
Abstract
Missense mutations in the TP53 (p53) gene have been linked to malignant progression. However, our in-silico analyses reveal that hepatocellular carcinoma (HCC) patients with mutant p53 (mutp53) have better overall survival compared to those with p53-null (p53null) HCC, unlike other cancer types. Given the historical use of sorafenib (SOR) monotherapy for advanced HCC, we hypothesize that mutp53 increases sensitivity to SOR, a multikinase inhibitor that induces endoplasmic reticulum (ER) stress. Here we show that mutp53 inhibits stress granule (SG) formation by binding to an ER stress sensor, PKR-like ER kinase (PERK), and a key SG component, GAP SH3 domain-binding protein 1 (G3BP1), contributing to increased sensitivity of SG-competent cells and xenografts to ER stress inducers including SOR. Our study identifies a unique vulnerability imposed by mutp53, suggesting mutp53 as a biomarker for ER stress-inducing agents and highlighting the importance of SG inhibition for cancer treatment.
Similar content being viewed by others


Introduction
Understanding the mechanisms behind chemotherapy resistance is vital for improving current treatment strategies. Stress granules (SGs), cytoplasmic, liquid-liquid phase-separated, non-membranous cytoplasmic foci, consisting of ribonucleoprotein (RNP) and mRNAs, are implicated in chemotherapy resistance. SGs form when protein translation initiation is inhibited1. SGs enhance drug resistance in cancer by sequestering mRNAs or proteins involved in pro-death signaling, like TRAF2 and RACK12,3. SGs are also linked to cellular quiescence4. Moreover, inhibition of translation initiation, which triggers SG formation, can free up to 30% of cellular ATP, conserving cellular energy and promoting survival during genotoxic stress5. Additionally, phosphorylation of eukaryotic initiation factor 2α (eIF2α) uniquely enhances the translation of proteins like the transcription factor ATF4, promoting the mRNA expression of pro-survival genes involved in autophagy, amino acid metabolism, protein synthesis, and oxidative stress response6. However, little is known about SG regulation in cancer, and there are currently no strategies that leverage SG inhibition to increase the efficacy of cancer therapy.
Tumor suppressor p53 is the most frequently mutated gene in human cancers (~50%), and the majority of p53 mutations are missense mutations in the DNA binding domain7. Unlike other tumor suppressors, these missense mutations not only cause the loss of wild-type p53 (wtp53) functions but also confer oncogenic gain-of-function (GOF) activities, independent of wtp53, that generally enhance malignant progression and drug resistance8,9. Clinically, p53 mutations are linked to cancer malignancy, metastasis, and therapy resistance10. Given its unique properties and high expression in cancers, missense mutant p53 (mutp53) is an ideal therapeutic target. However, directly targeting mutp53 itself has proven challenging11. One promising alternative strategy is to inhibit the signaling pathways or proteins modified by mutp53 that are essential for cell survival12. Identifying and targeting these vulnerabilities could accelerate the development of novel and effective therapeutic strategies for tumors expressing mutp53.
Our in-silico analysis of The Cancer Genome Atlas (TCGA) dataset unexpectedly revealed that patients with hepatocellular carcinoma (HCC) carrying missense mutp53 had better overall survival compared to those with p53null HCC, distinct from other cancer types. While combination chemotherapies are commonly used to treat many cancers, advanced HCC has historically been treated with sorafenib (SOR) as a first-line, single-agent therapy since 2007, until recent antibody-based therapies were approved13. We therefore hypothesized that SOR, a multikinase inhibitor that induces ER stress14, might have unrecognized benefits for treating mutp53-expressing tumors. Our in vitro and in vivo studies show that mutp53 inhibits ER stress-induced SG formation, likely contributing to increased sensitivity to SOR.
Results
The potential benefit of p53 mutations in HCC for increased SOR sensitivity
Missense mutp53 expression in tumors is generally associated with poor biological outcomes in patients15. However, its oncogenic activities are often mutation type- or context-dependent16. To examine whether the presence of missense mutp53 in tumors impacts the overall survival of cancer patients compared to p53null tumors, we used the cBioPortal for Cancer Genomics to analyze available TCGA datasets. These datasets, collected since the year 2006, cover various cancer types, including ovarian serous cystadenocarcinoma, head and neck squamous cell carcinoma, pancreatic adenocarcinoma, colorectal adenocarcinoma, invasive breast cancer, and hepatocellular carcinoma (HCC)17. Tumors with missense mutations in the p53 DNA-binding domain were grouped as missense, while those with truncating (nonsense, frameshift, in/dels, splice site) mutations or deep deletions were grouped as p53null. Although tumors with missense mutp53 tended to confer worse overall survival than p53null cases, these results were not significant (Fig. 1a and Supplementary Fig. S1a). Surprisingly, missense mutp53 conferred better overall survival to HCC patients compared to p53null patients (Fig. 1a). This is consistent with a report showing that HCC patients whose tumors have a p53 stop-gain mutation (nonsense mutation resulting in p53null status) treated with SOR tend to experience disease progression. However, those with non-synonymous p53 mutations (missense mutp53) show some response to SOR or achieve stable disease, resulting in a better prognosis compared to those with p53null18. Given that SOR was the first-line systemic drug for treating advanced HCC from the year 2007 until recently, we hypothesized that missense mutp53 could increase HCC sensitivity to SOR as a non-canonical mutp53 activity. To test this hypothesis, we knocked down mutp53 in Huh7 (p53Y220C), SNU449 (p53A161T), and HepG2 (p53wt) HCC cells (Fig. 1b and Supplementary Fig. S1b–d), or overexpressed p53R175H-3XFLAG in p53null Hep3B HCC cells (Fig. 1c). These cells with and without mutp53 or wtp53 were treated with SOR or several common chemotherapy agents including vincristine (VCR), doxorubicin (DRB), or cisplatin (CDDP), followed by Annexin V/PI staining and flow cytometry. Intriguingly, mutp53-expressing Huh7, SNU449, and Hep3B cells showed increased apoptosis compared to p53-knockdown or -null cells, following SOR and VCR treatment, but little difference was observed in cells treated with DRB and CDDP (Fig. 1b, c and Supplementary Fig. S1b, c). Knockdown of wtp53 in HepG2 cells did not significantly affect cell viability following SOR treatment (Supplementary Fig. S1d). These results suggest that mutp53 cooperates with SOR to promote cytotoxicity in HCC cells. Cleaved caspase-3 (CC3) staining and colony formation assays also confirmed that SOR treatment induced greater apoptosis and more efficiently inhibited colony formation in mutp53-expressing SNU449 and Huh7 cells compared to mutp53-knockdown cells (Fig. 1d, e and Supplementary Fig. S1e, f). Notably, following DRB and CDDP treatment, SNU449 cells expressing mutp53 formed more colonies than mutp53-knockdown cells, which was distinct from the results observed with SOR treatment (Fig. 1e). These data suggest that mutp53 enhances the sensitivity of HCC cells to SOR and VCR, which could explain why patients with mutp53-positive HCC show better overall survival than those with p53null tumors.

a Kaplan-Meier curves of indicated TCGA datasets. Overall survival of patients with missense mutp53 and p53null tumors. P-values were calculated using logrank test. b, c Annexin V/PI staining and flow cytometry using Huh7 (p53Y220C) cells with and without p53 knockdown (shp53 targeting p53 exon 7/8) or Hep3B cells with and without 3xFLAG-tagged p53R175H overexpression, treated with vehicle, SOR (25 µM for Huh7, 8 µM for Hep3B), VCR (25 µM), DRB (6 µM for Huh7, 1 µM for Hep3B), or CDDP (6 µM for Huh7, 12 µM for Hep3B) for 48 h. Graphs showing the percent of apoptosis (Q2 + Q4) and total cell death (Q1 + Q2 + Q4) with representative WB for p53, and β-tubulin using untreated cells (n = 3). The molecular weight markers (kDa) are shown on the right. d Immunofluorescence for cleaved caspase-3 (CC3, red) and DAPI (blue) using SNU449 (p53A161T) cells with and without p53 knockdown, treated with 12 µM SOR for 48 h. Graph showing the number of CC3+ cells in 200 randomly selected cells following SOR treatment (n = 3). Scale: 100 µm. e Colony formation assays using control and mutp53-knockdown SNU449 cells. Colonies were treated with SOR (12 µM), DRB (1 µM), or CDDP (12 µM) for 48 h and stained with crystal violet 4 days later. Graphs showing a percentage of the number of colonies relative to that in the vehicle control (n = 3). All experiments in (b–e) are from three independent experiments (n = 3). Mean ± S.E.M., two-tailed Student’s t-test. ns not significant. Source data are provided as a Source Data file.
Mutp53 enhances the sensitivity of HCC and other types of cancer cells to ER stress
To test whether mutp53 could sensitize other cancers to SOR and VCR, we exposed p53R156P-expressing KHOS/NP osteosarcoma cells to several chemotherapy drugs. Both control and mutp53-knockdown KHOS/NP cells somehow showed intrinsic resistance to SOR, even at 100 µM, so we did not use SOR to treat this cell line. However, control KHOS/NP cells showed higher extent of apoptosis in response to VCR compared to mutp53-knockdown cells, but less or similar sensitivity to DRB and CDDP (Fig. 2a). Consistently, immunofluorescence for CC3 and colony formation assays revealed that mutp53-expressing KHOS/NP cells showed higher sensitivity to VCR than did mutp53-knockdown cells (Fig. 2b, c).

a WB for p53 and GAPDH and Annexin V/PI and flow cytometry using control vector and mutp53-knockdown (shp53) KHOS/NP cells, treated with 25 µM VCR, 3 µM DRB, or 1.5 µM CDDP for 72 h. A graph showing the percent of apoptosis and total cell death (n = 3). Immunofluorescence for CC3 using control and mutp53-knockdown KHOS/NP cells, treated with 25 µM VCR (b) or 1 µM TG (e) for 48 h. Graphs showing the number of CC3+ cells in 200 cells (n = 3). Scale: 100 µm. Colony formation assays using control and mutp53-knockdown KHOS/NP cells, treated with 25 µM VCR for 48 h (c) or increasing concentrations of TG for 1 h (f). Graphs showing a percentage of the number of colonies compared to that in the vehicle control (n = 3). Annexin V/PI and flow cytometry using KHOS/NP cells with and without mutp53 knockdown (d) and Hep3B cells with and without p53R175H overexpression (g), following treatment with 1 µM TG for 48 h. Graphs showing the percent of apoptosis and total cell death (n = 3). All experiments are from three independent experiments. Mean ± S.E.M., two-tailed Student’s t-test. ns not significant. Source data are provided as a Source Data file.
To understand why SOR and VCR unexpectedly induced apoptosis more efficiently in mutp53-expressing cells than in p53null cells, we performed a thorough literature search to identify any shared characteristics of these drugs. This search identified both SOR and VCR as agents capable of inducing ER stress through PERK activation14,19. To examine whether mutp53 could promote ER stress-mediated cell death, we treated control and mutp53-knockdown KHOS/NP cells with a canonical ER-stress inducer, thapsigargin (TG), followed by Annexin V/PI and flow cytometry (Fig. 2d), CC3 staining (Fig. 2e), or colony formation assays (Fig. 2f). Consistent with the results of VCR treatment, mutp53-expressing KHOS/NP cells had more apoptotic cells with reduced colony formation than did mutp53-knockdown cells following TG treatment. Similarly, overexpression of p53R175H in p53null Hep3B HCC cells and OSC19 head and neck cancer cells resulted in increased apoptosis following TG treatment (Fig. 2g and Supplementary Fig. S2a). Together, these findings suggest that mutp53 facilitates ER stress-mediated apoptosis in multiple cancer types.
Mutp53, but not wtp53, inhibits SG formation
ER stress promotes cell death or cell survival through many downstream signaling pathways20. The formation of SGs following ER stress is associated with cell survival and drug resistance in cancer cells1,21,22. We hypothesized that mutp53 could inhibit ER stress-mediated SG formation, subsequently enhancing apoptosis. We used G3BP1 as a SG marker, since it colocalized with other SG markers, including T-cell intracellular antigen 1 (TIA1), poly(A)-binding protein (PABP), and eukaryotic initiation factor 4E (eIF4E) in Huh7 and KHOS/NP cells, following ER stress (Supplementary Fig. S3a). We also compared SG detectability by three markers, G3BP1, eIF4E, and/or PABP, in Huh7 and SNU449 HCC cells, which was similar between these three markers following SOR, although eIF4E detected SGs in the vehicle control group at higher levels than G3BP1 did (Supplementary Fig. S3b). Additionally, we confirmed little or no SG formation in Huh7 cells treated with CDDP and DRB (Supplementary Fig. S3c). We then examined the SG-forming potential of various parental cancer cell lines, detected by G3BP1, following treatment with different ER stress inducers (Table 1). Intriguingly, some cell lines were competent to SG formation, while others were reluctant to SG formation (e.g., MDA-MB-231, H1299, CAL33, FaDu). Also, some cells formed SGs only when treated with specific ER stress inducers. This could be because SOR (multi-kinase inhibitor) and VCR (mitotic destabilizer) have distinct molecular targets from TG, which may alter SG-forming potential. Notably, both TOV112D and CAL33 cells express p53R175H; however, TOV112D cells formed SGs by TG and SOR, while CAL33 cells were not SG-competent. Thus, p53 status appeared not to be the determining factor of SG-forming potential of the cell. The SG-forming potential is likely influenced by various factors, including the drug’s mechanism of action, the cellular context, and intrinsic factors such as the expression of proteins involved in SG formation, drug trafficking, and drug metabolism.
Next, we investigated how mutp53 could impact the SG-forming potential of SG-competent cells by treating control and mutp53-depleted KHOS/NP, PLC/PRF/5 (p53R249S, HCC), and SNU449 cells with ER stress inducers. Knockdown or knockout of mutp53 significantly increased the number of SG-positive cells following ER stress (Fig. 3a, b, and Supplementary Fig. S3d, e). Knockdown of mutp53 in Huh7 cells by two different p53 shRNAs targeting p53 exon7/8 and 3’UTR also increased the SG formation following VCR and SOR treatment (Supplementary Fig. S3f).

Immunofluorescence for G3BP1 and DAPI using control and p53-knockdown KHOS/NP (a), PLC/PRF/5 (b), or HepG2 (c) cells treated with vehicle, TG (1 µM, 1 h, a), or SOR (25 μM, 2 h, b, c). Scale: 10 µm. All graphs in this figure showing numbers of cells with G3BP1+ SGs out of 200 randomly selected cells. Cells were counted as being G3BP1-SG+ when they contained at least 5 cytoplasmic SGs (n = 3). WB for p53 and β-tubulin using untreated PLC/PRF/5 cells (b). d Immunofluorescence for G3BP1 and DAPI and a summarized graph (n = 3), using MEFs with different p53 status (p53+/+, p53-/-, p53R172H/R172H) treated with 50 µM SOR for 2 h or 15 µM TG for 1 h. Scale: 10 µm. e Immunofluorescence for G3BP1, p53, and DAPI and a summarized graph (n = 3) in p53null OSC19 cells expressing p53R175H and p53R248W (no tag), following treatment with TG (10 µM) for 1 h. Scale: 10 µm. WB of untreated cells for p53 and GAPDH. f Structure and domains of human p53, including transactivation (TA) domains, a proline rich domain (PRD), a DNA binding domain (DBD), a mutation at p53R248W, NLS and a mutation at p53K305N, NES and mutations at p53L348A/L350A, an oligomerization domain (OD), and a C-terminal domain (CTD). Immunofluorescence for G3BP1, p53, and DAPI and a summarized graph (n = 3) using OSC19 cells expressing vector, p53R248W, p53R248W/NLS, and p53R248W/NES treated with TG (10 µM) for 1 h. WB of untreated cells for p53 and Vinculin. Unmerged images are in Supplementary Fig. S3i. All experiments are from three independent experiments. Mean ± S.E.M., two-tailed Student’s t-test. ns not significant. Source data are provided as a Source Data file.
Notably, knocking down wtp53 in HepG2 and SJSA1 cells, as well as upregulating wtp53 using an MDM2 antagonist, Nutlin-3a, in SJSA1 cells, did not significantly alter the cells’ SG-forming potential (Fig. 3c and Supplementary Fig. S3g, h). Additionally, we determined the SG-forming potential of mouse embryonic fibroblasts (MEFs) with different p53 genotypes (p53+/+, p53-/-, p53R172H/R172H). Consistently, p53R172H/R172H MEFs formed fewer SGs compared to p53+/+ and p53-/- MEFs following ER stress (Fig. 3d). These results collectively indicate that mutp53, but not wtp53, suppresses ER stress-mediated SG formation, even in non-cancerous cells.
Furthermore, exogenous overexpression of two hotspot mutp53, p53R175H and p53R248W, in p53null OSC19 cells suppressed TG-induced SG formation (Fig. 3e). Given that SGs are cytoplasmic, non-membrane RNP assemblies, we inserted additional disrupting mutations in the nuclear localization signal (NLS) or nuclear export signal (NES) in p53R248W to generate constructs expressing NLS-mutated p53R248W/K305N (p53R248W/NLS) or NES-mutated p53R248W/L348A/L350A (p53R248W/NES), respectively (Fig. 3f). In OSC19 cells, p53R248W/NLS, which predominantly localized to the cytoplasm, inhibited SG formation more efficiently than did p53R248W, while p53R248W/NES, which was restricted to the nucleus, could not significantly inhibit SG formation (Fig. 3f and Supplementary Fig. S3i). These results suggest that the cytoplasmic portion of mutp53 suppresses SG formation.
Mutp53’s binding to PERK prevents the PERK-eIF2α interaction, inhibits phosphorylation of eIF2α, and contributes to the suppression of SG formation
During ER stress, PERK, a receptor tyrosine kinase in the ER membrane that acts as an ER stress sensor, dimerizes and autophosphorylates at multiple sites23. Subsequently, phosphorylated PERK (p-PERK) binds to and phosphorylates eIF2α, which inhibits translation initiation, the first step of SG formation. Hence, we investigated the effects of mutp53 on the PERK-eIF2α axis. Knockdown of mutp53 in SNU449 and Huh7 HCC cells resulted in a consistent and significant increase in eIF2α phosphorylation (p-eIF2α) and a modest increase in p-PERK under ER stress (Fig. 4a). Increased eIF2α phosphorylation was consistently observed in TOV112D and KHOS/NP cells following mutp53 knockdown; however, the increase in p-PERK levels by mutp53 knockdown was observe in TOV112D cells, but not in KHOS/NP cells (Supplementary Fig. S4a). Knockdown of wtp53 in U2OS cells had minimal effects on total protein levels and phosphorylation of PERK and eIF2α (Supplementary Fig. S4b).

a WB for the indicated proteins using control and p53-knockdown Huh7 and SNU449 (p53A161T) HCC cells treated with vehicle control or SOR. Mean values of densitometric analyses for p-PERK/total PERK and p-eIF2α/total eIF2α below images (n = 3). b Co-IP studies for p53 and PERK using Huh7 cells treated with vehicle or SOR. c PLA (red) for p53 and PERK, using control and p53-knockdown (shp53) Huh7 cells treated with vehicle or SOR. Scale: 10 µm. d Immunofluorescence for G3BP1 and DAPI and a summarized graph (n = 3) using Huh7 cells treated with or without SOR and/or a PERK inhibitor, GSK2606414 (PERKi, 800 nM, 2 h). Scale: 10 µm. e PLA (red) for PERK and eIF2α, using control and p53-knockdown (shp53) Huh7 cells treated with vehicle or SOR. Scale: 10 µm. Graph quantifying the number of cells positive for PERK-eIF2α interaction puncta out of 200 randomly selected cells (n = 3). Scale: 10 µm. All experiments are from three independent experiments. Mean ± S.E.M., two-tailed Student’s t-test. ns not significant. Source data are provided as a Source Data file.
To understand the mechanism by which mutp53 significantly inhibited phosphorylation of eIF2α, we examined the interaction of mutp53 with PERK using co-IP studies in Huh7 and TOV112D cells. Mutp53 bound both the native and phosphorylated forms of PERK regardless of ER stress (Fig. 4b and Supplementary Fig. S4c). Given that PERK is present in the ER membrane, we confirmed that mutp53 constitutively bound to PERK in the cytoplasm using PLA in Huh7 and KHOS/NP cells (Fig. 4c and Supplementary Fig. S4d). We did not observe any interaction between mutp53 and eIF2α in subsequent co-IP studies (Fig. 5a). These results suggest that mutp53 inhibits SG formation, at least partially, through its interaction with p-PERK, subsequently inhibiting eIF2α phosphorylation. Moreover, the PERK inhibitor GSK2606414 (PERKi) significantly reduced ER stress-induced SG formation with inhibition of eIF2α phosphorylation in Huh7 and KHOS/NP cells, where mutp53 knockdown had no effect (Fig. 4d and Supplementary Fig. S4e, f).

a Co-IP studies for p53, followed by WB for the indicated SG core proteins, using KHOS/NP cells treated with TG. MWM: molecular weight marker. b Co-IP studies for p53 and G3BP1 using SNU449 cells treated with SOR. c PLA (red) for G3BP1 and p53, using control and p53-knockdown (shp53) Huh7 cells treated with vehicle or SOR. Scale: 10 µm. d Co-IP studies for FLAG and p53 using Huh7 cells expressing FLAG-tagged full-length (Full) or ΔRGG G3BP1, followed by WB for FLAG and p53. e Immunofluorescence and WB (untreated) for the indicated proteins, using control, G3BP1-knockdown (shG3BP1), p53-knockdown (shp53), and G3BP1/p53-knockdown SNU449 cells treated with SOR. Graph showing the number of PABP-SG+ cells in 200 cells (n = 3). Scale: 10 µm. f Annexin V/PI staining using control, G3BP1-knockdown, p53-knockdown, and G3BP1/p53-knockdown SNU449 cells treated with vehicle or SOR. Graph showing the percentage of apoptotic cells. g Immunofluorescence for G3BP, A11 (anti-amyloid oligomer), and DAPI using control and mutp53-knockdown Huh7 cells treated with vehicle or SOR. Insets showing co-localization of G3BP+-SGs and A11-positive puncta (n = 3). Scale: 10 µm. Graph showing area of colocalization of A11-positive large puncta and G3BP-positive SGs (n = 75 across three independent experiments). Bar in (g) indicates median. h Oligomerization assays for G3BP1 using control and p53-knockdown Huh7 cells treated with vehicle or SOR. For protein crosslinking, cell lysates were treated with vehicle or BMH (1 mM, 30 min), followed by WB for G3BP1 and GAPDH. All experiments are from three independent experiments. Mean ± S.E.M., two-tailed Student’s t-test. ns not significant. Source data are provided as a Source Data file.
To further investigate the mechanism of mutp53-mediated inhibition of eIF2α phosphorylation, we examined the effect of mutp53 on the PERK-eIF2α interaction using PLA. Knockdown of mutp53 robustly increased the PERK-eIF2α interaction in the cytoplasm of Huh7 and SNU449 cells (Fig. 4e and Supplementary Fig. S4g). Consistent with increased eIF2α phosphorylation, mutp53 knockdown increased ATF4 protein levels, which was further increased under ER stress (Supplementary Fig. S4h). Together, these results suggest that mutp53’s binding to PERK prevents the PERK-eIF2α interaction and subsequently inhibits eIF2α phosphorylation and SG formation, which may contribute to increased sensitivity to ER stress. Additionally, we observed a modest increase in other ER stress signaling by mutp53 knockdown in SOR-treated Huh7 cells, as demonstrated by protein levels of p-IRE1, spliced XBP1s, and cleaved nuclear ATF6α (ATF6α-N) (Supplementary Fig. S4i). How this signaling coordinates with decreased SG formation and increased ER stress sensitivity by mutp53 warrants further investigation.
Mutp53’s binding to G3BP1 inhibits G3BP1 oligomerization and SG formation, which contributes to increased sensitivity to ER stress inducers
Next, we examined the effects of mutp53 on key SG core proteins, including eIFs and RNPs24. There was no significant change in the protein levels of eIF2α, eIF4E, eIF4G, eIF4A1, PABP, TIA1, and G3BP1 by mutp53 knockdown in KHOS/NP cells, regardless of the presence of ER stress (Supplementary Fig. S5a). However, co-IP studies revealed that mutp53 constitutively bound G3BP1, but not other SG core proteins, in KHOS/NP and SNU449 cells (Fig. 5a, b and Supplementary Fig. S5b). PLA also confirmed the constitutive interaction of mutp53 and G3BP1 in the cytoplasm of Huh7 and KHOS/NP cells (Fig. 5c and Supplementary Fig. S5c). Although no binding was detected between wtp53 and G3BP1 in SJSA1 cells, regardless of treatment with TG and/or Nutlin-3a (to increase wtp53 levels), a modest interaction between wtp53 and G3BP1 was observed using in vitro transcribed/translated proteins (Supplementary Fig. S5d, e). To clarify this result, we conducted PLA studies for wtp53 and G3BP1, along with immunofluorescence for these proteins, in Hep3B cells with doxycycline-inducible wtp53 (iwtp53). We further confirmed a modest interaction between these two proteins in the cytoplasm with no change in SOR-induced SG formation upon wtp53 induction (Supplementary Fig. S5f, g). The lack of SG suppression by wtp53 may be due to the scarce amount of wtp53 in the cytoplasm available to bind to G3BP1.
We also identified a domain in G3BP1 required for its binding to p53R175H by performing co-IP studies using in vitro transcribed/translated p53R175H and different deletion mutants of G3BP1 (Supplementary Fig. S5h). G3BP1 mutants with a deletion of the RGG domain at the C-terminus failed to bind to mutp53, suggesting that the RGG domain, which facilitates the interaction of G3BP1 with RNA25, is crucial for this binding. Co-IP and PLA analyses in Huh7 cells expressing FLAG-tagged full-length or ΔRGG G3BP1 further confirmed that ΔRGG G3BP1 could not bind to endogenous mutp53 (Fig. 5d and Supplementary Fig. S5i).
To determine the functional association between mutp53 and G3BP1, we examined the effects of G3BP1 knockdown on TG-induced SG formation (detected by PABP or eIF4E antibody) and cell viability in control and mutp53-knockdown cells. Increased PABP- or eIF4E-SG+ formation due to mutp53 knockdown was significantly reduced by simultaneous G3BP1 knockdown in both SNU449 and KHOS/NP cells, following SOR and TG treatment, respectively (Fig. 5e and Supplementary Fig. S5j). Moreover, SOR- and TG-induced apoptosis, which was significantly reduced by mutp53 knockdown, was restored by concomitant G3BP1 knockdown (Fig. 5f and Supplementary Fig. S5k, l). To further link SG formation and cell survival following ER stress, Hep3B cells overexpressing full-length G3BP1 or G3BP1-ΔRGG were treated with SOR, followed by SG detection and Annexin V/PI staining and flow cytometry (Supplementary Fig. S5m, n). Overexpression of full-length G3BP1, but not G3BP1-ΔRGG, promoted SG formation and cell survival, suggesting the critical role of the RGG domain for G3BP1-mediated SG formation and cell survival following ER stress. Collectively, these results suggest that the inhibition of SG formation by mutp53 through its binding to G3BP1 contributes to increased apoptosis following ER stress.
G3BP1 and other RNA binding proteins within the SG form large insoluble amyloid oligomers that phase-separate from the cytoplasm26. To examine the effects of mutp53 on G3BP1 oligomerization within SGs, we performed immunofluorescence studies using anti-G3BP1 and anti-amyloid oligomer A11 antibodies27, following treatment of control and mutp53-knockdown Huh7 and KHOS/NP cells with SOR and TG, respectively. In mutp53-expressing control cells, fewer A11-positive puncta and G3BP1-positive SGs were detected compared to mutp53-knockdown cells. Notably, more A11-positive puncta co-localized with G3BP-positive SGs in mutp53-knockdown cells (insets, Fig. 5g and Supplementary Fig. S5o). Furthermore, our bismaleimidohexane (BMH) crosslinking study confirmed that mutp53 knockdown promoted G3BP1 oligomerization in Huh7 cells (Fig. 5h). These data suggest that mutp53 binds to G3BP1 and inhibits G3BP1 oligomerization, thereby inhibiting SG formation.
The oligomerization domain of mutp53 is required for binding to G3BP1, inhibiting SG formation, and enhancing ER stress-mediated apoptosis
To determine the domain in mutp53 required for interaction with G3BP1, we generated several deletion mutants targeting the NLS, oligomerization domain (OD), and C-terminal domain (CTD) of 3xFLAG-tagged p53R175H, while preserving the NES (Fig. 6a, b). Hep3B cells expressing the full-length p53R175H and the deletion mutants were treated with SOR, followed by PLA and immunofluorescence for FLAG (p53) and G3BP1. Intriguingly, only the OD-deleted p53R175H (ΔOD), but not the full-length, NLS-, or CTD-deleted p53R175H, failed to bind to G3BP1 and inhibit SOR-induced SG formation (Fig. 6c, d and Supplementary Fig. S6a). The lack of binding of p53R175H ΔOD to G3BP1 was also confirmed by co-IP studies using Hep3B cells (Supplementary Fig. S6b). Moreover, in contrast to full-length p53R175H, p53R175H ΔOD failed to sensitize Hep3B cells to SOR and VCR, as detected by CC3 staining and Annexin V/PI & flow cytometry analyses (Fig. 6e, f). Together, these results indicate that the OD domain in mutp53 is required for the mutp53-G3BP1 interaction, inhibiting G3BP1’s oligomerization and subsequent SG formation, which contributes to increased apoptosis following ER stress.

a Deletion mutants of p53R175H in the NLS, OD, and CTD with 3xFLAG tags. b WB for p53 and GAPDH in untreated Hep3B (p53null) cells expressing vector control or each p53 construct. c PLA (red) for G3BP1 and mutp53 using Hep3B cells expressing vector, full-length p53R175H, and different p53R175H deletion mutants, following treatment with vehicle or SOR. Scale: 10 µm. d Immunofluorescence for G3BP1 (red), p53 (green), and DAPI (blue) and a summarized graph using Hep3B cells with different p53R175H deletion mutants, treated with SOR (n = 3). Scale: 10 µm. Unmerged images are in Supplementary Fig. S6a. e Immunofluorescence for CC3 and a summarized graph using Hep3B cells expressing vector, full-length (p53R175H), or OD-deleted p53R175H (ΔOD), treated with vehicle or SOR for 48 h (n = 3). Scale: 100 µm. f Annexin V/PI & flow cytometry using the same set of Hep3B cells as in (e), treated with vehicle, SOR (8 µM, 48 h) or VCR (25 µM, 48 h). Graph showing the percentage of apoptosis (n = 3). All experiments are from three independent experiments. Mean ± S.E.M, two-tailed Student’s t-test. ns: not significant. Source data are provided as a Source Data file.
Mutp53 enhances the sensitivity of HCC xenografts to SOR, accompanied by reduced SG formation and increased apoptosis
To conduct proof-of-concept studies, we examined whether mutp53 could inhibit SG formation and enhance SOR sensitivity in subcutaneous human HCC xenografts in mice. Immunocompromised mice bearing control and mutp53-knockdown Huh7 tumors (~60 mm3) were intraperitoneally injected with vehicle or SOR. Consistent with the in vitro results, mutp53-expressing control tumors responded well to SOR, while mutp53-knockdown tumors showed significantly reduced sensitivity to SOR (Fig. 7a). Immunohistochemistry (IHC) for p53 and CC3, as well as immunofluorescence for PABP and G3BP1, revealed increased CC3 positivity and a reduced frequency of SG formation in SOR-treated control tumors compared to mutp53-knockdown tumors (Fig. 7b, c). Additionally, we used another HCC cell line, PLC/PRF/5 (p53A161T), for the xenograft experiments and observed essentially the same results as in the Huh7 xenografts (Fig. 7d and Supplementary Fig. S7a). It should be noted that the mutp53-knockdown groups in both xenograft experiments exhibited a slightly slower tumor formation latency and tumor growth compared to the control group (Fig. 7a, d and Supplementary Fig. S7b). These results demonstrate proof of concept that mutp53 suppresses SG formation and enhances apoptosis in tumors during treatment with ER stress-inducing chemotherapy drugs (e.g., SOR), leading to efficient inhibition of the growth of mutp53-expressing tumors.

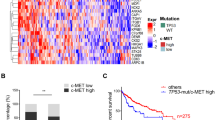
a Tumor growth curve of control and mutp53-knockdown Huh7 xenografts with representative tumor images. NSG mice bearing Huh7 tumors (~60 mm3) were treated with vehicle or 100 mg/kg SOR for once daily, 3 days/week for ~3 weeks. Mean ± S.E.M. (n = 6), Two-way ANOVA with Geisser-Greenhouse correction. b IHC for p53 and CC3 using Huh7 tumors at experimental endpoints. Graphs showing IHC scores, which were measured as the sum of average intensity and average extensity of CC3 (n = 3). *p < 0.05; unpaired student’s t-test. Scale: 100 µm. c Representative images of immunofluorescence for PABP and G3BP1 to detect SGs, using Huh7 tumors from (a). Scale: 10 µm. Graphs showing the number of PABP-SG+ or G3BP1-SG+ cells in 200 cells (n = 3). d Tumor growth curve of control and mutp53-knockdown PLC/PRF/5 xenografts with representative tumor images. The same set of experiments were done as in (a). Mean ± S.E.M. (n = 7), Two-way ANOVA with Geisser-Greenhouse correction. e Analyses of TCGA datasets of HCC. Overall survival of HCC patients with missense mutp53 and p53null was stratified according to G3BP1 mRNA expression levels (below or above the mean). Logrank test. All experiments in (b, c) are from three independent experiments. Mean ± S.E.M., two-tailed Student’s t-test. ns not significant. Source data are provided as a Source Data file.
Additionally, using TCGA datasets, we attempted to clinically validate the functional association of p53 mutation status with G3BP1, whose expression levels are generally correlated with SG-forming potential28. The same HCC cases as in Fig. 1a were further stratified by G3BP1 mRNA levels. Intriguingly, high G3BP1 mRNA expression was correlated with worse overall survival in patients with p53null HCC, while G3BP1 mRNA expression levels did not significantly impact the overall survival of patients with missense mutp53-carrying HCC (Fig. 7e). The poorest overall survival observed in p53null HCC cases with high G3BP1 expression may be due to high SG-forming potential, resulting in resistance to SOR. These results align with our finding that mutp53 inhibits G3BP1-mediated SG formation, leading to increased sensitivity to SOR.
Discussion
Canonical mutp53 GOF typically promotes cell survival and malignant progression. However, our study reveals that mutp53 suppresses ER stress-mediated SG formation primarily by binding to PERK and G3BP1, which likely contributes to increased apoptosis and tumor suppression under ER stress, highlighting a unique vulnerability imposed by mutp53 (Fig. 8). In SG-competent cells, the potential to form SGs influences whether cells survive or undergo apoptosis. Mutp53-mediated inhibition of SG formation following ER stress may override other mutp53 GOFs related to cell survival in this context, thereby promoting apoptosis. In SG-reluctant cells such as MDA-MB-231 and H1299 cells, the role of mutp53 in ER stress-mediated apoptosis may be diminished due to the lack of SG formation. Indeed, in these cells, mutp53 shows little impact on or modestly enhances cell survival following ER stress, likely due to its canonical GOF (Supplementary Fig. S8a–d). This finding aligns with a report by Sicari et al.29, which demonstrated a survival advantage of mutp53 during ER stress. Since Sicari et al. drew their conclusions using SG-reluctant MDA-MB-231 and H1299 cell lines, the discrepancy from our study likely hinges on the cellular ability to form SGs, underlining the significance of SG formation in determining biological outcomes under ER stress.

Mutp53 inhibits ER stress-mediated formation of SGs by interacting with PERK and G3BP1, which contributes to enhanced sensitivity to ER stress inducers like SOR. Created in BioRender. Iwakuma, T. (2025) https://BioRender.com/h97c210.
The primary mechanism by which mutp53 inhibits SG formation involves its ability to bind and inhibit two key proteins involved in ER stress-induced SG formation. Firstly, mutp53 binds to PERK, preventing the PERK-eIF2α interaction and thereby reducing PERK-mediated phosphorylation of eIF2α, which is crucial for inhibiting protein translation and initiating SG formation30. Secondly, mutp53 binds to the SG-nucleating protein G3BP1, inhibiting G3BP1’s oligomerization within SGs which is important for the liquid–liquid phase separation necessary to form SGs31.
While the SG competency of cells following ER stress is dependent on cellular context, knockdown of four different missense mutp53 (R156P, A161T, Y220C, and R249S), but not wtp53, increases SG formation in different cell lines, while overexpression of two hotspot mutp53 (R175H, R248W) inhibits SG formation in OSC19 and Hep3B cells. It remains unknown whether all missense mutp53 variants can suppress SG formation with similar efficiency. Given the significant role of the cytoplasmic portion of mutp53 in binding to PERK and G3BP1 and suppressing SG formation, the subcellular localization of mutp53 is likely a key determinant of its ability to inhibit SG formation. Although wtp53 neither binds to G3BP1 nor inhibits SG formation in SJSA1 cell, in vitro transcribed/translated wtp53 and G3BP1 proteins can still interact, and binding of wtp53 to G3BP1 is detected by PLA in the cytoplasm when wtp53 is transiently overproduced in Hep3B cells, consistent with a previous report32. However, wtp53 fails to inhibit SG formation even in Hep3B cells overexpressing wtp53. Thus, the protein levels of p53 in the cytoplasm and the specific type of p53 mutations may play key roles in p53-mediated SG inhibition.
The ability of mutp53 to inhibit SG formation may also depend on the type of cellular stress and the specific eIF2α kinases activated to induce SG formation. Since SG formation commonly requires G3BP1, mutp53 might still inhibit SG formation induced by other stresses, including reactive oxygen species, viral infection, and heat shock33. Notably, ER stress can activate both survival and death signaling pathways. The outcome−whether cells survive or die−likely depends on the extent and duration of ER stress, the type of ER stress inducers, cellular context, and the ability to form SGs. Hence, even in the presence of mutp53 in SG-competent cells, different experimental conditions and intrinsic factors could influence cellular outcomes following ER stress.
We observe that all six HCC cell lines examined, including Hep3B, SNU475, Huh7, HepG2, PLC/PRF/5, and SNU449, can form SGs following treatment with SOR (Table 1). Intriguingly, ER stress is associated with the development of various liver diseases, including HCC34, and altered expression of SG components is observed in HCC35. Due to the high SG-forming potential of HCC cell lines, HCC may be an ideal cancer type to exploit mutp53-mediated suppression of ER stress-induced SG formation. Thus, HCC patients with mup53 tumors could be preferentially given SOR treatment, as HCC patients with p53wt or p53null tumors may show intrinsic resistance to SOR due to high SG formation. We also demonstrate SG inhibition and increased ER stress sensitivity by mutp53 in other cancer types. This suggests that other mutp53-expressing, SG-competent tumors may also be sensitive to ER stress, warranting further study.
During tumorigenesis, tumor cells encounter various kinds of stress, including ER stress. In this context, mutp53 might promote ER stress-induced apoptosis by suppressing SG formation and hider tumor development. This could explain why mutations in p53 are not common in the early stages of tumorigenesis in certain cancer types. Thus, ER stress during early tumorigenesis might select against p53 mutations. Additionally, unlike established cell lines, mutp53 in some tumors may not accumulate sufficiently to suppress SG formation during ER stress. In such tumors, mutp53 would have little impact on SG formation and sensitivity to ER stress.
Lastly, a recent study by Wang et al.36 suggested that mutp53 GOF could not affect survival, metastasis, or drug response, which seems counterintuitive to numerous observations associated with mutp53 GOF, including our findings. Specifically, their conclusions are based on a doxycycline-inducible CRISPR-Cas9 p53 knockout system using multiple SG-competent and -reluctant cell lines, without overexpression or other mutp53 knockdown systems. They also did not expose cells to ER stress. Therefore, the main differences lie in cell lines used, experimental settings, and cellular contexts.
In summary, this is the first study to demonstrate that inhibiting ER stress-induced SG formation is a unique vulnerability imposed by mutp53. Our study also suggests that both mutp53 and the susceptibility of cells to SG formation are crucial determinants of the response to ER stress-inducing drugs. Moreover, our findings highlight an innovative therapeutic strategy that supports the use of the FDA-approved drug, SOR, to specifically target HCC carrying p53 mutations. Besides SOR in HCC, other ER stress inducers, such as VCR, may also be considered for treating other types of mutp53-expressing cancers, although this strategy is only applicable to SG-competent tumors. Notably, drugs that specifically target the ER stress pathway are not yet FDA-approved but are under clinical investigation37. Our findings may accelerate the development of ER stress-inducing drugs to target mutp53-expressing cancers.
Methods
Ethics approval
This study complies with all relevant ethical regulations of the Institutional Biosafety Committee (IBC00093) and the Institutional Animal Care and Use Committee protocol (IACUC00031) of the Children’s Mercy Research Institute that approved the study protocol.
Cell lines
The Hep3B cell line was gifted by Dr. Lisa Zhang at the University of Kansas Medical Center (KUMC), while the other HCC cell lines, including Huh7, SNU449, PLC/PRF/5, SNU475, and HepG2, were gifted by Dr. Steven A. Weinman at KUMC. Head and neck cancer cell lines, including OSC19, HN31, CAL33, and FaDu, were gifted from Dr. Sufi Thomas at KUMC. All other cell lines, including KHOS/NP (CRL-1544, ATCC), MG63 (CRL-1427, ATCC), U2OS (HTB-96, ATCC), SJSA1, TOV112D, MDA-MB-231, H1299, SW620, HT-29, HCT116, Panc-1, and PC3, have been routinely maintained in the Iwakuma laboratory at the Children’s Mercy Research Institute. Mouse embryonic fibroblasts (MEFs) were previously generated from p53R172H/R172H, p53-/-, or p53+/+ 13.5 days post-coitum embryos38. Hep3B cells expressing an inducible wtp53 (iwtp53) was generated by infecting a doxycycline-inducible wtp53 vector, in which the pLVX-tetOne-puro-GFP (171123, Addgene) vector was used to replace GFP with human p53 cDNA, followed by puromycin selection. All cell lines were authenticated annually or every 10 to 12 passages using STR profiling by the University of Arizona Genetics Core facility (Tucson, AZ, USA). All cell lines were tested for mycoplasma contamination by PCR and were treated with the mycoplasma removal agent (BUF035, BIO-RAD, Hercules, CA, USA), BM cyclin (10799050001, Millipore Sigma, St. Louis, MO, USA), and normocin (ant-nr-05, InvivoGen, San Diego, CA, USA) every 3–4 months.
Generation of lentiviral and retroviral vectors
HEK-293T cells were transfected with lentiviral and a retroviral envelope PMD2.G (12259, Addgene, Watertown, MA, USA) and a lentiviral packaging psPAX2 (12260, Addgene) or a retroviral packaging gag/pol (14887; Addgene) plasmids, along with indicated vector constructs (described below), using the jetPRIME transfection reagent (89129-924, Polyplus, Sébastien Brant, France) according to manufacturer’s directions. After 48 h, virus-containing supernatants were filtered through a 0.22 µm filter (229747; Celltreat, Pepperell, MA, USA). Virus was concentrated by >20,000 × g centrifugation for 1.5 h, followed by infection into target cells and subsequent antibiotic selection.
Gene knockdown and knockout for p53 and G3BP1
To knockdown p53 or G3BP1, cells were infected with a lentiviral vector shp53 pLKO.1 puro targeting p53 exon 7/8 (19119, Addgene), a pLVshRNA-Bsd-U6-hTP53 targeting p53 3’UTR (VectorBuilder), or a pGIPz vector encoding shRNA for G3BP1 (RHS4430-200239796, Horizon Discovery, Lafayette, CO, USA), while an empty lentiviral vector pCDH-CMV-MCS-EF1α-Puro (pCDH-EF-puro, CD510B-1, System Biosciences, Palo Alto, CA, USA) or a pGIPz nonsilencing shRNAmir lentiviral control vector (RHS4346, Horizon Discovery) was used as a negative control. p53-knockout cells were made by infection with a p53 sgRNA lentiviral vector (pXPR003-sgTP53-4, 118022, Addgene) and an adenoviral vector encoding GFP-Cas9 (1901, VECTOR BIOLABS, Malvern, PA, USA), followed by single colonization. Single cells were expanded and confirmed for p53 genetic deletion by genomic PCR and sequencing. Knockdown or knockout for target proteins was confirmed by western blotting (WB) and immunofluorescence using specific antibodies. Target sequences and primer sequences are found in Supplementary Table 1.
Generation of constructs encoding mutp53 with or without point mutations or deletions
Mutant p53 (p53R175H, p53R248W) cDNAs were subcloned into a lentiviral vector pCDH-EF-puro lentiviral vector or a retroviral vector pBabe-puro (1764, Addgene). To insert point mutations at K305N in the nuclear localization signal (NLS) or at L348A and L350A in the nuclear export signal (NES) in p53R248W, the QuikChange XL site-directed mutagenesis kit (200517, Agilent Technologies, Santa Clara, CA, USA) was used. To generate p53 deletion constructs in pCDH-EF-puro-p53R175H-3XFLAG vector, PCR primers were designed to amplify the regions from N-terminal ATG to the domains of interest with a silent mutation of a SacI site and from the end of the domain of interest with a silent mutation of a SacI site to a C-terminal 3X-FLAG, followed by ligation of two fragments of p53R175H and pCDH-EF-puro at generated SacI sites. All primers were purchased from Integrated DNA Technologies. The entire coding region was sequenced to confirm the appropriate mutations or deletions in p53R175H. These constructs were used to generate lentiviral vectors to be infected into cells, and the expression of mutant proteins was confirmed by WB and immunofluorescence using p53-specific antibodies. Target sequences and primer sequences are found in Supplementary Table 1.
Annexin V/propidium iodide (PI) staining & flow cytometry
Cells (60,000–100,000) seeded into each well of a 6-well plate were treated 48 h later with indicated agents for 24–72 h. Cells were harvested using a cell dissociation buffer, followed by Annexin V and PI staining using the Annexin V/PI flow cytometry cell staining kit (V13245, Thermo Fisher, Waltham, MA, USA) according to manufacturers’ directions. Cells were then analyzed using a BD LSR-Fortessa (BD Biosciences, Franklin Lakes, NJ, USA) flow cytometer. First, cells were gated according to forward scatter and side scatter to exclude cellular debris. Next, cells were gated for positive stain with PI or Annexin V Alexa Fluor 488 antibody. The left bottom quadrant (Q3) indicates live cells. The left top quadrant (Q1) indicates necrotic cells. The right bottom quadrant (Q4) indicates early apoptosis. The right top quadrant (Q2) indicates late apoptosis. Thus, apoptosis is the sum of early and late apoptosis (Q2 + Q4), while total cell death is the sum of necrosis and apoptosis (Q1 + Q2 + Q4).
Colony formation assay
Cells (500–3000 cells), depending on the cell lines, were seeded on a 6-well plate and allowed to grow to form colonies for 2–4 days. Colonies were then treated with indicated treatments for 48 h, followed by washing out three times with 1X PBS (41620004-2, Bioworld, Irving, TX, USA). The colonies were cultured for an additional 5–7 days and stained using crystal violet (C0775-100G, Millipore Sigma). Images were taken by the iBright FL1000 imaging system (Thermo Fisher, Waltham, MA, USA). Colonies greater than 2 mm in diameter were manually counted using FIJI (ImageJ 2) software.
Antibodies
Antibody information, including vendors, catalog numbers, clone numbers, assays used, and dilutions, is described in Supplementary Table 2. All antibodies are validated using cells with overexpression and/or depletion of the target proteins.
WB, co-immunoprecipitation (co-IP), and G3BP1 oligomerization assays
Cell pellets were resuspended in IP lysis buffer (PI87788, Thermo Fisher) for co-IP or Cellytic M Cell Lysis Reagent (C2978-250ML, Millipore Sigma) for WB containing protease inhibitors (11836153001, Roche, Basel, Switzerland), lysed by passage through a syringe or by vortexing, and spun at max speed at 4 °C for 20 min, followed by determination of protein concentrations of the supernatant using a BCA kit (PI23227, Thermo Fisher) with a multi-functional plate reader (Agilent BioTek Synergy H1, Winooski, VT, USA). After adjusting protein concentrations, samples for WB were mixed with 4x SDS sample buffer and heated at 95 °C for 5 min. Samples for co-IP were added to antibody-conjugated protein A/G Plus-Agarose beads (sc-2003, Santa Cruz Biotechnology) and rotated overnight at 4 °C, followed by washing with 1X PBS, incubation with SDS buffer (1–1.5X), and heating for 5 min at 95 °C. These heat-denatured samples for WB or co-IP were subjected to SDS PAGE using a pre-stained protein ladder (P7719L, New England BioLabs, Ipswich, MA, USA) as a molecular weight marker (MWM). Gels were transferred to PVDF membrane using transblot turbo apparatus (Biorad, Hercules, CA, USA) according to the manufacturer’s directions. Membranes were washed in TBS-T (0.02% Tween 20) and blocked in 4% non-fat dry milk or bovine serum albumin, followed by incubation with primary and secondary antibodies in blocking solution. After washing in TBST, membranes were dripped in methanol and then dried in the dark before imaging using the Azure Sapphire FL Biomolecular Imager (Redmond, WA, USA).
For G3BP1 oligomerization assays, cell lysates were incubated with 1 mM of BMH at 30 °C for 30 min to crosslink G3BP1, followed by SDS PAGE and WB for G3BP1.
Densitometry analysis
WB images were imported into the FIJI software, and same sized rectangles were used to calculate Mean Gray Value for the band of interest (total PERK, p-PERK, total eIF2α, p-eIF2α) and a background measurement. To invert the pixel density, each measurement (including background) was subtracted from 255, the maximum intensity value in an 8-bit gray-scale image. To calculate the inverted total PERK, the inverted band value for unphosphorylated PERK was added to the corresponding inverted band value for p-PERK. Next, inverted background measurements were subtracted from inverted band measurements to calculate net band values. For p-PERK and p-eIF2α, net bands were normalized to total PERK and eIF2α net bands, respectively.
Immunofluorescence, SG detection, and proximity ligation assay (PLA)
Cells (3000–5000) seeded on an 8-chambered LabTek slide (154941, Thermo Fisher) were treated with SG inducers or chemotherapy drugs for 1–2 h before SG detection. An MDM2 antagonist, Nutlin-3a (SML0580, Millipore Sigma), and a PERK inhibitor, GSK2606414 (17376, Cayman Chemical, Ann Arbor, MI, USA), were applied for 8 h and 2 h prior to staining, respectively. For immunofluorescence studies, cells were fixed with 4% paraformaldehyde (15710, Electron Microscopy Sciences, Hatfield, PA, USA) in 1X PBS at 4 °C for 20 min and permeabilized using 0.3% Triton X-100 (sc-29112, Santa Cruz Biotechnology, Dallas, TX, USA) in 1X PBS at 4 °C for 5 min. Blocking was conducted using 1.0% Bovine Serum Albumin (BSA, B005-100GM, Caisson Labs, Smithfield, UT, USA) in 0.3% Triton X-100 in 1X PBS for 1 h at room temperature. Cells were incubated with appropriate primary and secondary antibodies in blocking solution. After washing with 1X PBS, cells were mounted using Prolong AntiFade Gold Mountant with DAPI (P36941, Thermo Fisher). Images were analyzed using the all-in-one BZ-X800 microscope (Keyence, Osaka, Japan). To quantify the SG-forming potential of cells, we counted the number of SG-positive cells (at least 5 SG puncta in the cytoplasm) using FIJI.
For PLA, following permeabilization, cells were stained using Duolink In situ Red Starter Kit Mouse/Rabbit (DUO92101-1KT, Millipore Sigma) according to manufacturer’s directions. Briefly, cells were incubated with antibodies for two target proteins that were expected to co-localize at 4 °C overnight, followed by provided secondary antibodies conjugated with circle-forming oligonucleotides. When two proteins are located within 40 nm in cells, the oligonucleotides can be ligated and amplified, which can be detected with fluorescently labeled complementary oligonucleotide probes. Images were analyzed using the Keyence all-in-one BZ-X800 microscope.
In vitro transcription/translation
The cDNAs for G3BP1 and its deletion mutants were subcloned into pCR4Blunt-TOPO vector (T3, Thermo Fisher Scientific), while p53R175H and wtp53 cDNAs were subcloned into pCR-Blunt II-TOPO vector (Sp6 and T7, respectively, Thermo Fisher Scientific). The coded proteins were synthesized in vitro using the TnT® Quick Coupled Transcription/Translation System with an appropriate promoter (Promega, Madison, WI).
Tumor xenografts
Human HCC cells were detached using cell dissociation buffer (C5914-100ML, Millipore Sigma). Cells were resuspended in a 1:1 mixture of HBSS (14-175-095, Thermo Fisher) to growth factor reduced Matrigel (356231, Corning, Corning, NY, USA). Cancer cells (~2 × 106 cells/100 µl) were injected into the flank of NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) male and female mice (strain 005557, Jackson Laboratory, Bar Harbor, ME, USA) at around 6 weeks old. Tumors were allowed to grow to ~60 mm3 in size before intraperitoneally administering with vehicle or SOR at a dose of 100 mg/kg of body weight once daily three times/week for about 3 weeks. Tumor volume was three-dimensionally measured three times/week by a digital caliper. Mice were euthanized before tumors reached 1.5 cm in diameter or mice became moribund based on the approved protocol. At experimental endpoint, tumors were harvested and suspended in 10% neutral buffered formalin for 48 h. Tumors were then transferred to 70% ethanol and processed, paraffin-embedded, and sectioned by microtome at the KUMC or Children’s Mercy pathology core facility. Mice were housed in a pathogen-free barrier in Innorack IVS ventilated caging system (Innovive, San Diego, CA) with Innovive microisolator cages. Cages were provided with irradiated corn cob bedding (M-BTM-C8, Innovive, San Diego, CA.) and M-innorichment nesting sheets with acidified water (M-WB- 300) and irradiated rodent chow (5061 -Pico-Vac®Rodent Diet 20, Lab Diet Richmond, Indiana) provided ad libitum. The room was under a 12:12 light–dark cycle with room temperature maintained at 20.5 to −22.5 °C and humidity between 30–50%. All animal studies were performed in compliance with the IACUC protocol (IACUC00031) of the Children’s Mercy Research Institute.
Immunofluorescence of human HCC xenografts for G3BP1 and PABP
Tumors were obtained from xenografts. Formalin-fixed paraffin-embedded (FFPE) tissue sections on glass slides were heated at 50 °C for 5 min to melt paraffin and incubated twice for 5 min in xylene, followed by incubation in decreasing concentrations of ethanol for 3 min each: 100%, 96%, 90%, 90%, and 80%. After rinsing in cold, running tap water for 5 min, slides were boiled in citrate buffer (pH 6.0) for 5 min before steaming for an additional 20 min. After washing with 1X PBS 3 times, primary blocking solution, containing 1.0% horse serum and 0.3% Triton X-100 in 1X PBS, was applied to the slides for 30 min. Then, primary antibodies (PABP, G3BP1) in blocking solution were incubated on tumor sections in a humidity chamber overnight at 4 °C, followed by incubation with secondary antibodies for 2 h at room temperature. After washing, slides were mounted with Prolong AntiFade Gold Mountant and dried overnight before imaging on the all-in-one BZ-X800 microscope.
Immunohistochemistry (IHC) of human HCC xenografts for p53 and CC3
Deparaffinized and antigen-retrieved FFPE tumor sections were washed for 5 min in 1X PBS. Slides were then prepared using the ImmPRESS® Horse Anti-Mouse IgG PLUS Polymer Kit (MP-7802-15, Vector Laboratories, Newark, CA, USA) according to manufacturers’ instructions. For the primary antibody, p53 DO-1 mouse monoclonal antibody was used at a 1:200 dilution, while cleaved caspase-3 antibody was used at a 1:50 dilution. For visualization of staining, the ImmPACT DAB kit was used (SK-4105, Vector Laboratories). For counterstain, hematoxylin (H-3401-500, Vector Laboratories) was applied to slides for 5 min, then washed in 40 °C water for 5 min, followed by a wash in running tap water for 30 min. Finally, slides were cleared by two 10 s incubations in 95% EtOH, two 10 s incubations in 100% EtOH, and two 1 min incubations in xylene. Coverslips were then mounted using VectaMount Permanent Mounting Medium (H-5000-60, Vector Laboratories).
TCGA (The Cancer Genome Atlas) database analysis
TCGA datasets, which have been collected since 2006, were accessed via cBioPortal.org. Patient clinical data and p53 mutation status were downloaded for indicated datasets, and data was imported into Excel. Patients were stratified according to the p53 status: wtp53 (no variant), p53null (deletion, nonsense, frameshift, splice site mutations), or missense mutp53 (missense mutations). The p53 status was then paired to overall survival in months, and Kaplan-Meier curves were plotted, and p-value was calculated using GraphPad Prism 5 software (GraphPad Software, La Jolla, CA, USA).
For expression of G3BP1, the G3BP1 mRNA expression (Batch normalized from Illumina HiSeq_RNASeqV2) (log2(value + 1)) was paired to each patient’s p53 status and overall survival. The mean G3BP1 mRNA expression of this dataset was calculated and patients with relative G3BP1 mRNA expression at or below the mean were categorized as low G3BP1, while patients with relative G3BP1 expression above the mean were categorized as high G3BP1. Patients whose data was missing in one or more categories (p53 status, overall survival, or G3BP1 mRNA expression) were not included in the corresponding analysis.
Statistics and reproducibility
All experiments were performed independently at least three times with mean and S.E.M. calculated using GraphPad Prism 5, unless specifically mentioned in the figure legend. Statistical significance was analyzed by two-tailed Student’s t-test or two-way Analysis of Variance (ANOVA) with Geisser-Greenhouse correction using GraphPad Prism 5 as indicated in figure legends. Flow cytometry data was gated using BD FACSDiva software. P-values < 0.05 were considered statistically significant.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data supporting the findings of this study are available within the main text, main figures, supplementary information, and source data file. Source data are provided with this paper.
References
Zhan, Y. et al. Understanding the roles of stress granule during chemotherapy for patients with malignant tumors. Am. J. Cancer Res. 10, 2226–2241 (2020).
Arimoto, K., Fukuda, H., Imajoh-Ohmi, S., Saito, H. & Takekawa, M. Formation of stress granules inhibits apoptosis by suppressing stress-responsive MAPK pathways. Nat. Cell Biol. 10, 1324–1332 (2008).
Kim, W. J., Back, S. H., Kim, V., Ryu, I. & Jang, S. K. Sequestration of TRAF2 into stress granules interrupts tumor necrosis factor signaling under stress conditions. Mol. Cell Biol. 25, 2450–2462 (2005).
Khong, A., Matheny, T., Huynh, T. N., Babl, V. & Parker, R. Limited effects of m(6)A modification on mRNA partitioning into stress granules. Nat. Commun. 13, 3735 (2022).
Buttgereit, F. & Brand, M. D. A hierarchy of ATP-consuming processes in mammalian cells. Biochem. J. 312, 163–167 (1995).
Wortel, I. M. N., van der Meer, L. T., Kilberg, M. S. & van Leeuwen, F. N. Surviving stress: modulation of ATF4-mediated stress responses in normal and malignant cells. Trends Endocrinol. Metab. 28, 794–806 (2017).
Parrales, A. & Iwakuma, T. Targeting oncogenic mutant p53 for cancer therapy. Front. Oncol. 5, 288 (2015).
Alvarado-Ortiz, E. et al. Mutant p53 gain-of-function: role in cancer development, progression, and therapeutic approaches. Front. Cell Dev. Biol. 8, 607670 (2020).
Yamamoto, S. & Iwakuma, T. Regulators of oncogenic mutant TP53 gain of function. Cancers 11. https://doi.org/10.3390/cancers11010004 (2018).
Olivier, M. et al. The clinical value of somatic TP53 gene mutations in 1,794 patients with breast cancer. Clin. Cancer Res. 12, 1157–1167 (2006).
Zhu, G. et al. Mutant p53 in cancer progression and targeted therapies. Front. Oncol. 10, 595187 (2020).
Nishikawa, S. & Iwakuma, T. Drugs targeting p53 mutations with FDA approval and in clinical trials. Cancers 15. https://doi.org/10.3390/cancers15020429 (2023).
Llovet, J. M. et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 359, 378–390 (2008).
Adjibade, P. et al. Sorafenib, a multikinase inhibitor, induces formation of stress granules in hepatocarcinoma cells. Oncotarget 6, 43927–43943 (2015).
Robles, A. I., Jen, J. & Harris, C. C. Clinical outcomes of TP53 mutations in cancers. Cold Spring Harb. Perspect. Med. 6. https://doi.org/10.1101/cshperspect.a026294 (2016).
Kennedy, M. C. & Lowe, S. W. Mutant p53: it’s not all one and the same. Cell Death Differ. 29, 983–987 (2022).
Gao, J. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 6, pl1 (2013).
Tang, J. et al. Targeted sequencing reveals the mutational landscape responsible for sorafenib therapy in advanced hepatocellular carcinoma. Theranostics 10, 5384–5397 (2020).
Szaflarski, W. et al. Vinca alkaloid drugs promote stress-induced translational repression and stress granule formation. Oncotarget 7, 30307–30322 (2016).
Corazzari, M., Gagliardi, M., Fimia, G. M. & Piacentini, M. Endoplasmic reticulum stress, unfolded protein response, and cancer cell fate. Front. Oncol. 7, 78 (2017).
Reineke, L. C. & Neilson, J. R. Differences between acute and chronic stress granules, and how these differences may impact function in human disease. Biochem. Pharm. 162, 123–131 (2019).
Grabocka, E. & Bar-Sagi, D. Mutant KRAS enhances tumor cell fitness by upregulating stress granules. Cell 167, 1803–1813.e1812 (2016).
Cui, W., Li, J., Ron, D. & Sha, B. The structure of the PERK kinase domain suggests the mechanism for its activation. Acta Crystallogr D. Biol. Crystallogr 67, 423–428 (2011).
Kedersha, N. & Anderson, P. Mammalian stress granules and processing bodies. Methods Enzymol. 431, 61–81 (2007).
Sidibe, H., Dubinski, A. & Vande Velde, C. The multi-functional RNA-binding protein G3BP1 and its potential implication in neurodegenerative disease. J. Neurochem 157, 944–962 (2021).
Wolozin, B. & Ivanov, P. Stress granules and neurodegeneration. Nat. Rev. Neurosci. 20, 649–666 (2019).
Cascella, R. et al. The release of toxic oligomers from alpha-synuclein fibrils induces dysfunction in neuronal cells. Nat. Commun. 12, 1814 (2021).
Aulas, A. et al. G3BP1 promotes stress-induced RNA granule interactions to preserve polyadenylated mRNA. J. Cell Biol. 209, 73–84 (2015).
Sicari, D. et al. Mutant p53 improves cancer cells’ resistance to endoplasmic reticulum stress by sustaining activation of the UPR regulator ATF6. Oncogene 38, 6184–6195 (2019).
Adomavicius, T. et al. The structural basis of translational control by eIF2 phosphorylation. Nat. Commun. 10, 2136 (2019).
Yang, P. et al. G3BP1 is a tunable switch that triggers phase separation to assemble stress granules. Cell 181, 325–345.e328 (2020).
Kim, M. M., Wiederschain, D., Kennedy, D., Hansen, E. & Yuan, Z. M. Modulation of p53 and MDM2 activity by novel interaction with Ras-GAP binding proteins (G3BP). Oncogene 26, 4209–4215 (2007).
Chen, X. & Cubillos-Ruiz, J. R. Endoplasmic reticulum stress signals in the tumour and its microenvironment. Nat. Rev. Cancer 21, 71–88 (2021).
Malhi, H., Guicciardi, M. E. & Gores, G. J. Hepatocyte death: a clear and present danger. Physiol. Rev. 90, 1165–1194 (2010).
Dolicka, D., Foti, M. & Sobolewski, C. The emerging role of stress granules in hepatocellular carcinoma. Int. J. Mol. Sci. 22. https://doi.org/10.3390/ijms22179428 (2021).
Wang, Z. et al. Loss-of-function but not gain-of-function properties of mutant tp53 are critical for the proliferation, survival, and metastasis of a broad range of cancer cells. Cancer Discov. 14, 362–379 (2024).
Mahalingam, D. et al. A phase II, multicenter, single-arm study of mipsagargin (G-202) as a second-line therapy following sorafenib for adult patients with progressive advanced hepatocellular carcinoma. Cancers 11. https://doi.org/10.3390/cancers11060833 (2019).
Parrales, A. et al. DNAJA1 controls the fate of misfolded mutant p53 through the mevalonate pathway. Nat. Cell Biol. 18, 1233–1243 (2016).
Acknowledgements
This work was supported by NIH R01 CA214916 (TI), Braden’s Hope For Childhood Cancer (TI), Masonic Cancer Alliance (TI), and NIH P30 CA168524 (RAJ). We thank Dr. Martha Montello at the Medical Writing Center in Children’s Mercy Hospital for editing this manuscript. We also thank University of Kansas Medical Center investigators Dr. Lisa Zhang and Dr. Steven A. Weinman for providing HCC cell lines, as well as Dr. Sufi Thomas for providing head and neck cell lines. We thank Dr. Souneek Chakraborty and Dr. Mrinalini Bhosale for their help with mouse experiments. Finally, we thank Dr. Dan A. Dixon, Dr. Lane Christenson, Dr. Michael Wolfe, and Dr. Subhrajit Saha, who provided helpful input.
Author information
Authors and Affiliations
Contributions
E.T. wrote the manuscript, and T.I. finalized it. E.T., A.P., A.R., S.N., and S.O. performed experiments. E.T., A.P., A.R., D.A.D., S.O., and T.I. designed the research and analyzed the data. All authors reviewed and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Thoenen, E., Ranjan, A., Parrales, A. et al. Suppression of stress granule formation is a vulnerability imposed by mutant p53. Nat Commun 16, 2365 (2025). https://doi.org/10.1038/s41467-025-57539-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-57539-6
