
Abstract
Rechargeable sodium-chlorine (Na-Cl2) batteries show great promise in grid energy storage applications due to their high electrochemical performance. However, the use of highly corrosive thionyl chloride (SOCl2)-based electrolytes has severely hindered their real-world applications. Here we show a non-corrosive ester (methyl dichloroacetate) as a promising alternative to SOCl2, which can form a non-corrosive electrolyte with aluminum chloride and sodium bis(fluorosulfonyl)imide for high-performance rechargeable Na-Cl2 batteries. The resultant battery shows a reversible capacity of up to 1200 mAh g−1 at a current density of 100 mA g−1 calculated based on the mass of carbon with a discharge voltage of ~2.5 V, a wide temperature range from −40 to 80 °C, and long-term cycling stability of 700 cycles at −40 °C, which outperforms conventional rechargeable Na-Cl2 batteries and state-of-the-art Na metal batteries. The electrochemical performance and safety have been further extended to fibre batteries, which realize wearable applications of rechargeable Na-Cl2 batteries. Based on donor number and charge transfer as two key descriptors, we further propose the design principle of organic electrolytes for rechargeable Na-Cl2 batteries, which can fully unlock the designability and sustainability of organic solvents towards practical Na-Cl2 batteries.
Similar content being viewed by others

Introduction
The ever-growing demand for clean and renewable energy has driven the development of rechargeable batteries towards high energy density, high safety, and high sustainability1,2,3,4,5,6. Rechargeable Na–Cl2 battery which was developed by Dai et al. in 2021 showed advantageous element abundance and electrochemical performance among state-of-the-art Na batteries7,8,9,10,11,12,13,14,15 (Fig. 1a). Based on a Cl-based electrolyte composed of SOCl2, AlCl3, and F-based additives such as sodium bis(fluorosulfonyl)imide (NaFSI) and sodium trifluoromethanesulfonimide (NaTFSI), the resultant battery can deliver a maximum reversible capacity of 1200 mAh g−1 based on the mass of carbon with a discharge voltage of 3.5 V, making it a promising candidate in grid energy storage applications16,17. Considerable efforts have been devoted to improving their electrochemical performance17,18,19, however, the highly corrosive SOCl2 in electrolyte remains a serious problem for the production, operation, and recycling of the conventional Na–Cl2 battery15,20,21 (Fig. 1b). For example, the common battery packaging materials such as aluminum plastic film and stainless-steel case can be corroded by the SOCl2-based electrolyte, leading to inferior electrochemical performance and safety concerns. In addition, the relatively low boiling point of SOCl2 at 76 °C has inhibited the energy storage performance beyond room temperature22. Therefore, it is important to innovate the current SOCl2-based electrolyte towards low corrosivity, high thermal stability, and high electrochemical performance.

a Comparison of the theoretical specific energy of state-of-the-art Na and Li metal batteries7,8,9,10,11,12,13,14,15. The theoretical specific energy of Na–S, Na–O2, Na–I2 and Na–Cl2 batteries are calculated based on Na2S, NaO2, I2, and NaCl, respectively. More details are provided in Supplementary Table 1. b Photograph of a broken Na–Cl2 pouch cell with the leakage of SOCl2 electrolyte on a fresh leaf, leading to severe corrosion and toxic gas releasing. c Schematic illustration of the rechargeable Na–Cl2 battery based on an electrolyte comprised of AlCl3, NaFSI, and SOCl2. The high corrosivity and inferior thermal stability of SOCl2 demands alternatives such as organic solvents, but their compatibility with AlCl3 requires rational molecular structure design.
Non-corrosive organic solvents such as esters and ethers provide an attractive solution to the above issues23,24,25,26,27. Unfortunately, their compatibility with AlCl3 remains a major challenge28,29 (Fig. 1c). For instance, AlCl3 can react with a variety of cyclic ethers such as 1,3-dioxolane due to the ring-opening polymerization reaction. In addition, AlCl3 exhibits inferior solubility in some other common organic solvents such as tetrahydrofuran and acetonitrile, making it difficult to select proper organic solvents for electrolyte preparation. In our attempts to harness organic electrolytes for rechargeable Na–Cl2 battery, we notice that a non-corrosive chlorinated ester, i.e., methyl dichloroacetate (MDCA), can form a mild organic electrolyte with AlCl3 for non-corrosive and wide-temperature Na–Cl2 batteries, enabling grid and wearable energy storage applications. More importantly, we present the design principle of organic electrolytes based on charge transfer and donor number as two key descriptors, which provides new insights into the development of non-corrosive and high-performance Na–Cl2 batteries.
Results and discussion
Ester electrolytes for rechargeable Na–Cl2 batteries
While the electron-donating groups in the organic solvent improve the dissolution of AlCl3, too strong an interaction can lead to violent reactions (Fig. 2a). To test this hypothesis, we performed density functional theory simulations to analyze the electron density distributions of methyl acetate (MA), methyl dichloroacetate (MDCA), and methyl trichloroacetate (MTCA) based on the terminal groups with different chlorination degrees (Fig. 2b). With a non-chlorinated −CH3 group, MA shows the lowest minimum electrostatic potential (Emin) of −39.98 kcal mol−1 at the acyl oxygen atom, resulting in intensified coordination and violent reaction with AlCl3 (Fig. 2c); MTCA with a trichloro (–CCl3) group exhibits the highest Emin of −32.55 kcal mol−1, resulting in inferior AlCl3 solubility (Fig. 2c). In comparison, MDCA with a –CHCl2 group demonstrates a moderate Emin of −37.70 kcal mol−1, thus ensuring sufficient solubility for AlCl3 without undesirable side reactions (Fig. 2c), as verified by nuclear magnetic resonance (NMR) spectroscopy (Supplementary Fig. 1a).

a Schematic illustration of the interaction between Al3+ and C=O in ester regulated by the electron-donating and electron-withdrawing properties of the terminal group (R group in a). b Calculated electrostatic potential (ESP) maps of methyl acetate (MA), methyl dichloroacetate (MDCA), and methyl trichloroacetate (MTCA) based on –CH3, –CHCl2, and –CCl3 as the terminal group, respectively. c Photographs of the mixtures of 4 M AlCl3 and the corresponding esters in (b). MA shows a violent reaction with AlCl3, while MTCA exhibits poor solubility for AlCl3. The mixture of MDCA and AlCl3 forms a clear electrolyte without the undesirable side reactions.
The balance between coordination and non-reactivity for the mixture of AlCl3 and MDCA allows us to prepare electrolyte with the formula of 4 M AlCl3 in MDCA (denoted as AM electrolyte). In 13C NMR spectrum, the chemical shift of the C=O group in MDCA at 164.4 ppm was shifted down-field compared to bare MDCA, due to the weakened shielding effect indicating the coordination between Al3+ and MDCA30 (Supplementary Fig. 1a), which was further confirmed by the Raman spectra31 (Supplementary Fig. 1b). With the addition of 1 M NaFSI into AM electrolyte (denoted as ANM electrolyte), the signature peaks of MDCA were observed in the 13C NMR spectrum, and the up-field shift compared with that of AM electrolyte revealed the weakened interaction between MDCA and AlCl3 due to the coordination between Na+ and MDCA. In addition, the introduction of NaFSI could convert Al2Cl7− (~312 cm−1) into AlCl4− (~347 cm−1) as verified by Raman spectra, which was in good agreement with the Fourier-transform infrared (FT-IR) spectra (Supplementary Fig. 1c). Snapshots of the chemical structures of ANM electrolyte based on ab initio molecular dynamics (AIMD) simulations showed the presence of AlCl4− and [AlCl2·(MDCA)]+ in the electrolyte (Supplementary Fig. 2a). Radical distribution function (RDF) further elucidated the coordination environment at 2.11 Å and 1.82 Å corresponded to the Al−Cl and Al−O bonds in AlCl4− and [AlCl2·(MDCA)]+, respectively (Supplementary Fig. 2b).
The ANM electrolyte exhibited higher chemical stability compared to the AM electrolyte, as verified by the obvious color change after the immersion of a Na metal foil in the AM electrolyte overnight (Supplementary Fig. 3) 13C NMR spectroscopy further showed the decomposition of MDCA in the AM electrolyte with the Na metal, while the MDCA remained stable in the ANM electrolyte. Therefore, the incorporation of NaFSI in the ANM electrolyte can effectively suppress the chemical reaction between the electrolyte and Na metal negative electrode. Moreover, the elimination of Al2Cl7− in ANM electrolyte compared with that of AM electrolyte can further effectively suppress the Al deposition on the Na metal negative electrode32,33. The Na plating and stripping behaviors measured by Na||Al cells demonstrated that the ANM electrolyte showed long-term cycling stability of over 300 cycles (2100 h) at 0.15 mA cm−2 and 0.5 mAh cm−2 (Supplementary Fig. 4). In contrast, the Na||Al cell using the AM electrolyte showed drastically decreased Coulombic efficiency in less than 200 h. An average Coulombic efficiency of 94.33% was demonstrated in Aurbach test using a Na||Al cell with the ANM electrolyte (Supplementary Fig. 5). The enhanced Na plating and stripping stability could be attributed to the formation of a NaF-rich solid-electrolyte interphase layer with the presence of NaFSI in the ANM electrolyte34,35 (Supplementary Fig. 6).
Redox reactions of the positive electrode
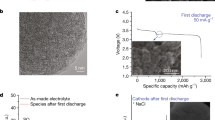
We further implemented the ANM electrolyte with a ketjenblack (KJ) positive electrode and Na metal negative electrode for battery preparation (see Section “Methods”). The resultant battery was first discharged at 150 mA g−1 to deliver a discharge capacity of 3102 mAh g−1 calculated based on the mass of KJ unless otherwise specified (Fig. 3a). The possible reactions during the first discharge involve the reduction of MDCA, with the formation of NaCl, HCCl2COAlCl2 and NaOCH3. The higher plateau at ~2.5 V corresponded to MDCA reduction with Na+ dissolving in the electrolyte by forming NaAlCl4 according to Eq. 1, without NaCl salt generated on the positive electrode as proved by X-ray diffraction (XRD) (Fig. 3b and Supplementary Fig. 7a). When Na+ became saturated, Eq. 1 terminated and NaCl salt began to deposit on the positive electrode, corresponding to a lower discharge plateau at ~1.8 V as demonstrated in Eq. 2, which was terminated when the positive electrode was fully covered by NaCl salt (Supplementary Fig. 7b).

a First discharge curve using the ANM electrolyte at a current density of 150 mA g−1. b XRD patterns of the positive electrodes in the as-prepared, discharged to 1000 mAh g−1, and fully discharged states. c 27Al NMR spectra of the ANM electrolyte before and after the first discharge. d Galvanostatic charge-discharge curves at the 50th cycle. The charge capacity and current density are 200 mAh g−1 and 150 mA g−1, respectively. e Cl K-edge XANES spectra of the positive electrodes in the first discharged and subsequent charged states. f FT-IR spectra of the positive electrodes in the as-prepared, discharged, and charged states. g In situ Raman spectra of the ANM electrolyte during a continuous discharge-charge process. TOF-SIMS depth profiling of the secondary ion fragments on the discharged (h) and charged (i) positive electrodes. j Schematic illustration of the possible reduction mechanism of MDCA during discharge.
After the first discharge, the formation of NaCl on the positive electrode was verified by XRD spectra and scanning electron microscopy (SEM) (Fig. 3b and Supplementary Fig. 8). High-resolution mass spectrometry (HRMS) profiles showed the formation of HCCl2COAlCl2 (m/z = 208.8479) from MDCA (Supplementary Fig. 9), which was further confirmed by the emerging peak at 95.56 ppm in 27Al NMR spectra (Fig. 3c). The calculated shift of 27Al NMR for HCCl2COAlCl2 was at 94.43 ppm via density functional theory (DFT), which was close to our experimental result (Supplementary Fig. 10). In addition, the FT-IR spectra of the discharged positive electrode revealed the formation of NaOCH3 after the first discharge36,37 (Supplementary Fig. 9b), indicating the reduction of MDCA on the positive electrode according to Eqs. 1 and 2.
The in situ formed NaCl on the positive electrode could enable subsequent oxidation during battery charge, accompanied with Na plating on the negative electrode as verified by the plating potential at ~−2.95 V versus Ag/AgCl in a three-electrode Swagelok cell (Supplementary Fig. 11). At a charge capacity of 200 mAh g−1, the resultant battery exhibited decent electrochemical reversibility at the 50th cycle (Fig. 3d). The reactions on the positive electrode involve NaCl/Cl2 redox (Supplementary Fig. 12) and cleavage/formation of C–O bond in MDCA during cycling. For instance, the oxidation of NaCl was confirmed by the significantly decreased signal of NaCl at the first charged state compared with that at the first discharged state in XRD (Supplementary Fig. 13a). Moreover, the Cl K-edge X-ray absorption near-edge structure (XANES) spectra (Fig. 3e) showed the absorption peak at 2824 eV corresponding to the formation of Cl2 during charging38,39, in agreement with the profiles of differential electrochemical mass spectrometry (DEMS) (Supplementary Fig. 13b, c). The detected C–Cl bond at the fully charged KJ positive electrode based on X-ray photoelectron spectroscopy (XPS) indicated the interaction between the generated Cl2 and carbon, suggesting the trapping of Cl2 inside the pores of the carbon40 (Supplementary Fig. 13d). The positive electrode-electrolyte interphase layer was further investigated using XPS, which showed the formation of NaF on the discharged positive electrode after 20 cycles (Supplementary Fig. 13e).
In addition, the redox reactions of MDCA had been verified by the decreased HCCl2COAlCl2 signal in 27Al NMR spectra during charging (Supplementary Fig. 13f), along with the weakened NaOCH3 peak in FT-IR spectra (Fig. 3f). To further confirm the reversible cleavage/formation of C−O bond in MDCA, we conducted in situ Raman spectroscopy and observed the disappeared C−O vibration (~395 cm−1) during discharging from 2.0 to 1.5 V41, as well as the regeneration of C−O vibration at the end of charging process (Fig. 3g), indicating the reversible consumption and re-generation of MDCA during battery cycling. Three-dimensional distributions of C−, NaCl−, Cl2−, CCl2COAlCl2−, and NaOCH2− were probed on the discharged positive electrode using time-of-flight secondary-ion mass spectrometry (TOF-SIMS), verifying the formation of NaCl, HCCl2COAlCl2, and NaOCH3 in the discharged state (Fig. 3h and Supplementary Fig. 14a). Besides, NaCl was mainly distributed on the positive electrode surface, while most of the reduction products of MDCA were found inside the positive electrode, suggesting that MDCA could diffuse into the carbon to form HCCl2COAlCl2 and NaOCH3. During charge, the oxidation of NaCl with the formation of Cl2 was verified by the decreased intensity of NaCl⁻ and enhanced intensity of Cl2⁻ according to TOF-SIMS (Fig. 3i and Supplementary Fig. 14b). In addition, the decreased intensities of HCCl2COAlCl2 and NaOCH3 further supported the proposed battery reactions of our rechargeable Na–Cl2 batteries according to Eq. 3 and 4 (Supplementary Fig. 15).
We thus proposed the possible reduction mechanism of MDCA in Fig. 3j. During discharge, the coordination with AlCl3 caused the activated complex to reduce on the positive electrode to form a radical anion intermediate (Int 1 in Fig. 3j), which underwent isomerization to produce an acyl radical intermediate (Int 2 in Fig. 3j), simultaneously releasing AlCl3 and NaOCH3. Int 2 could be further reduced to form an acyl carbanion intermediate (Int 3 in Fig. 3j) and then attacked AlCl3 to obtain the final product. The above process was further verified by introducing 2,2,6,6-tetramethylpiperidinyl-1-oxide (TEMPO) to the electrolyte as a radical trapping agent, and the TEMPO-Int 2 adduct (m/z = 268.0899) could be detected by HRMS (Supplementary Fig. 16).
Battery performance based on the ester-based electrolyte
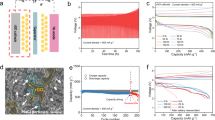
The electrochemical stability window of the ANM electrolyte was assessed using linear scan voltammetry, demonstrating stable oxidation stability up to 4.38 V at 25 °C (Supplementary Fig. 17). The rechargeable Na–Cl2 battery based on the ANM electrolyte showed decent electrochemical properties at room temperature, for instance, a maximum reversible capacity of 1200 mAh g−1 (1.4 mAh cm−2) based on the mass of carbon at an average discharge voltage of ~2.5 V, which was significantly higher than ~2.0 V based on the AM electrolyte (Fig. 4a). The charge curve exhibited a polarization effect at the end, indicating that the deposited NaCl during first discharge was almost consumed, as verified by the SEM image without observable NaCl crystals (Supplementary Fig. 18). With the deposited NaCl on the positive electrode of ~1500 mAh g−1 during the first discharge according to Eq. 2, the depth of charge reached 80% with a capacity of 1200 mAh g−1 for the subsequent charging process. Electrochemical impedance spectroscopy (EIS) showed a much lower charge-transfer resistance (Rct) of 75 Ω based on the ANM electrolyte compared with 7664 Ω based on the AM electrolyte, indicating that NaFSI can effectively facilitate charge transfer at the electrode-electrolyte interface42 (Fig. 4c and Supplementary Fig. 19 and Supplementary Table 2). Thus, the ANM electrolyte enabled an enhanced battery cycling stability of more than 200 cycles compared with less than 50 cycles using the AM electrolyte at 200 mAh g−1 and 1000 mA g−1 (Fig. 4b and Supplementary Fig. 20). The Coulombic efficiency above 100% in the initial cycles can be attributed to the additional reduction of MDCA due to the activation of KJ during NaCl oxidation over the initial cycles, similar with conventional Li/Cl2 batteries using the SOCl2-based electrolyte43. Once the active sites have been fully activated and passivated, the Coulombic efficiencies could be stabilized at ~100%. After 200 cycles in ANM electrolyte, the Na metal negative electrode exhibited a smooth surface without observable Na dendrite formation (Supplementary Fig. 21). In comparison, the use of methyl chloroacetate (MMCA) as the solvent for electrolyte preparation resulted in low Coulombic efficiencies and cycling stability (Supplementary Fig. 22a, b), which could be attributed to the inferior negative electrode-electrolyte interface, as verified by the irreversible Na plating and stripping in the Na||Al cell (Supplementary Fig. 22c).

a Galvanostatic charge-discharge curves of the rechargeable Na–Cl2 batteries using the AM and ANM electrolytes at 150 mA g−1. The charge capacity is 1200 mAh g−1, corresponding to an areal capacity of 1.4 mAh cm−2. b Cycling performance of the rechargeable Na–Cl2 batteries using the AM and ANM electrolytes at 1000 mA g−1. The specific charge capacity is 200 mAh g−1. c Nyquist plots of the Na–Cl2 batteries using the AM and ANM electrolytes. d Cycling performance of the rechargeable Na–Cl2 battery using the ANM electrolyte at 80 °C. The charge capacity and current density are 800 mAh g−1 (0.9 mAh cm−2) and 200 mA g−1, respectively. The inset shows the representative galvanostatic charge-discharge curves of the resultant battery at the 12th cycle. e Galvanostatic charge-discharge curves of the rechargeable Na–Cl2 battery using the ANMC electrolyte at increasing temperatures from −40 °C to 80 °C. The charge capacity and current density are 200 mAh g−1 and 150 mA g−1, respectively. f Cycling performance of the rechargeable Na–Cl2 battery using the ANMC electrolyte at −40 °C. The charge capacity and current density are 200 mAh g−1 and 150 mA g−1, respectively.
Distinguished from the high reactivity and low boiling point of SOCl2 that inhibits high-temperature battery performance, our ANM electrolyte exhibited cycling stability at 80 °C with a reversible capacity of 800 mAh g−1 (0.9 mAh cm−2) for 20 cycles (Fig. 4d). While the ANM electrolyte exhibited decent electrochemical performance over a broad temperature range from −20 °C to 80 °C (Supplementary Fig. 24a), we further introduced chloroform-d (CDCl3) as an efficient diluent to enhance the low-temperature performance44. With a volume ratio of MDCA: CDCl3 at 1:1, the obtained ANMC electrolyte showed an enhanced ionic conductivity of 3.36 mS cm−1 and a reduced viscosity of 2.55 mPa s at 25 °C, compared with 1.28 mS cm−1 and 18.25 mPa s of the ANM electrolyte (Supplementary Fig. 23). 13C NMR spectra confirmed the chemical stability of the ANMC electrolyte (Supplementary Fig. 24c). The introduction of CDCl3 had narrowed the 23Na spectrum line (Supplementary Fig. 24d), due to the reduced spin-lattice relaxation time originated from the heightened molecular motion. Therefore, the introduction of CDCl3 can facilitate Na+ ion transport in the electrolyte, thus benefiting the battery performance at low temperatures. The battery with the ANMC electrolyte afforded a wide temperature range from −40 °C to 80 °C (Fig. 4e), which is highly competitive among state-of-the-art conversion-type Na metal batteries17,45,46,47 (Supplementary Fig. 25a). The higher Coulombic efficiencies at high temperatures can be attributed to the enhanced MDCA reduction, as well as the facilitated Na+ transport and reaction kinetics (Supplementary Fig. 24b). In addition, it exhibited a long cycle life of more than 700 cycles at −40 °C (Fig. 4f), making it a promising competitor among low-temperature Na metal batteries48,49,50,51,52,53,54 (Supplementary Fig. 25b). With the use of CHCl3 as an alternative diluent, the resultant battery showed a decreased cycle life of 400 cycles (Supplementary Fig. 24e), which could be attributed to the higher bond energy of C−D bond compared to the C−H bond55. Such a wide temperature range can enable a variety of applications such as deep-sea and polar exploration, as demonstrated in a simulated desert area with a large temperature fluctuation (Supplementary Fig. 25c).
To validate the practicability of our rechargeable Na–Cl2 battery, we further investigated the battery retention performance in the fully charged state at 50 °C, which retained electrochemical performance during an overall retention time of 70 days, while the conventional Na–Cl2 battery using the SOCl2 electrolyte rapidly failed after only 27 h (Fig. 5a). This can be explained by the reduced corrosivity of our ester-based electrolyte compared to the highly corrosive electrolyte based on SOCl2, which caused severe corrosion to both Na metal negative electrode and packaging materials (Supplementary Fig. 26). At a negative/positive (N/P) ratio of 5:1 based on the mass of active materials, our rechargeable Na–Cl2 battery could retain more than 88% of the original capacity after 40 cycles, compared with the rapid battery failure at the 7th cycle based on the conventional SOCl2 electrolyte (Supplementary Fig. 27), thus verifying the decent electrochemical reversibility derived from our ANM electrolyte.

a Comparison of the retention capability of the rechargeable Na–Cl2 batteries using ANM and SOCl2 electrolytes. Voltage-time curves of the batteries were recorded in the fully charged states at 50 °C. The batteries were alternately cycled at 200 mAh g−1 and 150 mA g−1 to validate the electrochemical performance. b Photographs of the rechargeable Na–Cl2 pouch cells after 3-day retention at 25 °C. c Variations of the temperature and voltage of the Na–Cl2 pouch cell in the nail penetration test. The inset shows the temperature distribution of the cell right after the nail penetration test. d Cycling performance of rechargeable Na–Cl2 pouch cell using the ANM electrolyte at a charge capacity of 100 mAh and a current density of 200 mA g−1. e Cycling performance of the rechargeable Na–Cl2 fiber battery based on the ANMC electrolyte at increasing temperatures, such as −20, 0, 25, 50, and 80 °C. The charge capacity and current density are 200 mAh g−1 and 150 mA g−1, respectively.
We further fabricated rechargeable Na–Cl2 pouch cells using our ANM electrolyte, which demonstrated consistent cell thickness after 3-day retention at room temperature, in stark contrast to the severe volume expansion using the conventional SOCl2 electrolyte (Fig. 5b). Based on the headspace coupled to gas chromatography-mass spectrometry, sulfur dioxide, propane, chloromethane, and ethyl chloride were detected as the gas products (Supplementary Fig. 28), indicating the severe corrosions of the Na metal negative electrode and aluminum plastic film by the SOCl2 electrolyte. We further conducted nail penetration test using our Na–Cl2 pouch cell, which avoided short circuit and obvious heat generation (Fig. 5c). In addition, our ANM electrolyte showed mild reactivity with water, while the SOCl2 electrolyte reacted violently with water, releasing large amounts of heat and gases (Supplementary Video 1). In stark contrast to the severe corrosion of beef by the SOCl2 electrolyte from a broken pouch cell, our ANM electrolyte showed substantive less corrosive to the beef (Supplementary Fig. 29 and Supplementary Video 2). A Na–Cl2 pouch cell was produced with a first discharge capacity of 640 mAh (Supplementary Fig. 30), which could deliver a reversible capacity of ~100 mAh at 150 mA g−1 in the subsequent cycles for more than 20 cycles (Fig. 5d). We further extended the battery concept to fiber battery, based on a multi-walled carbon nanotube (MWCNT) as the positive electrode and a Na metal deposited MWCNT fiber as the negative electrode coupling with the ANMC electrolyte (Supplementary Fig. 31). The resultant fiber battery exhibited a wide temperature range from −20 °C to 80 °C (Fig. 5e), demonstrating their potential applications as wide-temperature, high-safety, and wearable power supplies3,56,57,58.
We further made a thorough comparison on the corrosion of the SOCl2- and MDCA-based electrolytes. Firstly, the conventional SOCl2-based electrolyte is corrosive to common packaging materials such as aluminum plastic film and stainless-steel coin cell case, and showed corrosivity to biological tissues such as beef, while our MDCA-based electrolyte showed significantly enhanced chemical stability to these materials. In addition, our ANM electrolyte demonstrated significantly higher Coulombic efficiencies and cycling stability in Na||Al cells compared with those based on conventional SOCl2 electrolyte (Supplementary Fig. 32). XPS profiles further showed the formation of a composite SEI comprised of NaF and organic chloride (Supplementary Fig. 33), which could efficiently suppress the corrosion of Na metal, thereby avoiding the use of substantial Na metal on the negative electrode towards practical full cells. Moreover, the conventional SOCl2-based electrolyte can generate a variety of corrosive and volatile products such as SCl2 and S2Cl2 during battery charge21, which may cause further corrosion or safety issues during battery operation. In contrast, the organic oxidation product of MDCA is less corrosive, suppressing electrolyte corrosion during long-term cycling. Notably, the trapping of Cl2 inside the pores of carbon could avoid its reaction with the other battery components, contributing to retention capability at the fully charged state at 50 °C using the MDCA-based electrolyte. Besides, the corrosion issue of Cl2 could be further eased by the immediate solidification of the MDCA-based electrolyte within seconds due to the rapid hydrolysis of AlCl3 (Supplementary Fig. 34), which could further suppress the leakage of Cl2 and benefit operation safety.
Design principles of organic electrolyte for rechargeable Na–Cl2 battery
While the ESPmin serves as a preliminary descriptor of the nucleophilic trend among similar molecules, as demonstrated by the influence of the varied chlorine substituents on methyl acetate, it cannot definitively predict the interactions of AlCl3 with different types of organic solvent59. To facilitate the development of organic electrolytes for rechargeable Na–Cl2 batteries, it is important to establish the electrolyte design principle. The primary criterion lies in the formation of a Lewis acid-base adduct between AlCl3 and organic solvent. This allows the electron deficiency of the Lewis acid to be filled by the donor’s lone pair electrons. Donor number (DN) describes the electron pair donation capability60, thus representing an important descriptor to evaluate the AlCl3 dissociation capability of the organic solvent (Fig. 6a). However, DN alone is not enough to fully describe the dissolution capability of the organic solvent. For instance, dichloroethane (DCE) has a DN of 0 which is very similar to 0.7 of acetyl chloride (AcCl), but only AcCl is capable of forming a Lewis acid-base adduct with AlCl3, while DCE exhibits poor solubility for AlCl3. Therefore, charge transfer (ΔN), defined as the fractional number of electrons transferring between the Lewis base and Lewis acid, is further introduced to evaluate the direction of charge transfer between AlCl3 and the organic solvent61 (Fig. 6a). For instance, a negative ΔN indicates electron transfer from the organic solvent to AlCl3, thus ensuring the formation of the Lewis acid-base adduct. However, if the ΔN is too negative, severe electron transfer from the organic solvent to AlCl3 would occur, leading to a violent reaction without the formation of organic electrolytes.

a Donor number (DN) and charge transfer (ΔN) of the common organic solvents and their compatibility with AlCl3. b Solvent diagram of DN versus ΔN according to density functional theory calculations. DME 1,2-dimethoxyethane, THF tetrahydrofuran, DOL 1,3-dioxolane, DMC dimethyl carbonate, DMSO dimethyl sulfoxide, AC acetone, GBL γ-butyrolactone, TEP triethyl phosphate, SL sulfolane, Py pyridine, PC propylene carbonate, EC ethylene carbonate, DCE dichloroethane, MMCA methyl monochloroacetate, AN acetonitrile, ArCl acetyl chloride. c Photographs of the mixtures of AlCl3 and various organic solvents. d, e Cycling stability and representative galvanostatic charge-discharge curves of the rechargeable Na–Cl2 battery using a nitrobenzene/AlCl3/NaFSI electrolyte. The current density and charge capacity are 500 mA g−1 and 200 mAh g−1, respectively.
Therefore, we propose the design principle of organic electrolytes for rechargeable Na–Cl2 batteries, using DN and ΔN as the key descriptors based on theoretical calculations and experimental results (Fig. 6b, c and Supplementary Table 3 and Supplementary Fig. 35). In Regions I and II of Fig. 6b, the organic solvents with high DN (>10) and relatively low ΔN values are prone to react with AlCl3, while those with low DN values in Region III demonstrate poor AlCl3 solubility (Fig. 6c). Therefore, the organic solvents in Region IV with low DN and moderate ΔN values hold promise to ensure the dissolution of AlCl3 without solvent decomposition. For verification, we selected nitrobenzene, acetyl chloride (ArCl) in Region IV as the organic solvents for electrolyte preparation (Supplementary Fig. 36), which showed decent solubility for AlCl3 without solvent decomposition. The resultant batteries showed decent electrochemical properties, for example, an average CE of 98.8% during 200 cycles with a reversible capacity of 200 mAh g−1 based on the nitrobenzene/AlCl3/NaFSI electrolyte (Fig. 6d, e). These results confirm the effectiveness of the design principle for organic electrolytes, and provide new insights into electrolyte innovation for next-generation rechargeable Na–Cl2 batteries (Supplementary Fig. 37).
Conclusions
In summary, we develop a family of non-corrosive ester-based electrolytes for rechargeable Na–Cl2 batteries. This overcomes the key challenges in high corrosivity and poor thermal stability of SOCl2-based electrolytes, thus enabling practical applications of rechargeable Na–Cl2 batteries. We verify that the −CHCl2 group in MDCA can ensure sufficient AlCl3 dissolution and suppress the undesirable reaction with AlCl3, realizing ester-based electrolytes for rechargeable Na–Cl2 batteries. Leveraging the non-corrosivity and thermal stability of our MDCA electrolyte, the resultant batteries exhibit a wide temperature range from −40 to 80 °C, and long-term cycling stability over 700 cycles at −40 °C, which significantly extend the electrochemical performance of the conventional Na–Cl2 batteries. We further present the design principle of organic electrolytes based on donor number and charge transfer as two key descriptors, which provide new insights for electrolyte development of rechargeable Na–Cl2 batteries. Our results not only harness organic electrolytes for non-corrosive and wide-temperature Na–Cl2 batteries towards practical applications but, in a broader context, establish a general electrolyte design principle to unlock the designability and sustainability of rechargeable Na–Cl2 batteries.
Methods
Materials
Methyl acetate (MA, 99.0%), methyl monochloroacetate (MMCA, 99.0%), methyl dichloroacetate (MDCA, 99.0%), methyl trichloroacetate (MTCA, 99.0%), nitrobenzene (99.0%), chloroform-D (CDCl3, 99.8%), tetrahydrofuran (THF, 99.0%), dimethyl sulfoxide (DMSO, 99.9%), acetone (AC, 99.5%), ethylene carbonate (EC, 99.9%), acetonitrile (AN, 99.9%), dichloroethane (DCE, 99.5%), 1,2-dimethoxyethane (DME, 99.0%), 1,3-dioxolane (DOL, 99.9%), dimethyl carbonate (DMC, 99.0%), γ-butyrolactone (GBL, 99.0%), triethyl phosphate (TEP, 99.0%), sulfolane (SL, 99.0%), pyridine (Py, 99.0%), propylene carbonate (PC, 99.9%) and nitromethane (99.0%) were purchased from Adamas Reagent (Shanghai). Anhydrous aluminum chloride (AlCl3, 99.0%) and acetyl chloride (ArCl, 99.0%) were purchased from TCI Development Co., Ltd. Thionyl chloride (SOCl2, 99.0%) was purchased from Energy Chemical (Shanghai). Sodium bis(fluorosulfonyl)imide (NaFSI, 99.9%) was purchased from Changde Dado New Material Co., Ltd. Ni foam (99.8%, 0.5 mm in thickness), ketjenblack (KJ, ECP-600JD), and PTFE emulsion binder (60 wt%, Daikin, D-210C) were purchased from Cyber Electrochemical Materials. Na metal cubes (99.7%) were purchased from Shanghai Aladdin Biochemical Technology Co., Ltd. All the organic solvents were dried using 4 Å molecular sieves, and the NaFSI salt was dried at 90 °C overnight in a vacuum oven before use. All the other chemicals were used as received without further purification.
Preparation of the electrolytes
All the electrolytes were prepared inside an argon-filled glove box with the contents of H2O and O2 both below 1 ppm. The AM electrolyte was prepared in a glass vial by dissolving 4 M AlCl3 in MDCA under stirring at 800 rpm for 1 h. The ANM electrolyte was prepared by adding 1 M NaFSI to the AM electrolyte, followed by stirring at 800 rpm for 2 h. The ANMC electrolyte was prepared by dissolving 4 M AlCl3 in the mixture of MDCA and CDCl3 (1:1 in volume), followed by adding 1 M NaFSI. The SOCl2 electrolyte was prepared by adding 4 M AlCl3 in SOCl2, followed by adding 1 M NaFSI under stirring at 800 rpm for 2 h. The nitrobenzene electrolyte was prepared by dissolving 8 M AlCl3 in nitrobenzene/CDCl3 (1:1 in volume), followed by adding 2 M NaFSI. The acetyl chloride electrolyte was prepared based on the same method by replacing nitrobenzene with acetyl chloride.
Preparation of the electrodes
To prepare the slurry of the positive electrode, ketjenblack (KJ) and PTFE (polytetrafluoroethylene, 60% aqueous dispersion) were mixed at a weight ratio of 9:1 in ethanol. The mixture was sonicated until the carbon materials were uniformly dispersed. The nickel (Ni) foams with a diameter of 14 mm were fabricated using a manual disk cutter (MTI, MSK-T-10), followed by cleaning in ethanol under ultrasonication for 30 min. One hundred and fifty microliters of the obtained slurry was repeatably dropped onto the Ni foam on an 80 °C hot plate after evaporation of the ethanol. The above process was repeated until the carbon mass loading reached 1–2 mg cm−2. The resultant positive electrodes were dried at 80 °C overnight in a vacuum oven, and further roll-pressed into a thinner electrode.
Preparation and characterization of the rechargeable Na–Cl2 batteries
All the batteries were assembled inside an argon-filled glove box with the contents of H2O and O2 both below 1 ppm. The Na metal cube was stored in kerosene before use. The Na metal negative electrode was prepared by cutting off the surface oxidation of a Na metal cube using a clean blade, followed by pressing the Na metal using a round plastic rod into a thin Na metal foil with a diameter of 14 mm and thickness of 0.45 mm. Two layers of glass fiber membrane (GF/D, Whatman) were used as the separator (diameter of 16 mm). 150 μL electrolyte was used for each cell. SS316 coin cell cases were used to prepare CR2032 coin cells, and a digital pressure controllable electric crimper (MTI, MSK-160E) was used for cell encapsulation. All the coin cells were encapsulated under an encapsulation pressure of 0.09 MPa. The Coulombic efficiency (CE) is calculated as the ratio of discharge capacity divided by the charge capacity in the preceding charge cycle. The Na–Cl2 fiber battery was fabricated based on a coaxial configuration, which was composed of a multi-walled carbon nanotube (MWCNT) fiber deposited with Na metal as the negative electrode, a MWCNT fiber as the positive electrode, the ANMC electrolyte, and a PP/Al2O3 separator. The assembled fiber battery was sealed inside a PTFE tube (outer diameter of 2.6 mm, inner diameter of 2 mm) using an organic silica gel (Kafuter, K-704). The electrochemical measurements were conducted at 25 °C in a thermostatic test chamber (Neware MHW-200). At least three cells were tested for a single electrochemical experiment, with statistical analysis of all the cells conducted and the most representative one presented in the plots. The low-temperature electrochemical characterizations were performed using a 0 °C freezer (Frestech Group), a − 20 °C freezer (Hicon Industry Co., Ltd), and a −40 °C freezer (Jiesheng Cryogenic Equipment Co., Ltd). The high-temperature electrochemical characterizations were performed using an electrothermal constant-temperature dry box (Yiheng Technology Co., Ltd.). The galvanostatic charge-discharge curves were measured using a Neware battery testing system (CT-4008Tn-5V50mA-164-U). A CHI660E electrochemical work station was used for the electrochemical impedance spectroscopy (EIS) measurements, using CR2032 coin cells with a Na metal foil (diameter of 14 mm) serving as both counter and reference electrodes, and a carbon loaded on Ni foam serving as the working electrode. It should be noted that the minor side reactions of the Na metal in the electrolyte may cause slight potential deviation from the ideal potential of Na metal. Prior to the EIS measurement, the cell was retained at the open-circuit voltage (OCV) condition for 60 min for stabilization. The EIS measurements were conducted under potentiostatic conditions, with the working electrode maintained at a predetermined constant potential. The frequency range was 0.01 to 105 Hz at an amplitude of 5 mV and 12 data points were collected per decade of frequency. Three-electrode Swagelok cell were conducted using a Na metal foil (diameter of 10 mm) served as the working electrode, Ag/AgCl as the reference electrode and carbon on Ni foam (diameter of 10 mm) as the counter electrode. Two hundred microliters electrolyte was used for the Swagelok cell and iR drop corrections were performed.
Characterizations
SEM images were obtained by field emission scanning electron microscopy (ZEISS, Gemini300) at an accelerating voltage of 5 kV. XRD patterns were recorded on a Rigaku X-ray diffractometer ARL Equinox at a rate of 2 ° min−1 over the range of 20–90° (2θ). Raman spectra were conducted on a Horiba LabRAM Solei Raman spectroscopy excited by a laser beam of 785 nm over the spectral range of 200–1000 cm−1. The tested electrolytes were sealed in a capillary. In situ Raman measurements were carried out using a spectroelectrochemical chamber (MicroElab) with a curve acquired every 5 minutes. XPS was performed on a Thermo ESCALAB 250Xl with a monochromatic Al Kα source (hν = 1486.6 eV) as the X-ray source. The XPS samples were disassembled in the glovebox. The positive electrodes and negative electrodes were rinsed with CHCl3 and 1,2-dimethoxyethane to remove the residual electrolyte, respectively, followed by vacuum treatment to remove the solvents. The obtained samples were then transferred to an Ar-filled chamber for XPS measurement without exposure to air. The vacuum degree of the analysis chamber was less than 5 × 10–9 Torr. The test area size is a circle with a diameter of 500 μm. All the binding energies were calibrated with C 1s peak (284.8 eV). ION-TOF TOF-SIMS 5 was used for TOF-SIMS measurement with the pressure of the analysis chamber below 1.1 × 10−9 mbar. Depth profiling was conducted using a Bi3+ ion beam of 30 keV (0.48 pA pulsed current) for organic imaging in a delay extraction mode. The sputtered Cs+ beam was 2 keV with a sputter raster of 250 × 250 μm2 and the analysis area is 50 × 50 μm2. HRMS were acquired on a Waters Micro mass quadrupole/time-of-flight (Q-ToF) Premier mass spectrometer (G2-XS/APGC, 50-2000 m/z) in both positive and negative ion modes. The solvent system was acetonitrile with a flow rate of 0.1 mL min−1. The data was analyzed by MassLynx V4.1 software. The gas products in pouch cells during retention were analyzed using headspace coupled to gas chromatography-mass spectrometry (Agilent 8890). The NMR experiment was performed at 25 °C on a Bruker Avance III 500 MHz liquid NMR spectrometer equipped with a Z-axis gradient filed (50 G/cm in maximum) BBFO probe. All the NMR tubes were dried in vacuum at 80 °C before use. In the glovebox, 400 μL of electrolyte was transferred into the NMR tube (5 mm high throughput). A coaxial internal insert that contains 100 μL NMR solvent (i.e., DMSO) was inserted into the NMR tube to ensure the analysis while preserving the original electrolyte. The NMR tubes were tightly sealed with their caps and the Parafilm tape. 13C NMR spectra were acquired with a spectral width of 239.5 ppm, an acquisition time of 1.08 s, a recycle delay of 1 s, and 512 number of scans. 27Al NMR spectra were acquired with a spectral width of 1009.6 ppm, an acquisition time of 0.25 s, a recycle delay of 1 s, and 32 number of scans. FT-IR spectra were recorded on a Nicolet 6700 FT-IR spectrometer in the 400–4000 cm−1 region at room temperature. Cl K-edge XANES spectra were carried out at 4B7A station of Beijing Synchrotron Radiation Facility (BSRF). The beamline was operated at 2.5 GeV with a maximum current of 250 mA using a Si (111) double-crystal monochromator. The spectrum of the standard NaCl sample was acquired using a total electron yield mode at a vacuum degree of 10−3 to 10−4 Pa. DEMS were conducted using a commercial quadrupole mass spectrometer (Linglu Instrument). A home-made Swagelok battery equipped with two poly(ether-ether-ketone) capillary tubes as the gas inlet and outlet was employed to detect the ion current of the gas product under argon purge. The battery was first flushed with Ar gas at a constant flow rate of 1 mL min−1 for 2.5 h to calibrate the baseline. The discharge/charge time was 74 minutes at a current density of 1.2 mA cm−2 with carrier gas flowing at 1 mL min−1. The DEMS results at the charge and discharge states mainly focused to reveal the variation of Cl2 during battery cycling. The electron multiplier was used to amplify the signal during tests. The ionic conductivities were measured using a commercial conductivity meter (Mettler Toledo FE38). The probe was calibrated to a standard solution (1413 μS cm−1) at 25 °C. Approximately 10 mL electrolyte was added to a vial and was allowed to equilibrate with the temperature for at least 20 min. Electrolyte viscosities were measured using an DHR 20 rheometer (TA Instruments) with a sheer rate of 300/s at 25 °C. DSC analysis was carried out using a differential scanning calorimeter (DSC250). Before analysis, 8 μL electrolyte in a sealed aluminum oxide pan was heated from 25 to 80 °C with a rate of 10 °C min−1 under N2 to erase the thermal history of the samples. While measuring the heat flow, the temperature was decreased to −60 °C and then increased to 80 °C with a rate of 10 °C min−1.
Ab initio molecular dynamics simulations
All the AIMD simulations were performed within the framework of density functional theory (DFT) using the projector-augmented-wave formalism in the Vienna Ab initio Simulation Package (VASP)62,63. The generalized gradient Perdew–Burke–Ernzerh of functional was adopted in the density functional theory calculations and the core electrons were described using the projector augmented wave method64,65. The plane-wave-energy cutoff was set at 450 eV and the K mesh was sampled at the Γ point only. According to the density of the mix electrolyte, we built a cell with 13.3 × 13.3 × 13.3 Å3 containing 10 MDCA, 4 AlCl3 and 1 NaFSI. Long-range van der Waals dispersion interactions were treated using the DFT-D2 method66. Each electrolyte system was equilibrated at 30 °C in the canonical ensemble (NVT) for 20 ps, wherein constant temperature conditions were maintained by is of a Nose–Hoover thermostat. A timestep of 0.5 fs was used to integrate the equations of motion.
Charge transfer calculations
Density functional theory (DFT) calculations were performed using the Gaussian 16 program package67. The ground state geometries of all the studied molecules were optimized and calculated using the B3LYP-D3(BJ)/6-311 G (d, p) level of theory68,69,70,71. Vibrational frequency analysis was performed at the same level of theory to confirm their positions at the minimum of the potential energy surface. In all cases, we obtained zero imaginary frequencies. To enhance the accuracy of the single-point energy, the stable conformations derived earlier were used as the initial structures for single-point calculations at the M06-2X/cc-pVTZ level72,73. Besides, the solvation effect was described with the implicit solvation model (Solvation Model Based on Density, SMD)74, and the relevant parameters were listed in Supplementary Table 4. Based on these results, the electrostatic potential (ESP) of the solvents were analyzed using Multiwfn75 and Visual Molecular Dynamics76,77. In addition, the charge transfer (ΔN) descriptor is attributed to the fractional number of electrons transferred between the molecules A and B and was defined as Eq. 561:
Where μA, μB and ηA, ηB are chemical potential and chemical hardness of A and B, respectively. The direction of charge/electron transfer is determined by the sign of ΔN, which the negative sign refers to the direction from A to B and a positive sign indicates the reverse direction. The chemical potential and chemical hardness can be expressed as Eqs. 6 and 7:
Where I is the ionization energy and A is the electron affinity of the system. In ΔSCF method, the ionization energy (I) and electron affinity (A) of the system can be calculated as Eqs. 8 and 9:
Where, \({E}_{N}\), \({E}_{(N+1)}\), \({E}_{(N-1)}\) represents the energy of neutral, anion, and cation systems, respectively.
NMR simulations
To calculate the 27Al spectrum of HCCl2COAlCl2, we first examined the thermodynamic stability of possible species in our electrolyte. As a preliminary test, we calculated the equilibria for 2HCCl2COAlCl2\(\,\rightleftharpoons\) [HCCl2COAlCl2]2 using the same theoretical methods applied previously.
Consistent with observations for AlCl3, our calculations indicated that the dimeric form of HCCl2COAlCl2 was thermodynamically favored in solution. The resonant species that was most likely detected by 27Al NMR spectroscopy was HCCl2COAlCl3−, which was used to calculate the chemical shift. The optimized structure was calculated at the MP2/pcSseg-2 level78,79 using GIAO method80, with AlCl4− as a reference.
Data availability
The data that support the findings of this study are available from the corresponding author upon request.
References
Armand, M. & Tarascon, J. M. Building better batteries. Nature 451, 652–657 (2008).
Larcher, D. & Tarascon, J. M. Towards greener and more sustainable batteries for electrical energy storage. Nat. Chem. 7, 19–29 (2014).
Ye, L. et al. A rechargeable calcium–oxygen battery that operates at room temperature. Nature 626, 313–318 (2024).
Wang, L., Zhang, Y. & Bruce, P. G. Batteries for wearables. Natl. Sci. Rev. 10, 1–2 (2023).
Yang, C. et al. Copper-coordinated cellulose ion conductors for solid-state batteries. Nature 598, 590–596 (2021).
Zhao, X., Zhao‐Karger, Z., Fichtner, M. & Shen, X. Halide‐Based Materials and Chemistry for Rechargeable Batteries. Angew. Chem. Int. Ed. 59, 5902–5949 (2020).
Cao, X. et al. Nanoflake-constructed porous Na3V2(PO4)3/C hierarchical microspheres as a bicontinuous cathode for sodium-ion batteries applications. Nano Energy 60, 312–323 (2019).
Zhang, H. et al. Prussian blue analogues with optimized crystal plane orientation and low crystal defects toward 450 Wh kg−1 alkali‐ion batteries. Angew. Chem. Int. Ed. 62, e202303953 (2023).
Zhang, H. et al. Long‐cycle‐life cathode materials for sodium‐ion batteries toward large‐scale energy storage systems. Adv. Energy Mater. 13, 2300149 (2023).
Wang, L. et al. Li-free cathode materials for high energy density lithium batteries. Joule 3, 2086–2102 (2019).
Kong, W. J. et al. From liquid to solid-state batteries: Li-rich Mn-based layered oxides as emerging cathodes with high energy density. Adv. Mater. 36, 2310738 (2023).
Zhang, L. et al. Metal–iodine batteries: achievements, challenges, and future. Energy Environ. Sci. 16, 4872–4925 (2023).
Xia, C., Black, R., Fernandes, R., Adams, B. & Nazar, L. F. The critical role of phase-transfer catalysis in aprotic sodium oxygen batteries. Nat. Chem. 7, 496–501 (2015).
Cao, W., Zhang, J. & Li, H. Batteries with high theoretical energy densities. Energy Stor. Mater. 26, 46–55 (2020).
Yuan, B. et al. Revitalizing chlorine–based batteries for low–cost and high–performance energy storage. Adv. Energy Mater. 14, 2303127 (2023).
Zhu, G. et al. Rechargeable Na/Cl2 and Li/Cl2 batteries. Nature 596, 525–530 (2021).
Xiang, L. et al. Ultrahigh‐rate Na/Cl2 batteries through improved electron and ion transport by heteroatom‐doped bicontinuous‐structured carbon. Angew. Chem. Int. Ed. 62, e202312001 (2023).
Ma, C. et al. Vertical‐channel cathode host enables rapid deposition kinetics toward high‐areal‐capacity sodium‐chlorine batteries. Small 20, 2310978 (2024).
Feng, W. et al. Iodine-induced self-depassivation strategy to improve reversible kinetics in Na-Cl2 battery. Nat. Commun. 15, 6904 (2024).
Hennesø, E. & Hedlund, F. H. Explosion of lithium–thionyl-chloride battery due to the presence of lithium nitride. J. Fail. Anal. Preven. 15, 600–603 (2015).
Xie, Z. et al. Rechargeable alkali metal–chlorine batteries: advances, challenges, and future perspectives. Chem. Soc. Rev. 53, 8424–8456 (2024).
Gangadharan, R., Namboodiri, P. N. N., Prasad, K. V. & Viswanathan, R. The lithium—thionyl chloride battery — a review. J. Power Sources 4, 1–9 (1979).
Yi, X. et al. Safe electrolyte for long-cycling alkali-ion batteries. Nat. Sustain. 7, 326–337 (2024).
Li, G.-X. et al. Enhancing lithium-metal battery longevity through minimized coordinating diluent. Nat. Energy 9, 817–827 (2024).
Ma, B. et al. Molecular-docking electrolytes enable high-voltage lithium battery chemistries. Nat. Chem. 16, 1427–1435 (2024).
Yu, Z. et al. Rational solvent molecule tuning for high-performance lithium metal battery electrolytes. Nat. Energy 7, 94–106 (2022).
Li, A.-M. et al. Methylation enables the use of fluorine-free ether electrolytes in high-voltage lithium metal batteries. Nat. Chem. 16, 922–929 (2024).
Utomo, N. W., Deng, Y., Zhao, Q., Liu, X. & Archer, L. A. Structure and Evolution of Quasi‐Solid‐State Hybrid Electrolytes Formed Inside Electrochemical Cells. Adv. Mater. 34, 2110333 (2022).
Canepa, P. et al. Elucidating the structure of the magnesium aluminum chloride complex electrolyte for magnesium-ion batteries. Energy Environ. Sci. 8, 3718–3730 (2015).
Gottlieb, H. E., Kotlyar, V. & Nudelman, A. NMR chemical shifts of common laboratory solvents as trace impurities. J. Org. Chem. 62, 7512–7515 (1997).
Sun, H. et al. A safe and non-flammable sodium metal battery based on an ionic liquid electrolyte. Nat. Commun. 10, 3302 (2019).
Lin, M.-C. et al. An ultrafast rechargeable aluminium-ion battery. Nature 520, 324–328 (2015).
Wang, G. et al. An efficient rechargeable aluminium–amine battery working under quaternization chemistry. Angew. Chem. Int. Ed. 61, e202116194 (2022).
Liu, T. et al. Recycled micro-sized silicon anode for high-voltage lithium-ion batteries. Nat. Sustain. 7, 1057–1066 (2024).
He, J. et al. Tuning the solvation structure with salts for stable sodium-metal batteries. Nat. Energy 9, 446–456 (2024).
Chusid, O., Ein Ely, E., Aurbach, D., Babai, M. & Carmeli, Y. Electrochemical and spectroscopic studies of carbon electrodes in lithium battery electrolyte systems. J. Power Sources 43, 47–64 (1993).
Seubold, F. H. Jr. Anionic hyperconjugation: the infrared spectra of sodium alkoxides. J. Org. Chem. 21, 156–160 (1956).
Vaudey, C. E. et al. Chlorine speciation in nuclear graphite studied by X-ray absorption near edge structure. J. Nucl. Mater. 418, 16–21 (2011).
Yang, C. et al. Aqueous Li-ion battery enabled by halogen conversion–intercalation chemistry in graphite. Nature 569, 245–250 (2019).
Zhu, G. et al. Shedding light on rechargeable Na/Cl2 battery. Proc. Natl. Acad. Sci. USA 120, e2310903120 (2023).
Mido, Y. & Hashimoto, M. Conformational isomerism of methyl dichloroacetate. An infrared, Raman and 35Cl NQR study. J. Mol. Struct. 131, 71–81 (1985).
Weng, S. et al. Temperature-dependent interphase formation and Li+ transport in lithium metal batteries. Nat. Commun. 14, 4474 (2023).
Zhu, G. et al. High-capacity rechargeable Li/Cl2 batteries with graphite positive electrodes. J. Am. Chem. Soc. 144, 22505–22513 (2022).
Lang, C. M., Kim, K. & Kohl, P. A. Catalytic additives for the reversible reduction of sodium in chloroaluminate ionic liquids. Electrochim. Acta 51, 3884–3889 (2006).
Kumar, A. et al. Sub-zero and room-temperature sodium–sulfur battery cell operations: A rational current collector, catalyst and sulphur-host design and study. Energy Stor. Mater. 42, 608–617 (2021).
Yang, X. et al. Upgrading cycling stability and capability of hybrid Na‐CO2 batteries via tailoring reaction environment for efficient conversion CO2 to HCOOH. Adv. Energy Mater. 14, 2304365 (2024).
Guo, C. et al. Hydrogen‐bonded organic framework for high‐performance lithium/sodium‐iodine organic batteries. Angew. Chem. Int. Ed. 61, e202213276 (2022).
Wang, C. et al. Extending the low-temperature operation of sodium metal batteries combining linear and cyclic ether-based electrolyte solutions. Nat. Commun. 13, 4934 (2022).
Zhong, S. et al. Molecular engineering on solvation structure of carbonate electrolyte toward durable sodium metal battery at −40 °C. Angew. Chem. Int. Ed. 62, e202301169 (2023).
Hu, L. et al. Restructuring electrolyte solvation by a versatile diluent toward beyond 99.9% coulombic efficiency of sodium plating/stripping at ultralow temperatures. Adv. Mater. 36, 2312161 (2024).
Hu, C. et al. Carbonate ester‐based sodium metal battery with high‐capacity retention at −50°C enabled by weak solvents and electrodeposited anode. Angew. Chem. Int. Ed. 63, e202407075 (2024).
Wang, S., Zhang, X.-G., Gu, Y., Tang, S. & Fu, Y. An ultrastable low-temperature na metal battery enabled by synergy between weakly solvating solvents. J. Am. Chem. Soc. 146, 3854–3860 (2024).
Yu, D. et al. Low‐temperature and fast‐charge sodium metal batteries. Small 20, 2311810 (2024).
Lv, X. et al. Construction of inorganic/organic hybrid layer for stable Na metal anode operated under wide temperatures. Small 19, 2300215 (2023).
Jurgeleit, H. C. & Wolfgang, R. Moderating and reactive collisions of hot hydrogen atoms: a study on isotope effects in methyl fluoride. J. Am. Chem. Soc. 85, 1057–1062 (1963).
Ye, L. et al. Deformation-tolerant metal anodes for flexible sodium-air fiber batteries. eScience 2, 606–614 (2022).
Zhang, Y. et al. Ultrahigh line-capacity and flexible graphene/carbon nanotube/tin oxide fibers as sodium ion battery anodes. Energy Stor. Mater. 48, 35–43 (2022).
Liao, M. et al. Industrial scale production of fibre batteries by a solution-extrusion method. Nat. Nanotech. 17, 372–377 (2022).
Wu, Y. et al. Electrostatic potential as solvent descriptor to enable rational electrolyte design for lithium batteries. Adv. Energy Mater. 13, 2300259 (2023).
Marcus, Y. The effectivity of solvents as electron pair donors. J. Solution Chem. 13, 599–624 (1984).
Sinha, S., Das, S., Roymahapatra, G. & Giri, S. Imidazolium based superalkalis as building block for Lewis base. Comput. Theor. Chem. 1210, 113639 (2022).
Kresse, G. & Furthmüler, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Perdew, J. P., Burke, K. & Ernzerhof, M. Errata: generalized gradient approximation made simple. Phys. Rev. Lett. 78, 1396 (1997).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Harl, J., Schimka, L. & Kresse, G. Assessing the quality of the random phase approximation for lattice constants and atomization energies of solids. Phys. Rev. B 81, 115126 (2010).
Frisch, M. J. et. al. Gaussian 16, Revision A.03 (Gaussian, Inc, Wallingford, CT, 2016).
Krishnan, R., Binkley, J. S., Seeger, R. & Pople, J. A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 72, 650–654 (1980).
Stephens, P. J., Devlin, F. J., Chabalowski, C. F. & Frisch, M. J. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 98, 11623–11627 (1994).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurateab initioparametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Grimme, S., Ehrlich, S. & Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 32, 1456–1465 (2011).
Zhao, Y. & Truhlar, D. G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 120, 215–241 (2007).
Kendall, R. A., Dunning, T. H. & Harrison, R. J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 96, 6796–6806 (1992).
Marenich, A. V., Cramer, C. J. & Truhlar, D. G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Chem. Phys. 113, 6378–6396 (2009).
Lu, T. & Chen, F. Multiwfn: a multifunctional wavefunction analyzer. J. Comput. Chem. 33, 580–592 (2011).
Zhang, J. & Lu, T. Efficient evaluation of electrostatic potential with computerized optimized code. Phys. Chem. Chem. Phys. 23, 20323–20328 (2021).
Lu, T. & Chen, F. Quantitative analysis of molecular surface based on improved Marching Tetrahedra algorithm. J. Mol. Graph. 38, 314–323 (2012).
Jensen, F. Segmented contracted basis sets optimized for nuclear magnetic shielding. J. Chem. Theory Comput. 11, 132–138 (2014).
Pritchard, B. P., Altarawy, D., Didier, B., Gibson, T. D. & Windus, T. L. New basis set exchange: an open, up-to-date resource for the molecular sciences community. J. Chem. Inf. Model 59, 4814–4820 (2019).
Wolinski, K., Hinton, J. F. & Pulay, P. Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculations. J. Am. Chem. Soc. 112, 8251–8260 (1990).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (22209108) and the Fundamental Research Funds for the Central Universities (23X010301599). The authors acknowledge the 4B7A station at the Beijing Synchrotron Radiation Facility.
Author information
Authors and Affiliations
Contributions
H.S. conceived and designed the project. Q.X. and S.T. prepared electrolytes and electrodes, fabricated rechargeable Na–Cl2 coin cells, and performed material and electrochemical characterizations. Q.X., Y.W., X. Zhang and M.L. fabricated and characterized rechargeable Na–Cl2 pouch cells and fiber batteries. S.T., N.L. and Y.S. performed the density functional theory simulations. Q.X., X. Zhao., S.G., B.Y., and L.M. performed in situ characterizations. S.T., Z.O., and S.W. conducted application demonstrations. All the authors discussed and analyzed the data. Q.X., S.T., M.S., Y.S., H.S., and H.P. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Chun Yuen Kwok, Xiangyu Zhao, and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xu, Q., Tang, S., Li, N. et al. Harnessing organic electrolyte for non-corrosive and wide-temperature Na-Cl2 battery. Nat Commun 16, 1946 (2025). https://doi.org/10.1038/s41467-025-57316-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-57316-5
This article is cited by
-
Noncorrosive organic electrolytes for wide-temperature metal-chlorine batteries
Science China Chemistry (2025)
