
Abstract
Overcoming immunosuppression in the tumor microenvironment (TME) is crucial for developing novel cancer immunotherapies. Here, we report that IL-16 administration enhances the polarization of T helper 1 (Th1) cells by inhibiting glutamine catabolism through the downregulation of glutaminase in CD4+ T cells and increases the production of Th1 effector cytokine IFN-γ, thus improving anti-tumor immune responses. Moreover, we find that establishing an IL-16-dependent, Th1-dominant TME relies on mast cell-produced histamine and results in the increased expression of the CXCR3 ligands in tumor-associated macrophages (TAM), thereby improving the therapeutic effectiveness of immune checkpoint blockade (ICB). Cancer patients exhibit impaired production of IL-16, which correlates with poorer prognosis. Additionally, low IL-16 production is associated with unresponsiveness to immunotherapy in cancer patients. Collectively, our findings provided new insights into the biological function of IL-16, emphasizing its potential clinical significance as a therapeutic approach to augment anti-tumor immunity and sensitize ICB-based cancer immunotherapy.
Similar content being viewed by others
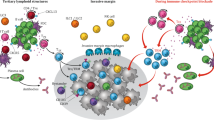


Introduction
In recent years, harnessing the host immune system to combat tumors has emerged as a promising strategy in cancer treatment1. Establishing effective anti-tumor responses requires the proper differentiation and function of tumor-infiltrating immune cells. A variety of immune cells populate the tumor microenvironment (TME), including myeloid cells (such as macrophages, dendritic cells, neutrophils, and mast cells) and lymphocytes (such as T cells and B cells). The proper functioning of tumor-infiltrating immune cells can exert immunosurveillance roles. For example, it is well recognized that a T helper 1 (Th1) -dominant immune microenvironment, characterized by the increased function of Th1 CD4+ T cells, activated CD8+ T cells, and M1-like macrophages, is essential for immune-mediated tumor eradication2,3. However, tumors evolve multiple mechanisms to re-educate immune cells into pro-tumor phenotypes, such as secreting high levels of immunosuppressive cytokines (e.g., IL-10, TGF-β)4 or creating a hypoxic environment5. In recent years, abnormal metabolic activity has also been identified as a crucial regulator of tumor-infiltrating immune cells. For example, the reduced availability of glucose impaired the anti-tumor function of Th1 cells, macrophages, and CD8+ cytotoxic T cells6,7,8,9. In addition to glucose, another key cellular energy source in the TME is glutamine, whose impact on tumor immunity has been attracting increasing attention. Blocking glutamine degradation into glutamate by glutaminase (GLS) inhibitor enhances anti-melanoma activity of T cells10. Importantly, the efficacy of GLS inhibitors has been tested in clinical trials in the treatment of some cancers, such as renal cancer and lung cancer11,12. Therefore, understanding the molecular determinants of immune suppression in the TME is crucial for creating an effective anti-tumor immune response.
Interleukin-16 (IL-16), also known as lymphocyte chemoattractant factor (LCF), is a less-studied cytokine first identified in 198213. IL-16 is produced by various types of immune cells, such as macrophages, T cells, B cells, mast cells, and eosinophils in a precursor form (pro-IL16, 631 amino acids)14, which is cleaved by caspase-3 to generate the mature, bioactive IL-16 (121 amino acids)15. CD4 is the natural receptor for IL-16, and it is predominantly expressed in CD4+ T cells. To date, the physiological and pathological functions of IL-16 remain poorly understood. Previous studies have indicated that IL-16 can exacerbate allergic asthma by promoting Th2 and Th17 cytokine production16. In multiple sclerosis, IL-16 was associated with enhanced Th1 cell inflammation17. It is also reported that IL-16 mediated the recruitment of Th1 cells18 or regulatory T cells (Treg)19. However, IL-16 does not influence the recruitment of CD4+ T cells during lung inflammation20. Interestingly, IL-16 has been reported to regulate the functions of cells that typically do not express the CD4 molecule, such as Langerhans cells21, monocytes22, or lung epithelial cells20, suggesting the potential existence of unidentified IL-16 receptor(s). Currently, the impact of IL-16 on tumor immunity remains unknown.
In this work, we uncover the key role of IL-16 in establishing a Th1 cell-mediated anti-tumor immune microenvironment by regulating glutamine metabolism, This effect further leads to the reprogramming of tumor-associated macrophages (TAM) toward an M1-like phenotype. In addition, the function of IL-16 in sensitizing tumor immunotherapy is also revealed. Hence, our study identifies IL-16 supplementation as a potential strategy for preventing tumor immune evasion and improving the efficacy of immunotherapy in cancer patients.
Results
IL-16 inhibits tumor growth in preclinical models and predicts a favorable prognosis in cancer patients
As a relatively understudied cytokine, the potential impact of IL-16 on tumor progression has long been overlooked. To investigate the changes in IL-16 levels in human cancers, we collected serum samples from patients diagnosed with breast cancer or lung cancer, and found that serum IL-16 concentrations were significantly lower in cancer cohorts compared to healthy individuals (Fig. 1a). Consistently, decreased tumoral expression of IL16 was observed in approximately half of The Cancer Genome Atlas (TCGA) cancer types (15 out of 31) including breast cancer (BRCA) and lung adenocarcinoma (LUAD) when compared to corresponding normal tissues (Fig. 1b and Supplementary Fig. 1), suggesting that the presence of tumors may inhibit the local or systemic production of IL-16. To gain a deeper understanding of the cell-specific changes of IL-16, we conducted single-cell RNA sequencing (scRNA-seq) using human breast cancer tissues and the corresponding normal tissues (Supplementary Fig. 2). As shown in Fig. 1c, high levels of IL16 were detected in immune clusters, including T cell clusters, B cell cluster, monocyte cluster and macrophage cluster, while tumor cells and stromal cells (endothelial cells and fibroblasts) expressed very low levels of IL16. In comparison to matched non-cancerous tissues, the most pronounced downregulation of IL16 was observed in macrophages/monocytes and, to a lesser extent, in T cells. Contrarily, the average expression of IL16 was increased in tumor-infiltrating mast cells and B cells, while no statistical significance was reached due to substantial intercellular variance (Fig. 1d).

a Serum levels of IL-16 in cancer patients were measured by ELISA. H, healthy control (n = 28); BC, breast cancer (n = 20); LC, lung cancer (n = 20). b The expression of IL-16 in tumor and corresponding normal tissues was analyzed using the integrated TCGA and the Genotype-Tissue Expression (GTEx) databases by Mann–Whitney U-test. BRCA = breast cancer, LUAD = lung adenocarcinoma. c, d Tumor tissues and the corresponding normal tissues from breast cancer patients (n = 6) were subjected to scRNA-seq and major cell clusters, and the cell-specific expression of IL16 was shown. Epic = epithelial cells, Mac = macrophages, Mono = monocytes, B = B cells, MC = mast cells, FB = fibroblasts, EC = endothelial cells (c); the levels of IL16 in various cell clusters were compared between tumor (T) and normal (N) tissues (d, left), the relative expression of IL16 (T/N) was calculated (d, right). e The correlations between IL16 expression and patient overall survival (OS) were analyzed in TCGA BRCA and LUAD cohorts. Patients were divided into IL16high and IL16low groups based on the optimal cut-off values. HR = hazard ratio. f, g E0071 cells were inoculated into the mammary fat pad of mice. When tumors were palpable, mice were intraperitoneally (i.p.) injected with PBS or IL-16 (1 μg/mouse, twice a week, n = 6/group). Tumor growth was monitored (f); tumors were weighed on day 22 (g). (h, i) LLC cells were subcutaneously (s.c.) inoculated into mice. When tumors were palpable, mice were i.p. injected with PBS or IL-16 (1 μg/mouse, twice a week, n = 5/group). Tumor growth was monitored (h); tumors were weighed on day 21 (i). j, k MMTV-PyMT spontaneous breast cancer mice were i.p. injected with PBS or IL-16 when tumors were palpable (n = 8/group). Tumor growth was monitored (j); tumors were weighed four weeks after IL-16 treatment (k). l E0071-bearing mice were administered with anti-IL-16 (αIL-16, 150 μg/mouse, twice a week, n = 4/group) or control IgG, tumor growth was monitored. Unpaired, two-tailed Student’s t-test (a, d, f–l), Mann–Whitney U-test (b), and log-rank test (e) were used. Data are presented as mean ± standard deviation (SD).
In terms of the prognostic potential of IL-16, we found that low IL16 expression was significantly correlated with inferior survival of patients in 10 out of 31 TCGA cancer types (32.2%), including breast cancer and lung cancer (Fig. 1e and Supplementary Fig. 3). The negative correlations between IL16 expression and patient survival were further validated in six breast cancer cohorts and four lung cancer cohorts in the Gene Expression Omnibus (GEO) datasets (Supplementary Fig. 4).
To explore the potential impact of IL-16 on tumor progression, we established murine E0771 breast cancer model and LLC lung cancer model. When the tumors were palpable, mice were administered recombinant IL-16. Compared to control mice, mice treated with IL-16 developed significantly smaller tumors in both models (Fig. 1f–i). In addition to the implanted tumors, IL-16 also significantly reduced the growth of spontaneously developed mammary tumors in MMTV-PyMT transgenic mice (Fig. 1j, k). Conversely, IL-16 neutralization by anti-IL-16 antibody (αIL-16) resulted in accelerated tumor growth (Fig. 1l). Thus, we identified IL-16 as a novel cytokine with potential antitumor properties.
IL-16 generates a Th1-dominant tumor immune microenvironment
To gain a deeper understanding of how IL-16 regulates tumor growth, we first compared the immune composition in the TME between IL-16-treated and control mice. Flow cytometry results demonstrated that the percentages of CD8+ tumor-infiltrating lymphocytes (TIL) were significantly increased by IL-16 administration (Fig. 2a). Furthermore, the CD8+ TILs from IL-16-treated mice expressed higher levels of granzyme B (GZMB) and perforin, indicating an increased cytotoxic function of CD8+ TILs (Fig. 2b). The percentages of total CD4+ TILs were unaffected by IL-16 (Fig. 2a). However, the proportions and numbers of Th1 cells (IFN-γ+CD4+ T cells) were significantly upregulated in IL-16-treated mice. Additionally, the percentages of CD4+ T cells expressing T-bet, a Th1-specific transcription factor23, were also elevated by IL-16 (Fig. 2c and Supplementary Fig. 5). Consistently, IL-16 treatment significantly enhanced the transcript levels of Prf1, Gzmb, and Ifng in tumor tissues (Fig. 2d). In contrast, the percentages of intratumoral Th1 cells were downregulated in αIL-16-treated mice (Fig. 2e). Compared to the enhanced Th1 response, the percentages of Th2 cells and Th17 cells were significantly reduced in the TME of IL-16-treated mice, while Treg percentages were comparable between the two groups (Supplementary Fig. 6a). Additionally, IL-16-treated mice had significantly higher percentages of NK cells in the TME, while the percentages of TAMs were similar between the two groups (Supplementary Fig. 6b, c). Consistently, IL-16 increased the percentages of Th1 cells and the expression of Prf1/Gzmb/Ifng in MMTV-PyMT tumors (Fig. 2f, g). Similar to breast cancer models, IL-16 treatment also facilitated a Th1-dominant TME in LLC tumors (Supplementary Fig. 7).

Tumor tissues were harvested 22 days after E0771 cell inoculation, (a) The percentages of CD8+ TILs and CD4+ TILs were analyzed by flow cytometry (gated on CD45+ live cells) (n = 6/group). b The percentages of GZMB+CD8+ and perforin+CD8+ T cells were analyzed by flow cytometry (gated on CD45+CD8+ live cells) (n = 6/group). c The percentages of IFN-γ+CD4+ cells (Th1 cells) and T-bet+CD4+ cells were analyzed (gated on CD45+CD4+ live cells) (n = 6/group). d The mRNA levels of indicated genes were analyzed by QPCR (n = 6/group). e E0771 mice were treated with αIL-16 or control IgG, the percentages of intratumoral Th1 cells were evaluated (n = 4/group). The percentages of Th1 cells (f) and the expression of indicated genes (g) in MMTV-PyMT tumors were evaluated (n = 8/group). h E0771 cells were inoculated into nude mice (n = 4/group), tumor growth was monitored. Mice were injected with anti-CD4 antibody (αCD4) to deplete CD4+ T cells before E0771 cell inoculation (n = 5/group), tumor growth was monitored (i), the percentages of intratumoral CD8+ T cells were analyzed by flow cytometry (j), the expression of GZMB and perforin in tumor tissues was evaluated by QPCR (k). l The correlation between serum IL-16 levels and IFN-γ levels was analyzed in cancer patients (n = 40). m The correlation between total IL-16 expression and the percentages of Th1 cells in tumor tissues of breast cancer patients (n = 6) was analyzed using scRNA-seq data. Unpaired, two-tailed Student’s t-test (a–g) and Spearman’s rank correlation test (l, m) were used. Data are presented as mean ± SD.
Next, we aimed to identify the primary cellular subpopulations responsible for the anti-tumor effect of IL-16. As shown in Fig. 2h, IL-16 failed to limit tumor progression in nude mice, underscoring the indispensable role of functional T cells. Given that the CD4 molecule is the natural receptor for IL-16, we wondered if the depletion of CD4-expressing cells would affect the function of IL-16. In the TME, CD4-positive cells were almost exclusively CD3+TCRβ+, confirming their T cell identity (Supplementary Fig. 8). As anticipated, in the absence of CD4-expressing cells, IL-16 was unable to restrain tumor growth (Fig. 2i). Importantly, IL-16 administration no longer influenced the percentages of intratumoral CD8+ T cells (Fig. 2j), as well as the expression of Gzmb and Prf1 in CD4+ cell-depleted mice (Fig. 2k), indicating that IL-16 enhances CD8+ T cell function by acting on CD4+ T cells.
In breast cancer and lung cancer patients, we observed a significant positive correlation between serum levels of IFN-γ and IL-16 (Fig. 2l). Our sc-RNA sequencing data also demonstrated a significant positive correlation between IL-16 expression and Th1 cell percentages in breast cancer patients (Fig. 2m). Hence, IL-16 supplementation is an effective approach to boost a Th1-dominant antitumor immunity.
IL-16 potentiates the differentiation of Th1 cells
To directly investigate the impact of IL-16 on Th1 cell function, we differentiated Th1 cells in the presence or absence of IL-16 and subsequently performed RNA sequencing. As shown in Fig. 3a, IL-16 treatment significantly downregulated 317 genes and upregulated 228 genes, respectively (Supplementary Table. 1). Among the upregulated genes, several Th1-associated markers were identified, such as Ifng, Tbx21, Il18r1, Irf1, which are enriched in Gene Ontology (GO) pathways related to Th1 cell differentiation or activation, and IFN-γ response (Fig. 3b and Supplementary Table. 2). The expression of T cell exhaustion markers, such as TIM3, CTLA-4, PD-1, and BTLA4, was not significantly changed by IL-16 (Supplementary Fig. 9). The IL-16-induced upregulation of IFN-γ and T-bet mRNA was confirmed by QPCR (Fig. 3c). Flow cytometry and enzyme-linked immunosorbent assay (ELISA) assays further demonstrated that IL-16 significantly enhanced the protein levels of IFN-γ in Th1 cells (Fig. 3d, e). The Th1-promoting effect of IL-16 was also observed in human peripheral blood mononuclear cells (PBMC)-derived CD4+ T cells (Fig. 3f). Contrarily, neutralization of autocrine IL-16 significantly reduced IFN-γ production in Th1 cells (Fig. 3g). The expression of Ifng in unpolarized Th0 cells was also slightly increased by IL-16 (Supplementary Fig. 10). On the other hand, IL-16 did not obviously alter the proliferation of Th1 cells, as evidenced by CFSE dilution assay (Fig. 3h). The differentiation of Th2 cells, Th17 cells, and Tregs was not significantly affected by IL-16 (Supplementary Fig. 11). Furthermore, IL-16 did not change the expression of GZMB and perforin in CD8+ T cells cultured in vitro (Fig. 3i), confirming its indirect impact on CD8+ T cells.

a, b Naive CD4+ T cells were magnetically sorted from mouse spleen and cultured under Th1 condition in the absence or presence of 50 ng/ml IL-16. Cells were harvested on day 5 and then subjected to RNA sequencing. The number of differentially expressed genes (DEGs, defined as p < 0.05, fold change > 1.5) was shown by volcano plot (a), GO enrichment analysis was performed using the IL-16-upregulated Th1 marker genes (b). c–e Naive CD4+ cells were cultured under Th1 condition in the absence or presence of 50 ng/ml IL-16 for 5 days, the mRNA levels of Ifng (IFN-γ-encoding gene) and Tbx21 (T-bet-encoding gene) in Th1 cells were evaluated by QPCR (c, n = 3/group), the percentages of IFN-γ+CD4+ cells (Th1 cells) were analyzed by flow cytometry (d, n = 4/group), the levels of IFN-γ in T-cell culture supernatant were analyzed by ELISA (e, n = 5/group). f Naive CD4+ T cells were sorted from human PBMCs and subsequently cultured under Th1 conditions in the absence or presence of 50 ng/ml IL-16. The levels of IFN-γ in T-cell culture supernatant were analyzed by ELISA (n = 5/group). g Th1 cells were differentiated in the presence of 10 μg/ml αIL-16 or control IgG, and the levels of IFN-γ in T-cell culture supernatant were analyzed by ELISA (n = 5/group). h Th1 cells were treated with PBS or 100 ng/ml IL-16 for 5 days, cell proliferation was evaluated by CFSE dilution assay (n = 4/group). i Naive CD8+ T cells were magnetically sorted from the mouse spleen and stimulated with 50 ng/ml IL-16 for 4 days, the expression of perforin and GZMB was evaluated by QPCR (n = 3/group). Unpaired, two-tailed Student’s t-test (c–g) was used. Data are presented as mean ± SD.
We then divided TCGA breast cancer patients and lung cancer patients into IL16high and IL16low groups based on their tumoral IL16 expression. The differentially expressed genes (DEG) between the two groups were highly enriched in GO pathways related to leukocyte activation, differentiation and function (Supplementary Fig. 12a). Immune cell infiltration analysis demonstrated that IL16 levels were significantly positively correlated with the infiltration of Th1 cells, CD8+ T cells, and NK cells, while significantly negatively correlated with the infiltration of Th2 cells in the TME (Supplementary Fig. 12b). These findings confirm the beneficial role of IL-16 in enhancing the Th1 response.
IL-16 facilitates Th1 cell differentiation by inhibiting glutaminolysis
Emerging evidence has indicated that metabolic reprogramming plays a fundamental role in immune cell function24. To explore the potential involvement of metabolic mechanisms in the function of IL-16, we conducted a metabolomic analysis on IL-16-treated Th1 cells. As shown in Fig. 4a, IL-16 treatment resulted in metabolic alterations in Th1 cells, with Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway “alanine, aspartate and glutamate metabolism” showing the most pronounced changes. Glutamine is the major cellular energy source in various tissue microenvironments, which is essential for the functions of infiltrating immune cells25,26. A decrease in intracellular glutamine levels or the catabolism of glutamine to glutamate impairs the differentiation of Th1 cells25,26. We observed that the levels of glutamine (Gln) and glutamate (Glu) were significantly upregulated and downregulated, respectively, in IL-16-exposed Th1 cells, resulting in an increased Gln/Glu ratio (Fig. 4b, c). Consistently, IL-16 administration increased the Gln/Glu ratio in tumor-infiltrating CD4+ T cells (Fig. 4d). Notably, the Th1-promoting effect of IL-16 was remarkably diminished when Th1 cells were cultured in glutamine-deprived medium (Fig. 4e, f), suggesting that IL-16 favors Th1 differentiation by modulating intracellular glutamine levels. Consequently, we screened a panel of genes related to glutamine metabolism using RNA-sequencing and observed that IL-16 significantly suppressed the expression of GLS, an enzyme that inhibits Th1 polarization by inducing glutaminolysis25 (Fig. 4g,h). In line with the RNA-sequencing data, we observed that Gls expression was significantly lower in intratumoral CD4+ T cells isolated from IL-16-treated E0771 mice (Fig. 4i). A GLS inhibitor, CB-839, recapitulated the Th1-promoting function of IL-16 (Fig. 4j). Furthermore, in E0771-bearing mice, administration of CB-839 achieved similar effects to IL-16 in reducing tumor growth and increasing Th1 cell percentages (Fig. 4k,l). Conversely, IL-16 neutralization increased Gls expression and decreased the Gln/Glu ratio in Th1 cells (Supplementary Fig. 13).

a Naive CD4+ T cells were cultured under Th1 condition in the absence or presence of 50 ng/ml IL-16 for 4 days. A Q300 metabolomics analysis was performed. The differential metabolites were subjected to KEGG enrichment analysis. b The differential metabolites were shown in a volcano plot. c The relative ratios of glutamine (Gln) to glutamate (Glu) were analyzed using metabolomics data (n = 3/group). d CD4+ TILs were sorted from PBS- or IL-16-treated E0771 mice, the relative Gln/Glu ratios were analyzed (n = 6/group). Control or IL-16-treated Th1 cells were differentiated in control or glutamine-free medium for 5 days (n = 3/group), the percentages of Th1 cells were evaluated by flow cytometry (e), and the levels of IFN–γ in T-cell culture supernatant were analyzed by ELISA (f). g The genes related to glutamine metabolism were analyzed in RNA-Sequencing data. h Scheme illustrating the GLS-mediated glutamine metabolism. Created in BioRender. Xiao, P. (2025) https://BioRender.com/e49a161. i CD4+ T cells were sorted from E0771 tumors, and the expression of Gls was evaluated by QPCR (n = 6/group). j Th1 cells were treated with a GLS inhibitor CB-839 (0.5 μM) for 5 days, and the percentages of Th1 cells were evaluated by flow cytometry (n = 4/group). E0771 mice were administered CB-839 (n = 6/group), tumor growth (k), and the percentages of intratumoral Th1 cells (l) were measured. E0771 mice were fed a control or glutamine-deficient diet (n = 5/group), tumor growth (m), and the percentages of intratumoral Th1 cells (n) were measured. Unpaired, two-tailed Student’s t-test (c–f, i–n) and ANOVA (m, n) were used. Data are presented as mean ± SD.
To further investigate the impact of glutamine catabolism on IL-16-mediated anti-tumor immunity, we fed mice a glutamine-deficient diet. Glutamine deprivation did not significantly alter tumor growth, but it did significantly decrease the percentages of intratumoral Th1 cells. Importantly, the limited glutamine availability dramatically blunted the comparative differences between control and IL-16-treated mice in terms of tumor growth and Th1 cell percentages. Consequently, the anti-tumor and Th1-promoting capabilities of IL-16 were remarkably compromised in mice fed a glutamine-deficient diet (Fig. 4m, n), indicating that IL-16 promotes a Th1-dominant anti-tumor immunity by inhibiting glutamine catabolism in CD4+ T cells.
GLS expression and its-mediated glutaminolysis have been reported to be suppressed by GSK3 signaling27,28,29, which is also required for the differentiation of Th1 cells30,31. We observed that the levels of phosphorylated GSK3 (p-GSK3, its inactive form) decreased upon IL-16 treatment in Th1 cells (Supplementary Fig. 14a). Consistently, CD4+ TILs from IL-16-treated mice expressed lower levels of p-GSK3 (Supplementary Fig. 14b), indicating that IL-16 enhanced the activation of GSK3 signaling. We then treated Th1 cells with CHIR-99021 (a GSK3 inhibitor). Notably, the capability of IL-16 in suppressing Gls expression and promoting IFN-γ production was compromised in the presence of CHIR-99021 (Supplementary Fig. 14c–f), corroborating the involvement of GSK3 signaling in the IL-16-induced Th1 response.
IL-16 administration shapes an anti-tumor macrophage phenotype via IFN-γ
Our previous study revealed that a Th1-dominant TME drives the functional reprogramming of M1-like anti-tumor macrophages. Indeed, we observed an increased expression of M1-marcrophage markers such as Tnfa, Cxcl9, Cxcl10 in E0771 tumors from IL-16-treated mice, while the expression of an M2-marcrophage marker Arg-1 was downregulated (Fig. 5a). A similar effect was also observed in MMTV-PyMT tumors (Fig. 5b). Consistently, the expression of M1 or M2 markers was significantly higher or lower, respectively, in TAMs isolated from IL-16-treated mice than those from control mice (Fig. 5c). However, neither did the expression of M1 markers nor the expression of M2 markers change when we added IL-16 to the M1 or M2 polarization conditions in vitro (Fig. 5d, e). Notably, in contrast to immunocompetent mice, IL-16 did not increase the tumoral expression of M1 markers in nude mice or in CD4+ T cell-depleted mice (Fig. 5f, g), implying that IL-16 reshapes TAM phenotypes by altering CD4+ T cell function in the TME.

a Tumor tissues from control or IL-16-administered mice were harvested 22 days after E0771 cell inoculation (n = 6/group), the levels of indicated genes were evaluated by QPCR. b The levels of indicated genes in MMTV-PyMT tumors were evaluated by QPCR (n = 8/group). c TAMs were magnetically sorted from E0771 tumors, and the levels of indicated genes were evaluated by QPCR (n = 6/group). d Peritoneal macrophages were differentiated into M1 macrophages with 1 μg/ml LPS + 20 ng/ml IFN-γ in the presence of 50 ng/ml IL-16 for 6 h, the levels of Tnfa, Cxcl9, Cxcl10 were evaluated by QPCR (n = 3/group). e Peritoneal macrophages were differentiated into M2 macrophages with 20 ng/ml IL-4 in the presence of 50 ng/ml IL-16 for 24 h, the levels of Arg-1 were evaluated by QPCR (n = 3/group). Tumor expression of Cxcl9 and Cxcl10 in E0771 nude mice (f, n = 4/group) or in CD4+ cell-depleted E0771 mice (g, n = 5/group) was evaluated by QPCR. h Scheme illustrating the preparation of TIF. Created in BioRender. Xiao, P. (2025) https://BioRender.com/e49a161. i Peritoneal macrophages were stimulated with TIF (50% v/v) from PBS- or IL-16-treated mice for 8 h in the presence of IgG or αIFN-γ (10 μg/ml), and the levels of indicated genes were evaluated by QPCR (n = 3/group). j Tumor expression of Cxcl9 and Cxcl10 in IFN-γ-neutralized E0771 mice was evaluated by QPCR (n = 4/group). k TAMs were isolated from PBS- or IL-16-treated E0771 tumors, then co-inoculated with E0771 cells into new recipient mice, and tumor growth was monitored (n = 5/group). l The correlations between IL16 expression and the levels of M1 macrophage marker genes in TCGA breast cancer and lung cancer patients were analyzed. Unpaired, two-tailed Student’s t-test (a–c, i), ANOVA (k), and Spearman’s correlation test (l) were used. Data are presented as mean ± SD.
To test this hypothesis, we collected tumor interstitial fluid (TIF) from IL-16-treated mice (IL16TIF) and control mice (CtrlTIF) as previously described (Fig. 5h)32,33. Macrophages treated with IL16TIF exhibited significantly higher expression of M1 markers than those treated with CtrlTIF. Importantly, the M1-promoting function of IL16TIF was significantly diminished in the presence of an IFN-γ-neutralizing antibody (αIFN-γ), suggesting that IL-16 enhances the M1 polarization of macrophages by increasing IFN-γ production in the TME. Conversely, IL16TIF reduced Arg-1 expression in macrophages, whereas this effect was unaffected by αIFN-γ (Fig. 5i). Moreover, when IFN-γ was neutralized in E0771-bearing mice, IL-16 was barely able to upregulate the expression of M1 markers (Fig. 5j).
Next, we isolated TAMs from PBS- or IL-16-treated E0771 tumors and then co-inoculated TAMs with E0771 cells into new recipient mice. In contrast to the pro-tumoral effect of PBS-treated TAMs, TAMs from IL-16-treated mice significantly delayed tumor growth (Fig. 5k). Consistently, in TCGA cohorts, IL16 expression showed significant positive correlations with a panel of M1 markers in breast cancer and lung cancer patients (Fig. 5l). Thus, IL-16 reprograms TAMs to an anti-tumor phenotype through potentiating a Th1-dominant TME.
IL-16 improves the effectiveness of immune checkpoint blockade therapy through TAM reprogramming
Given that IL-16 augmented the expression of CXCL9 and CXCL10, the two CXCR3 ligands that are required for anti-tumor immune responses following Immune Checkpoint Blockade (ICB)34,35, we next questioned if IL-16 could potentiate the therapeutic effectiveness of ICB. To this end, E0771 breast cancer or LLC lung cancer mice were treated with IL-16, anti-PD-L1 antibody (αPD-L1), or in combination. In both models, PD-L1 blockade only marginally reduced tumor development, while the co-administration of IL-16 plus αPD-L1 exhibited significantly greater anti-tumor capacity when compared to IL-16 or αPD-L1 monotherapy (Fig. 6a, b). In agreement, mice treated with αPD-L1 + IL-16 showed significantly higher levels of Ifng, Prf1, and Gzmb in the TME (Fig. 6c), indicating that IL-16 is required for the optimal induction of anti-tumor immunity following PD-L1 blockade. Importantly, the IL-16-mediated sensitization to PD-L1 blockade was compromised when mice were co-administered with NBI-74330, a CXCR3 inhibitor (Fig. 6d), suggesting that CXCR3 ligands are necessary for the therapeutic efficacy of αPD-L1 plus IL-16.

Mice inoculated with E0771 cells (a, n = 5 for PBS groups, n = 7 for IL-16 groups) or LLC cells (b, n = 4/group) were i.p. administered with IL-16 (0.8 μg), αPD-L1 (150 μg), or in combination, tumor growth was measured. c The expressions of indicated genes in E0771 tumors were evaluated by QPCR. d E0771 mice were administered with a CXCR3 inhibitor (NBI-74330) and co-treated with IL-16 + αPD-L1 therapy (n = 4/group); tumor growth was monitored. e The cell-specific expression of CXCL9 and CXCL10 in tumor tissues from breast cancer patients (n = 6) was analyzed using scRNA-seq. f E0771 mice were administered with clodronate liposomes to deplete TAMs (MφΔ) (n = 5 for Ctrl groups, n = 6 for MφΔ groups), followed by IL-16 + αPD-L1 treatment, tumor growth was measured. ANOVA (a–d, f) was used. Data are presented as mean ± SD.
Furthermore, our scRNA-seq data demonstrated that the expression of CXCL9 and CXCL10 was highly enriched in the macrophage cluster (Fig. 6e). Therefore, we depleted macrophages using clodronate liposomes in tumor-bearing mice. Remarkably, compared to control mice in which macrophage depletion attenuated tumor development, macrophage deficiency led to accelerated tumor progression in mice that received the IL-16 + αPD-L1 combination therapy (Fig. 6f). This differential outcome of macrophage depletion suggests that IL-16 + αPD-L1 treatment reprograms TAMs from a pro-tumor to an anti-tumor phenotype, thereby restraining tumor growth.
IL-16 determines the outcome of ICB therapies in cancer patients
To investigate the potential impact of IL-16 on ICB therapy in a clinical setting, we collected serum samples from lung cancer patients who received anti-PD-1 monoclonal antibody (αPD1) prior to the initiation of therapy. Notably, patients with progressive disease (PD, non-responders) had significantly lower serum IL-16 levels compared to patients with stable disease (SD), partial response (PR), and complete response (CR), which were classified as responders (Fig. 7a). When we divided patients based on their serum IL-16 levels, 100% PD patients were IL-16low, while in SD, PR and CR groups the proportions of IL-16low patients were only 58.3%, 40%, and 0%, respectively (Fig. 7b). The area under the receiver operating characteristic (ROC) curve was 0.895, indicating high accuracy of serum IL-16 in distinguishing between responders and non-responders (Fig. 7c). More importantly, IL-16high patients exhibited significantly longer survival than IL-16low patients (Fig. 7d). The positive correlation between IL-16 and CXCL9/CXCL10 levels in these patients indicates that IL-16 may sensitize ICB therapy through upregulating the two CXCR3 ligands (Fig. 7e).

a–e Serum samples of lung cancer patients responding or not responding to αPD-1 therapy were harvested before treatment, and the levels of IL-16 were evaluated by ELISA (a). PD, n = 4; SD, n = 12; PR, n = 15; CR, n = 4. The proportions of IL-16low (gray) and IL-16high (red) patients in PD, SD, PR, and CR groups were analyzed (b). ROC curve analysis indicated the role of serum IL-16 in predicting the responsiveness to αPD-1 therapy in lung cancer patients (c). The progression-free survival (PFS) was compared between IL-16low (n = 17) and IL-16high (n = 18) patients (d). Serum levels of CXCL9 and CXCL10 in lung cancer patients were evaluated by ELISA. The correlations between serum levels of IL-16 and CXCL9/CXCL10 were analyzed (e). f Cancer patients were divided into IL16high and IL16low groups based on the optimal cut-off values in six immunotherapy GEO datasets, PFS or OS was compared between the two groups. Unpaired, two-tailed Student’s t-test (a), log-rank test (d, f), and Spearman’s correlation test (e) were used. Data are presented as mean ± SD.
Strikingly, through exploring multiple immunotherapy datasets, we further discovered that high IL16 expression was associated with favorable survival in cancer patients who received αPD-1/PD-L1 therapy, including two non-small cell lung cancer cohorts36,37, one hepatocellular carcinoma cohort38, one head and neck cancer/melanoma/lung cancer mixed cohort39, and one melanoma cohort40. Additionally, high IL16 expression also predicted a better outcome in melanoma patients receiving adoptive T cell therapy (ACT)41 (Fig. 7f). These results strongly indicate that the combination of IL-16 and immune checkpoint blockade may be an effective therapeutic approach.
Mast cell-derived histamine is responsible for maintaining IL-16 expression in the TME
Finally, we aimed to identify the potential factors responsible for IL-16 downregulation in tumor hosts. Since both local and systemic IL-16 levels were decreased in cancer patients (Fig. 1), it can be hypothesized that specific secreted products in the TME influenced IL-16 expression in a paracrine and/or telecrine manner. Through screening the STITCH database, which predicts protein-compound interactions, we noticed that histamine, an endogenous metabolite, has a potential regulatory role on IL-16 (Fig. 8a). Indeed, histamine has been reported to trigger the expression or release of IL-1642,43. In line with the reduced IL-16 production, we observed significantly lower serum histamine concentrations in breast cancer and lung cancer patients compared to healthy individuals (Fig. 8b). Notably, there was a significant positive correlation between serum levels of histamine and IL-16 in cancer patients (Fig. 8c).

a Compound-protein network of IL-16 was analyzed using STITCH online tool (http://stitch.embl.de/). Created in BioRender. Xiao, P. (2025) https://BioRender.com/e49a161. b Serum histamine levels in cancer patients were evaluated by ELISA. H, healthy control (n = 28); BC, breast cancer (n = 20); LC, lung cancer (n = 20). c The correlation between serum IL-16 and histamine levels was analyzed in cancer patients. d Mice inoculated with E0771 cells were i.p. administered with histamine (2 mg/kg, twice a week), serum IL-16 levels were measured on day 21 (n = 5/group). e, f E0771 mice were i.p. administered with histamine (2 mg/kg), αIL-16 (150 μg), or histamine + αIL-16 (150 μg), tumor growth was monitored (e), the intratumoral percentages of Th1 cells were evaluated on day 21 (f) (n = 6 for IgG groups, n = 5 for αIL-16 groups). g The cell-specific expression of HDC was analyzed by scRNA-seq (left). HDC expression in tumor tissues and corresponding normal tissues of breast cancer patients (n = 6) was compared (right). h The expression of HDC in tumor and normal tissues was analyzed using the integrated TCGA and GTEx databases by Mann–Whitney U-test. i The correlations between the levels of IL16 and HDC were analyzed in TCGA datasets. j Peritoneal macrophages were stimulated with 10 μM histamine for 48 h, Il16 expression was evaluated by QPCR (n = 3/group). k Serum levels of IL-16 were evaluated in wild-type (WT) or mast cell-deficient (MCΔ) E0771 tumor-bearing mice by ELISA (n = 7/group). E0771 mice were administered with mast cell stabilizer DSCG (n = 6/group). Serum IL-16 levels (l), the percentages of intratumoral Th1 cells (m), and tumor growth (n) were measured. o Mast cells were treated with tumor interstitial fluid (50% v/v) for 48 h, Hdc expression was evaluated by QPCR (n = 3/group). Unpaired, two-tailed Student’s t-test (b, d, g, j–o), ANOVA (e, f), Mann–Whitney U-test (h), and Spearman’s correlation test (c, i) were used. Data are presented as mean ± SD.
To further explore the impact of histamine on IL-16-mediated immune regulation, tumor-bearing mice were treated with exogenous histamine. As shown in Fig. 8d, serum IL-16 levels were significantly elevated by histamine (Fig. 8d). Moreover, histamine administration significantly inhibited tumor growth and increased the percentages of intratumoral Th1 cells. Importantly, neutralization of IL-16 compromised both the anti-tumor efficacy and the Th1-promoting capacity of histamine (Fig. 8e, f), suggesting that IL-16 contributes to the therapeutic function of histamine.
Histamine synthesis is mediated by the enzymatic activity of histidine decarboxylase (HDC). Our scRNA-seq data indicated that HDC is almost exclusively expressed in the mast cell cluster, and the expression of HDC in mast cells was downregulated in tumor tissues compared to normal tissues (Fig. 8g). In agreement, tumor HDC expression was significantly decreased in 21 out of 31 TCGA cancer types (67.7%), and it exhibited a significant positive correlation with tumor IL16 expression in 24 out of 31 TCGA cancers (77.4%) (Fig. 8h, i and Supplementary Figs. 15, 16). Since the most pronounced IL16 downregulation was observed in TAMs (Fig. 1k), we stimulated macrophages with exogenous histamine. This treatment resulted in a significant upregulation of Il16 levels (Fig. 8j), indicating the direct effect of histamine on macrophage IL16 expression.
To investigate the impact of mast cells on IL-16 production, we inoculated E0771 cells into mast cell-deficient KitW-sh/W-sh (MCΔ) mice, in which histamine production is drastically decreased44,45. Importantly, we observed a significant reduction in serum IL-16 levels in MCΔ mice (Fig. 8k). However, tumor growth was not obviously affected by mast cell deficiency (Supplementary Fig. 17). We further treated mice with a mast cell stabilizer—disodium cromoglycate (DSCG), which prevents histamine release from mast cells. DSCG treatment significantly reduced serum IL-16 levels in breast cancer mice (Fig. 8l), suggesting that the reduced IL-16 production in tumor-bearing mice was at least partially attributed to the impaired histamine production by mast cells. Additionally, DSCG treatment decreased the percentages of intratumoral Th1 cells and accelerated tumor development (Fig. 8m, n). Finally, we cultured primary mast cells and stimulated them with tumor interstitial fluid. This treatment significantly downregulated Hdc expression in mast cells (Fig. 8o), implying a suppressive role of tumor-derived soluble factors in HDC expression.
Taken together, IL-16 fosters a Th1-dominant tumor immune microenvironment that not only facilitates tumor immune eradication but also overcomes resistance to ICB therapies (Fig. 9).

Created in BioRender. Xiao, P. (2025) https://BioRender.com/e49a161.
Discussion
Although IL-16 was discovered as early as 1982, the regulatory roles of this cytokine in various disease contexts are far less understood. Of note, the impact of IL-16 on anti-tumor immunity remains largely unclear. In this study, we delineate the role and importance of IL-16 in shaping the TME to augment anti-tumor immune responses. IL-16 inhibits tumor growth in preclinical murine breast cancer and lung cancer models and is associated with better prognosis across a panel of human cancers, implying a broad therapeutic potential of IL-16. Compared to many identified immunotargets from preclinical studies, IL-16 may have greater potential in terms of clinical application. Firstly, as an extracellular cytokine, IL-16 has good druggability and thus can be supplemented in a convenient way. Previous studies have reported the multifaceted impacts of IL-16 on immune regulation whilst yielding discrepant results. The precise effects of IL-16 remain a topic of debate and appear to vary depending on specific pathological contexts and experimental conditions16,18,19,20. In our study, IL-16 was administered systemically, which is a clinically relevant route. Therefore, although IL-16 is known to mediate T cell recruitment, its anti-tumor effect is unlikely attributed to the chemoattractive capacity since systemic administration cannot effectively generate a concentration gradient in the TME required for immune cell recruitment.
Secondly, the relatively specific action on CD4+ cells minimizes the off-target effects of IL-16. In fact, IL-16 has been reported to regulate cellular functions in a CD4-independent manner21,22,46, presumably due to the existence of alternative or yet unidentified IL-16 receptors47. Although our study found that IL-16 lost its anti-tumor function after the depletion of CD4+ cells, this finding does not rule out the possibility that IL-16 acts on a non-CD4 receptor, which is primarily expressed on CD4+ cells. Given that IL-16 has been reported to regulate the function of epithelial cells20, it is possible that IL-16 can directly affect the malignant behaviors of some tumor cells of epithelial origin. The inability of IL-16 to affect M1 marker gene expression in nude mice and isolated macrophages suggests that IL-16 does not directly act on macrophages. Instead, the IL-16-established Th1 TME reprograms TAMs to an anti-tumor phenotype. This functional switch results in the increased production of CXCR3 ligands (CXCL9 and CXCL10), which improve the therapeutic effectiveness of ICB by facilitating the recruitment of CXCR3+CD8+ T cells34. Additionally, CXCR3 ligands in murine tumors and the plasma of patients with melanoma have been identified as reliable biomarkers for clinical response to anti-PD-1 therapy48. From a clinical perspective, high expression of IL-16 is not only associated with favorable patient survival in multiple cancer types but also predicts better responsiveness in patients receiving immunotherapy, including PD-1/PD-L1 blockade and adoptive T cell transfer. Notably, although the transcript levels of IL-16 do not exhibit positive correlations with patient survival in some other TCGA cancer types, IL-16 may still play an anti-tumor role in these cancers, as post-transcriptional regulation mechanisms are also pivotal for the production of IL-16. For instance, the cleavage of IL-16 precursor by caspase-3 is required for generating bioactive IL-1615. Therefore, the prognostic significance of the protein levels of IL-16 warrants further exploration. On the other hand, despite a Th1-dominant TME generally favoring anti-tumor immunity in established tumors, the hyperactivation of Th1 cells may also lead to pro-tumor inflammatory responses, particularly in patients whose cancer was driven by chronic inflammation. The timing and dosage of IL-16 supplementation in cancer patients should be carefully considered. Additionally, Gao et al. reported that serum levels of IL-16 were elevated in colorectal cancer patients and gastric cancer patients49. Qin et al. also observed higher serum IL-16 levels in nasopharyngeal carcinoma patients compared to control subjects50. Whether IL-16 plays a regulatory role in these cancers requires further exploration. Surprisingly, in contrast to amplifying Th1 cell differentiation in the TME, IL-16 was found to suppress Th1 immunity in certain infectious diseases. For example, IL-16 knockout mice exhibited enhanced Th1 and CTL responses upon the infection of influenza A virus51. In addition, IL-16 facilitated mycobacterium infection by inhibiting macrophage phagosome conversion, which was mediated by IFN-γ52. Although the mechanisms for these conflicting results remain unidentified, the exact impacts of IL-16 on Th1 immunity are complex and dependent on specific pathological microenvironments. Therefore, the potential adverse effects of IL-16, such as exacerbating pathogen infection, should be considered in future clinical applications.
Although glucose and glutamine serve as primary sources of cellular energy in TME, the regulation of glutamine metabolism in tumor-infiltrating immune cells remains largely elusive. In this study, we identify IL-16 as a suppressor of glutamine catabolism in CD4+ T cells, thus providing new insights into the immunometabolism-mediated remodeling of the TME. To date, the mechanisms underlying the modulation of immune cell functions by glutamine metabolism are far from being fully understood. Whether glutamine acts as a signaling molecule to enhance Th1 polarization or its specific metabolites mediate this effect requires further exploration. Due to the indispensable role of glutamine in supporting the effector functions of immune cells, blocking its catabolism by CB-839 enhances anti-tumor immunity and improves the effectiveness of tumor immunotherapy10. Importantly, CB-389 (brand name Telaglenastat) has been utilized in the treatment of patients with solid tumors in clinical trials11,12, highlighting the promising clinical translatability of targeting glutamine metabolism. Further study identified GSK3 signaling mediated the IL-16-induced inhibition of glutaminolysis. However, this effect seems to be partial, as the inhibition of GSK3 compromised but did not fully abrogate the function of IL-16.
In this study, we reveal that impaired histamine production by mast cells is a contributing factor for IL-16 downregulation in the TME. The strong positive correlation between IL16 and HDC levels across multiple cancer types further validates the close relationship between histamine signaling and IL-16 production. Our data demonstrate that tumor interstitial fluid decreases Hdc expression in mast cells. However, the identification of the exact factors contributing to this effect requires further investigation. In contrast to DSCG, which accelerated the growth of tumor xenografts, mast cell deficiency did not have an obvious impact on tumor progression, despite a decrease in IL-16 production. To date, the precise role of mast cells in cancers remains controversial. Both pro-tumor and anti-tumor functions of mast cells have been proposed53,54,55. Therefore, it is plausible that the global ablation of mast cells causes multifaceted changes in the TME, and thus, the effects of IL-16 are masked. It is noteworthy to mention that the expression of IL-16 and HDC are increased in tumor tissues compared with corresponding normal tissues in a minority of TCGA cancers. This may be attributed to a higher infiltration of IL16-expressing or HDC-expressing cells in the TME than in non-cancerous tissues in these cancer types.
In summary, our present study identifies IL-16 as a promising cytokine candidate that could be exploited for cancer immunotherapy.
Methods
Mice and tumor models
Six-to-eight-week-old C57BL/6 mice were purchased from Gempharmatech (Nanjing, China, stock for mouse strain: #T064543). 2 × 106 E0771 breast cancer cells (American Type Culture Collection, #CRL-3461, Shanghai, China) were inoculated into the mammary fat pad of female mice. 5 × 105 Lewis lung carcinoma (LLC) cells (American Type Culture Collection, #CRL-1642, Shanghai, China) were inoculated subcutaneously (s.c.) into the left flank of female or male mice. In the nude mouse model, 8 × 105 E0771 cells were inoculated into the mammary fat pad of female Balb/c nude mice (Gempharmatech, stock for mouse strain: #D000521). In the spontaneous breast cancer model, female MMTV-PyMT transgenic mice were used (Gempharmatech, stock for mouse strain: #T004993). When tumors were palpable, mice were intraperitoneally (i.p.) injected with PBS or 1 μg IL-16 (Novus Biologicals, #NBP2-35104, Englewood, CO, USA) intraperitoneally (i.p.) twice a week. Tumor size was measured using a caliper. Tumor volume was calculated using the formula: length × width2/2. Tumor weight was measured on an analytical balance. The experimental and control mice were co-housed. Animal experiments were performed following protocols approved by the Zhejiang University Institutional Animal Care and Use Committee. According to the protocol, we euthanized mice by cervical dislocation when tumor growth exceeded 10% of the animal’s original body weight or with an average tumor diameter exceeding 2 cm. Tumor growth was monitored two to three times a week.
Mouse IL-16 administration
For IL-16 neutralization, 150 μg anti-IL-16 antibody (Biolegend, #519108, San Diego, CA, USA) was i.p. injected into tumor-bearing mice when tumors were palpable. Injection was performed twice a week.
T cell depletion
For CD4+ T cell depletion, 200 μg CD4 antibody (Biolegend, #100457) was i.p. injected into mice twice a week, starting from day 0 after tumor inoculation.
IFN-γ neutralization
For IFN-γ neutralization, 100 μg anti-IFN-γ antibody (BioXCell, #505847) was i.p. injected into tumor-bearing mice when tumors were palpable. Injection was performed twice a week.
Histamine administration
For histamine administration, 5 mg/kg histamine (Aladdin, #H111796, Shanghai, China) was i.p injected into tumor-bearing mice when tumors were palpable. Injection was performed twice a week.
PD-L1 blockade
For PD-L1 blockade, 150 μg anti-PD-L1 (BioXCell, #BE0101) was i.p injected into tumor-bearing mice when tumors were palpable. Injection was performed twice a week.
For CXCR3 inhibition
For CXCR3 inhibition, 5 mg/kg NBI 74330 (MedChemExpress, HY-15320) was i.p. injected into tumor-bearing mice when tumors were palpable. Injection was performed twice a week.
Human samples
Serum samples of breast cancer patients, non-small cell lung cancer (NSCLC) patients, and healthy donors were collected at Sir Run Run Shaw Hospital, Zhejiang University School of Medicine. Serum samples of NSCLC patients who received anti-PD1 therapy were collected at the Second Affiliated Hospital of Zhejiang University School of Medicine. The anti-PD1-treated NSCLC patients are driver-gene negative and are classified as clinical stage III or stage IV without surgical resection history. Serums were collected prior to the initiation of immunotherapy. Therapeutic responsiveness was assessed according to Response Evaluation Criteria in Solid Tumors (RECIST) v1.1 criteria. Experiments were approved by the Medical Ethics Committee of Sir Run Run Shaw Hospital, Zhejiang University School of Medicine (No. 2022-0256), and the Medical Ethics Committee of Second Affiliated Hospital, Zhejiang University School of Medicine (No. 2020-907). The basic information on cancer patients is provided in Supplementary Table. 3. Informed consents were obtained from all participants.
Flow cytometry
Tumor tissues were digested to obtain single cell suspensions as previously described2, then blocked with TruStain FcX™ PLUS (Biolegend, #156604) on ice for 10 min. Cells were stained with surface markers on ice for 20 min in staining buffer (PBS + 2.5% FBS + 1 mM EDTA), then washed with PBS. For intracellular staining, cells after surface staining were fixed in Fixation Buffer (Biolegend, #420801) at room temperature for 30 min. Thereafter, cells were permeabilized and stained in Intracellular Staining Permeabilization Wash Buffer (Biolegend, #421002) with added antibodies against intracellular antigens. For the detection of Th1 cells, the isolated single cells from tumors or in vitro-cultured T cells were stimulated with 1×Cell Activation Cocktail with Brefeldin A (Biolegend, 423304) for 5 h before staining. Flow cytometry was performed on a NovoCyteTM Flow Cytometer (Agilent, Santa Clara, CA, USA). The following antibodies or dye were used: CD45-APC/CY7 (cat#103116, 1:500), IFNγ-PE (cat#505808, 1:150), T-bet-AF647 (cat#644804, 1:200), F4/80-FITC (cat#123107, 1:200), CD11b-PE (cat#101207, 1:400), CD8-FITC (cat#100706, 1:200), CD4-PE/CY7 (cat#100422, 1:200), IL-17A-FITC (cat#506907, 1:150), IL-4-PE (cat#504103, 1:100), Foxp3-AF647 (cat#126407, 1:200), Zombie Violet™ Viability dye (cat#423114, 1:200). Gating strategies are shown in Supplementary Fig. 18. All antibodies or dye were purchased from Biolegend. For CFSE dilution, isolated CD4+ T cells were labeled with 2.5 μM CFSE (Thermo Fisher Scientific, #C34554, Waltham, MA, USA) at 20 °C for 10 min, then washed twice with PBS before seeding. Flow cytometry data were analyzed using FlowJo software (Tree Star, Ashland, OR, USA).
T cell isolation and differentiation
Murine naive CD4+ T cells were isolated from mouse spleen using EasySep™ Mouse Naive CD4+ T Cell Isolation Kit (STEMCELL, #19765, Cambridge, MA, USA) on an EasyEights™ EasySep™ Magnet (STEMCELL, #18103), then seeded in a round-bottom 96-well plate pre-coated with anti-CD3 (BioXCell, #BE0001-1) and anti-CD28 (BioXCell, #BE0015-5). For the differentiation of Th1 cells, the isolated naive CD4+ T cells were cultured in RPMI 1640 medium supplemented with 10% heat-inactivated fetal bovine serum (Serana, #S-FBS-SA-015, Brandenburg, Germany), 100 U/ml penicillin & 100 µg/ml streptomycin (Thermo Fisher Scientific, #15070063), 10 mM HEPES (Thermo Fisher Scientific, #15630106), 0.05 mM 2-mercaptoethanol (Thermo Fisher Scientific, #21985023), 10 ng/mL IL-12 (Peprotech, #577002), 5 μg/mL anti-IL-4 (Biolegend, #504122). On day 3, T cells were replenished with fresh media and cultured for an additional two days. For Th0 cells, isolated naive CD4+ T cells were cultured using the same condition as Th1 cells, except that no exogenous cytokine or antibody was added. For human Th1 differentiation, peripheral blood mononuclear cells from healthy donors were isolated through Ficoll density gradient centrifugation (Solarbio, #P8900, Beijing, China). Human naive CD4+ T cells were isolated using EasySep™ Human Naive CD4+ T Cell Isolation Kit (STEMCELL, #19555). The Th1 differentiation procedure is similar to that for murine Th1 cells, with the exception that human antibodies or cytokines were used: anti-CD3 (Biolegend, #317325), anti-CD28 (Biolegend, #302902), anti-IL-4 (Biolegend, #500838), IL-12 (Peprotech, 200-12H-10UG), IL-16 (Peprotech, 200-16A-10UG). CD8+ T cells were isolated from mouse spleen using EasySep™ Mouse Naive CD8+ T Cell Isolation Kit (STEMCELL, #19858), and cultured in plate-immobilized anti-CD3 and anti-CD28 in RPMI 1640 medium supplemented with 10% FBS, 100 U/ml penicillin & 100 µg/ml streptomycin (Thermo Fisher Scientific, #15070063), 5 ng/ml IL-2 (Biolegend, #575402), 10 mM HEPES (Thermo Fisher Scientific, #15630106), 0.05 mM 2-mercaptoethanol (Thermo Fisher Scientific, #21985023) for 4 days. For the isolation of tumor-infiltrating CD4+ T cells, single cell suspensions were obtained as described above. The isolation steps were conducted using biotin-conjugated anti-CD4 antibody (BioLegend, #100403) and EasySep™ Biotin Positive Selection Kit (STEMCELL, #17683) on an EasyEights™ EasySep™ Magnet (STEMCELL, #18103).
Single-cell RNA sequencing
Fresh breast cancer tissues and the corresponding normal tissues were collected at Harbin Medical University Cancer Hospital and preserved in the sCelLive® Tissue Preservation Solution (Singleron Biotechnologies, Nanjing, China) on ice immediately after surgery. Tissues were then rinsed with Hanks Balanced Salt Solution (HBSS) for 3 times, minced into small pieces, and digested in sCelLive® Tissue Dissociation Solution (Singleron Biotechnologies) at 37 °C for 15 min. The dissociated tissues were filtered through 40 μm cell strainers. The red blood cells were removed by GEXSCOPE® red blood cell lysis buffer (Singleron Biotechnologies) at room temperature for 6 min. The single-cell suspensions in PBS were loaded into microfluidic devices. ScRNA-seq libraries were constructed according to Singleron GEXSCOPE® protocol by GEXSCOPE® Single-Cell RNA Library Kit (Singleron Biotechnologies) and Singleron Matrix® Automated single-cell processing system (Singleron Biotechnologies). Individual libraries were diluted to 4 ng/µL and pooled for sequencing. Pools were sequenced on an Illumina NovaSeq 6000 System (San Diego, CA, USA) with 150 bp paired-end reads. Experiments were approved by the Medical Ethics Committee of Harbin Medical University Cancer Hospital (No. 20200305). Informed consents were obtained from patients. The basic information of cancer patients is provided in Supplementary Table. 1. Informed consents were obtained from all participants.
Single-cell RNA sequencing data analysis
The raw data and processed data were obtained from Singleron Biotechnologies. The demultiplexing, barcoded processing, gene counting, and aggregation were performed using the CeleScope (Singleron Biotechnologies) software. All scRNA-seq reads were aligned to the human reference genome (hg38), and the cell-by-gene count matrices were produced. The scRNA-seq expression profiles were analyzed using R package “Seurat”. The cells within a minimum of 500 genes expressed, total UMI counts > 1000, and the percentage of mitochondrial gene counts < 20% were kept for subsequent analysis. UMI counts were normalized for each cell by the total expression, multiplied by 106, then log-transformed.
Next, we discovered the variable genes by using the FindVariableFeatures function with the following parameters: selection.method = “vst”, nfeatures = 2000, and the ScaleData function was used to regress out variation due to differences in total UMIs per cell. Principal component analysis was used on the scaled expression profiles for the variable genes, and the top 30 principal components (PCs) were conducted for subsequent analysis. Cells were clustered by using FindClusters function. All cells were visualized by Uniform Manifold Approximation and Projection (UMAP) on the top 30 PCs. Cell types were assigned to each cluster by examining the upregulated genes with lowest Bonferroni-adjusted p values.
ELISA
Human IL-16 (#EH0178), CXCL9 (#EH0008) and CXCL10 (#EH0102) ELISA Kits were brought from FineTest (Wuhan, China), murine IL-16 ELISA Kit was brought from Sino Biological (China, #KIT51303, Beijing), human and mouse IFN-γ ELISA Kit was brought from Thermo Fisher Scientific (#88-7316-77, #88-7314-88). Histamine ELISA Kit was brought from Elabscience (Houston, #E-EL-0032, TX, USA). ELISA assays were performed according to the manufacturer’s instructions.
Metabolomics analysis
Th1 cells stimulated with PBS or IL-16 were subjected to metabolomics analysis by the Q300 Kit from Metabo-Profile Corp. (Shanghai, China)56,57. The targeted metabolites were quantified using ultra-performance liquid chromatography tandem mass spectrometry (UPLC-MS/MS, ACQUITY UPLC-Xevo TQ-S, Waters Corp., Milford, MA, USA). The raw data generated from UPLC-MS/MS were processed using iMAP platform (Metabo-Profile, Shanghai, China). Metabolite list was provided in Supplementary Table 4.
TAM isolation and co-inoculation
TAMs were isolated from single cell suspensions of E0771 tumors using EasySep™ Mouse F4/80 Positive Selection Kit (STEMCELL, #100-0659) on an EasyEights™ EasySep™ Magnet (STEMCELL, #18103) according to the manufacturer’s instructions. In TAM co-inoculation experiment, 1 × 106 TAMs were co-injected with E0771 cells into the mammary fat pad of mice.
Peritoneal macrophage culture
Peritoneal macrophages were isolated and cultured as previously described58.
Tumor interstitial fluid collection
Tumor interstitial fluid was harvested according to a previously described centrifugal method with minor modification32,33. Briefly, E0771 tumors were dissected and rinsed in PBS. Tumor interstitial fluid was collected through centrifuging tumor tissues on 20 μm filters at 110 × g for 10 min at 4 °C.
Macrophage depletion
Mice were i.p. injected with 100 μl clodronate liposomes (FormuMax Scientific, #F70101C-A, Sunnyvale, CA, USA) one day before tumor inoculation, followed by injections of 100 μL clophosome liposome twice a week.
Glutamine/glutamate measurement
The cellular contents of glutamine and glutamate were quantified using Glutamine/Glutamate-Glo™ Assay Kit (Promega, #J8021, Madison, WI, USA) according to the manufacturer’s instructions.
GLS inhibition
Glutaminase inhibitor CB-839 (200 mg/kg, MedChemExpress, #HY-12248) was given i.p. every three days from day 3 of tumor inoculation. For in vitro experiments, CB-839 (0.5 μM) was added to CD4+ T cell culture medium.
Glutamine deprivation
Mice were fed a glutamine-deficient diet or a control diet ordered from BIOPIKE (Beijing, China) one week before tumor inoculation. For in vitro glutamine deprivation, the isolated splenic CD4+ T cells were cultured in glutamine-free RPMI 1640 medium (Thermo Fisher Scientific) under the Th1 condition mentioned above.
Immunoblotting
Immunoblotting was performed as previously described58. The following antibodies were used: GSK-3α/β (Cell Signaling Technology, cat#5676 T, 1:1000, Danvers, MA, USA), p-GSK-3α/β (Cell Signaling Technology, cat#9331S, 1:1000, MA, USA).
RNA-sequencing
Control and IL-16-treated Th1 cells were lysed in Trizol reagent, sent to Biomarker Technologies (Beijing, China), and subjected to RNA-sequencing.
TCGA and GEO data analysis
For TCGA data analysis: High-throughput RNA sequencing data and clinical information were obtained from the official website of TCGA database (https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga). Patient survival, pan-cancer gene expression, gene correlation analysis, enrichment analysis, and immune infiltration analysis were performed using Xiantao Academic Platform (www.xiantao.love), which is embedded with R software and R packages for data analyzing.
For GEO data analysis: GSE93157, GSE100797, GSE126044, GSE140901, GSE78220, and GSE190266 datasets were obtained from GEO database (http://www.ncbi.nlm.nih.gov/geo). Using the R package “survminer”, the expression levels of IL16 in these datasets were categorized into high/low groups using the optimal cutoff of the prognostic model. The Kaplan-Meier survival curves were plotted to assess the prognostic status of the patients between groups using the R package “survival”.
QPCR
Total RNA from tumor tissues or in vitro-cultured T cells or macrophages was extracted using Trizol method, then reversely transcribed to cDNA using PrimeScript RT Master Mix (Takara, #RR036B, Japan). QPCR was performed using SYBR Green Reagent (CwBio, #CW0955M, Beijing, China). The primer sequences are listed in Supplementary Table. 5.
Statistical analysis
The comparison between two groups was performed using an unpaired, two-tailed Student’s t-test. The comparison between multiple groups was performed using Analysis of Variance (ANOVA). Patient survival was analyzed using log-rank test. The comparison of gene expression between tumor and normal tissues in the integrated TCGA and GTEx databases was analyzed using Mann–Whitney U-test. Correlation analysis was performed using Spearman’s rank correlation test. The animal experiments and the in vitro, cell-based experiments were repeated at least twice. All data are presented as mean ± standard deviation (SD). P < 0.05 was considered statistically significant.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The RNA-seq data generated in this study have been deposited in the GEO database under accession code GSE247579; the scRNA-seq data generated in this study have been deposited in the GEO database under accession code GSE254991. The GEO data used in this study are available in the GEO database under accession code GSE93157, GSE100797, GSE126044, GSE140901, GSE78220, GSE190266. All data are included in the Supplementary Information or available from the authors, as are unique reagents used in this article. The raw numbers for charts and graphs are available in the Source Data file whenever possible. Source data are provided with this paper.
References
Butterfield, L. H. & Najjar, Y. G. Immunotherapy combination approaches: mechanisms, biomarkers and clinical observations. Nat. Rev. Immunol. 24, 399–416 (2023).
Xiao, P. et al. Myeloid-restricted ablation of Shp2 restrains melanoma growth by amplifying the reciprocal promotion of CXCL9 and IFN-gamma production in tumor microenvironment. Oncogene 37, 5088–5100 (2018).
Borst, J., Ahrends, T., Babala, N., Melief, C. J. M. & Kastenmuller, W. CD4(+) T cell help in cancer immunology and immunotherapy. Nat. Rev. Immunol. 18, 635–647 (2018).
Tie, Y., Tang, F., Wei, Y. Q. & Wei, X. W. Immunosuppressive cells in cancer: mechanisms and potential therapeutic targets. J. Hematol. Oncol. 15, 61 (2022).
Yuan, X., Ruan, W., Bobrow, B., Carmeliet, P. & Eltzschig, H. K. Targeting hypoxia-inducible factors: therapeutic opportunities and challenges. Nat. Rev. Drug Discov. 23, 175–200 (2023).
Chang, C. H. et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 153, 1239–1251 (2013).
Ho, P. C. et al. Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses. Cell 162, 1217–1228 (2015).
Chen, S. et al. Tumor-associated macrophages are shaped by intratumoral high potassium via Kir2.1. Cell Metab. 34, 1843–1859 e1811 (2022).
Xu, J., Zhao, Y., Tyler Mertens, R., Ding, Y. & Xiao, P. Sweet regulation - The emerging immunoregulatory roles of hexoses. J. Adv. Res. S2090-1232, 00157 (2024).
Varghese, S. et al. The glutaminase inhibitor CB-839 (Telaglenastat) enhances the antimelanoma activity of T-cell-mediated immunotherapies. Mol. Cancer Ther. 20, 500–511 (2021).
Lee, C. H. et al. Telaglenastat plus everolimus in advanced renal cell carcinoma: a randomized, double-blinded, placebo-controlled, phase II ENTRATA trial. Clin. Cancer Res. 28, 3248–3255 (2022).
Riess, J. W. et al. Phase 1 trial of MLN0128 (Sapanisertib) and CB-839 HCl (Telaglenastat) in patients with advanced NSCLC (NCI 10327): rationale and study design. Clin. Lung Cancer 22, 67–70 (2021).
Center, D. M. & Cruikshank, W. Modulation of lymphocyte migration by human lymphokines. I. Identification and characterization of chemoattractant activity for lymphocytes from mitogen-stimulated mononuclear cells. J. Immunol. 128, 2563–2568 (1982).
Cruikshank, W. W., Kornfeld, H. & Center, D. M. Interleukin-16. J. Leukoc. Biol. 67, 757–766 (2000).
Zhang, Y. et al. Processing and activation of pro-interleukin-16 by caspase-3. J. Biol. Chem. 273, 1144–1149 (1998).
Li, C. et al. Interleukin-16 aggravates ovalbumin-induced allergic inflammation by enhancing Th2 and Th17 cytokine production in a mouse model. Immunology 157, 257–267 (2019).
Skundric, D. S., Cai, J., Cruikshank, W. W. & Gveric, D. Production of IL-16 correlates with CD4+ Th1 inflammation and phosphorylation of axonal cytoskeleton in multiple sclerosis lesions. J. Neuroinflamm. 3, 13 (2006).
Lynch, E. A., Heijens, C. A., Horst, N. F., Center, D. M. & Cruikshank, W. W. Cutting edge: IL-16/CD4 preferentially induces Th1 cell migration: requirement of CCR5. J. Immunol. 171, 4965–4968 (2003).
McFadden, C. et al. Preferential migration of T regulatory cells induced by IL-16. J. Immunol. 179, 6439–6445 (2007).
Smith, S. et al. IL-16/miR-125a axis controls neutrophil recruitment in pristane-induced lung inflammation. JCI Insight 3, e120798 (2018).
Stoitzner, P. et al. Interleukin-16 supports the migration of Langerhans cells, partly in a CD4-independent way. J. Investig. Dermatol. 116, 641–649 (2001).
Mathy, N. L., Bannert, N., Norley, S. G. & Kurth, R. Cutting edge: CD4 is not required for the functional activity of IL-16. J. Immunol. 164, 4429–4432 (2000).
Szabo, S. J. et al. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell 100, 655–669 (2000).
Leone, R. D. & Powell, J. D. Metabolism of immune cells in cancer. Nat. Rev. Cancer 20, 516–531 (2020).
Johnson, M. O. et al. Distinct regulation of Th17 and Th1 cell differentiation by glutaminase-dependent metabolism. Cell 175, 1780–1795 e1719 (2018).
Nakaya, M. et al. Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity 40, 692–705 (2014).
Momcilovic, M. et al. The GSK3 signaling axis regulates adaptive glutamine metabolism in lung squamous cell carcinoma. Cancer Cell 33, 905–921 e905 (2018).
Papadopoli, D., Pollak, M. & Topisirovic, I. The role of GSK3 in metabolic pathway perturbations in cancer. Biochim. Biophys. Acta Mol. Cell Res. 1868, 119059 (2021).
Liu, J. Q. et al. Glutaminolysis is required in maintaining immune regulatory functions in B cells. Mucosal Immunol. 15, 268–278 (2022).
Beurel, E. et al. Regulation of Th1 cells and experimental autoimmune encephalomyelitis by glycogen synthase kinase-3. J. Immunol. 190, 5000–5011 (2013).
Hill, E. V. et al. Glycogen synthase kinase-3 controls IL-10 expression in CD4(+) effector T-cell subsets through epigenetic modification of the IL-10 promoter. Eur. J. Immunol. 45, 1103–1115 (2015).
Sullivan, M. R. et al. Quantification of microenvironmental metabolites in murine cancers reveals determinants of tumor nutrient availability. Elife 8, e44235 (2019).
Cheng, J. et al. Cancer-cell-derived fumarate suppresses the anti-tumor capacity of CD8(+) T cells in the tumor microenvironment. Cell Metab. 35, 961–978 e910 (2023).
House, I. G. et al. Macrophage-derived CXCL9 and CXCL10 are required for antitumor immune responses following immune checkpoint blockade. Clin. Cancer Res. 26, 487–504 (2020).
Marcovecchio, P. M., Thomas, G. & Salek-Ardakani, S. CXCL9-expressing tumor-associated macrophages: new players in the fight against cancer. J. Immunother Cancer 9, e002045 (2021).
Cho, J. W. et al. Genome-wide identification of differentially methylated promoters and enhancers associated with response to anti-PD-1 therapy in non-small cell lung cancer. Exp. Mol. Med. 52, 1550–1563 (2020).
Limagne, E. et al. MEK inhibition overcomes chemoimmunotherapy resistance by inducing CXCL10 in cancer cells. Cancer Cell 40, 136–152.e112 (2022).
Hsu, C. L. et al. Exploring markers of exhausted CD8 T cells to predict response to immune checkpoint inhibitor therapy for hepatocellular carcinoma. Liver Cancer 10, 346–359 (2021).
Prat, A. et al. Immune-related gene expression profiling After PD-1 blockade in non-small cell lung carcinoma, head and neck squamous cell carcinoma, and melanoma. Cancer Res. 77, 3540–3550 (2017).
Hugo, W. et al. Genomic and transcriptomic features of response to Anti-PD-1 therapy in metastatic melanoma. Cell 168, 542 (2017).
Lauss, M. et al. Mutational and putative neoantigen load predict clinical benefit of adoptive T cell therapy in melanoma. Nat. Commun. 8, 1738 (2017).
Arima, M. et al. Expression of interleukin-16 by human epithelial cells. Inhibition by dexamethasone. Am. J. Respir. Cell Mol. Biol. 21, 684–692 (1999).
Gantner, F. et al. Histamine h(4) and h(2) receptors control histamine-induced interleukin-16 release from human CD8(+) T cells. J. Pharm. Exp. Ther. 303, 300–307 (2002).
Jain, S. et al. Melanotan II causes hypothermia in mice by activation of mast cells and stimulation of histamine 1 receptors. Am. J. Physiol. Endocrinol. Metab. 315, E357–E366 (2018).
Xu, X. et al. Neutrophil histamine contributes to inflammation in mycoplasma pneumonia. J. Exp. Med. 203, 2907–2917 (2006).
Fenster, C. P., Chisnell, H. K., Fry, C. R. & Fenster, S. D. The role of CD4-dependent signaling in interleukin-16 induced c-Fos expression and facilitation of neurite outgrowth in cerebellar granule neurons. Neurosci. Lett. 485, 212–216 (2010).
Blake, D. J., Martiszus, J. D., Lone, T. H. & Fenster, S. D. Ablation of the CD9 receptor in human lung cancer cells using CRISPR/Cas alters migration to chemoattractants including IL-16. Cytokine 111, 567–570 (2018).
Chow, M. T. et al. Intratumoral activity of the CXCR3 chemokine system is required for the efficacy of anti-PD-1 therapy. Immunity 50, 1498–1512 e1495 (2019).
Gao, L. B. et al. The association of interleukin-16 polymorphisms with IL-16 serum levels and risk of colorectal and gastric cancer. Carcinogenesis 30, 295–299 (2009).
Qin, X. et al. The association of interleukin-16 gene polymorphisms with IL-16 serum levels and risk of nasopharyngeal carcinoma in a Chinese population. Tumour Biol. 35, 1917–1924 (2014).
Jia, R., Liu, S., Xu, J. & Liang, X. IL16 deficiency enhances Th1 and cytotoxic T lymphocyte response against influenza A virus infection. Biosci. Trends 13, 516–522 (2020).
Su, H. et al. Mycobacterium tuberculosis hijacks host macrophages-derived interleukin 16 to block phagolysosome maturation for enhancing intracellular growth. Emerg. Microbes Infect. 13, 2322663 (2024).
Maciel, T. T., Moura, I. C. & Hermine, O. The role of mast cells in cancers. F1000Prime Rep. 7, 09 (2015).
Komi, D. E. A. & Redegeld, F. A. Role of mast cells in shaping the tumor microenvironment. Clin. Rev. Allergy Immunol. 58, 313–325 (2020).
Varricchi, G. et al. Are mast cells MASTers in cancer? Front. Immunol. 8, 424 (2017).
Hua, S. et al. Microbial metabolites in chronic heart failure and its common comorbidities. EMBO Mol. Med. 15, e16928 (2023).
Xu, H., Luo, H., Zhang, J., Li, K. & Lee, M. H. Therapeutic potential of Clostridium butyricum anticancer effects in colorectal cancer. Gut Microbes 15, 2186114 (2023).
Xiao, P. et al. Phosphatase Shp2 exacerbates intestinal inflammation by disrupting macrophage responsiveness to interleukin-10. J. Exp. Med. 216, 337–349 (2019).
Acknowledgements
This work was supported by National Natural Science Foundation of China (81930041, U22A20307 to QW; 82171730, 82371762 to PX; 81970484 to QC; 82272872 to TL), Natural Science Foundation of Zhejiang Province (LY22H160003 to WZ, LY24H160012 to PX), Natural Science Foundation of Heilongjiang Province of China: Distinguished Youth Program (JQ2023H004 to TL).
Author information
Authors and Affiliations
Contributions
P.X., Z.W., K.W., and Q.W. conceived the study. Z.W., P.X., T.L., XX, ZS, YL, KG, YD, YZ, PW, SL, M.S., B.C., and W.C. performed the experiments. Z.S., Y.L., and J.X. conducted the bioinformatics analysis. T.L., P.W., S.L., Z.W., Q.C., Y.D., and K.G. collected and processed clinical specimens. P.X., Z.W., N.A., Y.L., Y.H., H.G., and Q.W. wrote and revised the manuscript. P.X., Q.C., Q.W., T.L., Z.W., S.S., K.W., N.A., M.S., and H.L. provided financial, technical, or material support.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wen, Z., Liu, T., Xu, X. et al. Interleukin-16 enhances anti-tumor immune responses by establishing a Th1 cell-macrophage crosstalk through reprogramming glutamine metabolism in mice. Nat Commun 16, 2362 (2025). https://doi.org/10.1038/s41467-025-57603-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-57603-1
This article is cited by
-
Targeting glutamine metabolism as a potential target for cancer treatment
Journal of Experimental & Clinical Cancer Research (2025)
