displayed for all genotypes. d Venn diagram showing the intersection of genes downregulated (DR) in Kat6a–/– Kat6b+/+ vs. Kat6a+/+Kat6b+/+ embryos with those downregulated in Kat6a–/–Tg(Kat6b) samples vs. Kat6a+/+Kat6b+/+ embryos compared to those upregulated in Kat6a–/–Tg(Kat6b) samples vs. Kat6a+/+Kat6b+/+ embryos. e Top 20 gene ontology (GO) (BP) terms downregulated in Kat6a–/–Kat6b+/+ vs. Kat6a+/+Kat6b+/+ embryos and rescued in Kat6a–/–Tg(Kat6b) when compared to Kat6a+/+Kat6b+/+ embryos. GO terms enriched with p < 10–6 are shown. f Top 20 gene ontology (GO) (BP) terms upregulated in Kat6a–/–Kat6b+/+ vs. Kat6a+/+Kat6b+/+ embryos and rescued in Kat6a–/–Tg(Kat6b) when compared to Kat6a+/+Kat6b+/+ embryos. GO terms enriched with p < 10–6 are shown. g–i Specific gene families previously reported to be affected by loss of KAT6A17,18,44 were examined. Correction for multiple testing was conducted within gene family. FDRs for each comparison are shown above each bar. g–i Log2 fold-change in levels of HOX gene mRNA (g), DLX gene mRNA (h) and TBX gene mRNA (i) across pairwise comparisons Kat6a–/–Kat6b+/+ vs. Kat6a+/+Kat6b+/+ embryos, Kat6a–/–Tg(Kat6b) vs. Kat6a+/+Kat6b+/+ embryos and Kat6a–/–Tg(Kat6b) vs. Kat6a–/–Kat6b+/+ embryos. FDRs are displayed above or below the bars.
A total of 482 genes were differentially expressed in Kat6a–/–Kat6b+/+ vs. Kat6a+/+Kat6b+/+ E9.5 embryos with transcriptome-wide significance (FDR < 0.05); 245 genes were downregulated, and 237 genes were upregulated (Supplementary Data 2). Overexpression of Kat6b in Kat6a–/–Tg(Kat6b) embryos reduced the number of differentially expressed genes compared to Kat6a+/+Kat6b+/+ embryos to 285 genes, 165 downregulated and 120 upregulated (Fig. 2b; Supplementary Data 3). Other pairwise comparisons are shown in Supplementary Fig. 3a and Supplementary Data 4–7. A heat map shows that Kat6a–/–Tg(Kat6b) embryos clustered more closely with wild-type Kat6a+/+Kat6b+/+control embryos than Kat6a–/–Kat6b+/+samples (Fig. 2c).
Of the 245 genes downregulated in Kat6a–/–Kat6b+/+ vs. Kat6a+/+Kat6b+/+, only 28 genes were similarly downregulated in Kat6a–/–Tg(Kat6b) samples (Fig. 2d; Supplementary Data 2 and 3), while two of the rescued genes, (Cdx2, Hoxa10), were overcompensated, i.e., expressed at higher levels in Kat6a–/–Tg(Kat6b) vs. Kat6a+/+Kat6b+/+ E9.5 embryos.
The genes downregulated in Kat6a–/–Kat6b+/+ vs. Kat6a+/+Kat6b+/+ E9.5 embryos were enriched in gene ontology (BP) terms relating to embryonic development, embryo patterning and DNA binding transcription factors regulating these processes (Supplementary Fig. 3b). The genes that displayed a rescue of mRNA levels from downregulated in Kat6a–/–Kat6b+/+ vs. Kat6a+/+Kat6b+/+ E9.5 embryos to normal levels in Kat6b in Kat6a–/–Tg(Kat6b) vs. Kat6a+/+Kat6b+/+ E9.5 embryos were enriched for similar developmental processes (Fig. 2e, f). Genes upregulated in Kat6a–/–Kat6b+/+ vs. Kat6a+/+Kat6b+/+ E9.5 embryos showed no specificity to embryonic development (Supplementary Fig. 3c).
Among the rescued genes were the major families of embryonic patterning genes previously reported as downregulated in Kat6a–/– vs. Kat6a+/+ embryos; Hox genes17, Tbx genes18 and Dlx genes44. The major downregulation of Hox genes observed in Kat6a–/–Kat6b+/+ vs. Kat6a+/+Kat6b+/+ E9.5 embryos (FDR < 10−6 to 0.03 within gene family; Supplementary Fig. 3d, e; Data 8) was no longer present in Kat6a–/–Tg(Kat6b) vs. Kat6a+/+Kat6b+/+ E9.5 embryos (Fig. 2g), indicating that KAT6B can promote expression of Hox genes in the absence of KAT6A. Only Hoxb3 mRNA levels were not fully returned to wild-type levels, but still were significantly elevated in Kat6a–/–Tg(Kat6b) vs. Kat6a–/–Kat6b+/+ embryos (FDR = 10−5; Fig. 2g), indicating a partial rescue. Similarly, Dlx genes downregulated in Kat6a–/–Kat6b+/+ vs. Kat6a+/+Kat6b+/+ embryos (FDR = 6 × 10−5 to 0.004 within gene family; Supplementary Fig. 3d, e, Data 8) were no longer downregulated in Kat6a–/–Tg(Kat6b) vs. Kat6a+/+Kat6b+/+ (FDR = 0.2 to 1; Fig. 2h). Tbx genes were downregulated Kat6a–/–Kat6b+/+ vs. Kat6a+/+Kat6b+/+ embryos (FDR < 10−6 to 0.04 within gene family; Supplementary Data 8) and were upregulated in Kat6a–/–Tg(Kat6b) vs. Kat6a–/–Kat6b+/+, except Tbx15 (Fig. 2i; Supplementary Fig. 3d, e, Supplementary Data 8). This upregulation returned Tbx2, Tbx3 and Tbx5 mRNA levels in Kat6a–/–Tg(Kat6b) embryos to comparable levels to Kat6a+/+Kat6b+/+ embryos (FDR = 0.06 to 0.3). The mRNA levels of Tbx1 were significantly elevated in Kat6a–/–Tg(Kat6b) embryos above Kat6a–/–Kat6b+/+ embryos (FDR = 0.0006), but still lower than in Kat6a+/+Kat6b+/+ embryos (FDR = 0.006), indicating a partial rescue. Overall, 217 (89%) of 245 genes downregulated and 222 (94%) of the 237 upregulated genes in Kat6a–/–Kat6b+/+ vs. Kat6a+/+Kat6b+/+ E9.5 embryos were rescued by overexpression of Kat6b in Kat6a–/–Tg(Kat6b) vs. Kat6a+/+Kat6b+/+ embryos, indicating that KAT6B overexpression substantially restores expression of genes, which were reduced in the absence of KAT6A (Fig. 2d; Supplementary Fig. 4a, b). Examining the gene expression not rescued in more detail we found the majority of these 28 downregulated and 15 upregulated genes showed relatively small differential gene expression changes, less than 2-fold, (Supplementary Fig. 4c, d), which were associated with GO terms relating to transcription in the case of downregulated genes and cell adhesion in the case of upregulated genes (Supplementary Fig. 4e, f). Some 236 genes were uniquely differentially expressed in Kat6a–/–Tg(Kat6b) vs. Kat6a+/+Kat6b+/+ embryos (Supplementary Fig. 4g–i) and a comparatively small number of genes (75) were differentially expressed when embryos overexpressing Kat6b were compared to wild type (Supplementary Fig. 4j–l).
Kat6b overexpression restored transplantable haematopoietic stem cells that are absent in Kat6a –/– mice
KAT6A is essential for development of HSCs. Germline deletion of the Kat6a gene results in a complete failure to form definitive HSCs during foetal development42,43, and deletion of Kat6a in adult HSCs causes their complete loss49.
The numbers of HSCs in Kat6a+/+Kat6b+/+, Kat6a+/+Tg(Kat6b), Kat6a–/–Kat6b+/+ and Kat6a–/–Tg(Kat6b) foetal livers were assessed by flow cytometry (Fig. 3a, b). Analysis was performed at E14.5, at the peak of foetal haematopoiesis in mice50,51 and HSCs were identified as CD48– CD150+, as described52. As previously described42,43, Kat6a–/–Kat6b+/+ foetal livers showed a 95% reduction in cells with an HSC cell surface phenotype compared to wild-type Kat6a+/+Kat6b+/+ control foetal livers (p = 0.00007; Fig. 3b). Overexpression of Kat6b in Kat6a–/–Tg(Kat6b) foetuses elevated the number of HSC 11.5-fold, compared to Kat6a–/–Kat6b+/+ foetuses (p = 0.01; Fig. 3b), however, HSCs were still reduced in Kat6a–/–Tg(Kat6b) foetuses compared to Kat6a+/+Kat6b+/+ control foetuses (p = 0.04; Fig. 3b). Consistently, expression of the haematopoietic marker gene Kit50,53 was downregulated in Kat6a–/–Kat6b+/+ vs. Kat6a+/+Kat6b+/+ E9.5 embryos (FDR = 0.007; Fig. 3c), rescued in Kat6a–/–Tg(Kat6b) vs. wild-type embryos (FDR = 0.2) and upregulated in Kat6a–/–Tg(Kat6b) vs. Kat6a–/–Kat6b+/+ embryos (FDR = 6 × 10–5; Fig. 3c).

a Flow cytometry gating strategy for the assessment of haematopoietic stem cells (HSCs) in E14.5 foetal livers. HSCs were identified as the CD48–CD150+ cell population within the lineage marker negative (LIN–) cKIT and SCA1 positive cell population (LSK; gated on single, viable cells, lacking expression of lineage (LIN) markers B220, CD19, CD4, CD8, GR1 and TER119 and expressing SCA1 and cKIT). Representative plots for HSCs are shown for each genotype. b Total number of HSCs per foetal liver of N = 3 Kat6a–/–Kat6b+/+, N = 8 Kat6a+/+Kat6b+/+, N = 6 Kat6a–/–Tg(Kat6b) and N = 4 Kat6a+/+Tg(Kat6b) foetuses. c Log2 fold-change in Kit mRNA levels in E9.5 embryos as assessed by RNA-sequencing across the comparisons Kat6a–/–Kat6b+/+ vs. Kat6a+/+Kat6b+/+, Kat6a–/–Tg(Kat6b) vs. Kat6a+/+Kat6b+/+ and Kat6a–/–Tg(Kat6b) vs. Kat6a–/–Kat6b+/+ embryos. N = 4 embryos per genotype. The FDRs are shown above the bars. Data were analysed as described in the ‘methods’ section under RNA sequencing and analysis. d Percentage survival of irradiated recipient mice after transplantation of 1 × 106 foetal liver cells from N = 3 Kat6a–/–Kat6b+/+, N = 4 Kat6a–/–Tg(Kat6b), N = 3 Kat6a+/+TgKat6b) or N = 5 Kat6a+/+Kat6b+/+ E14.5 mouse foetuses; each foetal liver sample was transplanted into 3 lethally irradiated recipients. Data are displayed in a Kaplan-Meier plot and were analysed using a Mantel-Cox test. e Automated haematological analyser (ADVIA) assessment of red blood cells per μl peripheral blood in recipient mice of E14.5 foetal liver donor cells from N = 5 Kat6a+/+Kat6b+/+, N = 3 Kat6a+/+Tg(Kat6b), N = 4 Kat6a–/–Tg(Kat6b) or N = 3 Kat6a–/–Kat6b+/+ foetuses, at a time 3–4 weeks after transplantation when the recipient mice of Kat6a–/–Kat6b+/+ donor cells in each set reached the ethical end-point. Each foetal liver sample was transplanted into 3 recipient mice. Each circle represents the average of the recipient mice of an individual foetal liver donor. Data are displayed as mean ± s.e.m. and were analysed using a one-way ANOVA with Dunnett post-hoc correction. f Gating strategy for the assessment of peripheral blood of foetal liver transplant recipient mice. B cells were defined as B220+CD19+, T cells as CD4+ or CD8+, granulocytes as GR1hi MAC1+ and monocytes as GR1loMAC1+. Foetal liver donor-derived cells (CD45.1+) were distinguished from residual recipient cells (CD45.1/2+). g, h Contribution of foetal liver (donor)-derived cells to peripheral blood populations at 4 weeks (f) and 20 weeks (g) post transplantation of the recipients described in (e). i Gating strategy for the assessment of stem and progenitor cells in the bone marrow of foetal liver transplant recipient mice. Cell types as indicated on the plots were distinguished by CD48 vs. CD150, CD34 vs. CD16.32 or IL7Rα expression. (j–k) Contribution of foetal liver (donor)-derived cells to stem and progenitor populations in the bone marrow of foetal liver cell recipients at 20 weeks post-transplantation in CD48 vs. CD150 (i), CD34 vs CD16/32 (j) cell populations. l Gating strategy for the assessment of B cell progenitors in the bone marrow of foetal liver transplant recipient mice. Cells were gated on single, viable cells co-expressing B220 and CD19 and distinguished by cKIT, IgM and IgD expression to identify the cell populations indicated in the plots. m Contribution of foetal liver (donor)-derived cells to bone marrow B cell progenitors 20 weeks post-transplant. Circles represent individual foetuses (b) or the mean of three recipients of a single foetal liver donor (g, h, j, k, m). Data are presented as mean ± s.e.m. and were analysed using a one-way ANOVA (b) or two-way ANOVA with Tukey post hoc correction (g, h, j, k, m). HSCs, haematopoietic stem cells; LK; lineage marker negative cKIT+ cells; LSK, lineage marker negative SCA1+cKIT+ cells; HPC-1 (haematopoietic progenitor 1), HPC-2 (haematopoietic progenitor 2), MPP (multipotent progenitor), CLP (common lymphoid progenitor), CMP (common myeloid progenitor), GMP (granulocyte macrophage progenitors), MEP (megakaryocyte/erythroid progenitor). Granulo (granulocytes), Mono (monocytes); superscript: int, intermediate.
To assess the effect of KAT6B overexpression on the function of HSCs lacking KAT6A, 1 × 106 foetal liver cells were transplanted into irradiated recipients, as previously described43. Recipients of Kat6a–/–Kat6b+/+ foetal liver cells reached the ethical endpoint within 21 days (p < 10–6; Fig. 3d), exhibiting anaemia (p = 10–6; Fig. 3e) and minimal contribution of the foetal liver donor cells to the recipient peripheral blood (Supplementary Fig. 5a). In contrast, recipients of Kat6a–/–Tg(Kat6b) cells survived beyond 150 days, as did recipients of wild type or Kat6a+/+Tg(Kat6b) cells (Fig. 3d). At 4 weeks post-transplantation, Kat6a–/–Tg(Kat6b) donor cell contribution to peripheral blood cells was similar to Kat6a+/+Kat6b+/+ donor cells, except for a reduction in CD8 T cells (p = 0.03; Fig. 3f, g). At 20 weeks post-transplantation, contribution to all peripheral cell types was identical between Kat6a–/–Tg(Kat6b) and Kat6a+/+Kat6b+/+ donor cells (Fig. 3h). Interestingly, compared to wild type controls, Kat6a+/+Tg(Kat6b) donor cells showed an elevated contribution to CD8 T cells at 4 weeks, (p < 10–6; Fig. 3g) and 20 weeks (p = 0.00005; Fig. 3h), with a reduced contribution to CD4 T cells at 20 weeks (p < 10–6; Fig. 3h). In the bone marrow at 20 weeks post-transplantation, recipients of Kat6a–/–Tg(Kat6b), Kat6a+/+Kat6b+/+ and Kat6a+/+Tg(Kat6b) donor cells showed comparable contributions to all stem and progenitor cell populations, except for a small reduction (less than 10%) in Kat6a–/–Tg(Kat6b) contribution to the B220+CD19+, ProB and PreB cell populations (p < 10–6 to 0.002; Fig. 3i–m). No difference was observed between recipients of Kat6a+/+Kat6b+/+ and Kat6b+/+Tg(Kat6b) recipients across bone marrow populations analysed (Fig. 3i–m).
Kat6b overexpression rescued the anterior homeotic transformation seen in Kat6a –/– mice
Germline deletion of the Kat6a gene results in duplication of the first cervical vertebra, the atlas, accompanied by an extensive and complete homeotic transformation of 19 body segments. This anterior homeotic transformation is caused by a posterior shift of the anterior expression boundary and expression levels of Hox genes, including Hoxa3, Hoxa4, Hoxb3 and Hoxb417. Consistent with this previous work17 and the reduced Hox gene expression in E9.5 embryos shown here (Fig. 2g), Kat6a–/–Kat6b+/+ pups displayed an anterior homeotic transformation at E18.5 (Fig. 4a; Supplementary Fig. 6). In addition, we saw disrupted sternum sections, sternebrae and incorrect alignment of ribs at the sternum in Kat6a–/–Kat6b+/+ pups (Fig. 4b). Congruent with a rescue of the Hox gene expression profile at E9.5 (Fig. 2g), the vertebral segment identity, sternebrae structure and rib attachment were rescued in Kat6a–/–Tg(Kat6b) pups at E18.5 (Fig. 4a, b). The cervical vertebrae, sternebrae and rib attachment were normal in Kat6a+/+Kat6b+/+ and Kat6a+/+Tg(Kat6b) pups (Fig. 5a, b). No obvious effects of KAT6A or KAT6B genotype was observed on lumbar and sacral elements (Supplementary Fig. 7).

a, b Alizarin red (bone) and Alcian blue (cartilage) stained skeletal preparations of N = 9 Kat6a+/+Kat6b+/+, 1 Kat6a–/–Kat6b+/+, 4 Kat6a–/–Tg(Kat6b) and 5 Kat6a+/+Tg(Kat6b) E18.5 foetuses with labelled cervical vertebrae (a) and sternum (b), arrow indicates abnormal rib attachment (b). Note that embryos homozygous for this allele of Kat6a rarely survive until E18.5. Representative images are shown. Representative images of RNA/RNA whole-mount situ hybridisation to detect Hoxa5 (c) and Hoxc5 (d) mRNA in Kat6a+/+Kat6b+/+, Kat6a–/–Kat6b+/+, Kat6a–/–Tg(Kat6b) and Kat6a+/+Tg(Kat6b) E10.5 embryos, with anterior expression boundary indicated. Note no staining using the sense control probe (n = 3 wild type embryos). The 3–4 most cranial somites are indicated by a dot (c, d). The anterior expression boundary of Hoxa5 is indicated by a stippled line (c). The distance between the anterior boundary of the Hoxc5 expression domain in the neural tube and the caudal boundary of the otic vesicle is indicated by a bracket (d). The distance was measured and is displayed in the bar graph in (d). Hoxa5 N = 3, Kat6a+/+Kat6b+/+, N = 3, Kat6a–/–Kat6b+/+, N = 3, Kat6a–/–Tg(Kat6b) and N = 3, Kat6a+/+Tg(Kat6b) were used. Hoxc5 N = 4, Kat6a+/+Kat6b+/+, N = 4, Kat6a–/–Kat6b+/+, N = 4, Kat6a–/–Tg(Kat6b) and N = 4, Kat6a+/+Tg(Kat6b) were used. This experiment was repeated a second time with each probe with N = 3 embryos per genotype. Data are presented as means ± s.e.m. and were analysed by one-way ANOVA with Tukey’s multiple comparison test. Circles in the bar graph (d) represent individual embryos. 1b, mandibular region of the first pharyngeal arch; 2, second pharyngeal arch; At, atlas = 1st cervical vertebra, C1; Ax, axis = 2nd cervical vertebra, C2; C1 to C7, 1st to 7th cervical vertebrae; C1’ to C8’, 1st to 8th abnormal Kat6a–/–Kat6b+/+ cervical vertebrae, whereby C8’ is supernumerary; E eye, FB forebrain, FL forelimb, HB hindbrain, He heart, HL hindlimb, M manubrium, MB midbrain, o otic vesicle, S sternebrae, T1 1st thoracic vertebra (rib bearing). Xp xiphoid process, Xs xiphisternum. Scale bar = 1 mm (a, b), 600 µm (c), 680 µm (d).

a–e Examination of N = 7 Kat6a+/+Kat6b+/+, N = 1 Kat6a–/–Kat6b+/+, N = 5 Kat6a–/–Tg(Kat6b) and N = 6 Kat6a+/+Tg(Kat6b) foetuses at E18.5 during dissection and by histopathology. Note that embryos homozygous for this allele of KAT6a rarely survive until E18.5. a Lateral view of the head and neck of E18.5 Kat6a+/+Kat6b+/+, Kat6a–/–Kat6b+/+, Kat6a–/–Tg(Kat6b) and Kat6a+/+Tg(Kat6b) foetuses. The arrow indicates an underdeveloped lower jaw in the Kat6a–/–Kat6b+/+ mouse. b Ventral view of the palate of E18.5 Kat6a+/+Kat6b+/+, Kat6a–/–Kat6b+/+, Kat6a–/–Tg(Kat6b) and Kat6a+/+Tg(Kat6b) foetuses. The arrow indicates a cleft palate in the Kat6a–/–Kat6b+/+ mouse. c, d Images of the heart and aortic arch of Kat6a+/+Kat6b+/+, Kat6a–/–Kat6b+/+, Kat6a–/–Tg(Kat6b) and Kat6a+/+Tg(Kat6b) E18.5 foetuses (c). Traces of the aortic arch and arteries (d). e H&E sections of the hearts of Kat6a+/+Kat6b+/+, Kat6a–/–Kat6b+/+, Kat6a–/–Tg(Kat6b) and Kat6a+/+Tg(Kat6b) E18.5 foetuses. Arrowhead indicates the ventricular septal defect in Kat6a–/–Kat6b+/+ animals. AA aortic arch, LA left atrium, LCCA left common carotid artery, LSA left subclavian artery, LV left ventricle, P2 secondary palate, PP primary palate, RA right atrium, RCCA right common carotid artery, RSA right subclavian artery, RV right ventricle, VS ventricular septum. Scale bars are 2 mm (a), 1 mm (b, c), 500 μm (e).
To examine the pattern of Hox gene expression more closely we performed whole mount in situ hybridisation. We have previously shown that not only is Hox gene expression down regulated in embryos but also that the anterior boundary is shifted posteriorly, which leads to an anterior homeotic transformation17. We examined the expression patterns of Hoxa3, Hoxa5 and Hoxc5, as we have previously shown that the expression of these genes most clearly demonstrates the effect of Kat6a loss on the anterior boundary of Hox gene expression17,54. As previously reported, the Hoxa5 and Hoxc5 expression is shifted posteriorly and expressed at lower levels in Kat6a–/–Kat6b+/+ embryos (Fig. 4c, d; Supplementary Fig. 7). The anterior boundary and level of expression of both Hoxa5 and Hoxc5 in rescued in Kat6a–/–Tg(Kat6b) embryos to the wild type position. Interestingly, the anterior boundary of Hoxa5 expression in Kat6a+/+Tg(Kat6b) embryos is not different to wild type littermate embryos. Similarly, Hoxa3 expression, reduced and posteriorly shifted in Kat6a–/–Kat6b+/+ embryos, is restored in Kat6a–/–Tg(Kat6b) embryos to the wild type pattern of expression (Supplementary Fig. 7).
Kat6b overexpression rescued the cleft palate, cardiac and aortic arch defects observed in Kat6a –/– mice
Kat6a–/– mice have previously been shown to have cleft palate18,44, ventricular septum defects18,45 and aortic arch defects18. Congruent with these previous studies, cleft palate (Fig. 5a, b), aortic arch defects (Fig. 5c, d) and ventricular septal defects (Fig. 5e) were observed in Kat6a–/–Kat6b+/+ mice (Fig. 5a–e). Overexpression of Kat6b in Kat6a–/–Tg(Kat6b) mice rescued each of these major phenotypic anomalies (Fig. 5a–e).
Kat6b overexpression rescued the pattern of H3K23 acetylation at Kat6a target loci
Western blot analysis showed that loss of KAT6A function results in a global reduction in H3K23ac in E9.5 embryos and MEFs, and that this is restored in the Kat6a–/–Tg(Kat6b) condition. To examine specific genomic loci in more detail, in particular Hox clusters, Tbx and Dlx genes, we performed Cleavage Under Targets and Tagmentation (CUT&Tag) sequencing for H3K23ac. Since H3K23ac is one of the most abundant histone acetylation modifications and is widespread throughout the genome we used a Drosophila spike-in control to allow direct comparison between samples. Across samples H3K23ac showed a characteristic peak at the transcription start site (TSS) and a large difference between Kat6a–/– vs. Kat6b+/+ in a multidimensional scaling plot (Supplementary Fig. 8a–c; Data 9). As expected, H3K23ac levels were reduced across the genome at 16889 loci in Kat6a–/– samples compared to wild type controls (Fig. 6a; Supplementary Fig. 8d). This reduction in H3K23ac was completely reversed in cells prepared from Kat6a–/–Tg(Kat6b) embryos (Fig. 6a; Supplementary Fig. 8e–g; Data 10–12). Specifically examining all Hox genes we found that H3K23ac was reduced in Kat6a–/– cells compared to wild type and normal levels were restored in Kat6a–/–Tg(Kat6b) cells (Fig. 6b). Similarly at both Tbx and Dlx genes, H3K23ac was reduced in Kat6a–/– cells and restored in Kat6a–/–Tg(Kat6b) cells (Fig. 6c, d). Mapping individual reads across Hox, Tbx and Dlx loci (Fig. 6e–g) showed that H3K23ac was distributed across the gene body with distinctive peaks at promoters and other genomic features (Supplementary Fig. 8c). Unfortunately, we were unable to correlate the distribution of KAT6A or KAT6B with H3K23ac peaks as no CUT&Tag capable antibodies with the necessary specificity are available. The wild type pattern of peaks was faithfully reproduced in Kat6a–/–Tg(Kat6b) cells (Fig. 6e–g).

a–g CUT&Tag results detecting histone H3 lysine 23 acetylation in (H3K23ac) in primary mouse embryonic fibroblasts isolated from E14.5 Kat6a+/+Kat6b+/+, Kat6a–/–Kat6b+/+, Kat6a–/–Tg(Kat6b) and Kat6a+/+Tg(Kat6b) E10.5 foetuses. N = 4 foetuses per genotype. Data were analysed as described in the ‘methods’ section. A false discovery rate (FDR) of less than 0.05 was considered significant. a Venn diagram showing number of genes with reduced H3K23ac levels in the indicated samples compared to wild type controls. In comparison to wild type cells Kat6a–/– cells show a global reduction in H3K23ac which is restored to normal in Kat6a–/–Tg(Kat6b) cells. b–d Log2 fold changes in H3K23ac levels at HOX genes (b), TBX genes (c) and DLX genes (e). Note that the reduction in H3K23ac in Kat6a–/– MEFs is restored to normal in Kat6a–/–Tg(Kat6b) MEFs. The FDR is indicated above or below each bar. e–g Read depth plots of H3K23ac in the HOXA gene cluster (e), at the Dlx1/Dlx2 locus (f) and at the Tbx3/Tbx5 locus. Note that the characteristic distribution of H3K23ac in wild type cells is restored in Kat6a–/–Tg(Kat6b) cells.
Kat6b overexpression rescued the lethality of Kat6a –/– mice
Overexpression of KAT6B rescued histone acetylation, gene expression patterns, transplantable HSCs, segment identity defects, heart and aortic arch defects and cleft palate previously described in Kat6a–/–Kat6b+/+ mice. This demonstrates a complete rescue of all previously described developmental anomalies resulting from loss of KAT6A, including those that were incompatible with survival. To assess if perinatal lethality was rescued, Kat6a–/– Tg(Kat6b) mice were allowed to develop to birth and were monitored in early life.
Kat6a–/–Kat6b+/+mice die between E14.5-E18.5 depending on the genetic background17,18. Overexpression of Kat6b completely rescued this foetal to perinatal lethality. Kat6a–/–Tg(Kat6b) mice developed normally in the postnatal period, even compared to Kat6a heterozygous (Kat6a+/–) mice, which were notably runted in early life (Fig. 7a, b). Furthermore, Kat6a–/–Tg(Kat6b) mice showed normal weight gain over the first 3 weeks of life, compared to reduced weight gain observed in both male and female Kat6a+/– mice (p = 0.001 and 1 × 10–5; Fig. 7b). Remarkably, Kat6a–/– Tg(Kat6b) mice of both sexes reached adulthood and were healthy and fertile. Among the offspring of Kat6a–/–Tg(Kat6b) x Kat6a+/–Kat6b+/+ matings, Kat6a–/–Kat6b+/+ mice were completely absent (N = 0 of 94; p < 10–6; Fig. 7c). In contrast, Kat6a–/–Tg(Kat6b) mice were present at the expected Mendelian ratio at weaning (N = 32 of 94; p = 1; Fig. 7c). This demonstrates that KAT6B overexpression in the absence of KAT6A not only rescued the effects of the homozygous loss of Kat6a, but improved development in early life compared to Kat6a heterozygosity.

a Representative photographs of wild type, Kat6a+/–Kat6b+/+ and Kat6a–/–Tg(Kat6b) mice at postnatal day 7 (left) and Kat6a+/+Kat6b+/+ and Kat6a–/–Tg(Kat6b) mice at 12 weeks of age (right). b Weights of Kat6a+/+Kat6b+/+ (23 female; 16 male), Kat6a+/–Kat6b+/+ (16 female; 4 male), Kat6a–/–Tg(Kat6b) (4 female; 4 male) and Kat6a+/+Tg(Kat6b) (4 female; 4 male) mice from postnatal week 1 to week 12. Data presented as mean ± s.e.m. and were analysed using a two-way ANOVA with Holm-Sidak post-hoc correction. c Genotypes at 3 weeks of age of offspring (N = 94) of matings with the parental genotypes Kat6a+/–Kat6b+/+ x Kat6a–/–Tg(Kat6b). The observed genotype frequency was compared to the expected Mendelian frequencies, p values of the binomial probability testing if a genotype was observed differed from the expected frequency are shown (two sided). d–h Flow cytometry analysis of the bone marrow of N = 4 adult Kat6a+/+Kat6b+/+, 5 Kat6a–/–Tg(Kat6b) and 5 Kat6a+/+Tg(Kat6b) mice. Flow cytometry gating strategies defining haematopoietic cell population using cell surface markers as described in Fig. 4. Each circle represents an individual mouse. Data in (e, g, h) are presented as mean ± s.e.m. Each circle represents an individual mouse. Data were analysed using a two-way ANOVA with Tukey post-hoc correction. Abbreviations as defined in Fig. 4. Flow cytometry gating strategy (d) and analysis (e) of B cell progenitor populations in the bone marrow. Gating strategy (f) and analysis (g, h) of haematopoietic stem and progenitor population in the bone marrow.
Given the requirement for KAT6A in the adult haematopoietic system49,55, bone marrow stem and progenitor populations were assessed in adult Kat6a–/–Tg(Kat6b) mice at 12 weeks of age. All stem and progenitor populations showed similar frequencies as in wild-type control mice (Fig. 7d–h), indicating that haematopoietic development in adulthood is normal in Kat6a–/–Tg(Kat6b) animals.
Discussion
In this study we demonstrate that KAT6B, expressed at 4-fold above endogenous levels in Kat6a–/– mice, rescues the previously described developmental defects resulting from homozygous loss of KAT6A. Remarkably, Kat6a–/–Tg(Kat6b) mice are born, show normal development to adulthood and normal blood cell development in adulthood.
In addition to rescuing the anatomical defects resulting from loss of KAT6A, KAT6B overexpression reverted ~90% of the changes in gene expression caused by loss of KAT6A and re-established global acetylation levels at H3K9 and H3K23 in whole E9.5 embryos and H3K23 in MEFs. Examining locus-specific acetylation we found complete restoration of the normal pattern of H3K23ac down to the level of individual peaks within transcription units. The rescue of genes downregulated in Kat6a–/–Kat6b+/+ mice by KAT6B overexpression is particularly interesting, as KAT6B is not ordinarily required for expression of many KAT6A-dependent genes. In particular, while KAT6A is essential for the normal expression the Hox, gene families, KAT6B has no role in regulation of these genes and the axial skeleton develops normally without KAT6B. The capacity of overexpressed KAT6B to carry out the role of promoting their expression in the absence of KAT6A indicates that any target gene specificity of KAT6A arising from the differences in amino acid sequence between KAT6A and KAT6B, and perhaps affecting protein-protein interaction, can be overcome by higher levels of KAT6B.
In utero treatment with retinoic acid, an activator of Hox gene expression56,57, rescues the anterior homeotic transformation in Kat6a–/– mice, but not other developmental defects17. Indeed, in utero retinoic acid treatment causes cardiac defects in Kat6a+/– mice18. Conversely, the cardiac septum defect of Kat6a–/– mice was rescued by overexpressing the Tbx1 gene18. Body segment identity has been restored in Kat6a–/– mice by additionally deleting Bmi154. BMI1 is a polycomb repressor protein that represses Hox gene expression and, when deleted, causes a posterior homeotic transformation58,59. Combined deletion of Kat6a and Bmi1 genes was found to restore the respective anterior and posterior homeotic transformations observed in single mutant mice54, but did not rescue the haematopoietic defects of Kat6a–/– mice55. Collectively, these results suggest that KAT6A does not act as a binary on-off switch, but rather the level of KAT6A activity acts to balance the activity of repressors to generate an appropriate level of gene expression in a cell-type specific context.
Our data suggest that KAT6B overexpression in the absence of KAT6A may result in a more favourable stoichiometry of complex subunits to allow KAT6B to take on the role of KAT6A, compared to endogenous levels of KAT6B or KAT6B overexpression in the presence of KAT6A. In the absence of KAT6A, the limitation of auxiliary complex member availability may permit KAT6B, when overexpressed, to perform its own typical roles, as well as those of KAT6A, without spurious effects resulting from an overabundance of KAT6B. This is evident at the level of global histone acetylation, as acetylation levels at H3K9 and H3K23 in Kat6a–/–Tg(Kat6b) E9.5 embryos were similar to wild type controls, while these residues were hyperacetylated in Kat6a+/+Tg(Kat6b) samples.
Notwithstanding the profound rescue of the expression levels of genes downregulated in Kat6a–/–Kat6b+/+ embryos at E9.5 when Kat6b is overexpressed, some effects of the loss of KAT6A persisted. When only Tbx genes were considered (as opposed to transcriptome-wide analyses), Tbx1 and 15 were still significantly downregulated in Kat6a–/–Tg(Kat6b) embryos compared to wild type control embryos. However, Tbx1 was significantly increased in Kat6a–/–Tg(Kat6b) embryos compared to Kat6a–/–Kat6b+/+ samples. This partial rescue was sufficient to rescue the cardiac defects of Kat6a–/–Kat6b+/+ mice, consistent with the rescue of heart defects in Kat6a–/– mice by transgenic overexpression of Tbx118.
In addition to rescuing congenital defects resulting from loss of KAT6A, KAT6B overexpression restored the formation of definitive HSCs capable of reconstituting the haematopoietic system of irradiated recipient mice. Paralleling a previous report that loss of KAT6A reduces CD8 cell surface expression60, we found that KAT6B overexpression promoted CD8 T cell formation at the expense of CD4 T cell development in transplant recipient mice, and that this effect of overexpressed KAT6B was modulated by the loss of KAT6A.
Recently, inhibitors have been developed for both CBP/p30061 and KAT6A/KAT6B30 protein pairs with the view of developing novel cancer therapeutics. Currently, the KAT6A/KAT6B inhibitors are in clinical trials for the treatment of solid cancers31,32,62. In the process of developing these inhibitors it has become clear that it would be unlikely that any inhibitor would differentially inhibit one and not the other of each of these protein pairs. It is therefore relevant to determine the functional equivalence of the proteins within pairs. The combined deletion of these proteins only reveals how similar their functions are at endogenous expression levels. In contrast, our data on the rescue of the Kat6a–/– mice by overexpression of KAT6B indicate that KAT6B can replace KAT6A so completely that it can rescue the 100% lethality, producing healthy and fertile mice that lack KAT6A. These results suggest that simultaneous inhibition of both KAT6A and KAT6B function is likely to be beneficial in treating cancers dependent on KAT6 activity.
Methods
Mice
All animal experiments were conducted with approval of the WEHI Animal Ethics Committee and according to the Australian code for the care and use of animals for scientific purposes. Mice were kept in a 14 h light/10 h dark cycle. Noon of the day the vaginal copulation plug was first observed was defined as embryonic day 0.5 (E0.5).
Mouse alleles
The Kat6a null allele used in this study lacked exons 5 to 9 and has been described previously17. KAT6B overexpression transgenic mice were generated by microinjected into mouse pronuclei using bacterial artificial chromosome (BAC) pBACe3.6 clone RP23-360F23 to produce a germline founder. BAC clone RP23-360F23 includes the complete wild-type Kat6b gene, as well as 21 kb 5’ and 42 kb 3’ containing regulatory sequences. Seven copies of the BAC inserted into the mouse genome to result in an ~4-fold increase in Kat6b expression as described previously23. Mice were maintained on a FVB x BALB/c hybrid background as Kat6b overexpressing mice were not viable on inbred backgrounds. Mice were genotyped by genomic 3-way PCR for the Kat6a allele and by simple PCR to detect the SacB gene in the backbone of pBACe3.6 clone RP23-360F23 using the primers listed in Supplementary Table 1.
Primary mouse embryonic fibroblast (MEF)
MEFs were derived from E14.5 foetuses and grown in Dulbecco’s modified Eagle medium (Gibco, 11995) supplemented with 100 U/ml penicillin/streptomycin (Gibco, 15140122) and 10% foetal calf serum. Cells were cultured in at 37 °C, 5% CO2 and 3% O2. Cell counts were determined at each passage using a CountessTM cell counter (ThermoFisher).
Acid histone extraction
MEFs were washed in DPBS (Gibco, 14190144) containing 0.5 mM sodium butyrate (Sigma, B5887) and cOmplete™ EDTA-free protease inhibitor cocktail (Roche, 11873580001), scraped using a cell scraper (Fisher Scientific, 08-100-241) and collected by centrifugation (200 × g, 5 min). Whole E9.5 embryos were dissected and photographed under a dissecting microscope (Zeiss) and placed into a 1.5 ml Eppendorf tube containing 100 μl DPBS with 0.5 mM sodium butyrate (Sigma, B5887) and cOmplete™ EDTA-free protease inhibitor cocktail (Roche, 11873580001). Samples were lysed in Histone acid lysis buffer (10 mM HEPES pH 7.9, 1.5 mM MgCl2, 10 mM KCl and 0.5 mM DTT) for 30 min at 4 °C on a roller, collected by centrifugation (10,000 × g, 10 min) and resuspended in 0.2 M H2SO4. Samples were incubated on ice for 1–2 h before being dialysed in dialysis tubing (SpectrumTM Spectra/Por Dialysis Membrane Tubing; molecular weight cut-off 20 kDa; Fisher Scientific, 08-607-067) against 0.1 M acetic acid (Sigma, A6283) for 1 h at 4 °C and MQ-H2O overnight at 4 °C. Protein concentrations were determined using a bicinchoninic acid (BCA) assay (ThermoFisher, 23225).
Western immunoblotting
Acid extracted histones were run on 4–12% Bis-Tris gels (ThermoFisher, NP0322) and transferred onto nitrocellulose membranes (Licor, 926-31090). Membranes were blocked for 1 h at room temperature (RT) on a roller in blocking buffer (Intercept® (PBS); Licor, 927-70001) and probed with antibodies again H3K9ac (Epicypher, 13-0033; dilution 1:5000), H3K14ac (Abcam, ab52946; 1:1000) or H3K23ac (Millipore, 07-355; 1:5000) and pan H3 (Abcam, 10799; 1:5000) overnight at 4 °C. The following morning membranes were washed in PBS + 0.1% Tween-20 (Sigma, P1379) and incubated with goat anti-mouse IgG secondary (IRDye® 800 CW; LI-COR Biosciences 926-32210; 1:10,000) and goat anti-rabbit IgG (IRDye®; LI-COR Biosciences, 926-68071; 1:10,000) secondary antibodies for 1 h at RT on a roller. Samples were imaged and analysed using an automated western blot imager software (Odyssey Imager; LI-COR Biosciences).
Histology
E18.5 hearts were dissected, washed in PBS and fixed overnight in 10% neutral buffered formalin. Hearts were embedded in agarose to control orientation, then dehydrated and infused with paraffin and embedded for histological sectioning and haematoxylin and eosin (H&E) staining.
Skeletal preparations
Skin and internal organs were removed from E18.5 pups under a dissecting microscope (Zeiss). Pups were fixed in 4% PFA (Sigma, 158127) overnight at RT on a roller, briefly rinsed in 95% EtOH and stained overnight at RT in a solution containing 5 ml 0.4% Alcian Blue 8 GX (w/v), 5 ml glacial acetic acid, 70 ml 95% EtOH, 20 ml MQ-H2O and 100 µl 0.5% Alizarin red (w/v). Samples were washed in MQ-H2O. Soft tissues were dissolved in 2% (w/v) KOH (Sigma, 221473) in H2O for 24 h at RT on a roller. Following digestion, skeletal preparations were washed in 0.25% (w/v) KOH in H2O for 30 min at RT on a roller, followed by ascending concentrations of glycerol (20%, 33%, 50%) in 0.25% (w/v) KOH in H2O, for 1 h, 1 h and overnight, respectively at RT on a roller. Prepared skeletons were stored in 50% (w/v) glycerol (Sigma, G5516) in ddH2O.
Whole mount in situ hybridization
Embryos used for whole-mount in situ hybridisation were fixed in 4% paraformaldehyde overnight then dehydrated through a methanol series and stored at −20 °C. After genotyping selected embryos were rehydrated through a methanol series, washed in phosphate buffer saline/Tween20, then bleached in hydrogen peroxide, washed in phosphate buffer saline/Tween20, then treated with proteinase K which was stopped by the addition of 1 M glycine. Embryos were then prehybridised a solution of 50% Formamide/5x SSC ph4.5/ 1% SDS/ 50 µg/ml yeast RNA/ 50 µg/ml heparin for 1 h. Embryos were then transferred to a fresh aliquot of the hybridisation buffer containing in vitro transcribed (Roche 11175025910), digoxigenin-labelled cRNA of the Hox gene under investigation and incubated at 55 °C overnight. Then embryos were washed extensively, treated with RnaseA, blocking reagent (Roche 10057177103) and foetal bovine serum, incubated with alkaline phosphatase- labelled anti-digoxigenin antibody (Roche 11214667001) overnight, washed extensively, followed by alkaline phosphatase reaction with NBT-BCIP (Roche 11681451001) for colour development. Finally, embryos were washed and then cleared in glycerol63. Sense and antisense probes for Hoxa3, Hoxa5 and Hoxc5 have been previously described17,54.
Foetal liver transplantation
E14.5 foetal livers were dissected and passed through a 40 μm cell sieve (Corning, 431750). 1 × 106 cells, as determined using an automated haematology analyser (Advia 2120i, Siemens Healthineers), were injected into the tail vein of 3x irradiated recipients (2 × 550 rad, 3 h apart)49.
Flow cytometry
Erythrocytes were lysed by washing samples 2 × 10 ml in a hypotonic solution (150 mM NH4Cl, 0.1 mM EDTA, 12 mM NaHCO3, pH 7.2). Cells were resuspended in a FACS buffer (150 nM NaCl, 3.7 mM KCl, 2.5 mM CaCl22H2O, 1.2 mM MgSO4•7H2O, 0.8 mM K2HPO4, 1.2 mM KH2PO4, 11.5 mM HEPES, pH 7.4) supplemented with 2% foetal calf serum and stained with conjugated antibodies (Supplementary Table 2) for 1 h on ice. Samples were washed in 3–4 ml FACS buffer and analysed on a flow cytometer (BD LSRFortessaTM X-20, BD) at <7500 events/sec. Data were analysed using flow cytometry analysis software (FlowJo version 10.7, Tree Star Inc.). Cell surface markers used to identify individual cell types are shown in Supplementary Table 3.
RNA isolation and sequencing
Total RNA from whole E9.5 embryos was extracted using an RNA extraction kit (Qiagen RNeasy mini kit; Qiagen, 74104), according to the manufacturer’s instructions and including the optional DNaseI digestion step. RNA quality and quantity were assessed on an automated analyser (Tapestation 4200; Agilent, G2991BA), and 500 ng RNA used to generate libraries using a library construction kit (TruSeq RNA prep kit v2; Illumina, RS-122-2002), according to the manufacturer’s instructions. Samples were run on a sequencing machine (NextSeq 2000; Illumina) to give 66 bp paired end reads.
RNA sequencing analysis
Reads were aligned to the Mus musculus (mm39) genome using Rsubread64. Differential expression (DE) analyses were performed using the edgeR and limma software packages65. Library sizes were normalised using the trimmed mean of M-values (TMM) method66 and the surrogate variable approach67 was used to adjust for unwanted variation in the data. The false discovery rate (FDR) was controlled below 0.05. R version 4.2.2 was used for all analyses.
Cut&Tag detection of H3K23 acetylation
CUT&Tag sequencing was performed on 50,000 MEFs combined with 50,000 Drosophila melanogaster S2 cells, as described in Kaya-Okur et al.68, with minor modifications as described in ref. 69. All buffers were prepared fresh with complete EDTA-free protease inhibitors (Roche, 11873580001) and 0.5 mM sodium butyrate (Sigma, B5887). Per sample, 10 µl concanavalin-A beads (Bangs Laboratories, BP531) were washed twice in 10 volumes binding buffer (20 mM HEPES pH 7.5 (Sigma, 83264), 10 mM KCl (Sigma, 60142), 1 mM CaCl2 (Sigma, 21115) and 1 mM MnCl2 (Sigma, M1787) and resuspended in 10 µl binding buffer. Combined MEFs and S2 D. melanogaster cells were washed twice in 1 ml wash buffer (20 mM HEPES pH 7.5, 150 mM NaCl (Sigma, 71386), 0.5 mM spermidine (Sigma, S0266) and resuspended in 90 µl wash buffer. Concanavalin-A beads and samples were combined and incubated (RT, 10 min), immobilised using a magnetic rack (ThermoFisher, MR02) and beads resuspended in 100 µl ice-cold antibody buffer (wash buffer supplemented with 0.05 % (w/v) digitonin (Merck, 300410), 2 mM EDTA (Invitrogen, 15575020) and 0.1 % (w/v) BSA (Sigma, A8577) containing 1:100 H3K23ac (Millipore, 07-355) primary antibody. Samples were incubated overnight at 4 °C. The following morning, beads were resuspended in 100 µl wash buffer supplemented with 0.05% (w/v) digitonin containing 1:100 secondary antibody (Guinea pig anti-rabbit IgG, Antibodies Online, ABIN101961), incubated (RT, 1 h), washed thrice and resuspended in 100 µl ice-cold dig-300 buffer (20 mM HEPES pH 7.5, 300 mM NaCl, 0.5 mM spermidine and 0.01 % (w/v) digitonin), supplemented with 1 2.5 µl pAG-Tn5 (EpiCypher, 15-1017). Samples were incubated (RT, 1 h), washed thrice, resuspended in 100 µl tagmentation buffer (wash buffer supplemented with 0.01 mM MgCl2 (Sigma, 63069) and incubated at 37 °C for 1 h. Tagmentation was stopped using 3.34 µl 0.5 M EDTA, 1 µl 10% (w/v) SDS (Sigma, 71736), and 0.83 µl 20 mg/ml thermolabile proteinase K (NEB, P8111S), incubating at 37 °C for 1 h and 800 rpm, followed by heat inactivation at 55 °C for 10 min. DNA was extracted using Ampure XP beads (Beckman, A63880), eluting in 25 µl 10 mM Tris-HCl pH = 8.0 (Invitrogen, 15568025), 1 mM EDTA and 25 µg/ml RNAse A (ThermoFisher, EN0531) at 37 °C for 10 min. 10 µl sample DNA elutes were used to generate sequencing libraries, which were amplified using PCR for 13 cycles. PCR products were cleaned up using 30 µl Ampure XP beads and eluted in 25 µl 10 mM Tris-HCl pH 8.0. Cleaned libraries were analysed using High Sensitivity D1000 gels (Agilent, 5067- 5584) on an Agilent 2200 tape station. Libraries were sequenced using an Illumina NextSeq2000.
Analysis of CUT&Tag data
All samples were composed of Mus musculus (test) and Drosophila melanogaster RNA (spike-in for normalisation70). Furthermore, the transgenic BAC backbone samples contained the sacB gene. For alignment an index was built using Rsubread v2.18.071 containing the mouse genome (mm39), the Drosophila genome (R655) and the sacB gene sequence. All samples were aligned to this combined genome index using Rsubread’s align function reporting uniquely mapped reads only. All PCR duplicate reads were then marked using sambamba v0.6.6. The data was then summarised at both the species and mouse gene level. Data around mouse genes was summarised at −1 kbp upstream of the TSS to the TSS, transcription end site (TES) to +1kbp downstream of the TES, and the gene body (TSS to TES). Fragment counts were produced using Rsubread’s featureCounts function. Fragments were counted if they were not a PCR duplicate and non-chimeric. Mouse genes were identified using RefSeq annotation to the mm39 genome. The analysis of each region was restricted to protein coding genes with official gene symbols. Riken, Gm (predicted), and pseudogenes were also removed. To avoid sex-based biases, the Xist gene and Y-chromosome were removed. Analysis of each region was then conducted independently. Differential analyses were conducted using the limma65 and edgeR66 software packages, versions 3.60.4 and 4.2.1 respectively.
For each analysis, lowly abundant regions were filtered using edgeR’s filterByExpr function with default parameters. The samples were then normalised to Drosophila melanogaster content using the following method:
-
1.
Calculate total drosophila content for each sample.
-
2.
Divide the above by the total filtered mouse counts for that sample.
-
3.
Divide the resulting numbers by the product of all values calculated in step 2 to the power of (1/number of samples).
Differential analyses between genotypes were then conducted as follows.
TSS region: the data was transformed to log2-counts per million (CPM) and sample weights were calculated using limma’s arrayWeights function using the genebygene method72. Linear models were fit to each region and robust empirical bayes moderated t-statistics with a trended prior variance were then applied to identify differential regions (robust limma-trend pipeline with sample weights)73.
Gene body region: the data was transformed to log2CPM with associated precision weights using voom. Linear models were then fit to each genomic region, differences between groups were assessed using robust empirical bayes moderated t-statistics (robust limma-voom pipeline)73.
TES region: Limma’s voomWithQualityWeights function was applied to simultaneously transform the data to log2CPM with associated precision weights and estimate sample level weights74. Linear models were then fit to each genomic region, differences between groups were assessed using robust empirical bayes moderated t-statistics (robust limma-voomWithQualityWeights pipeline).
For each analysis the false discovery rate (FDR) was controlled below 5% using the Benjamini and Hochberg method.
Statistics
The statistical analysis methods for the RNA-sequencing and CUT&Tag data are provided under the RNA-sequencing analysis and Analysis of CUT&Tag data section. Other data are presented as mean ± s.e.m. In all graphs circles represent individual mice or the average of transplant recipients that received cells from a single foetal liver donor. Statistical analyses were performed in Prism Graphpad Version 8.3.1 for Mac (GraphPad Software) and R version 4.2.2. Statistical tests employed and the number of biological replicates are stated in the figure legends.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All RNA sequencing and CUT&Tag data has been deposited in the NCBI GEO database under accession numbers: GSE287243 and GSE287244 [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE287244]. Source data are provided with this paper.
References
Strahl, B. D. & Allis, C. D. The language of covalent histone modifications. Nature 403, 41–45 (2000).
Wang, Z. et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat. Genet. 40, 897–903 (2008).
Wang, Z. et al. Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell 138, 1019–1031 (2009).
Sheikh, B. N. & Akhtar, A. The many lives of KATs—detectors, integrators and modulators of the cellular environment. Nat. Rev. Genet. 20, 7–23 (2019).
Voss, A. K. & Thomas, T. Histone lysine and genomic targets of histone acetyltransferases in mammals. Bioessays 40, e1800078 (2018).
Thomas, T. & Voss, A. K. The diverse biological roles of MYST histone acetyltransferase family proteins. Cell Cycle 6, 696–704 (2007).
Neuwald, A. F. Landsman D. GCN5-related histone N-acetyltransferases belong to a diverse superfamily that includes the yeast SPT10 protein. Trends Biochem. Sci. 22, 154–155 (1997).
Arany, Z., Sellers, W. R., Livingston, D. M. & Eckner, R. E1A-associated p300 and CREB-associated CBP belong to a conserved family of coactivators. Cell 77, 799–800 (1994).
Xu, W., Edmondson, D. G. & Roth, S. Y. Mammalian GCN5 and P/CAF acetyltransferases have homologous amino-terminal domains important for recognition of nucleosomal substrates. Mol. Cell Biol. 18, 5659–5669 (1998).
Arany, Z., Newsome, D., Oldread, E., Livingston, D. M. & Eckner, R. A family of transcriptional adaptor proteins targeted by the E1A oncoprotein. Nature 374, 81–84 (1995).
Lundblad, J. R., Kwok, R. P., Laurance, M. E., Harter, M. L. & Goodman, R. H. Adenoviral E1A-associated protein p300 as a functional homologue of the transcriptional co-activator CBP. Nature 374, 85–88 (1995).
Soskine, M. & Tawfik, D. S. Mutational effects and the evolution of new protein functions. Nat. Rev. Genet. 11, 572–582 (2010).
Kasper, L. H., Lerach, S., Wang, J., Wu, S., Jeevan, T. & Brindle, P. K. CBP/p300 double null cells reveal effect of coactivator level and diversity on CREB transactivation. EMBO J. 29, 3660–3672 (2010).
Yamauchi, T. et al. Distinct but overlapping roles of histone acetylase PCAF and of the closely related PCAF-B/GCN5 in mouse embryogenesis. Proc. Natl. Acad. Sci. USA 97, 11303–11306 (2000).
Doyon, Y. et al. ING tumor suppressor proteins are critical regulators of chromatin acetylation required for genome expression and perpetuation. Mol. Cell 21, 51–64 (2006).
Ullah, M. et al. Molecular architecture of quartet MOZ/MORF histone acetyltransferase complexes. Mol. Cell Biol. 28, 6828–6843 (2008).
Voss, A. K., Collin, C., Dixon, M. P. & Thomas, T. Moz and retinoic acid coordinately regulate H3K9 acetylation, Hox gene expression, and segment identity. Dev. Cell 17, 674–686 (2009).
Voss, A. K. et al. MOZ regulates the Tbx1 locus, and Moz mutation partially phenocopies DiGeorge syndrome. Dev. Cell 23, 652–663 (2012).
Simo-Riudalbas, L. et al. KAT6B Is a tumor suppressor Histone H3 Lysine 23 acetyltransferase undergoing genomic loss in small cell lung cancer. Cancer Res. 75, 3936–3945 (2015).
Klein, B. J. et al. Histone H3K23-specific acetylation by MORF is coupled to H3K14 acylation. Nat. Commun. 10, 4724 (2019).
Lv, D. et al. Histone acetyltransferase KAT6A upregulates PI3K/AKT signaling through TRIM24 binding. Cancer Res. 77, 6190–6201 (2017).
Bergamasco, M. I. et al. KAT6B is required for histone 3 lysine 9 acetylation and SOX gene expression in the developing brain. Life Sci. Alliance 8, e202402969 (2025).
Bergamasco, M. I. et al. The histone acetyltransferase KAT6B is required for hematopoietic stem cell development and function. Stem Cell Rep. 19, 469–485 (2024).
Borrow, J. et al. The translocation t(8;16)(p11;p13) of acute myeloid leukaemia fuses a putative acetyltransferase to the CREB-binding protein. Nat. Genet. 14, 33–41 (1996).
Viita, T. & Cote, J. The MOZ-BRPF1 acetyltransferase complex in epigenetic crosstalk linked to gene regulation, development, and human diseases. Front. Cell Dev. Biol. 10, 1115903 (2022).
Sheikh, B. N. et al. MOZ regulates B-cell progenitors and, consequently, Moz haploinsufficiency dramatically retards MYC-induced lymphoma development. Blood 125, 1910–1921 (2015).
Trecourt, A. et al. The KAT6B::KANSL1 fusion defines a new uterine sarcoma with hybrid endometrial stromal tumor and smooth muscle tumor features. Mod. Pathol. 36, 100243 (2023).
Murati, A. et al. Variant MYST4-CBP gene fusion in a t(10;16) acute myeloid leukaemia. Br. J. Haematol. 125, 601–604 (2004).
Zack, T. I. et al. Pan-cancer patterns of somatic copy number alteration. Nat. Genet. 45, 1134–1140 (2013).
Baell, J. B. et al. Inhibitors of histone acetyltransferases KAT6A/B induce senescence and arrest tumour growth. Nature 560, 253–257 (2018).
Sharma, S. et al. Targeting KAT6A/KAT6B dependencies in breast cancer with a novel selective, orally bioavailable KAT6 inhibitor, CTx−648/PF-9363. Cell Chem. Biol. 30, 1191–1210.e20 (2023).
Mukohara, T. et al. Inhibition of lysine acetyltransferase KAT6 in ER(+)HER2(-) metastatic breast cancer: a phase 1 trial. Nat. Med. 30, 2242–2250 (2024).
Arboleda, V. A. et al. De novo nonsense mutations in KAT6A, a lysine acetyl-transferase gene, cause a syndrome including microcephaly and global developmental delay. Am. J. Hum. Genet. 96, 498–506 (2015).
Tham, E. et al. Dominant mutations in KAT6A cause intellectual disability with recognizable syndromic features. Am. J. Hum. Genet. 96, 507–513 (2015).
Clayton-Smith, J. et al. Whole-exome-sequencing identifies mutations in histone acetyltransferase gene KAT6B in individuals with the Say-Barber-Biesecker variant of Ohdo syndrome. Am. J. Hum. Genet. 89, 675–681 (2011).
Simpson, M. A. et al. De novo mutations of the gene encoding the histone acetyltransferase KAT6B cause Genitopatellar syndrome. Am. J. Hum. Genet. 90, 290–294 (2012).
Thomas, T., Voss, A. K., Chowdhury, K. & Gruss, P. Querkopf, a MYST family histone acetyltransferase, is required for normal cerebral cortex development. Development 127, 2537–2548 (2000).
Kraft, M. et al. Disruption of the histone acetyltransferase MYST4 leads to a Noonan syndrome-like phenotype and hyperactivated MAPK signaling in humans and mice. J. Clin. Investig. 121, 3479–3491 (2011).
Merson, T. D. et al. The transcriptional coactivator Querkopf controls adult neurogenesis. J. Neurosci. 26, 11359–11370 (2006).
Bergamasco, M. I. et al. Loss of KAT6B causes premature ossification and promotes osteoblast differentiation during development. Dev. Biol. 520, 141–154 (2025).
Bergamasco, M. I. et al. Increasing histone acetylation improves sociability and restores learning and memory in KAT6B-haploinsufficient mice. J. Clin. Investig. 134, e167672 (2024).
Katsumoto, T. et al. MOZ is essential for maintenance of hematopoietic stem cells. Genes Dev. 20, 1321–1330 (2006).
Thomas, T. et al. Monocytic leukemia zinc finger protein is essential for the development of long-term reconstituting hematopoietic stem cells. Genes Dev. 20, 1175–1186 (2006).
Vanyai, H. K. et al. MOZ directs the distal-less homeobox gene expression program during craniofacial development. Development 146, dev175042 (2019).
Vanyai, H. K., Thomas, T. & Voss, A. K. Mesodermal expression of Moz is necessary for cardiac septum development. Dev. Biol. 403, 22–29 (2015).
Perez-Campo, F. M., Borrow, J., Kouskoff, V. & Lacaud, G. The histone acetyl transferase activity of monocytic leukemia zinc finger is critical for the proliferation of hematopoietic precursors. Blood 113, 4866–4874 (2009).
Perez-Campo, F. M., Costa, G., Lie, A. L. M., Stifani, S., Kouskoff, V. & Lacaud, G. MOZ-mediated repression of p16(INK) (4) (a) is critical for the self-renewal of neural and hematopoietic stem cells. Stem Cells 32, 1591–1601 (2014).
Sheikh, B. N. et al. MOZ (MYST3, KAT6A) inhibits senescence via the INK4A-ARF pathway. Oncogene 34, 5807–5820 (2015).
Sheikh, B. N. et al. MOZ (KAT6A) is essential for the maintenance of classically defined adult hematopoietic stem cells. Blood 128, 2307–2318 (2016).
Ikuta, K. & Weissman, I. L. Evidence that hematopoietic stem cells express mouse c-kit but do not depend on steel factor for their generation. Proc. Natl. Acad. Sci. USA 89, 1502–1506 (1992).
Morrison, S. J., Hemmati, H. D., Wandycz, A. M. & Weissman, I. L. The purification and characterization of fetal liver hematopoietic stem cells. Proc. Natl. Acad. Sci. USA 92, 10302–10306 (1995).
Kiel, M. J., Yilmaz, O. H., Iwashita, T., Yilmaz, O. H., Terhorst, C. & Morrison, S. J. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell 121, 1109–1121 (2005).
Osawa, M., Hanada, K., Hamada, H. & Nakauchi, H. Long-term lymphohematopoietic reconstitution by a single CD34-low/negative hematopoietic stem cell. Science 273, 242–245 (1996).
Sheikh, B. N. et al. MOZ and BMI1 play opposing roles during Hox gene activation in ES cells and in body segment identity specification in vivo. Proc. Natl. Acad. Sci. USA 112, 5437–5442 (2015).
Sheikh, B. N., Metcalf, D., Voss, A. K. & Thomas, T. MOZ and BMI1 act synergistically to maintain hematopoietic stem cells. Exp. Hematol. 47, 83–97.e88 (2017).
Geelen, J. A. Hypervitaminosis A induced teratogenesis. CRC Crit. Rev. Toxicol. 6, 351–375 (1979).
Kessel, M. & Gruss, P. Homeotic transformations of murine vertebrae and concomitant alteration of Hox codes induced by retinoic acid. Cell 67, 89–104 (1991).
van der Lugt, N. M. et al. Posterior transformation, neurological abnormalities, and severe hematopoietic defects in mice with a targeted deletion of the bmi-1 proto-oncogene. Genes Dev. 8, 757–769 (1994).
van der Vlag, J. & Otte, A. P. Transcriptional repression mediated by the human polycomb-group protein EED involves histone deacetylation. Nat. Genet. 23, 474–478 (1999).
Newman, D. M. et al. Acetylation of the Cd8 locus by KAT6A determines memory T cell diversity. Cell Rep. 16, 3311–3321 (2016).
Lasko, L. M. et al. Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nature 550, 128–132 (2017).
Sommerhalder, D. H. E. et al. First-in-human phase 1 dose escalation study of the KAT6 inhibitor PF-07248144 in patients with advanced solid tumors. J. Clin. Oncol. 41, 1054–1054 (2023).
Thomas, T., Loveland, K. L. & Voss, A. K. The genes coding for the MYST family histone acetyltransferases, Tip60 and Mof, are expressed at high levels during sperm development. Gene Expr. Patterns 7, 657–665 (2007).
Liao, Y., Smyth, G. K. & Shi, W. The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 41, e108 (2013).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Leek, J. T. & Storey, J. D. Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLoS Genet. 3, 1724–1735 (2007).
Kaya-Okur, H. S. et al. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat. Commun. 10, 1930 (2019).
Wichmann, J. et al. Loss of TIP60 (KAT5) abolishes H2AZ lysine 7 acetylation and causes p53, INK4A, and ARF-independent cell cycle arrest. Cell Death Dis. 13, 627 (2022).
Orlando, D. A. et al. Quantitative ChIP-Seq normalization reveals global modulation of the epigenome. Cell Rep. 9, 1163–1170 (2014).
Liao, Y., Smyth, G. K. & Shi, W. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 47, e47 (2019).
Ritchie, M. E. et al. Empirical array quality weights in the analysis of microarray data. BMC Bioinform. 7, 261 (2006).
Law, C. W., Chen, Y., Shi, W. & Smyth, G. K. voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 15, R29 (2014).
Liu, R. et al. Why weight? Modelling sample and observational level variability improves power in RNA-seq analyses. Nucleic Acids Res. 43, e97 (2015).
Mah S. Y. Y. et al. ING4 and ING5 are essential for histone H3 lysine 14 acetylation and epicardial cell lineage development. Development 151, dev202617 (2024).
Yang, Y. et al. The histone lysine acetyltransferase HBO1 (KAT7) regulates hematopoietic stem cell quiescence and self-renewal. Blood 139, 845–858 (2022).
Acknowledgements
The authors would like to thank N. Blasch, L. Wilkins and K. Florides for excellent animal care; C. Burström for expert technical assistance; S. Wilcox, K. Weston, T. Nikolaou and the Walter and Eliza Hall Histology department for exceptional service. M.I.B. was supported by an Australian Government Postgraduate Award. This work was supported by the Australian National Health and Medical Research Council through Project Grant 1160517 to T.T., Ideas Grant 2010711 to T.T., Research Fellowships 1081421 to A.K.V. and 1154970 to G.K.S. and Investigator Grant 1176789 to A.K.V.; through the Independent Research Institutes Infrastructure Support Scheme; and by the Victorian Government through an Operational Infrastructure Support Grant.
Author information
Authors and Affiliations
Contributions
M.I.B. and T.T. carried out experiments, performed data analyses and drafted the manuscript. Y.Y. and B.N.S. carried out experiments. A.L.G. performed the bioinformatics data analysis supervised by G.K.S. A.K.V. and T.T. conceived the project, designed experiments, performed data analyses and drafted the manuscript. All authors read and contributed to the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The Thomas and Voss laboratories have received research funding from CTx CRC for work unrelated to this project. All other authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bergamasco, M.I., Yang, Y., Garnham, A.L. et al. KAT6B overexpression rescues embryonic lethality in homozygous null KAT6A mice restoring vitality and normal lifespan. Nat Commun 16, 1958 (2025). https://doi.org/10.1038/s41467-025-57155-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-57155-4
This article is cited by
-
A comprehensive review of histone modifications during mammalian oogenesis and early embryo development
Histochemistry and Cell Biology (2025)
