
Abstract
DNA–RNA hybrids triggered by double-strand breaks (DSBs) are crucial intermediates during DSB repair, and their timely resolution requires numbers of RNA helicases, including DEAD box 1 (DDX1). However, how these helicases are recruited to DSB-induced hybrids in time remains largely unclear. Here, we revealed that squamous cell carcinoma antigen recognized by T cells 3 (SART3) promotes DDX1 binding to DNA–RNA hybrids at DSBs for optimal homologous recombination (HR) repair. SART3 itself associates with DNA–RNA hybrids and PAR chains and accumulates at DSBs in both PARylation- and DNA–RNA hybrids-dependent fashion. SART3 also associates with DDX1 and is necessary for DDX1 enrichment at DSBs. The defective SART3-DDX1 association observed in cells expressing the cancer-associated variant SART3-R836W impairs not only the accumulation of DDX1, but also hybrid removal and HR efficiency. Moreover, SART3 promotes DNA end resection through enhancing USP15-BARD1 association and BRCA1-BARD1 retention. Together, our study reveals an role of SART3 in DSB repair, rendering SART3 a promising target for cancer therapy.
Similar content being viewed by others


Introduction
DNA double-strand breaks (DSBs) can seriously threaten cell survival1. To counteract the detrimental effects, cells have evolved two major DSB repair pathways, namely error-free homologous recombination (HR) and error-prone nonhomologous end-joining (NHEJ). Specifically, HR utilizes sister chromatids synthesized as templates to guide repair in S and G2 phases. As the early event to initiate HR, the CtIP-MRN complex removes a few dozen nucleotides from the 5′-strand end2. Then, Exo1 and Dna2 nucleases extend resection to generate long 3′ single-stranded DNA (ssDNA) tails which are quickly bound by replication protein A (RPA) and provide a platform to recruit repair factors3,4,5,6. Interestingly, R-loops and DNA–RNA hybrids are important intermediates that facilitate this process7.
R-loops are DNA–RNA hybrid-containing nucleic acids with important roles in regulating gene expression, chromatin structure and genomic stability8,9. Emerging evidence suggests that transient R-loops or DNA–RNA hybrids formed around DSBs protect the 3′ overhang from degradation, facilitating recruitment of HR proteins10,11. Importantly, as essential intermediates of HR, the DNA–RNA hybrids must be removed in time to ensure a precise repair process and maintain genome integrity7,12. Currently several nucleases, such as RNase H1 and H2, and several helicases, including Senataxin13, DEAD-box RNA helicase 1 (DDX1)14, DDX515, DDX1816, DDX2117, DHX918, and XRN219, have been reported to resolve DNA–RNA hybrids to promote DNA repair. Nevertheless, how these helicases are enriched at DSBs to resolve DNA–RNA hybrids in time remains largely unclear.
SART3, squamous cell carcinoma antigen recognized by T cells 3, also named Tip110, p110, or p110nrb, is an RNA-binding protein (RBP) that contains half-a-tetracopeptide repeats (HAT) in the N-terminus and two RNA recognition motifs (RRMs, RRM1/2) near the C-terminus20. SART3 is highly expressed in the nucleus of malignant tumor cell lines and a multitude of cancer tissues21. Beyond regulating gene expression and being a potential antigen for cancer immunotherapy20,22,23, SART3 also promotes activation of translesion DNA synthesis (TLS), a major DNA damage tolerance pathway, in an RNA-independent manner24. However, whether and how SART3 engages in DNA repair pathways remains unclear.
In this study, we uncovered an unexpected role of SART3 in promoting HR repair. Combined with systematic analyses of the mass spectrometry data from DNA‒RNA hybrid pull-down25 and S9.6 IP-mass18, we confirmed that SART3 is a DNA‒RNA hybrid-binding protein through pull-down assay. We found that SART3 is recruited to DSB sites in a PARylation- and DNA‒RNA hybrids-dependent manner. Depletion of SART3 compromises the focus formation of RPA32, BRCA1 and BARD1, leading to deficient DNA end resection and HR repair. Interestingly, the knockdown of SART3 upregulates the level of DNA‒RNA hybrids accumulated around DSBs. Mechanistically, SART3 enriched at DSBs not only promotes timely removal of DNA‒RNA hybrids by recruiting DDX1, but also stimulates the USP15-BARD1 interaction to facilitate end resection, thereby enhancing HR. Significantly, a cancer-associated SART3 mutation (SART3-R386W) fails to recruit DDX1 to DSBs.
Results
SART3 accumulates at DSBs through its association with DNA–RNA hybrids
To determine whether SART3 participates in DSB repair, we first examined whether GFP-SART3 could be recruited to laser-induced DNA damage sites. We found that GFP-SART3 rapidly accumulated at the damage sites and remained there for more than 5 min (Fig. 1a and Supplementary Fig. 1a). To further confirm that SART3 can be recruited to DSBs, we performed ChIP-qPCR to examine SART3 enrichment at I-SceI-induced break sites in U2OS-DR-GFP cells. We found that the occupation of SART3 around DSBs significantly increased upon I-SceI expression (Fig. 1b). Moreover, U2OS cells stably expressing GFP-SART3 that were pretreated with KU 55933 (ATM inhibitor) or NU7026 (DNA-PK inhibitor) showed no difference in the recruitment of GFP-SART3 compared to that of DMSO-treated cells (Supplementary Fig. 1b-e). However, pretreatment with a PARP inhibitor (PARPi) (ABT-888) significantly decreased the percentage of cells with GFP-SART3 accumulation at microirradiated sites compared to that of DMSO treatment (Fig. 1c and Supplementary Fig. 1b, f). Consistently, knockdown of PARP1, which accounts for more than 90% of PARylation activity after DNA damage, significantly suppressed the recruitment of SART3 to micorirradiated sites (Fig. 1d and Supplementary Fig. 1g). These data suggest that SART3 is recruited to DSBs in a PARylation-dependent manner.

a U2OS cells stably expressing GFP-SART3 were microirradiated. Cell images were captured at different time points (left). Error bars represent mean ± SEM of 10 independent measurements. Scale bar, 10 µm. b U2OS-DR-GFP cells were transfected with Flag or Flag-SART3 and then infected with lentivirus expressing I-SceI. ChIP assays were performed. c, d U2OS cells stably expressing GFP-SART3 were pretreated with ABT-888 (c) or transfected with siRNA (d), followed by microirradiation. The proportion of cells with SART3 accumulation was measured. e Schematic representation of SART3 domains. HAT half-a-tetracopeptide repeats. CC coiled-coil. NLS nuclear localization sequences. RRM RNA recognition motif. f U2OS cells were respectively transfected with a series of SART3 truncated constructs (T1, T2, T3, and T4) or full length (FL), followed by microirradiation. Representative images are shown (left). Scale bar, 10 µm. g, h U2OS cells expressing GFP-SART3 were pretreated with transcription inhibitor prior to microirradiation. Representative images are shown (right). Scale bar, 10 µm. i, j HEK293T cells transfected with the indicated constructs were harvested for incubation with biotin-DNA‒RNA hybrids (i) or S9.6 antibody (j). The proteins pulled down were analyzed by immunoblotting. k U2OS cells expressing GFP-SART3-WT or -YA1 were microirradiated. l U2OS-DR-GFP cells were transfected with the indicated constructs, followed by ChIP-qPCR as in (b). m Schematic illustrates the positions of the primers employed for ChIP-qPCR in U2OS-DR-GFP cells (top). Primer-1 and primer-2 are denoted by black and red arrow respectively. SceGFP: I-SceI-cleaved GFP. U2OS-DR-GFP cells stably expressing Flag-SART3 were transfected with GFP or GFP-RNase H1 construct followed by ChIP-qPCR as in (b). n Recombinant GST-SART3-WT and -YA1 mutant were purified and then transferred onto PVDF membrane, followed by incubation with biotin-PAR polymers and analyzed by immunoblotting. o U2OS cells transfected with GFP-SART3-WT or -YA1 were pretreated with ABT-888 for 2 h prior to microirradiation. The proportion of cells with SART3 accumulation were measured. Error bars represent mean ± SEM, N = 3 (b–d, g–h, k–m, o) or 4 (f) independent experiments, and p values were calculated using an unpaired two-tailed Student’s t test (no adjustment for multiple comparisons). Source data are provided as a Source Data file.
To examine which domain of SART3 is responsible for its recruitment to damage sites, U2OS cells transfected with SART3 full-length (FL) and several truncated GFP-SART3 constructs, i.e. T1, T2, T3 and T4, were exposed to microirradiation (Fig. 1e). The results showed that T3 and T4, containing the RRM1/2 domains, were recruited to laser-induced damage sites (Fig. 1f), which agrees with that RRM domain is a potential PAR-binding module26,27. Intriguingly, pretreatment of U2OS cells stably expressing GFP-SART3 with transcription inhibitors (DRB or ActD) dramatically suppressed the recruitment of GFP-SART3 to microirradiated sites (Fig. 1g, h). Through analyses of mass spectrometry data from DNA–RNA hybrid pull-down and S9.6 IP-mass18,25, we speculate that SART3 is a potential binding protein of DNA–RNA hybrids. We hypothesize that SART3 enrichment at damage sites depends on its association with the DNA‒RNA hybrids formed at DSBs. To test our hypothesis, we synthesized a biotin-labeled DNA–RNA hybrid to examine its association with SART3 by pull-down assay. Similar to DDX21, SART3 displayed high binding affinity with the hybrids but not dsDNA (Supplementary Fig. 1h). Additionally, the hybrid association of SART3 was mediated by its RRM domain (Supplementary Fig. 1i). To further dissect the key residues in SART3 for this association, we generated four SART3 mutants, i.e., Y845AY848A (YA1), Y748AY750A (YA2), F753A (FA1), and F804AF810A (FA2), by mutating the RRM consensus sequence ([RK]-G-[FY]-[GA]-[FY]-[ILV]-X-[FY] or [ILV]-[FY]-[ILV]-X-N-L) (Supplementary Fig. 1j)28. Only the YA1 mutant failed to be pulled down by the biotin- DNA–RNA hybrid (Fig. 1i). Co-IP using the S9.6 antibody further confirmed that SART3 WT, instead of YA1 or ΔRRM (T1) mutants, bound to DNA–RNA hybrids (Fig. 1j), suggesting that Y845 and Y848 in RRM2 are required for SART3 binding to DNA–RNA hybrids. Moreover, YA1, but not other mutants, exhibited a significant reduction in accumulation at laser-induced damage sites (Fig. 1k and Supplementary Fig. 1k, l). ChIP-qPCR assay also showed that the SART3-YA1 mutant could not be enriched at DSBs (Fig. 1l). Additionally, overexpression of the nuclear-localized RNase H1 or treatment with RNase H, which resolves DNA–RNA hybrids, significantly attenuated the enrichment of SART3 at damage sites (Fig. 1m and Supplementary Fig. 1m, n). These data indicate that disrupting the association of SART3 with DNA–RNA hybrids largely impairs its accumulation at DSBs.
Given that PARPi pretreatment inhibits the accumulation of SART3 at damage sites, we compared the PAR binding ability of SART3 WT and YA1 mutant. Both GST-SART3 WT and YA1 mutant exhibited similar binding affinity with PAR chains (Fig. 1n). Thus, the YA1 mutant retains its PAR binding ability despite losing its association with DNA‒RNA hybrids. Although PARPi pretreatment led to a significant reduction in the recruitment of SART3 WT to damage sites induced by microirradiation or I-SceI cleavage (Fig. 1o and Supplementary Fig. 1o, p), it failed to further reduce the downregulated recruitment of the SART3-YA1 mutant. These data suggest that PARylation and DNA–RNA hybrids induced by DSBs likely act in an epistatic manner to promote SART3 accumulation. Collectively, our results indicate that SART3 can be targeted to DSBs, which is, at least in part, mediated through its binding with the DNA–RNA hybrids formed at DSBs.
SART3 promotes DSB repair
Considering that SART3 can be recruited to DSBs, we next investigated whether it is involved in DSB repair. Under unperturbed conditions, depletion of SART3 resulted in an elevated level of γH2AX, which could be rescued by complementing with SART3 (Fig. 2a). Upon ETO exposure, the γH2AX signal increased notably at 0.5 h post-treatment, then gradually decreased following recovery (Fig. 2a, b). Notably, depletion of SART3 caused an obvious delay in DSB repair as reflected by a much slower decrease in γH2AX levels at later time points (3 and 8 h) compared to the siNC control. These data imply that SART3 participates in DSB repair. We then examined whether the knockdown of SART3 affects HR efficiency by using DR-GFP reporter cells. The data showed that the depletion of SART3 significantly reduced HR efficiency but had no effect on cell cycle distribution (Fig. 2c and Supplementary Fig. 2a–c). Moreover, complementing SART3 in SART3-depleted cells fully rescued the HR efficiency (Fig. 2c). In addition, depletion of SART3 sensitized cells to multiple DNA damage-inducing agents, including ETO, CPT, HU and Olaparib, which was fully rescued in cells complemented with SART3 (Fig. 2d–g and Supplementary Fig. 2d–f). Double knockdown of PARP1 and SART3 did not further sensitize cells to either CPT or ETO treatment compared to SART3 depletion alone (Supplementary Fig. 2g–i), suggesting that SART3 and PARP1 function within the same epistatic pathway in response to CPT- or ETO-induced cell killing. Taken together, these data suggest that SART3 promotes HR repair.

a, b U2OS cells stably expressing GFP or GFP-SART3 were transfected with siNC or siSART3-1, followed by treatment with DMSO or ETO (10 µM for 2 h, and allowed to repair 0.5 h). The levels of γH2AX were analyzed by immunoblotting (a). U2OS cells were transfected with siNC or siSART3-1/3, followed by treatment with DMSO or ETO (10 µM for 2 h) and further recovery for 0.5, 3 or 8 h. The samples were analyzed by immunoblotting using indicated antibodies. b The intensity of γH2AX was quantified by Image J software and normalized. c U2OS-DR-GFP cells stably expressing Flag-SART3 or Flag were transfected with siNC, siSART3-1 or siCtIP. 24 h later, cells were infected with I-SceI lentivirus. Percentage of GFP-positive cells was quantitated by FACS at 48 h after virus infection (top). The SART3 knockdown efficiency was verified by immunoblotting (bottom). d–g U2OS cells stably expressing either GFP-SART3 or GFP were transfected with siNC or siSART3, followed by treatment with indicated concentrations of ETO for 24 h (e), CPT for 1 h (f), HU for 2 h (g) or Olaparib for 48 h (h). The cells were further incubated for colony formation. SiSART3 indicates siSART3-1, if not specified. In (c–g), error bars represent mean ± SEM (N = 3 independent experiments), and p values were calculated using unpaired two-tailed Student’s t test (c) or one-way ANOVA analysis with Tukey test (d–g). Source data are provided as a Source Data file.
SART3 promotes DNA end resection
To understand how SART3 modulates HR repair, we explored the effects of depleting SART3 on the accumulation of several representative DDR factors upon ETO exposure. Depletion of SART3 compromised the enrichments of BRCA1, BARD1, and RPA32, which were fully rescued by supplementing with GFP-SART3 (Fig. 3a–c and Supplementary Fig. 3a–e). However, SART3 ablation did not affect the recruitment of RNF8 and RNF168 (Fig. 3d, e), suggesting that SART3 functions downstream of RNF168. We then determined whether it affects DNA end resection. SART3 ablation caused a remarkable reduction in RPA32 phosphorylation at S33 in the presence of CPT treatment compared to siNC control (Fig. 3f), while it did not reduce the CPT-induced accumulation of Top1cc (Supplementary Fig. 3f), which can trigger RPA phosphorylation29. We utilized the U2OS cell line stably integrating ER-AsiSI-HA to quantify the endogenous efficiency of DSB end resection30 (Fig. 3g and Supplementary Fig. 3g). The result showed that DNA end resection was significantly impaired in SART3-depleted cells compared to that in control cells (Fig. 3g).

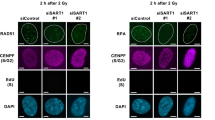
a–e U2OS, or U2OS cells stably expressing GFP or GFP-SART3 were transfected with siNC or siSART3. After 48 h, cells were treated with 10 µM ETO for 2 h and further recovered for 2 h. Immunofluorescence assays were performed using antibodies against BRCA1 (a), BARD1 (b), RPA32 (c), RNF8 (d), and RNF168 (e). Representative images are shown (top or left). The proportions of cells with foci were measured (bottom or right). Scale bars for overall and magnified images are respectively 50 and 10 µm. f U2OS cells stably expressing GFP or GFP-SART3 were transfected with siNC or siSART3 for 48 h, followed by treatment with 5 µM CPT for 2 h and further cultured for 3 h. Whole-cell lysates were harvested for immunoblotting with antibodies as indicated. g Cartoon illustrates a quantitative DNA resection assay (top). U2OS-ER-AsiSI cells transfected with the indicated siRNA were treated with 4-OHT. The gDNA was extracted and digested overnight followed by qPCR to measure DNA end resection (bottom). Immunoblotting verifies the knockdown efficiency of siRNAs (right). h U2OS cells were transfected with siNC or three different siRNAs targeting SART3, followed by incubation with 10 µM BrdU for 48 h. The cells were then treated with 5 µM CPT for 2 h and recovered for an additional 3 h. Immunofluorescence assays were performed using an antibody against BrdU. Denaturation was carried out using 2 M hydrochloric acid for 10 min. Representative images under native (non-denaturing) condition (left) and denatured condition (middle) are shown, and the proportion of cells with foci was quantified (right). Knockdown efficiencies of SART3 are examined by Western blotting (bottom-right). Scale bars for overall and magnified images are respectively 50 and 10 µm. In (a–e, g, h), error bars represent mean ± SEM (N = 3 independent experiments), and p values were calculated using unpaired two-tailed Student’s t test. Source data are provided as a Source Data file.
To further support that SART3 facilitates DNA end resection, SART3-depleted U2OS cells were labeled with 5-bromo-2′-deoxyuridine (BrdU), followed by ETO treatment. The results of immunofluorescence analysis showed that depletion of SART3 impaired BrdU foci under non-denaturing conditions, which reflects the ssDNA generated by DNA end resection (Fig. 3h), when the entire nuclear DNA was evenly labeled with BrdU as shown under denaturing conditions (Fig. 3h). We also examined whether depletion of SART3 affects the recruitment of CtIP or MRE11, two critical factors for end resection initiation. Although SART3 colocalized with CtIP and MRE11 at microirradiated damage sites, the knockdown of SART3 did not affect the recruitment of MRE11 and CtIP (Supplementary Fig. 3h–l), suggesting that SART3 regulates DNA end resection through other mechanisms. Interestingly, overexpression of Flag-RNase H1 or pretreatment with PARPi also impaired DNA end resection in siNC-treated cells, while their combination with SART3 depletion did not further decrease BrdU foci formation under non-denaturing conditions compared to SART3 depletion alone (Supplementary Fig. 3m, n). These results imply that SART3, DNA-RNA-hybrids, and PARylation function within the same epistatic pathway to promote DNA end resection. Notably, the knockdown of SART3 could reverse the resistance of BRCA1-deficient SUM149PT cells to PARPi caused by 53BP1 loss (Supplementary Fig. 3o), suggesting that SART3 also promotes HR downstream of end resection. Since SART3 can regulate gene expression, we then examined the protein levels of several DDR core factors in SART3-depleted cells. The results showed that the knockdown of SART3 had no effect on the protein levels of BRCA1, BARD1, CtIP, MRE11, RNF168, RAD51, and RPA32 (Supplementary Fig. 3p). Together, these results suggest that SART3 modulates end resection and later stages to promote HR.
SART3 recruits DDX1 to resolve DNA‒RNA hybrids at DSBs
Given that SART3 binds to DSB-induced DNA–RNA hybrids, which are transient intermediates to regulate DNA end resection and RPA recruitment10,11, we then examined whether SART3 loss affects the accumulation of DNA–RNA hybrids. As expected, CPT- or Irradiation (IR)-treated cells displayed an increase in hybrid accumulation (Fig. 4a, b and Supplementary Fig. 4a, b). Moreover, depletion of SART3 significantly upregulated the accumulation of DNA–RNA hybrids in the presence or absence of damage treatment when compared to siNC controls (Fig. 4a, b and Supplementary Fig. 4a, b). Furthermore, the damage-induced upregulation of hybrids could be completely abolished by treatment with RNase H (Fig. 4a, b and Supplementary Fig. 4a, b), an enzyme that removes DNA–RNA hybrids by degrading the RNA moiety of the structures. To further confirm that SART3 depletion promotes DNA–RNA hybrids accumulation at DSBs, we performed a DRIP-qPCR in U2OS-ER-AsiSI cells. The results showed that the depletion of SART3 led to an obvious enrichment of DNA–RNA hybrids recognized by S9.6 antibody around multiple AsiSI cutting sites, compared to the siNC control (Fig. 4c, d). Given that the RNase H1-D210N mutant is able to bind DNA–RNA hybrids but unable to degrade RNA moieties, we also performed R-ChIP analysis with U2OS-DR-GFP cells stably expressing V5-RNase H1-D210N to measure hybrid levels. Consistently, SART3 knockdown resulted in a substantial accumulation of the hybrids around I-SceI–induced DSBs (Fig. 4e). In addition, SART3 loss also upregulated the hybrid level at the NEAT1 transcription start site, a region prone to form the hybrids31 (Fig. 4e). Notably, similar to SART3 depletion, PARPi treatment also upregulated the level of DNA–RNA hybrids around DSBs (Supplementary Fig. 4c). Moreover, the combination of SART3 depletion with PARPi treatment failed to further increase the level of DNA–RNA hybrids compared to SART3 depletion or PARPi treatment alone. These results suggest that SART3-mediated removal of DNA–RNA hybrids is PARylation-dependent. Together, these results imply that SART3 is required for the timely dissolution of DNA–RNA hybrids once it is recruited by the DSB-induced structures.

a, b U2OS cells were transfected with siNC or siSART3. After 36 h, cells were treated with 5 µM CPT for 2 h, followed by S9.6 immunostaining. The numbers represent mean S9.6 intensity per nucleus measured by Cellprofiler (a). N = 281, 391, 364, 338, 301, 302, 308, 348, 330 correspond to the nine groups shown on the x-axis, based on three replicates. Error bars represent mean ± SD. P values were calculated using a two-tailed Mann–Whitney U test. Representative images are shown (b), with the areas highlighted by circles quantified for S9.6 intensity analysis. Scale bar, 10 µm. c Cartoon deciphers DR-IP (top). U2OS-ER-AsiSI cells were transfected with siNC or siSART3. After 48 h, cells were treated with 4-OHT followed by DRIP-qPCR analysis (bottom). d DRIP-qPCR analysis was performed as in (c) supplemented with RNase H during genome cleavage. The primers spanning the AsiSI-induced HR-1 break site and the actin exon region were used for qPCR. The yellow shaded area represents RNase H treatment. e R-ChIP-qPCR analysis of DNA‒RNA hybrids. Cartoon deciphers R-ChIP (top). U2OS-DR-GFP cells stably expressing V5-RNase H1-D210N mutant were transfected with siNC or siSART3. 24 h later, cells were infected with I-SceI lentivirus. After 36 h, cells were harvested followed by R-ChIP. The levels of DNA-RNA hybrids around DSBs were detected by qPCR. In (c–e), error bars represent mean ± SEM (N = 3 independent experiments), and p values were calculated using unpaired two-tailed Student’s t test. Source data are provided as a Source Data file.
To explore how SART3 regulates DNA‒RNA hybrid levels at DSBs, we performed TAP-MS to identify its binding partners by overexpressing SFB-SART3 in HEK293T cells. Several potential SART3-associated proteins are RNA helicases or nucleases, including DDX1, DDX5, DDX17, DDX21, and XRN2, which have been reported to inhibit DNA‒RNA hybrid levels14,15,17,19 (Supplementary Fig. 5a). Considering that knockdown SART3 upregulates the level of hybrids, we speculate that SART3 recruits some of these enzymes for the timely removal of DNA–RNA hybrids. To test our hypothesis, we first confirmed the interactions of SART3 with Flag-DDX1, Flag-DDX5, Flag-DDX21, Flag-DDX17, and GFP-XRN2 via Co-IP (Fig. 5a and Supplementary Fig. 5b–e). We then examined whether SART3 regulates the recruitment of these factors to damage sites. Our data showed that depletion of SART3 impaired the recruitment of DDX1, but not DDX5, DDX17, DDX21, and XRN2 (Fig. 5b and Supplementary Fig. 5f–k). Moreover, the depletion of DDX1 did not affect the recruitment of SART3 to sites of damage (Supplementary Fig. 5l, m).

a HEK293T cells transfected with the indicated constructs were lysed in the presence of RNase A, followed by immunoprecipitation and immunoblotting. b U2OS cells stably expressing GFP or GFP-SART3 were transfected with siNC or siSART3, followed by treatment with 5 µM CPT for 2 h and further recovery for 1 h. The cells were fixed and subjected to immunostaining and quantification analysis (right). The percentages of cells with more than 10 DDX1 foci were quantified. Scale bars for overall and magnified images are respectively 50 and 10 µm. c HEK293T cells transfected with GFP or GFP-SART3 were treated with CPT (5 µM, 2 h), followed by recovery and subsequent immunoprecipitation and immunoblotting. d U2OS cells stably expressing Flag or Flag-RNase H1 were transfected with siNC or siSART3. Cells were microirradiated and fixed, followed by co-immunofluorescence staining with anti-γH2AX and anti-DDX1 antibodies. Representative images are shown (left). Scale bar, 10 µm. e HEK293T cells expressing various of GFP-SART3 truncations were lysed in the presence of RNase A and Benzonase, followed by immunoprecipitation and immunoblotting. f HEK293T cells transfected with the indicated constructs were lysed for immunoprecipitation, followed by immunoblotting. g HEK293T cells transfected with the indicated constructs were harvested and incubated with biotin-DNA‒RNA hybrids. Pulled-down proteins were analyzed by immunoblotting, and GFP-SART3 levels were quantified by Image J, normalized to input SART3. h, i U2OS cells stably expressing GFP, GFP-SART3 (WT), GFP-SART3-R836W (h) or GFP-SART3-T4 (i) were transfected with siNC or siSART3. Cells were then treated with CPT, followed by co-immunofluorescence staining with anti-DDX1 and anti-γH2AX antibodies. The percentages of cells with more than 10 DDX1 foci were quantified (middle). Representative images are shown (left). Scale bars for overall and magnified images are respectively 50 and 10 µm. The knockdown efficiency of SART3 was determined by Western blotting (right). Error bars represent the mean ± SEM, N = 3 (b, h, i) or 4 (d) independent experiments, and p values were calculated using unpaired two-tailed Student’s t test. Source data are provided as a Source Data file.
Since DDX1 can resolve DNA‒RNA hybrids formed at DSBs to facilitate HR14, we hypothesize that SART3 recruits DDX1 to dissolve the hybrids. We further confirmed the SART3-DDX1 interaction by pull-down assay. The results showed that GST-SART3 bound to endogenous DDX1, and vice versa (Supplementary Fig. 5n, o). Moreover, the SART3-DDX1 association exhibited a dynamic pattern after CPT treatment, peaking at 1 h and decreasing to baseline at 2 h post-treatment (Fig. 5c). Notably, analogous to SART3 depletion, expression of RNase H1 impaired the enrichment of DDX1 at damage sites (Fig. 5d), suggesting that the recruitment of DDX1 also relies on DNA‒RNA hybrids. We then mapped the regions of DDX1 and SART3 responsible for their reciprocal association. The data showed that the RRM domain in SART3 and the helicase ATP-binding domain in DDX1 mediated the interaction (Fig. 5e and Supplementary Fig. 5p). Of great interest, among several SART3 recurrent missense variants identified from the Catalogue of Somatic Mutations In Cancer database (https://cancer.sanger.ac.uk), the R836W (a mutant of unknown clinical significance) and the R836A mutation were found to significantly inhibit the SART3-DDX1 association (Fig. 5f), with no obvious effect on SART3 binding to DNA‒RNA hybrids (Fig. 5g). Furthermore, like SART3 WT, the R836W mutant could be recruited to laser-induced damage sites, which could be markedly attenuated upon expression of RNase H1 (Supplementary Fig. 5q). In addition, the R836W mutant failed to restore the reduced DDX1 foci in SART3-depleted cells, while the T4 variant successfully rescued it (Fig. 5h, i). These results suggest that binding to SART3 is required for optimal DDX1 accumulation at sites of damage.
Next, we found that depletion of SART3 or DDX1 led to an increased S9.6 signal in the nucleoplasm of U2OS cells with or without CPT treatment (Fig. 6a and Supplementary Fig. 6a). However, simultaneous depletion of SART3 and DDX1 did not cause an obvious further increase of DNA‒RNA hybrid level compared with SART3 or DDX1 knockdown alone, as detected by either S9.6 immunofluorescence or DRIP-qPCR analysis (Fig. 6a, b and Supplementary Fig. 6a–c). These results suggest that SART3 and DDX1 regulate the accumulation of DNA‒RNA hybrids in an epistatic manner. Given that SART3 binds to both DDX1 and DNA‒RNA hybrids, we wondered whether SART3 could promote the association between DDX1 and DNA‒RNA hybrids for timely resolution of hybrids. We found that SART3 enhanced both the interaction between DDX1 and DNA‒RNA hybrids (Fig. 6c), and the DNA‒RNA-unwinding activity of DDX1 (Fig. 6d). In line with the fact that the R836W mutant but not the T4 variant, exhibits defects in both binding to DDX1 and promoting DDX1 foci formation (Fig. 5f–i), R836W but not T4 failed to restore the SART3 depletion-induced DNA‒RNA hybrids upregulation (Fig. 6e and Supplementary Fig. 6d–f). These results support that SART3 facilitates DDX1 binding to DNA–RNA hybrids to promote timely removal of the structures. Consequently, unlike SART3 WT, the R836W mutant failed to fully rescue the HR defect (Fig. 6f and Supplementary Fig. 6g). Notably, concurrent expression of RNase H1 in R836W-reconstituted cells even diminished the partial rescue effect caused by R836W (Fig. 6f and Supplementary Fig. 6g). Lastly, the R836W mutant only partially rescued the hypersensitivities to CPT and PARPi caused by SART3 loss (Fig. 6g, h). In line with the fact that DDX1 is not essential for DNA end resection, the R836W mutant retained the ability to fully rescue the end resection defect caused by SART3 depletion (Supplementary Fig. 6h). Together, our results suggest that SART3 promotes HR partially through recruiting DDX1 to DSBs to limit the accumulation of DNA‒RNA hybrids.

a U2OS cells were transfected with siNC, siSART3, siDDX1, or siSART3 and siDDX1, followed by CPT treatment. The samples then were performed immunostaining using S9.6 antibody. The numbers represent mean S9.6 intensity per nucleus measured by Cellprofiler. N = 263, 560, 570, 530, 322, 366, 410, 343, 371, 286, 332,414 correspond to the twelve groups shown on the x-axis, based on three replicates. Error bars represent mean ± SD. P values were calculated using a two-tailed Mann–Whitney U test. b U2OS-ER-AsiSI cells transfected with indicated siRNAs were treated with 4-OHT. The gDNA was extracted for DRIP-qPCR. The yellow shaded area represents RNase H treatment. c Purified DDX1 (15 µg) and increased amounts of SART3 (3.75, 7.5, 15, 30 µg) were incubated with 30 picomoles of biotin-DNA-RNA or biotin-dsDNA, followed by streptavidin-bead incubation. d The various amounts of DDX1 (0.05, 0.1, and 0.15 µg) were respectively incubated with FAM-labeled DNA-RNA substrates in the absence or presence of purified SART3 (0.2 µg). The samples were then examined by 12% native polyacrylamide gel for analysis. D 25 nt represents a DNA length of 25 nt, and R 25 nt represents an RNA length of 25 nt. The numbers represent the lane order. e U2OS-ER-AsiSI cells stably expressing GFP, GFP-SART3 (WT), GFP-SART3-R836W, GFP-SART3-YA1, or GFP-SART3-T4 were transfected with siNC or siSART3. After 48 h, cells were treated with 4-OHT followed by DRIP-qPCR analyses. f U2OS-DR-GFP cells stably expressing Flag, Flag-SART3 (WT), Flag-SART3-R836W or both Flag-SART3-R836W and Flag-RNase H1 were transfected with siNC or siSART3, followed by infection with I-SceI lentivirus. The percentage of GFP-positive cells was quantified by FACS analysis. g, h U2OS cells stably expressing GFP, GFP-SART3, or GFP-SART3-R836W were transfected with siRNA, treated with CPT for 24 h followed by CCK8 assay (g), or Olaparib for 48 h followed by colony formation assay (h). In (b), and (e–h), error bars represent mean ± SEM (N = 3 independent experiments), and p values were calculated using unpaired two-tailed Student’s t test (b, e, f) or one-way ANOVA analysis with Tukey test (g, h). Source data are provided as a Source Data file.
SART3 promotes USP15-BARD1 association and BARD1 deubiquitination
We then explored how SART3 loss impairs DNA end resection. It is known that USP15, a SART3 binding partner, can deubiquitinate BARD1 to maintain BRCA1/BARD1 retention at DSBs, thereby promoting end resection32,33. We wondered whether SART3 facilitates this event by modulating USP15-BARD1 association. We co-transfected GFP-USP15 and Myc-BARD1 in SART3-depleted HEK293T cells for Co-IP analysis. Consistent with the previous study33, CPT treatment enhanced the USP15-BARD1 association (Fig. 7a). Interestingly, SART3 depletion remarkably blocked the stimulatory effect. Moreover, SART3 knockdown also impaired the chromatin binding of USP15 and BARD1 after CPT treatment (Fig. 7b). These data suggest that SART3 facilitates the USP15-BARD1 interaction and BRAD1 retention at DSBs. Conversely, USP15 did not impact the recruitment of SART3 to the sites of damage (Supplementary Fig. 7a, b).

a HEK293T cells transfected with the indicated constructs were treated with CPT (5 µM, 2 h), followed by immunoprecipitation with anti-GFP agarose beads and immunoblotting with the indicated antibodies. Asterisks indicate non-specific bands. b U2OS cells transfected with siRNAs were treated with CPT and recovered for 0 h, 2 h, or 5 h, respectively. Triton-insoluble fractions (TIF) and whole-cell lysates (WCL) were harvested for immunoblotting. “R” stands for recovery. c, d HEK293T cell expressing GFP-SART3 and Myc-BARD1 were immunoprecipitated with anti-GFP beads, followed by immunoblotting with anti-Myc or anti-GFP (c). HEK293T cell lysates were immunoprecipitated with anti-BARD1 antibodies, followed by immunoblotting with antibodies against SART3 or USP15 (d). e Schematic representation of the SART3 truncated mutants (top). HEK293T cells co-transfected with Myc-BARD1 and GFP-SART3 truncations were harvested for Co-IP with anti-GFP beads, followed by immunoblotting with antibodies against Myc, GFP, or USP15. f HEK293T cells were co-transfected with GFP-USP15, Myc-BARD1, and Flag-SART3-WT or truncations. 36 h later, cells were treated with CPT, followed by Co-IP. g HEK293T cells transfected with the indicated siRNAs were co-transfected with HA-Ub, Flag-BARD1 and GFP-SART3-WT or truncations. 36 h later, cells were treated with CPT and subjected to denatured IP with anti-Flag agarose beads, followed by immunoblotting with the indicated antibodies. h, i U2OS cells transfected with siNC, siSART3, siUSP15-1, or both siSART3 and siUSP15-1 were treated with CPT (5 µM, 2 h) and further cultured for 2 h, followed by immunofluorescence with antibodies against BRCA1 or BARD1. Representative images are shown (left). Proportion of cells with BRCA1 or BARD1 foci was measured (right). Scale bars for overall and magnified images are respectively 50 and 10 µm. j U2OS-DR-GFP cells stably expressing Flag, Flag-SART3, Flag-SART3-ΔHAT4-7 or Flag-SART3-ΔRRM were transfected with either siNC or siSART3. After 24 h, cells were infected with I-SceI lentivirus. The percentage of GFP-positive cells was quantitated by FACS 48 h post I-SceI lentivirus infection. In (h–j), error bars represent mean ± SEM (N = 3 independent experiments), and p values were calculated using unpaired two-tailed Student’s t test. Source data are provided as a Source Data file.
To understand how SART3 promotes USP15-BARD1 association, we first co-transfected GFP-SART3 and Myc-BARD1 into 293T cells. Co-IP results showed their interaction (Fig. 7c) and their endogenous association (Fig. 7d). We further confirmed the direct association of SART3 with BARD1 or USP15 (Supplementary Fig. 7c, d). Moreover, CPT treatment promoted SART3-BARD1 association (Supplementary Fig. 7e). To determine which region of SART3 mediates its association with BARD1, we co-expressed Myc-BARD1 with a series of GFP-SART3 constructs in 293T cells. The Co-IP data showed that the SART3 T1 mutant (deletion of RRM1/2) but not T4 or T5, failed to bind BARD1 (Fig. 7e). Since the SART3 RRM1/2 domain also binds RNA, we examined whether the SART3-BARD1 interaction is mediated by RNA. RNase A treatment did not attenuate the SART3-BARD1 association (Supplementary Fig. 7f). Moreover, the SART3 YA1 and the R836W mutation did not affect the association (Supplementary Fig. 7g, h). These data suggest that RNA binding is not necessary for SART3-BARD1 association. We also found that the BARD1 N-terminus was responsible for its association with SART3 (Supplementary Fig. 7i). Consistent with the fact that the HAT domain of SART3 mediates its interaction with USP1534, we showed that the T1 but not T4 and T5 mutants associated with USP15 (Fig. 7e). We further dissected the HAT domain and found that the SART3-ΔHAT-4-7 mutant (deletion of HAT motifs 4–7) abolished its binding to USP15 (Supplementary Fig. 7j), suggesting that HAT (4–7) region of SART3 is critical for its interaction with USP15. Collectively, SART3 interacts with BARD1 and USP15 through the RRM1/2 and HAT (4–7) domains, respectively. Moreover, Co-IP results showed that depleting either USP15 or BARD1 did not impair the association of SART3 with BRAD1 or USP15 (Supplementary Fig. 7k, l). Therefore, we conclude that SART3 promotes the USP15-BARD1 interaction via respective association with USP15 and BARD1, both of which are required for the stimulatory effect of SART3 on USP15-BARD1 interaction after damage. Indeed, both SART3-ΔRRM (fails to bind BARD1) and SART3-ΔHAT4-7 (fails to bind USP15) truncations diminished the stimulatory effect after CPT treatment (Fig. 7f). Furthermore, as SART3 depletion, PARPi treatment also abolished the stimulatory effect of CPT exposure on USP15-BARD1 interaction. And combination of PARPi treatment with SART3 depletion did not cause a further inhibitory effect (Supplementary Fig. 7m), suggesting that SART3 promotes USP15-BARD1 association in a PARylation-dependent manner. Notably, the R836W mutation did not impair the stimulatory effect on the association of USP15-BARD1 (Supplementary Fig. 7n), which probably explains why the R836W mutant could partially rescue the HR defect and sensitivity to damage agents in SART3-depleted cells (Fig. 6f–h).
Given that USP15 can deubiquitinate BARD1 to maintain BRCA1/BARD1 retention at DSBs, we then explored the effect of SART3 on BARD1 deubiquitination and retention at DSBs. As expected, CPT treatment promotes BARD1 deubiquitination (Fig. 7g; lane 2 and 3). While SART3 depletion impaired this effect, leading to increased BARD1 ubiquitination (Fig. 7g; lane 3 and 4). The deubiquitination defect upon SART3 loss could be rescued by SART3 WT but not truncations, i.e., T4 and T1 (Fig. 7g; lane 3–7), supporting the notion that association with both USP15 and BARD1 is required for SART3 to promote BARD1 deubiquitination. Moreover, simultaneous depletion of SART3 and USP15 did not cause a further decrease in BRCA1 and BARD1 focus assembly compared to SART3 or USP15 single depletion after damage treatment (Fig. 7h, i, and Supplementary Fig. 7o). Consistently, both SART3-ΔHAT4-7 and ΔRRM mutations exhibited deficient HR efficiency (Fig. 7j and Supplementary Fig. 7p). These results suggest that SART3 facilitates BARD1 deubiquitination via promoting USP15-BARD1 association, therefore leading to BRCA1/BARD1 retention, and eventually DNA end resection at DSBs.
Binding to DNA–RNA hybrids is a prerequisite for SART3 functions in DSB repair
As mentioned above, the SART3-YA1 mutant exhibited defects in binding to DNA–RNA hybrids and in being recruited to DSBs (Fig. 1i–k), which prompted us to investigate its effects on cellular response to chemotherapeutic drugs. The results showed that the YA1 mutant failed to rescue cellular hypersensitivity to CPT and Olaparib, as well as HR defect caused by SART3 depletion (Fig. 8a–c). Moreover, the SART3-YA1 mutant also failed to restore the focus formation of RPA32, BRCA1, BARD1, and DDX1 (Fig. 8d), as well as DNA end resection efficiency (Fig. 8e). In addition, the YA1 mutant lost its ability to enhance the chromatin USP15-BARD1 interaction (Fig. 8f). Since end resection is also needed for microhomology-mediated end joining (MMEJ), another DSB repair pathway35,36, we then examined whether SART3 depletion will affect MMEJ activity. We found that depletion of SART3 significantly reduced the MMEJ activity, and complementing with SART3 WT, R836W mutant but not YA1 mutant could rescue the defect (Fig. 8g). Given that SART3 R836W mutant retains the normal end resection ability (Supplementary Fig. 6h) but exhibits defects in DDX1-mediated hybrid removal and HR efficiency (Fig. 6e, f), SART3-mediated DNA-RNA hybrids resolution may be specifically linked to HR but not MMEJ. Together, these results indicate that DNA–RNA hybrids-mediated SART3 accumulation at DSBs is necessary for its ability to promote optimal DSB repair. Notably, the SART3-YA1 fully restored the level of UVC-induced PCNA monoubiquitination as that of WT in SART3-depleted cells, suggesting that the YA1 mutation does not impair the function of SART3 in TLS (Fig. 8h).

a, b U2OS cells stably expressing GFP, GFP-SART3, or GFP-SART3-YA1 were transfected with siSART3 and treated with CPT for 24 h followed by CCK8 assay (a), or Olaparib for 48 h followed by colony formation assay (b). c U2OS-DR-GFP stably expressing Flag, Flag-SART3, or Flag-SART3-YA1 were transfected with siSART3, then infected with I-SceI lentivirus. The percentage of GFP-positive cells was quantitated by FACS (left). The specified proteins were examined by immunoblotting (right). d U2OS cells stably expressing GFP, GFP-SART3, or GFP-SART3-YA1 were transfected with siSART3, treated with ETO (RPA32, BRCA1 and BARD1) or CPT (DDX1) and further recovery for 2 h. Immunostainings was performed. The percentages of cells with more than 10 or 20 foci were quantified (bottom). Representative images are shown (top). Scale bars for overall and magnified images are respectively 50 and 10 µm. e U2OS-ER-AsiSI cells stably expressing GFP, GFP-SART3 or GFP-SART3-YA1 were transfected with the indicated siRNAs and treated with 4-OHT, followed by qPCR to measure DNA end resection (top). Immunoblotting verifies the knockdown of SART3 and CtIP (bottom). f HEK293T cells transfected with the indicated constructs were treated with CPT (5 µM, 2 h). Chromatin fractions were isolated for immunoprecipitation with anti-GFP beads, followed by immunoblotting with indicated antibodies. Asterisks indicate none-specific bands. CF-IP stands for Chromatin Fractions-IP. g U2OS-GFP-MMEJ cells stably expressing Flag, Flag-SART3, Flag-SART3-R836W, or Flag-SART3-YA1 were transfected with the indicated siRNAs followed by infection with I-SceI lentivirus. The percentage of GFP-positive cells was quantified by FACS analysis (top). The specified proteins were examined by immunoblotting (bottom). h U2OS cells stably expressing GFP, GFP-SART3, or GFP-SART3-YA1 were transfected with siSART3 for 48 h. Cells were then exposed to 15 J/m2 UVC and repaired for 4 h. The TIF and WCL were harvested and analyzed with the indicated antibodies. i Working model of how SART3 regulates HR repair. In (a–e), and (g) error bars represent mean ± SEM (N = 3 independent experiments), and p values were calculated using one-way ANOVA analysis with Tukey test (a, b) or unpaired two-tailed Student’s t test (c–e, g). Source data are provided as a Source Data file.
Discussion
SART3 is an RBP that functions in tumor antigenicity and regulation of gene expression37,38,39. Beyond these roles, our previous study revealed that SART3 participates in TLS in an RNA-binding-independent manner to protect cells from UV-induced DNA damage24. Here, we provide compelling evidence that SART3 promotes DSB repair. SART3 associates with DNA–RNA hybrids and PAR chains, and can be recruited to DSBs in a PARylation- and DNA‒RNA hybrids-dependent manner to promote HR. SART3 not only promotes DDX1 accumulation to facilitate timely removal of DNA‒RNA hybrids but also stimulates USP15-BARD1 interaction to enhance BARD1/BRCA1 retention at DSBs, thereby facilitating DNA end resection and HR (Fig. 8i). Depletion of SART3 can significantly impede DSB repair and sensitize tumor cells to multiple chemotherapeutic drugs.
DNA–RNA hybrids formed at DSBs need to be timely resolved by RNase H and helicases to enable recruitment of repair proteins7,11,12,13,14,15,16,17,18,40. However, how these helicases are recruited to DSBs-induced hybrids remains unclear. Our study reveals that SART3, which is recruited to DSBs in a DNA‒RNA hybrids-dependent manner, modulates the hybrids removal. SART3 likely forms a negative feedback loop with DNA–RNA hybrids, i.e. SART3 is being recruited by the hybrids to DSBs through its RRMs and in turn promoting the hybrids removal via recruiting DDX1. Specifically disrupting the SART3-DDX1 association but not SART3/DNA‒RNA-hybrids association by a cancer-associated variant, SART3-R836W, can abrogate DDX1 but not itself enrichment at DSBs. Given that PARPi exposure also inhibits SART3 accumulation at DSBs but fails to further decrease the accumulation of the YA1 mutant, PARylation and DNA–RNA hybrids at DSBs likely act epistatically to promote the accumulation of SART3 and its downstream factor DDX1 to regulate the hybrids resolution. In line with this notion, PARP1 depletion impairs the binding of DDX18 to R-loops16. Nevertheless, the detailed contributions of PARylation and DNA–RNA hybrids in SART3 recruitment warrant further exploration. Notably, SART3 loss in tumor cells also upregulates DNA‒RNA hybrid accumulation under unperturbed conditions, indicating that the stimulatory role of SART3 in hybrids unwinding is not limited to DSBs-induced structures. In addition to resolving DNA‒RNA-hybrids at DSB, DDX1 was also reported to converts RNA G-Quadruplex structures into R-Loops to facilitate the activation-induced cytidine deaminase targeting to immunoglobulin heavy-chain switch regions to promote class switch recombination41. Whether SART3 regulates this process deserves further exploration.
In addition to facilitating DDX1 recruitment to dissolve DNA–RNA hybrids, SART3 also promotes DSB end resection. We found that SART3 stimulates the USP15-BARD1 interaction and BARD1 deubiquitination, which can potentially enhance BARD1/BRCA1 retention at DSBs to facilitate end resection32,33. The finding that inhibition of SART3 binding to DNA‒RNA hybrids can abolish its stimulatory effect on USP15-BARD1 association indicates that SART3 accumulation at DSBs is a prerequisite for this effect. The dual functions of SART3 in DNA–RNA hybrid resolution and end resection act together to promote optimal HR and desensitize tumors to chemotherapeutics. Moreover, the observation that SART3 loss could reverse the PARPi resistance of BRCA1-deficient cells caused by 53BP1 loss suggests that SART3 can also modulate HR stages after end resection. A comprehensive understanding of the function of SART3 in HR deserves further investigation.
Importantly, our findings also highlight the functional defects of several RRM mutations in DDR. We found that the SART3-R836W mutation specifically disrupt the SART3-DDX1 association and abrogate the ability of SART3 to mediate DDX1 enrichment at DSBs to resolve DNA–RNA hybrids, leading to a HR defect. Given that the R836W also impairs TLS24, we speculate that, although currently annotated as a variant of uncertain significance, the R836W is likely pathogenic. Nevertheless, the R836W mutant still associates with DNA–RNA hybrids to accumulate at DSBs, retaining the ability to enhance USP15-BARD1 interaction and end resection. Therefore, the HR defect of R836W is mild. In contrast, the SART3-YA1 mutation disrupts the association of SART3 with DNA–RNA hybrids, and abrogates its enrichment at DSBs, causing a complete loss of HR. Moreover, unlike the SART3-R836A and R836W mutants, the SART3-YA1mutant does not impair TLS activation. These results raise a possibility that different functions of SART3 in DDR are distinctly regulated.
Together, we reveal that SART3 promotes HR repair, which is separated from its TLS function. Given that a potential synthetic lethality occur between HR and TLS pathways42, the functions of SART3 in DDR may explain its essential role in stem cell proliferation and embryonic development43. SART3-depleted U2OS cells also exhibit increased micronuclei formation24 and nuclear R-loops accumulation. Considering that nuclear R-loops not efficiently resolved may be converted to cytoplasmic hybrids to activate IRF3-mediated immune signaling and apoptosis8, and micronuclei can also trigger a cellular immune response44,45, high expression of SART3 in tumor cells likely promotes their escape from immune surveillance to facilitate carcinogenesis. Therefore, SART3 represents a promising target for cancer therapy.
Methods
Cell culture
Human U2OS (ATCC HTB-96) and HEK293T (ATCC CRL-3216) cells were obtained from the American Type Culture Collection (Rockville, MD, USA). U2OS-DR-GFP and U2OS-GFP-MMEJ cells were generously provided by Dr. Hailong Wang (Capital Normal University). U2OS cells stably expressing GFP-SART3 were prepared as previously described24. U2OS-ER-AsiSI cells were generated as described30 and cultured in Dulbecco’s modified Eagle’s medium (DMEM, without phenol red) supplemented with 10% fetal bovine serum (FBS) and 1 mg/mL puromycin. Other cell lines were cultured in DMEM (Gibco) with 10% FBS. All cells were grown in an atmosphere of 5% CO2 at 37 °C.
Plasmids
Full-length (FL) SART3 and its truncations have been described previously24. GFP-SART3-YA1, YA2, YA3, R742A, R836A, R746A and K768A mutants were generated by site-directed mutagenesis. GFP-USP15 was from Dr. Xingzhi Xu (Shenzhen University). Flag-DDX1 and Flag-DDX5 were generated by subcloning cDNAs into p2xFlag-CMV-14 vector (Sigma). Flag-DDX21 was generated by subcloning DDX21 (a gift from Dr. Lingling Chen at Chinese Academy of Sciences). The pBABE-HA-ER-AsiSI plasmid was a gift from Dr. Gaelle Legube (University of Toulouse).
Antibodies
The antibodies applied to this study include the following: Flag (F1804, Sigma); γH2AX (ab2893, Abcam); H3 (ab1791, Abcam); RPA32 (ab2175, Abcam); SART3 (ab36137, Abcam); V5 (ab15828, Abcam); PARP1 (sc-8007, Santa Cruz); GFP (sc-8334, Santa Cruz); DDX1 (sc-271438, Santa Cruz); BRCA1 (sc-6954, Santa Cruz); γH2AX (05-636, Millipore); RNF8 (09-813, Millipore); RNA168 (ABE367, Millipore); Top1cc (MABE1084, Millipore); pRPA32-S33 (A300-246A, Bethyl); BARD1 (A300-263A, Bethyl); USP15 (67557-1-Ig, Proteintech); DDX21 (10528-1-AP, Proteintech); DDX1 (CL594-67991, Proteintech); GST (10000-0-AP, Proteintech); Tubulin (AbM59005-37B-PU, Beijing protein innovation); PAR (ALX-210-890A-0100, Enzo life science); CtIP (61141, Active motif); Myc (HT101-02, TransGen Biotech); GFP (AE012, Abclonal); HA (902302, BioLegend); S9.6 (ENH001, Kerafast); MRE11 (NB100-142, Novus); 53BP1 (4937S, Cell Signaling Technology); BrdU (347580, BD-BDIS).
Short-interfering RNA (siRNA)
SiRNAs were purchased from GenePharma (Shanghai, China). SiRNA transfections in cells were carried out with RNAiMax (Invitrogen) according to the manufacturer’s instructions, which was followed by analysis at 48–72 h post-transfection.
Laser microirradiation
Laser microirradiation was performed using a pulsed nitrogen laser (Spectra-Physics; 365 nm, 10 Hz pulse) as previously reported46,47,48,49. For the kinetic analysis of SART3 recruitment to laser-induced damage sites, U2OS cells stably expressing GFP-SART3 were microirradiated and then imaged at 30 s intervals. The data are presented as means ± standard errors from 10 cells. To calculate the percentage of cells accumulating at laser irradiation sites, at least 20 cells expressing GFP-tagged proteins were counted per biological replicate. Standard errors were derived from three independent experiments. To examine the regulation of SART3 recruitment at laser-induced lesions, cells were pretreated with 10 µM ATM inhibitor (KU 55933), 20 µM DNA-PK inhibitor (NU7026), 50 µM PARP inhibitor (PARPi) (ABT-888) for 1 h, or 100 µM DRB (5,6-Dichloro-1-beta-Ribo-furanosyl Benzimidazole) for 3.5 h, or 2 µg/mL ActD (actinomycin D) for 2 h prior to microirradiation.
Chromatin immunoprecipitation (ChIP) and RNase H1-ChIP (R-ChIP) assays
ChIP was performed as previously described13 with minor modifications. Briefly, U2OS-DR-GFP cells were crosslinked with 1% formaldehyde for 15 min at room temperature and then terminated with 0.125 M glycine. Pelleted cells were resuspended in lysis buffer (50 mM PIPES pH 8.0, 85 mM KCl2, 0.5% NP-40) on ice for 15 min. The nuclei were enriched by low-speed centrifugation and resuspended in nuclear lysis buffer (50 mM Tris pH 8.0, 10 mM EDTA, 1% SDS). Chromatin was sonicated for 8 min (30 s on, 30 s off, high intensity) by Bioruptor (Diagenode), to obtain 500–1000 bp fragments. After sonication, samples were diluted 10-fold in ChIP dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris pH 8.0, 167 mM NaCl) and incubated with Flag magnetic, followed by pre-blocking with BSA, at 4 °C for 4 h. Magnetic samples then were washed once with low salt buffer (0.1% SDS, 1% NP-40, 1 mM EDTA, 50 mM Tris pH 8.0, 150 mM NaCl, 0.5% sodium deoxycholate), once with high salt buffer (0.1% SDS, 1% NP-40, 1 mM EDTA, 50 mM Tris pH 8.0, 500 mM NaCl, 0.5% sodium deoxycholate), and once with lithium chloride buffer (0.1% SDS, 1% NP-40, 1 mM EDTA, 50 mM Tris pH 8.0, 250 mM LiCl, 0.5% sodium deoxycholate). RNase H1-ChIP (R-ChIP) experiments were performed as previously described31. U2OS-DR-GFP cells expressing V5-RNase H1-D210N were crosslinked with 1% formaldehyde for 10 min and then terminated with 0.125 M glycine for 15 min at room temperature. The cell pellet was resuspended with cell lysis buffer (10 mM Tris pH 8.0, 10 mM NaCl, 0.5% NP-40) and incubated for 30 min on ice. Then the nuclear pellet was resuspended with nuclear lysis buffer and sonicated as above. The supernatants containing sheared chromatin were harvested and incubated with V5 pre-incubated Protein G magnetic beads in the buffer (0.1% sodium deoxycholate, 1% Triton X-100, 2 mM EDTA, 20 mM Tris pH 8.0, 167 mM NaCl) for overnight at 4 °C, followed by washing with wash buffer I (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris pH 8.0, 150 mM NaCl) three times, wash buffer II (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris pH 8.0, 500 mM NaCl) three times, wash buffer III (1% NP-40, 1 mM EDTA, 10 mM Tris pH 8.0, 250 mM LiCl, 1% sodium deoxycholate) once, and TE buffer once. Samples were then eluted with elution buffer (1% SDS and 0.1 M NaHCO3) and incubated overnight at 65 °C for crosslink reversal. The samples were then treated with RNase A and proteinase K for final DNA extraction. Immunoprecipitated DNA and input DNA were purified utilizing a DNA Clean & Concentrator Kit (Zymo Research, D4034) and subjected to qPCR. The primer sequences are included in Supplementary Data 1.
GST pull-down assay
Plasmids expressing GST fusion proteins were transformed into the E. coli DE3 strain. The bacteria were cultured to the log phase and protein expression was induced overnight at 16 °C supplemented with 0.2 mM isopropyl-β-D-thiogalactoside (IPTG). The cells were solubilized in NETN buffer (20 mM Tris pH 8.0, 200 mM NaCl, 0.5 mM EDTA, 0.5% NP-40) with 10 mM PMSF, 1 mM DTT, 1 mg/mL lysozyme, protease inhibitor, and sonicated on ice. The supernatant was incubated with precleared glutathione-Sepharose 4B beads (GE Healthare, 17-0756-01) for 4 h at 4 °C followed by extensive washing with NETN buffer and high salt NETN buffer (20 mM Tris pH 8.0, 400 mM NaCl, 0.5 mM EDTA, 0.5% NP-40). GST fusion proteins were immobilized on beads and incubated with cell lysates overnight at 4 °C. The beads were washed with NETN buffer and boiled in SDS‒PAGE loading buffer for analysis.
PAR binding assay
The PAR-binding assays were performed as previously described30. Briefly, the indicated protein was separated on an 8% SDS‒PAGE gel and then transferred to a PVDF membrane. The membrane was blocked with 5% skimmed milk at room temperature and then incubated with 10 nM PAR polymer (Trevigen, 4336-100-01) for 2 h at 4 °C. The PAR was detected by rabbit monoclonal anti-PAR antibody.
Cell cycle assay
U2OS cells were harvested after transfection with siNC or siSART3 for 48 h, followed by PBS wash and fixation using ice-cold 70% ethanol at −20 °C overnight. Then the cells were stained with FxCycleTM Propidium Iodide (PI)/RNase Staining Solution (F10797, Invitrogen) at room temperature for 30 min. Cell cycle distribution was analyzed using a FACS Calibur flow cytometer (Moflo XDP, Bechman). The gating strategy used in flow cytometry analysis is included in the Supplementary Fig. 8.
Immunofluorescence
Cells were seeded on cover glasses and subjected to the indicated treatments. To examine BARD1, RPA32, RNF8, and RNF168 foci formation, cells were pre-extracted with 0.25–0.5% Triton X-100 prior to fixation. To examine BrdU foci formation, cells were pre-extracted with buffer containing 20 mM HEPES, pH 7.4, 50 mM NaCl, 300 mM sucrose, 3 mM MgCl2, 0.5% Triton X-100 followed by fixation. To examine the foci formation of other DDR factors, cells were fixed in 4% paraformaldehyde (PFA) for 15 min and permeabilized in a 0.5% Triton X-100 solution for 5 min. Subsequently, cells were blocked in 5% bovine serum albumin (BSA) solution for 1 h at room temperature and incubated with primary antibodies overnight at 4 °C. The coverslips were washed with 0.2% Tween 20 in PBS (PBST) and incubated with Alexa Fluor 555 or 488 labeled secondary antibodies (Invitrogen) at room temperature for 1 h. S9.6 staining was performed as previously described50 with minor modifications. Cells were fixed with cold methanol for 10 min at −20 °C and then incubated with staining buffer (0.1% BSA in TBST) for 10 min at room temperature. RNase T1 (EN0541, Thermo Fisher), RNase III (M0245S, New England Biolabs), and/or RNase H were diluted into enzyme treatment buffer (3 mM MgCl2, 0.1% BSA, TBST) at a ratio of 1:200 and incubated with cells for 2 h at 37 °C. The coverslips were washed with staining buffer and incubated with blocking buffer (PBS containing 3% BSA and 2% goat serum) at 4 °C overnight. Subsequently, the cells were incubated with antibodies in blocking buffer at 4 °C overnight. The cells were then washed with PBS and incubated with Alexa Fluor 568 goat anti-mouse at 4 °C for at least 4 h. After washing with PBS, the coverslips were mounted onto glass slides using VECTA-SHIELD antifade medium containing DAPI. All the samples were visualized by a Leica DM5000 microscope. The average S9.6 intensity per nucleus was also assessed by CellProfiler.
Biotinylated DNA‒RNA hybrid pull-down assay
The biotinylated DNA‒RNA hybrid pull-down assay was performed as described previously25,51. The 5′ biotinylated DNA substrate was purchased from GENEray (Shanghai, China), and the RNA substrate was purchased from Genepharma (Shanghai, China). The sequences are included in Supplementary Data 1. Briefly, to generate DNA‒RNA hybrids, the DNA and RNA substrates were annealed in annealing buffer (1 mM EDTA, 10 mM Tris, pH 7.5) at a concentration of 10 µM by heating at 95 °C for 3 min, followed by slow cooling to room temperature. HEK293T cells transfected with the indicated plasmids were lysed using lysis buffer (20 mM Tris, pH 8.0, 137 mM NaCl, 10% glycerol, 1% NP-40, and 2 mM EDTA) supplemented with protease inhibitor and RNase inhibitor. Cell lysates were precleared with streptavidin beads (GE Healthcare) for 2 h at 4 °C. Thirty picomoles of biotinylated DNA‒RNA hybrid or dsDNA were incubated with precleared lysates or purified protein (digested with thrombin to remove the GST tag) solution overnight and then conjugated with streptavidin beads for 2 h at 4 °C. Beads containing protein complexes were washed three times with washing buffer (20 mM Tris, pH 7.5, 10 mM NaCl, 0.1% Tween 20). Eluted proteins were analyzed by SDS‒PAGE.
Colony formation assay
Cell survival assays after genotoxic reagent treatments were performed as described previously30. U2OS cells stably expressing GFP or GFP-SART3 were transfected with siNC or siSART3 for 24 h and reseeded in 6 cm dishes. After adhering to the dish, cells were incubated with the indicated concentrations of CPT for 1 h, PARPi (Olaparib) for 24 h, etoposide (ETO) for 24 h, and hydroxyurea (HU) for 2 h, and then further incubated in complete medium for two weeks. Colonies were fixed and counted.
HR and MMEJ assay
U2OS-DR-GFP or U2OS-GFP-MMEJ cells were infected with the indicated SART3-expressing retroviruses for 48 h. Then, these cell lines were transfected with the indicated siRNA for 24 h and further infected with the retrovirus expressing I-SceI for 36–48 h. Finally, the GFP-positive cells were analyzed by flow cytometry. Data from three independent experiments were analyzed for histograms. The gating strategy used in flow cytometry analysis is included in the Supplementary Fig. 8.
Co-immunoprecipitation (Co-IP)
HEK293T cells were transfected with the indicated siRNA and constructs for 48 h, followed by treatment with the indicated reagents. Cells were harvested and lysed with NETN buffer for immunoprecipitation. Finally, the immunoprecipitates were separated by SDS-PAGE and detected by immunoblotting with the indicated antibodies. For isolation of chromatin-fractions, cells were treated with indicated drugs followed by harvesting the Triton-insoluble fractions (TIF) as previously described24.
Tandem affinity purification and mass spectrometry (TAP-MS)
HEK293T cells transfected with SFB vector or SFB-SART3 (SFB: S-protein tag, Flag epitope tag, and streptavidin-binding peptide tag) were lysed with NETN buffer at 4 °C for 1 h. The supernatant was incubated with anti-Flag M2 beads at 4 °C for 4 h. The immunocomplexes were washed three times with NETN buffer, followed by elution with 3xFlag-peptide (200 µg/mL) for 2 h at 4 °C. The eluted supernatant was then incubated with GE streptavidin beads at 4 °C for 1 h. The immunocomplexes were washed and eluted with 1 mg/mL biotin (Sigma) at 4 °C for 1 h. The eluted product was then precipitated with trichloroacetic acid for analysis by mass spectrometry.
End resection assay
End resection assay was conducted as previously described30,49 with some modifications. U2OS-ER-AsiSI cells transfected with siNC or siRNA targeting SART3 or CtIP were treated with 300 nM 4-hydroxytamoxifen (4-OHT) for 4 h, followed by genomic DNA extraction with the QuickExtract DNA extraction kit (Epicentre). The level of DNA end resection adjacent to DSB1 (Chr 1:89231183) was quantified by qPCR using the equation 1/(2^(ΔCt-1) + 0.5)*10052. The primers information is provided in Supplementary Data 1.
DNA‒RNA immunoprecipitation (DRIP) qPCR (DRIP-qPCR)
DRIP assay was carried out as previously described13,53,54 with some modifications. Briefly, ER-AsiSI U2OS cells were treated with 400 nM 4-OHT for 2 h, then incubated with cytoplasmic cell lysis buffer (10 mM HEPES pH 8.0, 10 mM KCl, 1.5 mM MgCl2, 0.34 M sucrose, 0.1% Triton X-100, 10% glycerol, 1 mM DTT) to isolate nuclei. Total nucleic acids were extracted with a buffer containing 50 mM Tris pH 8.0, 5 mM EDTA, 1% SDS and Proteinase K at 37 °C for 6 h and recovered by phenol‒chloroform extraction and ethanol precipitation. Twenty micrograms of DNA were digested with restriction enzymes (50 units each of EcoRI, HindIII, BsrGI, XbaI) (NEB) in a 500 µl system, with or without RNase H, overnight at 37 °C. Fragmented DNA was purified and resuspended in TE buffer (10 mM Tris pH 7.5, 1 mM EDTA). Five micrograms of digests were immunoprecipitated overnight at 4 °C in 500 µl DRIP binding buffer (10 mM NaPO4 pH 7.0, 140 mM NaCl, 0.05% Triton X-100) with 4 µg S9.6 antibody (Kerafast, ENH001). After conjugating with 40 µL preblocked protein G Dynabeads (Invitrogen, 1004D) for 4 h at 4 °C, the DNA–RNA–antibody-bead complexes were washed three times with DRIP binding buffer. A total of 250 µl elution buffer (50 mM Tris pH 8.0, 10 mM EDTA, 0.5% SDS) and 5 U proteinase K were added to the DNA‒RNA-antibody-bead complexes and incubated for 45 min at 55 °C. DNA was purified utilizing a DNA Clean & Concentrator Kit (Zymo Research, D4034) and subjected to qPCR. The primer sequences were included in Supplementary Data 1.
In vitro hybrids unwinding assay
Unwinding assay was carried out as described previously55 with some modifications. His-DDX1 and GST-SART3 were respectively expressed in BL21 cells for purification. Specifically, His-DDX1 was purified and then eluted using imidazole. GST-SART3 was purified followed by thrombin cleavage. Unwinding assays were conducted in a total reaction volume of 20 µL, including 4 pmol of FAM-labeled DNA-RNA substrates and recombinant DDX1 protein (0.05, 0.1, and 0.15 µg) with or without 0.2 µg SART3, in the buffer that comprises 20 mM Tris-HCl (pH 7.5), 70 mM KCl, 2 mM MgCl2, 1.5 mM DTT, and 1 mM ADP. The reaction mixtures were incubated at 37 °C for 3 h followed by quench on ice. After treatment with Proteinase K at 55 °C for 1 h, the samples were electrophoresed through a 12% native polyacrylamide gel.
Statistics and reproducibility
Data analyses were performed using GraphPad Prism 8, Microsoft Excel, and SPSS, as applicable. Significant differences were determined using a two-tailed Mann–Whitney U test (fluorescence intensity). One-way ANOVA analysis with Tukey test (cell viability assays) or an unpaired two-tailed Student’s t-test (qPCR, ChIP, HR reporter, and other experiments). In all cases: ns, not significant. The data in Figs. 1i–j; 2b; 5a, c, e-g; 6c, d; 7a–g; 8f; Supplementary Figs. 1i; 5b–e, o–p; 7c–n are representative of 3 independent experiments.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The mass spectrometry proteomics data generated in this study have been deposited in ProteomeXchange Consortium via the PRIDE partner repository under the accession code PXD053595. Source data are provided with this paper.
References
Richardson, C. & Jasin, M. Frequent chromosomal translocations induced by DNA double-strand breaks. Nature 405, 697–700 (2000).
Cannavo, E. & Cejka, P. Sae2 promotes dsDNA endonuclease activity within Mre11-Rad50-Xrs2 to resect DNA breaks. Nature 514, 122–125 (2014).
Liu, T. & Huang, J. DNA end resection: facts and mechanisms. Genomics Proteom. Bioinform. 14, 126–130 (2016).
Mimitou, E. P. & Symington, L. S. Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature 455, 770–774 (2008).
Cejka, P. et al. DNA end resection by Dna2-Sgs1-RPA and its stimulation by Top3-Rmi1 and Mre11-Rad50-Xrs2. Nature 467, 112–116 (2010).
Niu, H. et al. Mechanism of the ATP-dependent DNA end-resection machinery from Saccharomyces cerevisiae. Nature 467, 108–111 (2010).
Brickner, J. R., Garzon, J. L. & Cimprich, K. A. Walking a tightrope: the complex balancing act of R-loops in genome stability. Mol. Cell 82, 2267–2297 (2022).
Crossley, M. P. et al. R-loop-derived cytoplasmic RNA-DNA hybrids activate an immune response. Nature 613, 187–194 (2023).
Petermann, E., Lan, L. & Zou, L. Sources, resolution and physiological relevance of R-loops and RNA-DNA hybrids. Nat. Rev. Mol. Cell Biol. 23, 521–540 (2022).
Liu, S. et al. RNA polymerase III is required for the repair of DNA double-strand breaks by homologous recombination. Cell 184, 1314–1329.e1310 (2021).
Ohle, C. et al. Transient RNA-DNA hybrids are required for efficient double-strand break repair. Cell 167, 1001–1013.e1007 (2016).
Marnef, A. & Legube, G. R-loops as Janus-faced modulators of DNA repair. Nat. Cell Biol. 23, 305–313 (2021).
Cohen, S. et al. Senataxin resolves RNA:DNA hybrids forming at DNA double-strand breaks to prevent translocations. Nat. Commun. 9, 533 (2018).
Li, L. et al. DEAD Box 1 facilitates removal of RNA and homologous recombination at DNA double-strand breaks. Mol. Cell. Biol. 36, 2794–2810 (2016).
Sessa, G. et al. BRCA2 promotes DNA-RNA hybrid resolution by DDX5 helicase at DNA breaks to facilitate their repair. EMBO J. 40, e106018 (2021).
Lin, W. L. et al. DDX18 prevents R-loop-induced DNA damage and genome instability via PARP-1. Cell Rep. 40, 111089 (2022).
Song, C., Hotz-Wagenblatt, A., Voit, R. & Grummt, I. SIRT7 and the DEAD-box helicase DDX21 cooperate to resolve genomic R loops and safeguard genome stability. Genes Dev. 31, 1370–1381 (2017).
Cristini, A., Groh, M., Kristiansen, M. S. & Gromak, N. RNA/DNA hybrid interactome identifies DXH9 as a molecular player in transcriptional termination and R-loop-associated DNA damage. Cell Rep. 23, 1891–1905 (2018).
Krishnan, R. et al. RNF8 ubiquitylation of XRN2 facilitates R-loop resolution and restrains genomic instability in BRCA1 mutant cells. Nucleic Acids Res. 51, 10484–10505 (2023).
Whitmill, A., Timani, K. A., Liu, Y. & He, J. J. Tip110: Physical properties, primary structure, and biological functions. Life Sci. 149, 79–95 (2016).
Yang, D. et al. Identification of a gene coding for a protein possessing shared tumor epitopes capable of inducing HLA-A24-restricted cytotoxic T lymphocytes in cancer patients. Cancer Res. 59, 4056–4063 (1999).
Wang, X. et al. K48-linked deubiquitination of VGLL4 by USP15 enhances the efficacy of tumor immunotherapy in triple-negative breast cancer. Cancer Lett. 28, 588 (2024).
Taniue, K. et al. LncRNA ZNNT1 induces p53 degradation by interfering with the interaction between p53 and the SART3-USP15 complex. PNAS Nexus 2, pgad220 (2023).
Huang, M. et al. RNA-splicing factor SART3 regulates translesion DNA synthesis. Nucleic Acids Res. 46, 4560–4574 (2018).
Wang, I. X. et al. Human proteins that interact with RNA/DNA hybrids. Genome Res. 28, 1405–1414 (2018).
Teloni, F. & Altmeyer, M. Readers of poly(ADP-ribose): designed to be fit for purpose. Nucleic Acids Res. 44, 993–1006 (2016).
Mamontova, E. M. et al. FUS RRM regulates poly(ADP-ribose) levels after transcriptional arrest and PARP-1 activation on DNA damage. Cell Rep. 42, 113199 (2023).
Maris, C., Dominguez, C. & Allain, F. H. The RNA recognition motif, a plastic RNA-binding platform to regulate post-transcriptional gene expression. FEBS J. 272, 2118–2131 (2005).
Pommier, Y., Nussenzweig, A., Takeda, S. & Austin, C. Human topoisomerases and their roles in genome stability and organization. Nat. Rev. Mol. Cell Biol. 23, 407–427 (2022).
Zhang, C. et al. Micropeptide PACMP inhibition elicits synthetic lethal effects by decreasing CtIP and poly(ADP-ribosyl)ation. Mol. Cell 82, 1297–1312.e1298 (2022).
Chen, J.-Y., Zhang, X., Fu, X.-D. & Chen, L. R-ChIP for genome-wide mapping of R-loops by using catalytically inactive RNASEH1. Nat. Protoc. 14, 1661–1685 (2019).
Long, L. et al. The U4/U6 recycling factor SART3 has histone chaperone activity and associates with USP15 to regulate H2B deubiquitination. J. Biol. Chem. 289, 8916–8930 (2014).
Peng, Y. et al. The deubiquitylating enzyme USP15 regulates homologous recombination repair and cancer cell response to PARP inhibitors. Nat. Commun. 10, 1224 (2019).
Timani, K. A., Liu, Y., Suvannasankha, A. & He, J. J. Regulation of ubiquitin-proteasome system-mediated Tip110 protein degradation by USP15. Int. J. Biochem. Cell Biol. 54, 10–19 (2014).
Wang, H. & Xu, X. Microhomology-mediated end joining: new players join the team. Cell Biosci. 7, 1–6 (2017).
Sfeir, A. & Symington, L. S. Microhomology-mediated end joining: a back-up survival mechanism or dedicated pathway? Trends Biochem. Sci. 40, 701–714 (2015).
Harada, K., Yamada, A., Yang, D., Itoh, K. & Shichijo, S. Binding of a SART3 tumor-rejection antigen to a pre-mRNA splicing factor RNPS1: a possible regulation of splicing by a complex formation. Int. J. Cancer 93, 623–628 (2001).
Liu, Y., Li, J., Kim, B. O., Pace, B. S. & He, J. J. HIV-1 Tat protein-mediated transactivation of the HIV-1 long terminal repeat promoter is potentiated by a novel nuclear Tat-interacting protein of 110 kDa, Tip110. J. Biol. Chem. 277, 23854–23863 (2002).
Liu, Y., Liu, J., Wang, Z. & He, J. J. Tip110 binding to U6 small nuclear RNA and its participation in pre-mRNA splicing. Cell Biosci. 5, 40 (2015).
Yang, S., Winstone, L., Mondal, S. & Wu, Y. Helicases in R-loop formation and resolution. J. Biol. Chem. 299, 105307 (2023).
de Almeida, C. R. et al. RNA helicase DDX1 converts RNA G-quadruplex structures into R-loops to promote IgH class switch recombination. Mol. Cell 70, 650–662.e658 (2018).
Pearl, L. H., Schierz, A. C., Ward, S. E., Al-Lazikani, B. & Pearl, F. M. Therapeutic opportunities within the DNA damage response. Nat. Rev. Cancer 15, 166–180 (2015).
Liu, Y. et al. TIP110/p110nrb/SART3/p110 regulation of hematopoiesis through CMYC. Blood 117, 5643–5651 (2011).
Krupina, K., Goginashvili, A. & Cleveland, D. W. Causes and consequences of micronuclei. Curr. Opin. Cell Biol. 70, 91–99 (2021).
Di Bona, M. & Bakhoum, S. F. Micronuclei and cancer. Cancer Discov. 14, 214–226 (2024).
Gong, J. et al. RBM45 competes with HDAC1 for binding to FUS in response to DNA damage. Nucleic Acids Res. 45, 12862–12876 (2017).
Yang, Y. et al. FANCD2 and REV1 cooperate in the protection of nascent DNA strands in response to replication stress. Nucleic Acids Res. 43, 8325–8339 (2015).
Yasuhara, T. et al. Human Rad52 promotes XPG-mediated R-loop processing to initiate transcription-associated homologous recombination repair. Cell 175, 558–570.e511 (2018).
Wu, W. et al. VGLL3 modulates chemosensitivity through promoting DNA double-strand break repair. Sci. Adv. 10, eadr2643 (2024).
Smolka, J. A., Sanz, L. A., Hartono, S. R. & Chédin, F. Recognition of RNA by the S9.6 antibody creates pervasive artifacts when imaging RNA:DNA hybrids. J. Cell Biol. 220, e202004079 (2021).
Nguyen, H. D. et al. Functions of replication protein A as a sensor of R loops and a regulator of RNaseH1. Mol. Cell 65, 832–847.e834 (2017).
Zhou, Y., Caron, P., Legube, G. & Paull, T. T. Quantitation of DNA double-strand break resection intermediates in human cells. Nucleic Acids Res. 42, e19 (2014).
Lu, W. T. et al. Drosha drives the formation of DNA:RNA hybrids around DNA break sites to facilitate DNA repair. Nat. Commun. 9, 532 (2018).
Yang, X. et al. m(6)A promotes R-loop formation to facilitate transcription termination. Cell Res. 29, 1035–1038 (2019).
Li, L., Monckton, E. A. & Godbout, R. A role for DEAD box 1 at DNA double-strand breaks. Mol. Cell. Biol. 28, 6413–6425 (2008).
Acknowledgements
The authors thank Drs. Gaelle Legube, Hailong Wang, Lingling Chen, Ceshi Chen, Zheng Tang, and Xingzhi Xu for reagents, Dr. Hongyan Shen for project assistance. This work was supported by National Natural Science Foundation of China (82330090 and 82341006 to C.G.), National Key R&D Program of China (2023YFA1801900 to T.T), National Natural Science Foundation of China (82030033 and 81921006 to T.T), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDA0460403 to C.G.), Postdoctoral Research Foundation of China (2021M703206 to X.M) and the State Key Laboratory of Membrane Biology.
Author information
Authors and Affiliations
Contributions
C.G. and T.T. designed and supervised the project; H.F. performed most of the experiments with help from M.H., H.W., H.Z., L.X., J.G., R.A., Y.G., Q.L., X.J. and X.M; M.H. initiated the project; H.F., M.H., T.T. and C.G. wrote and edited the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Li Lan and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Fu, H., Huang, M., Wu, H. et al. SART3 promotes homologous recombination repair by stimulating DNA-RNA hybrids removal and DNA end resection. Nat Commun 16, 2244 (2025). https://doi.org/10.1038/s41467-025-57599-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-57599-8
