
Abstract
Membrane contact sites between organelles are critical for the transfer of biomolecules. Lipid droplets store fatty acids and form contacts with mitochondria, which regulate fatty acid oxidation and adenosine triphosphate production. Protein compartmentalization at lipid droplet-mitochondria contact sites and their effects on biological processes are poorly described. Using proximity-dependent biotinylation methods, we identify 71 proteins at lipid droplet-mitochondria contact sites, including a multimeric complex containing extended synaptotagmin (ESYT) 1, ESYT2, and VAMP Associated Protein B and C (VAPB). High resolution imaging confirms localization of this complex at the interface of lipid droplet-mitochondria-endoplasmic reticulum where it likely transfers fatty acids to enable β-oxidation. Deletion of ESYT1, ESYT2 or VAPB limits lipid droplet-derived fatty acid oxidation, resulting in depletion of tricarboxylic acid cycle metabolites, remodeling of the cellular lipidome, and induction of lipotoxic stress. These findings were recapitulated in Esyt1 and Esyt2 deficient mice. Our study uncovers a fundamental mechanism that is required for lipid droplet-derived fatty acid oxidation and cellular lipid homeostasis, with implications for metabolic diseases and survival.
Similar content being viewed by others

Introduction
Lipid droplets (LDs) and mitochondria are critical regulators of lipid homeostasis and ATP production in eukaryotic cells1. LDs are dynamic organelles that store lipids as metabolic fuels and release fatty acids in times of energy demand via lipolysis. This is a highly regulated process involving a complement of proteins that temporarily transit or permanently reside on the surface of LDs2,3. The primary function of mitochondria is to meet the energy demands of cells by providing ATP through oxidative phosphorylation of metabolic substrates, including fatty acids. Both organelles are important for human health, as mutations in genes encoding LD and mitochondrial proteins are linked to metabolic diseases such as non-alcoholic fatty liver disease (e.g., PNPLA3, TM6SF2, MBOAT7, HSD17B13)4,5,6, type 2 diabetes (e.g., NDUFC2, COX7A2, HSL, CIDEA/B/C)7,8,9,10, lipodystrophies (e.g., PLIN1, BSCL1/AGPAT2, BSCL2/seipin, MFN2, CGI-58)11,12,13,14 and autosomal recessive fatty acid oxidation disorders (e.g., CPT1A, HADHA, ACADVL)15,16,17.
The interaction between LDs and mitochondria are increased during times of heightened energy demand, such as β3-adrenergic stimulation18,19 or exercise20, and during states of increased reliance on fatty acid oxidation, such as starvation21,22 and cold adaptation23,24. This has led to the view that close LD-mitochondria contact is essential for efficient β-oxidation and protection against the accumulation of cytosolic fatty acids and cellular lipotoxicity. In agreement with this, extensive LD and mitochondria contact is evident in metabolically active tissues including skeletal muscle25, heart26, liver27 and brown adipose tissue23,28 and proteins including PLIN523,29, SNAP2330, FATP4 (ACSVL4)31, VPS13C32, VPS13D33, and Rab8a34 appear to mediate LD-mitochondria interactions.
Although the importance of these organelles and their interaction is well recognized, we know little about the molecular machinery and processes by which fatty acids stored within LDs are transferred to mitochondria for their eventual oxidation. Protein-mediated lipid transport routes have been implicated in trafficking fatty acids at the membrane contact site of LDs and mitochondria and these proteins possess several important features including the capacity to extract lipids from a donor membrane; to shield lipids from the aqueous cytosolic environment, typically within their hydrophobic pockets; and to deliver lipids to a target membrane35. A recent study indicates that VPS13D meets these criteria and can facilitate fatty acid transfer at LD-mitochondria contact sites by mediating ESCRT-dependent remodeling of LD membranes33. While the details of proteins residing in the membrane contact sites of LDs and mitochondria are beginning to emerge, an unbiased inventory of proteins that reside at, or are temporarily recruited to these contact sites, is necessary to better understand this critical pathway of fatty acid trafficking and lipid metabolism.
Herein, we sought to unravel the mechanism of fatty acid transfer from their site of storage to their site of oxidation. Starting with unbiased proximity labeling proteomics in mammalian cells, we identified a multimeric protein complex containing ESYT1, ESYT2 and VAPB, which we further showed to localize to tripartite LD, mitochondria, and endoplasmic reticulum (ER) interaction sites. This complex appears to facilitate the transfer of fatty acids from LDs to mitochondria, thereby regulating efficient fatty acid oxidation and the maintenance of cellular lipid homeostasis.
Results
Proteins residing at lipid droplet-mitochondria contact sites
Lipid transfer between organelles requires spatial organization of proteins at lipid contact sites36. To identify and map proteins at LD-mitochondria contact sites that is known to reach 10 to 30 nm wide, we used BioID proximity labeling. This method takes advantage of the ability of BirA* to conjugate biotin onto local proteins37,38,39. We used small and monomer fluorescent proteins mScarlet40 and mTurquoise241 (unlike tetramaric GFP), and small targeting signal peptides (10–30 amino acids, Fig. 1a, b) to localize BirA* to either LDs or mitochondria. We also used multiple BirA* tag proteins to internally validate the interface proteome (Fig. 1. a, b, e). Initially, we engineered the C-terminal portion of BirA* to target LDs by fusing to the transmembrane domain of methyltransferase like protein AAM-B (also called METTL7B; AAMTMD-RFP-HA-BirA*) (Supplementary Fig. 1a), which was shown previously to specifically target constructs to LDs, but not to endoplasmic reticulum, as is the case for full length AAM-B (ER)42. This was confirmed by Airyscan super-resolution confocal microscopy staining with BODIPY (staining LDs), PLIN2 (constitutive LD localized protein), and the ER-specific protein calnexin. We show prominent ring like structures of BirA on the surface of LDs, with evidence of very limited staining on the ER (Supplementary Fig. 1c, d). We also targeted BirA* to the cytosolic side of the outer mitochondrial membrane using the targeting signal of FIS1 (Supplementary Fig. 1b). Mitochondrial localization of the FIS1TMD-CFP-FLAG-BirA* constructs were confirmed by Airyscan super-resolution confocal microscopy staining with TOMM20 (Supplementary Fig. 1e). Protein biotinylation was confirmed at the surface of organelles, with some diffuse biotin staining, which is likely indicative of on/off organellar interactions and/or protein movement within the cell (Supplementary Fig. 1f–h). All constructs were stably expressed in HepG2 cells.

a BioID targeted to the surface of LDs, containing the transmembrane domain of AAM-B, RFP (mScarlet), HA, and BirA* (AAMTMD-RFP-HA-BirA*). b BioID targeted to the surface of the outer membrane of mitochondria, containing the transmembrane domain of FIS1, CFP (mTurquoise2), FLAG, and BirA* (FIS1TMD-CFP-FLAG-BirA*). c Mass spectrometric analysis of biotinylated proteins purified from HepG2 cells stably expressing AAMTMD-RFP-HA-BirA*chimeras (LD-BioID). Plot compares BirA* to the negative control in HepG2 cells supplemented with biotin. Colored dots indicate proteins enriched using the three independent constructs (a–e). Proteins known to be enriched at LDs (ArtGAP1, SNX, FASN), peroxisomes (GOSAR1, ABCD3), endoplasmic reticulum (ESYT1, ESYT2, RTN4) and mitochondria (DNM1L, CSDE1, OCIAD1) are highlighted. d Mass spectrometric analysis of biotinylated proteins purified from HepG2 cells stably expressing Fis1TDM-CFP-FLAG-BirA* (Mito-BioID). Experiment and data analysis as indicated in (c). e Design of the Split-BioID approach, containing the transmembrane domain of FIS1, CFP, FLAG, and half of BirA*(BirAN*) targeted to the surface of the outer membrane of mitochondria (Fis1TMD-CFP-FLAG-BirAN*). The transmembrane domain of AAM-B anchors RFP, HA, and the other half of BirA*(BirAC*) targeted to the surface of LDs (AAMTMD-RFP-HA-BirA*). f Airyscan imaging of HepG2 cells stably expressing the Split-BioID. Merged image showing Fis1TMD-CFP-FLAG-BirAN* (magenta) and AAMTMD-RFP-HA-BirAC*(green). (representative of 4 independent experiments, scale bar, 2 μm). g Mass spectrometric analysis of biotinylated proteins purified from HepG2 cells stably expressing Split-BioID and negative controls (non-BirA*-transfected HepG2 cells). Colored dots indicate commonly enriched proteins in panels C & D. h Venn diagram showing the number of proteins enriched using Split-BioID, LD-BioID and mito-BioID approaches. i Dot-plot analysis of commonly enriched proteins using Split-BioID, LD-BioID and mito-BioID approaches. j Validation of proteins identified by BioID using an organelle coprecipitation assay of HepG2 cell lysate. LD-mitochondria fraction (LD+Mito), mitochondrial fraction (Mito), and cytosol fraction (Cyto) were examined by immunoblot. Representative of n = 3 biological replicates. For c, d, g and i, data analyzed with unpaired two-tailed Student’s t-test with FDR < 0.05, n = 4 biological replicates. See also Supplementary Fig. 1 and Supplementary Data 1. Created in BioRender. Keenan, S. (2025) https://BioRender.com/n75s805.
Biotinylated proteins were then recovered on streptavidin beads in the presence of 1% sodium dodecyl sulfate (SDS) and 6 M urea to eliminate nonbiotinylated protein-protein interactions, which were detected by mass spectrometry (MS) and quantified. HepG2 cells treated with biotin in the absence of BirA* were used as negative controls for all experiments. Enrichment of endogenous biotinylated proteins was not different between wildtype cells and BirA*-expressing cells (Supplementary Fig. 1i). Using the full length BirA* targeted to LDs, we identified 974 proteins (Fig. 1c), of which 174 and 307 were previously reported using APEX proximity labeling of LDs in Huh7 and U2OS epithelial cells, respectively43 (Supplementary Fig. 1j). We detected proteins known to reside at LDs (ArtGAP1, SNX, FASN), peroxisomes (GOSAR1, ABCD3), endoplasmic reticulum (RTN4, VAPB, ESYT1) and mitochondria (DNM1L, CSDE1, OCIAD1) (Fig. 1c). Using full length BirA* bait targeted to mitochondria we identified 625 proteins that reside near the mitochondria outer membrane (Fig. 1d).
We also employed a ‘split’ version of the promiscuous biotin ligase, BirA*, where inactive halves of the ligase are fused to two different bait proteins. When baits interact the halves come together to form an active BirA* that conjugates biotin onto proteins within 10–30 nm of the interaction site37,38,39,44,45. We targeted the N-terminal portion of BirA* to the cytosolic side of the outer mitochondrial membrane by fusing to the transmembrane domain of FIS146 (FIS1TMD-CFP-FLAG-BirA*N), and the C-terminal portion of BirA* was targeted to LDs by fusing to the transmembrane domain of methyltransferase like protein AAM-B (AAMTMD-RFP-HA-BirA*C) (Fig. 1e, f). Hence, when co-expressing these constructs BirA* is functional only at sites where LDs and mitochondria are in close proximity. Note that these experiments were conducted before the development of split-TurboID47, hence the use of split-BioID.
Using streptavidin conjugated horseradish peroxidase and western blotting, we confirmed that when expressed alone neither half is active, whereas co-expression leads to efficient protein biotinylation (Supplementary Fig. 1k). Using this approach, we identified 81 proteins localized to the LD-mitochondria interface in live cells (Fig. 1g). PCA analysis showed that while the proteomes identified using independent approaches are distinct (Supplementary Fig. 1l), 71 proteins were commonly enriched in all three proximity biotinylation experiments (Fig. 1h), many of which have been reported at other organelle contact sites, including the ER. This was unsurprising given the close association between LD-mitochondria-ER contact sites48,49. Given that three independent BirA* constructs were used for identification, we define these proteins as LD-mitochondria interface proteins. A complete list of all identified proteins can be found in Supplementary Data 1.
LD-mitochondria contact proteins exert diverse functions
Pathway enrichment analysis of the 71 high-confidence LD-mitochondria proteome using Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG)50,51 databases showed enrichment of proteins involved in protein processing at the ER, phagosome, endocytosis, and glycerophospholipid metabolism, indicating diverse and unappreciated functions at the LD-mitochondria interface (Supplementary Fig. 1m, n). We identified known proteins involved in lipid transport (e.g., ESYT1 and ESYT2), membrane docking and priming contacts (e.g., VAPB), metabolic sensors (e.g., RAB1A and RAB7, and their effector protein ARHGDIA) and membrane structural proteins, such as MBOAT7 (Fig. 1i). Enrichment of these proteins was confirmed at the LD-mitochondria interface using an organelle coprecipitation assay23 and immunoblot analysis (Fig. 1j).
ESYT1, ESYT2 and VAPB form homo- and hetero-oligomeric complexes
Among the most highly enriched proteins at the LD-mitochondria interface, we focused on ESYT1, which was previously reported to be localized to ER-mitochondria contact sites and to tether the plasma membrane to the ER to facilitate transfer of phospholipids via a highly conserved lipid binding SMP-domain (Synaptotagmin-like, Mitochondrial lipid binding Protein-domain)52,53,54. We then set out to determine whether ESYT1 forms a complex with other proteins at contact foci of LDs-mitochondria. 3HA-eGFP-ESYT1 was expressed in HepG2 cells under the PolG promoter to induce low level expression, where it had no effect on mitochondrial metabolism55. Possible protein-protein interactions were determined by HA-immunoprecipitation followed by label-free mass-spectrometry. As expected, ESYT1 was highly enriched (Fig. 2a, FDR p < 0.05) and efficiently co-isolated with known interactors RDH11, YIF1B, ARL6IP5, and EXOS656 (Supplementary Fig. 2a and Supplementary Data 2). ESYT2 and VAPB were also enriched with ESYT1 immunoprecipitation, both of which were also identified in our LD-mitochondria interface proteome (Fig. 1g, i and Supplementary Dataset 2). ESYT3 can form heterodimers and heteromultimers with ESYT1 and 2 but was not detected in either the BioID or immunoprecipitation experiments, likely owing to low abundance. Heterodimerization of ESYT1 and ESYT2 is documented57, however, VAPB interaction with ESYT1 and ESYT2 has not been investigated (Supplementary Fig. 2b, c). To validate these interactions, HA-tagged ESYT2 and VAPB were expressed in HepG2 cells and subjected to immunoprecipitation and mass spectrometry. The presence of ESYT1, ESYT2 and VAPB and other interactors were confirmed in pulldowns from the independent cell lines (Fig. 2b, c, Supplementary Data 2). Consistent with these results, immunoprecipitation and immunoblot analysis from HepG2 cells confirmed the interaction of endogenous ESYT1, ESYT2 and VAPB (Fig. 2d–f). Next, we interrogated protein-protein interactions from co-fractionation experiments performed on tissues from mice58. In agreement with our data, ESYT1-ESYT2-VAPB co-fractionated in high-molecular-weight pools from several tissues, including liver and muscle where ESYT1 and VAPB had identical profiles (Supplementary Fig. 2d).

a Volcano plot showing proteins associated with ESYT1. Data are from a label-free proteomic analysis of anti-HA immunoprecipitates from HepG2 cells stably expressing 3HA-eGFP-ESYT1 versus 3HA-EGFP. p-values of two-tailed Student’s t-test, n = 4 biological replicates. b, c Volcano plot showing proteins associated with ESYT2 and VAPB using HepG2 cells stably expressing 3HA-eGFP-ESYT2 and 3HA-eGFP-VAPB. Experiments and statistical analysis as indicated in (a). d–f Western blot analysis of ESYT1, ESYT2 and VAPB interactions following anti-HA immunoprecipitants from HepG2 cells stably expressing 3HA-eGFP-ESYT1, 3HA-eGFP-ESYT2, 3HA-eGFP-VAPB or empty vector (EV; 3HA-EGFP) and probed for endogenous proteins. MBOAT9 was used as a negative control. Representative of n = 3 biological replicates. g, h Lifetime maps (g) of the FLIM data acquisitions between eGFP-ESYT1, eGFP-ESYT2, and eGFP-VAPB (donor) with VAPB-mCH (acceptor) pseudo-colored according to the FRET palette defined in the phasor plot (h) (i.e., teal pixels = 0 % FRET while red pixels = 23% FRET) that reports hetero protein-protein interaction. i Hetero protein-protein interaction of ESYT1, ESYT2 and VAPB in HepG2 cells. From left to right columns: n = 21, 15, 16, 17, 18, 19 and 14 cells. j Fraction of heterocomplex between eGFP-ESYT1, eGFP-ESYT2, and eGFP-VAPB with VAPB-mCH. From left to right columns: n = 16, 13, 13, 15, 15, 18, and 17 cells. k Structure of ESYT1 in complex with ESYT2. Arrows- hydrophobic side chain residues at the interacting interface that are identified via in silico saturation mutagenesis and selected for further experimental validation of heterodimer formation. l Quantification of the fraction of pixels exhibiting FRET between eGFP-ESYT2 and wild type and mutant ESYT1-mCH (i.e., hetero protein-protein interaction) across multiple HepG2 cells, from left to right columns n = 5, 6, 8, 4, 6, 5, 4, 6 and 6 cells. Data analyzed using unpaired two-tailed t-tests. *P < 0.05. The box and whisker plots in 2i, j and l show the minimum, maximum and sample mean. In 2i, 2j and 2 l, *p < 0.05, unpaired t-test, two-sided. See also Supplementary Fig. 2 and Supplementary Data 2.
To determine in a living cell whether ESYT1-ESYT2 form a heterocomplex with VAPB, and investigate the stoichiometry of these three different subunits, we labeled these proteins with eGFP (eGFP-ESYT1, eGFP-ESYT2, eGFP-VAPB) or mCherry (mCH; ESYT1-mCH, VAPB-mCH) and employed fluorescence lifetime imaging microscopy (FLIM) of Förster resonance energy transfer (FRET) alongside fluorescence fluctuation spectroscopy (FFS) to spatially map hetero- versus homo-oligomer protein-protein interactions in HepG2 cells. The phasor approach to FLIM analysis of (FRET) between eGFP-ESYT1, eGFP-ESYT2 and eGFP-VAPB (donor molecules) with VAPB-mCH (acceptor molecule) (Fig. 2g, h), and eGFP-ESYT2 (donor molecule) with ESYT1-mCh (acceptor molecule) (Supplementary Fig. 2e), revealed that ESYT1 and ESYT2 form hetero- and homo-complexes in the absence and presence of forskolin stimulation. Specifically, it was found that the hetero-complex between ESYT1 and ESYT2 with VAPB is promoted by the protein kinase A (PKA)-activator forskolin and VAPB self-associates into a dimer or higher order oligomer independent of this stimulation (Fig. 2i). This result was orthogonally validated by FFS based approaches called cross raster image correlation spectroscopy (cRICS) (Fig. 2j) as well as number and brightness (NB) analysis (Supplementary Fig. 2f, g).
To further assess heterodimer formation of ESYT1 and ESYT2, we next performed in-silico saturation mutagenesis at the interface of modeled structures of ESYT1 and ESYT2 (Fig. 2k and Supplementary Fig. 2h). Here we assessed missense mutations based on their predicted change in binding affinity (ΔΔG), which indicates a more drastic effect on how ESYT1 interacts with ESYT2, as a guide for our experiments. Mutations of hydrophobic side chains of ESYT1 (F152, L250, I251) at the interface of ESYT1-ESYT2 to negatively charged amino acids (Glu and Asp) were ranked among the topmost to impact the formation of the heterodimer (Fig. 2k and Supplementary Fig. 2h). The phasor approach to FLIM analysis of FRET between eGFP-ESYT2 (donor) and ESYT1-mCH (acceptor) confirmed the presence of ESYT1-ESYT2 heterodimers and that heterodimer formation was enhanced by PKA activation (Fig. 2l). Consistent with the in-silico model, the phasor approach to ESYT1/ESYT2 FLIM-FRET analysis showed that double and triple residue mutations in ESYT1-mCherry (ESYT1L250E, I251E-mCH and ESYT1L250E, I251E, F152E-mCH) significantly reduced heterodimer formation with eGFP-ESYT2, both in presence and absence of PKA activation (Fig. 2l). Together, these data demonstrate the presence of ESYT1-ESYT2-VAPB multimeric complexes in mammalian cells that are regulated by β-adrenergic stimulation. Since β-adrenergic stimulation and PKA activation increases both lipolysis and LD-mitochondria contact18,59, this result collectively suggests that ESYT1-ESYT2-VAPB complex formation is functionally important at the LD-mitochondria interface.
ESYT1/2-VAPB complex forms at LD, mitochondria, and ER contact sites
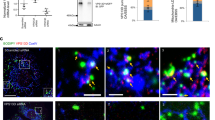
Previously identified proteins mediating LD and mitochondria tethering, including SNAP23, FATP4 (ACSVL4), VPS13 and VPS13D, and Rab8a are also localized at the interface of other cellular organellar contact sites, most notably the ER60,61,62,63,64,65,66,67. As ESYT1 and ESYT2 are predominantly localized to the ER where they form contact sites with the plasma membrane or mitochondria52,68, and in light of our identification of ESYT1/2 at the LD-mitochondria interface, we hypothesized that ESYT1, ESYT2, and VAPB complexes are in fact localized at a tripartite organellar interface consisting of LDs, mitochondria and ER. Consistent with this notion, Airyscan imaging and line scanning analysis showed that eGFP-ESYT1, eGFP-ESYT2 and eGFP-VAPB localize to the interface of LD-mitochondria-ER (Fig. 3a). Formation of tripartite organellar interface consisting of LDs, mitochondria and ER was also confirmed in HeLa and Hek293T cells (Supplementary Fig. 3a, b). We confirmed localization of endogenous ESYT1 and VAPB at the LD-mitochondria interface (Supplementary Fig. 3c). Since ESYT1, ESYT2 and VAPB localization has been well established in the ER, we next used the phasor approach to FLIM-FRET analysis to monitor eGFP-ESYT1/eGFP-ESYT2 (donor) interaction with VAPB-mCherry (acceptor) at LD-mitochondria contact sites in HepG2 cells before and after forskolin administration. We used an immunofluorescence mask defined by PLIN2-AF405 for lipid droplet and TOMM20-AF633 for mitochondrial localization (Fig. 3b). The interaction of ESYT1 or ESYT2 with VAPB was significantly increased at the LD-mitochondria interface after forskolin treatment, but not under non-stimulated conditions (as compared to whole cell FRET) (Fig. 3c). This indicates that ESYT1/2-VAPB complex recruitment to the LD-mitochondria interface is cellular signaling-dependent.

a Left panel: Single plane view of Airyscan imaging of HepG2 cells stably expressing eGFP-ESYT, eGFP-ESYT2 or eGFP-VAPB (blue). HepG2 cells were fixed and stained for LDs with HCS LipidTox red Neutral Lipid Stain (green), mitochondria with TOMM20 antibody (yellow), and ER with Calnexin antibody (magenta). White arrows highlight tripartite interfaces of LD, mitochondria, and ER. A total of 16 cells from four independent experiments were imaged and a representative of image is indicated. Large scale bars, 2 μm and inset scale bars 0.5 μm. Right panel: Line scan analysis of images presented to the left. A and B on x-axis corresponds to the line scan in panel a inset. b Left panel: Donor lifetime maps of the FLIM data acquisitions between donor eGFP-ESYT1, eGFP-ESYT2, and eGFP-VAPB with acceptor VAPB-mCH FRET pair pseudo-colored according to the palette defined in the phasor plot in (Fig. 2h) spatially map the hetero protein-protein interaction (red). Right panel: Identification of the LDs and mitochondrial interface based on PLIN2-AF405 and TOMM20-AF647 immunofluorescence intensity mask analysis. c Quantification of the fraction of ESYT1/VAPB (upper panel), ESYT2/VAPB (middle panel), and VAPB/VAPB interaction inside the LDs and mitochondrial interface defined by PLIN2-AF405 and TOMM20-AF647 intensity masks (panel B, right) versus whole cell protein interaction fraction with and without Forksolin stimulation across multiple HepG2 cells. ESYT1/VAPB -FSK n = 10; ESYT1/VAPB + FSK n = 7; ESYT2/VAPB -FSK n = 10; ESYT2/VFSK+fork n = 9; VAPB/VAPB -FSK n = 9; VAPB/VAPB + FSK n = 7 cells. *P < 0.05 paired t-test, one-tailed. See also Supplementary Fig. 3.
Loss of functional ESYT1, ESYT2 or VAPB leads to enlarged lipid droplets
The presence of the ESYT1-ESYT2-VAPB complex at the LD-mitochondria interface indicated a possible role in lipid metabolism. Accordingly, we determined the effects of CRISPR-Cas9-mediated depletion of ESYT1, ESYT2, or VAPB in HeLa cells (Fig. 4a). Because ESYT1 and ESYT2 can form both homo- and heterodimers, we also created an ESYT1-ESYT2 double knockout cell line (DKO). Airyscan imaging analysis showed that ESYT1, ESYT2, VAPB and the DKO deletion increased total cellular LD volume by 34-86% (Fig. 4b). This was due to increased LD size rather than LD number for ESYT1, ESYT2 and VAPB KO cells. For DKO, LD size was similar to control cells, but LD number was increased by 43% (Supplementary Fig. 4a, b). These findings were confirmed by transmission emission microscopy (Supplementary Fig. 4c).

a Representative western blot showing ESYT1, ESYT2 and VAPB protein in HeLa cells following CRISPR-Cas9 mediated knockout of ESYT1, ESYT2, VAPB and ESYT1 and ESYT2. GAPDH, loading control. * non-specific immunoreactive band. b 3D rendering images of Control and KO cells stained for LDs with HCS LipidTox Deep Red Neutral Lipid Stain (various colors) and mitochondria with TOMM20 antibody (red). Prior to staining ESYT1/2/VAPBKO and Control HeLa cells were treated with 400 μM fatty acids (oleic acid and palmitic acid; 2:1) for 4 h. Scale bars: 0.5 μm. c Quantification of volume occupied by LDs per cell from experiments shown in panel B. Control n = 35, ESYT1KO n = 44, ESYT2KO n = 42, VAPBKD n = 34, DKO n = 35 cells. d Lipidomic analysis showing triacylglycerol content in HeLa cells treated with 400 μM oleic acid and palmitic acid (2:1) for 4 h. n = 6 biological replicates. e, f HeLa cells were pulsed for 6 h with radiolabeled fatty acid (14C oleic acid) conjugated to 1% BSA and chased for 4 h in low glucose medium without fatty acids (starvation medium). Data indicates rate of 14C- oxidation (LD-derived fatty acid oxidation) and 14C remaining in triglyceride following the chase period. Bar graphs represent the mean ± SEM from 3 (e) and 4 (f) biological replicate experiments. g BODIPY C16 in LDs of cells that were pulsed with BODIPY C16 for 6 h, washed, and incubated in starvation medium for 4 h. n = 6 (VAPBKD, DKO) or 7 (Control, ESYT1KO, ESYT2KO) biological experiments. h Western blot showing steady state level of phosphorylated and total ATGL and HSL protein in cells. * denotes non-specific immunoreactive band. i Flux of 13C fatty acids into TCA cycle and non-mitochondria metabolites (n = 4 per group). The 13C-fatty acid mix contained myristic (0.2%), palmitoleic (9.4%), palmitic (38.9%), margaric (0.3%), linoleic (10.7%), oleic (26.9%), elaidic (1.6%), and stearic (1.6%) acid. For (c– i) bar graphs represent the mean ± SEM, *p < 0.05 vs. Control using one-way ANOVA with Bonferroni’s multiple comparison test. See also Supplementary Fig. 4 and Supplementary Data 3.
Given the differences in LD volume, we performed liquid chromatography tandem mass spectrometry lipidomics to determine whether components of the ESYT-VAPB complex impact acylglycerol abundance, which are the major lipid type in LDs. Cells were incubated in medium containing 500 µM fatty acid mixture (oleate and palmitate, 2:1 molar ratio) to enhance the detection of low abundance lipid species. Consistent with the changes in LD volume, acylglycerol abundance was increased in KO and DKO cell lines compared with control cells, including triglycerides (29-57%, except VAPBKO), diglyceride (32-56%) and monoglycerides (203-283%) (Fig. 4d and Supplementary Fig. 4d, e). Together, these data indicate that components of the ESYT-VAPB complex regulate LD metabolism.
ESYT1, ESYT2 and VAPB deficiency reduce lipid droplet-derived fatty acid oxidation
To test whether ESYT1, ESYT2 or VAPB regulates the oxidation of fatty acids derived from LDs, we used ‘pulse-chase’ experiments where cells were incubated with 14C-oleate for 6 h to load 14C-fatty acids into triglycerides contained in LDs, after which the metabolic fate of 14C was traced in the absence of extracellular 14C-fatty acids. The oxidation of fatty acids derived from LDs was reduced by 28-47% in ESTY1, ESYT2 and VAPB null cells, and by 67% in DKO cells (Fig. 4e). The amplified impairment of fatty acid oxidation in the DKO cells supports the notion that ESYT1 and ESYT2 form both homo- and heterodimers for fatty acid transfer. Consistent with these results, the 14C-fatty acid remaining in triglyceride at the end of experiments was increased in each of the KO cell lines (Fig. 4f). To further demonstrate the importance of ESYT1-ESYT2-VAPB as a mediator of LD-derived fatty acid oxidation, we incubated cells with fatty acid C16-BODIPY for 6 h, then assessed LD-BODIPY degradation via its green fluorescence 4 h later. The C16-BODIPY in LDs was increased by 27-47% in KO cell lines compared with control cells (Fig. 4g). The reduction in LD-derived fatty acid oxidation was unlikely to result from insufficient triglyceride lipolysis as inferred by similar contents and phosphorylation of activating serine resides of the rate-limiting lipolytic enzymes ATGL (Ser404) and HSL (Ser660) (Fig. 4h).
We performed orthogonal validation using 13C-isotopologue profiling in cells to track fatty acid carbons through the tricarboxylic acid (TCA) cycle and other non-mitochondria metabolic processes. Isotopic enrichment of 13C-fatty acids (16:0, 16:1, 18:0 and 18:1) was not significantly different in KO and control cells (Supplementary Fig. 4f). Upon entry to the mitochondria, fatty acids undergo β-oxidation which produces acetyl CoA composed of two 13C labels that can be further traced through the TCA cycle. In agreement with the radiolabel experiments, m + 2 isotopologue enrichment in TCA cycle metabolites such as citrate, fumarate, and malate were significantly reduced in single KO cell lines, with this effect further amplified in the DKO cells (Fig. 4i). Rerouting of 13C through TCA cycle anaplerotic pathways, including to aspartate and glutamate, was also reduced in KO cell lines. Isotopologue labeling was near undetectable in metabolites of carbohydrate and glutamine metabolism, indicating specificity of this response (Supplementary Fig. 4g). Together, these data demonstrate that each component of the ESYT1-ESYT2-VAPB complex is necessary for efficient LD-derived fatty acid oxidation in cells.
We next ruled out the possibility that ‘mitochondrial dysfunction’ could provide a mechanism for reduced fatty acid oxidation in KO cells. The decrease in LD-derived fatty acid oxidation was not related to changes in mitochondrial volume, as assessed by TOMM20 staining and the expression of proteins that regulate oxidative phosphorylation, or mitochondrial function, as assessed by measuring the rate of glucose oxidation by radiometric techniques or oxygen consumption rate in live cells (Supplementary Fig. 4h–l). LD-mitochondria contact is postulated to facilitate efficient fatty acid oxidation. The number of LDs in close contact with mitochondria (≤30 nm) was not different between control and ESYT1 and ESYT2 knockout cells, and LD-mitochondria contact was increased in VAPB and DKO cells, indicating that these proteins are unlikely to be important for physical tethering of LDs to mitochondria (Supplementary Fig. 4m). We also showed that the levels of other fatty acid transfer proteins previously identified at the LD-mitochondria interface33, including VPS13D, TSG101 and ESCR-III (Supplementary Fig. 4n, o), were increased in ESYT knockout cells compared with control cells.
SMP domain of ESYT1 and ESYT2 is required for trafficking of fatty acids from LDs to mitochondria
The SMP domain of ESYT1 and ESYT2 binds glycerophospholipids and transports these lipids between the ER and plasma membranes52,53,69. Having shown localization of ESYT1 and ESYT2 at the LD-mitochondria interface and the necessity of these proteins for LD-derived fatty acid oxidation, we reasoned that ESYT proteins could transfer fatty acids between organelles. Computational structural biology strategies were employed to test this possibility. Homology modeling was carried out in Schrodinger Maestro to model the ESYT1/ESYT2 heterodimer based on the experimental structure of ESYT2 asymmetrical dimer (PDB ID: 4P42). Two complexes were modeled, which differed in the chain within the ESYT2 homodimer substituted by ESYT1 (Fig. 5a), which was possible due to homology (39% sequence identity).

(a) Apolar channel (yellow) formed by ESYT1 or ESYT2 SMP domain in ESYT1/2 dimer complex. b-c Ligand docking analysis showing oleic acid inside apolar channel of ESYT1 as a lateral (b) and frontal (c) view. (d) Enrichment of oleic acid in 3HA-EGFP-ESYT2 cells (compared with empty vector, EV, 3HA-EGFP) following HA immunoprecipitation and mass spectrometry lipidomic analysis. Cells were incubated in 400 μM oleic acid for 4 h prior to lysing. *p < 0.05 vs EV by unpaired two-tailed t-tests. n = 4 biological replicates. Bar graphs represent the mean ± SEM. e-f A comparison of SMP domain and key residues substituted by heavy-side chain amino acid of ESYT1 that protrude into the apolar channel to obstruct fatty acid trafficking. g Airyscan images of HeLa cells showing lipid droplets in ESYT1KO cells without or with re-expression of wild type ESYT or mutations in the SMP domain. Cells were treated with 400 μM oleic acid and palmitic acid (2:1) for 4 h prior to staining. LD staining with HCS LipidTox Green (green) and DAPI (Blue). Scale bar: 3 μm. h Fatty acid oxidation in HeLa cells as described in G. n = 6 biological replicate experiments. i, j A comparison of WT SMP domain (i) and a residue substituted by heavy-side chain amino acid (I353Y; j) of ESYT2 that protrudes into the apolar channel. k Airyscan images of HeLa cells showing lipid storage in ESYT2KO cells in comparison with ESYT2KO cells with re-expression of ESYT2WT-eBFP or a mutation in the SMP domain (ESYT2I354Y-eBFP). Prior to staining cells were treated as in g for LD staining with HCS LipidTox Green Neutral Lipid (green) and nuclei staining with SYTOX Deep Red (Blue). Scale bar: 5 μm. (l) Fatty acid oxidation in HeLa ESYT2KO cells in comparison with rescued cell lines with expression of wild type or mutant ESYT2 on SMP domain. n = 4 biological replicates. In (h, i) * P < 0.05 using one-way ANOVA with Bonferroni’s multiple comparison test. Bar graphs represent the mean ± SEM. See also Supplementary Fig. 5.
Fragment screening analyses highlighted the presence of an apolar channel within both complexes (Fig. 5a), which corresponded to the apolar channel observed within Chain A of the ESYT2 homodimer. Within both complexes generated, and similar to the ESYT2 homodimer, the channel appears to be extended by the other subunit within the complex, suggesting that passage of fatty acids may occur when ESYT1 and ESYT2 form a larger complex with VAPB. The observed apolar channel was therefore considered as a point of fatty acid entry for further transport (Fig. 5b, c and Supplementary Fig. 5a–l) and was used as a search space for ligand docking of long chain fatty acids (palmitic acid, C16:0; oleic acid, C18:1; linoleic acid; C18:2) and other lipid types contained in LDs (triacylglycerol, phosphatidylcholine and cholesteryl oleate). In both cases, the fatty acids, which are released from LDs and are readily available for transport, docked and fitted within the apolar channel, whereas complex lipids such as tristearin (triacylglycerol) did not (Fig. 5a–c and Supplementary Fig. 5a–t). These results suggested an important functional role for the apolar channel in fatty acid binding. To directly test this possibility, we performed a lipidomic analysis of immunoprecipitated lipids from cells expressing ESYT2-HA or HA (empty vector). ESYT2 was enriched for oleic and linoleic acid (Fig. 5d). Notably, there was no enrichment of other lipids in ESYT-HA, including components of LDs such as triacylglycerols, cholesterol esters and phospholipids.
Further analysis identified residue pairs within the apolar channel of each model located at proposed lipid entry, midway through, and at the edge of the channel. Residues were tested for changes in protein stability upon mutation to larger residues. Within the ESYT1 apolar channel, mutations S172W, I294Y and S297Y resulted in moderate increases in stability while the local rigidification predicted possible impaired fatty acid transport (Fig. 5e-f and Supplementary Fig. 5u). Similarly, within the ESYT2 apolar channel, H231W at the entrance and I354Y located midway across the channel, conferred mild increases in protein stability which is predicted to block fatty acid entry or transport across the complex (Fig. 5i, j and Supplementary Fig. 5v).
In light of these predictions, we evaluated the requirement of ESYT1 and ESYT2 fatty acid transfer capacity for β-oxidation by creating point mutations designed to produce aromatic residues that block the lipid binding pocket in ESYT1 or ESYT2 (Fig. 5e, f, i, j). We expressed either eGFP-ESYT1WT or mutant eGFP-ESYT1S172W, eGFP-ESYT1I294Y or eGFP-ESYT1S297Y in ESYT1KO cells. Compared with ESYT1KO, eGFP-ESYT1WT reduced LD volume (Fig. 5g) and increased fatty acid oxidation (Fig. 5h), while expression of the ESYT1 mutants failed to rescue the ESYT1KO phenotype (Fig. 5g, h). Similarly, expression of ESYT2WT-eBFP in ESYT2KO cells decreased LD volume, but not LD number (Supplementary Fig. 5x, y), and increased fatty acid oxidation when compared to the parental ESYT2KO cells (Fig. 5l). Expression of ESYT2I354Y-eBFP had no effect on these parameters (Fig. 5k, l, Supplementary Fig. 5x, y). Together, these data demonstrate a role of ESYT proteins in fatty acid transport.
ESYT-VAPB complex proteins protect cells from lipotoxicity
Lipotoxicity refers to the detrimental effects of lipid metabolites on cellular functions in non-adipose tissues and is prevalent in metabolic diseases such as obesity and type 2 diabetes70,71. Lipotoxicity can result from excess fatty acid flux into cells and/or uncoupling of LD lipolysis and mitochondrial β-oxidation (Fig. 6a)70,72. To investigate whether components of the ESYT/VAPB complex are required to prevent cellular lipotoxicity, HeLa cells were incubated in medium containing 500 µM palmitate (complexed with albumin), a fatty acid known to induce stress signaling pathways and cell death72,73. ESYT1, ESYT2 and VAPB deletion resulted in marked remodeling of the cellular lipidome including increases in triglycerides, as well as lipids known to induce cell stress damage, such as diglycerides, ceramide, sphingomyelin, cardiolipin and lysophosphatidylcholine (Fig. 6b–f). In comparison to wildtype cells, endoplasmic reticulum stress was increased in all KO cell lines as indicated by increased phosphorylation of the UPR signal activators ATF6 (57–89%) and protein kinase R-like ER kinase (PERK, ~40%), and by reduced protein disulfide isomerase (PDI, ~30%) and binding immunoglobulin protein (BiP, 37–65%) levels (Fig. 6g). Activation of other stress-regulated protein kinases, such as c-Jun N-terminal kinase (JNK, 50–125%) and extracellular signal-regulated kinase (ERK, 63-95%), were also increased in KO cells (Fig. 6h). Sensitivity to palmitate-induced lipotoxic stress in KO cells extended to activation of pyroptosis, which is an inflammatory form of programmed cell death74. Specifically, ESYT1, ESYT2 and VAPB deletion resulted in activation of the canonical inflammasome pathway as indicated by increased caspase 1 cleavage, an increase in cleaved gasdermin D (GSDMD) and increased levels of the damage-associated molecular pattern high mobility group box 1 (HMGB1) (Fig. 6i). Re-expression of ESYT2WT, but not ESYT2S297Y, in ESYT2KO cells ameliorated JNK phosphorylation and GSDMD cleavage (Supplementary Fig. 5z). Taken together, these data demonstrate that components of the ESYT-VAPB complex are required to prevent lipotoxicity in cells.

a Schematic representation of fatty acid trafficking at LD, mitochondria and ER contacts in the presence of protein-mediated fatty acid transfer (efficient fatty acid trafficking) and with inefficient fatty acid trafficking mediated by disruption to the ESYT/VAPB complex. Created in BioRender. Keenan, S. (2025) https://BioRender.com/n75s805. b–f Lipidomic analysis showing abundance of lipids associated with lipotoxicity including diacylglycerol, ceramide, sphingomyelin (SM), cardiolipin, and lysophosphatidylcholine (LPC). Bar graphs represent the mean ± SEM from 6 biological replicate experiments. *p < 0.05 vs. Control using one-way ANOVA with Bonferroni’s multiple comparison test. g Representative Western blot and quantitation of ER stress marker proteins. Bar graphs represent the mean ± SEM from n = 4 biological replicate experiments of BIP, ATF5, p-PERK (Thr980), PERK and PDI. ATF5s/ATF6FL indicates ratio of protein content of short and full length ATF6. GAPDH was used as loading control. Data analysed using one-way ANOVA and a Kruskal-Wallis post hoc test. p < 0.05 vs. Control. (h) Representative Western blot and quantitation of p-ERK (Thr202/Tyr204) and ERK, p-JNK (Thr183/Tyr185) and JNK. GAPDH was used as a loading control. Bar graphs represent the mean ± SEM. (p-JNK/JNK n = 4 and p-Erk1/2 / ERK1/2 n = 5 independent experiments). Data analysed using one-way ANOVA and a Kruskal-Wallis post hoc test. *p < 0.05 vs. Control. i Representative Western blot and quantitation of pyroptosis markers with palmitic acid-induced stress in control and KO cells. In all experiments in this panel, HeLa cells were treated with palmitate (500 µM) conjugated to 1% BSA for 4 h. Bar graphs represent the mean ± SEM protein content of HMGB1 (n = 6 for Control, ESYT2KO, VAPBKD and DKO, n = 5 ESYT1KO), cleaved caspase 1 (n = 4), ratio of full length and splice variant of Gasdermin (n = 4) from independent experiments. GAPDH was used as loading control. Data analysed using one-way ANOVA and a Kruskal-Wallis post hoc test. *p < 0.05 vs. Control.
ESYT1 and ESYT2 regulate fatty acid oxidation in livers of mice
To determine the physiological relevance of ESYT1 and ESYT2 as a regulator of fatty acid oxidation in vivo, we studied mice lacking either Esyt1(Esyt1KO) or Esyt2 (Esyt1KO) in hepatocytes. To do this we employed CRISPR gene-editing and identified guide RNA’s (gRNA) targeting proximal regions of the mouse Esyt1 or Esyt2 gene. AAV constructs were generated expressing two gRNA sequences targeting Esyt1 or Esyt2 alongside a Cre-dependent mCherry to report AAV-transduced cells (AAV-gRNA-FLEX-mCherry, Fig. 7a). To target CRISPR-mediated excision of Esyt1 in hepatocytes, we crossed Alb-Cre with LSL-Cas9-GFP knock-in mice to generate Alb-Cre::LSL-Cas9-GFP mice, which specifically expressed Cas9 and GFP in hepatocytes. Hepatocyte-specific targeting and silencing of Esyt1 and Esyt2 was induced following administration of AAV-gRNA-FLEX-mCherry into adult Alb-Cre::LSL-Cas9-GFP mice, which was confirmed by mass spectrometry proteomics and immunohistochemistry (Fig. 7b and Supplementary Fig. 6a). Loss of Esyt2 reduced Esyt1 in hepatocytes, which is consistent with the notion that protein stability is often contingent on the presence of interacting partners. Loss of either Esyt1 or Esyt2 resulted in a ~50% reduction in fatty acid oxidation in isolated murine hepatocytes (Fig. 7c). There were no differences between groups for metabolic processes that influence LD volume including fatty acid uptake, storage of fatty acids in triglycerides and cholesterol esters, or de novo lipogenesis (Supplementary Fig. 6b, c). These data provide direct evidence that ESYT1 and ESYT2 are critical for fatty acid oxidation in vivo.

a Schematic outlining strategy for CRISPR-Cas9 mediated deletion of Esyt1 and Esyt2 in hepatocytes of mice. b Esyt1 and Esyt2 peptide abundance in liver of mice. Data are mean ± SEM. *P < 0.05 vs. WT using one-way ANOVA and a Kruskal-Wallis post hoc test. n = 5 Control, n = 3 ESYT1KO, n = 3 ESYT2KO (n = 5 ESYT1 peptide; n = 3 ESYT2 peptide), ESYT1KO (n = 3 for both ESYT1 and ESYT2 peptide), ESYT2KO (n = 3 for both ESYT1 and ESYT2 peptide). c Fatty acid oxidation in precision-cut liver slices from Control, Esyt1KO and Esyt2KO mice. Data are mean ± SEM. *P < 0.05 vs. WT using one-way ANOVA and a Kruskal-Wallis post hoc test. n = 8 Control, n = 5 ESYT1KO, n = 7 ESYT2KO. d dEsyt2 mRNA in WT and dEsytKO and dEsytFB-KO Drosophila. Data are mean ± SEM. *p < 0.05 vs. WT by unpaired two-tailed t-test. n = 3 per group. e Breeding strategy for the generation of fat body-specific dEsyt2 inhibition in Drosophila (left) and survival response of WT FB and dEsytFB-KO males under starvation (right), n = 100 flies per genotype. Data analyzed using Log-rank (Mantel-Cox) test. (f) Breeding strategy for the generation of global dEsyt inhibition in Drosophila (left) and survival response of WT and dEsytKO males under starvation (right), n = 100 flies per genotype. Data analyzed using Log-rank (Mantel-Cox) test. See also Supplementary Fig. 6. Created in BioRender. Keenan, S. (2025) https://BioRender.com/n75s805.
To provide further evidence that ESYT proteins are critical regulators of lipid metabolism in vivo, we silenced dEsyt in Drosophila. The Drosophila genome encodes one ESYT orthologue54,75 and silencing was induced in the whole body (dEsytdKO) and in the fat body (dEsytdFB-KO), which is a liver- and adipose-like tissue that stores triglyceride. dEsyt silencing was confirmed by qPCR (Fig. 7d). Triglycerides contained in the fat body are broken down by lipases and the liberated fatty acids provide the main source of energy during starvation76. In keeping with an important role of ESYT in regulating lipid metabolism and survival, deletion of dEsyt in Drosophila reduced survival in response to starvation (Fig. 7e, f). While altered lipid metabolism can result in changes to starvation resistance, the same is true for altered glucose metabolism or perturbations to cellular membrane function. Therefore, we cannot discount the effects of removing dEsyt on other processes that influence starvation resistance.
Discussion
Using unbiased proximity proteomics, we profiled the proteins residing at the interface of LDs and mitochondria. Within this proteome, we identified a multimeric protein complex consisting of ESYT1 and/or ESYT2 and VAPB, which facilitates mitochondrial oxidation of fatty acids derived from LDs and is essential to protect cells from lipotoxicity. We show that this complex resides at the interface of a tripartite organellar contact involving the LD, mitochondria and ER, and the interaction of these proteins is enhanced with β-adrenergic stimulation, a state characterized by increased lipolysis of triglycerides in LDs77. Deletion of ESYT/VAPB complex components reduces the oxidation of fatty acids derived from triglycerides stored in LDs, and results in the accumulation of enlarged LDs, which is a defining feature of cells in organisms with metabolic disease77,78. The reduction in fatty acid oxidation most likely results from loss of the lipid transfer function of ESYT1/2 and not from alterations in mitochondrial mass or function, proteins that regulate lipolysis (e.g., ATGL, HSL), or the tethering of LDs to mitochondria. The observation that fatty acid oxidation is decreased in the liver of mice with Esyt1 or Esyt2 deficiency reinforces the importance of this function. These findings constitute a molecular mechanism for fatty acid transfer in mammalian cells and identify a function for ESYT in regulating cellular lipid metabolism.
Although complimentary approaches to manipulate ESYT functions and multiple models revealed consistent findings on ESYT functions, our study has raised several considerations. Deletion of both ESYT1 and ESYT2 reduced LD-derived fatty acid oxidation by 67%, indicating that additional mechanisms regulate fatty acid trafficking between the LD and mitochondria. This is likely to include other proteins at LD-mitochondria contact sites, including a fatty acid transport process involving the coordinated actions of VPS13D and the endosomal sorting complex required for transport (ESCRT) protein tumor susceptibility 101 (TSG101), which is invoked with starvation33. While, these proteins were not identified in our proximity labeling of the LD-mitochondria proteome, immunoblot analysis showed increased abundance of these proteins with ESYT deletion. This indicates a compensatory mechanism to retain fatty acid trafficking between LDs and mitochondria in the absence of the complete ESYT-VAPB complex. Another complex consisting of PLIN5 and FATP479 was shown to enhance LD-mitochondria contact and increase fatty acid oxidation, however, direct evidence of fatty acid transfer by this complex requires confirmation. Taken together, these findings support a model whereby various protein complexes co-exist to coordinate membrane tethering and/or inter-organellar fatty acid transfer for efficient oxidation, and at least in the cell types examined herein, that the ESYT/VAPB complex is requisite for efficient fatty acid metabolism and to prevent cellular lipotoxicity. In addition, fatty acid transfer between organelles would require the conversion of fatty acid to fatty acyl-CoA esters80 for mitochondrial uptake. In this context, we identified ACSL3 in the split-BioID analysis. This protein has long chain acyl-coenzyme A synthetase activity and is likely to facilitate this function.
ESYT and VAPB proteins are localized at contact sites of several cellular organelles. ESYT has been reported at ER-plasma membrane and ER-mitochondria contact sites53,69,81,82, and both VAPB and ESYT1/2 are enriched in other proximity based proteomic mapping of LDs83 and the outer mitochondria membrane84. VAPB has docking and tethering functions85, most notably with the protein tyrosine phosphatase-interacting protein-51 (PTPIP51) to regulate ER-mitochondria contacts85. In these cases, VAPB anchors to the ER membrane using its C-terminal transmembrane domain while it interacts with the other proteins or protein complex using its N-terminal major sperm domain86. While our data show that ESTY1/2 and VAPB interact (i.e., by HA-affinity purification and mass spectrometry, immunoprecipitation and immunoblot analysis, and FLIM-FRET) and places the ESYT/VAPB complex at the interface of LD-mitochondria-ER, we have an incomplete understanding of how the components are organized (Fig. 8). We suggest that VAPB interacts with mitochondria, while ESYTs bind the ER and VAPB to interact with LDs to facilitate fatty acid transfer. Future studies using high temporal and nanoscale spatial resolution approaches will be required to provide further insights48,49,87.

Left panel: TEM image of HeLa cells showing an exploratory tripartite interaction among LDs, mitochondria and the ER. Right panel: Summarizing model of complex formation of ESYT1, ESYT2 and VAPB at the interface of these organelles. Created in BioRender. Keenan, S. (2025) https://BioRender.com/n75s805.
The prevailing view that mitochondria-LD coupling promotes flux of fatty acids from LDs for mitochondrial β-oxidation has been challenged by emerging evidence indicating the existence of metabolically distinct subpopulations of mitochondria that promote opposing functions of fatty acid oxidation or fatty acid synthesis and/or LD biogenesis. Some suggest that LD-associated mitochondria support higher rates of fatty acid oxidation22,33,79,88,89, while others indicate that LD-associated mitochondria have low fatty acid oxidation capacity and support LD expansion by providing pyruvate-derived ATP for triglyceride synthesis23,48. In these latter studies, fatty acids were shown to be selectively trafficked to and oxidized in mitochondria that were not in contact with LDs23,48. Our data using complimentary fatty acid tracing methods show that expression of wild type ESYT1/2, but not ESYT1/2 with impaired fatty acid binding capacity, is required for LD-derived fatty acid oxidation, which supports the premise that LD-associated mitochondria are important for cellular fatty acid oxidation.
There are several limitations to this study. While many lines of evidence strongly support a role for ESYTs in lipid transport, direct biochemical evidence or single-particle imaging for ESYT-mediated fatty acid transfer remains to be established. Additionally, alternative mechanisms could explain ESYT/VAPB effects on fatty acid oxidation, perhaps via phospholipid transfer from LDs to ER, and such possibilities require evaluation.
Impaired fatty acid metabolism and lipotoxicity are a common feature of metabolic diseases, including obesity, non-alcoholic fatty liver disease, diabetes, some cancers, and neurodegenerative diseases49,70,71,90,91,92. ESYT deletion results in susceptibility to cellular stress and lipotoxicity in cells, suggesting that impaired ESYT function may contribute to excessive lipid accumulation in tissues and disease pathogenesis. Further, our indexing of proteins residing at LD-mitochondria contact sites provides a valuable resource for further research that could lead to strategies to mitigate metabolic and other disease states.
Methods
Cell culture
HeLa, HepG2 and HEK293T cells were cultured in high glucose and GlutaMAX (Thermo Fisher Scientific; 11965092) supplemented with 10% fetal bovine serum (FBS; Cell Sera; AU-FBS/PG) and 1% Penicillin-Streptomycin (10,000 U/mL; Thermo Fisher Scientific; 15140122) at 37 °C in a 5% CO2 incubator. For starvation experiments, cells were incubated in DMEM low glucose (5.5 mM) (Thermo Fisher Scientific; 11054020) supplemented with glutamine for the indicated time.
Molecular cloning
Plasmids generated for this study were assembled using the Gibson assembly method93 and Q5® Site-Directed Mutagenesis Kit (E0554) from New England Biolabs (NEB). DNA was transformed into NEB® 5-alpha Competent E. coli cells via heat shock at 42 °C for 30 sec, followed by incubation for 2 min on ice and 1 h at 37 °C in Luria-Bertani (LB) on an Eppendorf Thermomixer shaker set to 300 rpm. Transformed bacteria were spread onto LB plates (containing either Ampicillin or Kanamycin) and incubated at 37 °C overnight. DNA was isolated from the resulting colonies using QIAprep Spin Miniprep Kit (Qiagen) and sequenced to verify the insertion of the respective DNA.
Transfections and transductions
All stable cell lines were produced by transducing with lentiviral or retroviral particles followed by either antibiotic selection or fluorescence-activated cell sorting. Lentiviral particles were produced by transfecting HEK293T cells in 6-well plates with 1.5 μg of vector and 1.0 μg and 0.5 μg of the packaging plasmids (i.e., proportions 3:2:1) using Lipofectamine LTX. The packaging plasmids for lentiviral transduction were psPAX2 (Addgene #122600) and pMD2.G (Addgene #12259). The packaging plasmids for retroviral transduction were pUMVC3 and pCMV-VSV-G plasmids (Addgene #8449 and 8454). The transfection media was removed after 16 h and virus-containing supernatants was collected after 48 h and filtered using a 0.45 μM PES filter (Merck). Target cells were transduced in 6-well plates using 500 μL of viral supernatant and 500 μL of fresh medium supplemented with 8 μg/mL of polybrene (Sigma-Aldrich). Selection with the appropriate antibiotic or fluorescence sorting was initiated after 48 h.
Generation of KO cells
ESYT1KO, ESYT2KO and VAPBKD HeLa cells were generated by the CRISPR/Cas9 system. The DNA oligonucleotide sequence (Star Method) was chosen as a guide RNA using CHOPCHOP. Two gRNAs oligos were designed and separately cloned into All-in-one plasmid system (pSPCas9(BB)-2A-GFP vector; Addgene #48138). HeLa cells transfected with the plasmids (GFP + ) were sorted into a 96 well plate using a FACS ARIA III (BD Biosciences) at a density of 1 cell per well. ESYT1, ESYT2 or VAPB deficiency was confirmed by immunoblot analysis and Sanger sequencing. The gRNAs used were: TGTTTTCCCTTACCGGGCGTCGG and CGCAAAACGCCATGTAGCTGAGG for ESYT1; AGTGGAACGGTGATTCGATTGGG and GACACGCTTACCTTTGGTACAGG for ESYT2; CAGCACGAGCTCAAATTCCG and TGAGCTCGTGCTGCGGCTCG for VAPB; and GGCTTCGCGCCGTAGTCTTA for control/scramble.
Immunoblot analysis
Cells were washed once with ice-cold PBS, centrifuged at 800 × g for 5 min at 4 °C, and lysed in RIPA buffer (50 mM Tris HCL, pH 7.4; 150 mM NaCl; 0.1% SDS; 0.5% sodium deoxycholate; 1% Triton X-100) supplemented with protease inhibitor cocktail (cOmplete, Roche) and phosphatase inhibitor (PhosSTOP, Roche) on ice. Cell lysates were clarified by centrifugation, and the protein concentration was determined using a BCA protein assay kit (Pierce, Thermofisher Scientific). After mixing with 5X Laemmli buffer, samples were subjected to 10% or 12% SDS-PAGE. Proteins were then transferred to a nitrocellulose membrane (0.45 µm, #1620264; Bio-Rad) followed by blocking with 5% (wt/vol) skim milk in TBS-T for 2 h at RT. Incubation with primary antibody was performed overnight at 4 °C followed by three washes for 5 min each in TBS-T. The following primary antibodies were used for immunoblotting: ESYT1 (ab118805, Abcam, 1:1,000), ESYT2 (HPA002132, Sigma-Aldrich, 1:1000), VAPB (ab241298, Abcam, 1:1000), RAB1 (ab302545, Abcam, 1:1,000), RAB7 (ab137029, Abcam, 1:1,000), RAB7 (ab137029, Abcam, 1:1,000), MBOAT7 (ab262944, Abcam, 1:1,000), PLIN2 (ab220738, Abcam, 1:1,000), GAPDH (ab9482, Abcam, 1:1,000), HMGB1 (ab18256, Abcam, 1:1,000), Gasdermin D (ab209845, Abcam, 1:1,000), Total OXPHOS (ab110413, Abcam, 1:1,000), ATGL (2138S, Cell Signaling Technology, 1:1000), HSL (4107 s, Cell Signaling Technology, 1:1000), BIP (3177S, Cell Signaling Technology, 1:1000), p-PERK (Thr980, 3179S, Cell Signaling Technology, 1:1000), PERK (3192S, Cell Signaling Technology, 1:1000), PDI (3501S, Cell Signaling Technology, 1:1000), p-JNK (Thr183/Tyr185, 9251S, Cell Signaling Technology, 1:1000), JNK (9252S, Cell Signaling Technology, 1:1000), p-ERK1/2 (Thr202/Tyr204, 9101S, Cell Signaling Technology, 1:1000), ERK1/2 (4695S, Cell Signaling Technology, 1:1000), cleaved caspase-1 (89332S, Cell Signaling Technology, 1:1000), ATF6 (70B1413., Enzo Life Sciences, 1:1000) and Streptavidin (SA10001, Thermo Fisher Scientific). Secondary antibodies were peroxidase-conjugated anti-rabbit used at a 1:5,000 dilution at RT for 2 h. The bound antibodies were detected by Clarity Western ECL Reagent (Bio-Rad) and visualized with Molecular Imager® ChemiDoc™ XRS+ (Bio-Rad). Western blots were quantified using Image Lab software (ver. 6.10 build 7, Bio-Rad).
In cell biotin labeling with BioID and Split-BioID and mass spectrometry analysis
For biotin labeling, we generated stably expressing full length BirA* (AAMTMD-RFP-HA-BirA* or Fis1TMD-CFP-FLAG-BirA*) and split BirA* (AAMTMD -RFP-FLAG-BirAN and BirA-C-HA-mTurquiose2CFP-Fis1TMD), using pBMN-Z vector (Addgene#1734, #36047), pcDNA3.1- PP1g -Flag - BirA* (AA2-140) (Addgene#86886), pcDNA3.1 - BirA* (AA141-321) - HA - NIPP1 (Addgene#86885), pPalmitoyl-mTurquoise2 (Addgene#36209) and pCytERM_mScarlet_N1(Addgene#85066). We used the first (E140/Q141) of several described splitting sites to generate the two BirA* fragments44,45. Cells were seeded at 50% confluency in a 10 cm petri dish the day prior to experiments. Biotin (10 mM) was diluted in complete media and added directly to cells to a final concentration of 50 μM and incubated at 37 °C for 16 h. Non-transfected HepG2 cells treated with 50 μM biotin for 16 h used as control. For both immunoblot and mass spectrometry experiments, labeling was stopped after the indicated time periods by transferring cells on plates to ice and washing gently with cold DPBS. Cells were dislodged by scrapping in DPBS containing 1 μM EDTA.
For BioID experiments, cell lysates were obtained by centrifugation and loaded consecutively on spin columns (Pierce) containing streptavidin-coated beads prewashed with lysis buffer 1 (Pierce) and biotinylated proteins were enriched94. The reduction of cysteine bonds was mediated by 5 mM Tris(2-carboxyethyl)phosphine (TCEP) for 30 mins at 37 °C and alkylation with 10 mM iodoacetamide. Beads were then resuspended in digestion buffer containing sequencing grade modified trypsin (Pierce) at 37 °C overnight. After quenching with 0.2% TFA, the samples were desalted by C18 reversed-phase spin columns according to the manufacturer’s instructions (Pierce) and eluted with 0.2% TFA and 80% acetonitrile (ACN). The eluted peptide sample was dried in a vacuum centrifuge and reconstituted to a final volume of 30 μl in 0.2% TFA and 1% acetonitrile (ACN). Analysis was performed on a Q-Exactive Plus mass spectrometer (Thermo Fisher Scientific) coupled to an Ultimate 3000 liquid chromatography system. Peptides were loaded on to an Acclaim C18 PepMap nano Trap x 2 cm, 100 µm I.D, 5 µm particle size, 300 Å pore size trap column (Thermo Fisher Scientific) at 15 µl/min for 3 min prior to switching the trap in line with the analytical column (Acclaim RSLC C18 PepMap Acclaim RSLC nanocolumn 75 μm × 50 cm, PepMap100 C18, 3 μm particle size 100 Å pore size; ThermoFisher Scientific). A 65 min 250 nl/min non-linear gradient (buffer A, 0.1% formic acid, 2% ACN and buffer B, 0.1% formic acid, 80% ACN) was used to separate peptides. Data were collected in positive mode using Data Dependent Acquisition using m/z 375–1400 as MS scan range, HCD for MS/MS of the 15 most intense ions with charge ≥ 2. Other instrument parameters were: MS1 scan at 70,000 resolution (at 200 m/z), MS maximum injection time 50 ms, AGC target 3E6, MS/MS resolution 17,500, MS/MS AGC target of 5E4, MS/MS maximum injection time 50 ms, minimum intensity was set at 5E3 and dynamic exclusion was set to 30 s.
Raw files were analyzed using the MaxQuant platform95 version 1.6.5.0 searching against the UniProt human database containing 20,399 reviewed, canonical entries (January 2019) and a database containing 246 common contaminants. Default search parameters were used with “Label free quantitation” set to “LFQ”. Trypsin/P cleavage specificity (cleaves after lysine or arginine, even when proline is present) was used with a maximum of 2 missed cleavages. Oxidation of methionine and N-terminal acetylation were specified as variable modifications. Carbamidomethylation of cysteine was set as a fixed modification. A search tolerance of 4.5 ppm was used for MS1 and 20 ppm for MS2 matching. False discovery rates (FDR) were determined through the target-decoy approach set to 1% for both peptides and proteins. Using the Perseus platform96 version 1.6.5.0, proteins group LFQ intensities were log2 transformed. Values listed as being “Only identified by site,” “Reverse,” or “Contaminants” were removed from the data set. Experimental groups were assigned to each set of quadruplicates and the rows filtered to contain at least one group where the number of valid values was 3. Using the “Subtract row cluster” function, the dataset was normalized using to the average abundance of cluster of endogenously biotinylated proteins containing ACACA, MCCC1, PC, and PCCA identified through hierarchical clustering. A modified two-sided t-test based on permutation-based FDR statistics96 was performed between each experimental group and the control, where significance was determined by an s0 factor of 1 at 5% FDR. To identify candidate interface proteins, we intersected the resulting lists from the proteomes of full-length LD-BioID, full-length mito-BioID, and split-BioID. We did not apply additional layers of filtering to avoid excluding important proteins involved in broader or complex processes, especially those that are less studied, have undefined localizations, or undergo dynamic transitions between cellular compartments.
Affinity purification of the interacting proteins
HepG2 cells stably expressing empty vector (3HA-eGFP, control), 3HA-eGFP-ESYT1, 3HA-eGFP-ESYT2 or 3HA-eGFP-VAPB were grown on 10 cm dishes. Cells were lysed in 0.75 ml lysis buffer (25 mM HEPES pH 7.4, 150 mM NaCl, 1 mM EDTA, 10% glycerol, 1% wt/vol n-dodecyl-β-D-maltoside) on ice for 30 min. The lysates were cleared by centrifuging at 18,000 × g for 15 min at 4 °C. An equal volume of cell lysates (1∼2 mg of protein) was mixed with 25 µl EZview™ Red Anti-HA Agarose beads (#E6679; Sigma-Aldrich). The protein bead mixture was gently rotated at 4 °C overnight, followed by washing three times with PBS and centrifuging at 16,000 × g for 3 min at 4 °C. For immunoblot analysis, immunoprecipitated proteins were eluted with 30 µl 2 × Laemmli sample buffer by vortexing at RT for 5 min then denatured at 95 °C for 5 min. The resultant samples were subjected to SDS-PAGE and immunoblotting. For proteomic analysis of the immunoprecipitated proteins, the cysteine bonds were reduced on beads with 5 mM Tris(2-carboxyethyl)phosphine (TCEP) for 30 mins at 37 °C followed by alkylation with 10 mM iodoacetamide. Beads were then resuspended in digestion buffer containing sequencing grade modified trypsin (Pierce) at 37 °C overnight. After quenching with 10% TFA, the samples were desalted by C18 reversed-phase spin columns according to the manufacturer’s instructions (Pierce). The eluted peptide sample was dried in a vacuum centrifuge and reconstituted to a final volume of 30 μl in 0.1% TFA and 1% CH3CN. Analysis was performed on a Q-Exactive mass spectrometer (Thermo Fisher Scientific) coupled to liquid chromatography system. The raw files were first searched by Maxquant and enriched proteins were analyzed by Perseus platform using the same search parameter as indicated above.
LD and mitochondria staining
Cells were treated with 500 µM oleate/palmitate (2:1) conjugated with 1% BSA for indicated times followed by 10 min fixation with Image-iT™ Fixative Solution (4% formaldehyde, methanol-free) at RT. After three washes with PBS, cells were permeabilized with 0.1% Triton X-100 for 10 min and blocked for 1 h at room temperature with 2% BSA in PBS. LDs were stained with HCS LipidTOX Deep Red Neutral Lipid Stain (1:1,000; Invitrogen) and mitochondria with anti-TOMM20 (Abcam) in PBS (2% BSA) for 1 h, followed by three washes in PBS at RT with light avoidance. Slides were mounted in Vectashield antifade aqueous mounting medium (Vector Laboratories).
Airyscan imaging
Images were acquired on a Zeiss LSM 880 confocal microscope with an Airyscan detector (Carl Zeiss). Data were collected using either 63x oil (1.4 NA) or 40x oil (1.3 NA) objective lens for most experiments. Samples were excited using laser lines of 405, 458, 488, 561, 594 and 633 nm and the emitted signal was detected on Airyscan detector using ‘SuperResolution’ (SR) mode. Where z-stacks were collected, the software-recommended optimal slice sizes were used. For all experiments, images were collected sequentially to minimize bleed through. To maintain clarity and uniformity throughout the paper, some images have been pseudocolored.
Deconvolution and 3D rendering
The raw Airyscan images containing 32-phase images were deconvolved with Huygens Professional Software (v22.10, Scientific Volume Imaging) using Array Detector deconvolution module. The deconvolved images were imported to Imaris software (v9.9, Oxford Instruments) and the whole cell, LD and mitochondria were reconstructed to 3D surfaces. To retain the structure detail, surfaces were created without applying smoothing. Using the whole cell mask, individual LDs and mitochondria surfaces were labeled per cell and the statistics were drawn per cell accordingly. The surface area, number, and volume were exported from the Statistics tool. In measuring LD-mito contact site, the distance between LDs and mitochondria was measured as the shortest distance between the organelle surfaces border and the distance <30 nm were considered as touching objects.
Fluorescence lifetime imaging microscopy of Förster resonance energy transfer
All live and fixed cell fluorescence lifetime microscopy (FLIM) measurements of Förster resonance energy transfer (FRET) were performed on an Olympus FV3000 laser scanning microscope coupled to a 488 nm pulsed laser operated at 80 MHz and an ISS A320 FastFLIM box. A 60x water immersion objective (1.2 NA) was used for all experiments, and the cells were imaged at 37 °C. Prior to acquisition of FLIM data in the donor channel (eGFP-ESYT1, eGFP-ESYT2, eGFP-VAPB) for FRET analysis with the acceptor channel (mCherry-VAPB), multi-channel intensity images were acquired from each selected HepG2 cell to verify that the FRET acceptor was present in excess of the donor (i.e., acceptor–donor ratio > 1), and in the case of fixed cell experiments where immunofluorescence against PLIN2 (AF405) and TOMM20 (AF647) was performed, to identify the localization of LD-mitochondria contacts sites for a masked FRET analysis of FLIM data. This involved sequential imaging of a two-phase light path in the Olympus FluoView software. The first phase was set up to image eGFP and mCherry via use of solid-state laser diodes operating at 488 and 561 nm, respectively, with the resulting signal being directed through a 405/488/561/633 dichroic mirror to two internal GaAsP photomultiplier detectors set to collect 500–540 nm and 600–700 nm. The second phase was set up to image AF405 and AF647 via use of solid-state laser diodes operating at 405 and 633 nm, respectively, with the resulting signal being directed through a 405/488/561/633 dichroic mirror to two internal GaAsP photomultiplier detectors set to collect 420–460 nm and 600–700 nm. Then in each HepG2 cell selected, a FLIM map of eGFP was imaged within the same field of view (256 × 256-pixel frame size, 20 µs/pixel, 90 nm/pixel, 20 frame integration) using the ISS VistaVision software. This involved excitation of eGFP with an external pulsed 488 nm laser (80 MHz) and the resulting signal being directed through a 405/488/561/633 dichroic mirror to an external photomultiplier detector (H7422P-40 of Hamamatsu) that was fitted with a 520/50 nm bandwidth filter. The donor signal in each pixel was then subsequently processed by the ISS A320 FastFLIM box data acquisition card to report the fluorescence lifetime of eGFP. All FLIM data were pre-calibrated against fluorescein at pH 9 which has a single exponential lifetime of 4.04 ns.
FLIM-FRET analysis
The fluorescence decay recorded in each pixel of an acquired FLIM image was quantified by the phasor approach to lifetime analysis97. As described in previously published papers98, this results in each pixel of a FLIM image giving rise to a single point (phasor) in the phasor plot, which when used in the reciprocal mode enables each point in the phasor plot to be mapped to each pixel of the FLIM image. Since phasors follow simple vector algebra, it is possible to determine the fractional contribution of two or more independent molecular species coexisting in the same pixel. For example, in the case of two independent species, all possible weightings give a phasor distribution along a linear trajectory that joins the phasors of the individual species in pure form. While in the case of a FRET experiment, where the lifetime of the donor molecule is changed upon interaction with an acceptor molecule, the realization of all possible phasors quenched with different efficiencies describes a curved FRET trajectory in the phasor plot that follows the classical definition of FRET efficiency98.
In the context of the FLIM-FRET experiments presented in this manuscript, the phasor coordinates (g and s) of the unquenched donor (eGFP-ESYT1, eGFP-ESYT2, eGFP-VAPB) were first determined independently in fixed and live HepG2 cells to enable definition of a baseline from which a FRET trajectory could be extrapolated and then used to determine the dynamic range of FRET efficiencies that describe FRET interaction with an acceptor (mCherry-VAPB). All FLIM-FRET quantification was performed in the SimFCS software developed at the LFD.
Fluorescence fluctuation spectroscopy (FFS)
All FFS measurements for Number and Brightness (NB) analysis and cross RICS were performed on an Olympus FV3000 laser scanning microscope coupled to an ISS A320 Fast FLIM box for fluorescence fluctuation data acquisition. For single channel NB FFS measurements, eGFP tagged plasmids were excited by a solid-state laser diode operating at 488 nm and the resulting fluorescence signal was directed through a 405/488/561 dichroic mirror to an external photomultiplier detector (H7422P-40 of Hamamatsu) fitted with an eGFP 500/25 nm bandwidth filter. For dual channel RICS FFS measurements (that enable cross RICS), the eGFP and mCherry plasmid combination, were excited by solid-state laser diodes operating at 488 nm and 561 nm, respectively, and the resulting signal was directed through a 405/488/561/640 dichroic mirror to two internal GaAsP photomultiplier detectors set to collect 500–540 nm and 620–720 nm, respectively.
All FFS data acquisitions employed a 60X water immersion objective (1.2 NA) and first involved selecting a 10.6 μm region of interest (ROI) within a HepG2 cell at 37 °C in 5% CO2. Then a single or simultaneous two channel frame scan acquisition was acquired (N = 100 frames) in the selected ROI with a pixel frame size of 256 × 256 (i.e., pixel size ~41 nm) and a pixel dwell time of 12.5 µs. These conditions resulted in a scanning pattern that was found to be optimal for simultaneous capture of the apparent brightness and mobility of the different eGFP and mCherry constructs being characterized by NB and cross RICS analysis; all of which was performed in the SimFCS software developed at the Laboratory for Fluorescence Dynamics (LFD).
Number and brightness (NB) analysis
The oligomeric state of the different eGFP-tagged plasmids investigated (i.e., eGFP-ESYT1, eGFP-ESYT2, and eGFP-VAPB) was extracted and spatially mapped throughout single channel FFS measurements via a moment-based brightness analysis that has been described in previously published papers99. In brief, within each pixel of an NB FFS measurement there is an intensity fluctuation \({{{\rm{F}}}}({{{\rm{t}}}})\) which has: (1) an average intensity \(\left\langle {{{\rm{F}}}}\left({{{\rm{t}}}}\right)\right\rangle\) (first moment) and (2) variance \({\sigma }^{2}\) (second moment); and the ratio of these two properties describes the apparent brightness (B) of the molecules that give rise to the intensity fluctuation. The true molecular brightness (ɛ) of the fluorescent molecules being measured is related to B by \({{{\rm{B}}}}={{{\rm{\varepsilon }}}}+1\), where 1 is the brightness contribution of a photon counting detector. Thus, if we measure the B of monomeric eGFP (Bmonomer = ɛmonomer + 1) under our NB FFS measurement conditions, then we can determine ɛmonomer and extrapolate the expected B of eGFP-tagged dimers (Bdimer = (2 x ɛmonomer) + 1) or oligomers (e.g., Btetramer = (4 x ɛmonomer) + 1), and in turn define brightness cursors, to extract and spatially map the fraction of pixels within a NB FFS measurement that contain these different species. These defined brightness cursors were used to extract the fraction of eGFP-ESYT1, eGFP-ESYT2, and eGFP-VAPB dimer and oligomer (i.e., number of pixels assigned Bdimer or Boligomer) within a NB FFS measurement and quantify the degree of protein homodimer and oligomer fractions across multiple cells. Artifact due to cell movement or photobleaching were subtracted from acquired intensity fluctuations via use of a moving average algorithm and all brightness analysis was carried out in SimFCS from the Laboratory for Fluorescence Dynamics.
RICS and cross RICS analysis
The fraction of interaction between the eGFP tagged ESYT1, ESYT2 and VAPB with mCherry tagged VAPB was extracted via application of the cross RICS functions described in previously published paper to the dual channel FFS measurements100. In brief, the fluorescence intensity recorded within each frame (N = 100) of each channel (i.e., CH1 and CH2) was spatially correlated via application of the RICS function, and spatially cross-correlated between channels (CC) via application of the cross RICS function, alongside a moving average algorithm (N = 10 frames) in both instances. Then the recovered RICS and cross RICS correlation profiles were fit to a 3D diffusion model and the amplitude versus decay of each fit recorded in the form of a G value. The ratio of the cross RICS amplitude (i.e., GCC) with the limiting channel RICS amplitude (i.e., GCH1 or GCH2) enabled the fraction of eGFP-ESYT1, eGFP-ESYT2, and eGFP-VAPB molecules interacting with VAPB-mCherry molecules to be extracted. All cross RICS analysis was carried out in SimFCS from the Laboratory for Fluorescence Dynamics.
Radiolabeled fatty acid tracer studies
To accumulate radiolabeled fatty acids in LDs, cells were treated for indicated times with 1 μCi/ml of [14C]-oleic acid (NEC317050C, PerkinElmer, Waltham, Massachussetts) and 500 μM oleic acid (Sigma#O1006) conjugated to 1% BSA (wt/vol) in low glucose DMEM (1 g per liter; Gabico#10567-014). Oxidation measurements were performed by trapping the released 14CO2 in 1 M sodium hydroxide in sealed system. Incompletely oxidized acid-soluble metabolites containing 14C and lipids were extracted from the total cell lysate mixed with 3 volumes of chloroform/methanol (2:1). The upper phase containing incompletely oxidized fatty acids were added to 2 mL scintillation cocktail and analyzed by liquid scintillation counting while the bottom organic phase was collected and dried.
Lipids were resuspended in chloroform/methanol (2:1) containing a standard mix of glyceryl tripalmitate (1.75 mg/ml; Sigma #T588), dipalmitin (0.875 mg/ml; Sigma #D2636), oleate (1.7 mg/ml; Sigma #O1006), and C24 ceramide (1 mg/ml, Sigma #43799). The mixture was spotted on 60-Å silica gel TLC plates (Whatmann, Partisil LK6D) and dried for several minutes at RT. Chloroform/methanol/water (65:30:5) solvent was used for initial separation until the solvent front reached ~30% of the plate. Next, after drying, plates were resolved in a solvent mixture of hexane/diethyl ether/acetic acid (70:30:1) until the solvent front reached 1 cm from the top of the plate. Lipid spots were then visualized by staining with 0.02% dichlorofluorescein (Sigma #D6665) and radioactive bands corresponding to different lipid standards were scraped and mixed in 2 mL scintillation cocktail and counted on a scintillation counter.
Fluorescent fatty acid tracer experiments
HeLa cells were seeded in 12 well plates in complete medium (DMEM Glutmax with 10% fetal bovine serum). Twenty-four hours after seeding, the cells were pulsed in low-glucose DMEM containing 1 μM BODIPY 558/568 C16 (Thermo Fisher Scientific, D3821). The cells were then washed three times with DPBS and chased for the indicated times in low-glucose medium. Fluorescence quantifications were performed at room temperature with a CLARIOstar plus fluorimeter (BMG LabTech).
Gas chromatography–mass spectrometry stable isotope labeling analysis
Cells were washed with Milli-Q water at 37 °C then snap-frozen by covering the plate in liquid nitrogen. Metabolites were extracted on ice by addition of 600 µl/well of methanol:chloroform (9:1 v/v), containing the internal standard, scyllo-inositol (16.6 µM). Cells were scraped and incubated on ice for 10 min. Samples were then centrifuged (5 min, 6000 g, 4 °C) to pellet precipitated proteins and the supernatants transferred to fresh Eppendorf tubes.
Cell extracts were transferred to vial inserts and evaporated to dryness under vacuum. Polar metabolites were derivatized online using an AOC 6000 autosampler robot (Shimadzu). Samples were first methoxyaminated by the addition of 25 µL methoxyamine (30 mg/mL in pyridine, 2 h, 37 °C, 750 rpm), followed by trimethylsilylation with 25 µL N, O-Bis(trimethylsilyl)trifluoroacetamide (BSTFA) + 1% trimethylchlorosilane (TMCS; 1 h, 37 °C, 750 rpm). Following a 1 µl splitless injection, metabolite profiles were acquired on a 2030 Shimadzu gas chromatograph and a TQ8050NX triple quadrupole mass spectrometer (Shimadzu, Japan). The mass spectrometer was tuned according to the manufacturer’s recommendations using tris-(perfluorobutyl)-amine (CF43). GC-MS was performed on a 30 m Agilent DB-5 column with 0.25 mm internal diameter column and 1 µm film thickness. The injection temperature (inlet) was set at 280 °C, the MS transfer line at 280 °C and the ion source adjusted to 200 °C. Helium was used as the carrier gas at a flow rate of 1 mL/min and the mass spectrometer was set to scan mode with a scan range of 50–500 m/z. The analysis of TMS samples was performed under the following oven temperature program; 100 °C start temperature, hold for 4 min, followed by a 10 °C min−1 oven temperature ramp to 320 °C with a following final hold for 11 min.
13C-derived carbon labeling was determined in key metabolites of the glycolytic and tricarboxylic acid (TCA) cycle via mass isotopomer peak shift analysis. Data files were imported into DExSI101 (V3.05), which enabled quantitation of mass isotopologues for relevant 13C-labeled metabolites. Fractional abundance data and mass isotopologue distributions were produced following correction for natural background isotopic abundance.
Lipidomic analysis
Lipidomic extraction
Briefly, 1 × 106 cells were lysed in water/methanol (1:3.5 v/v) with 10 µl internal standard (Splash Lipidomix (Cat no. 330707) Deuterated Ceramide Lipidomix (Cat no. 330713), Cholesterol-d7 (Cat no. 700041 P), Avanti Polar Lipids, USA). Then, 1 mL MTBE was added, and the mixture was shaken for 30 min at room temperature. Phase separation was induced by adding 0.25 mL of MS-grade water and centrifuged at 1,000 g for 20 min. The upper (organic) phase was collected and dried in a vacuum centrifuge. Samples were resuspended in 100 µl chloroform/methanol (1/9, v/v), centrifuged at 14,000 g for 10 min and transferred to glass LCMS vial.
Ultrahigh-performance liquid chromatography (UHPLC) and mass spectrometric (MS) analyses
Samples were analyzed by ultrahigh performance liquid chromatography (UHPLC) coupled to tandem mass spectrometry (MS/MS) employing a Vanquish UHPLC linked to an Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher Scientific, San Jose, CA, USA), with separate runs in positive and negative ion polarities. Solvent A was 6/4 (v/v) acetonitrile/water with 5 mM medronic acid and solvent B was 9/1 (v/v) isopropanol/acetonitrile. Both solvents A and B contained 10 mM ammonium acetate. 10 uL of each sample was injected into an Acquity UPLC HSS T3 C18 column (1 × 150 mm, 1.8 µm; Waters, Milford, MA, USA) at 50 °C at a flow rate of 80 μl/min for 3 min using 3% solvent B. During separation, the percentage of solvent B was increased from 3% to 70% in 5 min and from 70% to 99% in 16 min. Subsequently, the percentage of solvent B was maintained at 99% for 3 min. Finally, the percentage of solvent B was decreased to 3% in 0.1 min and maintained for 3.9 min.
All MS experiments were performed using an electrospray ionization source in positive mode at 3.5 kV and negative mode at 3.0 kV separately for each sample. The flow rates of sheath, auxiliary and sweep gases were 25 and 5 and 0 arbitrary unit(s), respectively. The ion transfer tube and vaporizer temperatures were maintained at 300 °C and 150 °C, respectively, and the ion funnel RF level was set at 50%. In the positive mode from 3 – 24 min, the top speed data-dependent scan with a cycle time of 1 s was used. Within each cycle, a full-scan MS-spectra were acquired firstly in the Orbitrap at a mass resolving power of 120,000 (at m/z 200) across an m/z range of 300–2000 using quadrupole isolation, an automatic gain control (AGC) target of 4e5 and a maximum injection time of 50 milliseconds, followed by higher-energy collisional dissociation (HCD)-MS/MS at a mass resolving power of 15,000 (at m/z 200), a normalized collision energy (NCE) of 27% at positive mode and 30% at negative mode, an m/z isolation window of 1, a maximum injection time of 35 milliseconds and an AGC target of 5e4. For the improved structural characterization of glycerophosphocholine (PC) lipid cations, a data-dependent product ion (m/z 184.0733)-triggered collision-induced dissociation (CID)-MS/MS scan was performed in the cycle using a q-value of 0.25 and a NCE of 30%, with other settings being the same as that for HCD-MS/MS. For the improved structural characterization of triacylglycerol (TG) lipid cations, the fatty acid + NH3 neutral loss product ions observed by HCD-MS/MS were used to trigger the acquisition of the top-3 data-dependent ion trap CID-MS3 scans in the cycle using a q-value of 0.25 and a NCE of 30%, with other settings being the same as that for HCD-MS/MS.
For identification and quantification of lipids and statistical analysis, LC-MS/MS data was searched through MS Dial 4.90. The mass accuracy settings are 0.005 Da and 0.025 Da for MS1 and MS2. The minimum peak height is 50000 and mass slice width is 0.05 Da. The identification score cut off is 80%. Post identification was done with a text file containing name and m/z of each internal standard. In positive mode, [M + H]+, [M + NH4]+ and [M + H-H2O]+ were selected as ion forms. In negative mode, [M-H]- and [M + CH3COO]- were selected as ion forms. All lipid classes available were selected for the search. PC, LPC, DG, TG, CE, and SM were identified and quantified at positive mode while Cer and CL were identified and quantified at negative mode. The retention time tolerance for alignment is 0.1 min. Lipids with maximum intensity <5-fold of average intensity in blank were removed. All other settings were default. All lipid LC-MS features were manually inspected and re-integrated when needed.
Triglyceride assessment
Triglyceride content was measured using an Infinity Triglycerides kit (Thermo Scientific #TR22421), following the manufacturer’s protocol. Briefly, lipids were extracted and dried as described above, resuspended in ethanol, then transferred into 96 well plates containing 200 μL Infinity Triglyceride reagent. A standard curve was generated using glyceryl trioleate (Sigma#92860) and the absorbance measured at 500 nm using a CLARIOstar plus fluorimeter (BMG LabTech).
In silico modeling and docking analyses
The ESYT1/ESYT2 complex was modeled using the experimental crystal structure of the ESYT2 dimer (PDB ID: 4P42)52, based on a sequence identity of 39% between homologs. As the ESYT2 dimer was asymmetric, two models were generated: one having ESYT1 replacing homodimer chain B (model 1) and the other replacing chain A (model 2). Models were built using Advanced Homology Modeling in Schrodinger Maestro v.11.4, after preprocessing of experimental structure 4P42 filled in missing atoms and residues in Prime. Both models were used for further analyses and lipid docking.
Initial fragment hotspot analysis using CCDC102 on both models identified an apolar groove within chain B, which was used along with SiteMap within Schrodinger to guide the search space for docking oleic acid, linoleic acid, palmitic acid, triacylglycerol, phosphatidylcholine and cholesteryl oleate. Lipid ligand files were initially obtained from PubChem, and subjected to ligand preparation using LigPrep, which assigns rotations within single bonds to permit the sampling of accurate ligand poses. Docking was carried out on both models (ESYT2 in model 1, ESYT1 in model 2) using Schrodinger Prime.
Within the lipid binding cavity, 9 residues within ESYT2 (model 1) and 7 residues within ESYT1 (model 2) were tested for stability changes upon mutation to larger residues, using mCSM-Stability103. Residues were chosen as pairs on opposite sides of the lipid channel entrance, middle and deeper ends, for their potential to block lipid entry upon mutation, where mutants were determined according to channel depth at residue position.
The in silico saturation mutagenesis at the protein interface for both model structures was carried out via mCSM-PPI2104, which uses a threshold of 5 Å distance from any other molecule in a 3D space to define interface residues for any given complex structure. Each interface residue is then mutated to each one of the other 19 standard amino acids, and served as input for a machine learning model that predicts changes in protein binding affinity.
Oxygen consumption measurements
The oxygen consumption rate (OCR) of HeLa cells (Control and KO cell lines) was measured using a Seahorse XF24 extracellular flux analyzer (Seahorse Bioscience). HeLa cells were seeded at 30,000 per well in 24-well XF plates the day before assay. Basal medium supplemented with 1 mM pyruvate, 10 mM glucose and 2 mM glutamine was added to each well of a Seahorse XF24 cell culture plate and incubated overnight at 37 °C. Cells were equilibrated for 1 h in XF assay medium in a non-CO2 incubator. OCR was monitored according to the manufacturer recommendations by sequential injections of oligomycin (1 μM), carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (1 μM) and rotenone/antimycin A (0.5 μM). Cells were harvested at the conclusion of OCR assessment and data normalized to cell number.
Mouse breeding and husbandry
All experimental protocols were approved by the University of Melbourne Anatomy & Neuroscience, Pathology, Pharmacology, and Physiology Animal Ethics Committee (Ethics #21228). To generate Alb-Cre:LSL-Cas9-GFP mice, hemizygous Alb-Cre mice (Strain #:003574) were bred with homozygous LSL-Cas9-GFP mice (Strain #:028551). In total, 20 male mice were used for experiments. All mice were housed at 22 °C and maintained on a 12 h light, 12 h dark cycle in cages with 2-5 mice per cage. Mice had ad libitum access to a standard chow diet (Specialty Feeds, WA, Australia) containing 23% energy from protein, 12% from fat, 59 % from carbohydrate and water or ad libitum access to a high-fat, high-sucrose diet (High Fat Rodent Diet SF04-001, 43 % energy from fat, Specialty Feeds, HFD) starting at 8–10 weeks of age for a total of 4 weeks. After 4 weeks of high fat feeding, mice were killed, and hepatocytes were isolated by collagenase digestion as previously described105.
AAV design and production
gRNAs for AAV-gScrambled-FLEX-mCherry (pAAV-U6>mScramble-GTGTAGTTCGACCATTCGTG)-CAG > LL:rev(mCherry):rev(LL):WPRE, AAV-Esyt-FLEX-mCherry (pAAV[-U6>mEsyt1[gRNA-TCCCTACGCGCTCGTCCGTGT]-U6>mEsyt1[gRNA-GGGGCACCAACAGTCGGTTA]-CAG > LL:rev(mCherry):rev(LL):WPRE) and AAV-Esty2-FLEX-mCherry (pAAV[-U6>mEsyt2[gRNA-ACAGCGCCAGCGCGCGGCAC]-U6>mEsyt2[gRNA-GGTCGCAGGCGCGCACTCCC]-CAG > LL:rev(mCherry):rev(LL):WPRE) viral vectors were cloned and packaged into the AAV-DJ steotype at a titre of > 2 × 1013 GC/ml (VectorBuilder, Chicago, IL). All AAV’s were injected intravenously at a concentration of 1 × 1012 genome copies per mouse in 100 µl.
Fatty acid metabolism in primary hepatocytes
Cell density was determined, and hepatocytes were plated at 500,000 cells per well in a 6-well plate (BD Falcon, #35043) and left to adhere for 4 h in adherence medium containing M199, Penicillin / Streptomycin (P/S), BSA 10%, dialysed FBS, dexamethasone (100 nM) and insulin (100 nM). The medium was replaced with low insulin (1 nmol/L) basal culture media containing M199, P/S and dexamethasone (100 nM) and incubated overnight at 37 °C. The next day, hepatocytes were incubated for 2 h with low-glucose DMEM Gluta-MAX (Life Technologies) containing 2% fatty acid-free BSA, 1 μCi/mL oleic acid (NEC317050UC, PerkinElmer) and 0.5 mmol/L oleic acid (Sigma-Aldrich). Fatty acid oxidation was calculated as the sum of complete oxidation to 14CO2 and incomplete oxidation (i.e., 14C-containing acid-soluble metabolites). Fatty acid uptake was calculated as the sum of fatty acid oxidation and fatty acids stored in complex lipids (i.e., 14C in the organic fraction of the lysed liver slices). Incorporation of 14C into triglyceride was measured by thin-layer chromatography as previously described106.
Identification of ESYT1 and ESYT2 by targeted mass spectrometry
Hepatocytes from Control, ESYT1KO and ESYT2KO mice were collected, snap frozen and stored at -80°C. Identification of ESYT1 and ESYT2 was performed using targeted mass spectrometry as previously described105. The peptide sequences used to detect ESYT1 (LLAETVAPAVR) and ESYT2 (ALALLEDEEQAVR) were unique to each protein.
Drosophila experiments
Breeding and husbandry
To generate fat-body dEsyt knockout flies (dEsyt2FB-KO), we crossed LppGal4, UAS-Cas9.P2 (Bloomington Drosophila Stock Center (BDSC) 67078) females with dEsyt2 gRNA expressing males (BDSC 83802). To generate whole-body dEsyt knockout flies (dEsytKO), we crossed Df(3 R)Exel7357/TM6B females (chromosomal deletion that includes Esyt2 and hereafter referred to as Df(Esyt2), BDSC 7948) with Mi(ET1)Esyt2MB02922/TM6C (a transposable element insertion that is a protein null allele75 and hereafter referred to as Esyt−, BDSC 23501) males. Of the resulting offspring we selected trans-heterozygote mutant animals. The controls used were wiUAS-Cas9.P2/+; LppGal4/+ heterozygotes (Fat-body control, WTFB-KO) or Df(3 R)Ecel7357/Tm6C,Sb (Whole-body control, WT). All flies were raised on standard molasses-based food at 25 °C.
For gene expression analysis, whole male adult flies (with heads removed) were snap frozen and stored at -80°C for later analysis. 10 flies were pooled to create each biological replicate. RNA was extracted with TRI reagent (Sigma-Aldrich) and reverse transcribed with iScript Reverse Transcriptase (Bio-Rad). Gene expression was determined by quantitative RT-PCR (BioRad CFX384 Touch™ Real-Time PCR System) using SYBR Green PCR Master Mix (QIAGEN). Results were analysed using BioRad CFX Manager™ software (BioRad). Esyt2 gene expression (Forward; 5’TATCTGGTGGGTTACATGGGC, Reverse; 5’CAGGATTACGTCCTTCTCGGA) was normalized to Actin-5 (Forward; 5’CGAAGAAGTTGCTGCTCTGG, Reverse; 5’AGAACGATACCGGTGGTACG) as the housekeeper gene. mRNA levels were determined using the 2−ΔΔCt method.
Starvation assay
For starvation experiments, male flies <36 h of age were transferred from normal media to vials containing 2 mL of starvation media (1% agar in water). Flies were kept at 25 °C and transferred to new vials every 48 h. To determine starvation resistance, 10 starvation vials with 10 flies each were monitored, and the number of dead flies were recorded every 12 h until all flies were dead.
Figure preparation
Figures were prepared using Microsoft PowerPoint (Microsoft 365 MSO). Images were edited with Image Lab (for Western blot), Image J (microscopy images). GraphPad Prism 9 (GraphPad Software) was used to create graphs and calculate statistical significance.
Statistical and reproducibility
Statistical analyses were performed using GraphPad Prism 9. All data was assessed for normal distribution using the D’Agostino & Pearson or Shapiro-Wilk test. For normally distributed data, differences between groups were assessed using two-tailed unpaired t-tests or one-way analysis of variance (ANOVA) with Bonferroni multiple comparison tests. For nonparametric data, differences between groups were assessed using a Kruskal-Wallis test with Dunn’s multiple comparison tests. Starvation resistance was analysed using a log-rank (Mantel Cox) test. Survivorship curves show the percentage of flies remaining alive as a function of time in hours. Differences were considered significant at p < 0.05. The number of technical replicates and/or biological replicates are described in each figure legend. No statistical method was used to predetermine sample size and no data were excluded from the analyses. The experiments were not randomized and the investigators were in most cases not blinded to allocation during experiments and outcome assessment.
Materials availability
All unique/stable reagents generated in this study are available from the lead contact with a completed materials transfer agreement.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All uncropped immunoblotting images can be found within the supplementary files. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD060912. Source data are provided with this paper.
References
Wang, S. et al. Lipolysis and the integrated physiology of lipid energy metabolism. Mol. Genet. Metab. 95, 117–126 (2008).
Renne, M. F., Klug, Y. A. & Carvalho, P. Lipid droplet biogenesis: a mystery “unmixing”? Semin Cell Dev. Biol. 108, 14–23 (2020).
Walther, T. C., Chung, J. & Farese, R. V. Jr. Lipid droplet biogenesis. Annu Rev. Cell Dev. Biol. 33, 491–510 (2017).
Buch, S. et al. A genome-wide association study confirms PNPLA3 and identifies TM6SF2 and MBOAT7 as risk loci for alcohol-related cirrhosis. Nat. Genet 47, 1443–1448 (2015).
Mahdessian, H. et al. TM6SF2 is a regulator of liver fat metabolism influencing triglyceride secretion and hepatic lipid droplet content. Proc. Natl Acad. Sci. USA 111, 8913–8918 (2014).
BasuRay, S., Wang, Y., Smagris, E., Cohen, J. C. & Hobbs, H. H. Accumulation of PNPLA3 on lipid droplets is the basis of associated hepatic steatosis. Proc. Natl Acad. Sci. USA 116, 9521–9526 (2019).
Albert, J. S. et al. Null mutation in hormone-sensitive lipase gene and risk of type 2 diabetes. N. Engl. J. Med. 370, 2307–2315 (2014).
Dahlman, I. et al. The CIDEA gene V115F polymorphism is associated with obesity in Swedish subjects. Diabetes 54, 3032–3034 (2005).
Olsson, A. H. et al. Two common genetic variants near nuclear-encoded OXPHOS genes are associated with insulin secretion in vivo. Eur. J. Endocrinol. 164, 765–771 (2011).
Helsley, R. N. et al. Obesity-linked suppression of membrane-bound O-acyltransferase 7 (MBOAT7) drives non-alcoholic fatty liver disease. Elife 8, e49882 (2019).
Rocha, N. et al. Human biallelic MFN2 mutations induce mitochondrial dysfunction, upper body adipose hyperplasia, and suppression of leptin expression. Elife 6, e23813 (2017).
Magre, J. et al. Identification of the gene altered in Berardinelli-Seip congenital lipodystrophy on chromosome 11q13. Nat. Genet 28, 365–370 (2001).
Gandotra, S. et al. Perilipin deficiency and autosomal dominant partial lipodystrophy. N. Engl. J. Med. 364, 740–748 (2011).
Agarwal, A. K. et al. AGPAT2 is mutated in congenital generalized lipodystrophy linked to chromosome 9q34. Nat. Genet 31, 21–23 (2002).
Sperk, A., Mueller, M. & Spiekerkoetter, U. Outcome in six patients with mitochondrial trifunctional protein disorders identified by newborn screening. Mol. Genet Metab. 101, 205–207 (2010).
Treacy, E. P. et al. Short-chain hydroxyacyl-coenzyme A dehydrogenase deficiency presenting as unexpected infant death: a family study. J. Pediatr. 137, 257–259 (2000).
Clayton, P. T. et al. Hyperinsulinism in short-chain L-3-hydroxyacyl-CoA dehydrogenase deficiency reveals the importance of beta-oxidation in insulin secretion. J. Clin. Invest 108, 457–465 (2001).
Gemmink, A. et al. Super-resolution microscopy localizes perilipin 5 at lipid droplet-mitochondria interaction sites and at lipid droplets juxtaposing to perilipin 2. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1863, 1423–1432 (2018).
Herms, A. et al. AMPK activation promotes lipid droplet dispersion on detyrosinated microtubules to increase mitochondrial fatty acid oxidation. Nat. Commun. 6, 7176 (2015).
Gemmink, A. et al. Decoration of myocellular lipid droplets with perilipins as a marker for in vivo lipid droplet dynamics: a super-resolution microscopy study in trained athletes and insulin resistant individuals. Biochim Biophys. Acta Mol. Cell Biol. Lipids 1866, 158852 (2021).
Nguyen, T. B. et al. DGAT1-dependent lipid droplet biogenesis protects mitochondrial function during starvation-induced autophagy. Dev. Cell 42, 9–21.e25 (2017).
Rambold, A. S., Cohen, S. & Lippincott-Schwartz, J. Fatty acid trafficking in starved cells: regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. Dev. Cell 32, 678–692 (2015).
Benador, I. Y. et al. Mitochondria bound to lipid droplets have unique bioenergetics, composition, and dynamics that support lipid droplet expansion. Cell Metab. 27, 869–885.e866 (2018).
Boutant, M. et al. Mfn2 is critical for brown adipose tissue thermogenic function. EMBO J. 36, 1543–1558 (2017).
Whytock, K. L., Shepherd, S. O., Wagenmakers, A. J. M. & Strauss, J. A. Hormone-sensitive lipase preferentially redistributes to lipid droplets associated with perilipin-5 in human skeletal muscle during moderate-intensity exercise. J. Physiol. 596, 2077–2090 (2018).
Kuramoto, K. et al. Perilipin 5, a lipid droplet-binding protein, protects heart from oxidative burden by sequestering fatty acid from excessive oxidation. J. Biol. Chem. 287, 23852–23863 (2012).
Shiozaki, M. et al. Closer association of mitochondria with lipid droplets in hepatocytes and activation of Kupffer cells in resveratrol-treated senescence-accelerated mice. Histochem. Cell Biol. 136, 475–489 (2011).
Yu, J. et al. Lipid droplet remodeling and interaction with mitochondria in mouse brown adipose tissue during cold treatment. Biochim. Biophys. Acta 1853, 918–928 (2015).
Kien, B. et al. Lipid droplet-mitochondria coupling via perilipin 5 augments respiratory capacity but is dispensable for FA oxidation. J. Lipid Res. 63, 100172 (2022).
Jagerstrom, S. et al. Lipid droplets interact with mitochondria using SNAP23. Cell Biol. Int. 33, 934–940 (2009).
Miner, G. E. et al. Perilipin 5 interacts with Fatp4 at membrane contact sites to promote lipid droplet-to-mitochondria fatty acid transport. Dev. Cell. 58,1250–1265.e6 (2022).
Ramseyer, V. D., Kimler, V. A. & Granneman, J. G. Vacuolar protein sorting 13 C is a novel lipid droplet protein that inhibits lipolysis in brown adipocytes. Mol. Metab. 7, 57–70 (2018).
Wang, J. et al. An ESCRT-dependent step in fatty acid transfer from lipid droplets to mitochondria through VPS13D-TSG101 interactions. Nat. Commun. 12, 1252 (2021).
Ouyang, Q. et al. Rab8a as a mitochondrial receptor for lipid droplets in skeletal muscle. Dev. Cell 58, 289–305.e286 (2023).
Wong, L. H., Gatta, A. T. & Levine, T. P. Lipid transfer proteins: the lipid commute via shuttles, bridges and tubes. Nat. Rev. Mol. Cell Biol. 20, 85–101 (2019).
Lev, S. Non-vesicular lipid transport by lipid-transfer proteins and beyond. Nat. Rev. Mol. Cell Biol. 11, 739–750 (2010).
Choi, C. R. & Rhee, H. W. Proximity labeling: an enzymatic tool for spatial biology. Trends Biotechnol. 40, 145–14 (2021).
Go, C. D. et al. A proximity-dependent biotinylation map of a human cell. Nature 595, 120–124 (2021).
Roux, K. J., Kim, D. I., Raida, M. & Burke, B. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol. 196, 801–810 (2012).
Bindels, D. S. et al. mScarlet: a bright monomeric red fluorescent protein for cellular imaging. Nat. Methods 14, 53–56 (2017).
Goedhart, J. et al. Structure-guided evolution of cyan fluorescent proteins towards a quantum yield of 93%. Nat. Commun. 3, 751 (2012).
Zehmer, J. K., Bartz, R., Liu, P. & Anderson, R. G. Identification of a novel N-terminal hydrophobic sequence that targets proteins to lipid droplets. J. Cell Sci. 121, 1852–1860 (2008).
Bersuker, K. et al. A proximity labeling strategy provides insights into the composition and dynamics of lipid droplet proteomes. Dev. Cell 44, 97–112.e117 (2018).
Schopp, I. M. et al. Split-BioID a conditional proteomics approach to monitor the composition of spatiotemporally defined protein complexes. Nat. Commun. 8, 15690 (2017).
De Munter, S. et al. Split-BioID: a proximity biotinylation assay for dimerization-dependent protein interactions. FEBS Lett. 591, 415–424 (2017).
Osellame, L. D. et al. Cooperative and independent roles of the Drp1 adaptors Mff, MiD49 and MiD51 in mitochondrial fission. J. Cell Sci. 129, 2170–2181 (2016).
Cho, K. F. et al. Split-TurboID enables contact-dependent proximity labeling in cells. Proc. Natl Acad. Sci. USA 117, 12143–12154 (2020).
Najt, C. P. et al. Organelle interactions compartmentalize hepatic fatty acid trafficking and metabolism. Cell Rep. 42, 112435 (2023).
Anastasia, I. et al. Mitochondria-rough-ER contacts in the liver regulate systemic lipid homeostasis. Cell Rep. 34, 108873 (2021).
Huang da, W., Sherman, B. T. & Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 (2009).
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003).
Schauder, C. M. et al. Structure of a lipid-bound extended synaptotagmin indicates a role in lipid transfer. Nature 510, 552–555 (2014).
Yu, H. et al. Extended synaptotagmins are Ca2 + -dependent lipid transfer proteins at membrane contact sites. Proc. Natl Acad. Sci. USA 113, 4362–4367 (2016).
Nath, V. R., Mishra, S., Basak, B., Trivedi, D. & Raghu, P. Extended synaptotagmin regulates membrane contact site structure and lipid transfer function in vivo. EMBO Rep. 21, e50264 (2020).
Chen, W. W., Freinkman, E., Wang, T., Birsoy, K. & Sabatini, D. M. Absolute quantification of matrix metabolites reveals the dynamics of mitochondrial metabolism. Cell 166, 1324–1337.e1311 (2016).
Szklarczyk, D. et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47, D607–D613 (2019).
Ge, J. et al. Stepwise membrane binding of extended synaptotagmins revealed by optical tweezers. Nat. Chem. Biol. 18, 313–320 (2022).
Skinnider, M. A. et al. An atlas of protein-protein interactions across mouse tissues. Cell 184, 4073–4089.e4017 (2021).
Varghese, M. et al. Adipocyte lipolysis affects Perilipin 5 and cristae organization at the cardiac lipid droplet-mitochondrial interface. Sci. Rep. 9, 4734 (2019).
Petkovic, M. et al. The SNARE Sec22b has a non-fusogenic function in plasma membrane expansion. Nat. Cell Biol. 16, 434–444 (2014).
Lang, A. B., John Peter, A. T., Walter, P. & Kornmann, B. ER-mitochondrial junctions can be bypassed by dominant mutations in the endosomal protein Vps13. J. Cell Biol. 210, 883–890 (2015).
Kumar, N. et al. VPS13A and VPS13C are lipid transport proteins differentially localized at ER contact sites. J. Cell Biol. 217, 3625–3639 (2018).
Yeshaw, W. M. et al. Human VPS13A is associated with multiple organelles and influences mitochondrial morphology and lipid droplet motility. Elife 8, e43561 (2019).
Milger, K. et al. Cellular uptake of fatty acids driven by the ER-localized acyl-CoA synthetase FATP4. J. Cell Sci. 119, 4678–4688 (2006).
Kuch, E. M. et al. Differentially localized acyl-CoA synthetase 4 isoenzymes mediate the metabolic channeling of fatty acids towards phosphatidylinositol. Biochim. Biophys. Acta 1841, 227–239 (2014).
Roland, J. T. et al. Rab GTPase-Myo5B complexes control membrane recycling and epithelial polarization. Proc. Natl Acad. Sci. USA 108, 2789–2794 (2011).
Cai, S. et al. In situ architecture of the lipid transport protein VPS13C at ER-lysosome membrane contacts. Proc. Natl Acad. Sci. USA 119, e2203769119 (2022).
Saheki, Y. et al. Control of plasma membrane lipid homeostasis by the extended synaptotagmins. Nat. Cell Biol. 18, 504–515 (2016).
Fernandez-Busnadiego, R., Saheki, Y. & De Camilli, P. Three-dimensional architecture of extended synaptotagmin-mediated endoplasmic reticulum-plasma membrane contact sites. Proc. Natl Acad. Sci. USA 112, E2004–E2013 (2015).
Unger, R. H. Lipotoxic diseases. Annu Rev. Med. 53, 319–336 (2002).
Ahammed, S. K. & Chowdhury, A. Insulin resistance and “lipotoxic liver diseases”. Trop. Gastroenterol. 34, 1–4 (2013).
Cunha, D. A. et al. Initiation and execution of lipotoxic ER stress in pancreatic beta-cells. J. Cell Sci. 121, 2308–2318 (2008).
Karaskov, E. et al. Chronic palmitate but not oleate exposure induces endoplasmic reticulum stress, which may contribute to INS-1 pancreatic beta-cell apoptosis. Endocrinology 147, 3398–3407 (2006).
Zeng, X. et al. Oleic acid ameliorates palmitic acid induced hepatocellular lipotoxicity by inhibition of ER stress and pyroptosis. Nutr. Metab. (Lond.) 17, 11 (2020).
Kikuma, K., Li, X., Kim, D., Sutter, D. & Dickman, D. K. Extended synaptotagmin localizes to presynaptic ER and promotes neurotransmission and synaptic growth in Drosophila. Genetics 207, 993–1006 (2017).
Baker, K. D. & Thummel, C. S. Diabetic larvae and obese flies-emerging studies of metabolism in Drosophila. Cell Metab. 6, 257–266 (2007).
Selen, E. S., Choi, J. & Wolfgang, M. J. Discordant hepatic fatty acid oxidation and triglyceride hydrolysis leads to liver disease. JCI Insight 6, e135626 (2021).
Merritt, J. L. 2nd, Norris, M. & Kanungo, S. Fatty acid oxidation disorders. Ann. Transl. Med. 6, 473 (2018).
Miner, G. E. et al. PLIN5 interacts with FATP4 at membrane contact sites to promote lipid droplet-to-mitochondria fatty acid transport. Dev Cell 58, 1250–1265.e6 (2023).
Li, L. O., Klett, E. L. & Coleman, R. A. Acyl-CoA synthesis, lipid metabolism and lipotoxicity. Biochim. Biophys. Acta 1801, 246–251 (2010).
Saheki, Y. & De Camilli, P. Endoplasmic reticulum-plasma membrane contact sites. Annu Rev. Biochem. 86, 659–684 (2017).
Sassano, M. L. et al. PERK recruits E-Syt1 at ER-mitochondria contacts for mitochondrial lipid transport and respiration. J. Cell Biol. 222, e202206008 (2023).
Mejhert, N. et al. The lipid droplet knowledge portal: a resource for systematic analyses of lipid droplet biology. Dev. Cell 57, 387–397.e384 (2022).
Hung, V. et al. Proteomic mapping of cytosol-facing outer mitochondrial and ER membranes in living human cells by proximity biotinylation. Elife 6, e24463 (2017).
Stoica, R. et al. ER-mitochondria associations are regulated by the VAPB-PTPIP51 interaction and are disrupted by ALS/FTD-associated TDP-43. Nat. Commun. 5, 3996 (2014).
Kors, S., Costello, J. L. & Schrader, M. VAP proteins—from organelle tethers to pathogenic host interactors and their role in neuronal disease. Front Cell Dev. Biol. 10, 895856 (2022).
Freyre, C. A. C., Rauher, P. C., Ejsing, C. S. & Klemm, R. W. MIGA2 links mitochondria, the ER, and lipid droplets and promotes De Novo lipogenesis in adipocytes. Mol. Cell 76, 811–825.e814 (2019).
de Almeida, M. E. et al. Acute exercise increases the contact between lipid droplets and mitochondria independently of obesity and type 2 diabetes. J. Physiol. 601, 1797–1815 (2023).
Talari, N. K. et al. Lipid-droplet associated mitochondria promote fatty-acid oxidation through a distinct bioenergetic pattern in male Wistar rats. Nat. Commun. 14, 766 (2023).
Goldberg, I. J., Trent, C. M. & Schulze, P. C. Lipid metabolism and toxicity in the heart. Cell Metab. 15, 805–812 (2012).
Lytrivi, M., Castell, A. L., Poitout, V. & Cnop, M. Recent insights into mechanisms of beta-cell lipo- and glucolipotoxicity in Type 2 diabetes. J. Mol. Biol. 432, 1514–1534 (2020).
Lee, Y. et al. Beta-cell lipotoxicity in the pathogenesis of non-insulin-dependent diabetes mellitus of obese rats: impairment in adipocyte-beta-cell relationships. Proc. Natl Acad. Sci. USA 91, 10878–10882 (1994).
Gibson, D. G. et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345 (2009).
Formosa, L. E. et al. Dissecting the roles of mitochondrial complex I intermediate assembly complex factors in the biogenesis of complex I. Cell Rep. 31, 107541 (2020).
Tyanova, S., Temu, T. & Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 11, 2301–2319 (2016).
Tyanova, S. et al. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740 (2016).
Digman, M. A., Caiolfa, V. R., Zamai, M. & Gratton, E. The phasor approach to fluorescence lifetime imaging analysis. Biophys. J. 94, L14–L16 (2008).
Hinde, E., Digman, M. A., Welch, C., Hahn, K. M. & Gratton, E. Biosensor forster resonance energy transfer detection by the phasor approach to fluorescence lifetime imaging microscopy. Microsc Res Tech. 75, 271–281 (2012).
Digman, M. A., Dalal, R., Horwitz, A. F. & Gratton, E. Mapping the number of molecules and brightness in the laser scanning microscope. Biophys. J. 94, 2320–2332 (2008).
Digman, M. A., Wiseman, P. W., Horwitz, A. R. & Gratton, E. Detecting protein complexes in living cells from laser scanning confocal image sequences by the cross correlation raster image spectroscopy method. Biophys. J. 96, 707–716 (2009).
Dagley, M. J. & McConville, M. J. DExSI: a new tool for the rapid quantitation of 13C-labelled metabolites detected by GC-MS. Bioinformatics 34, 1957–1958 (2018).
Radoux, C. J., Olsson, T. S., Pitt, W. R., Groom, C. R. & Blundell, T. L. Identifying interactions that determine fragment binding at protein hotspots. J. Med. Chem. 59, 4314–4325 (2016).
Pires, D. E., Ascher, D. B. & Blundell, T. L. mCSM: predicting the effects of mutations in proteins using graph-based signatures. Bioinformatics 30, 335–342 (2014).
Rodrigues, C. H. M., Myung, Y., Pires, D. E. V. & Ascher, D. B. mCSM-PPI2: predicting the effects of mutations on protein-protein interactions. Nucleic Acids Res. 47, W338–W344 (2019).
De Nardo, W. et al. Proteomic analysis reveals exercise training induced remodelling of hepatokine secretion and uncovers syndecan-4 as a regulator of hepatic lipid metabolism. Mol. Metab. 60, 101491 (2022).
Keenan, S. N. et al. Perilipin 5 S155 phosphorylation by PKA is required for the control of hepatic lipid metabolism and glycemic control. J. Lipid Res. 62, 100016 (2021).
Acknowledgements
This work was supported by funding from the Australian Research Council (Discovery Projects DP180102935 and DP210102358 to M.J.W., DP180101387 and DP21010298 to E.H. and J.L., Future Fellowship FT200100401 to E.H.); The Excellence in Diversity Fellowship (J.L.); Australian National Health and Medical Research Council (NHMRC Project Grant GNT2020078 to M.J.W., A.B.-G. and S.N.K.; GNT1140906 and Fellowship GNT2009732 to D.A.S.); Diabetes Australia (Y22G-WATM to M.J.W., S.N.K. and A.B.-G.); Department of Anatomy and Physiology ECR Seeding Grant (A.B-G.); The Excellence in Diversity Fellowship (J.L.); and the Jacob Haimson Beverly Mecklenburg Lectureship (E.H) and the Heart Foundation Future Leader Fellowship (M.R.). We thank Vanta Jameson for assistance with flow cytometry, and Rachel Templin and Georg Ramm for electron microscopy imaging. We thank Gio Fidelito, Olivia Lee and Ben Parker for assistance in graphical illustration and insightful discussions. This work was supported by the Melbourne Brain Centre Flow Cytometry Facility, Biological Optical Microscopy Platform (BOMP), Melbourne Mass Spectrometry and Proteomics Facility (MMSPF), Metabolomics Australia, and the Ramaciotti Centre for Structural Cryo-Electron Microscopy. The Novo Nordisk Foundation Center for Stem Cell Medicine is supported by Novo Nordisk Foundation grants (NNF21CC0073729). Graphical illustrations were created with BioRender.com (agreement number: OR27WIIE76).
Author information
Authors and Affiliations
Contributions
A.B-G., C.J.D., M.J.W. designed the research; A.B-G., C.J.D., S.N.K., J.L, E.C, S.N., D.P.D., V.K.N., N.A.S., C.H.M.R., S.P. performed research; E.C., H.T.N., D.A.S. analyzed data; G.H., M.R., G.D., E.H., D.B.A., T.Z. provided expertize and feedback; A.B-G., M.J.W. wrote the paper., All authors edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Dan Arking, Cathy S.-J. Fann, Chulhwan Kwak, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Bezawork-Geleta, A., Devereux, C.J., Keenan, S.N. et al. Proximity proteomics reveals a mechanism of fatty acid transfer at lipid droplet-mitochondria- endoplasmic reticulum contact sites. Nat Commun 16, 2135 (2025). https://doi.org/10.1038/s41467-025-57405-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-57405-5
This article is cited by
-
Interactions between lipid droplets and mitochondria in metabolic diseases
Lipids in Health and Disease (2025)
-
Metabolic reprogramming in hepatocellular carcinoma: an integrated omics study of lipid pathways and their diagnostic potential
Journal of Translational Medicine (2025)
-
The complex web of membrane contact sites in brain aging and neurodegeneration
Cellular and Molecular Life Sciences (2025)
-
Do fungi also undergo "ferroptosis"? A microscopic exploration of cellular death
Apoptosis (2025)
