
Abstract
Hypertrophic cardiomyopathy (HCM) affects approximately 600,000 people in the United States. Loss-of-function mutations in Myosin Binding Protein C3, MYBPC3, are the most common genetic cause of HCM, with the majority of mutations resulting in haploinsufficiency. To restore cardiac MYBPC3, we use an adeno-associated virus (AAV9) vector and engineer an optimized expression cassette with a minimal promoter and cis-regulatory elements (TN-201) to enhance packaging efficiency and cardiomyocyte expression. Rather than simply preventing cardiac dysfunction preclinically, we demonstrate in a symptomatic MYBPC3-deficient murine model the ability of AAV gene therapy to reverse cardiac hypertrophy and systolic dysfunction, improve diastolic dysfunction, and prolong survival. Dose-ranging efficacy studies exhibit restoration of wild-type MYBPC3 protein levels and saturation of cardiac improvement at the clinically relevant dose of 3E13 vg/kg, outperforming a previously published construct. These findings suggest that TN-201 may offer therapeutic benefits in MYBPC3-associated cardiomyopathy, pending further validation in clinical settings.
Similar content being viewed by others
Introduction
Mutations in MYBPC3, the gene encoding cardiac myosin-binding protein C (MYBPC3 or cMyBP-C), are the most common genetic cause of HCM1. HCM increases the risks of sudden cardiac death (SCD) and is a leading cause of SCD in children and young adults2,3,4,5. In primary literature, the reported prevalence of MYBPC3 mutations in patients with diagnosed HCM ranges from 17–26%1,6,7,8,9,10. MYBPC3-associated HCM, an autosomal dominant condition, is a progressive disease characterized by left ventricular (LV) hypertrophy, small LV volume, ventricular diastolic dysfunction, cardiac arrhythmias, and an imbalance between myocardial oxygen supply and demand. Disease burden may include dyspnea, exercise intolerance, atypical angina, syncope, ventricular arrhythmias, SCD, and heart failure. The Sarcomeric Human Cardiomyopathy Registry (SHaRe) identified risk factors for HCM severity based on genotype and lifetime burden of disease. Primary risk factors included an identified sarcomere mutation (including MYBPC3) and early onset of disease (before age 40 years)1.
Cardiac MYBPC3 protein has structural and functional roles in sarcomere biology, acting as a brake that limits actin-myosin cross-bridge interactions during cardiac contraction and relaxation11,12,13,14. MYBPC3 truncating mutations reduce MYBPC3 protein content, thereby enhancing maximal thin filament sliding velocity within the thick filament C zone. The pathophysiology of MYBPC3-associated HCM is clearly attributable to MYBPC3 protein deficiency. Myectomy samples from patients who have HCM with a MYBPC3 mutation had 40% lower MYBPC3 protein levels, with no detectable expression of truncated MYBPC3 from the mutant MYBPC3 gene, compared to normal control samples15,16,17,18,19. The lack of detectable truncated MYBPC3 suggests that either no protein was synthesized from the mutant MYBPC3 allele or, alternatively, that the truncated protein was susceptible to rapid degradation. Truncating variants account for 91% of pathogenic MYBPC3 genes and cause similar clinical severity and outcomes regardless of the location of the truncation within the protein product, consistent with locus-independent loss of function20. These data support a rationale for developing TN-201, a MYBPC3 gene replacement therapy, which is designed to correct both the pathogenic reduction in MYBPC3 in heterozygotes and the total loss of protein in homozygotes and compound heterozygotes21,22,23. The latter can exhibit severe dilatation and typically experience heart failure and death within the first year of life24.
Gene therapy is potentially curative and increasingly feasible for a myriad of genetic and chronic diseases. Although AAV currently represents the most tractable viral vehicle due to superior safety and manufacturability, the size constraints of the AAV genome (4.7 kb) frequently limit which therapeutic cargoes can be delivered and their expression25,26,27. Transgene expression is minimally dependent on the inclusion of two intact flanking ITR sequences, a promoter, and a polyadenylation signal. With additional accessory elements such as an intron and WPRE, standard cis-regulatory sequences used in AAV transgene expression cassettes can amount to ~1.2 kb without the promoter. This limits the ability to efficiently package large genes that require tissue-specific expression in AAV, including our primary therapeutic target MYBPC3 cDNA (3.825 kb).
The purpose of this study was to engineer an optimal AAV genomic cassette for MYBPC3 expression in cardiomyocytes and test its ability to rescue an in vivo MYBPC3-associated cardiomyopathy model. We demonstrated durable improvement in cardiac phenotypes, including reduction of cardiac hypertrophy, improvement in systolic and diastolic function, and extended survival in this murine model.
Results
Engineering a leaner AAV genomic cassette without loss of strength or specificity of cardiac expression
The size constraints of the AAV genome are highly restrictive for optimal expression of large therapeutic genes which require tissue-specific expression, such as MYBPC3 (ORF = 3.825 kb). After determining no major expression decreases occurred upon deletion of the WPRE element (540 bp) and minimization of sequences required for polyadenylation (Fig. S1), we focused our attention on minimizing a cardiac promoter without loss of strength or specificity for MYBPC3 gene replacement therapy.
We first established successful transgene expression and protein localization in MYBPC3-/- induced pluripotent stem cell-derived cardiomyocytes (iPSC-CMs) (Fig. 1a). We then tested multiple versions of the minimized cassette (compared to a 5.6 kb standard cassette) with different cardiac promoters, called pCard, driving expression of human MYBPC3 (Fig. 1b). The constructs were packaged into AAV, used to infect human MYBPC3-/- iPSC-CMs and their resultant protein expression ascertained by immunoblotting. Surprisingly, a smaller version of the pCard0 promoter, pCard1, drove the greatest expression (Fig. 1c, d; from the human cardiac human cardiac troponin T (TNNT2) promoter, consistent with previous iterations28,29; sequences in patent30 and Source Data File). Given the pCard0 construct was 4.9 kb, exceeding the size of the wild-type AAV genome, 4.7 kb, we considered that the pCard1 4.7 kb version might drive increased expression through improved packaging of the AAV genome. Although assessment of virally packaged pCard0 genomes did demonstrate greater heterogeneity than those of the pCard1 cassette (Fig. S2a), we also observed decreased protein expression of the pCard0 cassette compared to pCard1 when MYBPC3-/- iPSC-CMs were transiently transfected with the naked plasmids (Fig. S2b). Thus, the increased expression driven by pCard1 virus appears to be due to a combination of increased potency of the DNA sequence itself and less heterogeneity of viral packaging.

a In order to validate the assay system, we confirmed transgene expression resulted in proper localization in human MYBPC3-/- iPSC-CMs transduced with AAV:MYBPC3. Immunofluorescence analysis was performed seven days post-infection (scale bars, 25 µm). b Cassette schematic indicating the genomic size of a standard cassette and the alterations tested. c Human MYBPC3-/- iPSC-CMs were transduced with AAV6-packaged constructs encoding human MYBPC3 driven by various promoter versions of human cardiac troponin T (TNNT2) (pCard0, pCard1, and pCard2) and harvested 5 days post-infection (n = 1–2/condition). d A head-to-head comparison of the independently packaged versions of the pCard0 and pCard1 constructs. MYBPC3-/- iPSC-CMs were harvested five days post-infection with CR9-01-packaged constructs (n = 2/condition). e To determine if the optimized promoter also increased expression in vivo and whether selectivity was maintained, adult female mice were retro-orbitally injected with 4E13 vg/kg AAV9 encoding the pCard0 (n = 3) and pCard1 (n = 4) constructs. Heart, skeletal muscle (tibialis anterior), liver, and whole brain samples were harvested two weeks post-injection. P-value per two-sided Student’s t-test with 1.0% False Discovery Rate with Two-stage step-up (Benjamini, Krieger, and Yekutieli). Matched transduction (f), RNA (g), and protein (h) analysis in MYBPC3-/- iPSC-CMs demonstrated near-WT levels of protein expression at 1 vg/dg compared to parental wild-type control iPSC-CMs one-week post-infection. WT No Inf (n = 3), MYBPC3-/- No Inf (n = 2), MYBPC3-/- 1 K (n = 3), MYBPC3-/- 3 K (n = 3), WT GFP 100 K (n = 3), and WT GFP 100 K (n = 3). 1 K = multiplicity of infection of 1000, or 1000 vector genomes/cell. Data are shown as means ± SEM. Source data are provided as a Source Data file.
Critical to the utility of this shorter and stronger promoter is the retention of cardiac selectivity. We therefore packaged the pCard0 and pCard1 versions into AAV9 for assessment of strength and selectivity in vivo. Adult mice were retro-orbitally injected with 4E13 vg/kg and tissue samples from the heart, skeletal muscle, liver, and whole brain were harvested two weeks post-injection. RNA was extracted from all tissues and analyzed by qPCR with primers specific to human MYBPC3. Both viral preps resulted in robust RNA expression in the heart, with ~200× selectivity of expression in the heart over the brain and >1000× selectivity over skeletal muscle and liver (Fig. 1e). Thus, the boost in expression of the pCard1 construct was confirmed in vivo (>2× improvement in cardiac RNA expression) and, critically, this decrease in size did not affect the selectivity of expression in vivo. Thus, AAV9 packaging pCard1-MYBPC3 was selected for further development as a therapeutic candidate and henceforth designated TN-201.
To evaluate the expression strength of the TN-201 genomic cassette in human cardiomyocytes, homozygous mutant MYBPC3-/- iPSC-CMs were generated and the dose-responsiveness of TN-201 genomic cassette-mediated transgene expression was compared with MYBPC3 levels in WT isogenic control iPSC-CMs. To achieve the greatest levels of iPSC-CM infection for this transduction assessment, the TN-201 AAV genomic cassette was packaged into the AAV9 serotype variant CR9-01, which exhibits higher infectivity than AAV9 of cultured iPSC-CMs31,32. This enabled efficient utilization of the virus while maintaining the integrity of TN-201 AAV genomic cassette assessment. The CR9-01:hMYBPC3 treatment is henceforth denoted as TN-201* to distinguish it from TN-201. MYBPC3 transgene RNA and MYBPC3 protein levels in MYBPC3-/- iPSC-CMs were compared with endogenous levels of MYBPC3 RNA and MYBPC3 protein in WT iPSC-CMs one-week post-infection.
To precisely determine how these transgene expression evaluations mapped to transduction levels, matched vector genome (Fig. 1f), RNA (Fig. 1g), and protein (Figs. 1h and S2c) analyses were performed one-week post-infection. Absolute quantification of nuclear vector genomes indicated that the lowest dose administered (1 K TN-201*) resulted in ~1.6E5 vg/µg DNA, which is equivalent to ~1 vector genome per diploid genome (vg/dg). A matched assessment of MYBPC3 RNA and MYBPC3 protein at this dose indicated ~1.6× the endogenous level of MYBPC3 RNA and ~0.9× the endogenous level of MYBPC3 protein. Consistent with vector genome analysis, flow cytometry one-week post-infection with CR9-01:pCard1-GFP, a green fluorescent protein reporter, demonstrated that at the lowest dose used (1 K)~60% of iPSC-CMs were transduced (Fig. S2d), indicating an infectious multiplicity of infection equivalent to one based on the Poisson distribution33. Thus, transduction equivalent to approximately one vector genome per diploid genome drove near-WT levels of MYBPC3 RNA and MYBPC3 protein one-week post-infection. This strength of expression in hiPSC-CMs propelled us to move forward in attempting to treat an in vivo model of MYBPC3-associated cardiomyopathy.
Murine Mybpc3 -/- model for treatment of HCM
Mybpc3-/- mice were established on the C57BL/6 background as a model for gene replacement of MYBPC3 in severely affected HCM patients by a CRISPR-Cas9 paired gRNA deletion of exons one and two (Fig. 2a; please refer to “Methods” section). Heterozygous and homozygous animals were compared with WT controls for a number of cardiovascular function and morphology parameters. Heterozygous Mybpc3+/- mice showed no cardiovascular phenotype and were therefore not considered to be a useful model for efficacy studies. Both male and female homozygous mice exhibited severe deficits in cardiac function (Fig. 2b) and pronounced cardiac hypertrophy (Fig. 2c–f) as early as two weeks of age (Table S1). With body weight normalized LV mass 3–4 times that of WT siblings, increased posterior wall thickness or LVPW;d (1.6× WT siblings), and severe cardiac dysfunction (ejection fraction (EF): 35%–40%), this model closely mimics late-stage HCM, as well as in infants with compound heterozygous or homozygous mutations in the MYBPC3 gene. LV hypertrophy is the defining feature of all patient classes with wall thickness as a key prognostic marker for HCM and a major risk factor for cardiovascular mortality and SCD34. LV dilatation is more common in pediatric compound heterozygotes or homozygotes24,35,36,37,38. Due to the severe phenotype of the Mybpc3-/- mice, and the lack of any MYBPC3 protein, this is a challenging model to demonstrate efficacy.

a A null mutant allele of Mybpc3 was generated by excision of exons one and two using CRISPR-Cas9 and confirmed by demonstrating MYBPC3 depletion in an immunoblot from cardiac tissue (n = 4/condition). Echocardiography analysis of homozygous mutants, heterozygous mutants, and WT littermates demonstrated a significant decrease in cardiac systolic function in homozygous animals based on b ejection fraction (%) at two weeks of age. c Homozygous mice exhibited marked LV hypertrophy at two weeks of age as evidenced by their LV mass normalized by body weight and d increased left ventricular posterior wall thickness during diastole (LVPW;d). Left ventricular internal diameters during e systole (LVID;s) and f diastole (LVID;d) were also significantly increased in homozygotes at two weeks of age. WT (n = 10; 4 M/6 F), Mybpc3+/- (n = 7; 4 M/3 F) and Mybpc3-/- (n = 12; 6 M/6 F). Heterozygous mice were not significantly different from WT in any parameter. P-value per one-way ANOVA with Tukey’s multiple comparisons test. Data are shown as means ± SEM. Source data are provided as a Source Data file.
AAV9:mMybpc3 and TN-201 improved hypertrophy, cardiac dysfunction and premature lethality of Mybpc3 -/- mice
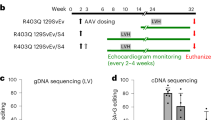
To test whether AAV treatment can improve Mybpc3-/- mice with cardiac dysfunction, and not merely prevent the onset of symptoms28, AAV9:mMybpc3, the mouse surrogate of TN-201 (also known as mTN-201), and TN-201 were administered to symptomatic Mybpc3-/- mice. Although AAV-mediated assessment of human MYBPC3 cDNA has previously been studied in human iPSC-CMs39, this experiment provided an opportunity for in vivo assessment of the human MYBPC3 activity in a mouse model. The mouse ortholog was utilized for the appropriate species-matched comparison since the human ortholog is only 88% identical at the amino acid level. Vehicle, AAV9:mMybpc3, or TN-201 were administered systemically (1E14 vg/kg) via retro-orbital intravenous (IV) injection to symptomatic 2-week-old Mybpc3-/- mice (9–12/group), when the EF averaged 35.2 ± 1.0% (versus 73.4 ± 0.9% in WT mice) and normalized LV mass was 4-fold above WT. An additional group of 12 WT littermates was administered vehicle as a control. Echocardiography was performed to assess cardiac function until week 54. Animal survival was followed up until animals were 20 months of age, at which time the remaining animals were euthanized.
Both treatments significantly improved LV hypertrophy, cardiac function, and lifespan relative to vehicle-treated Mybpc3-/- mice. As might be anticipated based on sequence divergence between the mouse and human genes, the species-matched treatment, AAV9:mMybpc3, produced larger improvements than the treatment with the human ortholog, TN-201. Both AAV9:mMybpc3 and TN-201 significantly decreased LV hypertrophy (Fig. 3a), with mean improvements of 5.1 ± 0.7 mg/g and 4.2 ± 0.8 mg/g for LV mass respectively, compared to vehicle-treated Mybpc3-/- mice at 31 weeks following viral delivery (Fig. 3b and Table S2). Treatment with AAV9:mMybpc3 increased EF by up to 26% above baseline over 6 weeks and then had a stable effect on EF up until the animals were 13 months of age, which was the age of the last echocardiography measurements. Animals treated with TN-201 had a 5% increase in EF over 10 weeks from baseline and then showed a slow decline in EF as they aged (Fig. 3c). Both AAV9:mMybpc3 and TN-201 resulted in superior cardiac function compared to vehicle-treated Mybpc3-/- mice, with mean EF improvements of 30 ± 1.7% and 18 ± 1.9%, respectively, at 31 weeks following viral delivery (Figs. 3d and S3).

a LV mass normalized to body weight progression over time and b at 31 weeks post-delivery. c EF progression and d EF at 31 weeks post-delivery. e Left ventricular posterior wall thickness in diastole (LVPW;d) and f QT interval at 31 weeks post-delivery. g Kaplan–Meier survival curve with animals followed out until 20 months of age. Median survival for Mybpc3-/- vehicle animals was 14 months, and all animals died by 16 months of age. For TN-201-treated animals, median survival was 16 months (i.e. 2 months lifespan extension from vehicle), and all animals died by 19 months of age. AAV9:mMybpc3 treatment extended lifespan, with only one animal euthanized due to skin lesions not related to heart failure by 20 months. WT (n = 12; 5 M/7 F), Mybpc3-/- Veh (n = 11; 5 M/6 F; 10 at 31 weeks), Mybpc3-/- AAV9:mMybpc3 (n = 12; 6 M/6 F) and Mybpc3-/- TN-201 (n = 9; 4 M/5 F). Data are shown as means ± SEM. WT mice were significantly different from all groups for all parameters, with the exception of AAV9:mMybpc3-treated animals for QT interval. P-value per one-way ANOVA with Tukey’s multiple comparisons test. Mantel–Cox test used for Kaplan–Meier survival curve analysis. Source data are provided as a Source Data file.
Notably, AAV9:mMybpc3 and TN-201 significantly improved posterior wall thickness by 0.19 ± 0.06 mm and 0.22 ± 0.07 mm, respectively, relative to vehicle (Fig. 3e) at 31 weeks post-delivery. This again indicated that both treatments improve LV hypertrophy. Similar to patients with MYBPC3-associated HCM40 and consistent with established mouse models41,42, Mybpc3-/- mice exhibit QT interval prolongation. Critically, both treatments significantly improved QT prolongation compared to vehicle-treated Mybpc3-/- mice (Fig. 3f) and lifespan was significantly increased in both treatment groups (Fig. 3g). AAV9:mMybpc3 treatment resulted in lifespan extension >6 months, with only one animal death prior to study-wide euthanasia, which was due to factors unrelated to heart failure. Even the TN-201 treatment group had their lifespans extended by approximately two months. Thus, species-matched treatment resulted in profound improvements in LV hypertrophy, EF, QT interval prolongation, and survival. Further, despite the sequence divergence between the mouse and human orthologs, TN-201 mediated significant efficacy in multiple parameters assessed in this mouse cardiomyopathy model.
AAV9:mMybpc3 improved cardiac function at a dose as low as 1E13 vg/kg
Given the proof-of-concept study was performed at 1E14 vg/kg, we moved forward with titration of AAV9:mMybpc3 to determine the dose-response relationship for cardiac improvement. We evaluated three single doses of AAV9:mMybpc3 in Mybpc3-/- mice. The treatments were given systemically at two weeks of age via retro-orbital injections, at 1E13 vg/kg, 3E13 vg/kg, or 1E14 vg/kg. As controls, the vehicle was injected into a group of wild type (WT) and a group of Mybpc3-/- animals.
AAV9:mMybpc3 was beneficial to the recovery of cardiac function at every dose tested, with dose-dependent improvements in LV mass (Fig. 4a), EF (Fig. 4b), and QT interval (Fig. 4c). The dose of 3E13 vg/kg yielded robust and highly significant improvements with decreased hypertrophy of 5.3 ± 0.8 mg/g LV mass and improved EF of 26 ± 3.0%. A similar trend was observed for improvement of QT prolongation (Fig. 4c). The lower dose of 1E13 vg/kg dose produced smaller improvements with decreased hypertrophy of 3.2 ± 1.3 mg/g LV mass and improved cardiac function as assessed by EF of 15 ± 3.7%. Indicative of treatment tolerability in this disease model, there were no differences in body weights among the treatment groups (Fig. 4d). Of note, animals treated with 3E13 and 1E14 vg/kg did not exhibit significant differences in improvement of hypertrophy, EF or QT interval prolongation, suggesting a plateau in effect. Thus, the clinically relevant dose of 3E13 vg/kg43,44 appeared to achieve near-maximal efficacy for saturation of cardiac benefit.

Dose-dependent improvement compared to vehicle-treated Mybpc3-/- mice in a LV mass normalized to body weight, b EF, and c QT interval at 31 weeks post-delivery. d No group differences in body weight at 31 weeks post-delivery. Data are shown as means ± SEM. WT mice were significantly different from all groups for all echocardiographic parameters, with the exception of 3E13 vg/kg and 1E14 vg/kg AAV9:mMybpc3-treated animals for QT interval. WT (n = 7; 4 M/3 F), Mybpc3-/- Veh (n = 7; 4 M/3 F), Mybpc3-/- 1E13 vg/kg (n = 4; 3 M/1 F), Mybpc3-/- 3E13 vg/kg (n = 6; 4 M/2 F), and Mybpc3-/- 1E14 vg/kg (n = 6; 4 M/2 F). P-value per one-way ANOVA with Tukey’s multiple comparisons test. Source data are provided as a Source Data file.
Further, to control for the effect of the AAV capsid proteins and other components of the vector expression cassette on the heart, AAV9 with the same cassette but encoding GFP (pCard1-GFP) was injected into Mybpc3-/- mice at the efficacious dose of 3E13 vg/kg. In contrast to AAV9:mMybpc3, echocardiographic and electrocardiographic assessments six weeks post-injection demonstrated no significant benefit of animals dosed with AAV9:pCard1-GFP compared to vehicle-treated animals in LV mass normalized to body weight, EF, posterior wall thickness in diastole, and QT interval (Fig. S4). This clearly shows that any beneficial effect observed is derived from the transgene itself.
Cardiac restoration of wild-type MYBPC3 protein levels
Given that animals treated with 3E13 and 1E14 vg/kg did not exhibit significant differences in improvement of hypertrophy, EF or QT interval prolongation, we hypothesized that both doses were adequate to restore wild-type levels of MYBPC3 protein to deficient mice. The kinetics and dose-responsiveness of transgene RNA and protein expression in Mybpc3-/- mice was assessed to match efficacy studies with AAV9:mMybpc3. Doses of 3E13 vg/kg and 1E14 vg/kg were administered systemically using retro-orbital IV injection at two weeks of age. Two weeks post-injection, homozygotes dosed with 3E13 and 1E14 vg/kg exhibited WT levels of cardiac MYBPC3 protein expression, as assessed by immunoblot (Fig. 5a and phosphorylation Fig. S5) and ELISA (Fig. 5b). Similar results were obtained six weeks post-injection, with dose-dependent Mybpc3 RNA expression observed (Fig. 5c). To determine the extent of cardiomyocyte transduction, immunohistochemistry for MYBPC3 four weeks post-injection was performed. Results indicated the ability to restore MYBPC3 protein expression to the majority of cardiomyocytes in Mybpc3-/- mice at both 3E13 and 1E14 vg/kg dose levels (Fig. 5d). Thus, cardiac restoration of wild-type MYBPC3 protein levels corresponded to the comparable efficacy observed between Mybpc3-/- animals dosed with 3E13 and 1E14 vg/kg (Fig. 4). Further, confocal imaged co-staining for α-actinin demonstrated proper sarcomeric incorporation in animals six weeks following viral dosing (Fig. 5e). To investigate the dose-dependent increase in transgene RNA without comparable increases in protein levels, we assessed transgene RNA and protein levels in a pilot safety evaluation of AAV9:mMybpc3 in naïve adult CD-1 mice. AAV9:mMybpc3 administered to WT mice was selected to achieve the greatest potential total levels of MYBPC3 protein expression. Retro-orbital IV injection at 10 weeks of age was used to deliver AAV9:mMybpc3 at 3E13 or 3E14 vg/kg, or vehicle control. AAV9:mMybpc3 was well tolerated in naïve CD-1 mice. Cardiac function and morphology were unaffected by treatment and similar among all groups, and body weight remained stable (Fig. S6). Interestingly, AAV9:mMybpc3 administered at 3E14 vg/kg to WT animals exhibited a 6.5-fold increase in Mybpc3 RNA (normalized to Gapdh) above vehicle-treated animals (Fig. 5f), but no statistically significant increase in MYBPC3 protein (Fig. 5f, g) based on ELISA. This observation of an increase in RNA without a proportional increase in protein is consistent with published findings that show transgenic mouse lines over-expressing Mybpc3 RNA do not have MYBPC3 protein levels greater than WT45,46. Taken together, these results explain the comparable restoration of cardiac MYBPC3 protein for 3E13 and 1E14 vg/kg, despite significant differences in transgene RNA. Further, these results highlight the attractive safety profile of MYBPC3 gene replacement therapy, given apparent homeostatic regulation of protein expression to near-WT levels in vivo.

Homozygous mice were dosed retro-orbitally IV with vehicle or the indicated doses (vg/kg) of AAV9:mMybpc3, and cardiac protein analyzed two weeks and six weeks post-injection. Restoration of WT levels of cardiac MYBPC3 protein upon dosing Mybpc3-/- mice at two weeks of age with 3E13 and 1E14 vg/kg based upon a immunoblot and b ELISA. c Mybpc3 transgene RNA two weeks and six weeks post-injection. WT (n = 3; 1 M/2 F), Mybpc3-/- Veh (n = 2; 2 F), Mybpc3-/- 3E13 vg/kg (n = 3; 1 M/2 F) and Mybpc3-/- 1E14 vg/kg (n = 3; 1 M/2 F). d Immunohistochemistry for MYBPC3 4 weeks post-injection. Homozygous mice were dosed retro-orbitally IV with vehicle or the indicated doses (vg/kg) of AAV9:mMybpc3 at two weeks of age. WT littermates were harvested, and samples processed simultaneously (n = 2/condition; 10× scale bars: 2 mm; 40× scale bars: 25 µm). e Confocal images for assessment of sarcomere structure in animals six weeks following viral dosing (n = 4/condition, 90× scale bars: 25 µm). f Adult WT male mice were dosed retro-orbitally IV with vehicle or the indicated doses (vg/kg) of AAV9:mMybpc3 and cardiac transgene expression was analyzed 10 weeks post-injection by qPCR for Mybpc3 RNA (normalized to Gapdh). Protein assessment in WT mice was performed by ELISA with equivalent total protein for each sample, as well as g immunoblot; Vehicle, Veh (n = 6), 3E13 vg/kg (n = 5) and 3E14 vg/kg (n = 6). P-value per one-way ANOVA with Tukey’s multiple comparisons test. Data are shown as means ± SEM. Source data are provided as a Source Data file.
AAV9:mMybpc3 improved cardiac diastolic dysfunction at 3E13 vg/kg
Homozygous mice exhibited diastolic dysfunction manifested by slowed maximal mitral E-wave velocity (MV E), slowed mitral annular e’ velocity, and prolonged isovolumic relaxation times (IVRT) at 14 weeks of age (Fig. 6 and Table S3). The decrease and prolongation in these parameters indicate heart muscle relaxation slowing. AAV9:mMybpc3 treatment was given systemically at two weeks of age via retro-orbital injections at 3E13 vg/kg. Improvement of diastolic dysfunction was assessed 12 weeks later. As a control, the vehicle was injected into a group of wild type (WT) and a group of Mybpc3-/- animals (n = 9/group). Importantly, AAV9:mMybpc3 was able to ameliorate diastolic dysfunction by improving these Doppler parameters in Mybpc3-/- mice. MV E treatment values were restored nearly to WT levels and were increased by 222 ± 44 mm/s relative to Mybpc3-/- vehicle. The other diastolic parameters e’ and IVRT were improved by 7.2 ± 1.1 mm/s and 7.2 ± 1.2 ms, respectively, relative to the vehicle. Taken together, the clinically relevant 3E13 vg/kg dose was able to ameliorate several diastolic dysfunction parameters associated with Mybpc3-/- mice and HCM, partially restoring proper heart muscle relaxation.

Homozygous mice were dosed retro-orbitally IV with vehicle or 3E13 vg/kg of AAV9:mMybpc3 at two weeks of age and assessed for diastolic dysfunction 12 weeks post-injection. a Representative tissue (top) and pulsed-wave transmitral (bottom) Doppler tracings. Mitral annular e’ velocity, maximal mitral E-wave velocity (E), and maximal mitral A-wave velocity (A) shown. b Quantitation of mitral E-wave velocity (MV E), c mitral annular e’ velocity, and d isovolumic relaxation time (IVRT) demonstrated dysfunction in Mybpc3-/- mice that was ameliorated by treatment, n = 9 (5 M/4 F)/group. P-value per one-way ANOVA with Tukey’s multiple comparisons test. Data are shown as means ± SEM. Source data are provided as a Source Data file.
Concomitant decreased markers of fibrosis and heart failure
Having established significant improvements in cardiac function with a dose as low as 1E13 vg/kg, we sought to understand the molecular signature of MYBPC3 restoration in the dynamic dose range. Vehicle or AAV9:mMybpc3 at doses of 1E13 and 3E13 vg/kg were administered systemically via retro-orbital IV injection to symptomatic two-week-old Mybpc3-/- mice. An additional group of WT littermates were administered vehicle as a control. Echocardiography was performed to assess cardiac function until Week 14 post-delivery, at which point cardiac tissue was harvested for characterization.
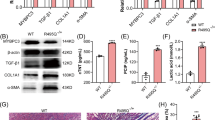
Consistent with characterization of other Mybpc3 mutant models47,48, we observed significant upregulation of cardiac markers of heart failure (Fig. 7a–c). Notably, the observed dose-dependency in echocardiography parameters (Fig. 4) was recapitulated in dose-dependent transcriptional reduction of cardiac markers of heart failure (Fig. 7a–c), such as a BNP (Nppb), where elevated levels are predictive clinically for HCM prognosis49. Further, consistent with MYBPC3-associated HCM patients50 and other Mybpc3 mutant models47,48,51, we observed mild cardiac fibrosis which was beneath robust detection by trichrome staining (representative trichrome Fig. S7 and whole heart images Fig. S8 for 1E14 vg/kg study), and was clearly detected by cardiac transcriptional analysis as elevation of Col3a1, Col4a1 and Postn transcripts (Fig. 7d–f). As with heart failure markers, significant decreases in fibrotic gene expression were consistently observed in the AAV9:mMybpc3 3E13 vg/kg dosed animals compared to vehicle-treated Mybpc3-/- mice, whereas animals treated with 1E13 vg/kg exhibited only trending decreases.

Transcriptional analysis of cardiac tissue from homozygous mice dosed retro-orbitally IV with vehicle or the indicated doses (vg/kg) of AAV9:mMybpc3 at two weeks of age and WT littermates for heart failure markers a Nppb, b Nppa, and c Myh7. Dose-dependent decreases in fibrotic marker expression in treated animals as assessed for d Col3a1, e Col4a1, and f Postn. Significant correlation between ejection fraction and MYBPC3 restoration as analyzed by transgene g RNA expression, h protein expression, and i cardiac transduction. Values are based on a simple linear regression model in GraphPad Prism. Analysis was performed 14 weeks post-injection. WT (n = 9; 5 M/4 F), Mybpc3-/- Veh (n = 10; 5 M/5 F), Mybpc3-/- 1E13 vg/kg (n = 7; 4 M/3 F), and Mybpc3-/- 3E13 vg/kg (n = 7; 3 M/4 F); preserved protein lysate unavailable for one 3E13 vg/kg animal. Data are shown as means ± SEM. P-value per one-way ANOVA with Tukey’s multiple comparisons test. Source data are provided as a Source Data file.
To precisely determine how cardiac improvement mapped to restoration of MYBPC3 expression, we analyzed EF as a function of cardiac transduction and transgene Mybpc3 expression in the individual Mybpc3-/- mice injected with AAV9:mMybpc3 at doses of 1E13 and 3E13 vg/kg. A dose-dependent increase in Mybpc3 RNA and MYBPC3 protein expression was observed at the level of cardiac tissue from individual treated mice, and this transgene expression was significantly correlated with the extent of cardiac improvement observed (Fig. 7g, h). Similarly, cardiac transduction exhibited dose-dependency which was positively correlated with EF (Fig. 7i). Importantly, this demonstrated that consistent with analysis of the TN-201 cassette in human iPSC-CMs (Fig. 1f, i), transduction of ≥1 vector genome per diploid genome (or ≥1.6E5 vg/µg DNA), as achieved with a dose of 3E13 vg/kg, resulted in MYBPC3 transgene expression equivalent to WT (Figs. 7h, i, and 5a). Further, consistent with the submaximal efficacy of 1E13 vg/kg (Fig. 4), protein restoration was equivalent to 0.51 ± 0.05 with respect to wild type.
Optimized construct outperforms published construct in late-stage Mybpc3 -/- mice
Rescue of function in symptomatic juvenile mice is, in the case of hypertrophic cardiomyopathy, more challenging than prevention of functional decline, as hypertrophic cardiomyopathy is a progressive disorder. As evidenced by the persistent slow decline and premature lethality of vehicle-treated Mybpc3-/- mice (Fig. 3), older animals exhibit more severe disease than juveniles. Therefore, we sought to test the utility of cassette improvements in a more challenging model, homozygous adults with more advanced disease. To this end, we performed a side-by-side experiment comparing our optimized AAV cassette with a previously published 5.4 kb expression cassette encoding Mybpc3 (Fig. 8a and detailed cassette comparison Fig. S9), which had proven sufficient to prevent hypertrophy when injected into homozygous neonates28.

a Homozygous mice with advanced disease (2.5 months of age) were injected retro-orbitally with 3E13 vg/kg or 1E14 vg/kg of AAV9 vector encoding Mybpc3 in the context of the published 5.4 kb cassette (denoted “Published”) or the TN-201 mouse surrogate (AAV9:mMybpc3, denoted mTN-201), or injected with vehicle control. Both cassettes utilize sequence from the human cardiac troponin T (TNNT2) promoter. b EF was measured to represent contractile ability at 27 weeks post-delivery. c LV mass was measured to represent hypertrophy and was normalized to body weight at 27 weeks post-delivery. WT (n = 8; 4 M/4 F), Mybpc3-/- Veh (n = 7; 3 M/4 F), Mybpc3-/- mTN-201 3E13 (n = 6; 2 M/4 F), Mybpc3-/- Published 3E13 (n = 7; 4 M/3 F), Mybpc3-/- mTN-201 1E14 (n = 7; 4 M/3 F), and Mybpc3-/- Published 1E14 (n = 6; 4 M/2 F). d Sustained cardiac transduction 20-months post-injection drove e sustained cardiac Mybpc3 RNA at ~4× levels of endogenous transcript and sustained restoration of wild-type protein levels in dosed homozygotes 20-months post-injection. MYBPC3 protein expression was assessed by ELISA with equivalent total protein for each sample, as well as immunoblot analysis of equivalent total protein, with MYBPC3 intensities normalized to WT (f, representative blot). n = 11 mice/group; 4 M/7 F for WT and 6 M/5 F for mMybpc3. P-value per one-way ANOVA with Tukey’s multiple comparisons test. Error bar: mean ± Standard Error of the Mean (SEM). Source data are provided as a Source Data file.
Homozygous mice with advanced disease (2.5 months of age) were injected retro-orbitally with 3E13 vg/kg or 1E14 vg/kg of AAV9 vector encoding Mybpc3 in the context of the published 5.4 kb cassette (denoted “Published”) or the TN-201 mouse surrogate (AAV9:mMybpc3, denoted mTN-201) or injected with vehicle control. Strikingly, the combined cis-regulatory modifications of our cassette resulted in significantly greater improvements in EF than the published cassette at 3E13 vg/kg (29% compared to 18%) and at 1E14 vg/kg (36% compared to 19%). Further, the optimized cassette exhibited significantly greater EF improvement at a dose of 3E13 vg/kg than the published cassette at 1E14 vg/kg or a 3.3× increase in dose (Fig. 8b). Improvements in cardiac hypertrophy exhibited a similar pattern, with the published cassette failing to have any significant effect on hypertrophy at the doses of 3E13 vg/kg and 1E14 vg/kg (Fig. 8c). In contrast, the optimized cassette resulted in more significant improvements compared to vehicle-treated Mybpc3-/- mice at both doses tested (≥3.0 mg/g). Thus, the optimized cassette outperformed the published cassette for improvements in hypertrophy and cardiac function and resulted in statistically significant benefit even in this model of advanced MYBPC3-associated cardiomyopathy.
Sustained cardiac transgene RNA and protein expression 20-months post-injection
Critical to the translational application of gene therapies is the durability of the AAV genome and therefore transgene expression in the cell type of interest, given current challenges of redosing. We therefore utilized the significant extension of lifespan afforded by AAV9:mMybpc3 in Mybpc3-/- mice to assess the persistence of cardiac transduction and transgene expression 20-months post-injection at a dose of 1E14 vg/kg. Consistent with durable efficacy findings for AAV9:mMybpc3 (Fig. 3), terminal cardiac analyses indicated sustained cardiac transduction (Fig. 8d), transgene RNA (Fig. 8e) and restoration of wild-type levels of MYBPC3 protein 20-months post-injection. (Fig. 8e, f).
Normalization of calcium handling abnormalities and relaxation defects in heterozygous iPSC-CMs
Finally, we sought to interrogate the utility of TN-201 in the heterozygous context using human iPSC-CMs. Despite highly variable penetrance of haploinsufficiency and contractile abnormalities in MYBPC3+/- iPSC-CMs, being dependent on media, maturation and co-culture conditions52,53,54,55, we observed the previously noted53,56 increased calcium amplitude of MYBPC3+/- iPSC-CMs compared to isogenic WT cells (Fig. 9a). This increase in calcium amplitude upon pacing was coupled with increased fluorescence rise rate (Fig. 9b). Importantly, addition of TN-201* was able to normalize this increase in fluorescence amplitude and rise rate when assessed one-week post-transduction (Fig. 9c, d), although we noted phenotypic muting upon even empty capsid transduction. Additionally, the mild decrease in MYBPC3 protein levels observed, as per52,53,54, was rescued based on immunoblotting and ELISA analysis (Fig. 9e, f). Importantly, utilizing engineered heart tissues (EHTs), we were able to observe expected defects in relaxation55,57 which were corrected upon transduction (Fig. 9g, h). Thus, despite the limitations of the MYBPC3+/- iPSC-CMs model, we were able to demonstrate rescue of the HCM hallmark relaxation defects.

MYBPC3+/- iPSC-CMs and their WT isogenic control iPSC-CMs were compared side-by-side for a Fluorescence Transient Amplitude (∆F/Fmin) and b Fluorescence Rise Rate ((∆F/Fmin)/s) using Fluo-4 AM dye on the Nautilai (CuriBio) fluorescence imager (n = 8/group). iPSC-CMs were stimulated at 10 volts, 3 ms, 1 Hz. As above, but with transduced iPSC-CMs one-week post-transduction 10k MOI of “Empty” (CR9-01: cassette without a transgene) or TN-201* for c Fluorescence Transient Amplitude (∆F/Fmin) and d Fluorescence Rise Rate ((∆F/Fmin)/s). WT Empty (n = 15), MYBPC3+/- Empty (n = 16), and MYBPC3+/- TN-201* (n = 16). MYBPC3 protein analysis of transduced iPSC-CMs one-week post-transduction by e immunoblotting [WT Empty (n = 10), MYBPC3+/- Empty (n = 8), and MYBPC3+/- TN-201* (n = 8)] and f ELISA (n = 10/group). Impaired time to 50% relaxation (RT50) (g) and impaired time to 90% relaxation (RT90) (h) of MYBPC3+/- engineered heart tissues was corrected with transduction; 10k MOI of “Empty” (CR9-01: cassette without a transgene) or TN-201*; WT Empty (n = 8), MYBPC3+/- Empty (n = 7), and MYBPC3+/- TN-201* (n = 7). P-value per one-way ANOVA with Tukey’s multiple comparisons test. Error bar: mean ± Standard Error of the Mean (SEM). Source data are provided as a Source Data file.
Discussion
The efficacy of TN-201 has been demonstrated in a Mybpc3-/- mouse model, which develops marked LV hypertrophy, poor cardiac function, and dilation at two weeks of age, resulting in death around one year of age, similar to patients with HCM with bi-allelic truncating or null mutations. Demonstrating efficacy in this model is challenging due to the severe phenotype of the Mybpc3-/- mice and the lack of any MYBPC3 protein. In these studies, a single dose of TN-201 or AAV9:mMybpc3 (mouse surrogate of TN-201 incorporating the mouse Mybpc3 gene instead of the human one) was systemically administered to Mybpc3-/- mice at doses ranging from 1E13 to 1E14 vector genomes per kilogram of body weight (vg/kg) at two weeks of age, after onset of cardiomyopathy. A single dose of AAV9:mMybpc3 in juveniles improved survival, cardiac systolic function, and hypertrophy in a dose-dependent manner, all of which were durable out to 13 months, the latest time point assessed by echocardiography. Diastolic dysfunction, an early hallmark of HCM58,59 was also ameliorated with AAV9:mMybpc3. Notably, sustained cardiac transgene RNA and MYBPC3 protein expression was observed 20-months post-injection of AAV9:mMybpc3 in Mybpc3-/- mice, the last time point measured. Critically, consistent with pioneering work in lentivirus60 and re-expression with an inducible TetOff system61, we were able to show with AAV that gene replacement in a MYBPC3-deficient mouse model could ameliorate symptoms, or provide preclinical therapeutic benefit, rather than simply preventing cardiac dysfunction when supplied perinatally28,62,63.
Differences between AAV9:mMybpc3 and TN-201 treatments in Mybpc3-/- mice are consistent with the sequence divergence between the mouse and human orthologs (88% amino acid identity). Further, differences may be due to the fact that rodents use α-myosin heavy chain (αMHC, or fast twitch myosin), rather than βMHC (or slow twitch myosin) in mature ventricular cardiomyocytes64,65. This may result in lower potency of the human MYBPC3 to restrict the activity of the more active fast twitch myosin14, potentially dampening its ability to reduce hypercontractility and hypertrophy over time in the murine model. Given the confounding effect of differing antibody affinity for the orthologs, we cannot rule out differences in ortholog stability in the mouse. Despite this sequence divergence, TN-201 mediated significant efficacy in multiple parameters assessed in this mouse cardiomyopathy model, providing proof of mechanism for gene replacement therapy.
Dose-ranging efficacy studies exhibited restoration of wild-type MYBPC3 protein levels and saturation of cardiac improvement at the clinically relevant dose of 3E13 vg/kg. In contrast, previous studies have been unable to restore wild-type MYBPC3 protein levels to homozygous mice28,62,63, even when dosing AAV within days of birth. Numerous clinical evaluations indicate that the frequency of safety events increases with dose66,67,68,69,70. Thus, the increased potency the Tenaya cassette was able to achieve is critical to the translational safety of the MYBPC3 gene replacement approach, as comparable improvement in cardiac function with the published cassette would require a >3-fold increase in dosage. Importantly, we also demonstrated that the cardiac transduction-transgene expression relationship observed in vivo in mouse cardiac tissue was recapitulated in human iPSC-CMs.
Also key to the translatability of the gene replacement approach is the consideration of transgene overexpression and any resultant potential safety concerns. These experiments generate two key findings that mitigate this concern: (1) naïve mice dosed with 3E14 vg/kg, 30× an efficacious dose, exhibited no alterations in cardiac function or body weight, and (2) a 7-fold increase in RNA without a proportional increase in MYBPC3 protein. This suggests exquisite post-transcriptional regulation of MYBPC3 protein expression and is consistent with transgenic mouse lines over-expressing Mybpc3 RNA exhibiting WT levels of MYBPC3 protein45,46. This may be due to the maintenance of precise stoichiometry of proteins bound in the sarcomere, with degradation of unincorporated sarcomeric components, such as excess MYBPC3, as a homeostatic regulatory mechanism.
A potential limitation of this work is the increased severity of our Mybpc3-/- model relative to established models. Our model has worsened cardiac dysfunction and LV hypertrophy, which may in large part be due to the different genetic background (129/Sv vs C57BL/6). Work in other heart failure models has shown that strain background can play a key role in phenotype71,72. There are also similarities between our model and established models, however. The MyBPC3t/t51 and cMyBP-C-/-47 mice both develop cardiac dysfunction and LV hypertrophy by juvenile age. Similar to ours, when aged, the cMyBP-C-/- mice have a decline in cardiac function48. The continued limitation of all these models has been their lack of true haploinsufficiency, which comprises the majority of MYBPC3 HCM patients. Our model and others quickly develop hypertrophy, dilation, and cardiac dysfunction, indicative of a combined HCM and DCM phenotype that in humans only develops in compound heterozygous children or a minority of heterozygous “end-stage” HCM adult patients.
In contrast to the homozygotes, the Mybpc3+/- mice do not develop the expected human phenotype, given that they exhibit normal47 or only slightly decreased protein levels48,73,74. Thus, the asymptomatic heterozygous mice are not a useful model for the 40% decrease and the resultant haploinsufficiency experienced in the human heterozygotes. We were able to restore WT expression to homozygous mutant mice at 3E13 vg/kg, providing evidence that TN-201 has the potential to restore the missing 40% of MYBPC3 protein expression in human heterozygous patients. Thus, the robust benefits in terms of survival, regression of hypertrophy and increases in EF seen in this severe disease model suggest that TN-201 may benefit patients across the full range of severity of MYBPC3-associated heart disease, pending further validation in clinical settings.
Methods
Ethical statement
Animal experiments were pre-approved by the Institutional Animal Care and Use Committee of Tenaya Therapeutics and were performed in compliance with the animal use protocol (AUP: 2020.007 and 2023.007) and the “Guide for the Care and Use of Laboratory Animals, 8th edition,” published by the US National Institutes of Health (ISBN-10: 0-309-15400-6). All ethical guidelines were adhered to whilst carrying out this study.
Study design
The number of biological replicates (n) per experiment is noted in each figure legend. Mice were randomized before being assigned to either vehicle or treatment groups based on a minimum of sex, age, and body weight. Ejection fraction was also used whenever possible. Whenever possible, every attempt was made to have equal representation of males and females per group. We have provided in the “Source Data” file an Excel tab delineating the precise animal number by sex for each study. Our research findings apply to both sexes. An individual blinded to the treatment performed and analyzed the echocardiography data. Mice were housed at 23–25 °C with light cycles of 12 h of light beginning at 6:00 a.m. and 12 h of dark beginning at 6:00 p.m., the humidity was 30–70%, and water was provided ad libitum. At the end of studies, mice were humanely euthanized by isoflurane inhalation or ketamine/xylazine intraperitoneal injection followed by cervical dislocation.
iPSC-CM generation
Tenaya’s Stem Cell Research Oversight Committee approved the study protocol (TT-2501) for iPSCs. The human iPSC lines used in this study were generated from a healthy male patient, WTC-11 (Coriell Institute: # GM25256). Informed written consent was obtained for this procedure. MYBPC3-/- and MYBPC3+/- iPSCs were generated by a CRISPR-Cas9 paired gRNA approach to remove exons one and two from the WTC-11 line which was modified with a blasticidin resistance gene at the MYH6 locus. Differentiations into iPSC-CMs were performed similarly to precedent75. Specifically, differentiation of iPSCs was induced through the Wnt modulation method with 7 μM glycogen synthase kinase 3 b (GSK3b) inhibitor CHIR99021 (Selleckchem) and 5 μM inhibitor of WNT production 2 (IWP2) (Sigma). Basal Media was composed of RPMI media supplemented with B27 (Thermo Fisher Scientific). Nineteen days after adding CHIR, iPSC-CMs were frozen in 90% fetal bovine serum with 10% dimethyl sulfoxide (DMSO). All differentiations undergo quality and purity assessments by cTnT flow cytometry (Fig. S10).
AAV production
AAV production was carried out in HEK293T cells76. HEK293T cells were seeded in a Corning HyperFlask (New York, NY) and triple transfected using a 2:1 PEI:DNA ratio (PEI Max) with a helper plasmid containing adenoviral elements (pHelper), a plasmid containing the Rep2 and Cap genes from the respective AAV serotype, and finally an ITR containing plasmid to be packaged. Three days following transfection, cells were harvested and lysed. Virus was purified using iodixanol ultracentrifugation and cleaned and concentrated in Hank’s Balanced Salt Solution (HBSS) + 0.001% Pluronic using a 100-kDa centrifuge column (Amicon, Darmstadt, Germany). AAV was titered using a PicoGreen assay.
Mouse model
Applied Stemcell Inc. (Milpitas, CA) was contracted for creation of Mybpc3 Exon1-Exon2 knockout mice, including generation of paired guide RNAs (GGCATCAAGCAGGCCACCCA and AAAGGTCAAGTTTGACCTCA) and utilization of the CRISPR/Cas9 system on a C57BL/6 background. Three male founders were obtained; TOPO cloning followed by sequencing was used to confirm the presence of the deletion in all founders. The germline transmission of the deletion was confirmed in F1 progeny by genotyping using forward primer (GAGAAGCCAGAGGACCAAGTG) and reverse primer (GGACCCTTCCTAGAACACCG, ~376 bp). The wild-type allele was assessed in parallel above forward primer and intronic reverse primer (GGAGCCAGGTCTCATGTGAA, ~656 bp). For genotyping, DNA was extracted from Viagen DirectPCR® Lysis Reagent (102-T) and proteinase K lysates by isopropanol precipitation. Three-primer PCR was run using Promega GoTaq® master mix. Breeders were maintained by backcrossing to C57BL/6 wild-type mice and experimental mice generated through timed heterozygous matings. Wild-type littermates were used as controls.
Cardiac function
Cardiac function was assessed by transthoracic echocardiography using high resolution micro-imaging systems (Vevo 3100 Preclinical Imaging System, VisualSonics). Briefly, anesthetized spontaneously breathing mice (1–3% isoflurane and 98.5–99% O2) were placed in the supine position on a temperature-controlled heating platform to maintain their body temperature at ~37 °C. Nair was used to remove hair and expose the skin to the probe. Parasternal short-axis M-mode tracings of left ventricle (LV) were recorded for LV mass and LV ejection fraction (EF) calculations. EF was used to determine systolic function, whereas LV mass was used to determine hypertrophy. Vevo Lab software 5.6.1 was used for analyses.
Simultaneous to echocardiography recordings, electrocardiography (ECG) leads were placed on animals in the Lead II position and ECG signals recorded with ADInstruments Powerlab. LabChart 8 software was used to assess QT intervals.
Cardiac transgene and transduction analyses
RNA was extracted from cardiac tissue using the miRNeasy mini Kit (Qiagen Sciences, 217004), cDNA synthesized (Invitrogen SuperScript III First-Strand Synthesis SuperMix for qRT-PCR) and analyzed by qRT-PCR using the indicated Taqman probes listed in Table S4.
For cardiac lysates, equal masses of tissue were homogenized in RIPA Lysis Buffer and 1× Protease Inhibitor. A Pierce™ BCA Protein Assay Kit was run to determine the precise protein concentration of each sample lysate. Equal protein amounts were loaded for Western Blotting and detected with mouse anti-MYBPC3 (sc-137180, Santa Cruz Biotechnologies) and rabbit anti-GAPDH (ab181602, Abcam). Imaging was performed on the Li-Cor Odyssey and analyzed using ImageJ (NIH) densitometry analysis. Detection by ELISA was performed with MyBPC3 DuoSet ELISA (R&D Systems, DY7439-05).
For cardiac vector genomes analysis, DNA was extracted using the DNeasy Blood and Tissue Kit (Qiagen Sciences, 69506). Absolute quantification of cardiac viral genomes per microgram of genomic DNA was assessed by qPCR using linearized standards across six orders of magnitude. Primers used are listed in Table S5.
Matched RNA, protein and transduction analysis in iPSC-CMs
As above, with cells being directly harvested from the well with Qiazol (Qiagen). DNA digestion was then performed using TURBO DNase (Invitrogen) and RNA was repurified using Quick-RNA MicroPrep kit (Zymo).
On-plate lysis was performed by harvesting iPSC-CMs 7-days post-infection with RIPA Lysis Buffer and 1× Protease Inhibitor, as above. Equal protein amounts were loaded for Western Blotting on a NuPage™ Tris-Acetate gel. Detection of MYBPC3 (sc-137180, Santa Cruz Biotechnologies) and Tubulin (ab7291, Abcam) was performed on the Li-Cor Odyssey.
For vector genomes analysis, cells were harvested directly from the well in Buffer ATL with proteinase K (Qiagen) then incubated at 56 °C for 10 min at 1000 RPM on a thermomixer. Lysates were then treated with 8 µL of RNase A (Qiagen) for 20 min at room temperature. DNA was extracted using equal volume of Phenol/Chloroform/Isoamyl alcohol (PCI) solution (25:24:1), with a chloroform back extraction prior to precipitation. DNA samples were quantified using the Qubit 1× dsDNA High Sensitivity Assay Kit (Life Technologies). To specifically quantify nuclear vector genomes, the DNA samples were normalized by concentration and treated with T5 exonuclease (New England Biolabs) at two different dilutions (1:5 and 1:50) to ensure enzyme was not limiting. T5 exonuclease is a single-stranded DNA endonuclease and double-stranded DNA exonuclease, thus T5 digest can be used to determine the fraction of vector genome signal that is nuclear or episomal63,64. For T5 treatment, 25 µL of each dilution was aliquoted for treatment and mock treatment. All samples were incubated at 37 °C for two hours before being heat inactivated at 70 °C for 10 min then analyzed by qPCR using 2× PowerUp™ SYBR™ Green Master Mix (Applied Biosystems) on a Quant Studio 7 (Applied Biosystems) with primers that specifically bind to the AAV genomic cassette, rather than transgene RNA, endogenous RNA or DNA. Quantification of nuclear viral genomes per microgram of genomic DNA was assessed by qPCR using linearized standards across six orders of magnitude. The lower limit of detection of the assay resulted in values of 1.92E3 as the background level of signal, equivalent to ~1% of that detected in MYBPC3-/- iPSC-CMs dosed at 1 K.
Transient transfection of iPSC-CMs
Confluent MYBPC3-/- iPSC-CMs were transiently transfected ten days after seeding using 500 ng of plasmid DNA in OPTI-MEMTM (ThermoFisher) per 24-well. 2 µl of ViaFectTM (Promega) in OPTI-MEMTM was used per 24-well. ViaFectTM and DNA solutions were then combined and complexes allowed to form for 20 min at room temperature. 50 µl transfection mixes were added dropwise to each 24-well and lysates harvested after seven days.
In vitro measurement of cytosolic calcium transients in 2D iPSC-CMs
Calcium transients were measured in iPSC cardiomyocytes 7-days post-infection, 14-days post plating, using Fluo-4 AM (Invitrogen) dye on the Nautilai (CuriBio) fluorescence imager. Cells were cultured in iCell Cardiomyocytes Maintenance Medium (Fujifilm). Fluo-4 AM was resuspended in DMSO and Pluronic F-127 (Invitrogen) before being added to Tyrode’s Solution (Boston BioProducts) with 1.5 mM CaCl2. Cells were removed from incubation, maintenance media was aspirated off, and the Tyrode’s Solution with dye was added to each well and returned to incubation for 30 min. Dye was removed and replaced with Tyrode’s Solution with 1.5 mM CaCl2 for 10 min in the incubation. Cells were then transferred to the Nautilai where they were stimulated at 10 volts, 3 ms, 1 Hz by the C-Pace EM Tissue Culture Interface (IonOptix) for 5 min. Calcium transients were then measured by the Nautilai system. Data was analyzed using the CuriBio Pulse cloud analysis software.
Engineered heart tissues
Engineered heart tissues (EHTs) were fabricated using the commercially available 12X stiffness Mantarray kit (Curi Bio), and manufacturer’s protocols were followed. In brief, casting wells arrayed in a 24-well plate format were pre-filled with 50 μL of a 6.4 U/mL thrombin solution, and the post lattice was inserted into these wells. WT or MYBPC3+/- hiPSC-CMs and human cardiac fibroblasts (HCFs) were then mixed with 5 mg/mL of fibrinogen at a ratio of 5 × 105 cardiomyocytes and 7.5 × 104 HCFs per tissue. This cell and fibrinogen solution was then added to the casting wells containing thrombin and thoroughly mixed around the inserted posts. Tissues were allowed to form for 80 min at 37 °C before being lifted out of the casting wells and placed into a new 24-well plate filled with fresh RPMI + B27 media. EHTs typically began beating within one week of casting, at which point culture media was switched to DMEM + B27 for the remainder of the study. AAV transduction occurred at 14-days post-casting and endpoint measurements were taken 9 days post-transduction.
Immunohistochemistry
The base of each heart was fixed overnight at 4 °C in 4% paraformaldehyde, then dehydrated with progressive sucrose exchanges until sinking in 30%. Hearts were embedded in Optimal Cutting Temperature (OCT) compound and sectioned. Tissue was permeabilized and stained with the M.O.M.® (Mouse on Mouse) Immunodetection Kit (Vector Labs, BMK-2202), mouse anti-MYBPC3 (sc-137180), and rabbit anti-alpha Actinin 2 (PA5-27863).
Flow cytometry
Cells were dissociated with TrypLE (Gibco), quenched, and assessed on a SH800 cell sorter (Sony Biotechnology).
Statistical guidelines
The number of biological replicates and animals for each experiment is indicated in the figure legends. Statistical analyses were performed using GraphPad Prism 9. P-values per one-way ANOVA with Tukey’s multiple comparisons test were utilized whenever more than two groups were being compared. Student’s t-test was used to analyze two unpaired groups. Significant differences were defined as P < 0.05. Error bars in all in vivo studies represent SEM.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All data supporting the findings described in this manuscript are available in the article and in the Supplementary Information. The source data are provided with this paper. Sequences are available in the Supplemental Sequence tab of the Source Data file. Source data are provided with this paper.
References
Ho, C. Y. et al. Genotype and lifetime burden of disease in hypertrophic cardiomyopathy: insights from the Sarcomeric Human Cardiomyopathy Registry (SHaRe). Circulation 138, 1387–1398 (2018).
Norrish, G. & Kaski, J. P. The risk of sudden death in children with hypertrophic cardiomyopathy. Heart Fail Clin. 18, 9–18 (2022).
Miron, A. et al. A validated model for sudden cardiac death risk prediction in pediatric hypertrophic cardiomyopathy. Circulation 142, 217–229 (2020).
Maron, B. J. & Maron, M. S. Hypertrophic cardiomyopathy. Lancet 381, 242–255 (2013).
Maron, B. J., Maron, M. S. & Semsarian, C. Genetics of hypertrophic cardiomyopathy after 20 years: clinical perspectives. J. Am. Coll. Cardiol. 60, 705–715 (2012).
Erdmann, J. et al. Mutation spectrum in a large cohort of unrelated consecutive patients with hypertrophic cardiomyopathy. Clin. Genet. 64, 339–349 (2003).
Kaski, J. P. et al. Prevalence of sarcomere protein gene mutations in preadolescent children with hypertrophic cardiomyopathy. Circ. Cardiovasc. Genet. 2, 436–441 (2009).
Millat, G. et al. Prevalence and spectrum of mutations in a cohort of 192 unrelated patients with hypertrophic cardiomyopathy. Eur. J. Med. Genet. 53, 261–267 (2010).
Lopes, L. R. et al. Novel genotype-phenotype associations demonstrated by high-throughput sequencing in patients with hypertrophic cardiomyopathy. Heart 101, 294–301 (2015).
Neubauer, S. et al. Distinct subgroups in hypertrophic cardiomyopathy in the NHLBI HCM registry. J. Am. Coll. Cardiol. 74, 2333–2345 (2019).
Stelzer, J. E., Dunning, S. B. & Moss, R. L. Ablation of cardiac myosin-binding protein-C accelerates stretch activation in murine skinned myocardium. Circ. Res. 98, 1212–1218 (2006).
Previs, M. J., Beck Previs, S., Gulick, J., Robbins, J. & Warshaw, D. M. Molecular mechanics of cardiac myosin-binding protein C in native thick filaments. Science 337, 1215–1218 (2012).
McNamara, J. W. et al. Ablation of cardiac myosin binding protein-C disrupts the super-relaxed state of myosin in murine cardiomyocytes. J. Mol. Cell. Cardiol. 94, 65–71 (2016).
Toepfer C. N., et al. Hypertrophic cardiomyopathy mutations in MYBPC3 dysregulate myosin. Sci. Transl Med. https://doi.org/10.1126/scitranslmed.aat1199 (2019).
Marston, S. et al. Evidence from human myectomy samples that MYBPC3 mutations cause hypertrophic cardiomyopathy through haploinsufficiency. Circ. Res. 105, 219–222 (2009).
van Dijk, S. J. et al. Cardiac myosin-binding protein C mutations and hypertrophic cardiomyopathy: haploinsufficiency, deranged phosphorylation, and cardiomyocyte dysfunction. Circulation 119, 1473–1483 (2009).
van Dijk, S. J. et al. Contractile dysfunction irrespective of the mutant protein in human hypertrophic cardiomyopathy with normal systolic function. Circ. Heart Fail. 5, 36–46 (2012).
McNamara, J. W. et al. MYBPC3 mutations are associated with a reduced super-relaxed state in patients with hypertrophic cardiomyopathy. PLoS ONE 12, e0180064 (2017).
O’Leary, T. S., Snyder, J., Sadayappan, S., Day, S. M. & Previs, M. J. MYBPC3 truncation mutations enhance actomyosin contractile mechanics in human hypertrophic cardiomyopathy. J. Mol. Cell. Cardiol. 127, 165–173 (2019).
Helms, A. S. et al. Spatial and functional distribution of MYBPC3 pathogenic variants and clinical outcomes in patients with hypertrophic cardiomyopathy. Circ. Genom. Precis. Med. 13, 396–405 (2020).
Prondzynski, M., Mearini, G. & Carrier, L. Gene therapy strategies in the treatment of hypertrophic cardiomyopathy. Pflug. Arch. 471, 807–815 (2019).
Carrier, L. Targeting the population for gene therapy with MYBPC3. J. Mol. Cell. Cardiol. 150, 101–108 (2021).
Helms, A. S., Thompson, A. D. & Day, S. M. Translation of new and emerging therapies for genetic cardiomyopathies. JACC Basic Transl. Sci. 7, 70–83 (2022).
Wessels, M. W. et al. Compound heterozygous or homozygous truncating MYBPC3 mutations cause lethal cardiomyopathy with features of noncompaction and septal defects. Eur. J. Hum. Genet. 23, 922–928 (2015).
Dong, J. Y., Fan, P. D. & Frizzell, R. A. Quantitative analysis of the packaging capacity of recombinant adeno-associated virus. Hum. Gene Ther. 7, 2101–2112 (1996).
Wu, Z., Yang, H. & Colosi, P. Effect of genome size on AAV vector packaging. Mol. Ther. 18, 80–86 (2010).
Kyostio-Moore, S. et al. The impact of minimally oversized adeno-associated viral vectors encoding human factor VIII on vector potency in vivo. Mol. Ther. Methods Clin. Dev. 3, 16006 (2016).
Mearini, G. et al. Mybpc3 gene therapy for neonatal cardiomyopathy enables long-term disease prevention in mice. Nat. Commun. 5, 5515 (2014).
Dutsch, A. et al. Phosphomimetic cardiac myosin-binding protein C partially rescues a cardiomyopathy phenotype in murine engineered heart tissue. Sci. Rep. 9, 18152 (2019).
Lombardi L. Gene therapy vectors for treating heart disease. U.S. patent US 2023/0372541 A1 (2023).
Cheng Z., Reid C. A. Adeno-associated virus with engineered capsid. U.S. patent US 2023/0220014 A1 (2023).
Wu, I. et al. AAV9:PKP2 improves heart function and survival in a Pkp2-deficient mouse model of arrhythmogenic right ventricular cardiomyopathy. Commun. Med . 4, 38 (2024).
Ellis, E. L. & Delbruck, M. The growth of bacteriophage. J. Gen. Physiol. 22, 365–384 (1939).
Spirito, P. et al. Magnitude of left ventricular hypertrophy and risk of sudden death in hypertrophic cardiomyopathy. N. Engl. J. Med. 342, 1778–1785 (2000).
Lekanne Deprez, R. H. et al. Two cases of severe neonatal hypertrophic cardiomyopathy caused by compound heterozygous mutations in the MYBPC3 gene. J. Med. Genet. 43, 829–832 (2006).
Xin, B., Puffenberger, E., Tumbush, J., Bockoven, J. R. & Wang, H. Homozygosity for a novel splice site mutation in the cardiac myosin-binding protein C gene causes severe neonatal hypertrophic cardiomyopathy. Am. J. Med. Genet. A 143A, 2662–2667 (2007).
Zahka, K. et al. Homozygous mutation of MYBPC3 associated with severe infantile hypertrophic cardiomyopathy at high frequency among the Amish. Heart 94, 1326–1330 (2008).
Marziliano, N. et al. A case of compound mutations in the MYBPC3 gene associated with biventricular hypertrophy and neonatal death. Neonatology 102, 254–258 (2012).
Prondzynski, M. et al. Evaluation of MYBPC3 trans-splicing and gene replacement as therapeutic options in human iPSC-derived cardiomyocytes. Mol. Ther. Nucleic Acids 7, 475–486 (2017).
Johnson, J. N. et al. Prevalence and clinical correlates of QT prolongation in patients with hypertrophic cardiomyopathy. Eur. Heart J. 32, 1114–1120 (2011).
Toib, A. et al. Remodeling of repolarization and arrhythmia susceptibility in a myosin-binding protein C knockout mouse model. Am. J. Physiol. Heart Circ. Physiol. 313, H620–H630 (2017).
Flenner, F. et al. Translational investigation of electrophysiology in hypertrophic cardiomyopathy. J. Mol. Cell. Cardiol. 157, 77–89 (2021).
Mendell, J. R. et al. Current clinical applications of in vivo gene therapy with AAVs. Mol. Ther. 29, 464–488 (2021).
Shen, W., Liu, S. & Ou, L. rAAV immunogenicity, toxicity, and durability in 255 clinical trials: a meta-analysis. Front. Immunol. 13, 1001263 (2022).
Yang, Q. et al. A mouse model of myosin binding protein C human familial hypertrophic cardiomyopathy. J. Clin. Invest. 102, 1292–1300 (1998).
Sadayappan, S. et al. Cardiac myosin-binding protein-C phosphorylation and cardiac function. Circ. Res. 97, 1156–1163 (2005).
Harris, S. P. et al. Hypertrophic cardiomyopathy in cardiac myosin binding protein-C knockout mice. Circ. Res. 90, 594–601 (2002).
Carrier, L. et al. Asymmetric septal hypertrophy in heterozygous cMyBP-C null mice. Cardiovasc. Res. 63, 293–304 (2004).
Geske, J. B., McKie, P. M., Ommen, S. R. & Sorajja, P. B-type natriuretic peptide and survival in hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 61, 2456–2460 (2013).
Hata, Y. et al. Clinicopathological and genetic profiles of cases with myocytes disarray-investigation for establishing the autopsy diagnostic criteria for hypertrophic cardiomyopathy. J. Clin. Med. https://doi.org/10.3390/jcm8040463 (2019).
McConnell, B. K. et al. Dilated cardiomyopathy in homozygous myosin-binding protein-C mutant mice. J. Clin. Invest. 104, 1771 (1999).
Helms, A. S. et al. Effects of MYBPC3 loss-of-function mutations preceding hypertrophic cardiomyopathy. JCI Insight. https://doi.org/10.1172/jci.insight.133782 (2020).
Seeger, T. et al. A premature termination codon mutation in MYBPC3 causes hypertrophic cardiomyopathy via chronic activation of nonsense-mediated decay. Circulation 139, 799–811 (2019).
De Lange, W. J. et al. cMyBP-C ablation in human engineered cardiac tissue causes progressive Ca2+-handling abnormalities. J. Gen. Physiol. https://doi.org/10.1085/jgp.202213204 (2023).
Pioner, J. M. et al. Slower calcium handling balances faster cross-bridge cycling in human MYBPC3 HCM. Circ. Res. 132, 628–644 (2023).
Kinnear, C. et al. Myosin inhibitor reverses hypertrophic cardiomyopathy in genotypically diverse pediatric iPSC-cardiomyocytes to mirror variant correction. Cell Rep. Med. 5, 101520 (2024).
Cohn, R. et al. A contraction stress model of hypertrophic cardiomyopathy due to Sarcomere mutations. Stem Cell Rep. 12, 71–83 (2019).
Ho, C. Y. et al. Echocardiographic strain imaging to assess early and late consequences of sarcomere mutations in hypertrophic cardiomyopathy. Circ. Cardiovasc. Genet. 2, 314–321 (2009).
Michels, M. et al. Diastolic abnormalities as the first feature of hypertrophic cardiomyopathy in Dutch myosin-binding protein C founder mutations. JACC Cardiovasc. Imaging 2, 58–64 (2009).
Merkulov, S., Chen, X., Chandler, M. P. & Stelzer, J. E. In vivo cardiac myosin binding protein C gene transfer rescues myofilament contractile dysfunction in cardiac myosin binding protein C null mice. Circ. Heart Fail. 5, 635–644 (2012).
Giles, J. et al. Recovery of left ventricular function following in vivo reexpression of cardiac myosin binding protein C. J. Gen. Physiol. 151, 77–89 (2019).
Li, J. et al. AAV9 gene transfer of cMyBPC N-terminal domains ameliorates cardiomyopathy in cMyBPC-deficient mice. JCI Insight 5, e130182 (2020).
Dominic, K. L. et al. The contribution of N-terminal truncated cMyBPC to in vivo cardiac function. J Gen. Physiol. https://doi.org/10.1085/jgp.202213318 (2023).
Yutzey, K. E. & Robbins, J. Principles of genetic murine models for cardiac disease. Circulation 115, 792–799 (2007).
Gacita, A. M. et al. Genetic variation in enhancers modifies cardiomyopathy gene expression and progression. Circulation 143, 1302–1316 (2021).
Verdera, H. C., Kuranda, K. & Mingozzi, F. AAV vector immunogenicity in humans: a long journey to successful gene transfer. Mol. Ther. 28, 723–746 (2020).
Bolt, M. W., Brady, J. T., Whiteley, L. O. & Khan, K. N. Development challenges associated with rAAV-based gene therapies. J. Toxicol. Sci. 46, 57–68 (2021).
Muhuri, M. et al. Overcoming innate immune barriers that impede AAV gene therapy vectors. J. Clin. Invest. https://doi.org/10.1172/JCI143780 (2021).
Ertl, H. C. J. Immunogenicity and toxicity of AAV gene therapy. Front. Immunol. 13, 975803 (2022).
Kishimoto, T. K. & Samulski, R. J. Addressing high dose AAV toxicity - ‘one and done’ or ‘slower and lower’? Expert Opin. Biol. Ther. 22, 1067–1071 (2022).
Barrick, C. J., Rojas, M., Schoonhoven, R., Smyth, S. S. & Threadgill, D. W. Cardiac response to pressure overload in 129S1/SvImJ and C57BL/6J mice: temporal- and background-dependent development of concentric left ventricular hypertrophy. Am. J. Physiol. Heart Circ. Physiol. 292, H2119–H2130 (2007).
Forte, E. et al. Dynamic interstitial cell response during myocardial infarction predicts resilience to rupture in genetically diverse mice. Cell Rep. 30, 3149–3163 e3146 (2020).
McConnell, B. K. et al. Comparison of two murine models of familial hypertrophic cardiomyopathy. Circ. Res. 88, 383–389 (2001).
Vignier, N. et al. Nonsense-mediated mRNA decay and ubiquitin-proteasome system regulate cardiac myosin-binding protein C mutant levels in cardiomyopathic mice. Circ. Res. 105, 239–248 (2009).
Yang, J. et al. Phenotypic screening with deep learning identifies HDAC6 inhibitors as cardioprotective in a BAG3 mouse model of dilated cardiomyopathy. Sci. Transl. Med. 14, eabl5654 (2022).
Reid, C. A., Boye, S. L., Hauswirth, W. W. & Lipinski, D. M. miRNA-mediated post-transcriptional silencing of transgenes leads to increased adeno-associated viral vector yield and targeting specificity. Gene Ther. 24, 462–469 (2017).
Acknowledgements
We thank members of the Tenaya Animal Care Team: Xiaomei Song, Cristina Dee-Hoskins, Kevin Robinson, Yolanda Hatter, Jessie Madariaga, and Carolina Gomez.
Author information
Authors and Affiliations
Contributions
L.M.L., W.G.T., T.H., and K.I. conceived the idea. L.M.L., A.G.S, W.G.T., and T.H. designed the studies. L.M.L., T.N.Q, E.L., and A.G. generated and maintained the mice. A.G.S. performed all echocardiographic measurements and analyses, with J.Y. assistance. S.S. and J.H. generated the iPSC line. A.B., M.C., and R.C. performed the iPSC-CM differentiations. C.F., T.W.C., N.R., S.J., Z.C., C.R., C.F., E.E., and O.C.T. generated the virus. C.A.L. and J.L. analyzed the virus for release. F.J. and W.S.P. oversaw viral production and release. Z.C. and C.R. performed the viral injections. L.M.L., A.G., E.L., T.N.Q., K.K., J.W., and J.H.T. performed all in vitro experiments and ex vivo and analyses. L.M.L., A.G.S., E.L., T.N.Q., A.G., K.K., J.W., J.H.T., J.M.L., K.I., W.G.T., and T.H. interpreted the results of the experiments. L.M.L. wrote the manuscript with the review and support from all authors.
Corresponding author
Ethics declarations
Competing interests
The authors of this publication declare the following competing interests: All authors of this publication were employed by Tenaya Therapeutics at the time of the study and hold or held equity in Tenaya Therapeutics. L.M.L. is an inventor on published U.S. Patent No.: US 2023/0372541 A1 held by Tenaya Therapeutics that covers gene therapy cassettes for treating heart disease.
Peer review
Peer review information
Nature Communications thanks Raffaele Coppini, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Greer-Short, A., Greenwood, A., Leon, E.C. et al. AAV9-mediated MYBPC3 gene therapy with optimized expression cassette enhances cardiac function and survival in MYBPC3 cardiomyopathy models. Nat Commun 16, 2196 (2025). https://doi.org/10.1038/s41467-025-57481-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-57481-7
This article is cited by
-
PLN-L31A/I40A for the treatment of inherited heart disease caused by PLN-R14del mutations
Acta Pharmacologica Sinica (2026)
-
Hypertrophic cardiomyopathy—an enigma unravelling
Indian Journal of Thoracic and Cardiovascular Surgery (2026)
-
Association of radiotherapy with atrial fibrillation-related gene expression in breast cancer patients: a study based on the TCGA-BRCA database
Discover Oncology (2026)
-
Cardiac troponin T promoters lead the way: optimizing cardiac specificity in AAV-mediated gene therapy
Gene Therapy (2025)
-
Hypertrophic cardiomyopathy
Nature Reviews Disease Primers (2025)
