
Abstract
Cyclin-dependent kinase 1 (CDK1) is the pivotal kinase responsible for initiating cell division. Its activation is dependent on binding to regulatory cyclins, such as CCNB1. Our research demonstrates that copper binding to both CDK1 and CCNB1 is essential for activating CDK1 in cells. Mutations in the copper-binding amino acids of either CDK1 or CCNB1 do not disrupt their interaction but are unable to activate CDK1. We also reveal that CCNB1 facilitates the transfer of copper from ATOX1 to CDK1, consequently activating its kinase function. Disruption of copper transfer through the ATOX1-CCNB1-CDK1 pathway can impede CDK1 activation and halt cell cycle progression. In summary, our findings elucidate a mechanism through which copper promotes CDK1 activation and the G2/M transition in the cell cycle. These results could provide insight into the acquisition of proliferative properties associated with increased copper levels in cancer and offer targets for cancer therapy.
Similar content being viewed by others


Introduction
Copper is an essential trace metal element for all living cells. It serves as a catalytic cofactor for enzymes involved in antioxidant defense, iron absorption, cellular respiration, and connective tissue maturation1,2. Substantial evidence indicates that cancerous cells require a higher level of copper compared to healthy cells3,4,5. Increased intracellular copper is considered to be closely associated with abnormal cell proliferation. Copper has been shown to act as a cofactor for mitochondrial cytochrome c oxidase, promoting ATP supply required for rapid cell division, as well as for ULK1/2, which activates autophagy for tumor survival and growth6,7. Notably, copper can activate MEK1 to promote BRAFV600E-mediated MAPK signaling8 and activate PDK1 to promote the PI3K-AKT pathway in tumor cells9. These signaling pathways have been recognized for their importance in cell proliferation by promoting cell cycle progression from the G1 to S stage10,11,12, highlighting the involvement of elevated copper in the cell cycle.
Cyclin-dependent kinase 1 (CDK1) is a conserved Ser/Thr protein kinase subunit, whose activation is essential for G2 to mitosis (G2/M) transition during cell cycle progression13,14,15. The activation of CDK1 relies on its physical association with the regulatory subunit CCNB1 (or cyclin B1)16,17,18. CCNB1 binding precedes and is required for the activating phosphorylation of Thr161 (T161) residue by CDK1 activating kinase (CAK) and specifies the complex translocation into nucleus19,20. Interestingly, recent reports indicate that copper ions are predominantly located in the perinuclear and nucleus areas during mitotic stages and can be converted to monovalent copper by the histone H3-H4 tetramer21,22. Elevated copper levels enhance the expression of CCNB1, CDC2, and genes associated with chromosome structure23,24. Copper deficiency leads to a substantial increase in nuclear DNA content25. These findings suggest the relevance of copper in the regulation of mitosis. In this study, we discovered that copper is essential for CCNB1-dependent CDK1 kinase activation, thereby triggering the G2/M progression in the cell cycle.
Results
Copper is crucial for cell cycle G2/M progression
To investigate the role of copper in cell mitosis, we knocked down CTR1 in HepG2 and HEK293T cells (Supplementary Fig. 1a), and synchronized the cells to the S stage using thymidine (TdR) (Supplementary Fig. 1b). The results revealed that CTR1 interference caused the majority of HepG2 or HEK293T cells to remain in the G2/M stage (Fig. 1a; Supplementary Fig. 1c). We then established HepG2 and HEK293T cell lines that stably expressed the FUCCI system, a cell cycle reporter26, in which a bright green fluorescence signal represents cells in the G2/M phase and a magenta signal indicates cells entering the G1 phase (Fig. 1b). Consistent with the flow cytometric data mentioned above, knockdown of CTR1 led to the arrest of the majority of cells at the G2/M stage, as shown by the bright green cells (Fig. 1c; Supplementary Fig. 1d). Additionally, introducing a defective mutant of the copper-binding sites of CTR1 (CTR1M154A) into HepG2 cells with silenced endogenous CTR1 also arrested numerous cells at the G2/M stage (Fig. 1d, e). Consistent with these findings, treatment of HepG2 cells with demetallation or copper chelation using tetrathiomolybdate (TTM, 50 μM) or tetraethylenepentamine (TEPA, 20 μM) resulted in the majority of cells being blocked at the G2/M stage; however, copper supplementation promoted the progression of these cells through G2/M stage in demetallized medium (Fig. 1f, g). These observations could be recapitulated in HEK293T cells (Supplementary Fig. 1e, f). Interestingly, the addition of zinc (Zn), iron (Fe), manganese (Mn), nickel (Ni), cobalt (Co), molybdenum (Mo), or magnesium (Mg) ions did not rescue the G2/M arrest of HepG2 cells cultured in a metal-free medium (Supplementary Fig. 1g). These findings indicate that copper plays a crucial role in cell cycle G2/M progression.

a Flow cytometric analysis of the impact of copper on cell cycle progression in CTR1 knocked-down HepG2 cells. After synchronization to the S phase by 2.5 mM TdR, cells were released and cultured for 12 h. The percentages of different cell cycle stages are quantified on the right. The same treatment condition was used hereafter, if not specifically noted. b The schematic representation of FUCCI reporter system. Magenta: G1 phase; bright green: G2/M phase. c FUCCI reporter assay assessing the cell cycle progression of CTR1 knocked-down HepG2 cells. Scale bars are 50 μm. d, e Examination of the effect of copper-binding deficiencies of CTR1 on cell cycle progression using flow cytometry and FUCCI assay. Scale bars are 50 μm. f, g Flow cytometry (f) and FUCCI reporter assay (g) analyzing copper’s impact on cell cycle progression of HepG2 cells under different treatments. The cells were synchronized to the S phase using TdR and cultured in metal-deprived medium without or with copper (-Metal or Cu, respectively), or cultured in regular medium containing 20 μM TEPA or 50 μM TTM for 12 h. Scale bars represent 50 μm. In (a, d, f), data are representative of three biologically independent experiments. Source data are provided as a Source Data file.
Copper binding is essential for CDK1 activation and G2/M progression
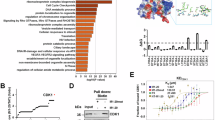
To investigate the mechanism through which copper regulates G2/M phase progression, we synchronized HepG2 cells to the G2 stage and performed protein mass spectrometry analysis to identify potential copper-binding proteins (CBPs) in the nucleus and cytoplasm (Supplementary Fig. 2a). Through an intersection analysis of the identified copper binding proteins and the proteins involved in the regulation of G2/M progression in the AmiGO database, we found that CDK1 was enriched in both the nucleus and cytoplasm samples (Fig. 2a, b). Immunoblot analysis of the proteins pulled down by copper-beads from nuclear and cytoplasmic extracts confirmed CDK1 as a copper-binding protein (Fig. 2c). When purified recombinant proteins were tested by pulldown and isothermal titration calorimetry (ITC) assay, CDK1 exhibited a copper-binding capability (Fig. 2d; Supplementary Fig. 2b). Copper typically forms coordination bonds with amino acids such as cysteine (C), histidine (H), and methionine (M)27. To analyze the copper-binding region of CDK1, we used the web-based server of MIB to predict its potential Cu binding sites28. In the predicted copper-binding region of CDK1, we individually mutated each vulnerable amino acid to alanine (A) and purified the recombinant proteins from bacteria to perform the pulldown assay (Supplementary Fig. 2c, d). In copper-pulldown assays, CDK1-H23A, M85A, and H120A mutants, but not others, showed a significantly reduced copper-binding ability compared to CDK1 WT (Fig. 2e), leading to the generation of a copper-binding deficient mutant, CDK1 CBM. In Inductively Coupled Plasma Optical Emission Spectrometry (ICP-OES) analysis, CDK1CBM exhibited a virtually lost copper-binding affinity compared to its wild-type (WT) protein counterpart (Fig. 2f).

a Intersection analysis of copper-binding proteins (CBPs) from G2-stage HepG2 cells and G2/M progression regulatory proteins compiled from the AmiGO database. Lysates from the cytoplasm or nuclei of HepG2 cells synchronized to the G2 phase were examined using mass spectrometry (MS) after copper-bead pulldown. b The filtered results are charted for copper-beads enriched proteins. c Pulldown assay to verify copper binding to CDK1 within the nuclear and cytoplasmic compartments of HepG2 cells. d In vitro pulldown assay to assess the copper-binding affinity of purified GST-CDK1, using GST-ATOX1 and GST as positive or negative controls, respectively. e Pulldown assay gauging copper binding to GST-CDK1 wild-type (WT) or its copper-binding deficient mutants. f ICP-OES-quantified copper-binding capacities of 10 μg purified recombinant CDK1WT and CDK1CBM proteins. (P-values indicated on top). g, h Examination of the effects of copper-binding deficiencies of CDK1 on cell cycle progression using flow cytometry and FUCCI assay after transfecting cells with CDK1 WT or CDK1CBM expression plasmids. The cells were synchronized to the S phase and then treated with 20 μM copper in the CBM groups, followed by flow cytometric analysis (g) or FUCCI-based assays (h). The same treatment condition was used hereafter, if not specifically noted. Scale bars represent 50 μm. i Analysis of H1.4 and p53 phosphorylation in cells with inducible CDK1 knockdown and transfected by HA-CDK1WT or HA-CDK1CBM. j Molecular modeling analysis of CDK1 protein structures to visualize the differences in conformation between WT and CBM mutants. k IP assay to examine interactions of transfected CDK1 WT or CBM mutant with endogenous CCNB1. In (f), unpaired two-tailed Student’s t-test was used for statistical analysis. In (f, g), data are representative of three biologically independent experiments. Source data are provided as a Source Data file.
To determine the role of copper binding to CDK1 in G2/M progression, we ectopically expressed CDK1WT and CDK1CBM in HepG2 and HEK293T cells while simultaneously knockdown of endogenous CDK1 was induced (Supplementary Fig. 2e). Introducing CDK1CBM resulted in the arrest of numerous synchronized cells at the G2/M phase, which could not be rescued by the addition of copper (Fig. 2g, h; Supplementary Fig. 2f, g).
As CDK1 is a key regulator of cell progression through the G2 phase and mitosis13,14,15, we examined the effects of copper on CDK1 kinase activity in HepG2 and HEK293T cells synchronized to the G2 stage using colchicine (Supplementary Fig. 2h). The cells were then cultured in the presence of copper or its chelator, TTM. Copper significantly enhanced the phosphorylation of p53 at S315 and H1.4 at T17 (Supplementary Fig. 2i), both of which are mediated by CDK129,30. In contrast, TTM exhibited clear opposite effects (Supplementary Fig. 2i). However, TTM and copper did not change the activated phosphorylation of CDK1 at T161, or the inhibitory phosphorylation at Tyr15 (Supplementary Fig. 2j). Consistently, cells with inducible knockdown of endogenous CDK1 and expressing HA-CDK1CBM showed significantly decreased phosphorylation of p53 and H1.4 compared to cells expressing ectopic HA-CDK1WT. Importantly, this effect could not be rescued by the addition of copper (Fig. 2i).
To determine whether mutations in the copper-binding sites of CDK1 cause significant alterations in protein structure or thermal fluctuations, we conducted molecular modeling analyses. The results showed that the point mutations in CDK1CBM did not significantly alter the secondary or tertiary structures or create detectable thermal fluctuations (Fig. 2j; Supplementary Fig. 2k). The thermal fluctuations level of each residue’s Cα in these regions of CDK1 revealed no significant changes in flexibility or rigidity, particularly in regions essential for CDK1 to carry out its kinase activity (Supplementary Fig. 2k). Similar results were obtained by comparing the average structure of the converged period for WT CDK1 and its CBM mutant with DSSP (Supplementary Fig. 2l). Notably, with endogenous CDK1 knockdown, ectopically expressed HA-CDK1WT and HA-CDK1CBM showed similar binding affinity to endogenous CCNB1 (Fig. 2k). These results collectively indicate the crucial role of copper binding in CDK1 activation and G2/M progression in cells.
Copper activates CDK1 independently of CCNB1 in vitro, but not in cells
CCNB1, as a regulatory protein for CDK1, plays a crucial role in promoting its kinase activity16,17. Therefore, we investigated whether CCNB1 is involved in copper-mediated CDK1 activation. In our in vitro kinase assay, the presence of both CCNB1 and CDK1 enhanced the phosphorylation of p53 and H1.4, and this effect was significantly amplified by the addition of copper (Fig. 3a; Supplementary Fig. 3a). Interestingly, copper also increased the CDK1-mediated phosphorylation of these substrates in the absence of CCNB1 (Fig. 3a; Supplementary Fig. 3a). When we removed all metals from the purified proteins (apo-state), CCNB1 had no significant effect on CDK’s kinase activity. However, the addition of copper resulted in a similar increase in p53 and H1.4 phosphorylation in both the CDK1/CCNB1 and CDK1 alone samples (Fig. 3b; Supplementary Fig. 3b). These findings suggest that copper can activate CDK1 independently of CCNB1 in vitro. Based on these results, we explored whether copper directly regulates CDK1 kinase function. In vitro kinase assays, increasing copper concentration led to a responsive enhancement of CDK1-mediated phosphorylation of H1.4 and p53 (Fig. 3c; Supplementary Fig. 3c). However, this effect was largely attenuated by copper chelators TTM and TEPA in a dose-dependent manner (Fig. 3d; Supplementary Fig. 3d-f). Meanwhile, purified recombinant CDK1CBM did not respond to copper in promoting phosphorylation of H1.4 and p53 in vitro (Fig. 3e), providing evidence that copper can activate CDK1 activity independent of CCNB1 in vitro.

a Analysis of H1.4 phosphorylation by incubation with CDK1 and CCNB1 alone or in combination, with or without 5 μM copper. The densitometric analysis of protein phosphorylation levels is shown on the blot. b Analysis of H1.4 phosphorylation with the same conditions as (a) using apo-CCNB1 and apo-CDK1, with densitometric analysis depicted on the blot. c, d Assessment of H1.4 phosphorylation using in vitro kinase assay, with H1.4 incubated with a gradient concentration of copper (c) or TTM (d). e Examination of CDK1WT and CDK1CBM’s ability to phosphorylate H1.4 and p53 in vitro in the presence or absence of copper through western blot analysis. f Evaluation of H1.4 and p53 phosphorylation in CCNB1-silenced cells treated with increasing concentrations of copper. g Co-IP analysis of CDK1 and CCNB1 interaction in lysates of HepG2 cells with CDK7 knockdown by CRISPR-Cas9. h Analysis of H1.4, p53, and CDK1-T161 phosphorylation in sgCDK7 cells treated with 20 μM copper. In (a–d), data are representative of three biologically independent experiments. Source data are provided as a Source Data file.
Previous studies have shown that CCNB1 binding is necessary for CDK1 activation in living cells16. In our experiments, we observed a significant decrease in p53 and H1.4 phosphorylation upon CCNB1 knockdown, and copper supplementation was unable to restore their phosphorylation levels (Fig. 3f). These results suggest the indispensability of CCNB1 in copper-mediated CDK1 activation in cells. Additionally, previous research has highlighted the essential role of CDK7 in facilitating the stable binding of CCNB1 to CDK1 through the phosphorylation activation of CDK1 at T161 (CDK1T161)31. Therefore, when CDK7 was knocked down in cells, both CCNB1 binding to CDK1 and CDK1T161 phosphorylation were significantly diminished (Fig. 3g, h). Importantly, even with the addition of copper, we observed no reinstatement of phosphorylation of H1.4 and p53 (Fig. 3h). Similar results were observed in cells expressing the CDK1T161A mutant (Supplementary Fig. 3g, h), indicating that CCNB1 binding is indispensable for the copper-mediated activation of CDK1 kinase activity in a cellular environment.
CCNB1 acts as a mediator for copper-activated CDK1 during the G2/M stage
Although the copper-induced activation of CDK1 does not rely on CCNB1 in vitro, it requires CCNB1 in cells. To further understand the role of CCNB1 in copper-mediated CDK1 activation, we conducted an intersection analysis of the identified proteins and the proteins involved in the regulation of G2/M progression in the AmiGO database, which revealed that CCNB1 is a potential copper-binding protein (Fig. 2b). Subsequent experiments confirmed that CCNB1 exhibited copper binding capabilities when tested with cell nucleus and cytoplasm lysates, as well as purified recombinant proteins by pulldown or ITC (Fig. 4a, b; Supplementary Fig. 4a). We generated CCNB1 mutants, individually mutating the vulnerable amino acids in the copper-binding region to alanine (A) (Supplementary Fig. 4b-d), and observed that CCNB1-M380A, M389A, H397A, and H409A mutants exhibited significantly reduced copper binding ability (Fig. 4c; Supplementary Fig. 4e). By introducing mutations in all four residues, we generated a copper-binding deficient CCNB1 mutant (CCNB1CBM) (Fig. 4d). ICP-OES analysis confirmed that CCNB1CBM lost its ability to bind copper compared to the wild-type (WT) proteins (Fig. 4e).

a Pulldown assay assessing copper binding to CCNB1 in cell nucleus and cytoplasm lysates. b Pulldown assay examining copper binding to purified GST-CCNB1; using GST-ATOX1 and GST as positive and negative controls, respectively. c Immunoblot detection of copper binding to GST-CCNB1 WT and its CBM mutants of GST-CCNB1-F2. d Pulldown assay testing copper binding to WT or its CBM of CCNB1 FL with putative copper-binding residues replaced by alanines. e ICP-OES-quantified copper-binding capacities of 10 μg purified recombinant CCNB1WT and CCNB1CBM proteins (P-values indicated on top). f Molecular modeling analysis of CCNB1 protein structures, with blue and red ribbons representing WT and CBM mutants, respectively. g Analysis of H1.4 and p53 phosphorylation in CCNB1 knockdown cells transfected with WT or its CBM type of HA-CCNB1 plasmids. All cells were synchronized to the S phase and treated with copper. h, i Effects of copper-binding deficiencies of CCNB1 on cell cycle progression evaluated using flow cytometry and FUCCI assay. Scale bars represent 50 μm. j ICP-OES-quantified copper contents of CCNB1 and CDK1 samples alone and after their incubation. Apo- and Cu-states of purified CCNB1 and CDK1 alone were directly tested. Upon incubation, when Cu-CCNB1 and apo-CDK1 were separated by SEC and analyzed for copper contents. k Western blot analysis of in vitro GST-p53 phosphorylation when incubated with CCNB1 and CDK1 in their apo- or Cu-states, or in the indicated combinations. In (e), unpaired two-tailed Student’s t-test was used for statistical analysis. In (e, f), data are representative of three biologically independent experiments. Source data are provided as a Source Data file.
Molecular modeling analyses demonstrated that these copper-binding site mutations cause negligible structural alterations (Fig. 4f) or thermal fluctuations in CCNB1 (Supplementary Fig. 4f, g). Similarly, DSSP analysis also demonstrated that the mutations had negligible effect on the secondary structures of the CCNB1 (Supplementary Fig. 4h). Additionally, compared to CCNB1WT, Flag-CCNB1CBM exhibited comparable affinity to CDK1 when endogenous CCNB1 was silenced (Supplementary Fig. 4i), indicating that disruption of the copper-binding sites did not affect their interaction. Subsequent experiments with inducible knockdown of endogenous CCNB1 revealed that ectopic CCNB1WT, but not CCNB1CBM, restored reduced p53 and H1.4 phosphorylation (Fig. 4g). Furthermore, in cells synchronized to the late S phase and with endogenous CCNB1 knockdown, CCNB1CBM blocked the cell cycle at the G2/M phase, which could not be rescued by copper addition (Fig. 4h, i; Supplementary Fig. 4j). These results strongly support the requirement of copper-CCNB1 binding for CDK1 activation and the G2/M transition in cells.
Considering our data, it is plausible to suggest that CCNB1 may act on delivering copper to CDK1, thereby promoting its activation. To validate this hypothesis, we employed a copper transfer test between CCNB1 and CDK1 proteins as described previously32,33,34. The proteins were prepared in copper-free (apo-) or copper-bound (Cu-) states. After incubating Cu-CCNB1 and apo-CDK1 at 4 °C for 30 minutes, the proteins were separated using size exclusion chromatography (SEC), and ICP-OES was performed to determine the copper contents in the eluate (Supplementary Fig. 4k). Eluted CCNB1 showed significantly lower copper levels than the original Cu-CCNB1, while eluted CDK1 exhibited markedly increased copper content compared to apo-CDK1 (Fig. 4j). In in vitro kinase assays, the combination of Cu-CCNB1 and apo-CDK1, similar to Cu-CDK1 alone, significantly increased p53 phosphorylation, whereas none of the assays with apo-CDK1, apo-CCNB1, or Cu-CCNB1 alone showed this kinase activity (Fig. 4k). These findings suggest that the physical interaction between CCNB1 and CDK1 facilitates copper transfer from CCNB1 to CDK1, with CCNB1 serving as a mediator for copper-activated CDK1. However, the source of copper on CCNB1 remains unidentified.
CCNB1 transfers copper from ATOX1 to CDK1, thereby activating its kinase activity and promoting G2/M progression
Copper enters the cells primarily through CTR1 and is then delivered to its targets by various copper chaperone proteins, such as ATOX1, CCS, and COX171. We performed co-IP studies to test the binding of Flag-CCNB1 to three HA-tagged copper chaperones, and found that ATOX1, but not CCS or COX17, was associated with CCNB1 (Fig. 5a). The interaction between CCNB1 and ATOX1 was further confirmed by in vitro protein pulldown assays (Fig. 5b; Supplementary Fig. 5a) and the co-IP assay of endogenous proteins (Fig. 5c). However, CDK1 did not show binding to ATOX1, CCS, or COX17 in co-IP tests and in vitro protein binding assays, suggesting that CDK1-associated copper is not transferred directly from these copper chaperones (Supplementary Fig. 5b, c). The subsequent bi-molecular fluorescence complementation (BiFC) analysis confirmed the interaction between ATOX1 and CCNB1 in a cellular environment (Fig. 5d; Supplementary Fig. 5d).

a IP analysis examining interactions of HA-tagged copper chaperone proteins with Flag-CCNB1 in HepG2 cells. In vitro pulldown assay using Ni agarose (b) and co-IP analysis (c) to evaluate CCNB1 and ATOX1 interaction. d BiFC analysis of CCNB1 and ATOX1 interaction. Scale bars represent 100 μm. e ICP-OES-quantified copper contents of purified proteins alone or after their incubations to assess the copper transfer. f Analysis of p53 phosphorylation after incubation with apo- or Cu-states of ATOX1, CCNB1, CDK1, or their different combinations as indicated. g, h Analysis of H1.4 and p53 phosphorylation in cells expressing shATOX1 (g) or expressing both shATOX1 and HA-ATOX1 WT or CBM (h), synchronized to the S phase and treated with copper. i, j Flow cytometry (i) and FUCCI (j) assay to assess the cell cycle progression of HepG2 cells with ATOX1 knockdown. k Mechanistic model of copper-promoting cell cycle progression by activating CDK1. In (i), data are representative of three biologically independent experiments. Source data are provided as a Source Data file.
Subsequently, we evaluated the potential of copper delivery from ATOX1 to CCNB1 or CDK1 using MEMO1 as a control to obtain Cu from ATOX133. Our experiments revealed that apo-CCNB1 incubated with Cu-ATOX1 showed marked copper increase compared to apo-CCNB1 alone (Fig. 5e). Conversely, Cu-ATOX1 incubation with CCNB1CBM or CDK1 did not lead to detectable copper loading to either protein, indicating that ATOX1 may directly deliver copper to CCNB1, but not to CDK1 (Supplementary Fig. 5e, f). Furthermore, Cu-ATOX1/apo-CCNB1/apo-CDK1 promoted p53 phosphorylation in vitro, similar to the effects of Cu-CCNB1/apo-CDK1 or Cu-CDK1 alone, while Cu-ATOX1/apo-CDK1 or other control groups did not show this activity (Fig. 5f). These findings suggested a copper transfer path of ATOX1-CCNB1-CDK1 to activate CDK1 kinase activity.
Subsequently, we evaluated the contribution of ATOX1 to CDK1 substrate phosphorylation in a cellular environment. ATOX1 knockdown or expression of the copper binding deficient mutant, ATOX1CBM, significantly decreased H1.4 and p53 phosphorylation in synchronized HepG2 cells (Fig. 5g, h). ATOX1 knockdown and introduction of ATOX1CBM led to the cells stalling at the G2/M phase (Fig. 5i, j; Supplementary Fig. 5g, h), which was consistent with a previous study35. This suggests the essential role of ATOX1-mediated copper delivery in CDK1 activation and G2/M transition. Taken together, we uncovered a copper transfer path involving ATOX1-CCNB1-CDK1, which promotes CDK1 kinase activity and drives G2/M progression in cancer cells (Fig. 5k).
Discussion
As a pivotal kinase and a master regulator of mitosis, CDK1 catalyzes the phosphorylation of several hundreds of mitotic proteins to coordinate cell cycle progression36. In this study, we unveiled a mechanism promoting CDK1 activation. Our findings reveal that CCNB1 acts as a copper transporter, facilitating the transfer of copper ions from its chaperone ATOX1 to CDK1. This copper acquisition leads to CDK1 activation and promotes G2/M cell cycle progression.
The involvement of metal ions in CDK1 kinase activity has been suggested in the investigation of CDC2 activity under EDTA treatment by Simanis and Nurse37. Among the potential metal ions, magnesium (Mg2+) has been considered due to its reported ability to stabilize ATP binding and enhance phosphotransferase activity of cAMP-dependent protein kinase (PKA) and CDK238,39,40. Interestingly, we observed that the loss of CDK1’s copper binding ability results in the inactivation of its function, and the restoration of G2/M progression in metal-deprived cells is specifically achieved through copper supplementation, not magnesium ions. These findings emphasize the crucial role of copper in CDK1 activation.
Using site-directed mutagenesis, this study identifies specific residues in CDK1 that are responsible for copper binding, underscoring the significance of these amino acids in CDK1 activity. However, the mutational data implies that residues are not located close to each other in the known structure for Cu binding. Copper binding to proteins typically occurs in regions with square planes, distorted octahedra, or tetrahedra geometries27,41. Therefore, the binding of copper to CDK1 may induce local structural changes in its domains, enhancing its functionality. Further investigation is needed to explore the structural aspects of this copper-induced modulation.
Previous research has emphasized the importance of CCNB1 in CDK1 activation through allosteric mechanisms17,18,42. However, our study reveals that CCNB1 serves as a copper transducer, facilitating the activation of CDK1 in cell. Cu activates CDK1 independent of CCNB1 in vitro, implicating its fundamental role in CDK1 activity. Unlike in vitro scenarios where copper can freely interact with CDK1, intracellular CCNB1 plays a crucial role in the controlled delivery of copper to CDK1. Meanwhile, the observation that copper is transferred from ATOX1 to CCNB1, as evidenced by their interaction, highlights the crucial role of this copper transfer pathway in cell mitosis. Furthermore, this study found that CDK1 binds to both Cu(I) and Cu(II) in vitro, both of which can affect its kinase activity. However, we do not yet have appropriate technologies to accurately determine the redox state of copper that activates CDK1 in a cellular environment. Considering that ATOX1 is mainly responsible for Cu(I) transfer in cells32,33, we speculate that the activity of cellular CDK1 may mainly depend on Cu(I).
This study provides valuable insights into the role of copper in cancerous cell mitosis. Substantial evidence has indicated that cancerous cells require a higher level of copper compared to healthy cells3,4,5. Simutaneously, high expressions of cyclin B1 and CDK1 are associated with more aggressive cancer phenotype and are considered major contributors to CDK1-driven G2/M transition in cancer cells43,44,45,46. Our finding reveals a direct link of increased intracellular copper to CDK1/CCNB1-driven cancerous cell mitosis. Disruption of copper transfer pathway via CTR1-ATOX1-CCNB1-CDK1 suppressed cancerous cell division. This finding could be confirmed by that knockout or knockdown of the main copper transporter CTR1 and the cytoplasmic/nucleic copper chaperone ATOX1 significantly inhibit tumor growth7,47,48,49.
In summary, our study provides innovative insights into the essential role of copper binding in CDK1 kinase activity, both in vitro and in cells. Furthermore, we reveal a function of CCNB1 as a copper mediator, facilitating CDK1 activation and promoting G2/M progression. These findings offer valuable knowledge for the design of CDK inhibitors that target copper incorporation and open new avenues for the development of effective strategies in cancer therapies.
Methods
Cell culture
HepG2 (CTCC-001-0014) and HEK293T (CTCC-001-0188) lines were purchased from Meisen CTCC. The cells were cultured in Gibco Dulbecco’s Modified Eagle Medium (DMEM, Gibco) supplemented with 10% fetal bovine serum (FBS, Meisen CTCC), 1% glutamine (Gibco) and 1% penicillin-streptomycin (Gibco) in a humidified atmosphere with 5% CO2 at 37 °C.
Chemicals and drugs
Tetrathiomolybdate (TTM), tetraethylenepentamine (TEPA), dimethyl sulfoxide (DMSO), cuprous chloride (CuCl), cupric Sulfate (CuSO4), magnesium chloride (MgCl2), zinc sulfate (ZnSO4), iron (II) sulfate (FeSO4), manganese chloride (MnCl2), nickel chloride (NiCl2), cobalt chloride (CoCl2) ions or ammonium molybdate ((NH4)2MoO4), isopropyl β-D-thiogalactoside (IPTG), colchicine, thymidine (TdR), and doxycycline (Dox) were also obtained from Sigma Aldrich, USA.
Plasmid construction, transfection and infection
The full-length human ATOX1, CTR1, CDK1, CCNB1, COX17 and CCS coding sequences were individually amplified using the cDNA mix from HepG2 or HEK293T cells and subcloned into the expression vector pcDNA3.1-3×HA. The coding sequences of the CDK1 and CCNB1 were subcloned into the overexpression vector pCDH-CMV-Flag (MiaoLing Plasmid Sharing Platform, China). Mutant forms of CDK1, CCNB1, CTR1 and ATOX1 expression plasmids were created by site-directed mutagenesis. The full-length human ATOX1, CDK1, or CCNB1 coding sequences were also subcloned into the pcDNA3.1-BiFC-A or pcDNA3.1-BiFC-B vectors, which were gifted by Dr. Ze Yu, Wenzhou Medical University. Tet-puro-pLKO-shCDK1, pLKO-shCCNB1 and pLKO-shCTR1 plasmids were created through subcloning of annealed specific primers (All sequences of oligonucleotides are listed in Supplementary Table 1). Cell transfection was carried out using Lipofectamine 2000 (Thermo Fisher Scientific, USA). CDK7 knockdown cells were generated using CRISPR-Cas9 technology. Lentiviral particles were packaged used to infect HEK293T or HepG2 cells. After 24 h, the media were replaced with fresh media containing puromycin (2 μg/mL), depending on employed lentiviral vectors.
Western blot analysis
Cells were lysed with cold RIPA buffer (Beyotime, China) containing a protease inhibitor cocktail (Bimake, USA) for 10 min. Protein concentration was determined using a BCA protein assay reagent (Beyotime, China). Equal amounts of total protein extract were loaded and separated by SDS-PAGE and transferred to nitrocellulose membranes. The membranes were then blocked with 5% non-fat milk or BSA and incubated with primary antibodies (All antibodies are listed in Supplementary Table 2) overnight at 4 °C with primary antibodies. After washing with TBST, the membranes were incubated with horseradish-peroxidase-conjugated secondary antibodies (1: 3000, Sigma Aldrich, USA). Immunoblotted protein bands were visualized using chemiluminescent substrate (ECL, Tanon™) and imaged using the Tanon Chemiluminescent Imager (Tanon, China).
Co-immunoprecipitation (co-IP)
HepG2 cells were lysed using Pierce IP Lysis Buffer (Thermo Scientific, USA) according to the manufacturer’s instructions. Primary antibodies (1-10 μg) were incubated with Dynabeads™ magnetic beads (1.5 mg) using the Dynabeads™ Protein G Immunoprecipitation Kit (Invitrogen, USA). The antigen-containing sample (100-1,000 µL) was added and incubated at room temperature for 10 min to allow binding. The beads-antibody-antigen complex was washed with washing buffer and analyzed by western blot.
Immunoprecipitation (IP) analyses
Cultured cells were transfected with HA-CDK1 WT or CBM plasmid and Flag-CCNB1 WT or CBM plasmid. Stable cell lines expressing Flag-CDK1 or CCNB1 were generated first and then transfected by HA-ATOX1, CCS, or COX17 plasmids. After 48 h, the cells were washed with pre-chilled PBS and lysed with BeyoLytic™ Mammalian Active Protein Extraction Reagent (Beyotime, China). The cell lysate was centrifuged, and the supernatant was incubated with anti-Flag or anti-HA magnetic beads (Bimake, USA) overnight at 4 °C. The beads were washed and then analyzed by western blot.
Recombinant protein purification
The pGEX-based vectors or His-based vectors carrying GST-CDK1, GST-CDK1 mutants, GST-CCNB1 WT (or FL), F1 and F2, GST-CCNB1 mutants, GST-ATOX1, GST-p53, GST-H1.4, His-CCNB1 and His-ATOX1 were expressed transformed into BL21 (DE3) E. coli. The bacteria were cultured and induced with IPTG to express the proteins at 16 °C. The recombinant proteins were purified using Glutathione resin (GenScript) or High Affinity Ni-Charged Resin (GenScript).
In vitro copper binding
Purified GST-CDK1 or its mutants, GST-CCNB1 WT, F1, F2 or its other mutants and GST-ATOX1 proteins (10 μg) were incubated with 50 μL Profinity IMAC resin (Bio-Rad, USA) charged with or without 100 mM copper (CuSO4) at 4 °C for 2 h. HepG2 cells were lysed, and the cell lysate was incubated with Profinity IMAC resin charged with or without copper at 4 °C. The precipitates were collected and analyzed by western blot50.
In vitro kinase assays
The 0.6 μg recombinant GST-p53 or GST-H1.4 and 1.4 μg GST-CDK1 or its mutants were incubated in 180 μL of kinase buffer (2 mM dithiothreitol (DTT), 25 mM Tris-HCl pH 7.5, 0.5 mM Na3VO4, 25 mM β-glycerol phosphate (β-GP), 120 μM ATP with or without 20 mM MgCl2) for 30 min at 22 °C. The reaction was conducted in the presence or absence of 2.5 molar equivalents of copper (CuSO4) with a six-step titration of TTM ranging from 0-100 μM or TEPA ranging from 0 to 50 μM, and a fixed concentration of 5 μM Cu. The mixed sample was analyzed by western blot using p53, p-p53, H1.4, p-H1.4, CDK1 and CCNB1 antibodies.
In vitro protein pulldown assay
Recombinant fusion proteins of GST-ATOX1, His-CCNB1, GST-CDK1 and His-ATOX1 were purified as previously described. Approximately 100 µg of GST-Vector or GST-ATOX1 was incubated with His-CCNB1, or His-SUMO control and His-CCNB1 was incubated with GST-ATOX1, or His-SUMO control and His-ATOX1 was incubated with GST-CDK1 in a 1 mL binding buffer system (50 mM Tris pH 7.5, 100 mM NaCl, 0.25% Triton X-100, 35 mM β-ME) for 4 h at 4 °C. Subsequently, 40 µL of pre-equilibrated glutathione agarose or Ni agarose was added, and the samples were incubated together at 4 °C for 2-4 h with gentle rocking. The samples were then centrifuged at 200 g for 5 min, washed 5 times, and then analyzed by western blot.
Isothermal titration calorimetry (ITC)
ITC experiments were performed with an ITC200 instrument (MicroCal). In a typical run, 20 automated injections of 2 µL CuSO4 with 120 s breaks between injections were made at 25 °C and 1,000 rpm stirring speed in a low feedback mode. The protein concentration was 10 µM, while the CuSO4 concentration in the syringe was 150 µM. The buffer was PBS, and the buffers for the syringe Cu and the protein sample were identical. Data integration, fitting and evaluation were performed using the software OriginTM 7 with the ITC200 plugin provided by MicroCal/Malvern Panalytical.
Bimolecular fluorescence complementation (BiFC) assay
To explore the interaction between ATOX1 and CCNB1 or CDK1, BiFC assays were performed51. HepG2 cells were transfected by BiFC constructs encoding the N- and C-terminal Venus fused to the CCNB1, CDK1 or ATOX1 genes. Positive and negative controls were transfected in parallel. After 24 h, the cells were washed, stained with Hoechst, and observed under a fluorescence microscope (Leica DMI3000B, Germany).
Cell cycle synchronization assay
For S phase synchronization, HEK293T cells were treated with 2.5 mM TdR for 24 h (first block), and then incubated with fresh medium for 9 h (first release). Subsequently, cells were subjected to another round of TdR treatment (second block). HepG2 cells were treated with 2.5 mM TdR for 28 h, followed by incubation with fresh medium for 3 h. Additionally, HepG2 and HEK293T cells were treated with 0.7 or 1 μg/mL colchicine for 7 h to synchronize the cells to G2 phase.
Flow cytometric analysis
Cells in S phase were treated with 20 μM Cu, Zn, Fe, Mn, Ni, Co, Mo or Mg salt, 20 μM TEPA, or 50 μM TTM after replacing the medium. The cells of all treatment groups (expressing CDK1 WT or CBM in with inducible CDK1 knockdown, CCNB1 WT or CBM with CCNB1 knockdown, ATOX1 WT or CBM with ATOX1 knockdown, CTR1 WT or CBM with CTR1 knockdown, shNC or shATOX1 and shNC or shCTR1) were individually collected and tested by a DNA Content Quantitation Assay Kit (Solarbio), and then subjected to cell-cycle analysis using a flow cytometer (Beckman, USA). In addition, synchronized cells in G2 phase were also collected and subjected to the same flow cytometer for cell-cycle analysis.
Fluorescent ubiquitination-based cell cycle indicator (FUCCI) assay
The FUCCI assay26 was employed to visualize cell cycle phases of HepG2 and HEK293T cells. PCR amplification was conducted from the pBOB-EF1-FastFUCCI template, followed by homologous recombination with the LV3 overexpression vector to generate plasmids of LV3-mKO2-hCdt1 (green) and mAG-hGem (magenta). After the lentiviruses were packaged, cells were infected with the lentiviral particle-containing medium to obtain cells incorporating the FUCCI reporter system. Subsequently, cell cycle synchronization was performed, followed by treatment with Cu, TTM and TEPA in the different cell lines, or transfection by CDK1 WT or CBM expressing plasmids into shCDK1 cells, or transfection by CCNB1 WT or CBM expressing plasmids into shCCNB1 cells, or transfection by CTR1 WT or CBM expressing plasmids into shCTR1 cells. The progression of the cell cycle phase was observed and photographed under a fluorescence microscope (DeltaVision).
Size exclusion chromatography (SEC)
SEC was performed using a Superdex™ 75 10/300 GL column (Cytiva) and an ÄKTA purifier (Cytiva)32. Purified proteins in DTT-PBS buffer were loaded onto a 5×4 mL desalting column (Biosharp) with ultrasound-degassed DTT-free buffer. To prepare the Cu-form proteins, the fully reduced DTT-free apo-CDK1, apo-CCNB1 WT or CBM, apo-MEMO1 and apo-ATOX1 proteins were immediately incubated with copper (CuCl) for 1 h. Excess copper was then removed using a desalting column. Both apo- and Cu-proteins were prepared using PBS at a concentration ratio of 6: 1 and volume ratio of 1: 1 and incubated at 4 °C for 30 min. Samples were injected using a 1 mL syringe and a 100 µL sample loop. The flow rate was set to 0.5 mL/min, and protein elution was monitored by absorption at 280 nm. Fractions were analyzed for copper contents using inductively coupled plasma-optical emission spectrometry (ICP-OES). A BACTRON™ Anaerobic/Environmental Chamber was used to establish anaerobic conditions, and O2 levels were measured using the Intell Instruments Pro AR8100 Oxygen Detector.
Inductively coupled plasma-optical emission spectrometry (ICP-OES)
10 µg of recombinant proteins was acidified by 2 mL of concentrated nitric acid and incubated at 120 °C for 4 h. After acidic digestion, samples were diluted with 2% nitric acid and analyzed for copper content using ICP-OES (Optima 8300) in kinetic energy discrimination mode against a calibration curve of known copper concentrations.
Molecular dynamics (MD)
MD simulations were conducted for CDK1, CCNB1 and their corresponding mutants. The GROMACS MD package52 (version 2018) and the AMBER99SB-ILDN force field53 were employed for all MD simulations. Four cubic boxes with native and mutated CDK1 (9 × 9 × 9 nm3) or CCNB1 (8 × 8 × 8 nm3) that were obtained from PDB ID 4Y72 were initially constructed and filled with TIP3P water models. CDK1 and CCNB1 molecules carried +1e and +3e net charges, respectively, and thus 1Cl- and 3Cl- were accordingly inserted as counterions for CDK1, CCNB1 and their mutants. A cutoff distance of 1.0 nm was set for short-range electrostatic and van der Waals interactions. Long-distance electrostatic interactions were calculated using the Particle Mesh Ewald (PME) method54. The LINCS algorithm was employed for covalent bond length constraints. The V-rescale thermostat and C-rescale barostat were utilized for temperature and pressure couplings, respectively. Energy minimizations were performed using the steepest descent algorithm. Subsequently, the systems were equilibrated in NPT ensembles for 0.1 ns (T = 298 K and P = 1.0 bar), with the positional restraints applied to the protein molecules. A simulation of 100 ns was carried for each system after NPT equilibrium and the timestep for the simulations was 2 fs. The convergence of the system was determined according to the energy, the RMSD of the protein molecule and the fluctuation of the secondary structures. The trajectories of all proteins were analyzed and visualized with the GROMACS MD package (version 2018) and VMD software, respectively55. The initial and final configurations of CDK1 WT, CBM or CCNB1 WT, CBM are shown in Supplementary Data 1.
Mass spectrometry (MS) and data analysis
HepG2 cells were synchronize to the S phase and then released into the G2 phase after 3 h. Subsequently collected cells were performed nuclear and cytoplasmic separation with the NE-PERTM Nuclear and Cytoplasmic Extraction Reagent Kit (Beyotime, China), followed by binding to a Cu-charged resin for 2 h at 4 °C. The protein was then eluted and the lysate was subjected to SDS-PAGE. The strip was cut into 1 mm3 cubes, followed by in gel enzymatic digestion (trypsin was purchased from Sigma of a proteomics grade, protein: trypsin ratio = 20: 1) and desalination of the resulting peptide using a C18 column, redissolved in 0.1% formic acid solution (Thermo Scientific, USA). Redissolved samples were injected into an Easy column (SenSil C18-AQ, Fresh Bioscience, China) and separated using the Easy nLC 1200 NL HPLC liquid phase system (Thermo Scientific, USA). Mobile phase A consisted of a 0.1% formic acid aqueous solution, while mobile phase B was an 80% acetonitrile solution (Thermo Scientific, USA) containing 0.1% formic acid. The samples were gradient-elution at 250 nL/min for 75 min, and then analyzed by mass spectrometry using Q-Exactive HF (Thermo Scientific, USA). The ion source voltage was set to 2.4 kV, and both the precursor ions and their secondary fragments were detected and analyzed using the high-resolution Orbitrap. The MS1 scans at 300-1650 m/z was acquired in the Orbitrap XL at 45,000 resolution setting; MS2 scans at 100 m/z was obtained in the Orbitrap XL at 30,000 resolution setting. The data acquisition mode used the data-dependent acquisition (DDA) procedure. Briefly, after the MS1 scans, the first 20 peptide precursor ions with the highest signal intensity were selected to enter the HCD collision pool for fragmentation with 32% of the fragmentation energy, and MS2 scans was also performed in sequence. To enhance the effective utilization of the mass spectrometer, the automatic gain control (AGC) was set to 2e5, with a maximum injection time of 100 ms, and the dynamic exclusion time of the tandem mass spectrometry scan was set to 30 s to avoid repeated scanning of precursor ions. In addition, the ion transfer tube temperature was 320°C with an isolation window of 1.4 m/z.
All raw files were searched with Proteome Discoverer (v. 2.2 Thermo Fisher Scientific) against a database of human proteins (Uniprotkb_taxonomy_id_9606_2024_09_20.fasta, from March 13, 2018, containing 204,411 sequences). IT-generated HCD and CID spectra were searched using the Sequest HT search algorithm with 10 ppm and 0.6 Da mass tolerances for the precursor and fragment ions, respectively. Similarly, high-resolution OT-generated HCD spectra were searched with a 10 ppm and 0.05 Da mass tolerance. The database search was limited to only fully tryptic peptides, with a maximum of two missed cleavages and a minimum of 6 amino acid residues for the peptide sequence length using the SEQUEST: Xcorr as the scoring software. As a variable modification, oxidation (M) and acetyl (K) were selected, and the quantitative method used label-free quantification. At least one high-confidence peptide sequence was required for protein identifications, and the data presented in this study are at the 1% false discovery rate (FDR) level for proteins and peptides. Data were derived from three biologically independent experiments.
Statistical analysis
For all experiments, unless otherwise indicated, the sample sizes (n) consisted of three biologically independent experiments. Data were presented as means ± standard error of mean (SEM). Unpaired two-tailed Student’s t-test was performed for statistical analysis in the Microsoft Excel or GraphPad Prism 7 software. Protein quantification was performed using Image J. Values of P < 0.05 were considered statistically significant.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The raw files were searched with a database of human proteins (Uniprotkb_taxonomy_id_9606_2024_09_20.fasta, from March 13, 2018, containing 204,411 sequences, https://www.uniprot.org/taxonomy/9606). The structural information of CDK1 and CCNB1 proteins were obtained from Protein Data Bank (PDB ID 4Y72). The mass spectrometry data generated in this study have been deposited in the ProteomeXchange Consortium via the PRIDE database and can be obtained with accession code PXD054518. Source data are provided with this paper.
References
Kim, B. E., Nevitt, T. & Thiele, D. J. Mechanisms for copper acquisition, distribution and regulation. Nat. Chem. Biol. 4, 176–185 (2008).
Chen, L., Min, J. & Wang, F. Copper homeostasis and cuproptosis in health and disease. Signal Transduct Target Ther 7, 378 (2022).
Tang, D., Kroemer, G. & Kang, R. Targeting cuproplasia and cuproptosis in cancer. Nat. Rev. Clin. Oncol. 21, 370–388 (2024).
Gupte, A. & Mumper, R. J. Elevated copper and oxidative stress in cancer cells as a target for cancer treatment. Cancer Treat. Rev. 35, 32–46 (2009).
Ge, E. J. et al. Connecting copper and cancer: from transition metal signalling to metalloplasia. Nat. Rev. Cancer 22, 102–113 (2022).
Ishida, S., Andreux, P., Poitry-Yamate, C., Auwerx, J. & Hanahan, D. Bioavailable copper modulates oxidative phosphorylation and growth of tumors. Proc. Natl. Acad. Sci. USA 110, 19507–19512 (2013).
Tsang, T. et al. Copper is an essential regulator of the autophagic kinases ULK1/2 to drive lung adenocarcinoma. Nat. Cell Biol. 22, 412–424 (2020).
Brady, D. C. et al. Copper is required for oncogenic BRAF signalling and tumorigenesis. Nature 509, 492–496 (2014).
Guo, J. et al. Copper Promotes Tumorigenesis by Activating the PDK1-AKT Oncogenic Pathway in a Copper Transporter 1 Dependent Manner. Adv. Sci (Weinh). 8, e2004303 (2021).
Hume, S., Dianov, G. L. & Ramadan, K. A unified model for the G1/S cell cycle transition. Nucleic Acids Res 48, 12483–12501 (2020).
Meloche, S. & Pouysségur, J. The ERK1/2 mitogen-activated protein kinase pathway as a master regulator of the G1- to S-phase transition. Oncogene 26, 3227–3239 (2007).
Chambard, J. C., Lefloch, R., Pouysségur, J. & Lenormand, P. ERK implication in cell cycle regulation. Biochim. Biophys. Acta 1773, 1299–1310 (2007).
Gautier, J., Matsukawa, T., Nurse, P. & Maller, J. Dephosphorylation and activation of Xenopus p34cdc2 protein kinase during the cell cycle. Nature 339, 626–629 (1989).
Draetta, G. & Beach, D. Activation of cdc2 protein kinase during mitosis in human cells: cell cycle-dependent phosphorylation and subunit rearrangement. Cell 54, 17–26 (1988).
Santamaría, D. et al. Cdk1 is sufficient to drive the mammalian cell cycle. Nature 448, 811–815 (2007).
Labbé, J. C. et al. MPF from starfish oocytes at first meiotic metaphase is a heterodimer containing one molecule of cdc2 and one molecule of cyclin B. EMBO J 8, 3053–3058 (1989).
Evans, T., Rosenthal, E. T., Youngblom, J., Distel, D. & Hunt, T. Cyclin: a protein specified by maternal mRNA in sea urchin eggs that is destroyed at each cleavage division. Cell 33, 389–396 (1983).
Strauss, B. et al. Cyclin B1 is essential for mitosis in mouse embryos, and its nuclear export sets the time for mitosis. J. Cell Biol. 217, 179–193 (2018).
Li, J., Meyer, A. N. & Donoghue, D. J. Nuclear localization of cyclin B1 mediates its biological activity and is regulated by phosphorylation. Proc. Natl. Acad. Sci. USA 94, 502–507 (1997).
Hagting, A., Jackman, M., Simpson, K. & Pines, J. Translocation of cyclin B1 to the nucleus at prophase requires a phosphorylation-dependent nuclear import signal. Curr. Biol. 9, 680–689 (1999).
McRae, R., Lai, B. & Fahrni, C. J. Subcellular redistribution and mitotic inheritance of transition metals in proliferating mouse fibroblast cells. Metallomics 5, 52–61 (2013).
Attar, N. et al. The histone H3-H4 tetramer is a copper reductase enzyme. Science 369, 59–64 (2020).
dos, Santos, N. V., Matias, A. C., Higa, G. S., Kihara, A. H. & Cerchiaro, G. Copper Uptake in Mammary Epithelial Cells Activates Cyclins and Triggers Antioxidant Response. Oxid. Med. Cell. Longev. 2015, 162876 (2015).
Huster, D. et al. High copper selectively alters lipid metabolism and cell cycle machinery in the mouse model of Wilson disease. J. Biol. Chem. 282, 8343–8355 (2007).
Renault, E. & Deschatrette, J. Alterations of rat hepatoma cell genomes induced by copper deficiency. Nutr. Cancer 29, 242–247 (1997).
Sakaue-Sawano, A. et al. Genetically Encoded Tools for Optical Dissection of the Mammalian Cell Cycle. Mol. Cell 68, 626–640.e5 (2017).
Tainer, J. A., Roberts, V. A. & Getzoff, E. D. Protein metal-binding sites. Curr. Opin. Biotechnol. 3, 378–387 (1992).
Lu, C. H. et al. MIB2: metal ion-binding site prediction and modeling server. Bioinformatics 38, 4428–4429 (2022).
Kreis, N. N. et al. Restoration of the tumor suppressor p53 by downregulating cyclin B1 in human papillomavirus 16/18-infected cancer cells. Oncogene 29, 5591–5603 (2010).
Arion, D., Meijer, L., Brizuela, L. & Beach, D. cdc2 is a component of the M phase-specific histone H1 kinase: evidence for identity with MPF. Cell 55, 371–378 (1988).
Larochelle, S. et al. Requirements for Cdk7 in the assembly of Cdk1/cyclin B and activation of Cdk2 revealed by chemical genetics in human cells. Mol. Cell 25, 839–850 (2007).
Chen, L. et al. APEX2-based Proximity Labeling of Atox1 Identifies CRIP2 as a Nuclear Copper-binding Protein that Regulates Autophagy Activation. Angew. Chem. Int. Ed. Engl. 60, 25346–25355 (2021).
Zhang, X. et al. Memo1 binds reduced copper ions, interacts with copper chaperone Atox1, and protects against copper-mediated redox activity in vitro. Proc. Natl. Acad. Sci. USA 119, e2206905119 (2022).
Grasso, M. et al. The copper chaperone CCS facilitates copper binding to MEK1/2 to promote kinase activation. J. Biol. Chem. 297, 101314 (2021).
Matson et al. Copper Chaperone Atox1 Interacts with Cell Cycle Proteins. Comput. Struct. Biotechnol. J. 16, 443–449 (2018).
Petrone, A., Adamo, M. E., Cheng, C. & Kettenbach, A. N. Identification of Candidate Cyclin-dependent kinase 1 (Cdk1) Substrates in Mitosis by Quantitative Phosphoproteomics. Mol. Cell. Proteomics 15, 2448–2461 (2016).
Simanis, V. & Nurse, P. The cell cycle control gene cdc2+ of fission yeast encodes a protein kinase potentially regulated by phosphorylation. Cell 45, 261–268 (1986).
Knape, M. J. et al. Divalent metal ions control activity and inhibition of protein kinases. Metallomics 9, 1576–1584 (2017).
Hallenbeck, P. C. & Walsh, D. A. Autophosphorylation of phosphorylase kinase. Divalent metal cation and nucleotide dependency. J. Biol. Chem. 258, 13493–13501 (1983).
De, Bondt, H. L. et al. Crystal structure of cyclin-dependent kinase 2. Nature 363, 595–602 (1993).
Adman, E. T. Copper protein structures. Adv. Protein Chem. 42, 145–197 (1991).
Brown, N. R. et al. CDK1 structures reveal conserved and unique features of the essential cell cycle CDK. Nat. Commun. 6, 6769 (2015).
Niméus-Malmström, E. et al. Cyclin B1 is a prognostic proliferation marker with a high reproducibility in a population-based lymph node negative breast cancer cohort. Int. J. Cancer 127, 961–967 (2010).
Aaltonen, K. et al. High cyclin B1 expression is associated with poor survival in breast cancer. Br. J. Cancer 100, 1055–1060 (2009).
Malumbres, M. & Barbacid, M. Cell cycle, CDKs and cancer: a changing paradigm. Nat. Rev. Cancer 9, 153–166 (2009).
Asghar, U., Witkiewicz, A. K., Turner, N. C. & Knudsen, E. S. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat. Rev. Drug Discov. 14, 130–146 (2015).
Jin, J. et al. Copper enhances genotoxic drug resistance via ATOX1 activated DNA damage repair. Cancer Lett 536, 215651 (2022).
Yu, Z. et al. Blockage of SLC31A1-dependent copper absorption increases pancreatic cancer cell autophagy to resist cell death. Cell Prolif 52, e12568 (2019).
Wang, J. et al. Inhibition of human copper trafficking by a small molecule significantly attenuates cancer cell proliferation. Nat. Chem. 7, 968–979 (2015).
Turski, M. L. et al. A novel role for copper in Ras/mitogen-activated protein kinase signaling. Mol. Cell. Biol. 32, 1284–1295 (2012).
Han, Y. et al. In vivo imaging of protein-protein and RNA-protein interactions using novel far-red fluorescence complementation systems. Nucleic Acids Res 42, e103 (2014).
Abraham, M. J. et al. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1, 19–25 (2015).
Lindorff-Larsen, K. et al. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 78, 1950–1958 (2010).
Essmann, U., Perera, L. & Berkowitz, M. L. The origin of the hydration interaction of lipid bilayers from MD simulation of dipalmitoylphosphatidylcholine membranes in gel and liquid crystalline phases. Langmuir 11, 4519–4531 (1995).
Humphrey, W., Dalke, A. & Schulten, K. VMD: visual molecular dynamics. J. Mol. Graph 14, 33–38 (1996).
Acknowledgements
We are grateful for the valuable advices and discussions from Dr. Yusheng Liu at Northeast Forestry University, and Dr. Falong Lu at The Institute of Genetics and Developmental Biology of the Chinese Academy of Sciences. We also acknowledge Dong Gao at Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences for revising the manuscript. This work was financially supported by the National Natural Science Foundation of China (32072801 and U22A20369 to C.B.T, 32302822 to H.F.Y, and 32172350 to Z.Z.), the Fundamental Research Funds for the Central Universities (2572023CT09 to C.B.T.).
Author information
Authors and Affiliations
Contributions
C.B.T. conceived and designed the study. J.W. performed experiments and data analysis with assistance from other authors. D.Y. conducted cell cycle and in vitro kinase assays. H.F.Y., W.R., and S.D. contributed to mass spectrometry analyses and experimental support. Z.G. performed the ITC tests in vitro. J.J. and Y.N. participated in the cell cycle analyses and SEC assays. S.Z. generated and characterized the CDK1 and CCNB1 knockdown cells and contributed to experimental work. Z.Z. assisted with the analysis of protein structure. J.W., H.F.Y., and C.B.T. drafted the manuscript, with input from other co-authors. X.M. and G.S. provided discussions and critical review of the manuscript. All authors reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, J., Yang, D., Yu, HF. et al. Copper is essential for cyclin B1-mediated CDK1 activation. Nat Commun 16, 2288 (2025). https://doi.org/10.1038/s41467-025-57538-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-57538-7
This article is cited by
-
Tumor cell cycle regulation: integrated perspective of stage characteristics, regulatory networks, and signaling pathway intervention strategies
Molecular Biomedicine (2026)
-
Balancing between cuproplasia and copper-dependent cell death: molecular basis and clinical implications of ATOX1 in cancer
Journal of Experimental & Clinical Cancer Research (2025)
