
Abstract
Biomass valorization is a way to promote the ‘waste-to-wealth’ concept, which is a pre-requisite condition for a future sustainable lifestyle. The direct utilization of natural polymer for value-added materials should be prioritized. With this object, we demonstrate a facile and economical method to prepare chitin-derived supramolecular nanowires-stabilized single-atom sites Pt catalysts (SS-Pt-CSNs). A comprehensive characterization shows the structure of single-atom Pt coordinated with the organic supramolecular (ligand-type) entity, which is both flexible (like in homogenous catalyst) and robust (like in heterogenous catalyst). Such hybrid characteristics of these SS-Pt-CSNs materials exhibit excellent catalytic performance for chemo-selective hydrogenation of various unsaturated bonds with a selectivity of more than 90:1 and a high turnover number (TON) of 121,350. Mechanistic studies demonstrate that both empty coordination sites of Pt and dynamic B-doping are the key factors for exceptional efficiency and high selectivity.
Similar content being viewed by others


Introduction
The mutual transformation between inorganic and organic matter in nature is the fundamental basis for the regular operation of the earth. This also comes with a cost: for example, with an annual production of ∼100 billion tons, chitin is the world’s second most abundant biomass widely existing in nature1,2. The seafood processing industries produce approximately 6–8 million tons3 of crab, shrimp, and lobster shells worldwide each year. The waste shells are often dumped in landfills or the sea, resulting in a massive waste of resources3. Remarkably, this biomass-derived natural product is sustainable, biodegradable, non-toxic, and inexpensive. Hence, the direct utilization of natural biomass towards a more profitable material is condictio sine qua non for a sustainable future and circular economy4,5,6. Chitin bears naturally fixed nitrogen in the form of amide functionality7,8, which provides more flexibility to utilize it as catalytic support for heterogeneous catalysis9,10,11,12,13: chitin poses coordination sites (cf. amide, hydroxyl, and ether groups), which can be high binding affinity for transition-metal ions. It also contains long carbohydrate chains and intermolecular hydrogen bonds as a stable polymer support7,14.
In modern catalytic science, both homogeneous and heterogeneous catalysis are complementary in nature. The complicated and chemo-selective organic synthesis could elegantly be performed through the elaborate design of ligands in homogeneous catalysis15,16. Heterogeneous catalysis is often unable to deliver precise control over selectivity, although a durable catalyst lifetime is needed for any potential. To bridge this knowledge gap, i.e., designing a hybrid catalyst comprising the advantages from superiorities of the homogeneous and heterogeneous catalyst is the need of the hour. On this aspect, single-atom materials are considered promising candidates in numerous catalytic reactions17,18,19,20,21,22,23,24. However, precise control over selectivity is still an obstacle in using single-atom metal catalysts25,26,27. To overcome this limitation, an appropriate design of the catalytic environment is an extremely crucial factor, which demands both flexibility and robustness in the surrounding micro-environment of the single metal sites. We envision that the advantage of chitin might be an alternative for the preparation of single-atom materials. Herein, we deliver a facile and economical method to prepare chitin supramolecular-stabilized single-atom sites Pt catalysts (SS-Pt-CSNs) through a strong metal-organic polymer coordinative effect28 (Fig. 1).

The supramolecular nanowires were fabricated by dissolving the shrimp powders in the NaOH/urea system and emulsion polymerization. After freeze drying, the nanowires were impregnated with H2PtCl6 solution, and a thermal activating process in an Ar atmosphere was used to obtain SS-Pt-CSNs. Further treatment with borane switched the catalytic selectivity.
Since the chemoselective and controllable catalytic hydrogenation of organic molecules with multiple unsaturated moieties is a formidable challenge in the fine chemical industry29, our chitin supramolecular nanowires-stabilized single-atom Pt catalyst is employed to hydrogenate various unsaturated bonds (C=C, C=O, C≡C, –NO2) containing α,β-unsaturated carbonyl compounds, remote unsaturated bonds and even natural products. The current study achieves the tunable hydrogenation of 31 substrates to generate 62 products in up to 99% yields with excellent chemo-selectivity (>90:1) and a high TON (121,350) by using the SS-Pt-CSNs catalyst.
Results
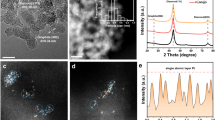
The procedure of catalyst preparation was summarized in the methods and materials preparation (Fig. 1). Scanning electron microscopy (SEM) (Fig. 2a) and N2 adsorption showed the three-dimensional network and hierarchical pore structures with the large specific surface area of SS-Pt-CSNs, respectively (Supplementary information and Figs. S1e-g and 2). The original structure of the chitin nanowires remained intact, which was confirmed by Fourier transform infrared spectrometer (FT-IR), thermogravimetric analysis (TGA), and powder X-ray diffraction (XRD)30,31,32 (Figs. S3–5). These results collectively demonstrated the potential coordination and anchoring ability of CSNs. Because no Pt nanoparticles were observed by the transmission electron microscopy (TEM) (Fig. 2b), high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images were performed to exhibit the uniform dispersion of Pt single atom (Fig. 2c). In the atomic-resolution STEM energy-dispersive spectroscopy spectra and elemental mapping images, Pt, N, and O elements were always adjacently distributed (Fig. 2d). The slight shift of N 1s and O 1s peaks in the X-ray photoemission spectroscopy (XPS) spectra also indicated the possible coordination situation. The oxidation state of SS-Pt-CSNs was determined to be Pt2+ according to the XPS spectra (Fig. S6 and 7). Meanwhile, the X-ray absorption near-edge structure (XANES) showed the Pt L3-edge intensity of SS-Pt-CSNs lay between Pt foil, H2PtCl6, PtO2, and Pre-Pt4+-CSNs (Fig. S8) suggesting the positively charged state of single-atom Pt sites was +2 valence (Fig. 2e). In addition, the average Pt atomic environment in SS-Pt-CSNs consisted of N/O coordination shells with average coordination numbers of 3.0, which was verified by extended X-ray fine structure (EXAFS) (Fig. 2f, g, Fig. S9a, and Table S1). Meanwhile, the wavelet-transformed (WT) EXAFS graphs of Pt samples were obtained to get more information about the k and R space resolutions of the scattering atoms (Fig. S9b-e). The WT EXAFS counterplots of SS-Pt-CSNs exhibited a maximum at ∼3.9 Å−1 attributed to the Pt−N/O path, similar to those of the first shell of PtO2 without Pt-Pt bonds at ~8.2 Å−1. The above analyses showed the unique configuration of SS-Pt-CSNs, i.e., isolated Pt atoms were coordinated with the surrounding N and O atoms.

a SEM morphology, b TEM, c HAADF-STEM, and d elemental mapping images of SS-Pt-CSNs, corresponding to carbon (yellow) (d1), nitrogen (blue) (d2), oxygen (red) (d3) and platinum (green) (d4). e Pt L3-edge XANES. f Fourier-transformed EXAFS spectra. g The fitting curve of SS-Pt-CSNs.
Density functional theory (DFT) calculations were used to deduce four coordination models based on the XAS results and supramolecular chitin structure. EXAFS results suggested that three O/N atoms might surround a Pt single atom, and we also took four O/N atoms into consideration. As shown in Fig. S10, PtN1O2-CSNs (Fig. S10a), PtO3-CSNs (Fig. S10b), PtN1O3-CSNs (Fig. S10c), and PtO4-CSNs (Fig. S10d) exhibited theoretical coordination energies of −0.98, −0.64, −0.71, and −0.55 eV, respectively. PtN1O2-CSNs posed the most stable structure in coordination. Furthermore, the adsorption and dissociation of H2 were also calculated for the four models (Fig. S11). Herein, the ∆G decrease of PtN1O2-CSNs was the largest (−2.75 eV), which indicated this model was active for hydrogenation. Meanwhile, the solid-state 13C-NMR spectrums (Fig. 3a, b) indicate that acetylamino groups can capture atomic Pt by π-coordination33,34, which is attributed to the slight shift of the C sp2 peak from 173.7 ppm in the metal-free state to 174.6 ppm with single-atom Pt sites. The interaction between Pt and chitin nanowires promotes electron transfer, inducing anti-bonding orbital occupation of Pt–H. As presented in Fig. 3c, the energy level of the Pt–H bond orbital increases to the unsaturated occupation of the anti-bonding states, due to the decreasing of valence electrons (higher oxidation state) in the higher d-band center. It is obviously shown in integral crystal orbital Hamilton populations (ICOHP). The effect of hydrogen adsorption strength has a stronger characterization on the Pt-N1O2 model. In order to support the conclusion above, we further calculated the surface electrostatic potential to analyze the coordinative micro-environment of the PtN1O2-CSNs structure (Fig. 3e). The electrostatic potential of CSNs-250 was −0.31 e− at an electron-rich state. However, the electrostatic potential of SS-Pt-CSNs was significantly changed to 0.24 e+ by introducing single-atom Pt sites, changing to an electron-deficient state. In order to evaluate the redox activity, charge analysis was performed. From the state of CSNs-250 to SS-Pt-CSNs, the differential charge density (Δρ) could fully display the degree of electron distribution. In Fig. 3f, the obvious electronic perturbations occurring on the Pt-N1O2 structures indicated that an empty coordination site existed in SS-Pt-CSNs. So, the electron-deficient Pt site surrounded by vast electrons might have a strong ability for adsorbing and splitting H2.

a, b Solid-state 13C-NMR spectra of the CSNs-250 and SS-Pt-CSNs. c The d-band center of single Pt atom as a function of valence electron number. d The change in the gipps free energy of adsorption hydrogen (ΔGH*), crystal orbital hamilton population (COHP), and integral COHP (ICOHP) of Pt–H bond for H* on single Pt atom as a function of d-band center. e Surface electrostatic potential of the CSNs-250 and PtN1O2-CSNs: the potential contour was scaled to −0.5 (red) and +0.5 (blue), kBTe−1 (where kB is the Boltzmann constant, T is temperature, and e is the charge of an electron). (f) Δρ diagrams of SS-Pt-CSNs with the electric field around Pt-N1O2 bond.
Since SS-Pt-CSNs have a good ability to absorb, split, and bind H2, we select the hydrogenation of cinnamyl aldehyde (CAL) as the model reaction during the initial optimization. In previous reports, the catalytic hydrogenation of CAL might afford different products, such as C=C double bond hydrogenation product 3-phenylpropanal (HCAL), C=O double bond hydrogenation product cinnamyl alcohol (COL), and over hydrogenation product 3-phenylpropanol (HCOL). Firstly, the hydrogenation of CAL was investigated using iPrOH as solvent under 30 bar H2 at 80 °C (Fig. S12a and Table S2). No hydrogenation product was obtained without a catalyst or with only CSNs as catalysts. H2PtCl6, the catalyst precursor for preparing SS-Pt-CSNs, only gave a 10% yield of COL. Commercially available 5.0 wt% Pt/C afforded a mixture of HCAL, COL, and HCOL in yields of 52%, 6%, and 18%, respectively. To our delight, When we used 1.2 wt% SS-Pt-CSNs as a catalyst, a 90% yield of HCAL was obtained without COL or HCOL generation. Increasing the Pt loading to 2.4 wt% and 4.8 wt% led to the formation of Pt Nanoparticles (Fig. S13c, d), which delivered an adverse effect: periodic yield decrease, but the selectivity of HCAL still remained. Changing the calcination temperature from 250 °C to 800 °C also generated Pt nanoparticles (the Pt and other element content tested by ICP-OES or XPS) (Fig. S13b and Table S3). The yield of HCAL further dropped by using 3.0 wt% Nano-Pt-CSNs (Ar-800). Pt nanoparticles were observed as well by changing the calcination atmosphere from Ar to H2 (Fig. S13a), and three kinds of hydrogenation products were all obtained by using 1.2 wt% Nano-Pt-CSNs (H2-250) as a catalyst. Other metal catalysts (1.2% Pd-CSNs (Ar-250), 1.2% Au-CSNs (Ar-250), and 1.2% Ru-CSNs (Ar-250) prepared by the same procedure were also tested, and SS-Pt-CSNs was still the best with high yield and selectivity (Fig. S12b). Considering turnover frequency (TOF) and selectivity, the SS-Pt-CSNs are significantly superior to commercial and most reported catalysts (Table S4).
Nearly all works on CAL selective hydrogenation focused on obtaining a single hydrogenation product, i.e., HCAL or COL. We have already achieved the highly selective hydrogenation of CAL to HCAL using 1.2 wt% SS-Pt-CSNs as a catalyst. So, we were curious if the selective hydrogenation of CAL to COL could be achieved using the same catalyst. Some references reported that Lewis acid catalysts could activate the carbonyl group and improve the reaction selectivity35,36,37,38. So, we added a catalytic amount of BH3·THF (12.5 mol%) into the reaction system above to test the reaction selectivity (Fig. S14a and Table S5). No hydrogenation product was obtained without any catalyst or with CSNs as a catalyst. H2PtCl6 gave a 7% yield of COL and a 3% yield of HCAL. Pt/C indeed afforded more COL in 37% yield than the reaction system without BH3·THF, but a 21% yield of HCAL was obtained at the same time. When using 1.2 wt% SS-Pt-CSNs as a catalyst 95% yield of COL was obtained without HCAL or HCOL formation, which was the exact oppositely selective to the reaction system without BH3·THF. The yield was dropped again while using 2.4 wt% Nano-Pt-CSNs and 4.8 wt%. Nano-Pt-CSNs catalysts, but with good selectivity. 3.0 wt% Nano-Pt-CSNs (Ar-800) not only led to a decreased yield of COL, but also promoted the HCAL generation. A similar situation was also observed by using 1.2 wt% Nano-Pt-CSNs (H2-250) as the catalyst. 1.2% Pd-CSNs (Ar-250) gave COL and HCAL at the same time, while both 1.2% Au-CSNs (Ar-250) and 1.2% Ru-CSNs (Ar-250) delivered worse yields of COL than Pt-CSNs (Fig. S14b). SS-Pt-CSNs also show the priority of reactivity and selectivity among the most reported catalysts (Table S6).
In order to test the stability and durability of SS-Pt-CSNs, the catalyst recycling experiments were successfully conducted up to five times without any noticeable influence on conversion or selectivity (Fig. S15). For instance, an 87% yield of HCAL (HCAL/(COL + HCOL) > 87:6) and a 93% yield of COL (COL/(HCAL + HCOL) > 93:5) were obtained for the fifth time, respectively. The robustness of SS-Pt-CSNs material led to no variation in reactivity and selectivity (Fig. S16–21). To emphasize the efficiency of our catalyst, TOF was also calculated. The average TOF values of the low conversion level (10-20%) for the selective hydrogenation of the ‘C=C’ and ‘C=O’ double bonds from CAL were 0.59 S−1 and 0.77 S−1, respectively (Fig. S22). Notably, this Pt-based single-atom
catalyst shows outstanding catalytic activity with a TON as high as 121,350 by flow chemistry technique, representing the best performance yet in all heterogeneous CAL hydrogenations (Table S4).
To further demonstrate the compatibility of SS-Pt-CSNs, a series of unsaturated bonds were investigated (Fig. 4). Cinnamyl aldehyde and its various phenyl derivatives, such as p-Me, p-NMe2, o-OMe, m-F, and m-CF3, were all transformed to the corresponding 3-phenylpropanals (2a–2f) and cinnamyl alcohols (3a–3f) smoothly. Heterocyclic furan was also compatible with these two catalytic systems (2g and 3g). 2-Me, 2-Ph and 3-Ph substituted C=C double bond did not influence the hydrogenation of C=C double bonds (2h–2j) and C=O double bonds (3h–3j). Replacing the aldehyde group of cinnamyl aldehyde with acetyl or benzoyl group was tolerated in our reaction conditions (2k, 2l, 3k, and 3l). Not only α, β-unsaturated carbonyl compounds containing aryl groups, but also alkyl groups were studied. (E)-But-2-enal, (E)-hex-2-enal, and 3-methylbut-2-enal gave the target aldehydes in 82–99% yields. (2m–2o) and alcohols in 82–94% yields (2m–2o), respectively. Pent-1-en-3-one cyclohex-2-en-1-one, isophorone, and testosterone and testosterone afforded the desired products in satisfactory yields (2p–2s, 3p–3s). Besides α, β-unsaturated carbonyl compounds, we also focused on other substrates containing multiple unsaturated moieties. Remote unsaturated carbonyl compounds, such as pent-4-enal, hex-5-en-2-one, 6-methylhept-5-en-2-one, and cyclohexadec-7-en-1-one were transformed into alkyl groups in 90%, 87%, 87% and 85% yields (2t–2w), to alcohol in 90%, 85%, 98%, and 77% yields (3t–3w). Moreover, complex natural products, such as citronellal, dehydroepiandrosterone, and estrone derivative were all access to the corresponding selective products in satisfied yields (2x–2z, 3x–3z).

Reaction conditions: 1 (0.4 mmol), H2 (30 bar), SS-Pt-CSNs (5.0 mg), BH3·THF (12.5 mol% based on 1 for the hydrogenation of C=O bonds, none for the hydrogenation of C=C bonds), iPrOH (3.0 mL), 80 °C, 4 h. Yields were determined by isolated or gas chromatography analysis and calibrated with biphenyl as the internal standard. The compound 4 represents the hydrogenation of C=C and C=O bonds without an aromatic core or O-heterocyclic group.
Remarkably, natural products containing both conjugated and remote unsaturated bonds, such as D(+)-carvone and progesterone afforded the corresponding products in satisfied yields (2aa, 2ab, 3aa, and 3ab). At last, other types of unsaturated bonds were investigated. SS-Pt-CSNs achieved the chemoselective hydrogenation of 4-nitrobenzaldehyde. Nearly equivalent of 4-aminobenzaldehyde was obtained without BH3·THF (2ac), and 86% yield of 4-nitrobenzyl alcohol was obtained with the help of BH3·THF (3ac). 4-Nitrostyrene and 4-nitro-phenylacetylene as substrates gave 4-ethylnitrobenzene in 80% and 83% yields (2ad and 2ae), as well as 4-amino-styrene and 4-amino-phenylacetylene produced in 96% (3ad) and 90% yields (3ae), respectively. Notably, most substrate examples gave selectivities more than 90:1, which showed the good activity and selectivity of our SS-Pt-CSNs for CAL hydrogenation.
In order to gain more insights into the mechanism of the reaction, several control experiments were conducted. The single-atom Pt catalyst is crucial for the selective hydrogenation of C=C bonds, and hydrogen came from H2 gas rather than iPrOH (Fig. S23). The addition of BH3·THF has indeed switched the selectivity of the hydrogenation reaction (Fig. S24), and no hydrogenation product was obtained with BH3·THF as the sole catalyst. The hydrogen also came from H2 gas rather than iPrOH or BH3·THF. We further investigated the effect of boranes on SS-Pt-CSNs. Firstly, SS-Pt-CSNs were mixed with BH3·THF and stirred at 80 °C for 1 h. The obtained SS-Pt-CSNs (B) were added to the reaction without any external BH3·THF. 84% yield of COL with COL/(HCAL + HCOL) = 84:9 was obtained, which is the same as adding SS-Pt-CSNs and 12.5 mol% BH3·THF simultaneously. In addition, the TEM picture showed no nanoparticles in SS-Pt-CSNs (B) (Fig. S25). These results indicated that BH3·THF might affect the supramolecular nanowires and change the catalytic environment of single-atom Pt sites. Then, we directly studied the catalyst after the standard hydrogenation reaction of C=O bonds (SS-Pt-CSNs (B)) through some related characterizations. To identify the changes in active sites of catalysts, temperature-programmed reduction (TPR) experiments for H2 were carried out (Fig. 5a). When the temperature increased by about 300 °C, a certain amount of decomposition occurred in the CSNs-250, SS-Pt-CSNs, and SS-Pt-CSNs (B) as the same results of TG measurement. Only one broad signal of H2-TPR for SS-Pt-CSNs, whereas CSNs-250 did not give any peak, suggesting that the single Pt atoms were the only active sites for H2 dissociation. However, the SS-Pt-CSNs (B) was reduced between 300 °C to 400 °C and showed two states at 325 and 373 °C, suggesting the two interfacial active sites with supports. We used XPS to confirm and probe the chemical states of B elements. Compared to NaBH4 and Na2B4O7, Fig. 5b presented the obvious B 1s line of SS-Pt-CSNs (B), in which the peak could be further differentiated into two characteristic peaks set at 190.7 and191.9 eV corresponding to B–C and B–O bonding. The spectra indicated that BH3·THF interacted with the hydroxyl group in the supporting chain during the reaction. To further confirm the states of B species (Fig. 5c), the 11B NMR spectrum revealed that the –OH group of the chitin chain could capture BH3·THF and affect the catalytic environment around single-atom Pt sites. Hence, A plausible mechanism and DFT calculation (Supplementary Data 1 and Fig. S26) of the chemoselective hydrogenation between C=C and C=O bonding using the SS-Pt-CSNs catalyst was shown in Fig. 5d. In the hydrogenation of the C=C bond process, SS-Pt-CSNs first activated H2 to generate Pt–H species. Then, the C=C bond of 1a was inserted into Pt–H species and underwent protonation to afford the desired product 2a. In the hydrogenation of the C=O bond process, BH3 first reacted with the –OH groups of SS-Pt-CSNs to give B species as Lewis acid sites. Then, this composite catalyst (single-atom Pt sites and B species) activated H2 to generate Pt–H species. Next, B species coordinated with the carbonyl group of 1a and made the C=O bonds near Pt–H species. At last, the C=O bond was inserted into Pt–H species and underwent protonation to afford the desired product 3a.

a H2-TPR profiles of CSNs-250, SS-Pt-CSNs, and SS-Pt-CSNs (B) in the temperature range of 100 to 400 °C. b The high-resolution B 1s XPS pattern of NaBH4, Na2B4O7, and SS-Pt-CSNs (B). c The 11B-NMR pattern of H3BO3, NaBO3·4H2O, BH3·THF, CSNs-250, SS-Pt-CSNs and CSNs-250 (B) and SS-Pt-CSNs (B). d Proposed mechanism of the hydrogenation cycles between with BH3·THF or without BH3·THF.
Discussion
In summary, we have prepared biowaste-derived supramolecular nanowires-stabilized single-atom Pt catalyst from cheap and readily available biowaste (Chitin). The recyclable catalyst precisely controlled the product selectivity (average selectivities > 90:1) in the hydrogenation of α,β-unsaturated carbonyl compounds with broad substrate scope. Notably, this catalyst achieved extremely high TON (121,350) in such a hydrogenation reaction. The catalytic behavior could be attributed to the hybrid nature of the synthesized material, which exhibits both flexibility and robustness. DFT calculations showed that the empty coordination site of Pt and dynamic B-doping are the key factors for its exceptional efficiency and high selectivity. We believe this work will stimulate the scientific community to search for new waste-derived materials for the chemical industry.
Methods
Materials
Chitin powder was purchased from Golden-Shell Biochemical Co. Ltd. (Zhejiang, China). The chitin powders were purified with NaOH, HCl, and NaClO2 aqueous to remove the protein, mineral, and pigments, respectively. Chloroplatinic acid hexahydrate (H2PtCl6·6H2O, Pt ≥ 37.5%, Aladdin). Commercial platinum-carbon catalyst (Pt/C, 5 wt%, Aladdin), Cinnamaldehyde (95%, Aladdin), and sodium borohydride (Sinopharm Chemical Reagent Co., Ltd). All the other reagents such as isopropanol, methanol, toluene, tert-butyl alcohol, etc. were gained from various merchant resources. Unless otherwise noted, materials were obtained from commercial suppliers and used without further purification. Thin-layer chromatography (TLC) employed glass 0.25 mm silica gel plates. Flash chromatography columns were packed with 200–300 mesh silica gel.
Preparation of chitin supramolecular nanowires (CSNs)
By freeze-thaw process three times, 6 g chitin powders were added to the 100 g mixture of NaOH/Urea/H2O (11:4:85) aqueous solution under −30 °C to generate a transparent chitin solution. Then, the mixture of 250 g isooctane and 16.5 g Span 85 was firstly placed into a 1 L three-necked flask with a mechanical stirrer in an ice bath, followed by the addition of prepared chitin solution. After 1 h, with high-speed stirring, the 9 g Towen 85 was added to this mixture. The hybrid emulsion was kept stirring for 1 h in an ice bath and secondly heated in a water bath at 80 °C for 8 min to obtain regenerated microspheres made of chitin nanowires through neutralizing with hydrochloric acid (6 M). Finally, the obtained microspheres were washed with deionized water and ethanol several times, solvent-exchanged with tert-butyl alcohol (tBuOH), and lyophilized for later use.
Preparation of chitin supramolecular nanowires-anchored single-atom sites Pt catalysts (SS-Pt-CSNs)
Three hundred milligrams of the above chitin porous microspheres were immersed in 100 mL deionized water and the suspension was sonicated for 10 min at room temperature. Subsequently, H2PtCl6 [9.56 mg, 0.0185 mmol of 1.2 wt% Pt4+-CSNs; 19.12 mg, 0.037 mmol of 2.4 wt% Pt4+-CSNs; 38.24 mg, 0.074 mmol of 4.8 wt% Pt4+-CSNs] was dissolved in 6.0 mL H2O was added dropwise the suspension by fast stirring for 3 h. The resulting products were collected by filtration washed with deionized water and tert-butyl alcohol (tBuOH) and dried at freeze drying overnight. After drying, 1.2 wt% Pt4+-CSNs were transferred to activate at 250 °C under an argon atmosphere for 4 h, the 1.2 wt% Pt4+-CSNs were called SS-Pt-CSNs (Ar-250). The 1.2 wt% Pt4+-CSNs were transferred to activate at 250 °C under a hydrogen atmosphere for 4 h, it is called Nano-Pt-CSNs (H2-250). The 1.2 wt% Pt4+-CSNs were transferred to calcinate at 800 °C under an argon atmosphere for 2 h, called Nano-Pt-CSNs (Ar-800). The 2.4 wt% Pt4+-CSNs were transferred to activate at 250 °C under an argon atmosphere for 4 h, it is called 2.4% Nano-Pt-CSNs (Ar-250). The 4.8 wt% Pt4+-CSNs were transferred to activate at 250 °C under an argon atmosphere for 4 h, it is called 4.8% Nano-Pt-CSNs (Ar-250).
General procedure for chitin supramolecular nanowires-anchored precious metal catalysts (1.2% M-CSNs, M = Pd, Ru, and Au)
Three hundred milligrams of the above chitin porous microspheres were immersed in 100 mL acetone and the suspension was sonicated for 10 min at room temperature. Subsequently, Pd(OAc)2 [7.60 mg 0.0338 mmol of 1.2 wt% Pd2+-CSNs] was dissolved in 6.0 mL acetone and was added dropwise the suspension by fast stirring for 3 h. The resulting products were collected by filtration washed with deionized water and tert-butyl alcohol (tBuOH) and dried at freeze drying overnight. After drying, 1.2 wt% Pd2+-CSNs were transferred to activate at 250 °C under an argon atmosphere for 4 h, the 1.2 wt% Pd2+-CSNs were called 1.2% Pd-CSNs (Ar-250). Subsequently, the metal precursors change to RuCl3·xH2O [7.39 mg 0.0356 mmol of 1.2 wt% Ru3+-CSNs] and HAuCl4 [7.53 mg 0.0183 mmol of 1.2 wt% Au3+-CSNs] in 6.0 mL deionized. To repeat the same procedure, the 1.2 wt% Ru3+-CSNs and 1.2 wt% Au3+-CSNs were called 1.2% Ru-CSNs (Ar-250) and 1.2% Au-CSNs (Ar-250).
Characterization instrument
Scanning electron micrograph (SEM) images were recorded with a HITACHI 5-4800 microscope (Tokyo, Japan) at an accelerating voltage of 5 kV. Transmission electron microscopy (TEM) images were observed on a JEM-2010 (HT) electron microscope (JEOL, Japan) with an accelerating voltage of 200 kV. The size distribution of Metal nanoparticles was determined by a Mastersizer 2000 laser particle size analyzer (Malvern, UK). Scanning transmission electron microscopy (STEM) imaging and energy-dispersive X-ray spectroscopy (EDX) mapping were acquired on a JEOL JEM-ARM200F microscope operated at 200 kV with a Schottky cold-field emission gun at Wuhan University. The metal loading contents were determined using an inductively coupled plasma-optical emission spectrum (ICP-OES), Prodigy 7 (Leeman Labs Inc., USA). Surface area and pore size distribution were evaluated using nitrogen adsorption (Micromeritics, AsAp2020, U.S.A.). The specific surface area was calculated from the nitrogen adsorption isotherm using the Brunauer-Emmett-Teller (BET) equation and the pore size distribution using the density functional theory (DFT) model. X-ray photoelectron spectroscopy (XPS) was collected on a VG Multi Lab 2000 system with a monochromatic A1 Kα X-ray source (Thermo VG Scientific). Infrared spectroscopy was carried out using a Fourier transform infrared (FT-IR) spectrometer (model 1600, PerkinElmer Co., U.S.A.). X-ray absorption measurements were acquired in transmission mode at beamline (44 A in Taiwan Photon Source) at National Synchrotron Radiation Research Center (NSSRC) in Taiwan. A pure metal foil spectrum was acquired simultaneously with each measurement for energy calibration. Multiple scans were taken to reduce the noise. GC yields were recorded with a Shimadzu GC-2014 gas chromatograph instrument with the FID detector and biphenyl was added as an internal standard. 1H and 13C NMR data were recorded with Bruker Advanced II (400 MHz) spectrometers with tetramethylsilane as an internal standard. All chemical shifts (δ) were reported in ppm and coupling constants (J) in Hz. All chemical shifts are reported relative to tetramethylsilane and d-solvent peaks (77.00 ppm, chloroform), respectively. High-resolution mass spectra (HRMS) were measured with Waters Premier GC-TOF MS, and the accurate masses were reported for the molecular ion ([M]+). High-resolution mass spectra (HRMS) were measured with a Thermo Fisher LTQ Orbitrap Elite, and the accurate masses were reported for the molecular ion ([M + H]+). 11B NMR data of catalysts were recorded with Bruker AVANCE NEO 400 MHz spectrometers.
Chemoselective hydrogenation reactions
Hydrogenation of C=C double bonds
Selective hydrogenation tests were carried out in a 200 mL Teflon-lined stainless-steel autoclave. As shown in Fig. S1a-d, a 5.0 mL vial fitted with a normal magnetic stirring bar (4 × 2 mm) and septum cap. Single-atom Pt catalyst (5.0 mg), substrate (0.4 mmol), and 3.0 mL isopropanol (IPA) were added to the vial. Then, the vial was placed in a 200 mL high-pressure autoclave and flushed three times with 30 bar of hydrogen. The autoclave was pressurized with 30 bar of hydrogen placed into a stainless-steel block and heated at the desired temperature (80 °C). After the reaction, the autoclave was quickly cooled down at room temperature and vented. Then, the liquid was analyzed by gas chromatograph using diphenyl as an internal standard. Control experiments with Nano-Pt-SCNs (H2-250), Nano-Pt-CSNs (Ar-800), commercial Pt/C, homogeneous H2PtCl6 catalysts, blank chitin microspheres, and various percentages of catalysts were performed under the same conditions. The catalysts were separated by centrifugation for a recyclability test.
Hydrogenation of C=O double bonds
Selective hydrogenation tests were carried out in a 200 mL Teflon-lined stainless-steel autoclave. A 5.0 mL vial fitted with a normal magnetic stirring bar (4 × 2 mm) and septum cap. Single-atom Pt catalyst (5.0 mg), substrate (0.4 mmol), 12.5 mol% BH3·THF, and 3.0 mL isopropanol (IPA) were added into the vial. Then, the vial was placed in a 200 mL high-pressure autoclave and flushed three times with 30 bar of hydrogen. The autoclave was pressurized with 30 bar of hydrogen placed into a stainless-steel block and heated at the desired temperature (80 °C). After the reaction, the autoclave was quickly cooled down at room temperature and vented. Then, the liquid was analyzed by gas chromatograph using diphenyl as an internal standard. Control experiments with Nano-Pt-SCNs (H2-250), Nano-Pt-CSNs (Ar-800), commercial Pt/C, homogeneous H2PtCl6 catalysts, blank chitin microspheres, and various percentages of catalysts were performed under the same conditions. The catalysts were separated by centrifugation for a recyclability test.
Data availability
The data generated in this study are provided in the Supplementary Information. Source data are provided in this paper. Supplementary Data 1 includes Cartesian coordinates. Data supporting the findings of this manuscript are also available from the corresponding author upon request.
References
Kim, S. K. Chitin, Chitosan, Oligosaccharides and Their Derivatives: Biological Activities and Applications (CRD Press, 2011).
Yan, N. & Chen, X. Don’t waste seafood waste. Nature 524, 155–157 (2015).
Food and Agriculture Organization of the United Nations. The State of World Fisheries and Aquaculture (FAO, 2014).
Chen, X., Song, S., Li, H., Gözaydın, G. & Yan, N. Expanding the boundary of biorefinery: organonitrogen chemicals from biomass. Acc. Chem. Res. 54, 1711–1722 (2021).
Mondelli, C., Gozaydin, G., Yan, N. & Perez-Ramirez, J. Biomass valorisation over metal-based solid catalysts from nanoparticles to single atoms. Chem. Soc. Rev. 49, 3764–3782 (2020).
Xu, C., Nasrollahzadeh, M., Selva, M., Issaabadi, Z. & Luque, R. Waste-to-wealth: biowaste valorization into valuable bio(nano)materials. Chem. Soc. Rev. 48, 4791–4822 (2019).
Lu, L. J. et al. Carbon nanofibrous microspheres promote the oxidative double carbonylation of alkanes with CO. Chem 4, 2861–2871 (2018).
Rinaudo, M. Chitin and chitosan: properties and applications. Prog. Polym. Sci. 31, 603–632 (2006).
Jagadeesh, R. V. et al. MOF-derived cobalt nanoparticles catalyze a general synthesis of amines. Science 358, 326–332 (2017).
Friend, C. M. & Xu, B. Heterogeneous catalysis: a central science for a sustainable future. Acc. Chem. Res. 50, 517–521 (2017).
Fu, Q. & Bao, X. H. Heterogeneous catalysis confined microenvironment for catalysis control. Nat. Catal. 2, 834–836 (2019).
Huang, X. Q. et al. Heterogeneous catalysis confined microenvironment for catalysis control. Science 348, 1230–1234 (2015).
Gao, C. B., Lyu, F. L. & Yin, Y. D. Encapsulated metal nanoparticles for catalysis. Chem. Rev. 121, 834–881 (2021).
Song, J. W. et al. Processing bulk natural wood into a high-performance structural material. Nature 554, 224–228 (2018).
Chen, G. et al. Ligand-accelerated enantioselective methylene C(sp3)-H bond activation. Science 353, 1023–1027 (2016).
Wang, P. et al. Ligand-accelerated non-directed C–H functionalization of arenes. Nature 551, 489–493 (2017).
Shao, X. Z. et al. Iridium single-atom catalyst performing a quasi-homogeneous hydrogenation transformation of CO2 to formate. Chem 5, 693–705 (2019).
Cui, X. J., Li, W., Ryabchuk, P., Junge, K. & Beller, M. Bridging homogeneous and heterogeneous catalysis by heterogeneous single-metal-site catalysts. Nat. Catal. 1, 385–397 (2018).
Zhao, D. et al. Atomic site electrocatalysts for water splitting, oxygen reduction and selective oxidation. Chem. Soc. Rev. 49, 2215–2264 (2020).
Yang, X. F. et al. Single-atom catalysts: a new frontier in heterogeneous catalysis. Acc. Chem. Res. 46, 1740–1748 (2013).
Wang, Y., Wang, D. S. & Li, Y. D. Rational design of single-atom site electrocatalysts: from theoretical understandings to practical applications. Adv. Mater. 33, 2008151 (2021).
Tang, C. et al. Tailoring acidic oxygen reduction selectivity on single-atom catalysts via modification of first and second coordination spheres. J. Am. Chem. Soc. 143, 7819–7827 (2021).
Qiao, B. T. et al. Single-atom catalysis of CO oxidation using Pt-1/FeOx. Nat. Chem. 3, 634–641 (2011).
Liu, P. X. et al. Photochemical route for synthesizing atomically dispersed palladium catalysts. Science 352, 797–800 (2016).
Zhao, X. J. et al. Thiol treatment creates selective palladium catalysts for semihydrogenation of internal alkynes. Chem 4, 1080–1091 (2018).
Chen, X. W. et al. Regulating coordination number in atomically dispersed Pt species on defect-rich graphene for n-butane dehydrogenation reaction. Nat. Commun. 12, 2664 (2021).
Tan, Y. et al. ZnAl-hydrotalcite-supported Au25 nanoclusters as precatalysts for chemoselective hydrogenation of 3-nitrostyrene. Angew. Chem. Int. Ed. 56, 2709–2713 (2017).
Lee, S. H. et al. k. Dynamic metal-polymer interaction for the design of chemoselective and long-lived hydrogenation catalysts. Sci. Adv. 6, b7369 (2020).
Zhang, L. L., Zhou, M. X., Wang, A. Q. & Zhang, T. Selective hydrogenation over supported metal catalysts: from nanoparticles to single atoms. Chem. Rev. 120, 683–733 (2020).
Duan, B. et al. Highly biocompatible nanofibrous microspheres self-assembled from chitin in NaOH/urea aqueous solution as cell carriers. Angew. Chem. Int. Ed. 54, 5152–5156 (2015).
Xu, D. et al. High-flexibility, high-toughness double-cross-linked chitin hydrogels by sequential chemical and physical cross-linkings. Adv. Mater. 28, 5844–5849 (2016).
Huang, J., Zhong, Y., Zhang, L. & Cai, J. Extremely strong and transparent chitin films: a high-efficiency, energy-saving, and “green” route using an aqueous KOH/urea solution. Adv. Funct. Mater. 27, 1701100 (2017).
Heux, L., Brugnerotto, J., Desbrieres, J., Versali, M. F. & Rinaudo, M. Solid state NMR for determination of degree of acetylation of chitin and chitosan. Biomacromolecules 1, 746–751 (2000).
Pei, X. et al. Ultra-small Pd clusters supported by chitin nanowires as highly efficient catalysts. Nano Res. 11, 3145–3153 (2018).
Titze, M., Heitkämper, J., Junge, T., Kästner, J. & Peters, R. Highly active cooperative lewis acid—ammonium salt catalyst for the enantioselective hydroboration of ketones. Angew. Chem. Int. Ed. 60, 5544–5553 (2021).
Ding, Y. et al. A heterogeneous metal-free catalyst for hydrogenation: lewis acid–base pairs integrated into a carbon lattice. Angew. Chem. Int. Ed. 57, 13800–13804 (2018).
Gilkey, M. et al. Mechanistic insights into metal lewis acid-mediated catalytic transfer hydrogenation of furfural to 2-methylfuran. ACS Catal 5, 3988–3994 (2015).
Ma, M. et al. Creation of surface frustrated Lewis pairs on high-entropy spinel nanocrystals that boosts catalytic transfer hydrogenation reaction. Chem. Eng. J. 470, 144291 (2023).
Acknowledgements
This work was supported by the National Key R&D Program of China (no. 2021YFA1500100) (A.L.), the National Natural Science Foundation of China (22031008) (A.L.), the Key Project of Natural Science Foundation of Guizhou Province (no. ZK[2023] Key 025) (X.P.), the Science Foundation of Wuhan (2020010601012192) (A.L.), and the National Natural Science Foundation of China (91963109) (A.L.).
Author information
Authors and Affiliations
Contributions
Y.L., L.L., X.P., and A.L. conceived and designed the project. Y.L. and X.P. performed the initial discovery and optimization of test conditions. Y.L. and X.J. prepared the catalysts. Y.L., L.L., X.P., and A.D.C. performed mechanistic studies and all computational experiments. D.Y. and J.-L.C. performed the XAS test and analyzed the data. Y.L., L.L., W.L., X.P., and A.L. wrote the manuscript with input from all other authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Qingxin Guan, and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, Y., Lu, L., Jiang, X. et al. Dynamic boron-doping switched chitin-based single-atom Pt catalyst for chemo-selective hydrogenation. Nat Commun 16, 2296 (2025). https://doi.org/10.1038/s41467-025-57434-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-57434-0
