
Abstract
Heteroatom-doped polyaromatic hydrocarbons show great potential for advancing photoelectric materials. SVI=N doping, characterized by soft-hard atom binding, donor-acceptor transmission, and chiroptical tuning, provides a powerful approach for further optimizing the performance and functionality of these materials. However, the introduction of chiral sulfur(VI) has been a formidable challenge due to the intricate enantioselective discrimination and embedded linkages with the heteroatoms in the polyaromatic systems. Herein, we establish an enantioselective Pd-catalyzed desymmetrization of diaryl sulfoximines and sulfondiimines to access the chiral SVI=N-doped heterocycles with high yields and enantioselectivities. The flexibility and rigidity of the molecule has a distinct effect on the enantioselectivity. The split aromatic compounds exhibit C–H···π interactions involving the additive TMCPA with the ligand and the S-aryl motif, producing the (R)-configuration, while combined aromatic compounds exhibit the opposite (S)-configuration due to the restricted bond rotation. The photophysical and chiroptical study demonstrates an SVI=N-doped carbazole-based polyaromatic heterocycle with intense double absorption peaks and a favorable luminescence dissymmetry factor.
Similar content being viewed by others
Introduction
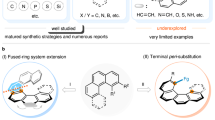
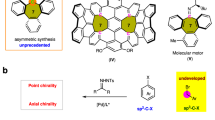
Polycyclic aromatic hydrocarbons (PAHs) with unique optical and electrical properties are commonly used in organic light-emitting diodes (OLEDs)1,2,3, organic field effect transistors (OFETs)4, and organic solar cells (OSCs)5,6. Embedding heteroatoms (such as B, N, S, O, and P) into the π-conjugated frameworks of PAHs produces unexpected opportunities for optoelectronics advancement by providing an adjustable band gap, high electrochemical activity, and desirable charge polarization7,8,9. The replacement of the C=C bond with an isoelectronic B–N unit improved the stability, produced tunable energy levels, and amplified photoluminescence quantum yields (PLQYs)10,11. PVI=N-doped phenanthrenes with enhanced PLQYs were achieved via the introduction of PVI chirality, showing significant potential in circularly polarized luminescent materials (Fig. 1a)12. Sulfur-decorated PAHs, characterized by soft-atom and electron-rich properties, serve as valuable n-type semiconductors13 and can readily oxidize into p-type SVI (sulfonyl) semiconductors14, providing intriguing ambipolar transport characteristics and high resonance energy. The incorporation of the SVI motif enables the solubility and intermolecular interactions of PAHs and increases the processability of the materials15. Sulfoximines16 and sulfondiimines17, as aza-replacement of sulfones, provide customizable N-handles for fine-tuning the reactivity and functionality, concurrently bestowing chirality upon sulfur (Fig. 1b). These pioneering studies show the great potential of unexploited chiral SVI=N-embedded PAHs with attractive photophysical and chiroptical properties. Great contributions have been made to the construction of sulfur stereogenic centers18,19,20,21,22,23,24. However, a difficult challenge remains in differentiating similar aromatic compounds stereoselectively, and a π-extension system is rare in chiral naphthalene-type heterocycles. Powerful C–H activation25,26,27,28,29,30 is a straightforward strategy for heteroatom doping with polycyclic aromatics7,8,9. In contrast to C-31,32, Si-33,34, and P-chirality35,36,37, the construction of S(VI)-chirality poses notable challenges due to the presence of S=N double bond (Fig. 1c and Fig. S7). First, the spatial configuration of S=N bond leads to a longer distance between the coupled carbon atoms, particularly in rigid frameworks (4.87 Å), enabling C–H activation more difficult. Second, the polarizability of S=N bond, especially in the second S=N bond, imparts a high electron density to the nitrogen (−1.177), which can potentially coordinate with palladium and disrupt aryl coordination38. Along with our exploration of sulfoximine chemistry39,40,41, we herein present a Pd-catalyzed desymmetrization for the construction of chiral SVI=N-doped polyaromatic heterocycles, producing an (R)-configuration with the assistance of C–H···π interactions of TMCPA in split aromatics and an (S)-configuration originating from impeded C–S bond rotation in combined aromatics; moreover, promising photophysical and chiroptical properties were achieved from these polyaromatic heterocycles (Fig. 1d).

a Incorporation of B–N, PV=N, and SVI=N bonds. b Sulfoximine and sulfondiimine as aza-analogs of sulfone. c Challenges in the Pd-catalyzed C–H activation of S=N-containing compounds. d Pd-catalyzed enantioselective desymmetrization of diaryl sulfoximines and sulfondiimines involving split (acyclic) and combined (cyclic) aromatics (this work).
Results
We began enantioselective C–H coupling with symmetric diaryl sulfoximine (1a) for the chiral dibenzothiazine oxide 2a with Pd(OAc)2 as a catalyst (Table 1). Chiral ligands play a crucial role in controlling the sulfur(VI) stereogenic center. Compared to L1, an electron-rich monodentate phosphine ligand L2 produced the cyclization product in 9% yield and 38% enantiomeric excess (ee) (Table 1, entries 1–2). A series of electron-donating bidentate ligands, including N/P- and P/P-based motifs, were investigated (entries 3–7). Chiraphos (L5) provided a moderate yield and a diminished enantiomeric excess of 2a, while a rigid ligand (L7) caused improved enantioselectivity. However, increasing the steric hindrance in the coordinating environment (L8) substantially attenuated the ee (entry 8). The impact of solvents was equally important (entries 9-13). Dimethylformamide (DMF) enabled the complete conversion of 1a but produced an almost racemic product (entry 9). The protonated byproduct from 1a was exclusively observed in a less bulky protonic solvent (isopropylalcohol (iPrOH), entry 10). Notably, tert-amyl alcohol (tAmylOH) and 1,2-dichloroethane (DCE) enhanced the stereoselectivity (entries 11 and 12). The yield of product 2a was significantly increased without compromising the optical purity of these mixed solvents (entry 13). Interestingly, replacing pivalic acid (PivOH) with tetramethylcyclopropylcarboxylic acid (TMCPA) substantially promoted both the reaction efficiency and enantioselectivity (entry 16). In the absence of the additive, a large decrease in the yield was observed, but the ee only slightly decreased (entry 17). These results demonstrated the pivotal role of the acid additives in facilitating the concerted metalation-deprotonation (CMD) process.
A comprehensive investigation was conducted with the optimized reaction conditions for the enantioselective synthesis of chiral SVI=N-heterocycles via the desymmetrization of the diaryl sulfoximines (Fig. 2). Electron-rich and electron-poor substituents at diverse aromatic positions tethered to sulfur(VI) were well tolerated, producing annulated sulfoximines in excellent yields and ee values (2a-2g). The absolute (R)-configuration of 2a was further confirmed via X-ray diffraction analysis. Heteroaryl sulfoximines containing thienyl and pyridinyl moieties were shown to be efficient in this enantioselective desymmetrization process (2 h and 2i). When the cyclic substrate dibenzothiophene oxide 1j was used, exceptional enantioselectivity was obtained for the corresponding product (2j, >99% ee); here, the structural rigidity of the substrate reversed the stereoselectivity (2a vs. 2j) with the (S)-configuration, which was confirmed via X-ray diffraction analysis. Highly enantioenriched pentacyclic sulfoximines were produced when the five-membered adaptor was replaced with a six-membered ring with the oxygen/nitrogen/carbonyl groups in dibenzothiophene oxide (2k-2m). The expanding of the conjugated system successfully increased the enantiomeric purity of the sulfur(VI)-chiral center (2n). In this cyclization strategy, excellent compatibility was exhibited with the N-substituted aromatic of sulfoximines possessing electronically diverse groups at different positions (2o-2u). Condensed and heteroaryl-derived sulfoximines efficiently underwent enantioselective cyclization (2v and 2w). Notably, a double-site carbazole-based substrate produced corresponding product 2x in >99% ee with excellent diastereoselectivity. Furthermore, the current desymmetrization strategy was successfully extended to the diaryl sulfondiimines, producing the challenging dibenzothiazine imides (4a-4o) in excellent yields and enantiomeric excesses (up to 99% ee). In comparison to sulfoximine 1g, sulfondiimine 3e with an electron-donating group (–OMe) contained a mixture of para- and ortho-positions (4e1 and 4e2). The diverse protecting groups on nitrogen, such as –Ns (4-nitrobenzene sulfonyl), –Cs (4-cyanobenzene sulfonyl), and –Boc (t-butyloxy carbonyl), displayed excellent reaction efficiency and stereoselectivity (4f-4h). The absolute configuration of 4o was confirmed via X-ray diffraction analysis as a representative example of a sulfondiimine product.

aConditions: 1 or 3 (0.15 mmol), Pd(OAc)2 (6 mol%), L7 (6.6 mol%), TMCPA (30 mol%), K2CO3 (1.1 equiv), tAmylOH/DCE (6/1), 100 °C, N2, 8 h (for sulfoximines 1) or 12 h (for sulfondiimines 3). bPd(OAc)2 (8 mol%), L7 (8.8 mol%). cPd(OAc)2 (10 mol%), L7 (11 mol%), TMCPA (40 mol%). dK2CO3 (3 equiv). eK2CO3 (2 equiv.). Ns = 4-nitrobenzene sulfonyl. Cs = 4-cyanobenzene sulfonyl.
The parallel kinetic resolution of the diaryl sulfoximines and sulfondiimines bearing diverse para-substituents was also evaluated under standard conditions (Fig. 3a). Excellent stereoselectivities were achieved with ee values greater than 97% in most cases. When opposing electronic groups were present on either side of the aromatic ring, the enantioselective annulation slightly favored the electron-deficient substituents (2y and 4p). To highlight the practical synthetic utility of the structurally novel chiral SVI=N-doped heterocycles, the derivatization of sulfoximine and sulfondiimine was further performed (Fig. 3b). The process began with the lithium-halogen exchange of 2-bromobiphenyl 5, followed by the nucleophilic addition of 2m to form the corresponding alcohol intermediate 6a. Then, an intramolecular Friedel−Crafts reaction produced sulfoximine 6b with a fluorenyl-based spiro skeleton; here, the absolute (S)-configuration of 6b was confirmed via X-ray diffraction analysis. Notably, the gram-scale synthesis of cyclic sulfondiimine 4h was accomplished with an enhanced yield of 95% while retaining stereochemical fidelity. Subsequently, the Suzuki coupling of 4h with phenylanthracenyl boronic acid 7 provided extended conjugated product 8a, in which free sulfoximine 8b was liberated via trimethyl silyl iodide (TMSI)/triethylamine (Et3N) treatment.

a Parallel kinetic resolution. b further derivatization of the S = N-doped heterocycles.
With synthesized S=N-doped heterocycles in hand, the photophysical characteristics of 2c, 2l, 2x, and 4m were investigated. The UV‒Vis spectra revealed a maximum absorption peak at approximately 280 nm for 2c and 2x and a higher absorption peak at approximately 365 nm for 2l and 4m. Due to the expansive conjugate system, 2x displayed an additional absorption feature at 388 nm (Fig. 4a). At the maximum excitation wavelength, sulfoximines 2c, 2l, and 2x exhibited broadened fluorescence emission bands, with 2x showing the farthest emission wavelength at 532 nm. However, 4m with an N=S=N motif displayed a noticeable reduction in fluorescence intensity (Fig. 4b). The absolute quantum yield was determined to be 0.12 for 2x in CH2Cl2. Moreover, the chiroptical properties of 2x with two chiral sulfur (VI) centers were studied via electronic circular dichroism (ECD) and circularly polarized luminescence (CPL) spectroscopy; here, the enantiomers of (R, R)-2x and (S, S)-2x showed evident mirror images from 250 nm to 490 nm in their CD spectra (Fig. 4c). The mirror-image spectrum in Fig. 4d showed the CPL activity of compound 2x, and the luminescence dissymmetry factor (glum) of (R, R)-2x was measured to be -0.0022 at 563 nm (Fig. 4e, f). These results highlighted the significant application potential of the SVI=N-heterocycles in advancing enantiomeric photoelectric materials, such as light-emitting diodes (LEDs), organic photovoltaics and chiroptical devices12,42.

a Absorption spectra of 2c, 2 l, 2x, and 4 m in DCM (1.0 × 10−5 M). b Emission spectra of 2c, 2l, 2x, and 4m in DCM (1.0 × 10−5 M). c CD spectra of (R, R/S, S)-2x in DCM (1.0 × 10−5 M). d CPL spectra of (R, R/S, S)-2x in DCM (1.0 × 10−5 M) excited at 388 nm. e glum values wavelength curves for (R, R/S, S)-2x. f Structure, fluorescence image of CH2Cl2 irradiated at 365 nm, and optical values for (R, R)-2x.
To clarify the reaction mechanism, several mechanistic experiments were conducted. When the mono-oxide and dioxide derivatives of ligand L7 were tested43,44, products 2a and 2j were obtained in low yield or not observed at all (Fig. 5a). These results suggest that the diphosphine is the active ligand in the current transformation, rather than its oxidized forms. Parallel kinetic isotope effect (KIE) experiments45 with substrates 1aa and 1aa’ revealed a kH/kD ratio of 2.7 (Fig. 5b), indicating that C–H activation is involved in the rate-determining step.

a Control experiments for mono-oxide and dioxide derivatives of ligand L7. b Parallel kinetic isotope effect experiments.
Based on the experimental results and the reaction model for a bidentate ligand46, we next investigated the origins of the enantioselectivity in substrate 1a using density functional theory (DFT) calculations. The chiral Pd(0) catalyst undergoes oxidative addition with 1a, followed by ligand exchange with TMCPA to form intermediate INT1 (Fig. 6a). Two possible modes of C–H activation were considered due to the square planar geometry of Pd(II). In the first mode, bidentate coordination of the phosphine ligand leads to outer-sphere C–H bond activation. In the second mode, monodentate coordination of the phosphine ligand leaves a coordination site for the carboxylate anion, enabling inner-sphere C–H activation. Computational results indicate that the energy barrier for inner-sphere C–H activation is at least 9.1 kcal/mol lower than that for outer-sphere C–H activation (Fig. 6b). In the outer-sphere C–H bond activation transition states, steric hindrance exists between the ligand and the S-aryl group, regardless of which aryl C–H bond is activated. To mitigate the steric repulsion, the resulting C–Pd bond is elongated. In TS3-(R)-A and TS3-(R)-B, the C–Pd bond lengths are 2.50 Å and 2.52 Å, respectively, compared to 2.30 Å in TS3-(R). In contrast, the inner-sphere C–H bond activation transition state TS3-(R) not only lacks steric hindrance but also benefits from favorable π···π, C–H···O, and C–H···π non-covalent interactions, confirmed by independent gradient model based on Hirshfeld partition (IGMH) analysis47. The bidentate phosphorus-coordinated INT1 isomerizes to INT2 with the aryl fragment of sulfoximine coordinated to palladium. Subsequent concerted metalation-deprotonation via TS3 generates the palladacycle intermediate INT4, which undergoes reductive elimination (TS5, 12.3 kcal/mol), irreversibly producing the reduced palladium(0) complex INT6 with product coordination. The DFT-computed free energy profile suggests that C–H activation has the highest energetic barrier, with an overall barrier of 29.8 kcal/mol, which is consistent with the results of parallel KIE experiments (Fig. 5b).

a The enantioselective C–H annulation of substrate 1a were investigated at the level of ωB97M-V/def2-TZVPP-SMD(2-methyl-2-propanol)//B3LYP-D3(BJ)/6-31G(d)-LANL2DZ. b The optimized structures and their relative Gibbs free energies of inner-/outer-sphere C–H activation transition states.
To elucidate the origin of the opposite enantioselectivity observed in split and combined substrates, we conducted a detailed study on the enantiodetermining step (C–H activation). For split substrate 1a, INT1 can rotate around the S–C and N–C bonds prior to C–H activation, generating two sets of diastereomeric transition states (Fig. 7a). In TS3-(R)/TS3-(S)-iso1, the oxygen atom of sulfoximine is distal to the Pd plane, whereas it is proximal in TS3-(S)/TS3-(R)-iso1. TS3-(R)/TS3-(S)-iso1 can isomerize to TS3-(S)/TS3-(R)-iso1 via C–S bond rotation. Among the transition states producing the R-enantiomer, TS3-(R)-iso1 is 6.7 kcal/mol higher in energy than TS3-(R) for two reasons. First, in TS3-(R)-iso1, the sulfoximine undergoes substantial distortion to maintain the Pd’s square planar geometry. This distortion is reflected in the highlighted dihedral angle ϕ(OSNC), which measures 105.9° in TS3-(R)-iso1 compared to 69.2° in the DFT-optimized geometry. Second, TS3-(R) benefits from multiple favorable noncovalent interactions between the phosphine ligand, substrate, and TMCPA anion (Fig. 6b). Among the transition states that produces the S-enantiomer, TS3-(S) is 4.5 kcal/mol lower in energy than TS3-(S)-iso1, stabilized by favorable π···π and C–H···O noncovalent interactions, despite increased distortion (99.4° vs 81.0° in TS3-(S)-iso1). The energy difference is also attributed to steric hindrance in TS3-(S)-iso1 involving TMCPA, the tolyl fragment of sulfoximine, and the methoxy group of ligand. Therefore, TS3-(R) and TS3-(S) are the key enantioselectivity-determining transition states, with TS3-(R) being 1.7 kcal/mol lower in energy, which is consistent with experimental observations. The additional C–H···π interactions between TMCPA and aryl groups of the ligand and substrate facilitate enantiodiscrimination in TS3-(R) (Fig. S8 and S9), explaining why different acid additives affect enantio-selectivity experimentally (Table 1, Entries 13-17). When C–S bond rotation is restricted, only TS3-(S) and TS3-(R)-iso1 are accessible, leading predominantly to form S-enantiomer, as TS3-(S) is 5 kcal/mol lower in energy than TS3-(R)-iso1. When combined substrates are used, the conformational restriction efficiently inhibits the C–S bond rotation feasible in split molecules, thus reversing the enantioselectivity to favor the S-enantiomer. Computational studies on combined substrate 2j shows consistent results with split substrates, confirming that restricting C–S bond rotation leads to S-enantiomer as the major product (ΔΔGǂ = 2.9 kcal/mol, Fig. 7b). Interestingly, analysis of the TS7-(R) and TS7-(S) structures reveals that enantioselectivity is driven by π···π stacking between the ligand and the N-aryl fragment, independent of acid additives. To further validate this chiral induction model, we conducted the reaction under standard conditions without TMCPA, relying on the acetate anion from palladium acetate for C–H activation. The reaction produced the target product with >99% ee, fully supporting the reliability of the computational model.

a Enantioselectivity-determining transition states and their relative Free energies for split substrate 1a. b origins of enantioselectivity for combined substrate 1j.
Discussion
In conclusion, a Pd-catalyzed enantioselective C–H annulation of sulfoximines and sulfondiimines was successfully established with the assistance of TMCPA. This strategy provides a robust pathway for accessing chiral SVI = N-embedded heterocycles with excellent yields and enantioselectivities. The π-extension of the sulfoximines and sulfondiimines was efficiently achieved via the current strategy. Modulating the rigidity of the diaryl sulfoximines enabled the formation of an SVI = N-heterocycle with an inverted stereoconfiguration. The concerted Pd-catalyzed deprotonation was identified as the rate- and enantio-determining step. DFT studies revealed that distinct enantioselectivities emerged from the C–H···π interactions of TMCPA in split sulfoximines and from the π-π stacking between the ligand and the N-aryl motif in combined sulfoximines. Further study of the chiral SVI = N-doped π-conjugated materials is in progress in our laboratory.
Methods
General reaction procedure
A 25 mL oven-dried Schlenk tube was charged with diaryl sulfoximines 1 or sulfondiimines 3 (0.15 mmol, 1.0 equiv), Pd(OAc)2 (6 mol%), (R,R,R,R)-MeO-BIBOP L7 (6.6 mol%), TMCPA (30 mol%), K2CO3 (1.1 equiv), and a stirring bar. The tube was fitted with a N2 balloon, and a mixture of 1 mL tAmylOH/DCE (6/1) was added. Subsequently, the reaction mixture was transferred to 100 oC and stirred for 8–12 h. Upon cooling to room temperature, the reaction mixture was diluted with DCM, filtered, and concentrated under reduced pressure. The resulting residue was subjected to purification by column chromatography, affording the corresponding product.
Data availability
The X-ray crystallographic coordinates for the structures reported in this study have been deposited at the Cambridge Crystallographic Data Centre (CCDC), under deposition number CCDC 2287655 (2a), CCDC 2287657 (2j), CCDC 2287651 (4o), and CCDC 2287663 (6b). These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif. The authors declare that all other data supporting the findings of this study are available within the article and Supplementary Information files and are also available from the corresponding author on request. Computational data are available on Figshare: https://doi.org/10.6084/m9.figshare.27291531.
References
Reineke, S. et al. White organic light-emitting diodes with fluorescent tube efficiency. Nature 459, 234–238 (2009).
Gather, M. C., Kçhnen, A. & Meerholz, K. White Organic Light-Emitting Diodes. Adv. Mater. 23, 233–248 (2011).
Hong, G. et al. A Brief History of OLEDs—Emitter Development and Industry Milestones. Adv. Mater. 33, 2005630 (2021).
Wang, C., Dong, H., Hu, W., Liu, Y. & Zhu, D. Semiconducting π-Conjugated Systems in Field-Effect Transistors: A Material Odyssey of Organic Electronics. Chem. Rev. 112, 2208–2267 (2012).
Burlingame, Q. et al. Intrinsically Stable Organic Solar Cells under High-Intensity Illumination. Nature 573, 394–397 (2019).
Ding, P., Yang, D., Yang, S. & Ge, Z. Stability of Organic Solar Cells: toward Commercial Applications. Chem. Soc. Rev. 53, 2350–2387 (2024).
Stępień, M., Gońka, E., Żyła, M. & Sprutta, N. Heterocyclic Nanographenes and Other Polycyclic Heteroaromatic Compounds: Synthetic Routes, Properties, and Applications. Chem. Rev. 117, 3479–3716 (2017).
Hirai, M., Tanaka, N., Sakai, M. & Yamaguchi, S. Structurally Constrained Boron‑, Nitrogen‑, Silicon‑, And Phosphorus-Centered Polycyclic π‑Conjugated Systems. Chem. Rev. 119, 8291–8331 (2019).
Borissov, A. et al. Recent Advances in Heterocyclic Nanographenes and Other Polycyclic Heteroaromatic Compounds. Chem. Rev. 122, 565–788 (2022).
Giustra, Z. X. & Liu, S.-Y. The State of the Art in Azaborine Chemistry: New Synthetic Methods and Applications. J. Am. Chem. Soc. 140, 1184 (2018).
Li, W. et al. BN-Anthracene for High-Mobility Organic Optoelectronic Materials through Periphery Engineering. Angew. Chem. Int. Ed. 61, e202201464 (2022).
Deng, H. et al. Palladium-Catalyzed Stereospecific C–P Coupling toward Diverse PN-Heterocycles. Chem 8, 569–579 (2022).
Jiang, W., Li, Y. & Wang, Z. Heteroarenes as High Performance Organic Semiconductors. Chem. Soc. Rev. 42, 6113–6127 (2013).
Huang, T.-H., Lin, J. T., Chen, L.-Y., Lin, Y.-T. & Wu, C.-C. Dipolar Dibenzothiophene S,S-Dioxide Derivatives Containing Diarylamine: Materials for Single-Layer Organic Light-Emitting Devices. Adv. Mater. 18, 602–606 (2006).
Shu, C. et al. Mixed-Linker Strategy for the Construction of Sulfone-Containing D-A-A Covalent Organic Frameworks for Efficient Photocatalytic Hydrogen Peroxide Production. Angew. Chem. Int. Ed. 63, e202403926 (2024).
Lücking, U. Sulfoximines: A Neglected Opportunity in Medicinal Chemistry. Angew. Chem. Int. Ed. 52, 9399–9408 (2013).
Passia, M. T., Schöbel, J.-H. & Bolm, C. Sulfondiimines: Synthesis, Derivatisation And Application. Chem. Soc. Rev. 51, 4890–4901 (2022).
Zhang, X., Ang, E. C. X., Yang, Z., Kee, C. W. & Tan, C.-H. Synthesis of Chiral Sulfinate Esters by Asymmetric Condensation. Nature 604, 298–303 (2022).
Huang, S. et al. Organocatalytic Asymmetric Deoxygenation of Sulfones to Access Chiral Sulfinyl Compounds. Nat. Chem. 15, 185–193 (2023).
Liao, M. et al. Enantioselective Sulfinylation of Alcohols and Amines by Condensation with Sulfinates. Chem 10, 1541–1552 (2024).
Sun, Y. & Cramer, N. Enantioselective Access to S-Chiral 1,2-Benzothiazines by CpxRh(III)-Catalyzed C‒H Functionalization of Sulfoximines. Angew. Chem. Int. Ed. 57, 15539–15543 (2018).
Zhou, T. et al. Efficient Synthesis of Sulfur-Stereogenic Sulfoximines via Ru(II)-Catalyzed Enantioselective C−H Functionalization Enabled by Chiral Carboxylic Acid. J. Am. Chem. Soc. 143, 6810–6816 (2021).
Hirata, Y. et al. Cobalt(III)/Chiral Carboxylic Acid-Catalyzed Enantioselective Synthesis of Benzothiadiazine-1-oxides via C−H Activation. Angew. Chem. Int. Ed. 61, e202205341 (2022).
van Dijk, L. et al. Data Science-Enabled Palladium-Catalyzed Enantioselective Aryl-Carbonylation of Sulfonimidamides. J. Am. Chem. Soc. 145, 20959–20967 (2023).
Yang, J.-M. et al. Regio-Controllable [2+2] Benzannulation with Two Adjacent C(sp3)–H Bonds. Science 380, 639–644 (2023).
Strassfeld, D. A., Chen, C.-Y., Park, H. S., Phan, D. Q. & Yu, J.-Q. Hydrogen-bond-acceptor Ligands Enable Distal C(sp3)–H Arylation of Free Alcohols. Nature 622, 80–86 (2023).
von Münchow, T., Dana, S., Xu, Y., Yuan, B. & Ackermann, L. Enantioselective Electrochemical Cobalt-Catalyzed Aryl C–H Activation Reactions. Science 379, 1036–1042 (2023).
Zhou, L. et al. Synthesis of Planar Chiral Ferrocenes via Enantioselective Remote C–H Activation. Nat. Chem. 15, 815–823 (2023).
Guo, S.-M. et al. A C–H Activation-Based Enantioselective Synthesis of Lower Carbo[n]helicenes. Nat. Chem. 15, 872–880 (2023).
Li, J.-J. et al. Atroposelective Remote meta-C–H Activation. Chem 9, 1452–1463 (2023).
Albicker, M. R. & Cramer, N. Enantioselective Palladium-Catalyzed Direct Arylations at Ambient Temperature: Access to Indanes with Quaternary Stereocenters. Angew. Chem. Int. Ed. 48, 9139–9142 (2009).
Savary, D. & Baudoin, O. Enantioselective Pd0-Catalyzed C(sp2)–H Arylation for the Synthesis of Chiral Warped Molecules. Angew. Chem. Int. Ed. 60, 5136–5140 (2021).
Shintani, R., Otomo, H., Ota, K. & Hayashi, T. Palladium-Catalyzed Asymmetric Synthesis of Silicon-Stereogenic Dibenzosiloles via Enantioselective C–H Bond Functionalization. J. Am. Chem. Soc. 134, 7305–7308 (2012).
Sato, Y., Takagi, C., Shintani, R. & Nozaki, K. Palladium-Catalyzed Asymmetric Synthesis of Silicon-Stereogenic 5,10-Dihydrophenazasilines via Enantioselective 1,5-Palladium Migration. Angew. Chem. Int. Ed. 56, 9211–9216 (2017).
Lin, Z.-Q., Wang, W.-Z., Yan, S.-B. & Duan, W.-L. Palladium-Catalyzed Enantioselective C–H Arylation for the Synthesis of P-Stereogenic Compounds. Angew. Chem. Int. Ed. 54, 6265–6269 (2015).
Xu, G., Li, M., Wang, S. & Tang, W. Efficient Synthesis of P-Chiral Biaryl Phosphonates by Stereoselective Intramolecular Cyclization. Org. Chem. Front. 2, 1342–1345 (2015).
Lin, Y., Ma, W.-Y., Sun, Q.-Y., Cui, Y.-M. & Xu, L.-W. Catalytic Synthesis of Chiral Phosphole Oxides via Desymmetric C–H Arylation of o-Bromoaryl Phosphine Oxides. Synlett 28, 1432–1436 (2017).
Mukherjee, K. et al. Kinetic Resolution of Sulfur-Stereogenic Sulfoximines by Pd(II)–MPAA Catalyzed C–H Arylation and Olefination. Chem. Sci. 12, 14863–14870 (2021).
Zeng, D., Ma, Y., Deng, W.-P., Wang, M. & Jiang, X. Divergent Sulfur(VI) Fluoride Exchange Linkage of Sulfonimidoyl Fluorides And Alkynes. Nat. Synth. 1, 455–463 (2022).
Zeng, D., Ma, Y., Deng, W.-P., Wang, M. & Jiang, X. The Linkage of Sulfonimidoyl Fluorides and Unactivated Alkenes via Hydrosulfonimidoylation. Angew. Chem. Int. Ed. 61, e202207100 (2022).
Zhao, S., Zeng, D., Wang, M. & Jiang, X. C-SuFEx Linkage of Sulfonimidoyl Fluorides and Organotrifluoroborates. Nat. Commun. 15, 727 (2024).
Albano, G., Pescitelli, G. & Di Bari, L. Chiroptical Properties in Thin Films of p-Conjugated Systems. Chem. Rev. 120, 10145–10243 (2020).
Ji, Y. et al. Mono-Oxidation of Bidentate Bis-phosphines in Catalyst Activation: Kinetic and Mechanistic Studies of a Pd/Xantphos-Catalyzed C–H Functionalization. J. Am. Chem. Soc. 137, 13272–13281 (2015).
Ladd, C. L. & Charette, A. B. Access to Cyclopropyl-Fused Azacycles via a Palladium-Catalyzed Direct Alkenylation Strategy. Org. Lett. 18, 6046–6049 (2016).
Simmons, E. M. & Hartwig, J. F. On the interpretation of Deuterium Kinetic Isotope effects in C–H Bond Functionalizations by Transition-Metal Complexes. Angew. Chem. Int. Ed. 51, 3066–3072 (2012).
Vyhivskyi, O., Kudashev, A., Miyakoshi, T. & Baudoin, O. Chiral Catalysts for Pd0-Catalyzed Enantioselective C–H Activation. Chem. Eur. J. 27, 1231–1257 (2021).
Lefebvre, C. et al. Accurately extracting the signature of intermolecular interactions present in the nci plot of the reduced density gradient versus electron density. Phys. Chem. Chem. Phys. 19, 17928–17936 (2017).
Acknowledgements
The authors are grateful for the financial support provided by NSFC (22125103 (X.J.) and 22371075 (M.W.)), Top-Notch Young Talents Program of China (202312A797 (M.W.)), STCSM (22JC1401000 (X. J.)), and the Fundamental Research Funds for the Central Universities.
Author information
Authors and Affiliations
Contributions
X.J. and M.W. conceived the idea and supervised the whole project. M.W., D.Z., and X.Z. designed and carried out the experiments, and conducted the DFT calculations. X.J., M.W., and D.Z. discussed the results, contributed to the writing of the manuscript, and commented on the manuscript. All authors approved the final version of the manuscript for submission.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Bing-Feng Shi, and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zeng, D., Zhang, X., Wang, M. et al. Asymmetric S=N-embedded polyaromatic construction via enantioselective Pd-catalyzed C–H activation. Nat Commun 16, 3046 (2025). https://doi.org/10.1038/s41467-025-57251-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-57251-5
