
Abstract
Mutations that accumulate in the human male germline with age are a major driver of genetic diversity and contribute to genetic diseases. However, aging-related male germline mutation rates have not been measured directly in germline cells (sperm) at the level of individuals. We developed a study design in which we recalled 23 sperm donors with prior banked samples to provide new sperm samples. The old and new sequential sperm samples were separated by long timespans, ranging from 10 to 33 years. We profiled these samples by high-fidelity duplex sequencing and demonstrate that direct high-fidelity sequencing of sperm yields cohort-wide mutation rates and patterns consistent with prior family-based (trio) studies. In every individual, we detected an increase in sperm mutation burden between the two sequential samples, yielding individual-specific measurements of germline mutation rate. Deep whole-genome sequencing of sequential sperm samples from two individuals followed by targeted validation measured remarkably stable mosaicism of clonal mutations that likely arose during embryonic and germline development, suggesting that age did not substantially impact the diversity of spermatogonial stem cell pools in these individuals. Our application of high-fidelity and deep whole-genome sequencing to sequential sperm samples provides insight into aging-related mutation processes in the male germline.
Similar content being viewed by others


Introduction
Human evolution and genetic diseases are both linked to the rate at which new (de novo) mutations occur in the germline1. Based on “trio” studies that sequenced parents and their offspring, the de novo mutation rate in humans has been estimated to be between 1.1 and 1.3 × 10−8 mutations per base pair per generation for single-nucleotide substitutions, which are the most common and well-studied DNA alterations2,3,4,5,6,7,8. Notably, ~80% of de novo mutations arise in the paternal (male) germline, and for every additional year of paternal age, offspring inherit ~1.3–1.5 more de novo mutations2,3,5,8,9,10. This increase in de novo mutations with paternal age has been associated with a higher risk of developmental disorders in the offspring of older men, including achondroplasia, Apert syndrome, Marfan syndrome, autism spectrum disorders, progeria, and other disorders4,11,12,13,14,15,16. Collectively, these data point to the male germline, namely spermatogonial stem cells and the mature sperm they generate, as the main source of de novo mutations in humans, and they indicate that spermatogonial stem cells accumulate mutations linearly with age17. The male germline is also distinguished by having the lowest age-related mutation rate of any cell type in the body other than oocytes, which has further motivated studies of its mutation processes9,17,18.
Most age-related mutations in sperm are present at very low mosaicism (i.e., in a very low fraction of sperm) that is below the detection limits of standard DNA sequencing, and consequently, they are only measurable by specialized DNA sequencing methods such as high-fidelity duplex sequencing and microdissection of clonally related cells17,19,20,21,22. To date, these high-fidelity sequencing methods have estimated age-related mutation rates in sperm and other tissues by profiling cohorts of individuals spanning a range of ages, where each individual is sampled at only a single or at closely spaced timepoints17,23. However, this approach can only measure the average mutation rate across a cohort of individuals, and it cannot measure the mutation rate of each individual. Indeed, age-related mosaic mutation rates have yet to be measured systematically at the level of individuals in any tissue, as this requires sequential biological samples separated by decades, which are challenging to collect. Despite this challenge, measurement of individual-specific mutation rates could elucidate variability in the accumulation of mutations with age among individuals, which would otherwise not be apparent in studies that profile only one sample per individual.
Importantly, sperm is one of the only biological tissues that is stored frozen for decades. Here, we introduce a study design in which we recontact sperm donors with available prior frozen samples to provide new semen samples, which enables the collection of sequential sperm samples separated by long timespans for each individual. We profile these unique specimens using high-fidelity duplex sequencing to directly measure individual-level germline mutation rates. Additionally, we perform high-depth whole-genome sequencing (WGS) in a subset of individuals, with validation by targeted duplex sequencing, since mutations that arise early in embryonic or germline development are detectable by WGS19,24. These higher-level mosaic mutations that occur early in development are implicated in recurrent de novo genetic diseases within families, but it remains uncertain how their mosaicism varies in the sperm of individual men over long periods of time19,25,26.
Results
Measurement of individual male germline mutation rates across decades
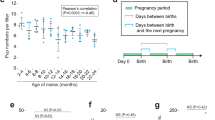
We recruited 23 prior sperm donors who had at least one specimen stored frozen for more than 10 years at Cryos or California Cryobank, two of the oldest and largest sperm banks in the world (Fig. 1a and “Methods”). Each participant provided a new sperm sample as well as blood (Cryos) or saliva (California Cryobank) and completed a medical history, fertility, and exposure questionnaire to aid interpretation of potential outlier results (Supplementary Data 1 and 2 and “Methods”). The median timespan between the sperm samples collected at the first timepoint (T1) and the second timepoint (T2) was 15.9 years (range 10.2–33.4) (Fig. 1b and Supplementary Data 1). The median age at T1 was 27.3 years old (range 21.5–36.9), and the median age at T2 was 42.7 years old (range 33.5–67.3) (Fig. 1b and Supplementary Data 1). We profiled mosaic mutations in all 69 samples (T1 and T2 sperm and T2 blood/saliva from each individual) using NanoSeq, a duplex sequencing method with single-molecule fidelity21, and each individual’s blood/saliva sample was separately sequenced to allow filtering of non-mosaic variants (“Methods”). We obtained a total of 8063 gigabases of NanoSeq sequencing data (average 117 gigabases per sample) for mosaic mutation analysis (Supplementary Data 3). In sperm, this yielded an average of 3.8 × 109 high-fidelity interrogated base pairs per sample (~1.2 sperm haploid genome equivalents) (Supplementary Data 4). Due to the large timespans between samples of each individual, we performed a fingerprinting analysis using inherited germline variants that confirmed that all the NanoSeq samples of each individual derived from the same individual (“Methods” and Supplementary Fig. 1a). Across all sperm samples, we detected a total of 3580 substitution and 339 insertion/deletion (indel) mosaic mutations (Supplementary Data 4 and 5).

a Schematic of study design. For each individual, sperm were profiled from timepoints T1 and T2, with the timespan between T1 and T2 ranging from 10 to 33 years. Either blood or saliva were collected at T2 along with a questionnaire. Illustrations created in BioRender. Evrony, G. (2025) https://BioRender.com/r30q384. b Ages of individuals at T1 and T2, ordered top-to-bottom by the age at T1. See Supplementary Data 1 for details. c Substitution mutation burdens (mutations per base pair (bp)) measured by NanoSeq for all sperm samples (T1 and T2). Dots and error bars show point estimates and their Poisson 95% confidence intervals. The dashed line and shaded region show a weighted least-squares linear regression and its 95% confidence interval. P values for the intercept and slope are from the linear regression (two-sided t test). See Supplementary Data 4 for complete burden statistics. d Sperm indel mutation burdens (mutations per base pair (bp)) measured by NanoSeq for all sperm samples (T1 and T2). Dots and error bars show point estimates and their Poisson 95% confidence intervals. The dashed line and shaded region show a weighted least-squares linear regression and its 95% confidence interval. P values for the intercept and slope are from the linear regression (two-sided t test). See Supplementary Data 4 for complete burden statistics and Supplementary Fig. 1b for individual-specific plots. e Sperm substitution mutation burdens measured by NanoSeq, with each colored line representing a different individual where the line connects the mutation burdens at T1 and T2. The dashed line shows a weighted least-squares linear regression. f Individual-specific sperm substitution mutation rates between T1 and T2 for all profiled individuals. Thick and thin horizontal lines indicate the median and the 1st/3rd quartiles, respectively.
Before analyzing individual-specific germline mutation rates, we analyzed the relationship between mutation burden and age across all the sperm samples in our cohort. The burden of sperm mosaic substitution mutations measured by high-fidelity duplex sequencing exhibited a linear increase with age at a rate of 5.74 × 10−10 substitutions per base pair (bp) per year (95% confidence interval: 4.92 × 10−10–6.55 × 10−10; P < 10−15; burden doubling time for a 20-year-old individual: 21.6 years) (Fig. 1c and Supplementary Data 4). The confidence interval of this mutation rate overlaps the mutation rate calculated from a prior study of paternally derived de novo mutations in 2976 trios10 (4.93 × 10−10 mutations per bp per year; 95% confidence interval: 4.76 × 10−10–5.09 × 10−10; burden doubling time for a 20-year-old individual: 25.4 years) (“Methods”). The slightly higher mutation rate in our data, though not statistically significant, may be due to the former trio study utilizing an approximate estimate of the number of base pairs interrogated per genome2,10, compared to our approach that quantifies an exact number of interrogated base pairs for each sample (“Methods”). The burden of sperm mosaic indel mutations also increased linearly with age, with a mutation rate of 2.59 × 10−11 indel mutations per bp per year (95% confidence interval: 7.73 × 10−12–4.41 × 10−11; P = 0.006), with the larger confidence interval relative to the substitution rate due to the low number of detected indels (Fig. 1d and Supplementary Data 4). This yields a relative substitution to indel mutation rate of 22.2, which is similar to an estimate of 18.0 from a prior trio study9, though both these estimates are likely significant overestimates as the prior study and our study require filters for many artifact-prone tandem repeat regions that may harbor true mosaic indels. Overall, these results demonstrate that direct profiling of sperm by high-fidelity duplex sequencing reliably measures age-related mutation accumulation in the male germline.
Next, we analyzed individual-specific germline mutation rates. Notably, every individual in our study exhibited an increase in sperm substitution mutation burden with age, i.e., the mutation burden at T2 was higher than the burden at T1 (Fig. 1e and Supplementary Data 4). Similarly, although indel mutation burdens had larger errors in their estimates due to the very low indel burdens in sperm, indel burdens also increased between T1 and T2 in 19 of 23 individuals (Supplementary Fig. 1b and Supplementary Data 4). Together, these data demonstrate that our study design utilizing sequential sperm samples separated by long timespans can measure individual-specific mutation accumulation with age.
The median substitution mutation rate across the individuals—calculated for each individual as the difference in mutation burden between T2 and T1 divided by the timespan between T2 and T1—was 5.3 × 10−10 mutations per bp per year (Fig. 1e, f). One individual, SPM-1536, had an outlier high mutation rate estimate of 1.48 × 10−9 (Grubbs test, P = 0.0005) (Fig. 1e, f, Supplementary Fig. 1c, and Supplementary Data 4). This individual’s history is notable for an undescended testicle, diagnosed and repaired at 10 years old (Supplementary Data 2). To further characterize individual-specific mutation rates, we calculated the residuals of each individual’s substitution mutation burden at T1 and T2 relative to the cohort-wide linear regression and plotted the rank of the residuals at T1 versus T2 (Supplementary Fig. 1d). This plot showed that SPM-1536 (the above mutation rate outlier) had a relatively low mutation burden at T1 and a relatively high mutation burden at T2, and that SPM-1514 had relatively high mutation burdens at both T1 and T2, while SPM-1515 had relatively low mutation burdens at both timepoints (Supplementary Fig. 1d).
We also analyzed the age-related mosaic substitution burdens of blood and saliva samples collected at T2. For blood samples, we did not see an association of mutation burdens with age (P = 0.13), and there was a nonzero y-intercept (Supplementary Fig. 1e). These results are likely due to our frozen blood samples limiting us to profiling whole blood mutation burdens rather than profiling a single short-lived blood cell type such as granulocytes that are more reflective of the hematopoietic stem cell compartment21. Prior studies have found variability in mutation rates among blood cell types27,28, so variability in cell type proportions among whole blood samples—especially longer-lived blood cell types—may have contributed to the variability in mutation burdens. Consequently, we also did not observe a statistically significant relationship of substitution mutation burdens between sperm and blood samples that were collected at the same timepoint T2 (P = 0.12) (Supplemental Fig. 1f). In contrast, the mutation burdens of saliva samples correlated well with age (P = 0.0007) and with mutation burdens in sperm that were collected at the same T2 timepoint (P = 0.009) (Supplementary Fig. 1e, f).
Mutational spectra
The mutational spectrum of the sperm mosaic substitution mutations (histogram of the number of mutations in each possible trinucleotide context29) and of the indel mutations that we identified by duplex sequencing were highly concordant with the spectra of paternally derived de novo mutations from a prior trio study10 (Fig. 2a, b; cosine similarities = 0.97 and 0.92 for substitutions and indels, respectively). These results further support the accuracy of direct high-fidelity duplex sequencing of sperm to reveal male germline mutations compared to the prior gold standard of trio studies.

a Mutational spectra of unique sperm mosaic substitution mutations (i.e., removing mosaic mutations detected more than once within the same sample) and paternally derived de novo mutations from the trio study by Halldorsson et al.10. The spectrum from our study was corrected for trinucleotide context opportunities (i.e., the distribution of trinucleotides in analyzed bases versus the human reference genome) (“Methods”). See Supplementary Fig. 2 for the mutational spectra of each individual sperm sample. b Mutational spectra of unique sperm mosaic indel mutations and paternally derived de novo mutations from the trio study by Halldorsson et al.10. c, d Fraction of mutation types for substitutions (c) and indels (d) for all sperm mutations and after combining sperm samples into three different age groups (20 ≤ age < 30 years old, 30 ≤ age < 40 years old, and age ≥ 40 years old). See Supplementary Fig. 3a, b for spectra of each age group. e Fraction of mutations attributed to COSMIC signatures SBS1 and SBS5 for sperm mutations after combining them into three different age groups (20 ≤ age < 30 years old, 30 ≤ age < 40 years old, and age ≥ 40 years old). Above each bar is the cosine similarity of the spectrum reconstructed from the component signatures to the original spectrum.
We analyzed whether mutational spectra and mutational signatures (mutational processes that produce the mutational spectra29) vary with age. Since the mutational spectra of individual sperm samples contained too few mutations for reliable analysis (Supplementary Fig. 2), we combined sperm samples into three age groups: 20–30, 30–40, and ≥40 years old at the time of collection. The spectra and fractional distributions of mutation types were similar between the age groups (Fig. 2c, d and Supplementary Fig. 3a, b). We then performed mutational signature analysis for substitutions by fitting the spectra to COSMIC30 signature SBS1 while extracting one additional signature SBSA that was highly similar to COSMIC signature SBS5 (Supplementary Fig. 3c; cosine similarity = 0.98). Note, there were insufficient indels for reliable signature analysis. Subsequent fitting of the spectra to COSMIC signatures SBS1 and SBS5 showed that these accounted for 19% and 81% of mutations, respectively, and that these two signatures alone can accurately reconstruct the original spectra (Fig. 2e), consistent with prior studies7,9,31. Furthermore, the fraction of mutations attributed to these signatures was similar across age groups (Fig. 2e), suggesting that the rates of these clock-like mutational processes do not vary with age in the male germline.
Clonal mosaicism
While the above high-fidelity duplex sequencing accurately calls mosaic mutations in single molecules regardless of their mosaicism level, including both “clonal” (present in multiple cells) and “non-clonal” (present in only one cell) mosaic mutations, this method’s shallow genome coverage per sample precludes sensitive identification of “clonal” mosaic mutations that are present in a higher fraction of sperm. Notably, prior studies have identified evidence of selection with age in the male germline for mutations in cancer-related genes, especially in the RAS/MAPK pathway, suggesting that these mutations drive clonal expansions in spermatogonial stem cells32. Importantly, pathogenic clonal sperm mutations undergoing selection contribute to de novo genetic diseases24,33. An increase in clonal mosaicism with age has been identified in blood (also known as clonal hematopoiesis), skin, esophagus, and other tissues34,35,36—representing an increase in the size of some stem cell clones and a reduction in the effective stem cell population size. However, one point of contrast for the analogous process of “clonal spermatogenesis” compared to clonal hematopoiesis, is that spermatogonial stem cell clones likely have a maximum possible size, even in the presence of driver mutations, due to the spatial limitation of clones to a single seminiferous tubule among hundreds of active seminiferous tubules37,38. At the same time, spermatogonial stem cells and seminiferous tubules variably decline in function with age among men39,40, which could feasibly increase the fraction of sperm produced by each remaining functional spermatogonial stem cell clone.
As deep WGS can detect mutations with as low as 1% mosaicism19, it can enable detection of clonal mosaic mutations in sperm. One prior study evaluated sperm clonal mosaicism by deep WGS in 12 young and 5 old individuals, including sequential samples up to 1 year apart, but it did not identify an increase in the number or the mosaicism of clonal mutations with age19. This result may be due to the spatial limitation on aging-related expansion of spermatogonial stem cell clones such that they cannot increase to sufficient size to be detectable by deep WGS. Alternatively, it may be that variability among men in the aging-related decline of spermatogonial stem cells and seminiferous tubules could put some mens’ aging-related clonal spermatogenesis within the detection range of deep WGS, but that detecting this in the setting of these variable processes may require profiling sequential sperm samples across longer timespans. In addition, deep WGS can detect mosaic mutations that occurred during early embryonic and germline development19, which are more likely to be present at higher mosaicism (i.e., in multiple seminiferous tubules) than aging-related mutations. Profiling these early-occurring mutations—especially passenger/non-driver mutations—in sequential sperm samples separated by long timespans would allow assessment of the stability of the fraction of sperm produced by these clones as a proxy for aging-related changes in the number of sperm-producing spermatogonial stem cells. For example, as subsets of seminiferous tubules gradually cease sperm production with age, the mosaicism levels of early developmental mutations could feasibly change.
To assess whether sequential sperm samples spanning long timespans enable the detection of aging-related changes in spermatogonial stem cell clonality, we performed high-depth PCR-free WGS (average genome coverage 318X) of sperm from timepoints T1 and T2 from two individuals (SPM-1047 and SPM-1564) and their blood/saliva samples from T2 (Supplementary Data 3 and 4 and “Methods”). SPM-1047 was chosen due to the relatively long timespan between T1 and T2 (22.1 years) and the availability of sufficient DNA for deep PCR-free WGS. SPM-1564 was chosen due to the individual harboring a known pathogenic clonal sperm mutation in the TSC2 gene (p.Q90X) that we hypothesized could affect clonal spermatogenesis if a second hit mutation occurred in TSC241. We called mosaic mutations by adapting and optimizing for further specificity a computational pipeline previously validated for deep WGS analysis of somatic mosaicism in the human brain42 (“Methods”). Due to the higher false-positive rate in calling mosaic indels in WGS data42,43, we focused our analysis on mosaic substitutions.
In the four sperm samples (SPM-1047 and SPM-1564 T1 and T2), we called 283 mosaic clonal substitutions, which corresponded to 193 unique substitutions when counting mutations identified in both T1 and T2 once (Supplementary Data 4 and 6). The number of mosaic clonal substitutions called per sperm sample ranged from 46 to 99, which is comparable to the 9 to 62 mosaic substitutions called per sample in a prior high-depth WGS study of sperm19. We then determined which mutation calls were true positives using high-coverage targeted duplex sequencing (targeted NanoSeq)44. We successfully designed hybridization probes for 187 of the 193 mutation loci, and sequencing achieved an average 1998 duplex read coverage (Supplementary Data 3). Of these, 105 mutations were validated by duplex reads in at least one of the respective individual’s sperm samples (Supplementary Data 7). The mutational spectrum of the validated clonal mutations resembled the spectrum we obtained by our non-targeted duplex sequencing of all sperm samples, albeit with a relatively lower cosine similarity score of 0.83 likely due to the lower number of mutations (Fig. 3a). To maximize accuracy in calling presence/absence of mutations in samples, we used in all subsequent analyses the 104 of 105 validated mutations that had mean duplex coverage >500, and we used in analyses the mosaicism levels measured by targeted duplex sequencing reads. While the genome coverage of non-targeted NanoSeq is low, 2 and 6 of the validated clonal sperm mutations found in SPM-1047 and SPM-1564, respectively, were also detected in the non-targeted NanoSeq data (Supplementary Data 7).

a Mutational spectrum of unique clonal mosaic substitution mutations identified by deep WGS and validated by targeted NanoSeq in sperm samples (timepoints T1 and T2 of SPM-1047 and SPM-1564) compared to the spectrum of mutations identified by non-targeted NanoSeq across all sperm samples in this study (reproduced from Fig. 2a). See Supplementary Data 6 and 7 for details of all mutations identified by WGS and targeted NanoSeq. b Number of clonal mosaic substitution mutations identified by deep WGS and validated by targeted NanoSeq in sperm samples. c Histogram of the fraction of validated clonal substitution mutations in each sample in different bins of mosaicism as measured by targeted NanoSeq. The parentheses and square brackets of mosaicism bins indicate exclusion and inclusion, respectively, of the bin’s limits. See Supplementary Data 7 for mosaicism values of all mutations. d Number of unique clonal substitution mutations detected by targeted NanoSeq in different combinations of the three samples of each individual: T1 sperm, T2 sperm, and blood (SPM-1047) or saliva (SPM-1564). The numbers in each box indicate the number of mutations in that category. e Comparison of the mosaicism as measured by targeted NanoSeq of clonal substitution mutations in sperm between T1 and T2 for mutations identified in at least one timepoint. The dashed line shows a linear regression with associated R2 value and P value (two-sided t test). f Absolute difference in mosaicism from T1 to T2 as measured by targeted NanoSeq (calculated as T2 mosaicism – T1 mosaicism) for clonal substitution mutations identified in sperm in at least one timepoint.
Notably, the number of mosaic clonal substitutions in sperm was remarkably stable with age in both SPM-1047 and SPM-1564 (Fig. 3b). The number of clonal mutations in SPM-1564 was the same in T1 and T2, and there was only one additional clonal mutation detected in SPM-1047 at T2 compared to T1. In addition, SPM-1564 sperm at T1 had 86% more clonal mutations than SPM-1047 sperm at T1 even though these individuals had similar T1 ages (Fig. 3b). While each individual’s clonal substitutions had a similar mosaicism distribution between T1 and T2, SPM-1564 had a larger percentage of mutation calls with high (> 10%) mosaicism (16/134 = 12% in SPM-1564 versus 0/73 = 0% in SPM-1047; P = 0.005; Chi-squared test), and this cluster of high mosaicism mutations in SPM-1564 was present at both T1 and T2 (Fig. 3c). Overall, while limited by the small number of identified mutations and profiled individuals, these results are consistent with age-related clonal spermatogenesis occurring at mosaicism levels below the detection limit of deep WGS in both individuals. Further, they suggest a higher burden of early developmental mutations in individual SPM-1564 in lineages that produced the germline.
Clonal sperm mutations shared between different combinations of each individual’s T1 and T2 sperm and blood/saliva samples correspond to different approximate timings of the mutations. For example, clonal mutations detected in all three samples of an individual must have occurred during early embryonic development. On the other hand, clonal mutations detected in sperm at both T1 and T2 but not in blood/saliva may have occurred later during germline development. Clonal sperm mutations that were not detected in blood/saliva had lower mosaicism levels in sperm than clonal sperm mutations that were also detected in blood/saliva (P < 10−11, two-sided Mann–Whitney test; Supplementary Fig. 4). In addition, only 6/36 (17%) and 6/37 (16%) of unique clonal sperm mutations detected in SPM-1047 T1 and T2, respectively, were also identified in the individual’s non-sperm (blood) sample, and likewise, these fractions were 13/67 (19%) in both SPM-1564 T1 and T2 (Fig. 3d). These results suggest that most clonal sperm mutations that arise during development occur after the germ cell lineage separates from somatic lineages.
Our study design allowed us to evaluate the stability of clonal mosaicism levels over long timespans. Of the clonal sperm mutations detected in SPM-1047 and SPM-1564, 103/104 (99%) were identified at both T1 and T2 (Fig. 3d). The only mutation detected at only one timepoint (chr7:98991712 C>T) was detected by deep WGS only at T2, and it was detected in 2/2264 targeted duplex reads at T2, but not in 2078 duplex reads at T1 (Supplementary Data 6 and 7). While this may be an aging-related clonal mutation, it is also possible that even greater targeted duplex sequencing depth would have detected this mutation at T1. The mosaicism levels of clonal sperm mutations were also remarkably correlated between T1 and T2 (R2 = 0.99; P < 0.001), with 99/104 (95%) of these mutations changing their absolute mosaicism level from T1 to T2 by less than 1% (Fig. 3e, f and Supplementary Figs. 5 and 6). SPM-1564’s pathogenic TSC2 mutation also did not significantly alter its mosaicism, with 11% and 13% mosaicism in sperm T1 and T2, respectively; this is likely because a second hit is required in TSC2 or in other genes of its associated signaling pathway to drive clonal expansion41. Overall, our results indicate that mosaic germline mutations that arise during early development are highly stable over long periods of time. With the caveat of our small number of profiled individuals, this result further suggests that even as the function of spermatogonial stem cells and seminiferous tubules declines with age, the fractional contribution of early developmental clones to sperm production is not significantly affected.
We then examined whether any cancer-related genes (Supplementary Data 8) had clonal mutations across the sperm samples. This yielded only the known TSC2 pathogenic mutation in SPM-1564 sperm at T1 and T2. No other validated clonal sperm mutation was protein-altering. We also examined whether any genes were recurrently mutated (>1 mutation) across sperm samples. This yielded only GLI2, which had two different mutations in SPM-1564, one of which was detected in both T1 and T2 sperm as well as saliva and another that was detected only in T1 and T2 sperm (Supplementary Data 6). However, both GLI2 mutations were intronic with a CADD score of 0, indicating they very likely have no functional impact. These data suggest that any putative genes driving clonal spermatogenesis that are recurrently mutated within and across individuals, as is observed in epithelial tissues35,36, have driver mutations with mosaicism <1%, which is below the detection limit of deep WGS.
Discussion
Here, we profile individual-specific mutation rates and patterns in the male germline using sequential sperm samples separated by long timespans. While the collection of sequential samples separated by long timespans is challenging for any tissue, we demonstrate an approach to achieve this for sperm and that high-fidelity sequencing of such sequential samples can measure individual-specific mutation rates. This suggests that this approach could be scaled in the future to systematically explore modifiers of mutation rates in the germline and in other tissues. Though our study did not focus on recruiting men with fertility-related conditions, one individual in our cohort with a relatively long history (10 years) of an undescended testicle had an outlier high mutation rate. However, since the low number of mutations per sample incurs a relatively high error in individual mutation rate estimates (Supplementary Fig. 1c), measurement of mutation rates of more individuals with this condition are warranted to determine if this condition explains our finding. Additionally, one individual in our cohort had Fabry disease and a low sperm count (SPM-1072) but did not have an outlier mutation rate (Supplementary Data 2 and 4), suggesting variability in how fertility-related conditions may affect mutation rates. Notably, while this study was under review, another study reported in a pre-print the identification of elevated mutation burdens in most, though not all oligozoospermic men including profiling of paired longitudinal sperm samples in 8 men45. Further high-fidelity sequencing studies of sperm from infertile men will help elucidate variability in mutation rates both among and within fertility-related conditions.
We find that direct high-fidelity duplex sequencing of purified sperm yields mutation rates and patterns fully consistent with prior trio studies. Trio analyses previously estimated germline mutation rates at the level of individuals by profiling de novo mutations in multiple children of the same parents, and these estimates increase in accuracy as the number of offspring and the timespan between offspring increase7,8. These studies have identified inter-family differences in age-related de novo mutation rates supported by multivariate modeling7,8,46 with a comparable distribution to our estimates (ratio of 90th to 10th percentile rates = 2.5 in a trio study8, compared to 2.9 in our study), suggesting the existence of parental effects on mutation rate beyond just age. However, this likely common variability in mutation rates among individuals has a lower magnitude than the extreme outliers caused by DNA repair defects and chemotherapy exposure identified by a recent larger trio study9. Importantly, in contrast to trio sequencing, direct sequencing of mosaic mutations in sperm is not limited by the number of offspring or the timespan between them, so it could be utilized in the future as a clinical assay either pre- or post-conception and longitudinally to deeply assess individual risk for de novo genetic diseases24,25. Further, the fidelity of our measurements suggests that duplex sequencing of sperm in humans may be well-suited for assessing the potential mutagenic impact of environmental exposures on the germline.
Our deep WGS profiling of sequential sperm samples in two individuals, followed by validation using targeted duplex sequencing, showed a highly stable number of clonal mutations and clonal mosaicism levels with age, suggesting that age-related clonal spermatogenesis occurs at mosaicism levels below the detection limit of deep WGS. This is consistent with the highly polyclonal spatial arrangement of sperm production38 and a prior deep WGS study that profiled individuals with samples spanning closely spaced (< 1 year) timepoints19. Our finding that spermatogonial lineages marked by early developmental mutations have remarkably stable mosaicism over long timespans further suggests that, at least in these individuals, any age-related dysfunction of spermatogonial stem cells and/or seminiferous tubules were sufficiently spatially random as to not significantly alter the fractional sperm contribution of these lineages. The very high cost of deep WGS limited the number of samples we could profile by this approach, and larger-scale studies are needed to corroborate our findings. We postulate that deep WGS paired with targeted duplex sequencing validation in sequential sperm samples of even older men, who will have a greater decline in sperm production, may identify changes in the mosaicism of spermatogonial clones marked by early developmental mutations. Importantly, while our data show that clonal mosaic mutations that arise during early development have stable mosaicism over long periods of time, this is likely not the case for mosaic mutations occurring later in life that undergo positive selection.
Our study has a few limitations. First, currently feasible sequencing depth due to sequencing costs limits the number of mutations we detect per sample. This adds measurement error to our mutation rate estimates and precludes analysis of mutational spectra in individual samples. As sequencing costs continue to decrease, direct sequencing of sperm will yield increasingly accurate and comprehensive measurements. Second, more definite conclusions about the stability of mutation rates and patterns over adult life and about variability among individuals are limited by our relatively small cohort size and our limitation to two timepoints per individual. This motivates future studies to profile more timepoints in larger cohorts, and these future studies may have implications for counseling individuals regarding the risk of de novo genetic disease risk in offspring. For example, a recent trio study identified individuals with outlier germline mutation burdens due to chemotherapy and DNA-repair deficiency9. Profiling sequential samples from such individuals using direct sperm sequencing would illuminate the time course of these processes. A third limitation is that, while our high-fidelity sequencing reliably detects mosaic mutations regardless of mosaicism level, it has shallow genome coverage, and our deep WGS profiling has high genome coverage but its fidelity is limited to detecting mutations with >1% mosaicism. Therefore, these two methods are not sensitive for detection of clonal mutations with <1% mosaicism that likely account for most mutations under positive selection in the germline. Detecting these mutations comprehensively will require the application of high-fidelity duplex sequencing approaches that achieve both high genome coverage and high fidelity, either by targeting specific genomic regions (e.g., targeted duplex sequencing44) or by dramatically lowering sequencing costs of non-targeted duplex sequencing. Indeed, during the review of this manuscript, two pre-printed studies demonstrated targeted duplex sequencing of sperm that revealed novel genes with mutations under positive selection in the male germline45,47. Further, newer versions of duplex sequencing enable interrogation of any genomic region44, in contrast to our use of restriction enzyme fragmentation in non-targeted duplex sequencing that captures ~30% of the genome.
It is not known whether variability in germline mutation rates among individuals correlates with mutation rates in somatic tissues. To the extent that an individual’s complement of genetic variants in DNA repair and DNA metabolic genes explains some of this variability, we speculate that a correlation may be identified by future larger-scale studies between germline and somatic mutation rates. If confirmed, this may enable germline mutation rate measurements to assess the risk of somatic mutation diseases such as cancer. High-fidelity DNA sequencing is revolutionizing our understanding of mosaic mutation processes. Future studies that couple this powerful technology with novel study designs will continue to yield new insights into the mechanisms and modifiers of mutation in the human body.
Methods
Research subjects
All research subjects were enrolled in a study approved by the New York University Grossman School of Medicine Institutional Review Board in compliance with all relevant ethical regulations. We queried the databases of Cryos and California Cryobank to identify previous sperm donors who still had at least one specimen stored for more than 10 years. Donors were contacted by a project manager by telephone call or written communication, and if they confirmed interest in the study, they were referred to a secure online consent form managed by New York University. Following consent, subjects completed an online medical history, fertility, and exposure questionnaire (Supplementary Data 9), and the sperm bank was notified that the subject is consented for collection of new sperm and blood/saliva samples. Subjects received nominal monetary compensation for participation. Later in the study, the links between the sperm bank de-identified subject IDs and this study’s de-identified subject IDs were deleted. Subjects consented to sharing their data via a controlled-access database.
Semen samples at both sperm banks were obtained by masturbation in a private room into a sterile cup. Ejaculates were kept at room temperature briefly to allow liquefaction. The motility of timepoint T1 sperm samples at both sperm banks and of timepoint T2 sperm samples at California Cryobank were estimated manually by microscopy. Blood was collected by Cryos in EDTA tubes. Saliva was collected by California Cryobank using Oragene OGR-600 saliva collection kits (DNA Genotek).
Purification of sperm
After semen collection by the sperm bank, prior to freezing in liquid nitrogen, sperm were purified from semen samples from timepoint T1 of individuals SPM-1072, SPM-1511, SPM-1514, SPM-1515, and SPM-1536, and all semen samples from timepoint T2 of all individuals using density gradient centrifugation48. All other semen samples collected at timepoint T1 were frozen without washing.
Subsequently, at New York University, for sperm samples that already underwent density gradient centrifugation prior to freezing at the sperm bank, we performed one additional density gradient centrifugation and two post-density gradient washes as previously described49 using ORIGIO gradient 40/80 buffer (Cooper Surgical, 84022010) and Origio sperm wash buffer (Cooper Surgical, 84050060) reagents. For semen samples that were frozen at the sperm bank without a wash, we performed an initial density gradient centrifugation followed by one post-density gradient wash (resuspending the washed sperm in 350 µL of Origio sperm wash buffer) as previously described48, and then we immediately performed a second density gradient centrifugation and two post-density gradient washes as previously described48. Therefore, every final sperm sample underwent two density gradient centrifugations. We have previously shown that this method of sperm purification removes detectable somatic cell contamination using a methylation assay interrogating four loci that are differentially methylated in sperm versus somatic cells48,50.
DNA extraction
DNA was extracted from sperm with our previously described method48 that utilizes Buffer RLT (Qiagen), Bond-Breaker TCEP Solution (Thermo Fisher Scientific), bead homogenization (20 Hz for 30 s), QIAamp DNA Mini Kit (Qiagen), and treatment with Monarch RNase A (New England Biolabs). The concentration and quality of all DNA samples were measured using a NanoDrop instrument (Thermo Fisher Scientific), a Qubit 1× dsDNA HS Assay Kit (Thermo Fisher Scientific), and a Genomic DNA ScreenTape TapeStation Assay (Agilent). DNA was stored at −20 °C.
DNA was extracted from blood samples using the Magattract HMW DNA Kit (Qiagen), per the manufacturer’s protocol for whole blood.
DNA was extracted from saliva samples using the QIAamp DNA Mini Kit (Qiagen) per the manufacturer’s protocol for body fluids and including an RNase A treatment step.
NanoSeq library preparation and sequencing
NanoSeq libraries were prepared as previously described21 with 50 ng of DNA input and using the HpyCH4V restriction enzyme (New England Biolabs) for fragmentation. NanoSeq libraries for mosaic mutation profiling were made with 0.6 fmol of library molecules input into the PCR step. NanoSeq libraries for inherited germline variant profiling (used as a filter during the analysis of mosaic mutations) were made with between 3 and 12 fmol of library molecules input into the PCR step, depending on the amount available. Libraries were sequenced with 150 bp paired-end reads using a NovaSeq 6000 instrument (Illumina). We filtered low-quality tiles from two sequencing runs using the bbamp v39.05 filterbytile tool (https://sourceforge.net/projects/bbmap) using default settings and a reference ‘dump’ file calculated from all samples of the same lane. See Supplementary Data 3 for sequencing and library metrics.
Whole-genome library preparation and sequencing
Two PCR-free libraries for deep whole-genome sequencing were prepared for each sample to provide the sufficient library for deep sequencing. Each library used 425–1000 ng of input DNA diluted in 45 µL of nuclease-free water (NFW), with the amount of input DNA depending on the amount of DNA that could be input into this volume. We then added 5 µL of 10× shearing buffer (100 mM Tris pH 8, 500 mM NaCl) to the DNA. Next, we fragmented DNA in a 96 AFA-TUBE TPX plate (Covaris, product 520291) in an R230 instrument (Covaris) with settings: temperature = 10 °C, iterations = 5, treatment duration = 10 s, peak incident factor = 280 W, duty factor = 25%, and cycles per burst = 50.
End repair of DNA was then performed by adding 7 µL of NEBNext Ultra II End Prep Reaction Buffer (NEB) and 3 µL of NEBNext Ultra II End Prep Enzyme Mix (NEB), and then incubating at 20 °C for 30 min followed by 65 °C for 30 min. Adapters were ligated by then adding 5 µL of xGen UDI-UMI adapter (IDT), 30 µL NEBNext Ultra II Ligation Master Mix (NEB) and 1 µL NEBNext Ligation Enhancer (NEB), followed by incubation at 20 °C for 15 min. Finally, libraries were purified and adapter dimers removed using a 0.8× magnetic (SPRI) bead to library volume ratio cleanup, the two library preparations for each sample were combined, and then libraries were further purified with a 0.9× magnetic bead to library volume ratio cleanup.
Libraries were sequenced with 150 bp paired-end reads using a NovaSeq 6000 instrument (Illumina). See Supplementary Data 3 for sequencing metrics.
Targeted NanoSeq library preparation and sequencing
Targeted NanoSeq libraries were prepared as previously described44 with 400 ng of DNA input and using Covaris (sonication) for fragmentation with the following modifications: (1) Covaris fragmentation to a target size of 220 bp was performed in a 55 µL volume in a 96 microTUBE Plate (Covaris cat. # 520078) on an R230 instrument (Covaris) with settings: repeat iterations = 20, repeat duration = 10 s, peak incident power = 450, duty factor = 25%, cycles per burst = 600. (2) Fragmented DNA proceeded without a bead purification directly to end repair in a volume of 60 µL, which was then purified with a bead-to-sample volume ratio of 2×. (3) The 60 µL volume of ligated DNA was purified by adding 60 µL of water and 96 µL of beads. (4) qPCR calculations showed that none of the libraries exceeded our target of 4000 haploid genomic equivalents of ligated DNA, so we input the entire volume of ligated library into the pre-hybridization indexing PCR that was performed in a total volume of 66 µL with seven cycles. The PCR reactions were purified once with a bead-to-sample volume ratio of 1×. (5) Hybridization was performed with a probe panel synthesized by Twist Bioscience. We submitted for panel design all 193 mosaic mutations that were detected by WGS in at least one sperm sample, as well as 12 randomly selected heterozygous germline variants for downstream confirmatory sample fingerprinting, of which 6 mosaic mutation loci and 1 germline variant failed Twist’s design filters and were not included in the panel. (6) Two libraries were multiplexed per hybridization with an input of between 850 and 975 ng per library. (7) We used the Twist Standard Hybridization Reagent Kit v2 and followed Twist’s protocol except that we used NEBNext Ultra II Q5 Master Mix (NEB) for the post-hybridization PCR (18 cycles). Libraries were sequenced with 150 bp paired-end reads using a NovaSeq X instrument (Illumina). See Supplementary Data 3 for sequencing and library metrics.
NanoSeq analysis
Primary data processing
NanoSeq primary data processing was performed using the NanoSeq analysis pipeline v.3.5.5 (https://github.com/cancerit/NanoSeq)21 for chromosomes 1–22, X, and Y (hg38 reference genome). We used the following non-default settings— ‘cov’ step: --exclude chrM,%random,chrUn_%,%_alt,chrEBV,HLA%; ‘var’ step: -c 0, ‘indel’ step: -c 0 --t3 135 --t5 10. In the ‘dsa’ step, we used the SNP mask ‘SNP.sorted.GRCh38.bed.gz’ supplied by the creators of the NanoSeq analysis pipeline (link in the above GitHub page) and a NOISE mask that we created by combining the following regions in hg38: (a) k50 Umap mappability51 < 0.95; (b) ENCODE hg38 blacklist (accession ENCFF356LFX); (c) Segmental duplications, centromeres, and telomeres obtained from the UCSC genome browser52; (d) Satellite repeats obtained from the RepeatMasker track (repClass=Satellite) of the UCSC genome browser; (e) gnomAD53 v3.1.2 indels with any of AF, AF_hom, or AF_het allele frequency tags ≥ 0.01 (including 1 base prior to each deletion for proper filtering by the NanoSeq pipeline); and (f) gnomAD v3.1.2 substitutions with any of AF, AF_hom, or AF_het allele frequency tags ≥0.001 and FILTER != ‘PASS’. This NOISE mask removes a total of 484 Mb (~15.7%) of the genome. We include this NOISE mask as Supplementary Data 10.
Following primary data processing, we confirmed that VerifyBamID254 calculated a FREEMIX value < 0.001 for all samples, which excludes significant contamination from an unrelated sample.
Fingerprinting
To further confirm concordance between each individual’s blood/saliva and sperm NanoSeq samples, we performed a fingerprinting analysis. Briefly, we counted the number of high-quality inherited germline variants (in the blood/saliva inherited germline sample) that were absent from blood/saliva and sperm NanoSeq mosaic mutation libraries. In the absence of false-positive germline variant calls, we would expect none of the high-quality variant calls from the inherited germline sample to be absent from the same individual’s mosaic samples, assuming the variant locus had sufficient sequencing depth in the mosaic sample.
Initial inherited germline variant calls were made from each inherited germline sample’s ‘neat’ CRAM file (‘neat’ CRAM files contain one random representative read pair from each duplex molecule read family; generated by the NanoSeq pipeline randomreadinbundle tool) using bcftools55 v1.15.1 mpileup with options ‘-B -Q 25 -q 10 -a FORMAT/AD,FORMAT/DP’, followed by bcftools call -m -v, bcftools +fill-tags -- -t 'FORMAT/VAF', and bcftools view -m2 -M2 -v snps -g ^miss. These germline variant calls were further filtered to exclude those with VAF < 0.3, read depth <20, or ‘QUAL’ value < 30.
For each individual’s initial inherited germline variant calls, we genotyped those loci jointly across the ‘neat’ CRAM files of all mosaic samples of all individuals using bcftools mpileup with options ‘-B -q 10 -a FORMAT/AD,FORMAT/DP’, followed by bcftools call -m, bcftools +fill-tags -- -t 'FORMAT/VAF'. We then applied the following filters to remove residual false-positive inherited germline variant calls: (1) filters recommended by the authors of bcftools (http://www.htslib.org/workflow/filter.html): bcftools filter -e “MQBZ < -(3.5 + 4*(FORMAT/DP)/QUAL) || RPBZ > (3 + 3*(FORMAT/DP)/QUAL) || RPBZ < -(3 + 3*(FORMAT/DP)/QUAL) || SCBZ > (2.5 + (FORMAT/DP)/30)”; (2) bcftools filter --SnpGap 3; (3) variants overlapping the ‘NOISE mask’ regions described above; (4) only substitutions with two alleles called (-v snps -m2 -M2); (5) variants that are not substitutions or substitutions that do not have exactly 2 alleles called (including the reference allele); (6) ‘QUAL’ value ≤ 500 or VAF ≤ 0.4; (7) variants within 2 bp of ‘Simple Repeats’56 annotated in the UCSC hg38 genome browser; (8) variants in the tandem repeats annotation used by the HipSTR tool57 (https://github.com/HipSTR-Tool/HipSTR-references/raw/master/human/hg38.hipstr_reference.bed.gz); (9) T > G variants, which had an elevated frequency of false-positives; (10) variants in repetitive sequences as quantified by the Brain Somatic Mosaicism Network pipeline repeat.2.py tool (https://github.com/bsmn/bsmn-pipeline/blob/master/utils/repeat.2.py), specifically, filtering variants with either ‘repeat_n’ or ‘repeat_length’ ≥3 at the variant position or with either repeat_n’ or ‘repeat_length’ ≥4 at either of the adjacent bases; (11) variants with another variant call within 20 bp using the GATK v4.4 VariantFiltration tool58 with options ‘--cluster-window-size 20 --cluster-size 2’. Finally, for each inherited germline/mosaic sample pair, we counted the number of the filtered high-quality inherited germline variants that had a total (REF and ALT) read depth ≥8 and ALT read depth 0 in the mosaic sample.
Data analysis
To facilitate analysis of NanoSeq results, we created an R59 package called NanoSeqTools (https://github.com/evronylab/NanoSeqTools), which loads all the NanoSeq pipeline output files required for downstream analysis. NanoSeqTools also utilizes BEDTools v2.31.160 and code from indelwald61. NanoSeqTools also corrects substitution mutation burdens for differences in trinucleotide distributions between the reference genome and the final interrogated base pairs, as previously described21, and these mutation burdens were used in our analyses. Weighted least-squares linear regressions were performed using the ‘lm’ function in R, with the ‘weights’ parameter set to 1/[raw mutation counts] for each sample. Additional R packages used in the analysis were tidyverse, outliers, ggbeeswarm, ggpubr, and RColorBrewer.
Mutational signature analysis
Mutational spectra and signature analysis were performed for unique mutations, i.e., counting only once those mutations detected more than once in a sample, which was 42 non-unique/3580 total substitution mutations and 5 non-unique/339 total indel mutations. Prior to analysis, mutational spectra of substitutions were also corrected for trinucleotide context opportunities21. Specifically, we calculated the fraction of each trinucleotide out of all trinucleotides across the human reference genome, after reverse complementing trinucleotides so that the central base is a pyrimidine. For each sample, we then calculated the trinucleotide fractional distribution across bases interrogated by NanoSeq using the NanoSeq pipeline results.cov.bed.gz output file. Then, for each sample, each trinucleotide context’s substitutions count was corrected for trinucleotide opportunities by multiplying its raw mutation count by that context’s fractional distribution in the full genome divided by its fractional distribution in the sample’s interrogated bases. Mutational spectra of substitutions and indels were tabulated with indelwald61 and sigfit62, respectively, and spectra were plotted with sigfit.
Mutational signature analysis was performed with sigfit62 after combining sperm mosaic substitutions into three age groups (20 ≤ age < 30 years old, 30 ≤ age < 40 years old, and age ≥40 years old) and correcting for trinucleotide context opportunities. Since COSMIC SBS1 is more challenging to extract in the setting of a small number of samples due to its lower level63, we initially used the ‘fit_extract_signatures’ function of sigfit to fit SBS1 (from the COSMIC SBS v.3.4 catalog) while simultaneously extracting additional signatures de novo. This showed that high reconstruction accuracies were achieved by extracting one additional signature, which most closely matched COSMIC SBS5 as described in the main text. The final analysis therefore fit the mutational spectra directly to COSMIC signatures SBS1 and SBS5.
Analysis of prior de novo mutation studies
Paternally phased de novo mutation burdens were calculated from data published in a prior study by Halldorsson et al. of 2976 trios10, using additional methodological details obtained from the associated study by Jonsson et al.2. Specifically, child and parent ages were obtained from Halldorsson, et al.’s aau1043_datas7.tsv supplementary file, and de novo mutation counts were obtained from the study’s aau1043_datas5_revision1.tsv supplementary file.
Paternally phased de novo substitution mutation burdens (mutations per base pair) were first calculated for each child as [total number of paternally phased de novo substitution mutations]/[fraction of the child’s de novo substitution mutations that were either paternally or maternally phased, which corrects for each child’s phasing rate] × [Jonsson et al’s correction factor of 1.009 that accounts for its false positive and negative rate]/[Jonsson et al’s interrogated genome size of 2,682,890,000]2,10. We then performed a linear regression in R of the de novo substitution mutation burdens versus paternal age using the ‘lm’ function, and we obtained 95% confidence intervals using the ‘confint’ function.
Whole-genome sequencing analysis
Deep whole-genome sequencing data was analyzed using a custom pipeline that was based on a mosaic mutation pipeline developed by the Brain Somatic Mosaicism Network (BSMN)42 (https://github.com/bsmn/bsmn-pipeline) that incorporates rigorous multi-step filtering of sequencing and alignment artifacts.
Sequencing reads were split into chunks and aligned to the hg38 version of the reference genome with bwa mem64 with option ‘-M’, followed by conversion to BAM format and sorting with sambamba65 and merging of chunks with samtools merge55. Next, we marked optical duplicates with Picard MarkDuplicates66 with option ‘-OPTICAL_DUPLICATE_PIXEL_DISTANCE 2500’. We then performed indel realignment using: a) GATK v3.767 RealignerTargetCreator with options ‘-known Mills_and_1000 G_gold_standard.indels.hg38.vcf.gz -known Homo_sapiens_assembly38.known_indels.vcf.gz’ (these and other GATK-related resource files specified below were obtained from the online GATK resource bundle); b) GATK v3.7 IndelRealigner with options ‘-known Mills_and_1000G_gold_standard.indels.hg38.vcf.gz -known Homo_sapiens_assembly38.known_indels.vcf.gz -targetIntervals [intervals file generated from the prior RealignerTargetCreator step]’. We then performed base quality score recalibration using: (a) GATK v3.7 BaseRecalibrator with options ‘-knownSites Mills_and_1000G_gold_standard.indels.hg38.vcf.gz -knownSites Homo_sapiens_assembly38.known_indels.vcf.gz -knownSites dbsnp_146.hg38.vcf.gz‘; (b) GATK v3.7 PrintReads with options ‘--disable_indel_quals -BQSR [output file of the prior BaseRecalibrator step]’. The final output BAM file was converted to CRAM format with samtools. Average coverage depth was calculated with GATK v4.4 DepthOfCoverage for regions in the wgs_calling_regions.hg38.interval_list file obtained from the online GATK resource bundle.
Substitution and indel variants were called from the above final CRAM file in the following steps: (a) GATK v4.4 HaplotypeCaller separately for each chromosome with options ‘-ERC GVCF -A StrandBiasBySample -ploidy [20% × average coverage calculated for representative chromosome 10]’. Note, this ploidy level was optimized by the BSMN pipeline to detect mosaic mutations; (b) GATK v4.4 GenotypeGVCFs separately for each chromosome with option ‘-ploidy [20% × average coverage calculated for representative chromosome 10]’; (c) GATK v4.4 GatherVcfs to combine the calls of all the chromosomes; (d) GATK v4.4 IndexFeatureFile to index the prior VCF file; (e) GATK v4.4 VariantRecalibrator with options ‘--resource:hapmap,known=false,training=true,truth=true,prior=15.0 hapmap_3.3.hg38.vcf.gz --resource:omni,known=false,training=true,truth=true,prior=12.0 1000G_omni2.5.hg38.vcf.gz --resource:1000G,known=false,training=true,truth=false,prior=10.0 1000G_phase1.snps.high_confidence.hg38.vcf.gz --resource:dbsnp,known=true,training=false,truth=false,prior=2.0 dbsnp_146.hg38.vcf.gz -an DP -an QD -an FS -an SOR -an MQ -an MQRankSum -an ReadPosRankSum --mode SNP --max-gaussians 4 --maximum-training-variants 5000000 -tranche 100.0 -tranche 99.9 -tranche 99.0 -tranche 90.0’; (f) GATK v4.4 ApplyVQSR of the VCF file produced by GatherVcfs with options ‘--mode SNP -ts-filter-level 99.0 --recal-file [recal file produced by the prior VariantRecalibrator step] --tranches-file [tranches file produced by the prior VariantRecalibrator step]’; (g) GATK v4.4 VariantRecalibrator of the VCF file produced by the prior ApplyVQSR step with options ‘-resource:mills,known=false,training=true,truth=true,prior=12.0 Mills_and_1000G_gold_standard.indels.hg38.vcf.gz -resource:dbsnp,known=true,training=false,truth=false,prior=2.0 dbsnp_146.hg38.vcf.gz -an QD -an DP -an FS -an SOR -an MQRankSum -an ReadPosRankSum --mode INDEL -tranche 100.0 -tranche 99.9 -tranche 99.0 -tranche 90.0 --max-gaussians 4’; (h) GATK v4.4 ApplyVQSR of the VCF file produced by the prior VariantRecalibrator step with options ‘--mode INDEL -ts-filter-level 99.0 --recal-file [recal file produced by the prior VariantRecalibrator step] --tranches-file [tranches file produced by the prior VariantRecalibrator step]’.
For the purpose of later filtering, we called copy number variants from the above final CRAM file with CNVpytor68 as follows: (a) cnvpytor -T [reference genome fasta] -rd [CRAM file]; (b) cnvpytor -his 100; (c) cnvpytor -stat 100; (d) cnvpytor -partition 100; (e) cnvpytor -call 100.
We then performed multi-step filtering of the called variants as follows:
-
Exclude variants that are not assigned FILTER = ‘PASS’ by the VariantRecalibrator steps.
-
Exclude substitution variants that are within 5 bp of an indel using bcftools filter with option ‘--SnpGap 5’.
-
Exclude variants that are in gnomAD v3.1.253 with allele frequency >0.001 using the bcftools annotate command and a gnomAD annotation file that contains CHROM, POS, REF, and ALT columns.
-
Exclude variants that are not PASS (i.e., not equal to ‘P’) in the 1000 genomes strict mask available at https://github.com/abyzovlab/CNVnator/raw/master/ExampleData/1KG.20160622.strict_mask.hg38_GRCh38.fa.gz. This was performed by extracting the masked base symbol from the mask for each variant call position with samtools faidx and then annotating variant calls with bcftools annotate.
-
We used the somatic_vaf.2.py script from the BSMN pipeline to annotate each variant with a binomial test (where # of trial successes = # of ALT reads and # of trials = # of total reads, with alternative hypothesis = smaller than expected # of successes) P value reflecting the probability that its variant allele frequency is by chance significantly different from 0.5 (the variant allele frequency expected for an inherited germline variant) and with the number of reads supporting the variant call that have mapping quality ≥ 20 and base quality ≥20. We then exclude variants with binomial P value > 1 × 10−6 or when there are less than three reads supporting the variant.
-
Exclude variants whose +/− 1 kilobase span is genotyped by CNVpytor with a copy number ≥2.5, since those regions’ higher copy number would have inherited germline variants with a lower variant allele frequency.
-
We used the strand_bias.2.py script from the BSMN pipeline to: (a) exclude variants that do not have at least one read in each strand with mapping quality ≥20 and base quality ≥20 supporting the variant, and, (b) exclude variants that have both strand bias Poisson and strand bias Fisher’s exact test P values < 0.05.
-
We used the repeat.2.py script from the BSMN pipeline to exclude variants that are in regions with short tandem repeat sequences, specifically variants with repeat_n value ≥ 4 or repeat_length value ≥ 10.
-
Exclude variants that are present in the BSMN panel of normal reference, which is a panel of variants found in >5/2504 PCR-free-sequenced whole-genome sequencing samples.
-
Exclude variants within the SNP and NOISE mask regions used for NanoSeq filtering (described in the ‘NanoSeq analysis’ section).
-
Exclude variants within 2 bp of ‘Simple Repeats’ annotated in the UCSC hg38 genome browser or that are within the tandem repeats annotation regions used by the HipSTR tool (described above in the ‘Fingerprinting’ section).
-
Exclude variants that pass all the above filters, but that within a 20 bp window there are >1 variants, using the GATK v4.4 VariantFiltration tool with options ‘--cluster-window-size 20 --cluster-size 2’.
-
Exclude variants that pass all the above filters, but which are present in the pre-filtering variant calls of any sample of the same individual with binomial test (where # of trial successes = # of ALT reads and # of trials = # of total reads, with alternative hypothesis = smaller than expected # of successes) P value > 1 × 10−6.
-
Two read-depth-based filters that examined read support for variants in the final CRAM files of each sample as quantified by the bcftools mpileup command with options ‘-d 9999 -B -a FORMAT/AD, FORMAT/DP’: (a) exclude variants detected in ≤10 reads that were also detected in ≥ 1 read in any one of the WGS samples of another individual, and exclude variants detected in >10 reads that were also detected in ≥ 2 reads in any one of the WGS samples of another individual; (b) exclude variants with <5 supporting reads whose trinucleotide context contains ‘TT’ (after collapsing to central pyrimidine context). These trinucleotide contexts are enriched for artifacts based on manual review of sequencing reads and based on outlier peaks in the mutational spectrum compared to the expected spectrum from our NanoSeq data and prior de novo mutation trio study data10. Variants that passed these two read-depth-based filters in at least one sample of an individual were retained in other samples of the individual even if they failed these filters to facilitate analyses of bona fide mosaic mutations across samples of the individual.
-
Variants that passed all the above filters in a subset of an individual’s samples were recovered from other samples of the individual if in those other samples the variant did not pass all the filters but was detected in ≥3 reads per the above bcftools mpileup data.
Subsequently, we analyzed single base substitutions and annotated them using ANNOVAR69 with GENCODE v43 gene annotations, gnomAD v3.1.2, and CADD scores70. We calculated each mutation’s mosaicism (i.e., % of cells with the mutation) as 2× VAF for blood and saliva samples and equal to the VAF for sperm samples. Plots were created in R with tidyverse71 and ggplot72 packages.
For analyses and plots of genes with >2 mutations, we manually reviewed the raw reads of these mutations (N = 19 mutations) in IGV and excluded 9 of these mutations from analyses and plots of frequently mutated genes, because they are likely artifacts based on their detection at low-level in reads of samples of a different individual than the individual in which the variant was called by the above pipeline.
Note, the mutational spectrum of SPM-1047 blood WGS closely resembled the duplex sequencing spectrum of all blood samples (cosine similarity = 0.92; N = 956 unique mutations), while the spectrum of SPM-1564 saliva WGS had a markedly different spectrum than that obtained by duplex sequencing of all saliva samples (cosine similarity = 0.23; N = 467 unique mutations) with a high fraction of C>T mutation calls (91.4%), likely due to DNA damage from the reagent used to preserve saliva. Therefore, this saliva sample was used only to assess the presence of sperm mutations identified by WGS in non-sperm tissue.
Targeted NanoSeq analysis
Targeted NanoSeq primary data processing was performed using the NanoSeq analysis pipeline21 as described above for standard NanoSeq data, except with minimum AS-XS = 10, maximum matched normal VAF = 0.1, minimum matched normal coverage = 25, and for the samples of each individual using as the matched normal reference the corresponding targeted NanoSeq ‘neat’ CRAM file from either blood (SPM-1047) or saliva (SPM-1564). For each sample, we then ran the snv_merge_and_vaf_calc.R script from the NanoSeq analysis pipeline21 to calculate the number of times each mutation was detected (‘TIMES_CALLED’ in the resulting VCF file). We further extracted: (1) the duplex read coverage of all targeted mutations in the panel using the NanoSeq pipeline’s results.cov.bed.gz files, since the NanoSeq pipeline only calculates this data for sites in which a mutation was detected; (2) read coverage and mutation read counts for all targeted mutations in the panel in each sample’s ‘neat’ CRAM file; (3) non-duplex read coverage and non-duplex mutation read counts for all targeted mutations in the panel, calculated as the above ‘neat’ CRAM values minus the above duplex values. The mean duplex coverage was calculated for each mutation across all samples of the individual with the mutation.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Sequencing data generated in this study have been deposited at the NCBI database of Genotypes and Phenotypes (dbGaP) under accession code phs003716.v1.p1. The data are available under restricted access for genetic privacy and consent reasons, and access can be obtained by researchers for IRB-approved studies by application via the dbGaP website in a process that usually takes less than 1 month.
Code availability
The source code for the NanoSeqTools analysis package is available at https://github.com/evronylab/NanoSeqTools.
References
Wang, S. et al. Per-nucleus crossover covariation and implications for evolution. Cell 177, 326–338.e316 (2019).
Jónsson, H. et al. Parental influence on human germline de novo mutations in 1548 trios from Iceland. Nature 549, 519 (2017).
Shendure, J. & Akey, J. M. The origins, determinants, and consequences of human mutations. Science 349, 1478 (2015).
Kong, A. et al. Rate of de novo mutations and the importance of father’s age to disease risk. Nature 488, 471–475 (2012).
Kessler, M. D. et al. De novo mutations across 1,465 diverse genomes reveal mutational insights and reductions in the Amish founder population. Proc. Natl. Acad. Sci. USA 117, 2560–2569 (2020).
Wong, W. S. W. et al. New observations on maternal age effect on germline de novo mutations. Nat. Commun. 7, 10486 (2016).
Rahbari, R. et al. Timing, rates and spectra of human germline mutation. Nat. Genet. 48, 126–133 (2016).
Sasani, T. A. et al. Large, three-generation human families reveal post-zygotic mosaicism and variability in germline mutation accumulation. eLife 8, e46922 (2019).
Kaplanis, J. et al. Genetic and chemotherapeutic influences on germline hypermutation. Nature 605, 503–508 (2022).
Halldorsson, B. V. et al. Characterizing mutagenic effects of recombination through a sequence-level genetic map. Science 363, eaau1043 (2019).
Deciphering Developmental Disorders, S. et al. Prevalence and architecture of de novo mutations in developmental disorders. Nature 542, 433 (2017).
Crow, J. F. The origins, patterns and implications of human spontaneous mutation. Nat. Rev. Genet. 1, 40–47 (2000).
O’Roak, B. J. et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485, 246 (2012).
Risch, N., Reich, E. W., Wishnick, M. M. & McCarthy, J. G. Spontaneous mutation and parental age in humans. Am. J. Hum. Genet. 41, 218–248 (1987).
Aitken, R. J., De Iuliis, G. N. & Nixon, B. The sins of our forefathers: paternal impacts on de novo mutation rate and development. Annu. Rev. Genet. 54, 1–24 (2020).
Goriely, A. & Wilkie, A. O. Paternal age effect mutations and selfish spermatogonial selection: causes and consequences for human disease. Am. J. Hum. Genet. 90, 175–200 (2012).
Moore, L. et al. The mutational landscape of human somatic and germline cells. Nature 597, 381–386 (2021).
Xia, B. et al. Widespread transcriptional scanning in the testis modulates gene evolution rates. Cell 180, 248–262.e221 (2020).
Yang, X. et al. Developmental and temporal characteristics of clonal sperm mosaicism. Cell 184, 4772–4783.e4715 (2021).
Salazar, R. et al. Discovery of an unusually high number of de novo mutations in sperm of older men using duplex sequencing. Genome Res. 32, 499–511 (2022).
Abascal, F. et al. Somatic mutation landscapes at single-molecule resolution. Nature 593, 405–410 (2021).
Schmitt, M. W. et al. Detection of ultra-rare mutations by next-generation sequencing. Proc. Natl. Acad. Sci. USA 109, 14508 (2012).
Ren, P., Dong, X. & Vijg, J. Age-related somatic mutation burden in human tissues. Front. Aging 3, 1018119 (2022).
Breuss, M. W. et al. Unbiased mosaic variant assessment in sperm: a cohort study to test predictability of transmission. eLife 11, e78459 (2022).
Breuss, M. W. et al. Autism risk in offspring can be assessed through quantification of male sperm mosaicism. Nat. Med. 26, 143–150 (2020).
Bernkopf, M. et al. Personalized recurrence risk assessment following the birth of a child with a pathogenic de novo mutation. Nat. Commun. 14, 853 (2023).
Machado, H. E. et al. Diverse mutational landscapes in human lymphocytes. Nature 608, 724–732 (2022).
Xing, D., Tan, L., Chang, C.-H., Li, H. & Xie, X. S. Accurate SNV detection in single cells by transposon-based whole-genome amplification of complementary strands. Proc. Natl. Acad. Sci. USA 118, e2013106118 (2021).
Koh, G., Degasperi, A., Zou, X., Momen, S. & Nik-Zainal, S. Mutational signatures: emerging concepts, caveats and clinical applications. Nat. Rev. Cancer 21, 619–637 (2021).
Alexandrov, L. B. et al. The repertoire of mutational signatures in human cancer. Nature 578, 94–101 (2020).
Spisak, N., de Manuel, M., Milligan, W., Sella, G. & Przeworski, M. The clock-like accumulation of germline and somatic mutations can arise from the interplay of DNA damage and repair. PLoS Biol. 22, e3002678 (2024).
Maher, G. J., Goriely, A. & Wilkie, A. O. M. Cellular evidence for selfish spermatogonial selection in aged human testes. Andrology 2, 304–314 (2014).
Breuss, M. W., Yang, X. & Gleeson, J. G. Sperm mosaicism: implications for genomic diversity and disease. Trends Genet. 37, 890–902 (2021).
Mitchell, E. et al. Clonal dynamics of haematopoiesis across the human lifespan. Nature 606, 343–350 (2022).
Martincorena, I. et al. Somatic mutant clones colonize the human esophagus with age. Science 362, 911–917 (2018).
Martincorena, I. et al. High burden and pervasive positive selection of somatic mutations in normal human skin. Science 348, 880–886 (2015).
Lim, J. et al. Selfish spermatogonial selection: evidence from an immunohistochemical screen in testes of elderly men. PLoS ONE 7, e42382 (2012).
Wood, K. A. & Goriely, A. The impact of paternal age on new mutations and disease in the next generation. Fertil. Steril. 118, 1001–1012 (2022).
Nie, X. et al. Single-cell analysis of human testis aging and correlation with elevated body mass index. Dev. Cell 57, 1160–1176.e1165 (2022).
Paniagua, R., Nistal, M., Sáez, F. J. & Fraile, B. Ultrastructure of the aging human testis. J. Electron Microsc. Tech. 19, 241–260 (1991).
Blair, J. D., Hockemeyer, D. & Bateup, H. S. Genetically engineered human cortical spheroid models of tuberous sclerosis. Nat. Med. 24, 1568–1578 (2018).
Wang, Y. et al. Comprehensive identification of somatic nucleotide variants in human brain tissue. Genome Biol. 22, 92 (2021).
Ha, Y.-J. et al. Comprehensive benchmarking and guidelines of mosaic variant calling strategies. Nat. Methods 20, 2058–2067 (2023).
Lawson, A. R. J. et al. Somatic mutation and selection at epidemiological scale. Preprint at https://www.medrxiv.org/content/10.1101/2024.10.30.24316422v1 (2024).
Kunisaki, J. et al. Sperm from infertile, oligozoospermic men have elevated mutation rates. Preprint at https://www.medrxiv.org/content/10.1101/2024.08.22.24312232v1 (2024).
Goldmann, J. M. et al. Differences in the number of de novo mutations between individuals are due to small family-specific effects and stochasticity. Genome Res. 31, 1513–1518 (2021).
Neville, M. D. C. et al. Sperm sequencing reveals extensive positive selection in the male germline. Preprint at https://www.medrxiv.org/content/10.1101/2024.10.30.24316414v1.full.pdf (2024).
Agarwal, A., Gupta, S. & Sharma, R. In Andrological Evaluation of Male Infertility: A Laboratory Guide (eds Agarwal, A. et al.) 101–107 (Springer International Publishing, 2016).
Liu, M. H. et al. DNA mismatch and damage patterns revealed by single-molecule sequencing. Nature 630, 752–761 (2024).
Jenkins, T. G., Liu, L., Aston, K. I. & Carrell, D. T. Pre-screening method for somatic cell contamination in human sperm epigenetic studies. Syst. Biol. Reprod. Med. 64, 146–155 (2018).
Karimzadeh, M., Ernst, C., Kundaje, A. & Hoffman, M. M. Umap and Bismap: quantifying genome and methylome mappability. Nucleic Acids Res. 46, e120 (2018).
Nassar, L. R. et al. The UCSC Genome Browser database: 2023 update. Nucleic Acids Res. 51, D1188–D1195 (2022).
Chen, S. et al. A genomic mutational constraint map using variation in 76,156 human genomes. Nature 625, 92–100 (2024).
Zhang, F. et al. Ancestry-agnostic estimation of DNA sample contamination from sequence reads. Genome Res. 30, 185–194 (2020).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. GigaScience 10, giab008 (2021).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
Willems, T. et al. Genome-wide profiling of heritable and de novo STR variations. Nat. Methods 14, 590 (2017).
Van der Auwera, G. A. et al. From FastQ data to high-confidence variant calls: the genome analysis toolkit best practices pipeline. Curr. Protoc. Bioinforma. 43, 11.10.11–11.10.33 (2013).
R-Core-Team. R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, Vienna, Austria, 2023).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Stammnitz, M. R. et al. The evolution of two transmissible cancers in Tasmanian devils. Science 380, 283–293 (2023).
Gori, K. & Baez-Ortega, A. sigfit: flexible Bayesian inference of mutational signatures. Preprint at https://www.biorxiv.org/content/10.1101/372896v2 (2020).
Cagan, A. et al. Somatic mutation rates scale with lifespan across mammals. Nature 604, 517–524 (2022).
Heng, L. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. Preprint at https://arxiv.org/abs/1303.3997 (2013).
Tarasov, A., Vilella, A. J., Cuppen, E., Nijman, I. J. & Prins, P. Sambamba: fast processing of NGS alignment formats. Bioinformatics 31, 2032–2034 (2015).
Broad Institute. Picard toolkit. Broad Institute, GitHub repository (2019).
Van der Auwera, G. A. & O’Connor, B. D. Genomics in the Cloud: Using Docker, GATK, and WDL in Terra (O’Reilly Media, 2020).
Suvakov, M., Panda, A., Diesh, C., Holmes, I. & Abyzov, A. CNVpytor: a tool for copy number variation detection and analysis from read depth and allele imbalance in whole-genome sequencing. GigaScience 10, giab074 (2021).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164 (2010).
Rentzsch, P., Witten, D., Cooper, G. M., Shendure, J. & Kircher, M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 47, D886–D894 (2018).
Wickham, H. et al. Welcome to the Tidyverse. J. Open Source Softw. 4, 1686 (2019).
Wickham, H. ggplot2: Elegant Graphics for Data Analysis (Springer New York, 2009).
Acknowledgements
The authors thank the research subjects who participated in this study. This work was supported by a grant from the Eunice Kennedy Shriver National Institute of Child Health and Human Development (R21HD105910; G.D.E. and J.E.S.). Sequencing performed at the New York University (NYU) Grossman School of Medicine Genome Technology Center was supported in part by the National Cancer Institute (P30CA016087) and a National Institutes of Health Shared Instrumentation Grant (1S10OD023423-01). The computational work was supported in part by the New York University Information Technology High Performance Computing resources, services, and staff expertise, and by the New York University Grossman School of Medicine High Performance Computing Core. J.E.S. has been supported by the Damon Runyon Cancer Research Foundation, the Vinney Family Scholars Award, and the Bristol Myers Squibb Foundation.
Author information
Authors and Affiliations
Contributions
J.E.S. and G.D.E. conceived the project and supervised the study. A.-B.S. and C.B. contributed input to the study design. E.L., A.-B.S., S.M., and P.C. recruited research subjects. C.A.L. and M.H.L. processed biological samples, with assistance from B.M.C., U.C., and S.P. G.D.E., M.G.-P., and N.J. prepared sequencing libraries. A.S. performed data analysis with input from G.D.E. and J.E.S. A.S., J.E.S., and G.D.E. wrote the manuscript with input from A.D.B. All co-authors contributed to the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
E.L. and A.-B.S. are employees of Cryos International Sperm and Egg Bank. S.M. and P.C. performed this work while employees of California Cryobank/CooperSurgical, Inc. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Anne Goriely and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Shoag, J.E., Srinivasa, A., Loh, C.A. et al. Direct measurement of the male germline mutation rate in individuals using sequential sperm samples. Nat Commun 16, 2546 (2025). https://doi.org/10.1038/s41467-025-57507-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-57507-0
