
Abstract
Three-dimensional genome organization orchestrates recombination and transcription of immunoglobulin heavy chain (Igh) genes. The structure of wild-type (WT) alleles includes a prominent architectural stripe that extends from a cluster of CTCF binding elements at the 3′ end of the locus (3′CBE), suggesting interactions of this end with sequences throughout the 2 Mb Igh TAD. Here we elucidate interplay between regulatory elements located in the 3′Igh domain (260 kb) that impact the stripe. The CTCF-lacking intronic enhancer, Eµ, promotes stripe formation and tethers sub-TADs between flanking CTCF-bound 3′CBE and IGCR1. Substituting Eµ with an EF1α promoter in different orientations partially recapitulates epigenetic features of WT Igh alleles, including active histone modifications, sub-TAD formation and interactions with the 3′CBE, but does not restore VDJ recombination. Loss of IGCR1 increases the stripe while inverting the 3′CBE redirects the stripe away from the Igh locus. However, inverted 3′CBE continue to serve as a boundary against aberrant activation of genes outside the Igh domain by Eµ. Our observations provide insights into mechanisms by which regulatory elements modulate chromatin structure and stripe formation.
Similar content being viewed by others


Introduction
Three-dimensional organization of the genome facilitates cell lineage-appropriate gene expression. Topologically associated domains (TADs) are megabase-sized genomic regions within which DNA interactions are more frequent than with sequences beyond1,2,3,4. TAD boundaries are largely cell type invariant and enriched for binding of the ubiquitously expressed architectural protein CTCF5,6,7,8. Chromatin fine structure within TADs shows additional cell specific heterogeneity that is contributed in part by interactions between gene promoters and regulatory sequences9,10. Enhancers are defined as cis-regulatory sequences that regulate gene transcription from promoter distal genomic regions11,12,13,14. They are marked by highly accessible chromatin, selective histone modifications and p300/CBP binding. TAD boundaries facilitate interactions between correct enhancer/promoter combinations by minimizing inter-TAD interactions15,16,17. High depth conformation capture studies have also revealed the presence of architectural stripes. These represent contact of one chromosomal region, referred to as the stripe anchor, with many sites over an extended genomic interval18. Mechanisms by which architectural stripes are generated are an area of active investigation19,20,21.
Cohesin-dependent loop extrusion is a major mechanism for communication between distant genomic regions22,23,24,25. In our current understanding, cohesin loads at selected sites in the genome and pulls chromatin strands until it encounters CTCF bound in a specific orientation26,27,28. Extrusion stalls at such encounters resulting in loops between oppositely oriented CTCF-bound sites. It is reasonable to hypothesize that enhancer/promoter interactions are also facilitated by loop extrusion. This could happen in different ways. For example, some enhancer/promoter combinations may have appropriately oriented CTCF sites that stall extrusion leading to enhancer/promoter contact. Indeed, many promoters and enhancers have closely associated CTCF sites29. For others, cohesin-dependent extrusion could bring distal sequences in spatial proximity so that promoter- or enhancer-bound factors interact. This mechanism has been termed ‘nudging’30,31. Alternatively, cohesin-dependent extrusion may stall at promoters and enhancers that lack CTCF binding, leading to interactions between DNA-bound factors. The last model implies that promoters and enhancers can mediate three-dimensional genome organization, much like properties attributed to CTCF-bound genomic sites. This model is supported by recent studies that attribute cohesin stalling characteristics to RNA polymerase II32. However, enhancer-dependent cohesin stalling and its impact on chromatin organization are relatively understudied.
Antigen receptor genes of the adaptive immune system undergo genomic rearrangements to assemble functional genes33,34. Immunoglobulin heavy chain (Igh) genes are assembled by DNA rearrangements that juxtapose a variable (VH), a diversity (DH), and a joining (JH) gene segment into an exon that encodes the antigen binding domain of antibody heavy chains. The order of rearrangements is fixed. DH to JH rearrangements occur first to create DJH recombined alleles, followed by VH rearrangements to DJH junctions to produce VDJH recombined alleles35. Three cis-regulatory elements control structure and rearrangements of Igh genes. The intronic enhancer, Eµ, was the first identified tissue-specific enhancer and was initially proposed to activate Igh promoters in mature B cells36. However, genomic deletion of Eµ reduces Igh rearrangements in developing progenitor (pro-) B cells well before Igh gene assembly is completed37,38. These observations indicate that Eµ also serves as a recombination enhancer, in part by inducing epigenetic changes and chromatin accessibility during V(D)J recombination39. Eμ also configures 3-dimensional locus structure by interacting with two CTCF-bound regulatory sequences to form two sub-TADs at the 3′ end of the Igh locus (3′ Igh) (Fig. 1a)40. A 60 kb domain forms between Eµ and the intergenic control region 1 (IGCR1). This domain contains most of the diversity (DH) gene segments and we have previously proposed that exclusion of VH gene segments from this domain drives the order of Igh rearrangements40. Disrupting Eµ/IGCR1 interactions by mutating or deleting IGCR1 results in Eµ interacting with the next available CTCF-bound site, located approximately 90 kb further 5′41,42 within proximal VHs. In this configuration, that most 3′ VH gene segments are brought into special proximity of the recombinase-rich recombination center (RC), disrupting the order of Igh rearrangements. Eµ also interacts with a cluster of CTCF-binding sites (3′CBE) located at the very 3′ end of the locus43 (Fig. 1a) that anchors a stripe (3′CBE stripe) that extends well over 1 Mb into the Igh locus44,45. Deletion of 3′CBE leads to altered VDJ and class switch recombination46,47. Of the three sequences that configure the 3′ Igh domain, it is noteworthy that only Eµ does not bind CTCF. Thus, the 3′ Igh domain serves as a case study for mechanistic dissection of interplay between CTCF-binding and non-binding regulatory sequences in mediating chromatin architecture and function.

a Schematic representation of the Igh locus highlighting Eµ (red oval, indicated by black arrow), IGCR1 (purple oval) and 3′CBE (blue and purple). Gene segments (VH, DH and JH) are indicated as colored rectangles. The 3′ Igh domain comprises 260 kb between IGCR1 and 3′CBE. Proximal and distal refer to variable gene segments (VH) close or far away from 3′ Igh domain, respectively. b, c cHi-C profiles of the 3′ Igh domain on wild-type (WT) and Eµ-deficient (Eµ−/−) Igh alleles. The genomic interval of the 3′ Igh domain (b, chr12: 113200001-113500000, mm10) and entire Igh locus (c, chr12: 113200001-116000000, mm10) are illustrated for both WT (left panel) and Eµ−/− (middle panel) alleles. Heatmaps indicate normalized interaction frequencies. Noteworthy interacting regions are demarcated by a black dash rectangle. High cross-linking frequency reflecting loops between Eµ and 3′CBE (Eµ/3′CBE) or IGCR1 (Eµ/IGCR1) are indicated by black ovals. Difference interaction map between WT and Eµ−/− Igh alleles is shown on the right. Color code represents decreased (blue) or increased (red) interactions on Eµ−/− alleles. The cHi-C analysis of clone Eµ−/− #1 is presented in the middle panel, whereas data from clone Eµ−/− #2 is depicted in Supplementary Fig. 1c. cHi-C data were visualized with software cooltools. Experiment was independently repeated twice with similar results. d Virtual 4C representation of cHi-C analysis using the 3′CBE as the viewpoint. Interacting reads within the 20 kb 3′CBE region (chr12:113210001-113230000) were extracted from cHi-C data (c) for WT and Eµ−/− alleles and mapped within the entire Igh locus (chr12:113200001-116000000). Differential 4C analysis, contrasting Eµ−/− and WT Igh alleles, highlights alterations in 3′CBE-associated interactions, denoted by increased (red) or reduced (blue) connections. IGCR1 and Eµ are indicated by black arrows and the 3′CBE anchor is indicated by the red arrow. ChIP-Seq profiles of CTCF and Rad21 derived from WT or Eµ−/− pro-B cell lines are shown above virtual 4C maps. Experiment was independently repeated twice with similar results. See also Supplementary Fig. 1, Data 1, 2.
In this study, we combined epigenetic, transcriptional, and recombinational assays of WT and mutated Igh alleles to uncover structure-function relationships between sequences that configure the locus for the first step of gene assembly (Supplementary Data 1). We demonstrate that deletion of Eµ leads to partial fusion of two sub-TADs located within 3′ 260 kb of the Igh TAD, indicating that Eµ serves as a boundary element. Eµ also regulates the intensity of the 3′CBE stripe, reflecting its impact throughout the 2.8 Mb locus. Analysis of Igh alleles with Eµ substituted by the EF1α promoter show that sub-TAD formation arises from enhancer-bound proteins, while modulation of the 3′CBE stripe is a consequence of enhancer-initiated bi-directional transcription. By altering each CTCF-associated TAD anchor corresponding to IGCR1 and 3′CBE, we identified sub-TADs with unidirectionally oriented CTCF-binding sites and demonstrated that stripe direction, but not boundary formation, depends on the orientation of CTCF sites. Cumulatively, our studies elaborate mechanisms by which CTCF binding and CTCF non-binding regulatory sequences cooperatively establish functional chromatin domains.
Results
For these studies we focused on the roles of Eµ, IGCR1 and the 3′CBE in configuring the 3′ Igh domain. We started with an Abelson virus-transformed recombinase deficient cell line (carrying a point mutation in the catalytic domain of Rag1) that has unrearranged Igh alleles. We generated mutated alleles using CRISPR/Cas9 and evaluated locus conformation (by capture Hi-C or Hi-ChIP), transcriptional status (by directional RNA-Seq), CTCF/Rad21 binding and histone modifications (by ChIP-Seq) and VDJ recombination after ectopic expression of Rag1. Abelson virus-transformed (Abl) cell lines are widely utilized for elucidating molecular mechanisms underlying Igh chromatin structure and recombination41,43,48,49,50,51,52. While earlier studies have shown that Abelson virus transformation affects Igh locus compaction44,53, there is no evidence that it impacts chromatin structure at the scale of hundreds of kilobases, such as within the 3′ Igh domain. Moreover, in cells used in our analyses the 3′CBE stripe, a reflection of long-range interactions, extended 1.5 Mb into the VH region (see below), indicating their suitability for studying interactions within this region.
Contributions of Eµ
Using capture Hi-C (cHi-C) assay to probe Igh locus structure, we visualized two sub-domains in the 260 kb region between IGCR1 and 3′CBE reflecting Eμ interactions with each end (Fig. 1b left). A previously noted stripe extending from 3′CBE into the locus was also clear in cHi-C assays reflecting long-distance interactions into the variable domain (Fig. 1c, left, black arrow). Deletion of Eµ (Supplementary Fig. 1a, b) led to partial fusion of the two sub-domains, resulting in increased interactions of the 60 kb DH-containing region with sequences 3′ of Eμ (Fig. 1b and Supplementary Fig. 1c, middle and right, black dash rectangle). We found that intensity of the 3′CBE stripe was also reduced in the absence of Eµ (Fig. 1c, Supplementary Fig. 1d and e, and Supplementary Data 2). Representation of the cHi-C data as a differential virtual 4C map using 3′CBE as the anchor further substantiated reduced long-distance interactions with 3′CBE on Eµ-deficient alleles (Fig. 1d). This was not due to altered CTCF or cohesin recruitment throughout the Igh locus (Fig. 1d). We conclude that Eμ imposes sub-structure to a CTCF-flanked 3′ Igh domain by serving as a boundary element between two sub-TADs and accentuates long-range loop extrusion of unrearranged Igh alleles. The sub-TAD between Eμ and IGCR1, contains the most frequently used DH gene segments and the RC, within which the first steps of Igh gene rearrangements occur.
Structural compensation of Eµ by the EF1α promoter
Enhancers can modulate chromatin structure by recruiting chromatin modifying activities11,54,55, seeding bulky nucleoprotein complexes or by expressing enhancer-associated RNAs (eRNA). Eµ is a source of bi-directional transcripts on unrearranged alleles56. Sense-oriented transcripts, that are stabilized by splicing and polyadenylation, comprise the dominant RNA species in pro-B cells whereas antisense transcripts (of undefined length) are expressed at much lower levels (Fig. 2a, b). To evaluate the contribution of transcriptional direction to Eµ-dependent chromatin structure, we replaced Eµ with the EF1α promoter in both orientations (Supplementary Fig. 2a–c). EF1αFor directs transcription toward Cµ (sense orientation), whereas EF1αRev directs transcription toward DH-JH region (antisense orientation) (Fig. 2a). Introduction of the promoter induced H3K4me3 and H3K27ac regardless of orientation (Fig. 2b), whereas RNA expression corresponded closely with promoter orientation (Fig. 2a, b). Sense-oriented transcripts from EF1αFor were appropriately spliced and expressed at comparable levels to WT, whereas EF1αRev induced higher levels of antisense transcripts compared to WT alleles (Fig. 2b). EF1α promoter substitution in both orientations lowered CTCF binding at IGCR1, however, Rad21 binding was reduced to a greater extent on EF1αRev alleles (Fig. 2b). Neither orientation of EF1α promoter affected CTCF/Rad21 binding at the 3′CBE. Though transcription and activating histone modifications of Eµ-deficient alleles were restored by EF1α promoter insertions, VDJ recombination was not (Supplementary Fig. 2d). These observations indicate that locus structure is necessary but insufficient to induce recombination. One possibility is that the EF1α promoter failed to recruit recombinase proteins and reconstitute a functional RC. Thus, replacement of Eµ by EF1α promoter recapitulated some, but not all, features of WT Igh alleles.

a Transcriptional analysis of WT, Eµ-deficient, and EF1α promoter replacement Igh alleles. Eµ generates bi-directional (sense and antisense) RNA. Eµ was replaced by the EF1α promoter in two different orientations. EF1αFor directs transcription toward the Cµ (sense orientation), whereas EF1αRev directs transcription toward DH-JH region (antisense orientation). RT-qPCR was performed using primer pairs S1–S5 to assess the transcription profiles of various Igh alleles as indicated. Transcription levels relative to WT are shown as the bar graphs. The data are presented as mean ± SEM from three independent experiments. Results from two different EF1αFor and EF1αRev clones (#1 and #2) are shown. Pair primers P1–P4, P2–P4, and P3–P4 were used for DJH rearrangement analysis of WT, Eµ-deficient, and EF1α promoter replacement alleles (see Fig. 3c). The Experiment was independently repeated three times with similar results. Source data are provided as a Source Data file. b Transcriptional and epigenetic features of Eµ−/− and EF1α promoter replacement Igh alleles. Directional RNA-seq and ChIP-seq analysis of Eµ manipulated cell lines are indicated. The 3′ Igh domain extending from IGCR1 to the 3′CBE is displayed. IGCR1, DQ52-Cµ and Eμ loci are highlighted by black, red and green rectangles, respectively. β-actin locus (chr5:142,890,001-142,940,000) was used as a control (right). Experiment was independently repeated twice with similar results. See also Supplementary Fig. 2, Data 1, and 6.
Both orientations of EF1α promoter restored the 3′ sub-TAD (between Eμ and 3′CBE) and associated interactions of Eμ/EF1α region with 3′CBE (Fig. 3a middle, quantified in the bar graph, and Supplementary Fig. 3a). However, neither orientation restored interactions of the region with IGCR1 (Fig. 3a, quantified in the bar graph, and Supplementary Fig. 3a). We infer that orientation-independent features of EF1α promoter, such as restoration of 3′ sub-TAD, reflect extrusion stalling by promoter bound proteins. By analogy we propose that Eμ′s ability to block cohesin extrusion is based on the nucleoprotein complex formed on the enhancer. To investigate whether extrusion stalling by Eμ involves cohesin loading and/or release, we assessed the occupancy of NIPBL and WAPL in WT and various Eμ mutations using ChIP-seq. We found both NIPBL and WAPL were enriched at Eμ, but not EF1α-substituted Igh alleles, suggesting that Eμ plays a unique role in regulating cohesin loading and unloading (Fig. 2b).

a, b cHi-C analysis of the 3′ Igh domain (a) and entire Igh locus (b). Interactions within the 3′ Igh domain are depicted as a heatmap for WT (left panel) and EF1α promoter replaced (middle panel) Igh alleles. Difference interaction maps between WT and EF1α promoter replaced Igh alleles are shown on the right. Heatmaps indicate normalized interaction frequencies. Color code represents decreased (blue) or increased (red) interactions on the EF1α promoter replaced Igh alleles. Interactions involving Eµ or EF1α with 3′CBE (Eµ or EF1α/3′CBE) or IGCR1 (Eµ or EF1α/IGCR1) were quantified using capture Hi-C data, as described in the “Methods” section. Ratios of EF1αFor #1 or EF1αRev #1 to WT signals are graphed, with WT normalized to 1. The data are presented as the mean, with each data point representing results from two independent experiments. The cHi-C analysis of clone EF1αFor #1 and EF1αRev #1 are presented in the middle panel, whereas data from clone EF1αFor #2 and EF1αRev #2 are depicted in Supplementary Fig. 3a. cHi-C data were visualized with software cooltools. Experiment was independently repeated twice with similar results. Source data are provided as a Source Data file. c Virtual 4C representation of cHi-C analysis using the 3′CBE as the viewpoint. Interacting reads within the 20 kb 3′CBE region (chr12:113210001-113230000) were extracted from cHi-C data (b) and mapped within the entire Igh locus (chr12:113200001-116000000). Differential 4C analysis, contrasting Eµ−/− #1, EF1αFor #1, EF1αRev #1 and WT Igh alleles, highlights alterations in 3′CBE-associated interactions, denoted by increased (red) or reduced (blue) connections. Experiment was independently repeated twice with similar results. See also Supplementary Fig. 3, Data 1–3.
We also observed several orientation-dependent effects of EF1α promoter that we attribute to transcription. First, the 5′ boundary of the 3′ sub-TAD (between EF1α and 3′CBE) was sharper on EF1αRev alleles compared to EF1αFor alleles (Fig. 3a). Second, we observed increased interactions of 3′CBE with EF1α within the 3′ sub-TAD on EF1αFor alleles whereas such interactions were reduced on EF1αRev alleles (Fig. 3a bar graph, Supplementary Data 3). Third, we found that intensity of the 3′CBE stripe was reduced on EF1αFor alleles compared to WT alleles; by contrast, EF1αRev alleles retained a WT-like stripe (Fig. 3b, Supplementary Fig. 1e and 3b, and Supplementary Data 2). Virtual 4C visualization of the Hi-C data highlighted distinct chromatin states induced by different orientations of EF1α promoter (Fig. 3c). We observed reduced interactions of the 3′CBE extending more than a Mb into the VH domain on both Eµ-deficient and EF1αFor alleles (Fig. 3c, blue) whereas these interactions were comparable on WT and EF1αRev alleles (Fig. 3c, Supplementary Fig. 1e and Supplementary Data 2). We conclude that long-distance interactions of 3′CBE are facilitated by Eμ and EF1αRev, but not by EF1αFor. We hypothesize that this is, in part, regulated by antisense transcripts (directed away from 3′CBE) initiated at Eμ or EF1αRev.
Contributions of IGCR1
We previously showed that Eµ looped to the next available CTCF-binding site located near the most proximal VH gene segments in the absence of IGCR142,48. To systematically probe the contributions of IGCR1 to locus conformation we carried out anti-CTCF and anti-Rad21 Hi-ChIP in WT and IGCR1−/− pro-B cells (Fig. 4a). On WT alleles, IGCR1 contacts extended 5′ into the proximal VH region, revealing prominent interactions with a cluster of VH gene segments closest to the 3′ Igh domain (Fig. 4b, black boxes). In addition, IGCR1 interacted with Eμ and the 3′CBE. Sub-TADs were revealed on both sides of IGCR1, giving it all the hallmarks of a classical boundary element. Weaker interactions were evident between proximal VH genes and Eµ, perhaps reflecting alleles that had escaped Eµ sequestration by IGCR1 (Fig. 4b, red boxes). As previously shown using lower resolution assays, Eµ interacted strongly with proximal VH genes (VH5-4, VH2-2, VH5-2, and VH81X) (Fig. 4b, red boxes) in the absence of IGCR1, creating a 180 kb sub-TAD (Eµ-proximal VH). We surmise that multiple ‘corner dots’ (Fig. 4b, red boxes) in this sub-TAD reflect Eμ interactions with multiple VH gene segment and their associated CTCF-binding sites. Presumably, these interactions occur in different cells.

a Schematic depiction of the Igh locus indicating localization of IGCR1 (purple oval and black arrow) between DH gene segments and proximal VH genes. Small yellow ovals within IGCR1 denote two CTCF-binding sites oriented in opposite directions. A 4 kb region covering two CTCF-binding sites of IGCR1 was deleted with CRISPR/Cas9 technology and termed as the IGCR1−/− alleles. b, c CTCF and Rad21 Hi-ChIP analysis. Hi-ChIP profiles of CTCF and Rad21 are shown for the 3′ Igh domain (chr12: 113200001-113800000, mm10) (b) and the entire Igh locus (c) in WT alleles (left panel) and IGCR1-deficient alleles (middle panel). Heatmaps indicate normalized interaction frequencies. The 3′CBE-associated stripe is indicated by black arrows (b, c). Interactions between proximal VH (VH5-4, VH2-2, VH5-2, and VH81X) and Eμ or IGCR1 are indicated by red or black boxes, respectively (b). Two distinct sub-TAD regions, VH1-15-IGCR1 and VH1-15-VH81X, are marked by black and light blue triangles, respectively (c). VH1-15 is located at 5′ of the VH enhancer EVH210. The data are representative of two biological replicate experiments. Hi-ChIP data were visualized with software cooltools. Experiment was independently repeated twice with similar results. See also Supplementary Fig. 4, Data 1 and 4.
A stripe anchored at IGCR1 was lost in IGCR1-deficient alleles, the 3′CBE stripe was also reduced in the Eμ-proximal VH interval in the absence of IGCR1 (Fig. 4b, right, black arrow). Our interpretation is that formation of the Eμ-proximal VH sub-domain precludes interaction of these VH genes with 3′CBE. Beyond proximal VH genes, the 3′CBE stripe increased (Fig. 4b, c, black arrow, Supplementary Fig. 4a and Supplementary Data 4) in intensity reminiscent of stronger stripe formation on EF1αRev alleles. We propose that the common phenotype of IGCR1-deficient and EF1αRev alleles arises from loss of IGCR1-mediated impediment to cohesin extrusion on IGCR1-deficient alleles, and increased transcription-dependent extrusion on EF1αRev alleles.
A sub-TAD with multiple CTCF-associated looping interactions that encompassed about half the VH gene segments was maintained on IGCR1-deficient alleles (Fig. 4c left and middle, black triangles). However, its 3′ boundary shifted from IGCR1 to more diffuse locations within the proximal VH genes on IGCR1-deficient alleles (Fig. 4c, middle, blue triangles). Creation of the domain on WT alleles utilizing unidirectionally oriented CTCF-binding sites amongst VH genes, with one oppositely oriented site at IGCR1 is readily explained by current models of loop extrusion. However, its persistence in the absence of IGCR1 demonstrates (a) abundant CTCF-associated interactions between similarly oriented sites within this domain (Supplementary Fig. 4b, c) and (b) a 5′ domain boundary that is established by currently unknown mechanisms. Our observations highlight functional distinctions between CTCF-binding elements (such as IGCR1 versus proximal VH) and reveal their interaction with CTCF-lacking regulatory regions (such as Eµ) in the structural organization of Igh alleles.
Aberrant rearrangements on IGCR1-deficient alleles
Tampering with Eµ/IGCR1 interaction has functional consequences. Guo et al. noted premature VH to DH recombination in the absence of IGCR141. We found that VDH recombination occurred by either deletional or inversional mechanisms on IGCR1-mutated alleles (Supplementary Fig. 5a)40, leading us to consider the possible fates of such VDH recombined alleles that occur in the absence of IGCR1. Products of VH to DH rearrangements (VDH) by deletion or inversion retain functional DH-associated RSSs (Fig. 5a, b, labeled 2 and 4). Such alleles could rearrange by deletion to a JH RSS (producing VDJH recombined alleles) or by inversion to an upstream VH RSS generating aberrant VDVH recombined products (Fig. 5a, b, labeled 3 and 5). We probed for the latter possibility using genomic DNA from an IGCR1-deficient pro-B cell line (Fig. 5a) or bone marrow primary pro-B cells (Fig. 5b) as templates for PCR amplification. In both cases we detected the low level of predicted VDVH recombination products that were confirmed by cloning and sequencing (Supplementary Fig. 6a, b). We also obtained evidence for Igh alleles that carried two recombination products (VDH and DJH) on one allele, albeit with low efficiency (Fig. 5c, Supplementary Figs. 5a–c and 6c). We conclude that various forms of aberrant rearrangements reduce the efficiency of functional Igh rearrangements on IGCR1-deficient alleles.

a, b VDVH rearrangement on IGCR1-deficient alleles. VDVH rearrangements are observed from pro-B cell lines expressing Rag2 (a) or from bone marrow pro-B cells (b) in the context of IGCR1 deletion. VDVH rearrangements were analyzed using indicated primer pairs F1-F2 (a) and F3-F4 (b). VHQ52 and VH7183 denote distinct proximal VH gene families, housing 15 or 17 VH genes, respectively. VH2-2 and VH81X belong to VHQ52 and VH7183 gene families, respectively (also refer to Supplementary Fig. 6b). F1-F2 and F3-F4 indicate VH2-2 -DH-VH81X and VHQ52-DH-VH7183 rearrangements (purple circle), respectively. ROSA26 serves as the loading control. The data presented are representative of two biological replicate experiments. Experiment was independently repeated twice with similar results. c Both VDH and DJH rearrangements on the same IGCR1-deficient alleles. The 2F1 cell line, deficient in Rag2, harbors a DSP2.2a-JH2 rearrangement on one allele and a DQ52-JH1 rearrangement on the other allele (also refer to Supplementary Fig. 5b). VH81X recombined to the 5′ and 3′ ends of the DSP2 gene segment through inversion (labeled 2) or deletion (labeled 3) on IGCR1-deficient alleles with DJH rearrangement were analyzed using indicated primer pairs. Inversions (P1-P2) and deletions (P1-P3) of VH81X-DSP2 rearrangements are depicted in Rag1/2-infected WT or IGCR1-deficient 2F1 cell lines (red rectangle). VH81X is the first functional VH gene, while DSP2 (including DSP2.2a) and DQ52 are DH genes. ROSA26 serves as the loading control. Experiment was independently repeated twice with similar results. See also Supplementary Figs. 5 and 6, Data 6.
Contribution of 3′CBE
3′CBE is the third tether that configures the 3′ Igh domain43,53. Its deletion has a small effect on Igh rearrangments46 and partially reduces class switch recombination in mature B cells47. Chromatin structural roles of the 3′CBE have not been fully explored. We deleted or inverted a 12 kb 3′CBE region containing 10 CTCF-binding sites in pro-B cells (Fig. 6a, Supplementary Fig. 7a, b) and assayed the effects by anti-CTCF Hi-ChIP. Deleting 3′CBE (3′CBE−/−) reduced the associated stripe and interactions of the region with Eµ and IGCR1 (Fig. 6b). Instead, proximal VH genes (VH2-2, VH5-2, and VH81X), IGCR1 and Eµ interacted with appropriately oriented CTCF-bound sites further downstream (Fig. 6b, red rectangle), which correlated with increased Crip1 RNA levels (Fig. 6c).

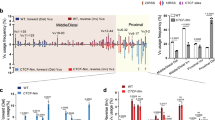
a Schematic map of Igh locus highlighting the presence of 3′CBE (denoted by a blend of blue and purple ovals). b CTCF Hi-ChIP profiles for the genomic interval chr12:112800001-113700000, mm10, are illustrated for WT alleles (left panel) and 3′CBE-deficient (3′CBE−/−) or inverted alleles (3′CBEInv #1) (middle panel). Heatmaps indicate normalized interaction frequencies. Interactions involving Eµ with 3′CBE (Eµ/3′CBE, black arrow) were quantified using CTCF Hi-ChIP data, as described in the “Methods” section, and presented after normalization to WT alleles as the mean, with each data point representing results from two independent experiments. VH2-2, VH5-2, and VH81X are proximal VH genes. Tmem121 and Crip1 are denoted by red arrows. The WT and inverted 3′CBE stripes are distinguished by green and orange arrows, respectively. The Hi-ChIP analysis of clone 3′CBE−/− and 3′CBEInv #1 are presented in the middle panel, whereas data from clone 3′CBEInv #2 is depicted in Supplementary Fig. 8a. The Experiment was independently repeated twice with similar results. Source data are provided as a Source Data file. c Transcriptional analysis of 3′CBE-deficient or inverted Igh alleles. Genome browser tracks showing CTCF ChIP-Seq and directional RNA-Seq data are presented for WT, 3′CBE−/−, and 3′CBEInv #1 and 3′CBEInv #2. CTCF ChIP-Seq data were extracted from CTCF Hi-ChIP experiments. On the right, RT-qPCR results from WT, deficient, or inverted 3′CBE Igh alleles are displayed. Two sets of Crip1 primers were utilized: Crip1 (a) and Crip1 (b). Sense transcript initiated at Eµ is used as a control. The data are presented as mean ± SEM from three independent experiments. CTCF ChIP-Seq and directional RNA-Seq experiments (left) were independently repeated twice with similar results. Source data are provided as a Source Data file. d CTCF Hi-ChIP profiles for the genomic interval chr12: 112500001-11680000, mm10, are provided for WT, 3′CBE-deficient, or 3′CBE-inverted alleles. The WT and inverted 3′CBE stripes are distinguished by green and orange arrows, respectively. The “+” indicates an extended configuration. Specifically, “A+” represents the extended TAD A. The Experiment was independently repeated twice with similar results. See also Supplementary Fig. 7, 8, Data 1, 5, 6.
Inverting 3′CBE generated a strong stripe away from the Igh locus and reduced the stripe into the locus (Fig. 6b, d, Supplementary Fig. 8a, b). A residual stripe into the Igh locus on 3′CBEInv alleles may reflect low level cohesin stalling at ‘incorrectly’ oriented CTCF sites. Inversion of 3′CBE also led to reduced interactions of Eµ with 3′CBE (Fig. 6b, bottom left and middle, Supplementary Fig. 8c and Supplementary Data 5). However, the inverted 3′CBE continued to act as a boundary element and prevented out-of-locus interactions of IGCR1 and Eµ with downstream genes. Accordingly, Crip1 gene expression was not much altered on 3′CBEInv alleles compared to WT alleles (Fig. 6c). Loss, but not inversion, of 3′CBE merged the 3′ sub-TAD A with the Igh locus to form a larger sub-TAD A+ that extended until IGCR1 (Fig. 6d). We conclude that orientation of CTCF-binding sites at the 3′CBE determines stripe orientation but not boundary formation.
Discussion
Genome-wide studies have revealed general principles of chromatin folding that include organization into compartments, TADs and loops57,58,59,60. Because folding achieves distinct functional outcomes at different loci, understanding how these forces cooperate to establish appropriate configurations must come from probing locus-specific mechanisms61,62. Here we carried out a systematic structure/function analysis of the Igh locus focusing on three regulatory elements (Eµ, IGCR1 and 3′CBE) in the 3′ Igh domain that regulate DNA recombination and transcription. We found that the CTCF-lacking enhancer Eµ serves as a boundary element within a CTCF bounded TAD and accentuates the stripe anchored by 3′CBE. By substituting the enhancer with an active promoter, we partially separated its role in sub-TAD formation from that in stripe extension. In contrast to Eμ, CTCF-binding IGCR1 attenuates the 3′CBE stripe, indicating mutually antagonistic effects of Eμ and IGCR1. Lastly, we show that direction of the 3′CBE stripe has a relatively minor role in its boundary function. Implications of these observations are discussed below.
Eμ plays an essential role in B lymphocyte development by regulating Igh gene assembly and expression. We propose that Eµ functions as a sub-TAD boundary by stalling cohesin extrusion. Unlike CTCF-mediated stalling sites, however, we did not observe cohesin accumulation at Eμ. We surmise this is because cohesin is efficiently removed from the genome by Eμ-associated WAPL. One mechanism by which the small (60 kb) 5′ sub-TAD forms is by blocking cohesin loaded between IGCR1 and Eμ by each regulatory element. Similarly, cohesin loaded between Eμ and 3′CBE stalls at these elements to generate the larger (200 kb) sub-TAD. This mechanism of establishing the structure of the 3′ Igh domain implies that Eμ is an orientation-independent extrusion stalling element. Alternatively, NIPBL accumulation at Eμ suggests it may recruit cohesin, thereby initiating loop extrusion bi-directionally until terminated by CTCF bound to IGCR1 and 3′CBE. The two models are not mutually exclusive, and sub-TAD formation on EF1α-substituted alleles indicates that Eμ is not essential for cohesin recruitment to the 3′Igh domain. While Eμ is necessary for processes like V(D)J recombination and cohesin stalling, our data do not address its sufficiency for these functions. Whether cohesin blocking ability is a property of all enhancers and promoters remains to be determined. Association of p300/CBP, a marker of many enhancers, with loop anchors suggests this is very plausible2,3. We hypothesize that enhancer/promoter communication between elements that lack proximal CTCF motifs, or have CTCF at only one location, may proceed similarly.
Eμ may block loop extrusion by interfering with cohesin progression because of the assembled nucleoprotein complex or by activating transcription. We demonstrate that replacing Eµ with various orientations of the EF1α promoter recapitulates some aspects of Eµ function. This includes features such as H3K4me3 and H3K27ac modifications, transcriptional activity, the delineation of neighboring sub-TADs, and interactions with 3′CBE. However, the EF1α promoter does not facilitate VDJ recombination, highlighting a distinct and irreplaceable role for Eµ in B cell development. Based on the observation that replacing Eμ with EF1α promoter in either orientation restored sub-TAD structure to the 3′ Igh domain, we propose that nucleoprotein complex formation is sufficient to stall cohesin movement. Such a complex could involve only enhancer binding proteins and associated co-factors or include RNA polymerase II as recently proposed32. We note some caveats with this interpretation. First, we cannot rule out that RNA synthesis by RNA polymerase II is a part of the blocking mechanism63,64,65,66. Active transcripts would be initiated irrespective of promoter orientation in our enhancer/promoter swap experiments. Second, though each orientation of the EF1α promoter drives strong transcription in the expected direction, it is possible that low levels of transcripts in the opposite direction are sufficient to block extrusion. Nevertheless, our observations support the idea that enhancers and promoters can gain proximity by stalling loop extrusion.
Architectural stripes detected by Hi-C remain an enigmatic feature of chromatin structure. Their visualization represents higher-than-background interaction of a limited genomic region (the stripe anchor) with many sites across a larger region. In highlighting this feature, Vian et al. proposed that stripes occur when the cohesin complex loads near CTCF-binding sites, such that extrusion is prevented in one direction18. Unidirectional extrusion in the other direction thus ‘reels in’ sequences that contact the stripe anchor and are revealed as a Hi-C stripe. Only parts of this provocative model have been experimentally tested. By investigating stripe formation with several genetically altered Igh alleles, we added several insights into mechanisms by which a stripe is generated and its functions. First, we experimentally validated the prediction that orientation of CTCF sites at the anchor determines stripe direction. Concurrently, we discovered that stripe direction contributed little to boundary functions of the associated CTCF sites. Second, we demonstrated that transcriptional orientation impacts stripe intensity. Cohesin movements have been previously connected with RNA polymerase activity32. Our analysis of EF1α promoter substituted Igh alleles directly demonstrates that transcriptional orientation, but not the enhancer/promoter-associated nucleoprotein complexes, regulate stripe intensity. Third, analysis of IGCR1-deficient alleles revealed that stripe intensity could be modulated to varying degrees along its length. Specifically, the 3′CBE stripe was strengthened beyond proximal VHs but weakened between Eμ and proximal VH gene segments on IGCR1-deficient alleles. We reasoned that diminution of the stripe in precisely the region (proximal VH-Eµ) that is marked by strong corner dots (Fig. 4b, red rectangle) on IGCR1-deficient alleles suggests that looped domains serve as barriers to stripe formation. In the model of stripe anchors serving as unidirectional extrusion initiators, it is difficult to explain the weakening of a stripe near the anchor and its strengthening several hundred kbs away. We hypothesize that stripe anchors might also be strong terminators of extrusion initiated variably across a genomic region. In this view, the absence of IGCR1 may permit a greater proportion of extrusion events initiated in the 5′ region of the locus to progress until 3′CBE on alleles that lack Eμ-proximal VH interactions. The extent to which our observations are generalizable genome-wide remains to be determined.
Methods
Detailed methods were expanded in the Supplementary Information.
Cell lines
WT, RAG2−/−, IGCR1−/−(1), IGCR1−/−(2) were kindly provided by David G. Schatz (Yale University) and Frederick W. Alt (Harvard Medical School), respectively. IGCR1−/− and 2F1 were from our lab as previously described42. These cell lines were Abelson virus-transformed pro-B cell line and cultured in RPMI medium with 10% fetal bovine serum (FBS), 1× MEM non-essential amino acids solution, and 1 × 2-Mercaptoethanol at 37 °C in a 5% CO2 humidified atmosphere. RAG2−/−, 2F1, IGCR1 Δ(#1), IGCR1 Δ(#2), IGCR1−/−(1), IGCR1−/−(2) pro-B cell lines were generated from mouse strain 129/Sv with endogenous RAG2 deficiency. 2F1 pro-B cell line was derived from RAG2−/− pro-B cell line and contained DQ52-to-JH1 rearrangement in one allele and DSP2.2a-to-JH2 rearrangement in the second by transient transfection of a RAG2 expression vector. IGCR1 Δ(#1) and IGCR1 Δ(#2) were derived from 2F1 with the deletion of IGCR1by CRISPR-Cas9 system, described below. IGCR1−/−(1) and IGCR1−/−(2) pro-B cell lines carried mutation of two CTCF-binding elements at the IGCR1 locus. WT pro-B cell line and its derivative cell lines IGCR1−/−, Eµ−/− #1, Eµ−/− #2, EF1αFor #1, EF1αFor #2, EF1αRev #1, EF1αRev #2, 3′CBE−/−, 3′CBEInv #1 and 3′CBEInv #2 expressed a catalytically inactive RAG1 together with endogenous RAG2 from mouse strain C57BL/6 J. IGCR1−/− (IGCR1 deletion), Eµ−/− #1 (Eµ deletion), Eµ−/− #2 (Eµ deletion), EF1αFor #1 (replacement of Eµ with forward orientation of EF1α promoter), EF1αFor #2 (replacement of Eµ with forward orientation of EF1α promoter), EF1αRev #1 (replacement of Eµ with reverse orientation of EF1α promoter), EF1αRev #2 (replacement of Eµ with reverse orientation of EF1α promoter), 3′CBE−/− (3′CBE deletion), 3′CBEInv #1 (3′CBE inversion) and 3′CBEInv #2 (3′CBE inversion) were generated with CRISPR-Cas9 system, described below.
Plasmids
gRNAs were cloned into lentiCRISPR v2 (Addgene, #52961) or pSpCas9(BB)-2A-GFP (PX458) (Addgene, # 48138) according to Feng Zhang (Massachusetts Institute of Technology) lab protocol. Forward or reverse EF1α promoter donor pGEM®-T Vector: These two donor plasmids were generated with two different steps. Step one: 4.3 kb DNA fragment covering Eµ was inserted into pGEM®-T Vector (Promega, #A3600). Step two: 1.2 kb EF1α promoter DNA fragment was amplified from plasmid pHIV-Zsgreen (Addgene, #18121). Eµ DNA was replaced with EF1α promoter DNA. The plasmid sequenced was confirmed by Sanger Sequencing provided by Azenta Life Sciences.
Antibodies
Antibodies information were listed below: Anti-trimethyl-Histone H3 (Lys4) antibody from MilliporeSigma (#07-473) (2 µl per 1 million cells), histone H3K27ac antibody from Active Motif (#39133) (2 µl per 1 million cells), anti-CTCF antibody from Abcam (#ab70303) (4 µl per 1 million cells for ChIP-Seq and 2.5 µl per 5 million cells for Hi-ChIP), anti-Rad21 antibody from Abcam (#ab992) (2 µl per 1 million cells for ChIP-Seq and 2.5 µl per 5 million cells for Hi-ChIP), anti-NIPBL antibody from Bethyl Laboratories (#A301-779A) (4 µl per 2 million cells) and anti-WAPL antibody from Proteintech (#16370-1-AP) (4 µl per 2 million cells).
Pro-B cell lines generation
Eµ−/− #1, Eµ−/− #2, IGCR1 Δ(#1) and IGCR1 Δ(#2): These pro-B cell lines were generated with lentiviral CRISPR-Cas9 systems as previously described40,42. EF1αFor #1, EF1αFor #2, EF1αRev #1, EF1αRev #2, 3′CBE−/−, 3′CBEInv #1, and 3′CBEInv #2: These pro-B cell lines were generated with the electroporation method. These pro-B cell lines were confirmed by PCR, southern blot, and high throughput sequencing.
Capture Hi-C
The capture Hi-C was carried out as previously described (Qiu et al.40). Hi-C was performed using the Arima Hi-C Kit (Arima Genetics Inc. Cat#A510008), and for each sample, 1 × 106 cells were used. Hi-C libraries were hybridized to Igh probes (mm10, chr12: 113,201,001–116,030,000,) designed and manufactured by Agilent (Agilent Technologies Inc.) as specified by the manufacturer. Libraries were uniquely barcoded and pooled to be sequenced on an Illumina NovaSeq instrument (2 × 150) to a depth of 100–200 million reads. See Supplementary Data 1.
Hi-ChIP
Hi-ChIP was performed using the Arima Hi-C+ Kit (Arima Genetics Inc. Cat# A101020) precisely following the manufacturer’s protocols (Hi-ChIP, Arima-Hi-C+ documents A160168 v00 and library preparation, A160169 v00). For each sample, 5 × 106 cells were used. Libraries were uniquely barcoded and pooled to be sequenced on an Illumina NovaSeq instrument to an average depth of 200 million reads. See Supplementary Data 1.
ChIP-seq
1 × 106 cells were cross-linked using 1% formaldehyde (Sigma) by adding 27 µl from a 37% stock buffer and incubating for 10 min at room temperature, quenched with 125 mM of glycine and lysed in lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris-HCl, PH 8). Chromatin was sonicated with Bioruptor (Diagenode, 30 s on/ 30 s off, 15 min on time) and diluted 10× with dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris-HCl, PH 8.0, 167 mM NaCl), and followed by immunoprecipitation with specific antibodies overnight at 4 °C. Add 30 µl magnetic protein A beads and incubate 2 h at 4 °C. Beads bound chromatin was washed with one time of low salt buffer (0.1% SDS, 1% Triton X-100, 150 mM NaCl, 2 mM EDTA, 20 mM Tris-HCl pH 8.0), high salt buffer (0.1% SDS, 1% Triton X-100, 500 mM NaCl, 2 mM EDTA, 20 mM Tris-HCl pH 8.0), LiCl buffer (0.25 M LiCl, 1% NP-40, 1% sodium Deoxycholate, 1 mM EDTA and 10 mM Tris-Cl, PH 8) and two times of TE buffer (10 mM Tris-HCl). After the last wash, resuspend the beads in 109 µl of TE, add 6 µl of 10% SDS, 5 µl of 5 M NaCl and incubate at 65 °C for 4 h. Then add 2.5 µl of 20 mg/ml proteinase K and incubate at 55 °C for 2 h. Isolate DNA using AMPure XP beads according to the manual.
For ChIP experiments targeting NIPBL (antibody: Bethyl A301-779A) and WAPL (antibody: Proteintech 16370-1-ap), we adapted the protocol from Bossen et al.67, with a few modifications. Two million pro-B cells were initially cross-linked with 1.5 mM EGS at room temperature for 15 min with continuous rotation. This was followed by fixation with 1% formaldehyde for 15 min at room temperature, also with rotation, and quenched using 200 mM glycine. After washing twice with PBS, cells were then resuspended in a buffer containing 1% SDS and incubated for 10 min on ice before sonication. Please note we extended the shearing time to a total of 20 min. Other steps followed the original protocol without further deviations.
The ChIP-seq libraries were prepared using the TruSeq Nano DNA Low Throughput Library Prep Kit (Illumina, Cat#20015964) according to the manufacturer’s protocol. Samples were indexed using the TruSeq DNA Single Indexes Set A (Illumina, Cat#20015960) and pooled for sequencing.
Genomic DNA-free RNA extraction
Genomic DNA free RNA was extracted from pro-B cell lines with RNeasy Plus Mini Kit (Qiagen, Cat# 74134) and RNase-Free DNase (Qiagen, Cat# 79254). 4 × 106 cells were used.
RT-qPCR
Genomic DNA free RNA was extracted from pro-B cell lines WT, Eµ−/− #1, EF1αFor #1, EF1αFor #2, EF1αRev #1, EF1αRev #2 as above described. 1 µg RNA was used to generate cDNA with SuperScript™ IV First-Strand Synthesis System (Thermo Fisher Scientific, # 18091050) with random hexamers according to the manufacturer protocol. Approximately 1/40 of the reverse-transcription-generated cDNA was analyzed with iTaq Universal SYBR. Primers that were used for PCR are provided in Supplementary Data 6. Normalization was performed with two different normalizations. First step was to normalize with internal control GAPDH. Second step was to normalize with the WT pro-B cell line.
Directional RNA-Seq
Directional RNA-Seq experiments were performed with two steps: rRNA depletion (PerkinElmer, Cat# NOVA-512961) and rapid directional RNA-Seq (PerkinElmer, Cat# NOVA-5198-01) according to manufacturer protocol. 1 µg total RNA was used for rRNA depletion. Checked RNA pattern before and after rRNA depletion with High Sensitivity RNA ScreenTape Analysis (Agilent Technologies). rRNA depleted RNA with RIN score higher than 9 was used for directional RNA-Seq. Quantity DNA concentration with Qubit™ 1× dsDNA High Sensitivity (HS) and Broad Range (BR) Assay Kits. Examine libraries with High Sensitivity DNA ScreenTape Analysis. Libraries were sequenced with NovaSeq SP flow cell 2 × 150 bp. See Supplementary Data 1.
RNA-free genomic DNA extraction
RNA-free genomic DNA was extracted from mouse total bone marrow or pro-B cell lines with DNeasy Blood & Tissue Kit (Qiagen, # 69504) and RNase A (Qiagen, # 19101).
DJH recombination
WT, Eµ−/− #1, EF1αFor #1, EF1αFor #2, EF1αRev #1 and EF1αRev #2 were infected with lentivirus containing plasmid pHIV-RAG1-IRES-puro. 2F1, IGCR1 Δ(#1), and IGCR1 Δ(#2) were infected with lentivirus containing dual plasmid pHIV-RAG2-IRES-puro and pHIV-RAG1-IRES-BFP. Lentivirus containing RAG2 infected RAG2−/−, IGCR1−/−(1) and IGCR1−/−(2) were described before42. Lentivirus preparation was described as above. Pro-B cell lines were infected with fresh lentivirus containing RAG1 or along with RAG2. BFP or puromycin positive cells were harvested for fluorescence-activated cell sorting (FACS) analysis and genomic DNA isolation on day 28 after infection. Fivefold serial dilutions of genomic DNA (200 ng, 40 ng, 8 ng) were used to perform PCR to analyze DJH rearrangements. Primers used in this assay were listed in Supplementary Data 6. Primers flanking the ROSA26 gene were used as a loading control under the same conditions.
Bioinformatic analysis
Capture Hi-C and Hi-ChIP analysis
Capture Hi-C and Hi-ChIP reads were aligned to the mm10 genome and processed using Juicer1 to generate.hic files. Juicer was run with the flags “-g mm10 -s Arima” for WT experiments. For mutant experiments, mutant genomes created by editing the mm10 genome sequence FASTA file by replacing the 743 bp region chr12:113427140-113427882 with the forward or reverse EF1α promoter sequence and deleting the 436 bp region chr12: 113427883-113428318. Mutant genome FASTA files were provided to Juicer using the “-z” flag when processing reads from mutant experiments. To compare WT and mutant experiments, capture Hi-C and Hi-ChIP data were normalized in two ways: (1) by down-sampling each experiment to the same number of total contacts and (2) by down-sampling each experiment to the same number of contacts within the Igh locus.
For Capture Hi-C experiments, contacts between regions within 2000 bp were removed prior to down-sampling and heatmaps were balanced using the “balance” command from Cooler68. Difference maps were calculated by subtracting the WT contacts from the mutant contacts for both Capture Hi-C and Hi-ChIP. Capture Hi-C heatmaps were visualized using cooltools and are shown in log scale with 5000 bp resolution bins (Figs. 1b, c, 3a, b, Supplementary Figs. 1c, d and 3a, b). For Hi-ChIP experiments, contacts within 2000 bp were removed in some cases (Fig. 6b, d, Supplementary Fig. 6c, d) and not in others (Fig. 4b, c). Hi-ChIP heatmaps were visualized using cooltools and are shown in linear scale with 5000 bp (Figs. 4b, 6b and Supplementary Fig. 6c) and 10,000 bp (Figs. 4c, 6d, Supplementary Fig. 1d and 6d) resolution bins. Detailed information is listed at Supplementary Data 1.
3′CBE stripe calculation
The coordinates for the 3′CBE stripe anchor location are chr12:113215304-113225303. For cHi-C data (Figs. 1 and 3), the VH locus (chr12:113578001-115178000) was segmented into four adjacent 600 kb segments, with the read counts for each segment being calculated. Subsequently, the ratios of reads for the Eµ-deficient and EF1α promoter replacement conditions were determined in relation to WT (Supplementary Fig. 1e). For CTCF or Rad21 Hi-ChIP (Fig. 4), The read counts of the VH locus (chr12:113628001-115178000), excluding the proximal VH region, were summed (Supplementary Fig. 4a). Counts number information is listed in Supplementary Data 2 and 4.
Loop quantification
To quantify the interaction frequency for loops between Eµ/EF1α, 3′CBE and IGCR1 in capture Hi-C (Fig. 3a) and Hi-ChIP (Fig. 6b) experiments, the number of contacts between these regions was counted after the experiments were down-sampled to the same number of contacts. The following mm10 coordinates were used to quantify interactions: Eµ/EF1α chr12:113426062-113429061, 3′CBE chr12:113215304-113225303, and IGCR1 chr12:113484501-113488500. Reads number was listed in Supplementary Data 3 and 5.
Virtual 4C analysis
To perform virtual 4C, capture Hi-C data were first converted from .hic to .mcool format using hic2cool (https://github.com/4dn-dcic/hic2cool). Capture Hi-C contacts were binned at 10 kb resolution and the number of contacts between each 10 kb bin and the 3′CBE (defined as the region chr12:113,210,000–113,220,0000) was calculated using Cooler68. To compare mutant data with WT data, the log2 ratio was calculated for each 10 kb bin. Virtual 4C data were visualized using IGV69.
ChIP-seq analysis
ChIP-seq reads were aligned to the mm10 genome using bwa-mem2. For mutant experiments, a bwa-mem2 index was created for each mutant genome sequence and bwa-mem2 was used to align reads to the mutant genome. Aligned reads were converted to bigWig format using deepTools bamCoverage with the flags “—normalizeUsing CPM –ignoreDuplicates –minMappingQuality 30 –binSize 1”. To compare single-end and paired-end ChIP-seq experiments, paired-end reads were processed as single-end reads. To compare ChIP-seq experiments with different read lengths, reads were trimmed to 61 bp using trimmoatic70. ChIP-seq data were visualized using IGV69. CTCF and Rad21 Hi-ChIP reads were also processed using this approach to generate ChIP-seq–like tracks that were visualized with IGV. Detailed information is listed at Supplementary Data 1.
RNA-seq analysis
RNA-seq reads were aligned to the mm10 genome using STAR71. Aligned reads were converted to bigwig format and filtered by strand orientation using deepTools72 bamCoverage with the flags “—normalizeUsing CPM —filterRNAstrand forward” or “—normalizeUsing CPM –filterRNAstrand reverse”. RNA-seq data were visualized using IGV69. Detailed information is listed at Supplementary Data 1.
Statistical analysis
Statistical analysis was performed using t-test. Significance levels are indicated by ‘**’ for p < 0.01 and ‘ns’ for p > 0.05.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data supporting the findings of this study are available from the corresponding authors upon request. All genome-wide datasets generated in this study have been deposited in the NCBI database under Gene Expression Omnibus portal (GSE241661 [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE241661], GSE241667 [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE241667], GSE241668 [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE241668], GSE241740 [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE241740], GSE280719 [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE280719]). Source data for the figures and Supplementary Figures are provided as a Source Data file. Source data are provided with this paper.
References
Anderson, E. C. & Nora, E. P. Setting new boundaries with transcription and CTCF. Nat. Genet. 52, 1003–1004 (2020).
Beagan, J. A. & Phillips-Cremins, J. E. On the existence and functionality of topologically associating domains. Nat. Genet. 52, 8–16 (2020).
Haws, S. A., Simandi, Z., Barnett, R. J. & Phillips-Cremins, J. E. 3D genome, on repeat: higher-order folding principles of the heterochromatinized repetitive genome. Cell 185, 2690–2707 (2022).
Ortabozkoyun, H. et al. Members of an array of zinc-finger proteins specify distinct chromatin boundaries. Mol. Cell. 84 https://doi.org/10.1016/j.molcel.2024.08.007 (2024).
Davidson, I. F. et al. CTCF is a DNA-tension-dependent barrier to cohesin-mediated loop extrusion. Nature 616, 822–827 (2023).
Zhang, H. et al. Chromatin structure dynamics during the mitosis-to-G1 phase transition. Nature 576, 158–162 (2019).
Wulfridge, P. et al. G-quadruplexes associated with R-loops promote CTCF binding. Mol. Cell https://doi.org/10.1016/j.molcel.2023.07.009 (2023).
Zhang, H. et al. CTCF and R-loops are boundaries of cohesin-mediated DNA looping. Mol. Cell 83, 2856–2871.e2858 (2023).
Barajas-Mora, E. M. et al. Enhancer-instructed epigenetic landscape and chromatin compartmentalization dictate a primary antibody repertoire protective against specific bacterial pathogens. Nat. Immunol. 24, 320–336 (2023).
Bhat, K. H. et al. An Igh distal enhancer modulates antigen receptor diversity by determining locus conformation. Nat. Commun. 14, 1225 (2023).
Furlong, E. E. M. & Levine, M. Developmental enhancers and chromosome topology. Science 361, 1341–1345 (2018).
Kvon, E. Z., Waymack, R., Gad, M. & Wunderlich, Z. Enhancer redundancy in development and disease. Nat. Rev. Genet. 22, 324–336 (2021).
Field, A. & Adelman, K. Evaluating enhancer function and transcription. Annu. Rev. Biochem. 89, 213–234 (2020).
Robson, M. I., Ringel, A. R. & Mundlos, S. Regulatory landscaping: how enhancer-promoter communication is sculpted in 3D. Mol. Cell 74, 1110–1122 (2019).
Oh, S. et al. Enhancer release and retargeting activates disease-susceptibility genes. Nature 595, 735–740 (2021).
Hsieh, T. S. et al. Enhancer-promoter interactions and transcription are largely maintained upon acute loss of CTCF, cohesin, WAPL or YY1. Nat. Genet. 54, 1919–1932 (2022).
de Wit, E. & Nora, E. P. New insights into genome folding by loop extrusion from inducible degron technologies. Nat. Rev. Genet. 24, 73–85 (2023).
Vian, L. et al. The energetics and physiological impact of cohesin extrusion. Cell 175, 292–294 (2018).
Kraft, K. et al. Serial genomic inversions induce tissue-specific architectural stripes, gene misexpression and congenital malformations. Nat. Cell Biol. 21, 305–310 (2019).
Huang, H. et al. CTCF mediates dosage- and sequence-context-dependent transcriptional insulation by forming local chromatin domains. Nat. Genet. 53, 1064–1074 (2021).
Batut, P. J. et al. Genome organization controls transcriptional dynamics during development. Science 375, 566–570 (2022).
Davidson, I. F. & Peters, J. M. Genome folding through loop extrusion by SMC complexes. Nat. Rev. Mol. Cell Biol. 22, 445–464 (2021).
Oldenkamp, R. & Rowland, B. D. A walk through the SMC cycle: from catching DNAs to shaping the genome. Mol. Cell 82, 1616–1630 (2022).
Hoencamp, C. & Rowland, B. D. Genome control by SMC complexes. Nat. Rev. Mol. Cell Biol. https://doi.org/10.1038/s41580-023-00609-8 (2023).
Gabriele, M. et al. Dynamics of CTCF- and cohesin-mediated chromatin looping revealed by live-cell imaging. Science 376, 496–501 (2022).
Rowley, M. J. & Corces, V. G. Organizational principles of 3D genome architecture. Nat. Rev. Genet. 19, 789–800 (2018).
Liu, Y. & Dekker, J. CTCF-CTCF loops and intra-TAD interactions show differential dependence on cohesin ring integrity. Nat. Cell Biol. 24, 1516–1527 (2022).
Rekaik, H. et al. Sequential and directional insulation by conserved CTCF sites underlies the Hox timer in stembryos. Nat. Genet. 55, 1164–1175 (2023).
Chen, H., Tian, Y., Shu, W., Bo, X. & Wang, S. Comprehensive identification and annotation of cell type-specific and ubiquitous CTCF-binding sites in the human genome. PLoS ONE 7, e41374 (2012).
Schoenfelder, S. & Fraser, P. Long-range enhancer-promoter contacts in gene expression control. Nat. Rev. Genet. 20, 437–455 (2019).
Blobel, G. A., Higgs, D. R., Mitchell, J. A., Notani, D. & Young, R. A. Testing the super-enhancer concept. Nat. Rev. Genet. 22, 749–755 (2021).
Banigan, E. J. et al. Transcription shapes 3D chromatin organization by interacting with loop extrusion. Proc. Natl Acad. Sci. USA 120, e2210480120 (2023).
Liu, C., Zhang, Y., Liu, C. C. & Schatz, D. G. Structural insights into the evolution of the RAG recombinase. Nat. Rev. Immunol. 22, 353–370 (2022).
Nair, L. et al. Mechanism of noncoding RNA-associated N(6)-methyladenosine recognition by an RNA processing complex during IgH DNA recombination. Mol. Cell 81, 3949–3964.e3947 (2021).
Kumari, G. & Sen, R. Chromatin interactions in the control of immunoglobulin heavy chain gene assembly. Adv. Immunol. 128, 41–92 (2015).
Banerji, J., Olson, L. & Schaffner, W. A lymphocyte-specific cellular enhancer is located downstream of the joining region in immunoglobulin heavy chain genes. Cell 33, 729–740 (1983).
Perlot, T., Alt, F. W., Bassing, C. H., Suh, H. & Pinaud, E. Elucidation of IgH intronic enhancer functions via germ-line deletion. Proc. Natl Acad. Sci. USA 102, 14362–14367 (2005).
Afshar, R., Pierce, S., Bolland, D. J., Corcoran, A. & Oltz, E. M. Regulation of IgH gene assembly: role of the intronic enhancer and 5’DQ52 region in targeting DHJH recombination. J. Immunol. 176, 2439–2447 (2006).
Subrahmanyam, R. et al. Localized epigenetic changes induced by DH recombination restricts recombinase to DJH junctions. Nat. Immunol. 13, 1205–1212 (2012).
Qiu, X. et al. Altered 3D chromatin structure permits inversional recombination at the IgH locus. Sci. Adv. 6, eaaz8850 (2020).
Guo, C. et al. CTCF-binding elements mediate control of V(D)J recombination. Nature 477, 424–430 (2011).
Qiu, X. et al. Sequential enhancer sequestration dysregulates recombination center formation at the IgH Locus. Mol. Cell 70, 21–33.e26 (2018).
Ba, Z. et al. CTCF orchestrates long-range cohesin-driven V(D)J recombinational scanning. Nature 586, 305–310 (2020).
Hill, L. et al. Wapl repression by Pax5 promotes V gene recombination by Igh loop extrusion. Nature 584, 142–147 (2020).
Ma, F. et al. Three-dimensional chromatin reorganization regulates B cell development during ageing. Nat. Cell Biol. 26, 991–1002 (2024).
Liang, Z. et al. Contribution of the IGCR1 regulatory element and the 3’Igh CTCF-binding elements to regulation of Igh V(D)J recombination. Proc. Natl. Acad. Sci. USA 120, e2306564120 (2023).
Zhang, X., Yoon, H. S., Chapdelaine-Williams, A. M., Kyritsis, N. & Alt, F. W. Physiological role of the 3’IgH CBEs super-anchor in antibody class switching. Proc. Natl. Acad. Sci. USA 118, e2024392118 (2021).
Jain, S., Ba, Z., Zhang, Y., Dai, H. Q. & Alt, F. W. CTCF-binding elements mediate accessibility of RAG substrates during chromatin scanning. Cell 174, 102–116.e114 (2018).
Hu, J. et al. Chromosomal loop domains direct the recombination of antigen receptor genes. Cell 163, 947–959 (2015).
Zhang, Y. W. et al. Molecular basis for differential versus V(D)J joining mechanisms. Nature https://doi.org/10.1038/s41586-024-07477-y (2024).
Zhang, Y. et al. The fundamental role of chromatin loop extrusion in physiological V(D)J recombination. Nature 573, 600 (2019).
Zhang, X. F. et al. Fundamental roles of chromatin loop extrusion in antibody class switching. Nature 575, 385 (2019).
Dai, H. Q. et al. Loop extrusion mediates physiological Igh locus contraction for RAG scanning. Nature 590, 338–343 (2021).
Zuin, J. et al. Nonlinear control of transcription through enhancer-promoter interactions. Nature 604, 571–577 (2022).
Balsalobre, A. & Drouin, J. Pioneer factors as master regulators of the epigenome and cell fate. Nat. Rev. Mol. Cell Biol. 23, 449–464 (2022).
Bolland, D. J. et al. Antisense intergenic transcription precedes Igh D-to-J recombination and is controlled by the intronic enhancer Emu. Mol. Cell Biol. 27, 5523–5533 (2007).
Zhang, D. et al. Alteration of genome folding via contact domain boundary insertion. Nat. Genet. 52, 1076–1087 (2020).
Emerson, D. J. et al. Cohesin-mediated loop anchors confine the locations of human replication origins. Nature 606, 812–819 (2022).
Dequeker, B. J. H. et al. MCM complexes are barriers that restrict cohesin-mediated loop extrusion. Nature 606, 197–203 (2022).
Xu, J. et al. Subtype-specific 3D genome alteration in acute myeloid leukaemia. Nature 611, 387–398 (2022).
Chen, L. F. et al. Structural elements promote architectural stripe formation and facilitate ultra-long-range gene regulation at a human disease locus. Mol. Cell 83, 1446–1461.e1446 (2023).
Ortabozkoyun, H. et al. CRISPR and biochemical screens identify MAZ as a cofactor in CTCF-mediated insulation at Hox clusters. Nat. Genet. 54, 202–212 (2022).
Zhang, S., Ubelmesser, N., Barbieri, M. & Papantonis, A. Enhancer-promoter contact formation requires RNAPII and antagonizes loop extrusion. Nat. Genet. 55, 832–840 (2023).
Barshad, G. et al. RNA polymerase II dynamics shape enhancer-promoter interactions. Nat. Genet. 55, 1370–1380 (2023).
Canzio, D. et al. Antisense lncRNA transcription mediates DNA demethylation to drive stochastic protocadherin alpha promoter choice. Cell 177, 639–653.e615 (2019).
Oh, H. J. et al. Jpx RNA regulates CTCF anchor site selection and formation of chromosome loops. Cell 184, 6157–6173.e6124 (2021).
Bossen, C. et al. The chromatin remodeler Brg1 activates enhancer repertoires to establish B cell identity and modulate cell growth. Nat. Immunol. 16, 775–784 (2015).
Abdennur, N. & Mirny, L. A. Cooler: scalable storage for Hi-C data and other genomically labeled arrays. Bioinformatics 36, 311–316 (2020).
Robinson, J. T. et al. Integrative genomics viewer. Nat. Biotechnol. 29, 24–26 (2011).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Ramirez, F. et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165 (2016).
Acknowledgements
We thank Frederick W. Alt (Harvard Medical School) for generously providing IGCR1-mutated cell lines and sorted bone marrow pro-B cells from IGCR1-deficient mice. This research was supported by the Intramural Research Program of the National Institute on Aging.
Author information
Authors and Affiliations
Contributions
X.Q., F.M., N.O., and R.S. conceived and designed the research. X.Q., F.M., H.D., F.Z.B., N.C., and A.H.B. generated reagents and conducted experiment design and execution, data collection and data analysis. N.O., J.F., and D.S. performed bioinformatics analysis. C.D. and C.N. performed FACS sorting. X.Q., F.M., N.O., and R.S. interpreted the data and results, and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Patrick Collins and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ma, F., Ollikainen, N., Du, H. et al. Interplay between CTCF-binding and CTCF-lacking regulatory elements in generating an architectural stripe at the Igh locus. Nat Commun 16, 2148 (2025). https://doi.org/10.1038/s41467-025-57373-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-57373-w
