
Abstract
Numerous patients with rheumatoid arthritis (RA) manifest severe syndromes, including elevated synovial fluid volumes (SF) with abundant immune cells, which can be controlled by TNF/JAK inhibitors. Here, we apply single-cell RNA sequencing (scRNA-seq) and subsequent validations in SF from RA patients. These analyses of synovial tissue show reduced density of SF-derived pathogenic cells (e.g., SPP1+ macrophages and CXCL13+CD4+ T cells), altered gene expression (e.g., SPP1 and STAT1), molecular pathway changes (e.g., JAK/STAT), and cell-cell communications in drug-specific manners in samples from patients pre-/post-treated with adalimumab/tofacitinib. Particularly, SPP1+ macrophages exhibit pronounced communication with CXCL13+CD4+ T cells, which are abolished after treatment and correlate with treatment efficacy. These pathogenic cell types alone or in combination can augment inflammation of fibroblast-like synoviocytes in vitro, while conditional Spp1 knocking-out reduces RA-related cytokine expression in collagen-induced arthritis mice models. Our study shows the functional role of SF-derived pathogenic cells in progression and drug-specific treatment outcomes in RA.
Similar content being viewed by others

Introduction
Rheumatoid arthritis (RA) is a prevalent inflammatory autoimmune disease, characterized by joint swelling, progressive bone destruction, and irreversible joint deformity1. Altered cellular compositions with aberrant molecular characteristics are present in the synovial tissue (ST), and the increased possibility of the presence and volume of synovial fluid (SF) in the joint cavity are positively related to the disease activity of RA2,3,4,5,6. Multiple pro-inflammatory cytokines and chemokines are released by the inflammatory ST of RA patients, attracting and activating immune cells (e.g., macrophages, dendritic cells (DCs), and T cells) to infiltrate the joint through various signaling pathways (e.g., TNF signaling and the JAK/STAT pathway), thus releasing more inflammatory factors to induce further inflammatory cascade amplification and persistent joint injury through cytokine-receptor communications7. Therefore, immunosuppressants or inhibitors targeting inflammatory-related proteins are considered as disease-modifying antirheumatic drugs (DMARDs), allowing the achievement of disease control1,8. Although incurable, around 20–40% of RA patients can achieve ≥70% improvement in disease activity score remission (ACR70), which is used as surrogates of low disease activity, after 6 months of treatment with conventional DMARDs (cDMARDs, e.g., methotrexate). Additional around 20% of methotrexate-insufficient responders can achieve ACR70 with biologic DMARDs (bDMARDs, e.g., adalimumab) or targeted-synthetic DMARDs (tsDMARDs, e.g., tofacitinib) through targeting the tumor necrosis factor (TNF) or its downstream JAK-STAT pathway, respectively8,9,10,11,12. However, even with the criteria of ACR20, which indicates an improvement of 20% and is the minimal required response after 6 months of treatment with DMARDs, some MTX-insufficient RA patients with poor responses are still widely reported8,13,14. Moreover, the 2021 ACR guideline for RA treatment make recommendations based on specific patient groups (e.g., non-TNF inhibitor bDMARDs or tsDMARDs for RA patients with heart failure15), and the US FDA recommends using JAK inhibitors for RA patients after TNF inhibitor failure due to the higher incidence of major adverse cardiovascular events and cancer induced by tofacitinib in elderly patients16,17. However, the incidence of adverse events is very low, and the safety issue of tofacitinib in all elderly patients is still controversial18. Therefore, it is important to reveal the altered molecular immunoprofiles after DMARDs treatment and the underlying mechanisms of severe RA.
It is well reported that RA pathogenesis and treatment outcomes are attributed to different cell types (e.g., fibroblasts, macrophages, and T cells) as well as their communications in the joint microenvironment, which may be induced by multiple factors (e.g., gut microbiota)19,20,21,22,23,24,25,26,27,28. For instance, macrophages are the main producers of pathogenic cytokines, such as TNF and interleukin-629,30, which are associated with stimulation of fibroblast-like synoviocytes (FLS) and osteoclast activity31. Peripheral helper T cells can induce activation of local B cells and antibody production in RA patients32. However, previous cytological investigations relying on traditional approaches (e.g., flow cytometry) have hampered the ability to systematically explore the damaging role of immune cells in RA and their communications at the single-cell level due to the low throughput. With the development of single-cell transcriptome sequencing (scRNA-seq), multiple studies have delineated the cellular transcriptional profile of different cell components of synovial tissue from RA patients and revealed some specific cell subtypes that may contribute to RA pathogenesis, development, and treatment outcomes19,20,21,22,23,24,25,26,33,34,35,36. For instance, FAPα+THY1+/THY1+HLA-DRAhi fibroblasts are potential key mediators of RA pathogenesis and induce severe and persistent inflammatory arthritis20,37. The proportion of MerTKpos and MerTKnegSPP1pos macrophages is associated with risk of disease flare after treatment cessation22. However, biopsy of ST is an invasive approach and is not routinely performed for RA diagnosis and post-treatment evaluation in clinical practice. Instead, noninvasively obtained SF may partially reflect the characteristics of immune cells in ST, such as the high overall overlap rate of patient-specific T cell clones between ST and SF revealed through TCR sequencing38. Recently, clonally expanded synovial CXCL13high peripheral helper T cells in SF of RA were identified through scRNA-seq with sorted CD4+ T cells21. Overall, although multiple scRNA-seq-based studies have been performed for RA, most of them focused on ST, which is difficult to conduct paired comparisons between pre- and post-treatment to reveal the dynamic changes. On the other hand, the impact of biologic DMARDs/targeted synthetic DMARDs on RA remains to be elucidated to distinguish between the responder and non-responder at single cell resolution, which may provide evidence for the precision prescription of RA.
In this study, we conduct scRNA-seq, bulk-sequencing, flow cytometry, ELISA, multiplex immunofluorescence (mIF), and in vitro activation assays with SF in RA and osteoarthritis (OA) to explore the dynamic immunoprofiles of cell components and their communications impacted by the treatment with adalimumab/tofacitinib, in order to reveal the cellular/molecular basis of individualized RA pathogenesis and treatment outcomes.
Results
Single-cell transcriptome profiling of synovial fluid (SF) cells in RA patients
To comprehensively define the transcriptional atlas of SF cells from RA patients, who are methotrexate insufficient responders, we performed scRNA-seq on 3 samples from treatment-naive osteoarthritis (OA) patients (n = 3) and 12 paired samples from RA patients (n = 6) before and after one month of treatment with TNF/JAK inhibitors (referred to as RA-BT and RA-AT) (Fig. 1a). Additional samples from 18 OA patients and 38 RA patients were used as validation through bulk RNA-sequencing, flow cytometry (FACS), ELISA, and/or scRNA-seq (Fig. 1a and Supplementary Data 1–3). In order to explore the cellular characteristics with different treatment strategies, six RA patients were randomly treated with the TNF inhibitor (i.e., adalimumab, n = 3) or the JAK inhibitor (i.e., tofacitinib, n = 3) (Fig. 1a). Both strategies for one month significantly reduced disease activity in RA patients according to DAS28-/SDAI-based disease activity reduction (Supplementary Fig. 1a–c). With 34 patients with available ACR20 information, a total of 22 patients achieved the minimal required response for improvement at the first month post-treatment (these early responders were referred to as ACR20_Y), while the rest 12 were not (referred to as ACR20_N) (Supplementary Fig. 1d, e and Supplementary Data 4). Significantly higher cell density was observed in SF from RA compared to OA patients (Supplementary Fig. 1f, and Supplementary Data 5), suggesting a more severe immune aberration of SF from RA.

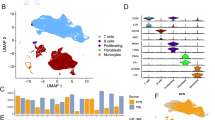
a Overview of the workflow and discovery/validation cohorts. OA osteoarthritis, RA rheumatoid arthritis, s single sample, p paired samples. Created in BioRender. Xu, H. (2025) https://BioRender.com/o60h131. b Visualization of nine main clusters across 63,035 cells using t-distributed stochastic neighbor embedding (tSNE). BT before treatment, AT after treatment. c Dot plot illustrating the expression level of marker genes across SF clusters. The dots’ size and color spectrum indicate positive percentage and average expression (log1p transformed) of particular markers genes in each cell type, respectively. d Quantification of absolute cell count and relative proportions of distinct cell clusters in SF among patients. The horizontal coordinates depict cell number and proportions, the vertical coordinates depict patients. e Density of macrophage clusters among different groups. Density is calculated by multiplying the total cell density in each SF sample by the proportion of macrophage cluster in the sample. The p-values were calculated using a two-sided Wilcoxon test. OA-BT: n = 3, RA-BT: n = 6, and RA-AT: n = 6. solid squares, patients didn’t receive b/tsDMARDs treatment; hollow circles, adalimumab treated; solid circles, tofacitinib treated. Data are presented as mean values ± SEM. f Deconvolution of bulk RNA-seq data for eight major cell clusters across all samples based on canonical marker genes. The p-values were calculated using a two-sided Wilcoxon test, comparing OA-BT (n = 5), RA-BT (n = 14), and RA-AT (n = 10). The box is bounded by the first and third quartile with a horizontal line at the median and whiskers extend to the maximum and minimum value. g Left: Representative flow cytometry (FACS) dot plots of CD64 and CD11b expression in SF cells. Right: Comparison of FACS-based macrophage proportions. Data are presented as mean ± SD. p-values were calculated using a two-sided Student’s t test. h Deconvolution of published bulk data (GSE55235) for macrophages in ST samples from OA (n = 10) and RA (n = 10) patients. The box is bounded by the first and third quartile with a horizontal line at the median and whiskers extend to the maximum and minimum value.
After stringent quality filtering and batch effect correction of scRNA-seq data (Supplementary Fig. 1g), 63,035 cells from SF were clustered into nine main cell types with an unsupervised approach (Fig. 1b), defined by well-established canonical marker genes (Fig. 1c). Due to the fragile characterization and low RNA content of neutrophils39,40, the annotated neutrophil cluster was excluded in the subsequent analysis. Fibroblasts, the main components in STs according to previous reports25,37, were low in proportion and mainly derived from OA patients, whereas macrophages (46.4%), DCs (17.1%), and T cells (31.5%) were the predominant components in the SF of RA (Fig. 1d). Although the proportions of DCs and macrophages were consistently enriched in SF from OA and RA patients respectively, higher density was only observed in macrophages from RA-BT than that from OA patients and significantly decreased after treatment (Fig. 1e, and Supplementary Fig. 2a–c). A similar trend in the proportion of cell types was verified in the validation cohort through either bulk transcriptome profiles or subsequent deconvolution analysis in validation cohort (5 OA-BT, 10 pairs of RA-BT/RA-AT, and 4 single RA-BT samples) (Fig. 1f). Macrophages and NK/T cells were significantly increased in RA patients compared to OA patients, but no significant difference was observed in RA-BT vs. RA-AT and ACR20_N vs. ACR20_Y (all comparisons between ACR20_N vs. ACR20_Y were conducted using the data at baseline) in all cell types (Fig. 1f and Supplementary Fig. 2d). A higher proportion of macrophages in RA patients and DCs/fibroblasts in OA patients can also be verified through flow cytometry (FACS) (Fig. 1g and Supplementary Fig. 2e–i), and a higher proportion of macrophages can be validated in ST according to the deconvolution score based on public transcriptome data (Fig. 1h).
Altered gene expression and pathways in RA and impact of DMARDs treatment
We first profiled the well-established RA-related genes in each cell type. Consistent with prior scRNA-seq-based findings in RA synovial tissue37,41,42, IL6, IL1A, IL12A, and IL12B exhibited low positive expression rate, while JAK1/JAK3 exhibited higher overall expression than TNF (Fig. 2a). Moreover, expression of JAK1/JAK3 was significantly higher in RA compared to that in OA and decreased after treatment with DMARDs (Fig. 2a, b). Interestingly, tofacitinib but not adalimumab can more specifically inhibit JAK1/JAK2/JAK3 expression in T cells and JAK3 expression in macrophages (Fig. 2b), which can be supported by the decreased expression of JAK3 post-treated with tofacitinib/adalimumab by using public bulk transcriptomic data (Supplementary Fig. 3a–e), suggesting higher regulatory effects of tofacitinib than adalimumab on JAK signaling in these cell types at transcriptional level possibly through a negative feedback loop43.

a Heatmap depicting the expression positivity of well-reported RA-related genes in the major cell types. The color spectrum signifies the ratio of cells that exhibit positive gene expression. b Dot plot illustrating the pairwise comparisons of expression levels of well-reported RA-related genes in different cell clusters. The p-values were calculated using a two-sided Wilcoxon test. The dot size and color spectrum indicate q-value (-log10 transformed) and fold change (log2 transformed) of gene expression, respectively. c Feature plot showing the count of differentially expressed genes (DEGs) determined by a two-sided Wilcoxon test. d Volcano plot displaying the DEGs in macrophages. The adjusted p-values were calculated using a two-sided Wilcoxon test. N.S., non-significant (p > 1 × 10−10); Sig, significant (p < 1 × 10−10); Overlapped-up and -down indicate the intersecting DEGs significantly up- and down-regulated in RA-BT group (p < 1 × 10−10), respectively. e, f Enriched gene ontology (GO) and gene set enrichment analysis (GSEA) pathways for the overlapped-up genes in macrophages from the RA-BT group. p-values were calculated by the one-sided Permutation test. g Volcano plot displaying the DEGs in T cells, similar to d. h Enriched GO pathways for the overlapped-up genes in T cells from RA-BT. i, j DEGs in macrophage and T cell clusters between the ACR20_N (n = 2882 macrophages; n = 985 T cells) and ACR20_Y (n = 12,658 macrophages; n = 5904 T cells) groups. The p-values were calculated by the two-sided Wilcoxon test, ****p < 2.2 × 10−16. The box is bounded by the first and third quartile with a horizontal line at the median and whiskers extend to the maximum and minimum value.
Next, differentially expressed genes (DEGs) were determined by comparing the transcriptome profile in each cell type of RA-BT vs. OA-BT, as well as RA-BT vs. RA-AT in overall patients and patients treated with adalimumab/tofacitinib, respectively (Fig. 2c). In macrophages, a series of genes encoding RA-related cytokines/chemokines (e.g., SPP1, CXCL2, and CXCL3) were significantly overexpressed in RA and downregulated after treatment (Fig. 2d and Supplementary Fig. 3f), thus contributing to enrichment of the overlapped DEGs among these comparisons in multiple pathways, including inflammatory response and chemokine signaling pathways according to GO and GSEA analysis (Fig. 2e, f). Similarly, immune response-related DEGs and enriched pathways were observed in T cells (Fig. 2g, h, and Supplementary Fig. 3g). On the other hand, these DEGs can also contribute to the response efficacy, such as higher expression of STAT1 in macrophages and GZMA/GZMK in T cells from ACR20_Y than those from ACR20_N (Fig. 2i, j).
The role of macrophage subtypes in disease severity of RA
To further characterize the role of macrophages in RA pathogenesis and treatment, we defined six transcriptionally distinct macrophage subtypes, including IFN-activated SPP1+ macrophages (expressing high levels of SPP1, CCL2, and STAT1), and S100A12+ macrophages (expressing high levels of inflammation-triggering alarmins S100A8/9/12) (Fig. 3a, b). All macrophage subtypes were highly enriched in RA compared to that in OA, and CCL4+ macrophages are predominately identified in patient RA_5 (Fig. 3c and Supplementary 4a), who has the lowest remission rate (Supplementary Data 1). SPP1+ macrophages were conspicuously abundant in RA patients, and the densities but not proportions of these cells were significantly reduced after treatment (Fig. 3d and Supplementary Fig. 4b), which can be validated by deconvoluting our bulk sequencing data of SF (Supplementary Fig. 4c). The number of DEGs varied across different comparisons and cell subtypes, while SPP1+ macrophages exhibited the most prolific altered molecular features in RA-BT with tofacitinib treatment (Supplementary Fig. 4d). Moreover, the proportion of SPP1+ macrophages was positively correlated with DAS28 score (Fig. 3e), which is consistent with the reduction of SPP1+ macrophages after methotrexate treatment in previous report22, suggesting SPP1+ macrophages may be the shared target of different DMARDs to control RA symptoms. Although the proportion was not significantly decreased after treatment, expression of SPP1 and CCL2 is positively correlated (Supplementary Fig. 4e) and substantially reduced towards that in OA after treatment with adalimumab/tofacitinib (Fig. 3f and Supplementary Fig. 4f). A higher baseline and more dramatic increase in expression of SPP1 were observed in SPP1+ macrophages than in other cell subtypes (Supplementary Fig. 4g). Given that both osteopontin (OPN, encoded by SPP1) and CCL2 are secretory proteins and predominately expressed in macrophages, the protein levels of OPN and CCL2 in SF estimated by ELISA were significantly higher in RA than those in OA, but were not decreased after treatment, possibly because of the stability of these proteins44,45 (Fig. 3g). Moreover, the OPN level was positively correlated with DAS28 and SDAI index (Fig. 3h and Supplementary Fig. 4h), while higher levels of OPN and CCL2 were enriched in ACR20-based early responders (Fig. 3i). Besides, the top DEGs between ACR20_Y and ACR20_N were key factors involved in RA pathogenesis and treatment outcome in both SPP1+ macrophages and S100A12+ macrophages (e.g., FOS and IRF1) (Supplementary Fig. 4i).

a tSNE plot of macrophage subclusters. b Heatmap of the marker gene expression for each macrophage subtype. c Histogram depicting the distribution of each patient with OA and RA among different subclusters of macrophages. d Density of SPP1+/S100A12+ macrophages among patient groups. Density is calculated by multiplying the total cell density in each SF sample by the proportion of SPP1+/S100A12+ macrophage cluster in the sample. The p-values were computed using the two-sided Wilcoxon test for comparisons between OA-BT (n = 3), RA-BT (n = 6), and RA-AT (n = 6). solid squares, patients didn’t receive b/tsDMARDs treatment; hollow circles, adalimumab treated; solid circles, tofacitinib treated. Data are presented as mean values ± SEM. e Correlation of SPP1+ macrophages proportion and DAS28. hollow circles, adalimumab treated; solid circles, tofacitinib treated. p-values were calculated using two-sided Pearson correlation test. f The different expression levels of SPP1 and CCL2 in SPP1+ macrophage among different groups. p-values were calculated by the two-sided Wilcoxon test for comparisons between OA-BT (n = 1715 cells), RA-BT (n = 10,207 cells), and RA-AT (n = 5059 cells), ****p < 2.2 × 10−16. The box is bounded by the first and third quartile with a horizontal line at the median and whiskers extend to the maximum and minimum value. g Different level of secreted osteopontin (encoded by SPP1) and CCL2 evaluated by enzyme-linked immunosorbent assay (ELISA) in SF. solid squares, patients didn’t receive b/tsDMARDs treatment; hollow circles, adalimumab treated; solid circles, tofacitinib treated. OA-BT: n = 10; RA-BT: n = 27; RA-AT: n = 12. The p-values were calculated by the two-sided Wilcoxon test. Data are presented as mean values ± SEM. h Pearson’s correlation analysis of secreted osteopontin levels and DAS28 in validation cohort. p-values were calculated using two-sided Pearson correlation test. i Comparison of the levels of osteopontin and CCL2 in ACR20_Y (n = 12) vs. ACR20_N (n = 8). The p-values were calculated by the two-sided Wilcoxon test. Data are presented as mean values ± SEM.
To establish the pathogenic role of these two types of SF-derived macrophages, we cocultured them with FLS. Given that SPP1 and S100A12 are not membrane proteins, we sorted their representative CD36+CD62L- (representing SPP1+) and CD62L+ (representing S100A12+) macrophages (CD11b+CD64+) labeled by membrane markers through FACS (Supplementary Fig. 5a, b), which can be validated by geometric mean46, quantitative PCR (qPCR) in SF samples and multiplex immunofluorescence (mIF) in ST samples from RA patients (Supplementary Fig. 5a, e, and Supplementary Data 6). In vitro coculture experiments were conducted to activate RA biopsy-derived FLS cells, which were derived from the ST of RA patients and confirmed by morphology and CD55 expression (Supplemental Fig. 5f). After coculturing in a direct and indirect system, the expression of proinflammatory cytokines (e.g., IL6) and cartilage-mediators (e.g., MMP1) in FLS cells were significantly stimulated by both cell types compared to FLS alone or coculture with double negative macrophages (i.e., CD36-CD62L- cells) (Fig. 4a and Supplementary Fig. 5g), suggesting regulatory modulation of FLS by SPP1+/S100A12+ macrophages. To further support the important role of SPP1+ macrophage in RA pathogenesis, we constructed a collagen-based RA mouse model by using Spp1flox/floxLyz2-Cre+ conditional knockout mice (referred to as Spp1-cKO), in which Spp1 is specifically deleted in myeloid cells (e.g., macrophages) (Fig. 4b). Although the RA symptoms (e.g., redness and swelling of paws) is not severe with C57 background mice47, which would require a possible back cross to DBA background in the future, we observed less severe swelling of paws from Spp1-cKO compared to their littermates of with Spp1flox/floxLyz2-Cre- genotype (refer as to Spp1-WT) (Fig. 4c). A delayed onset of arthritis but not a generally less severe arthritis was observed in the Spp1-cKO mice (Fig. 4d). More importantly, the expression of IL6 and MMPs of the whole lysate of paws from Spp1-cKO mice are significantly lower than that in Spp1-WT (Fig. 4e). These findings highlighted the potential pathogenic role of SPP1+ macrophages in RA pathogenesis.

a Upper left: schematic representation of coculture experiments involving sorted macrophages and fibroblast-like synoviocytes (FLS); Lower left: representative FACS plot of CD11b+CD64+CD36+CD62L- cell (representing SPP1+ macrophage), CD11b+CD64+CD62L+ cell (representing S100A12+ macrophage), and CD11b+CD64+CD36-CD62L- cell (representing double negative macrophage) from SF of RA patients (n = 10); Right: mRNA expression of IL-6, MMP-1, MMP−2 and MMP-13 in FLS, FLS + SPP1+ macrophage, FLS + S100A12+ macrophage, FLS + double negative macrophage through direct coculture system and Trans-well-based coculture system; Data are presented as mean ± SEM. The p-values were calculated by the two-sided Wilcoxon test. FLS fibroblast-like synoviocytes. Created in BioRender. Xu, H. (2025) https://BioRender.com/m45b690. b Schedule of injection with type II collagen (CII), Freund’s complete adjuvant (CFA), Freund’s incomplete adjuvant (IFA) and sample collection. Created in BioRender. Xu, H. (2025) https://BioRender.com/r59d731. c Representative images of wrist joint. d Clinical score of collagen-induced arthritis (CIA) between Spp1-cKO and Spp1-WT mice. n = 5 per group. p-values were calculated by the two-sided Student’s t test, *p < 0.05, **p < 0.01. Exact p-values for days 29, 31, 33, and 35 were 0.037, 0.0085, 0.049, and 0.048, respectively. e mRNA expression of II6, Mmp13, Mmp1a in hind paw of Spp1-cKO (n = 5) and WT (n = 5) mice. Data are presented as mean ± SEM. The p-values were calculated by the two-sided Wilcoxon test.
Functional characteristics of SPP1 +/S100A12 + macrophages
We employed gene set variation analysis (GSVA) to determine the functional characteristics of macrophages, and evaluate their contributions to RA. As expected, SPP1+ and C1QC+ macrophages exhibited preferential signatures of M1 and M2 respectively, whereas S100A12+ macrophages exhibited superior signatures of immature inflammatory monocytes (Fig. 5a). The M1 signature scores were significantly lower in OA and RA-AT than those in RA-BT, specifically for SPP1+/S100A12+ macrophages after adalimumab/tofacitinib treatment (Fig. 5b). The altered M1 signature scores can only be validated in OA vs. RA-BT but not RA-BT vs. RA-AT with bulk sequencing results, possibly because the M1 signature was decreased specifically in SPP1+/S100A12+ macrophages (Fig. 5c). On the other hand, a significantly higher M1 signature score in SPP1+/S100A12+ macrophages was observed in ACR20_Y than that in ACR20_N (Fig. 5d). Besides the classical macrophage signatures, we also conducted hallmark enrichment in each macrophage subtype, revealing the high presentation of RA-related signaling in SPP1+/S100A12+ macrophages, including IL6/JAK/STAT3, IFN-α/IFN-γ response signaling, and angiogenesis pathways (Fig. 5e and Supplementary Fig. 6a). The activity of most of these pathways in SPP1+/S100A12+ macrophages was high in RA-BT and decreased towards those in OA after treatment with adalimumab/tofacitinib (Supplementary Fig. 6b). Moreover, activity of these pathways was predominately higher in ACR20_Y than that in ACR20_N (Supplementary Fig. 6c).

a Heatmap of the monocyte, M1, and M2 enrichment scores. The wide range of hues symbolizes the pathway activities. b Differences in M1 signature scores of SPP1+/S100A12+ macrophages. p-values were calculated by the two-sided Wilcoxon test, ****p < 2.2 × 10−16. c Enrichment of the M1 signature using in-house bulk RNA-seq data for OA-BT (n = 5), RA-BT (n = 14), and RA-AT (n = 10). p-values were calculated by the two-sided Wilcoxon test. The box is bounded by the first and third quartile with a horizontal line at the median and whiskers extend to the maximum and minimum value. d M1 scores of SPP1+/S100A12+ macrophages in ACR20_N and ACR20_Y. p-values were calculated by the two-sided Wilcoxon test, ****p < 2.2 × 10−16. e Heatmap displaying the single sample gene set enrichment analysis (ssGSEA) results of hallmark gene sets. f Evolutionary trajectory of SPP1+ macrophage in RA patients treated with tofacitinib. Upper, pseudotime curve and activation trajectory; Middle, trajectory and histography of three states; Lower, Different dynamic evolution trajectories between ACR20_Y and ACR20_N. g Dynamic activities of SCENIC-based transcription factors in SPP1+/S100A12+ macrophages. The dot size and color spectrum indicate q-value (-log10 transformed) and log2 transformed fold change in expression levels of the transcription factors, respectively. p-values were calculated by the two-sided Wilcoxon test. h–i Representative images of multiplex immunofluorescence (mIF) and box plots showing CD68+SPP1+STAT1+ and F4/80+SPP1+STAT1+ cells in ST samples derived from OA/RA patients and control/CIA mice, respectively. DAPI (blue), SPP1 (red), STAT1 (green), CD68 and F4/80 (yellow), CD68+SPP1+STAT1+ and F4/80+SPP1+STAT1+ (white arrows). Scale bars: 20 μm. Box plot illustrating the proportion of CD68+SPP1+STAT1+ and F4/80+SPP1+STAT1+ cells to total cells in patients with OA (n = 3) versus RA (n = 3) and control mice (n = 3) versus CIA mice (n = 3), respectively. Data are presented as mean ± SD. The p-values were determined by a two-sided Student’s t test, *p < 0.05, **p < 0.01. The exact p values CD68+SPP1+STAT1+ = 0.018, F4/80+SPP1+STAT1+ = 0.036. j Pearson’s correlation of transcription factor expression levels and DAS28/SDAI.
To further reveal the commonalities and particularities of adalimumab/tofacitinib for RA relief, we tracked the molecular events of SPP1+ macrophages along the cell-fate transition during treatment. For tofacitinib, cells were divided into three states, and two distinct progression routes were revealed along the pseudo-time paths from State1/2 to State3, cells of which were highly enriched in RA-AT. Interestingly, cells from ACR20_Y and ACR20_N of RA-AT were enriched in State 3 and State2, respectively (Fig. 5f). Expression of genes involved in multiple pathways was reduced (e.g., inflammatory response and JAK-STAT cascade) along the progression routes (Supplementary Fig. 7a), suggesting a possible transition blockade of these signaling pathways in ACR20_N (Fig. 5f). For adalimumab, the progression trajectory was more complicated, with shared and distinct genes/pathways along the pseudo-time paths as observed in tofacitinib (Supplementary Fig. 7b, c). Nevertheless, since only two ACR20_N were included with available matched pre-/post-treated samples, validation in independent cohorts with a large sample size is warranted in the future.
Next, we assessed the regulatory network inference of SPP1+/S100A12+ macrophages by using SCENIC. Several transcription factors were differentially expressed, including RA-enriched (e.g., STAT1 and NFKB1) and OA-enriched (e.g., KLF2 and EGR1), which were diminished to a varied extent after treatment with either drug (Fig. 5g). To validate this finding, we conducted mIF on 12 ST samples from human or mice, revealing that the proportions of STAT1+SPP1+ macrophages were significantly higher in RA vs. OA patients and CIA vs. control mice, suggesting the consistency of the increased STAT1 expression in SPP1+ macrophages between SF and ST (Fig. 5h, i). Drug specificity was noticed, such as the tofacitinib-induced increase of KLF2 in SPP1+ macrophages and the decrease of HIF1A in S100A12+ macrophages, respectively. Interestingly, STAT1 was the only transcription factor that exhibited a positive correlation of its mean expression in SPP1+/S100A12+ macrophages with the DAS28/SDAI index of RA patients (Fig. 5j and Supplementary Fig. 7d) and also contributed to the response efficacy (Supplementary Fig. 4i). Given activation of STAT1 through JAK-based phosphorylation is a key molecular event in RA pathogenesis48, our finding suggested the potential role of STAT1 expression in specific macrophages (e.g., SPP1+/S100A12+ macrophages) in RA progression, which is consistent with previous observations in ST through immunohistochemistry staining49.
Potential role of NK/T subtypes in RA pathogenesis and treatment outcomes
Given that T cells were highly involved in RA pathogenesis, we performed subclustering for NK/T cells to further delineate the pathogenic programs. Based on the specific markers, a total of ten subtypes were identified, including two CD4+ T cell subtypes (i.e., CXCL13+CD4+ T cells and Tregs) and five CD8+ T cell subtypes (e.g., effector memory T cells, Tem) with varied proportions (Fig. 6a, b, and Supplementary Fig. 8a). Among all cell subtypes, only CXCL13+CD4+ T cells were significantly more abundant in RA than OA in terms of proportion (Fig. 6c and Supplementary Fig. 8b), which can be validated by deconvoluting our bulk sequencing data of SF (Fig. 6d) and public data of ST50 (Fig. 6e). Consistently, mIF assay with ST samples revealed a significant increase in CXCL13+CD4+ T cells in RA vs. OA patients and CIA vs. control mice, suggesting that the shift cell proportion of such cell type in ST from RA patients can be reflected by SF samples (Fig. 6f, g). Moreover, the density of CXCL13+CD4+ T cells significantly decreased after treatment (Fig. 6h). Interestingly, the classic exhaustion-related genes were highly enriched in CXCL13+CD4+ T cells, such as HAVCR2, TIGIT, LAG3, and PDCD151,52 (Fig. 6b). Indeed, CXCL13+CD4+ T cells have been highlighted as the most expanded T cell clones in SF and defined as PDCD1+ peripheral helper/follicular helper T cells in ST in RA21,37. Also, a recent study has revealed that expression of CXCL13 and exhaustion-related genes can effectively label tumor-reactive CD4+/CD8+ T cells, which is responsible for recognizing tumor-specific-antigens53,54. Given that RA is an autoimmune disease resulting from an autoimmune response to self-antigens, we considered CXCL13+CD4+ T cells as the self-antigen-related effector cells. Therefore, we examined the expression levels of both CXCL13 and exhaustion-related genes in CXCL13+CD4+ T cells, observing significantly higher expression of these genes in RA, which may decrease after treatment with adalimumab/tofacitinib (Supplementary Fig. 8c). This finding can be validated in our bulk cohort with enrichment signatures of CXCL13+CD4+ T cells, particularly exhausted cells estimated by dysfunction score (i.e., enrichment score of CXCL13, CTLA4, LAG3, PDCD1, TIGIT, and HAVCR2) (Fig. 6i). Interestingly, tofacitinib exhibited significantly higher efficiency than adalimumab to down-regulate the dysfunction score of CXCL13+CD4+ T cells (Fig. 6i). These findings highlighted the potential important role of CXCL13+CD4+ T cells in RA pathogenesis and treatment outcomes.

a t-SNE plot for T cell subclusters. b Violin plot displaying the expression levels of marker genes. c, d Proportion and deconvolution score of CXCL13+CD4+ T in samples with scRNA-seq (OA-BT: n = 3, RA-BT: n = 6, RA-AT: n = 6) and in-house bulk RNA-seq (OA-BT: n = 5, RA-BT: n = 14, RA-AT: n = 10), respectively. p-values were calculated by the two-sided Wilcoxon test. Data are presented as mean values ± SEM. e Deconvolution score of CXCL13+CD4+ T in published bulk data with ST samples (GSE12021, OA: n = 10, RA: n = 12; Zhang et al., OA: n = 15, RA: n = 9). p-values were calculated by the two-sided Wilcoxon test. Data are presented as mean values ± SEM. The box is bounded by the first and third quartile with a horizontal line at the median and whiskers extend to the maximum and minimum value. f, g mIF and box plots showing CD3+CD4+CXCL13+ cells in ST samples derived from patients and mice, respectively. DAPI (blue), CD3 (white), CD4 (cyan), CXCL13 (pink), and CD3+CD4+CXCL13+ (white arrows). Scale bars: 20 μm. Box plot illustrating the proportion of CD3+CD4+CXCL13+ cells to total cells in patients with OA (n = 3) versus RA (n = 3) and control mice (n = 3) versus CIA mice (n = 3), respectively. Data are presented as mean ± SD. The p-values were determined by a two-sided Student’s t test, **p < 0.01, ***p < 0.001. Exact p values OA/RA = 0.00087, Control/CIA = 0.0027. h Density of CXCL13+CD4+ T cluster. The p-values were calculated by the two-sided Wilcoxon test. OA-BT: n = 3; RA-BT: n = 6; RA-AT: n = 6. Data are presented as mean values ± SEM. i Left: Enrichment scores of the dysfunction markers in in-house bulk RNA-seq data. Right: fold change (log2 transformed) of markers’ expression. OA-BT: n = 5, RA-BT: n = 14, RA-AT: n = 10, respectively. The box is bounded by the first and third quartile with a horizontal line at the median and whiskers extend to the maximum and minimum value. p-values were calculated by the two-sided Wilcoxon test. Data are presented as mean values ± SEM. j Pearson’s correlation of CD8+ Tem1 proportion and DAS28. p-values were calculated using two-tailed Pearson correlation test.
On the other hand, we also estimated the correlation of proportions of all T cell subtypes with DAS28-based disease activity, revealing that only a subtype of CD8+ T effector memory (CD8+ Tem1) exhibited statistical significance (Fig. 6j, and Supplementary Fig. 8d), which can be validated through deconvoluting CD8+ Tem1 scores with our bulk sequencing data (Supplementary Fig. 8e).
Molecular characteristics of T cell subtypes
To further profile the molecular characteristics of the T cell subtypes, we first performed DEG analysis in each cluster. The results showed that CXCL13+CD4+ T cells had the most prolific altered molecular features in RA-BT compared to those in OA-BT and RA-AT in terms of DEG number (Fig. 7a). A total of 311 intersecting genes were identified to be highly expressed in CXCL13+CD4+ T cells of RA-BT, including JAK3, STAT3, JUNB, and IRF1 (Supplementary Fig. 9a). After comparing RA-BT and RA-AT, we found that the JAK_STAT and the T cell receptor signaling pathway can be downregulated by both inhibitors, whereas several pathways of immune-related functions were specifically enriched in RA-BT after tofacitinib treatment, such as cytokine and chemokine signaling pathways (Fig. 7b).

a Feature plot displaying the number of DEGs in each T cell subcluster among different samples. p-values were calculated by the two-sided Wilcoxon test. b Positively enriched GSEA pathways for CXCL13+CD4+ T cells in RA-BT. The enriched pathways in the RA-BT were divided into three groups: red, blue, and purple, corresponded to the shared, adalimumab-specific, and tofacitinib-specific, respectively. c Heatmap showing enrichment scores of ssGESA-based hallmark gene sets from the MSigDB database for each T cell subcluster. d Violin plots showing the level of activity in key pathways across different groups. The box is bounded by the first and third quartile with a horizontal line at the median and whiskers extend to the maximum and minimum value. p-values were calculated by the two-sided Wilcoxon test. ns p > 0.05, **p < 0.01, ****p < 0.0001. Exact p values and cell numbers in each group are provided in the Source data. e Upper: schematic representation of coculture experiments involving sorted CD4+ T cells and fibroblast-like synoviocytes (FLS); Lower: mRNA expression of IL-6, MMP-1, MMP-2 and MMP-13 in FLS, FLS + CD4+ T cells through direct coculture system (n = 10) and Trans-well-based coculture system (n = 9); Data are presented as mean ± SEM. Statistical significance was determined by the two-sided Wilcoxon test. *p < 0.05,****p < 0.0001. Created in BioRender. Xu, H. (2025) https://BioRender.com/o49j725.
Next, we performed ssGSEA-based hallmark enrichment analysis in each NK/T subtype, revealing the overall clustering of Treg, CD8+ Tem1, and CXCL13+CD4+ T cells among all pathways (Fig. 7c), suggesting their potential pathogenic role and communications in RA-related molecular events. For instance, high activity of both Treg and CXCL13+CD4+ T cells was found in RA-related pathways (e.g., TNF signaling and JAK-STAT signaling), as well as CD8+ Tem1 in the IFN-α/γ response (Fig. 7c). We further explored the impact of treatment on specific pathways and found that tofacitinib had a greater inhibitory role in JAK/STAT3 and IFN-α/γ pathways, whereas adalimumab exhibited greater impact on the TNF signaling pathway in Treg, CD8+ Tem1, and CXCL13+CD4+ T clusters (Fig. 7d), which is consistent with the direct targets of these two different DMARDs. The DEG analysis was also performed in CD8+ Tem1 and Treg cells, exhibiting enriched RA-related pathways in these two T cell subtypes, such as the JAK/STAT signaling pathway in Treg cells (Supplementary Fig. 9b–d). Interestingly, the activities of RA-related pathways and some immune-related DEGs can distinguish ACR20_Y from ACR20_N, including CXCL13 expression in CXCL13+CD4+ T cells (Supplementary Fig. 10). Moreover, we also conducted coculture assay by directly and indirectly incubating FLS with CD4+ T cells, which predominately consist of CXCL13+CD4+ T and Treg cells according to the scRNA-seq results (Fig. 6a). Interestingly, enhanced expressions of IL6 and MMPs in FLS are also significantly stimulated after coculturing with CD4+ T cells, supporting the potential pathogenic role of these T cells in RA (Fig. 7e).
Given that multiple pathways were altered in pathogenic T cell subtypes, their contribution to RA may be orchestrated by transcription factors. Therefore, we applied SCENIC analysis to find the key transcription factors in CD8+ Tem1, Treg, and CXCL13+CD4+ T cells. Shared and specific alterations of transcription factors were observed among different cell subtypes and DMARDs treatments, such as the higher IRF1 expression in RA-BT, which can be inhibited after treatment with adalimumab/tofacitinib. Drug specificity was also revealed, such as adalimumab-specific inhibition of KLF2 towards OA-BT in all three cell subtypes and tofacitinib-specific activation of BATF in CD8+ Tem1 (Fig. 8a). Of these transcription factors, expression of both ETV7 and IRF1 in CXCL13+CD4+ T cells and EOMES in Tregs were positively correlated with the DAS28/SDAI index (Fig. 8b, c). Interestingly, higher ETV7 expression in RA-BT than that in OA/RA-AT was specifically identified in CXCL13+CD4+ T cells compared to other cell subtypes, while expression changes of IRF1 were also identified in pathogenetic macrophage subtypes (e.g., SPP1+/S100A12+ macrophages) (Supplementary Fig. 4g). Moreover, IRF1 expression in CXCL13+CD4+ T cells was also significantly higher in ACR20_Y than that in ACR20_N (Supplementary Fig. 10a), possibly because IRF1 can be involved in RA pathogenesis and treatment outcomes by primarily targeting IFN and the JAK_STAT pathway55.

a Dot plot displaying the dynamic activity of SCENIC-based transcription factors in CD8+ Tem1, Treg, and CXCL13+CD4+ T. p-values were calculated by the two-sided Wilcoxon test. The dot size and color spectrum indicate the q-value (-log10 transformed) and gene expression fold change (log2 transformed) of each transcriptional factor, respectively. b, c Pearson’s correlation analysis of transcription factor expression levels and DAS28/SDAI index in CD8+ Tem1, Treg, and CXCL13+CD4+ T. p-values were calculated using two-tailed Pearson correlation test. *p < 0.05, **p < 0.01. d Heatmap of CellphoneDB-based cell-cell communication counts among CD8+ Tem1, Treg, and CXCL13+CD4+ T cell. e Heatmap of the fold change in CellphoneDB-based cell-cell communication counts after treatment with adalimumab or tofacitinib. f Predicted and detailed ligand-receptor communications among CD8+ Tem1, Treg, and CXCL13+CD4+ T cell. p-value were calculated using a one-sided permutation test in CellPhoneDB. Dot size and color spectrum indicate the p-value (-log10 transformed) and mean expression (log2 transformed) of ligand and receptor, respectively.
To explore the possible crosstalk among these three pathogenic T cell subtypes, we performed CellphoneDB analysis and found significantly more ligand-receptor communications in RA-BT than those in OA-BT, which were diminished after treatment except communications between Treg and CXCL13+CD4+ T cells (Fig. 8d). Interestingly, tofacitinib may have greater effects by diminishing the communications among these pathogenic T cell subtypes (Fig. 8e). In detail, most RA-enriched ligand-receptor pairs were involved in immune response and lymphocyte activation and can be blocked in a shared (e.g., FAM3C_CLEC2D in the CD8+ Tem1 and CXCL13+CD4+ T pair) and tofacitinib-specific (e.g., SPP1_PTGER4 in the CXCL13+CD4+ T and CD8+ Tem1 pair) manner. However, adalimumab-specific blockade is rare among the communications (Fig. 8f), suggesting tofacitinib may have a greater effect in blocking communications among T cells.
Communications between macrophage and pathogenic T cell subtypes
Since the potential important roles of both macrophages and T cells in RA pathogenesis and treatment outcomes have been highlighted and functional macrophage-T cell communications are wildly delineated in immunity56,57,58,59, we further investigated the communications between all macrophage subtypes and T cell subtypes, particularly focused on the interactions among potential pathogenic cell subtypes (i.e., SPP1+/S100A12+ macrophages, CD8+ Tem1, Treg, and CXCL13+CD4+ T cells) through CellphoneDB analysis. The overall number of communications is higher in RA-BT than that in OA, which is consistent with the CellChat-based pairwise comparison (Supplementary Fig. 11a, b). Interestingly, SPP1+ macrophages exhibited the most prolific communications with all T cell subtypes, particularly with CXCL13+CD4+ T cells (Fig. 9a). Increased interactions of these cells can be validated through mIF assay on ST samples derived from OA and RA patients, as well as control and CIA mouse models (Fig. 9b, c). Tofacitinib also exhibited greater effects to diminish the communications between pathogenic T cell and macrophage subtypes (Fig. 9d). Potential signal transduction associated with leukocyte cell-cell adhesion (e.g., ICAM1_ITGAL), immune response (e.g., TNF_FAS), T cell activation (e.g., SPP1_CD44 and SPP1_PTGER4), and cell migration (e.g., PLXNB2_SEMA4D) were involved (Fig. 9e). Interestingly, although the number of communications among different cell types is still much higher in RA-AT compared to that in OA, dramatic decreases were observed after treatment, and more prolific initial communications predicted a better response in terms of ACR20 (Fig. 9a), suggesting tofacitinib/adalimumab may partially exert their therapeutic effects through blocking the communications between macrophages and T cells. Although both drugs can decline the overall number of communications between SPP1+/S100A12+ macrophages and pathogenetic T cells (Fig. 9d), shared (e.g., CCL7_CCR5 in SPP1+ macrophages with all three T cell subtypes), tofacitinib-specific (e.g., TNF_VSIR in SPP1+ macrophages and CXCL13+CD4+ T pair), and adalimumab-specific (e.g., IL15_IL15R in SPP1+ macrophage and CXCL13+CD4+ T pair) effects were identified (Fig. 9e and Supplementary Fig. 11c). In addition, a series of ACR20_Y-specific and ACR20_N-specific interactions were also noticed (Supplementary Fig. 11d–g), suggesting the possible determination role of the initial communications in treatment efficacy.

a Heatmap of CellphoneDB-based ligand-receptor communication counts among all macrophage and T cell subclusters. b, c Representative images of mIF and box plots showing the interactions between SPP1+ macrophages (patients: CD68+SPP1+ cells; mice: F4/80+SPP1+ cells) and CXCL13+CD4+ T cells (CD3+CD4+CXCL13+ cells) in ST samples derived from OA (n = 3)/RA (n = 3) patients and control (n = 3)/CIA (n = 3) mice, respectively. within 15 μm from the CD3+CD4+CXCL13+ T cells were regarded as potential interaction partners. The interaction degree was measured by the proportion of these potential interacting macrophages relative to the total cell population. DAPI (blue), SPP1 (red), CD3 (white), CD4 (cyan), CXCL13 (pink), CD68 and F4/80 (yellow), CD68+SPP1+ and F4/80+SPP1+ (red arrows), and CD3+CD4+CXCL13+ (green arrows). Scale bars: 10 μm. Data were presented as mean ± SD. The p-values were determined by a two-sided Student’s t test, with **p < 0.01 and ***p < 0.001. Exact p values OA/RA = 0.00037, Control/CIA = 0.0078. d Heatmap of the fold change in communication counts. e Predicted and detailed CellphoneDB-based ligand-receptor communications. p-value were calculated using a one-sided permutation test in CellPhoneDB. Dot size and color spectrum indicate the p-value (-log10 transformed) and mean expression (log2 transformed) of ligand and receptor, respectively. f iTALK-based Cytokine-chemokine communications among pathogenic macrophage and T cell subclusters. g Left: schematic representation of coculture experiments involving sorted macrophages, CD4+ T cells and fibroblast-like synoviocytes (FLS); Right: mRNA expression of IL-6, MMP-1, MMP-2 and MMP-13 in FLS, FLS + SPP1+ macrophage, FLS + CD4+ T cells, FLS + SPP1+ macrophage+CD4+ T cells through direct coculture system and Trans-well-based coculture system (n = 3); Data are presented as mean ± SEM. Statistical significance was determined by one-way ANOVA followed by Tukey’s multiple comparisons test for multi-group comparisons. ns p > 0.05, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. The exact p values are provided in the Source data. FLS, fibroblast-like synoviocytes. Created in BioRender. Xu, H. (2025) https://BioRender.com/g62b838.
Given that RA is an inflammatory autoimmune disease, we focused on the potential crosstalk signaling molecules at the cytokine and chemokine levels through iTALK analysis. Not surprisingly, crosstalk between SPP1+/S100A12+ macrophages and pathogenetic T cell subtypes was more prolific in RA-BT and decreased towards OA after treatment (Fig. 9f). For instance, SPP1+/S100A12+ macrophages strongly expressed CCL2 and CXCL16 and acted on T cell subtypes in RA patients via binding to CCR5 and CXCR6, which was dramatically diminished after treatment (Fig. 9f). Moreover, these communications also exhibited slight differences between the ACR20_Y and ACR20_N in terms of the initial samples from RA patients (Supplementary Fig. 12a), which may partially explain the individualized treatment outcomes. In order to further determine the ligands that caused the transcriptional phenotypes of pathogenic T subtypes in RA, we compared the differential expression between pathogenetic T cell subtypes from RA-BT and those from OA-BT and selected the highly expressed genes in RA-BT as the potential downstream targets for NicheNet analysis60. We noticed that some ligands (e.g., SPP1, and CCL7) from SPP1+/S100A12+ macrophages were predicted to trigger the phenotype of pathogenic T subtypes, which is consistent with the observation described above (Fig. 9e, f, and Supplementary Fig. 12b). In turn, T subtypes may modulate the macrophages through the ligands (e.g., CCL4, and CD28) (Supplementary Fig. 12c), which was consistent with the findings in CellphoneDB-based communications (Fig. 9e, f, and Supplementary Fig. 11c). In conclusion, these results highlighted the pathogenic approach of macrophage-T cell communications to RA.
Due to the small sample size of the discovery cohort, we performed scRNA-seq on an additional 4 RA-BT samples from patients treated with tofacitinib (n = 2)/adalimumab (n = 2), including one ACR20_N and three ACR20_Y. A total of 14,563 cells were left for subsequent analysis with the same filtering criteria as the discovery cohort. A high proportion of pathogenic cell types (e.g., SPP1+/S100A12+ macrophages) were also identified (Supplementary Fig. 13a, b) with consistent differentially expressed genes (e.g., STAT1, IRF1, and FOS), altered hallmark pathways in these cell types, and higher M1 pathway activity in ACR20_Y (Supplementary Fig. 13c–h). Moreover, more prolific cell-cell communications were also observed between the pathogenic macrophage subsets and T cells in ACR20_Y (Supplementary Fig. 13i). These validations further supported our findings in the discovery cohort.
Experimentally, we cocultured FLS with both SPP1+ macrophages and CD4+ T cells from two RA patients with enough available cells with direct and indirect systems and compared the IL6 and MMPs expression with those in matched coculture assay of FLS with SPP1+ macrophages or FLS with CD4+ T cells. Interestingly, significantly higher expression was observed in combination of three cell types (Fig. 9g and Supplementary Fig. 12d), suggesting a potential synergistic effects of these two pathogenic cells on stimulating RA-related inflammatory.
Despite the consistency of previous studies on ST with our findings on SF, it is also important to conduct a pairwise comparison between ST and SF in terms of compositions and cell-cell communications to establish the role of SF to reflect ST, which is considered as the central player of RA1. Therefore, scRNA-seq and subsequent comparisons with 46,002 filtered cells were performed in matched ST, SF, and peripheral blood from one RA patient. Interestingly, SPP1+ macrophages were abundant in the SF and ST samples but nearly absent in peripheral blood (Supplementary Fig. 14a–d). Moreover, interaction pairs among these pathogenic cell types are more consistent in ST and SF compared to those in peripheral blood, particularly SPP1+ macrophage-related interactions (Supplementary Fig. 14e, f). These findings suggested that SF, but not peripheral blood, may reflect the enrichment of particular pathogenic cell types and their communications in ST.
Heterogeneity of DCs, osteoclasts and fibroblasts
Besides macrophages and NK/T cells, the heterogeneity of DCs, osteoclasts, and fibroblasts was also investigated through subcluster analysis. For DCs, four subtypes were divided in terms of canonical markers (Supplementary Fig. 15a, b), only moDC exhibited a significantly higher proportion among all DCs in RA than that in OA but not decreased after b/tsDMARDs treatment, whereas no significance was observed in terms of density (Supplementary Fig. 15c, d). Moreover, moDC also exhibited higher JAK-STAT and TNF signaling (Supplementary Fig. 15e). For osteoclasts, these cells are mainly derived from OA (496 out of 520) (Fig. 1d). Although CD14+ osteoclasts (expressing a high level of TGFB1) out of three subtypes were predominantly enriched in RA-BT, such a difference may be biased due to the small number of osteoclasts in RA-BT (Supplementary Fig. 15f–h). Finally for the fibroblast, we combined the fibroblasts from all SF samples as well as one ST sample to perform subtype classification according to the canonical markers61. A total of six subtypes were characterized, including three and two were derived from the lining and sublining layers, respectively (Supplementary Fig. 16a, b). Different subtypes were clustered together regardless of their derivation from either ST or SF based on hierarchical clustering analysis, indicating the similarity of fibroblasts from ST and SF in terms of activity (Supplementary Fig. 16c). Interestingly, fibroblasts from RA were predominately enriched in lining layer-related fibroblast subtypes, exhibiting predominate expression of canonical markers for lining (e.g., PRG4) and near-absent expression of canonical markers for sublining (e.g., THY1) (Supplementary Fig. 16d, e). The proportion of activated lining fibroblasts, which specifically express cartilage mediators (e.g., MMP1) as we described above, was higher in SF from RA-BT than that in OA and decreased after treatment in RA-AT (Supplementary Fig. 16d), suggesting that most fibroblasts in SF are derived from the lining layer, probably through detaching from the synovium, thus reflecting the fibroblasts-related disease activity status of ST. However, further validations with larger sample size are warranted in the future due to the small cell number of these cell types.
Discussion
Several studies have investigated the characteristics of synovial tissues in RA patients, particularly at the single cell level, highlighting the important role of multiple cell types (e.g., fibroblasts and immune cells) and the related mechanisms in RA pathogenesis and progression1,21,22,23,35,37. However, due to the potential heterogeneity and, more importantly, clinical unavailability of multiple ST samples from the same RA patients, longitudinal cellular/molecular changes after treatment with different DMARDs have not been systematically investigated. The cells in SF are mainly derived from ST and may reflect its local immune microenvironment, making investigation of SF an alternative way to explore the underlying cellular/molecular changes during RA treatment. Therefore, we provided the first glance at the impact of b/tsDMARD on heterogeneity and transcriptional profile of SF cells through scRNA-seq and subsequent validations (e.g., flow cytometry, ELISA, bulk sequencing, and in vitro activation assay) by using SF samples before and after treatment, aiming to provide a molecular basis for precision medicine of RA. Overall, our findings show that the immunological microenvironment of SF in RA patients is markedly more communicative among the pathogenic macrophages and T cell components via ligand-receptor pairs, to participate in the pathogenesis and b/tsDMARDs based treatment outcomes of RA (Fig. 10).

The left and right panels represent SF from RA patients before treatment (RA-BT) and after treatment (RA-AT), respectively. In the immunological milieu of SF from patients with RA, activity of various molecular pathways (e.g., JAK/STAT pathway) were increased. Pathogenic macrophages and T cell subtypes produced pro-inflammatory molecules (e.g., CCL2). ligand-receptor interactions were enhanced between pathogenic macrophage subsets (such as SPP1+/S100A12+) and T cell subsets (CD8+ Tem1, Treg, CXCL13+ CD4+ T cells), along with inter-crosstalk among the three T cell subtypes. These interactions may play a role in the development and advancement of RA and can be blocked in either shared or drug-specific manner. The lighter color of cells in RA-AT indicates lower level of activation of inflammatory genes and RA pathogenic pathways within these cells compared to those in RA-BT. Created in BioRender. Xu, H. (2025) https://BioRender.com/s60q947.
Consistent with the observation in ST samples, several inflammatory and cytotoxic immune cell types were enriched in SF samples, including SPP1+/S100A12+ macrophages, and CXCL13+CD4+ T cells22,32,37,62, suggesting a similarity of ST and SF in terms of functional cell components. In addition, both the proportion of SPP1+ macrophages and SPP1-encoded OPN in SF are positively related to DAS28-based severity, which is consistent with the correlation between SPP1 expression in ST and DAS2822, while a higher pre-treated OPN level in SF predicts better treatment outcomes in terms of ACR20. Interestingly, germline variants in SPP1 have also been revealed to contribute to RA susceptibility63,64, possibly through epigenetically upregulating SPP1 expression in macrophages. Mechanistically, the involvement of SPP1 in inflammation has been well established, including elevating the levels of glycolytic enzymes, cytoskeletal proteins, and integrins, inducing an activated migratory phenotype, and specifically contributing to bone damage and the activation of pro-inflammatory immune cells attracted to the SF65,66,67. Moreover, we first experimentally cocultured the SPP1+ macrophages with FLS cells and observed significantly increased RA-related cytokines. Therefore, our results highlighted the involvement of SPP1+ macrophage and OPN from SF in RA pathogenesis and severity. On the other hand, CXCL13+CD4+ T cells were also enriched in SF and ST of RA patients compared to that in OA, thus may interact with B and plasma cells to induce the pathogenesis of RA, particularly anticitrullinated peptide antibody-positive subtype32,68. Moreover, CXCL13+ T cells played the central role in responding to checkpoint inhibitor treatment for cancer by identifying the neoantigens and stimulating the T cell response53,54. Such molecular mechanisms may be shared by RA to induce the aberrant autoimmune response, thus highlighting the possible bifunction of CXCL13+CD4+ T cells to affect both B cell and T cell responses during RA onset and progression. Besides the potentially pathogenic cell types identified in previous studies, cell-cell communications among different cell types in RA were also reported in ST19,62, which is consistent with our findings.
Importantly, pathologically examining the inflammatory macrophages in the sublining of ST samples can facilitate RA diagnosis (particularly in patients with monoarthritis and/or seronegative for RF/CCP antibodies), as well as treatment outcome evaluation. ST biopsy is an invasive approach, thus is not routinely detected clinically. On the other hand, compared to the heterogeneity of ST and the unfeasibility of longitudinally obtaining ST biopsy at different time points, which is traumatic for patients, SF is more homogenous in cell components and flexible to obtain through a minimally invasive approach. Our analysis indicates the similarity of the pathogenic cell types from ST with those from SF but not blood. Therefore, we collected two SF samples from each patient before and after treatment to reflect the states of the pathogenic cells in ST samples, which facilitate investigation on the dynamic changes of the cell components and the cell-cell communications, as well as their correlation with ACR20_based treatment outcomes after administration with adalimumab/tofacitinib. For instance, some of the functional cell components may be downregulated (e.g., SPP1+ macrophage) after treatment, while some components were not impacted at all (e.g., S100A12+ macrophage), partially explaining the possible underlying cellular mechanisms that DMARDs exert clinical function in RA, but patients would experience relapse after DMARDs discontinuation, respectively65. Despite the fact that the proportions of the pathogenic cell types were not dramatically reduced after treatment, the total cellularity and activation of immune-related pathways (e.g., inflammatory response, TNF signaling, and JAK-STAT signaling) was significantly controlled, which is consistent with the improvement of clinical symptoms. Moreover, higher expression of inflammatory genes (e.g., STAT1 and FOS) were identified in ACR20-based early responders, which is not significantly related to the disease activity at the start (Supplementary Data 4), suggesting the potential prognostic role of these genes. Interestingly, JAK3 expression was also downregulated in macrophages and T cells, particularly post-treatment with tofacitinib. It has been demonstrated that tofacitinib can inhibit the JAK-STAT pathway through direct binding to the kinase domain of JAK69. Therefore, the downregulation of JAK expression identified in our findings may be induced through negative feedback of the JAK-STAT signaling. Moreover, although scRNA-seq has been performed on sorted CD4+ T cells from SF in previous reports, highlighting the importance of CXCL13+CD4+ T cells in RA21, unsorted cells can further facilitate revealing the enriched cell-cell communications among T cell subtypes as well as the potential crosstalk between T cells and macrophages in RA, according to our study. Interestingly, we highlighted the prolific communications between SPP1+/S100A12+ macrophages and CXCL13+CD4+ T cells, which are over-presented in ACR20-based early responders and dramatically abolished after treatment. Moreover, tofacitinib exhibited a greater impact on decreasing the number of cell-cell communications than adalimumab, suggesting JAK inhibitors may be more efficient to control inflammatory particularly in RA patients with synovial fluid. Since non-responders to tofacitinib and adalimumab are not perfectly overlapped, and these two drugs have distinct advantages and disadvantages15,16,17,18, our results provide the potential molecular and cellular basis of a precise prescription strategy.
On the other hand, we investigated the cellular characteristics of SF with different ACR20 statuses at the first month post-treatment, because most patients would achieve joint swelling and pain relief after 3 or 6 months post-treatment, leading to no or limited volume of SF. Enrolling more patients to conduct comparisons between early responders and non-responders in terms of the status of pathogenic cell types at baseline would be important to establish their prognostic value. Recently, a significantly reduction was observed in RA patients receiving tofacitinib compared to those receiving adalimumab at 3 months but not 9 months post-treatment70, suggesting the possible quicker effect of tofacitinib, which may be attributed to modulation of multiple aspects of the immune response mediated by JAK signaling71, or the response kinetics of these two drugs72. Therefore, the differences between tofacitinib and adalimumab identified in our study may be induced by the different response steps and would not be observed after reaching the therapeutic goal.
Our studies have some limitations, which should be noted to avoid overinterpretation and addressed in future studies. First, we only got matched ST and SF from one patient, leading to limited exploration for consistency and some pathogenic cell components enriched in ST (e.g., fibroblast and FLS7,73), as well as co-enrichment analysis to verify the potential interactions between pathogenic immune cells and fibroblasts. Second, our major findings can be verified in independent cohorts through experimental approaches. However, validation at the protein level is lacking due to the limited protein for a particular assay (e.g., western blot), and the sample size in our study is relatively small, arguing RA patient cohorts with a larger sample size for validation, especially on the different treatment outcomes between tofacitinib and adalimumab. On the other hand, ACR20_Y have the trend of higher RA-related variables (e.g., CRP) than that of ACR20_N, impact of the baseline should be considered if significance would be reached with larger sample size. Third, post-treatment SF samples were obtained after one month of treatment because the volume of SF would sharply decrease after three months of treatment for most patients. Whether the proportion of certain cell components and the related molecular pathways would be altered after longer treatment remains to be elucidated. Finally, SF can only be obtained from some of the RA patients, particularly those with severe syndrome, thus inducing the possible bias of our findings in SF-enriched patients and limiting the potential clinical value in patients with unavailable SF samples.
In summary, we systematically profile the single-cell transcriptome of SF cells from pre-/post-treated RA patients, revealing that the proportion, cell-cell communications, and molecular pathways of several main pathogenic cell components (e.g., SPP1+ macrophages and CXCL13+CD4+ T cells) are involved in RA pathogenesis, progression, and treatment outcomes. We demonstrate that bDMARDs and tsDMARDs induce different subsequent molecular events and signaling (e.g., crosstalk between SPP1+ macrophages and CXCL13+CD4+ T cells) (Fig. 10).
Methods
Enrollment of patients
In this study, forty-four patients fulfilling the American College of Rheumatology 2010 revised criteria for RA74 were randomly enrolled prospectively from West China Hospital of Sichuan University, and underwent arthrocentesis of the knee to obtain SF. Twenty-one patients with OA attending arthroplasty were included as controls. The study protocol was approved by the Ethics Committee of the West China Hospital, Sichuan University (2020-1151). All participants provided signed informed consent. Demographic, clinical and laboratory evaluations of all patients with RA are summarized in Supplementary Data 1–4). Blood tests, including assays of C-reactive protein (CRP), rheumatoid factor (RF), anti-cyclic citrullinated peptide (CCP) antibody, were performed and recorded at the Department of Division of Laboratory Medicine of West China Hospital. The 28-joint disease activity score (DAS28) >3.2 at baseline is required for all patients with RA. The level of disease activity was categorized as follows: a DAS28 score of less than 2.6 indicated remission; 2.6 to less than 3.2, low disease activity; 3.2–5.1, moderate disease activity; and more than 5.1, high disease activity75. All patients are MTX-insufficient responders and have high/moderate disease activity at baseline. Also, they did not received glucocorticoids and any biologic DMARDs (bDMARDs, e.g., adalimumab)/targeted-synthetic DMARDs (bDMARDs, e.g., tofacitinib) within three months before enrollment. For discovery cohort, RA patients were treated with adalimumab (40 mg every 2 weeks) or tofacitinib (5 mg twice a day) for 1 month after enrollment, they were also given a steady dose of methotrexate (MTX) at the same time. After 1 month, patients with RA receive the second arthrocentesis of the knee to obtain the SF from the same joint. Same criteria were applied to patients enrolled in validation cohort except that some patients were not treated with adalimumab/tofacitinib, whose samples were only used for baseline analysis (Supplementary Data 1). Synovial fluid sampling in validation cohort was conducted before b/tsDMARDs treatment. In addition, some RA patients (n = 34) were assessed for meeting 20% improvement in American College of Rheumatology response criteria (ACR20)76 at 1 month.
Single-cell dissociation of SF cells
The patient’s knee joint fluid was extracted under aseptic conditions, and hyaluronidase was added to the fresh SF sample obtained at a final concentration of 100 U/ml and incubated at 37 °C for 30 min. SF samples from each patient were obtained from the same joint longitudinal to avoid potential bias. To prepare single-cell suspensions for sequencing needs, the fresh SF samples were centrifuged at 800 × g for 10 min, the supernatant was collected and stored at −80 °C for ELISA assay, while the cell pellets were suspended by 1 ml red blood cell lysate to lyse the red blood cells and incubated on ice for six minutes, followed by centrifugation at 800 × g for five minutes. Next, 800 μl of pre-chilled PBS (SH30256.01, Cytiva) and 300 μl of pre-chilled debris removal solution (reagent for cell debris removal through density gradient centrifugation) were added to suspend the cell pellets. After gently adding additional 800 ul PBS and centrifuging at 3000 × g for 10 min, the liquid will be divided into three phases. The top two phases were discarded, 2 ml PBS were added to wash the lower phase of pellets once, centrifugation was performed at 800 × g for 5 min to remove contaminants and obtain pure cells. Finally, we collected a 20 μl aliquot for cell counting after discarding the supernatant and resuspending by adding 500 μl PBS. All centrifugation procedures were finished at 4 °C.
Library preparation for scRNA-seq
The cell suspension was placed onto 30 v3 chemistry-equipped Chromium microfluidic chips, and a 10× Chromium Controller (10× Genomics) was used to barcode the samples. Then, using components from a Chromium Single Cell 30 v3 reagent kit (10× Genomics), sequencing libraries were created using RNA from the barcoded cells after reverse transcription, in accordance with the manufacturer’s instructions. Sequencing was carried out using Illumina in accordance with the manufacturer’s instructions (NovaSeq6000). Single cell library preparation and subsequent next generation sequencing was performed by Novogene Co., Ltd. All the scRNA-seq data has passed the quality control (Supplementary Data 7)
Single cell RNA-seq data processing
We processed the scRNA-seq data mostly using the workflow that we had already constructed77,78,79,80,81,82. Briefly, using Cell Ranger (Version 4.3.0) software’s default settings, we aligned and quantified FASTQ files produced by 10× Genomics against the GRCh38 human reference genome (Version 6.1.2). The Read10X function of the Seurat package was used to read the output of Cell Ranger. Using Doubletfinder, with a doublet rate of 0.075, potential doublets were removed from each sample. Next, we chose the RenameCells method to distinctly label each cell and applied the Merge function to integrate all of the individual objects into a single aggregate object. In order to collect the high-quality cells for further research, we selected additional filtering parameters to weed out any empty oil beads, mortality, and doublet cells. Cells with less than 200 or more than 4000 identified genes, as well as those with more than 15% mitochondrial content, were excluded. The ‘LogNormalize’ global scaling normalization strategy was used with a scale factor of 10,000 to obtain equal overall gene expression in each cell. The top 2000 highly variable genes were also retrieved using the FindVariableFeatures technique. Principle component analysis (PCA) was used to reduce the expression matrix’s dimensionality, and the first 30 PCs were selected to regress out the undesired sources of variation with the ScaleData function. Sample batch effect correction was carried out using the RunFastMNN function83, which is based on a multi-canonical correlation analysis algorithm. For clustering study, the FindNeighbors and FindClusters functions with the built-in Louvain method were chosen. Nine distinct major clusters were identified and visualized using the t-SNE method. The non-parametric Wilcoxon rank sum test with Bonferroni correction, which was included in the FindAllMarkers function (adjusted p-value (Bonferroni) < 0.01, and PCT > 0.25), was used to determine the highly expressed genes in each cluster. Following that, using the recognized canonical marker genes taken from published literature, we annotated all cell populations. Additionally, we used a similar procedure to perform a subcluster analysis for each major cell type, respectively. Given the exceptional instability of neutrophils during processing and their almost exclusive origin from one individual, we immediately eliminated this cell type. The proportion of each cluster/subcluster was estimated by dividing the count of cells within each specific cluster/subcluster by the overall cell count in the respective sample. This ratio was then analyzed to ascertain statistical differences across various groups. To visualize gene co-expression in single cells, we calculated the geometric means of the expression levels of the two genes of interest. The geometric mean values were then displayed on the tSNE embedding, with colors representing the geometric mean expression of the targeted genes.
Bulk RNA-seq data processing
The centrifuged SF cells from each RA patient were collected for RNA exaction using the Plus Micro Kit (74034; Qiagen). After quality control (RIN > 7), full-length cDNA and sequencing libraries were prepared and used to carry out bulk RNA sequencing. The 150 bp paired-end read sequencing data were produced using the Illumina NovaSeq6000 and saved in the FASTQ format in accordance with the manufacturer’s instructions. Transcriptome library preparation and subsequent next generation sequencing was performed by Sequanta Technologies Co., Ltd. High quality sequence reads were aligned to the human hg38 reference transcriptome using the Kallisto (Version 0.50.0) as described previously84,85.
Deconvolution analysis
To further evaluate the accuracy of cell proportion in single cell sequencing data, we used bseqsc R package to infer and estimate the frequency of each cell population in the validation samples. We applied marker genes of 8 main clusters for deconvolution analysis (Macrophages: CD68, CD163, FCGR1A, VCAN, CD80, CD86; DCs: FCER1A, CD1C, CLEC10A, CD1E, HLA–DRA, HLA–DQA1, HLA–DQB1, HLA–DPA1, PLD4; T cells: CD3D, CD2, CD3E, CD3G, IL7R; NK cells: NKG7, KLRD1, KLRC1, KLRB1, KLRF1; B cells: CD79A, MS4A1, CD19; Plasma: JCHAIN, TCF4, PLD4, BCL11A; Osteoclasts: MMP9, CTSK, APOE, MMP14; Fibroblasts: PRG4, CLU, MMP3, LUM, COL1A2, COL3A1). The raw counts of scRNA-seq, marker genes list, cell type annotation, and sample data were input to create cell type-specific reference profiles with bseqsc_basis function. Then, using the bseqsc_proportions function, the bulk mRNA sequencing data was used to deconvolve. For better visualization of the results, ggplot2 was selected to show the percentage of clusters in each sample.
Trajectory analysis
To investigate dynamic biological processes of SPP1+ macrophages with drug intervention, we applied the Monocle (Version 2.22.0) algorithm86. First, we selected the NewCellDataSet function to create a new object for the monocle with count data of the included cells. Then, the estimateSizeFactors and estimateDispersions function were used to assist us in normalizing for differences in mRNA recovered across cells. Next, we chose the signature genes, which expressed in at least 10% cells and with a p < 0.01 calculated by the differentialGeneTest function, to establish the trajectory progress. Using differentialGeneTest with the “fullModelFormulaStr” option “sm.ns (Pseudotime)”, pseudotime-dependent genes were identified, and smooth expression curves were produced using plot_pseudotime_heatmap function. On the pseudotime axis, the frequency of scattered cells in various groups was examined using the ggridges program (Version 0.5.4).
SCENIC analysis
To investigate the particular transcription factors (TFs) of macrophage and T cell subclusters, the Single-Cell rEgulatory Network Inference and Clustering analysis (SCENIC) tool (Version 1.3.1) was employed. First, we filtered out noisy genes with low positive rates or low expression levels using the geneFiltering program’s default parameters. Second, the intersection between the remaining genes and the Rcis Target databases was maintained. Third, we used Gene Network Inference using Ensemble of Trees to reconstruct the gene regulatory network in accordance with the expression matrix (GENIE3). By calculating the significance score between each TF and its possible target genes, we developed the TF-potential target gene co-expression module. Relationships with significance scores below the default threshold as well as modules with gene counts below 50 were removed. Fourth, to further confirm the actual regulatory relationship between TF and putative target genes, we examined the TF-binding motif sequence on potential target genes in accordance with the cisTarget Human motif database. Finally, we measured the activity of the Regulons using AUCell, and visualized the data using the heatmap and ggplot2 tool.
Enrichment analysis
We used the FindAllMarkers function (min.pct = 0, and logfc.threshold = 0) of Seurat package to calculate q-value to demonstrate the differential expression of all genes in each cell cluster separately. Genes with q-values < 0.05 were considered as DEGs. In order to characterize the pertinent mechanism of RA pathogenesis, the enrichGO functions in the clusterProfiler package was used to calculate the GO biological processes with overexpressed DEGs of clusters or groups. In addition, the fgsea function from clusterProfiler package was applied to explore the enriched pathways. We also used gene set variation analysis (GSVA) to investigate some particular gene sets for cell subclusters, setting “technique” to “ssgsea” and “kcdf” to “Gaussian”. Gene sets associated to M1 and M2, for instance, were used to calculate the M1 and M2 scores in macrophages, respectively. In order to establish the potential characteristics of various cells, we also employed the enrichIt function from the escape R package for enrichment analysis using 50 hallmark gene sets in the MSigDB databases. Results were visualized by pheatmap and ggplot2 software.
Cell-cell interaction analysis
CellphoneDB87 and CellChat88 software was used to infer cell-cell communications among different cell clusters. For CellphoneDB, a raw count matrix and an extracted annotation file from a Seurat object were used as the input for “statistical analysis” function. We calculated the number of the total interactions between cell clusters, and illustrated in terms of different comparisons (e.g., OA-BT vs. RA-BT, RA-BT vs. RA-AT, ACR20_Y vs. ACR20_N). For CellChat, gene expression data of SF was imported using the “createCellChat” function according to the official workflow. Several functions, such as “identifyOverExpressedGenes,” “identifyOverExpressedInteractions,” and “computeCommunProb,” were employed to identify significant cell-cell interactions among the analyzed cell clusters. To compare interactions between RA-BT/ACR20_Y and OA-BT/RA-AT/ACR20_N, we utilized the “compareInteractions” function. The Circlize package was used to demonstrate the communications between certain cell clusters of interest in more depth, while the pheatmap tool was used to display the frequency of ligand-receptor couplings across all cell clusters. The dot plot function and ggplot2 package were used to plot the probable communication strengths of various ligand-receptor pairings. In addition, ITALK (Version 0.1.0) was used to investigate cytokine and chemokine-level intercellular modulation. A raw count matrix and an extracted annotation file from a Seurat object were used as the input. For visual clarity with the LRPlot function, we only displayed the top 120 communications between macrophages and T cells as determined by the iTalk res function.
NicheNet analysis
To infer active ligands and their gene regulatory effects on pathogenic cell types of RA, NicheNet software60 was implemented. For CD8+ Tem1, we included top 100 genes (mitochondrial and sex-specific genes are not included) that were significantly higher expressed in RA-BT compared to OA-BT. The same process was also applied to Treg and CXCL13+CD4+ T subclusters. A total of 209 concatenated genes were identified as potential targets. Next, we investigated the background profiles and possible ligands, for genes with a positive rate of more than 10% in any of the five pathogenic cell types, respectively. In order to create the expressed ligand-receptor communications and determine the ligand activity, sender and target cells were also added. The potential intensity of regulation between the ligand and target genes was calculated using the active ligand target links tool. The pheatmap software was used for visualization.
ELISA
The relevant protein level in the SF supernatant of RA and OA patients was assessed using the Human OPN ELISA Kit (E-EL-H1347; Elabscience) and the Human MCP-1 ELISA Kit (E-EL-H6005; Elabscience). The kits and samples were set to room temperature (RT) for 20 min. 100 μl of standards or samples were added to each pre-coated well and incubated at 37 °C for 90 min. After draining the liquid from each well, we added 100 μl of Biotinylated Detection Ab working solution, and incubated the mixture for 60 min at 37 °C. Next, the wells were wash by wash buffer for three times, followed by adding 100 μl of HRP Conjugate working solution and incubating at 37 °C for 30 min. After five times of wash with wash buffer, 90 μl of Substrate Reagent was added and incubated at 37 °C for 15 minutes. Finally, we added 50 μl of Stop Solution and measured the optical density (OD) at 450 nm. All of the data were calculated using ELISACalc.
Flow cytometry analysis
After centrifugation at 800 × g for 10 min, the cells from fresh SF samples were resuspended with PBS (100 μl for each 1 × 106 cells). For live-dead gating, Fixable Viability Stain 700 (564997; BD Bioscience) was applied to cell suspension (1:1000) and incubated for 15 min at room temperature in dark. After PBS washes twice, cell surface staining was performed in PBS with antibodies mixture and incubated for 30 min in dark at 4 °C. Next, wash cells twice and fix cells with BD Cytofix/cytoperm solution (554714; BD Bioscience) for 20 min. Finally, the cells were suspended with 300ul PBS for detection. The FACSymphony A5 (BD Bioscience) was used to perform the cells assay and the FlowJo application was used to examine all of the data shown here. The antibodies in this study were: CD45 (368503; Biolegend), CD11b (301329; Biolegend), CD64 (305014;Biolegend), CD11c (337220; Biolegend), and CD55 (311308; Biolegend). Detailed information of the antibodies can be found in Supplementary Data 8.
Immunofluorescent staining of synovial tissue
Multiplex immunofluorescence (mIF) staining was performed using the 6-Color Manual IHC Kit (AFIHC026; Aifang Biological) with the standard protocol we described previously78,79. Briefly, tissues were sliced into 4 µm sections and heat in the retrieval solution of EDTA or citric acid to recover antigens.
For co-colocalization of SPP1+ and CD36+CD62L-macrophages (CD68+), as well as S100A12+ and CD62L+macrophages (CD68+) in the ST from RA patient, all antibodies against CD68 (AF20022, Aifang Biological, 1:3000, TYR-520), SPP1(AF03532, Aifang Biological, 1:2000, TYR-570), CD36 (66395-1-Ig, Proteintech, 1:3000, TYR-780), CD62L (222511, ZEN-BIOSCIENCE, 1:2000, TYR-690), and S100A12 (14959, Cell Signaling Technology, 1:2000, TYR-620) were evaluated via immunohistochemistry.
For co-colocalization of SPP1+ macrophages and CXCL13+CD4+ T cell in the ST from RA and OA patients, all antibodies against CD68 (AF20022, Aifang Biological, 1:3000, TYR-690), SPP1(AF03532, Aifang Biological, 1:2000, TYR-620), CD3 (85061S, Cell Signaling Technology, 1:200, TYR-780), CD4 (ab133616, Abcam, 1:400, TYR-480), CXCL13 (ab246518, Abcam, 1:500, TYR-570) and STAT1 (14994T, Cell Signaling Technology, 1:1000, TYR-520) were evaluated via immunohistochemistry.
For co-colocalization of SPP1+ macrophages and CXCL13+CD4+ T cell in the ST from CIA and control mice (DBA/1J background), all antibodies against F4/80 (ab100790, Abcam, 1:1000, TYR-690), SPP1 (ab218237, Abcam, 1:500, TYR-620), CD3 (99940S, Cell Signaling Technology, 1:200, TYR-780), CD4 (25229S, Cell Signaling Technology, 1:200, TYR-570), CXCL13 (ab199043, Abcam, 1:500, TYR-480) and STAT1 (14994T, Cell Signaling Technology, 1:500, TYR-520) were evaluated via immunohistochemistry. Detailed information of the antibodies can be found in Supplementary Data 8.
Slides were scanned on a fluorescent microscope (NIKON ECLIPSE C1). The data analyses were performed with QuPath software (Version 0.5.0). In delving into the intricate cellular interactions within the synovial immune microenvironment, cells located within a 15 μm radius of a target cell are regarded as potential interaction partners.
Isolation of primary fibroblast-like synoviocytes (FLS) and coculture with sorted macrophages and CD4 + T cells
According to the well-established protocol89, knee ST samples were obtained from RA patients who underwent total knee replacement at the West China Hospital of Sichuan University. Primary FLS was isolated by digesting ST with 0.5 mg/ml collagenase VIII (C2139, Sigma-Aldrich) in RPMI-1640 medium (11875093, Gibco) in the incubator shaker for 2 h. Dissociated cells were grown to confluency in RPMI-1640 supplemented with 10% fetal bovine serum (FBS, A5669701, Gibco), 1% penicillin-streptomycin (SV30010, Cytiva) before passaging. Expression of CD55 was assessed by flow cytometry and passages between 3 to 5 were used for coculture experiment.
Single-cell suspensions of pooled human SF cells were stained with anti-CD11b (101205; Biolegend), anti-CD64 (305014; Biolegend), anti-CD36 (336206; Biolegend), and anti-CD62L (304844; Biolegend) antibodies. S100A12+ macrophages were sorted as CD11b+CD64+CD62L+, SPP1+ macrophages were sorted as CD11b+CD64+CD62L-CD36+, and the Double Negative macrophages were sorted as CD11b+CD64+CD62L-CD36- on a MoFlo Astrios EQ (BECKMAN COULTER). CD4+ T cells from human SF were enriched with MojoSort™ Human CD4 Nanobeads (480014, biolegend) following the manufacturer’s instructions.
For direct coculture, sorted SPP1+/S100A12+/Double Negative macrophages or CD4+ T cells from RA patients were cocultured with FLS at a ratio of 1:9 in 6-well cell-culture plates in a final volume of 2 ml complete medium. After 48 h, the levels of IL-6, MMP-1, MMP-2, and MMP-13 in the coculture precipitation were measured by quantitative polymerase chain reaction (qPCR). Trans-well based coculture was also employed as indirect coculture assay. FLS were plated in a 12-well culture plate in 1.5 ml of complete medium. To create a coculture system at a 1:9 ratio, we added 0.5 ml of complete medium containing SPP1+/S100A12+/Double Negative macrophages or CD4+ T cells to Trans-well inserts with 0.4-μm pore size and placed them in the wells.
To demonstrate that SPP1+ macrophages can affect FLS by activating pathogenic CD4+ T cells, we directly and indirectly co-cultured the aforementioned cells with FLS in a 1:1:9 ratio as described above.
Quantitative PCR
Total RNA was isolated from the mouse paws or cells using the PaPure Total RNA Kit (R4011-03; Magen). cDNAs were synthesized using the All-In-One 5X RT MasterMix (G592; abm). qPCR was performed using the ChamQ SYBR qPCR Master Mix (Q311-02; Vazyme) on a CFX96 Real-Time System (BIO-RAD). Gene expression was calculated using the 2ΔΔCt method and was normalized to GAPDH expression. The human primers applied for qPCR analysis were as follows: GAPDH: F (forward), 5′- CACATGGCCTCCAAGGAGTAA, R (reverse), 5′-TGAGGGTCTCTCTCTTCCTCTTGT; IL6: F, 5′-TGAGGAGACTTGCCTGGTGA, R, 5′-TTGGGTCAGGGGTGGTTATT; MMP1: F, 5′-CACGCCAGATTTGCCAAGAG, R, 5′-GTCCCGATGATCTCCCCTGA; MMP2: F, 5′-TGGAAGTCTGTGTTGTCCAGAG, R, 5′-TGATTTGAAGCCAAGCGGTC; MMP13: F, 5′-ACTGAGAGGCTCCGAGAAATG, R, 5′-GAACCCCGCATCTTGGCTT; SPP1: F, 5′-GAAGTTTCGCAGACCTGACAT, R, 5′-GTATGCACCATTCAACTCCTCG; S100A12: F, 5′-CTTCCACCAATACTCAGTTCGG, R, 5′-GCAATGGCTACCAGGGATATG. The mouse primers applied for qPCR analysis were as follows: Actin: F, 5′-CGTTGACATCCGTAAAGACC, R, 5′-AACAGTCCGCCTAGAAGCAC; Il6: F, 5′-CCAGAAACCGCTATGAAGTTCC, R, 5′-TTGTCACCAGCATCAGTCCC; Mmp1a: F, 5′-CTATTTCAAAGGCAGCAAAGTATGG, R, 5′-GTCTCTTCCTCACAAACAGCAG; Mmp13: F, 5′-GGTCCAGGCGATGAAGACCCC, R, 5′-GGGTGCAGGCGCCAGAAGAA;
Mice
Male DBA/1J mice (8 weeks old, 20–25 g) were purchased from HuaFuKang Co., Ltd. (Beijing, China). CRISPR-Cas9-based Spp1flox/flox conditional knockout and Lyz2-Cre mice on C57BL/6J background were purchased from GemPharmatech (China). Spp1flox/floxLyz2-Cre+ (Spp1-cKO) and Spp1flox/floxLyz2-Cre- (Spp1-WT) would be bred through mating these two strains. Spp1-cKO mice result in deletion of exon 2 to exon 8 (including the whole coding region of Spp1) of Spp1 gene in myeloid cells (e.g., macrophages). Spp1-WT mice were used as control. Same breeding and treatment strategies were applied to the Spp1-cKO and Spp1-WT mice. Male mice (C57BL/6J background) were experimented at 8-10 weeks. Mice were genotyped by PCR. For the Spp1flox+/- the following primers were Used: F:5’ GGTCCCTGTGTGATAACACAGACTC 3’ R: 5’ CTCTCCACCTCTCATCCTTAGCAAG 3’. For the presence of the Lyz2-Cre, we used as primers: F: 5’ GAACACACCTGGAAGATGCTCC 3’ and R: 5’ CATCCTTGGCACCATAGATCAGG 3’. Mice were housed under a 12-hour light/dark cycle with food and drinking water provided ad libitum. Room temperature was maintained within the range of 22–24 °C and relative humidity ranged between 45-55%.
CIA model
We dissolved chick type II collagen (20011; Chondrex) in 5 mL of acetic acid (0.05 M), followed by emulsification with complete Freund’s adjuvant (CFA) (7001; Chondrex) or incomplete Freund’s Adjuvant (IFA) (7002; Chondrex). For DBA/1J background mice, on day 0, the mice received an intradermal injection of 0.1 mL CII + CFA emulsion. On day 21, they were restimulated with an intradermal injection of 0.1 mL of CII + IFA emulsion. Finally, on day 42, the mice were euthanized, and knee joints were collected for mIHC. For C57BL/6 background mice, on day 0, the mice received an intradermal injection of 0.15 mL CII + CFA emulsion. On day 21, they were restimulated with an intradermal injection of 0.15 mL of CII + IFA emulsion. Finally, on day 51, the mice were euthanized by cervical dislocation, and hind paws were collected for qPCR.
Mice were scored on the arthritis index by two independent observers every 2 days. The scoring standard is 0-4 different grades: 0 point, no local redness and swelling; 1 point, the phenomenon of swelling of the knuckles appears; 2 points, there is a slight swelling of the ankle or wrist joint; For 3 points, the whole foot and paw appear severely swollen; 4 points, the paws appear stiff or deformed. All protocols were approved by the Biomedical Ethics Committee of the West China Hospital, Sichuan University (20230515007).
Statistical analysis
Continuous data were analyzed statistically using R, and they were reported as mean standard deviation (SD). Wilcoxon tests, both unpaired and paired, were used to determine which differences were statistically significant. Differences were considered significant if their adjusted p-value (Bonferroni) was less than 0.05.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All raw and processed data from scRNA-seq and bulk sequencing have been deposited in the Genome Sequence Archive in National Genomics Data Center90, China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences (access ID: HRA003842 and HRA008066) that are publicly accessible at https://ngdc.cncb.ac.cn/gsa-human. All other data are available in the article and its Supplementary files or from the corresponding author upon request. Source data are provided with this paper.
Code availability
The bioinformatic analysis code has been uploaded into the github (https://github.com/Xiaxy-XuLab/RA-SF, https://doi.org/10.5281/zenodo.14651008).
References
Smolen, J. S. et al. Rheumatoid arthritis. Nat. Rev. Dis. Prim. 4, 18001 (2018).
Masuko-Hongo, K. et al. Long-term persistent accumulation of CD8+ T cells in synovial fluid of rheumatoid arthritis. Ann. Rheum. Dis. 56, 613–621 (1997).
Hatano, Y. et al. Macrophage inflammatory protein 1 alpha expression by synovial fluid neutrophils in rheumatoid arthritis. Ann. Rheum. Dis. 58, 297–302 (1999).
Petersen, J., Ingemann-Hansen, T. & Halkjaer-Kristensen, J. S. Spontaneous and induced immunoglobulin secretion by synovial fluid B lymphocytes in rheumatoid arthritis. Ann. Rheum. Dis. 43, 140–145 (1984).
Sheldon, P. J., Papamichail, M. & Holborow, E. J. Studies on synovial fluid lymphocytes in rheumatoid arthritis. Ann. Rheum. Dis. 33, 509–514 (1974).
Davis, M. J., Denton, J., Freemont, A. J. & Holt, P. J. Comparison of serial synovial fluid cytology in rheumatoid arthritis: delineation of subgroups with prognostic implications. Ann. Rheum. Dis. 47, 559–562 (1988).
McInnes, I. B. & Schett, G. Pathogenetic insights from the treatment of rheumatoid arthritis. Lancet 389, 2328–2337 (2017).
Aletaha, D. & Smolen, J. S. Diagnosis and Management of Rheumatoid Arthritis: A Review. JAMA 320, 1360–1372 (2018).
Pappas, D. A. et al. Comparative effectiveness of first-line tumour necrosis factor inhibitor versus non-tumour necrosis factor inhibitor biologics and targeted synthetic agents in patients with rheumatoid arthritis: results from a large US registry study. Ann. Rheum. Dis. 80, 96–102 (2021).
Combe, B. et al. Filgotinib versus placebo or adalimumab in patients with rheumatoid arthritis and inadequate response to methotrexate: a phase III randomised clinical trial. Ann. Rheum. Dis. 80, 848–858 (2021).
Fitton, J., Melville, A. R., Emery, P., Nam, J. L. & Buch, M. H. Real-world single centre use of JAK inhibitors across the rheumatoid arthritis pathway. Rheumatology 60, 4048–4054 (2021).
Taylor, P. C. et al. Baricitinib versus Placebo or Adalimumab in Rheumatoid Arthritis. N. Engl. J. Med. 376, 652–662 (2017).
Kearsley-Fleet, L. et al. Biologic refractory disease in rheumatoid arthritis: results from the British Society for Rheumatology Biologics Register for Rheumatoid Arthritis. Ann. Rheum. Dis. 77, 1405–1412 (2018).
Buch, M. H., Eyre, S. & McGonagle, D. Persistent inflammatory and non-inflammatory mechanisms in refractory rheumatoid arthritis. Nat. Rev. Rheumatol. 17, 17–33 (2021).
Fraenkel, L. et al. 2021 American College of Rheumatology Guideline for the Treatment of Rheumatoid Arthritis. Arthritis Care Res. 73, 924–939 (2021).
Ytterberg, S. R. et al. Cardiovascular and Cancer Risk with Tofacitinib in Rheumatoid Arthritis. N. Engl. J. Med. 386, 316–326 (2022).
Winthrop, K. L. & Cohen, S. B. Oral surveillance and JAK inhibitor safety: the theory of relativity. Nat. Rev. Rheumatol. 18, 301–304 (2022).
Sepriano, A. et al. Safety of synthetic and biological DMARDs: a systematic literature review informing the 2022 update of the EULAR recommendations for the management of rheumatoid arthritis. Ann. Rheum. Dis. 82, 107–118 (2023).
Kuo, D. et al. HBEGF(+) macrophages in rheumatoid arthritis induce fibroblast invasiveness. Sci. Translational Med. 11, eaau8587 (2019).
Croft, A. P. et al. Distinct fibroblast subsets drive inflammation and damage in arthritis. Nature 570, 246–251 (2019).
Argyriou, A. et al. Single cell sequencing identifies clonally expanded synovial CD4(+) T(PH) cells expressing GPR56 in rheumatoid arthritis. Nat. Commun. 13, 4046 (2022).
Alivernini, S. et al. Distinct synovial tissue macrophage subsets regulate inflammation and remission in rheumatoid arthritis. Nat. Med. 26, 1295–1306 (2020).
Udalova, I. A., Mantovani, A. & Feldmann, M. Macrophage heterogeneity in the context of rheumatoid arthritis. Nat. Rev. Rheumatol. 12, 472–485 (2016).
Carmona-Rivera, C. et al. Synovial fibroblast-neutrophil interactions promote pathogenic adaptive immunity in rheumatoid arthritis. Sci. Immunol. 2, eaag3358 (2017).
Weyand, C. M. & Goronzy, J. J. The immunology of rheumatoid arthritis. Nat. Immunol. 22, 10–18 (2021).
Floudas, A. et al. Distinct stromal and immune cell interactions shape the pathogenesis of rheumatoid and psoriatic arthritis. Ann. Rheum. Dis., 81, 1224–1242 (2022).
Zaiss, M. M., Joyce Wu, H. J., Mauro, D., Schett, G. & Ciccia, F. The gut-joint axis in rheumatoid arthritis. Nat. Rev. Rheumatol. 17, 224–237 (2021).
Zhou, B. et al. Induction and Amelioration of Methotrexate-Induced Gastrointestinal Toxicity are Related to Immune Response and Gut Microbiota. EBioMedicine 33, 122–133 (2018).
Firestein, G. S. & McInnes, I. B. Immunopathogenesis of Rheumatoid Arthritis. Immunity 46, 183–196 (2017).
Kurowska-Stolarska, M. & Alivernini, S. Synovial tissue macrophages: friend or foe? RMD open 3, e000527 (2017).
Hannemann, N., Apparailly, F. & Courties, G. Synovial macrophages: from ordinary eaters to extraordinary multitaskers. Trends Immunol. 42, 368–371 (2021).
Rao, D. A. et al. Pathologically expanded peripheral T helper cell subset drives B cells in rheumatoid arthritis. Nature 542, 110–114 (2017).
Stephenson, W. et al. Single-cell RNA-seq of rheumatoid arthritis synovial tissue using low-cost microfluidic instrumentation. Nat. Commun. 9, 791 (2018).
Mizoguchi, F. et al. Functionally distinct disease-associated fibroblast subsets in rheumatoid arthritis. Nat. Commun. 9, 789 (2018).
Cheung, P., Khatri, P., Utz, P. J. & Kuo, A. J. Single-cell technologies - studying rheumatic diseases one cell at a time. Nat. Rev. Rheumatol. 15, 340–354 (2019).
Culemann, S. et al. Locally renewing resident synovial macrophages provide a protective barrier for the joint. Nature 572, 670–675 (2019).
Zhang, F. et al. Defining inflammatory cell states in rheumatoid arthritis joint synovial tissues by integrating single-cell transcriptomics and mass cytometry. Nat. Immunol. 20, 928–942 (2019).
Musters, A. et al. In Rheumatoid Arthritis, Synovitis at Different Inflammatory Sites Is Dominated by Shared but Patient-Specific T Cell Clones. J. Immunol. 201, 417–422 (2018).
Shaul, M. E. & Fridlender, Z. G. Tumour-associated neutrophils in patients with cancer. Nat. Rev. Clin. Oncol. 16, 601–620 (2019).
Zilionis, R. et al. Single-Cell Transcriptomics of Human and Mouse Lung Cancers Reveals Conserved Myeloid Populations across Individuals and Species. Immunity 50, 1317–1334.e1310 (2019).
Walker, J. G. et al. Characterisation of a dendritic cell subset in synovial tissue which strongly expresses Jak/STAT transcription factors from patients with rheumatoid arthritis. Ann. Rheum. Dis. 66, 992–999 (2007).
Rossi, J. F., Lu, Z. Y., Jourdan, M. & Klein, B. Interleukin-6 as a therapeutic target. Clin. Cancer Res. 21, 1248–1257 (2015).
Philips, R. L. et al. The JAK-STAT pathway at 30: Much learned, much more to do. Cell 185, 3857–3876 (2022).
Lanteri, P., Lombardi, G., Colombini, A., Grasso, D. & Banfi, G. Stability of osteopontin in plasma and serum. Clin. Chem. Lab. Med. 50, 1979–1984 (2012).
Leemasawatdigul, K. & Gappa-Fahlenkamp, H. Effect of storage conditions on the stability of recombinant human MCP-1/CCL2. Biologicals J. Int. Assoc. Biol. Standardization 39, 29–32 (2011).
Edalat, S. G. et al. Molecular maps of synovial cells in inflammatory arthritis using an optimized synovial tissue dissociation protocol. iScience 27, 109707 (2024).
Ramos, M. I. et al. Absence of Fms-like tyrosine kinase 3 ligand (Flt3L) signalling protects against collagen-induced arthritis. Ann. Rheum. Dis. 74, 211–219 (2015).
Kasperkovitz, P. V. et al. Activation of the STAT1 pathway in rheumatoid arthritis. Ann. Rheum. Dis. 63, 233–239 (2004).
Walker, J. G. et al. Changes in synovial tissue Jak-STAT expression in rheumatoid arthritis in response to successful DMARD treatment. Ann. Rheum. Dis. 65, 1558–1564 (2006).
Zhang, R. et al. RNA-seq and Network Analysis Reveal Unique Chemokine Activity Signatures in the Synovial Tissue of Patients With Rheumatoid Arthritis. Front. Med. 9, 799440 (2022).
Rui, S., et al. The landscape of TIGIT target and clinical application in diseases. MedComm. Onconol. 1, e18 (2022).
Chen, L. & Flies, D. B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 13, 227–242 (2013).
Liu, B., Zhang, Y., Wang, D., Hu, X. & Zhang, Z. Single-cell meta-analyses reveal responses of tumor-reactive CXCL13(+) T cells to immune-checkpoint blockade. Nat. Cancer 3, 1123–1136 (2022).
Lowery, F. J. et al. Molecular signatures of antitumor neoantigen-reactive T cells from metastatic human cancers. Science 375, 877–884 (2022).
Bonelli, M. et al. IRF1 is critical for the TNF-driven interferon response in rheumatoid fibroblast-like synoviocytes: JAKinibs suppress the interferon response in RA-FLSs. Exp. Mol. Med. 51, 1–11 (2019).
Underhill, D. M., Bassetti, M., Rudensky, A. & Aderem, A. Dynamic interactions of macrophages with T cells during antigen presentation. J. Exp. Med. 190, 1909–1914 (1999).
Guerriero, J. L. Macrophages: Their Untold Story in T Cell Activation and Function. Int. Rev. Cell Mol. Biol. 342, 73–93 (2019).
Doherty, T. M. T-cell regulation of macrophage function. Curr. Opin. Immunol. 7, 400–404 (1995).
Phan, A. T., Goldrath, A. W. & Glass, C. K. Metabolic and Epigenetic Coordination of T Cell and Macrophage Immunity. Immunity 46, 714–729 (2017).
Browaeys, R., Saelens, W. & Saeys, Y. NicheNet: modeling intercellular communication by linking ligands to target genes. Nat. Methods 17, 159–162 (2020).
Smith, M. H. et al. Drivers of heterogeneity in synovial fibroblasts in rheumatoid arthritis. Nat. Immunol. 24, 1200–1210 (2023).
Wu, X. et al. Single-cell sequencing of immune cells from anticitrullinated peptide antibody positive and negative rheumatoid arthritis. Nat. Commun. 12, 4977 (2021).
Gazal, S. et al. Identification of secreted phosphoprotein 1 gene as a new rheumatoid arthritis susceptibility gene. Ann. Rheum. Dis. 74, e19 (2015).
Juge, P. A. et al. SPP1 rs9138 variant contributes to the severity of radiological damage in anti-citrullinated protein autoantibody-negative rheumatoid arthritis. Ann. Rheum. Dis. 73, 1840–1843 (2014).
Kurowska-Stolarska, M. & Alivernini, S. Synovial tissue macrophages in joint homeostasis, rheumatoid arthritis and disease remission. Nat. Rev. Rheumatol. 18, 384–397 (2022).
Kahles, F., Findeisen, H. M. & Bruemmer, D. Osteopontin: A novel regulator at the cross roads of inflammation, obesity and diabetes. Mol. Metab. 3, 384–393 (2014).
MacDonald, L. et al. COVID-19 and RA share an SPP1 myeloid pathway that drives PD-L1+ neutrophils and CD14+ monocytes. JCI Insight 6, e147413 (2021).
Volkov, M., van Schie, K. A. & van der Woude, D. Autoantibodies and B Cells: The ABC of rheumatoid arthritis pathophysiology. Immunological Rev. 294, 148–163 (2020).
Hodge, J. A. et al. The mechanism of action of tofacitinib - an oral Janus kinase inhibitor for the treatment of rheumatoid arthritis. Clin. Exp. Rheumatol. 34, 318–328 (2016).
Deakin, C. T. et al. Comparative Effectiveness of Adalimumab vs Tofacitinib in Patients With Rheumatoid Arthritis in Australia. JAMA Netw. open 6, e2320851 (2023).
van Vollenhoven, R. F. et al. Tofacitinib or adalimumab versus placebo in rheumatoid arthritis. N. Engl. J. Med. 367, 508–519 (2012).
Fleischmann, R. et al. Efficacy and safety of tofacitinib monotherapy, tofacitinib with methotrexate, and adalimumab with methotrexate in patients with rheumatoid arthritis (ORAL Strategy): a phase 3b/4, double-blind, head-to-head, randomised controlled trial. Lancet 390, 457–468 (2017).
Bottini, N. & Firestein, G. S. Duality of fibroblast-like synoviocytes in RA: passive responders and imprinted aggressors. Nat. Rev. Rheumatol. 9, 24–33 (2013).
Aletaha, D. et al. 2010 Rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. 62, 2569–2581 (2010).
Anderson, J. et al. Rheumatoid arthritis disease activity measures: American College of Rheumatology recommendations for use in clinical practice. Arthritis Care Res. 64, 640–647 (2012).
Felson, D. T. et al. American College of Rheumatology. Preliminary definition of improvement in rheumatoid arthritis. Arthritis Rheum. 38, 727–735 (1995).
Luo, H. et al. Characterizing dedifferentiation of thyroid cancer by integrated analysis. Sci. Adv. 7, eabf3657 (2021).
Luo, H. et al. Pan-cancer single-cell analysis reveals the heterogeneity and plasticity of cancer-associated fibroblasts in the tumor microenvironment. Nat. Commun. 13, 6619 (2022).
Cao, M. et al. Characterization of immature ovarian teratomas through single-cell transcriptome. Front. Immunol. 14, 1131814 (2023).
Yin, D. et al. Novel insights into HBV‐hepatocellular carcinoma at single‐cell sequencing. MedComm Oncol. 2, e60 (2023).
Mao, W. et al. Pan-cancer single-cell landscape of drug-metabolizing enzyme genes. Pharmacogenetics Genomics 34, 217–225 (2024).
Zhou T. et al. The role of long non‐coding RNA Maternally Expressed Gene 3 in cancer‐associated fibroblasts at single cell pan‐cancer level. Interdiscip. Med. 2, e20240018 (2024).
Haghverdi, L., Lun, A. T. L., Morgan, M. D. & Marioni, J. C. Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat. Biotechnol. 36, 421–427 (2018).
Bray, N. L., Pimentel, H., Melsted, P. & Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 34, 525–527 (2016).
Wang, X. et al. Age-, sex- and proximal-distal-resolved multi-omics identifies regulators of intestinal aging in non-human primates. Nat. Aging 4, 414–433 (2024).
Trapnell, C. et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 32, 381–386 (2014).
Efremova, M., Vento-Tormo, M., Teichmann, S. A. & Vento-Tormo, R. CellPhoneDB: inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat. Protoc. 15, 1484–1506 (2020).
Jin, S. et al. Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 12, 1088 (2021).
Qin, Y. et al. Age-associated B cells contribute to the pathogenesis of rheumatoid arthritis by inducing activation of fibroblast-like synoviocytes via TNF-α-mediated ERK1/2 and JAK-STAT1 pathways. Ann. Rheum. Dis. 81, 1504–1514 (2022).
Database Resources of the National Genomics Data Center, China National Center for Bioinformation in 2022. Nucleic Acids Res. 50, D27–D38 (2022).
Acknowledgements
This study was supported by the National Key R&D Program of China (No. 2021YFA1301203(H.X.), 2016YFC0905002(H.X.)), National Natural Science Foundation of China (81973408(H.X.)), the China Postdoctoral Science Foundation (2022M712274(X.X.)), Sichuan Science and Technology Program (2023NSFSC1704(X.X.), 2024YFFK0062(Q.B.X.), 2025ZNSFSC1745(C.J.H.), 2025ZNSFSC0046(H.X.), 2025ZNSFSC0638(G.Y.)), 1.3.5 Project for Disciplines of Excellence, West China Hospital, Sichuan University (ZYYC23025 (H.X.), ZYYC23013 (L.Z.D.)). We thank Zhang Yanjing from Core Facilities of West China Hospital for assisting DNA quality control. We thank Mrs. Jingjing Ran for assisting in cell culture.
Author information
Authors and Affiliations
Contributions
G.Y., H.X., and Q.B.X. designed and supervised the project. X.Y.X., Z.X.R., H.L., H.N.C., W.H.Z., L.B.H., M.X.Q., and Y.S. analyzed the data. X.Y.X., Y.L.W., Y.Y.S., L.L.W., F.Z., W.Z., H.X., and Q.B.X interpreted the data. C.J.H., Z.N.X., Y.Q., Y.P.H., Y.J.B., B.C. and G.Y. collected the clinical samples and information. C.J.H and Z.N.X., and Y.L.W. performed experiments and conducted related analysis. C.J.H. and X.Y.X. draw the diagrams. C.J.H., X.Y.X., H.X., and Q.B.X. wrote and revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Diego Kyburz and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xia, X., He, C., Xue, Z. et al. Single cell immunoprofile of synovial fluid in rheumatoid arthritis with TNF/JAK inhibitor treatment. Nat Commun 16, 2152 (2025). https://doi.org/10.1038/s41467-025-57361-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-57361-0
This article is cited by
-
A single-cell RNA-seq dataset of synovial fluid from rheumatoid arthritis treated with TNF-α/JAK inhibitor
Scientific Data (2025)
-
Timepoint of aspiration can impact diagnostic of PJI - synovial fluid analyses exhibit a high intraindividual variation in periprosthetic joint infections of the knee
Archives of Orthopaedic and Trauma Surgery (2025)
-
Single-cell transcriptomic analysis reveals gut microbiota-immunotherapy synergy through modulating tumor microenvironment
Signal Transduction and Targeted Therapy (2025)
