
Abstract
The pursuit of advanced battery chemistries with enhanced energy density necessitates the exploration of new materials, a process intricately tied to synthesis science. Despite the promise of O3-type sodium oxygen anionic redox cathodes as high-capacity materials, their development has been severely hindered by a lack of understanding regarding synthetic mechanisms. Here, we elucidate the pivotal role of atmospheric conditions, particularly oxygen content, in the synthesis of such materials by synchronizing multiple operando characterization techniques to monitor changes in both solid and gaseous components. Utilizing the O3-Na[Li1/3Mn2/3]O2 system as a model, we demonstrate that a low oxygen environment is essential and the reaction is highly complex as evidenced by multiple oxygen uptake and release processes, resulting in numerous intermediates. This behavior contrasts sharply with Na-Mn-O and Li-Mn-O ternary systems which show less significant oxygen dynamics, underscoring the unique reaction mechanism within the Na-Li-Mn-O system. We further adopt a dynamic controlled atmosphere approach to modulate oxygen concentration and demonstrate successful synthesis of Ti-substituted NaLi1/3Mn2/3-xTixO2 materials, all exhibiting capacities surpassing 190 mAh g-1. Our findings highlight the importance of the atmospheric conditions for the synthesis of oxide cathode materials and these fundamental insights unlock avenues for developing novel high-energy-density sodium-ion battery chemistries.
Similar content being viewed by others

Introduction
Advancements in synthesis serves as a cornerstone for all facets of materials science and engineering. For instance, the controlled fabrication of nano-sized or even single-atom materials provides numerous opportunities for designing novel catalysis materials with improved efficiency and selectivity1,2,3. This progress is also evident in materials for electrochemical energy storage applications4,5,6,7,8,9,10. The synthesis methods for layered cathode materials in lithium-ion batteries have continuously evolved as the nickel content increases to achieve higher energy density11,12. Specifically, the lithium precursor has changed from lithium carbonate to lithium hydroxide13,14, and the calcination process has transitioned from single-step to multi-step9,15,16, and importantly, the calcination atmosphere has adopted pure oxygen instead of air17,18.
The evolutions of synthesis conditions are intricately guided by the thermodynamics and kinetics that govern the desired synthesis reactions, all aiming at achieving the production of a pure target product. An illustrative example involves the increase in the oxygen content in the calcination atmosphere as the nickel content increases in LiNixMnyCo1-x-yO2 (NMC)-type cathodes, leading to the oxidation state of nickel in the final lithiated product approaching trivalent17. To facilitate this reaction, a pure oxygen environment is imperative, ensuring a strong oxidizing environment that minimizes the presence of divalent nickel, which may lead to the formation of detrimental Li/Ni anti-site defects17. Additionally, the synergy between reaction thermodynamics and kinetics is evident in the synthesis of P2-type sodium transition metal oxides for application in sodium-ion batteries where the formation of various metastable species is observed, preceding the eventual attainment of thermodynamically stable compounds10,19. Despite extensive efforts devoted to developing theoretical frameworks predicting emerging materials and their synthesis conditions, substantial challenges persist in synthesizing compounds predicted to be thermodynamically stable, whose formation pathways are intricately intertwined with diverse metastable compounds that are computationally difficult to be fully captured20,21,22,23.
In this work, we use O3-Na[Li1/3Mn2/3]O2, a promising O3-type oxygen anionic redox (OAR) cathode material for sodium-ion batteries, to demonstrate the complex reaction mechanism in a ternary system featuring two alkaline metals, both of which can form various oxide species with manganese, and importantly, the pivotal role of the synthesis atmosphere in determining the reaction pathways during the synthesis. We highlight that, despite their attractive properties such as high capacity and good cycling stability demonstrated by Na[Li1/3Mn2/3]O224, manganese-based O3-type OAR sodium cathodes are significantly scarcer21,24 as compared to the P2 polymorphs25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44. The key issue lies in the lack of fundamental understanding into the synthesis reactions that leads to the inaccessibility of O3-type phase pure compounds. O3-NaLi1/3Mn2/3O2 is predicted to be thermodynamically stable45; however, Wang et al. demonstrated that using conventional atmosphere, namely argon, vacuum, air or oxygen, the product contains no or a limited amount of O3-Na[Li1/3Mn2/3]O2 with various impurities, irrespective of the choice of lithium and sodium precursors and calcination temperature24. The authors obtained high purity product only by purging the tube furnace with argon flow for 45 min, whereas a longer purging or continuous argon flow leads to impurities24. These results clearly indicate the high complexity in the synthesis towards O3-Na[Li1/3Mn2/3]O2 and the oxygen content in the atmosphere, essentially the oxygen chemical potential, is a critical factor that yet remains unexplored, resulting in limited progress in O3-type anionic redox sodium cathodes.
Here, we demonstrate that the high purity O3-Na[Li1/3Mn2/3]O2 is accessible by fine-tuning the calcination atmosphere with 1–2% oxygen in argon (or nitrogen), and fully uncover how oxygen chemical potential influences the reaction mechanism through a series of operando characterization techniques. Our operando gas chromatography (GC) quantifies the dynamic oxygen release-uptake-release behavior during the synthesis, supported by intermediate compounds such as α-NaMn(III)O2 and Na3Mn(V)O4 captured by operando X-ray diffraction. The results demonstrate that the synthesis of O3-Na[Li1/3Mn2/3]O2 requires a low oxygen content environment to oxidize initial low Mn-valance intermediates and subsequently avoid over-oxidizing to high Mn-valance impurities. This complex reaction scheme differs sharply from those of the two ternary systems, i.e., Na-Mn-O and Li-Mn-O, highlighting the unique mechanism when both the two alkaline metal ions are present. Based on these fundamental insights, we propose a dynamic controlled atmosphere (DCA) approach starting with an oxidizing environment followed by an inert atmosphere. With this method, we successfully synthesize O3-Na[Li1/3Mn2/3]O2 and O3-type Ti-substituted NaLi1/3Mn2/3-xTixO2 (x = 1/18, 1/9 and 1/6) as high-capacity OAR cathode materials for sodium-ion batteries.
Results
Optimizing the calcination atmosphere
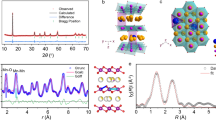
The honeycomb-ordered O3-Na[Li1/3Mn2/3]O2 is isostructural to Li[Li1/3Mn2/3]O2 (i.e., Li2MnO3), a classic oxygen-redox active cathode material, with the Li in the alkaline metal layer fully replaced by Na (Fig. 1a). First principles calculations (see methods for computational details) were first applied to identify proper composition for in-depth synthesis investigations by calculating the mixing enthalpy of structures with various degrees of Na-to-Li substitution. The results (Fig. 1b) show that the honeycomb-ordered Na[Li1/3Mn2/3]O2 (x = 0.75, x in NaxLi1-xMn0.5O1.5) has the lowest mixing enthalpy, indicating that it is thermodynamically favorable for O3-stacking structures, which is in good agreement with the previous report45. We thereby utilized Na[Li1/3Mn2/3]O2 for further synthesis investigations.

a Illustration of the honeycomb-ordered Li[Li1/3Mn2/3]O2 and Na[Li1/3Mn2/3]O2 structure. Structural model, C2/m. b Formation energies of mixing enthalpy for NaxLi1-xMn0.5O1.5. The vertical dashed line indicates the NaLi1/3Mn2/3O2 stoichiometry where x is 0.75. The ground states at each Na content are highlighted as red filled circles. c XRD patterns of the synthesis products under atmospheres containing various oxygen contents, namely 0% (pure Ar), 1%, 2%, 5%, 21%, and 100% (pure O2), respectively. Note the 0%, 21% and 100% O2 samples contain additional impurities which cannot be indexed by major structural databases. Source data are provided as a Source Data file.
Initial attempts of the synthesis were conducted under the most conventional atmospheres used for the solid-state synthesis of layered oxide cathodes, namely pure oxygen, air and inert conditions such as argon. The choice of precursors, i.e., Na2O2, Li2O and MnO2, is adopted from the previous work by Wang et al., except for the use of Mn(IV)O2 instead of Mn(III)2O3 due to the fact that the use of MnO2 leads to purer products (Supplementary Fig. 1; Fig. 1c). The oxidation state of manganese, therefore, is +IV in both the precursor and final product. The proposed reaction (reaction 1) is oxygen-releasing, suggesting an inert atmosphere such as argon would be suitable to obtain the desired product. The corresponding XRD results (Fig. 1c), however, suggests otherwise.
The product obtained in Ar contains only a minor amount of a O3 phase, but instead is dominated by α-Na1-δMnO2 and Li2MnO3. The existence of α-Na1-δMnO2 suggests that the atmosphere during the calcination is insufficient to sustain the Mn at +IV state. Unfortunately, simply increasing the oxygen content to 21% and 100% does not result in the formation of phase pure O3-Na[Li1/3Mn2/3]O2, instead, leads to various different impurities such as P3 structured phase (P3-NaxLiyMn1-yO2 (x < 1), denoted as P3-NLMO; Fig. 1c). These results collectively show that the conventional gas environments are not able to directly produce the target O3-Na[Li1/3Mn2/3]O2. In specific, an inert atmosphere leads to impurities with lower-than-target Mn valence while highly oxidizing conditions favor the formation of P3-type phases. This also indicates that the synthesis of O3-Na[Li1/3Mn2/3]O2 is sensitive to the calcination atmosphere, which requires dedicate tunning of the oxygen content.
We further selected three additional conditions, namely 1%, 2% and 5% oxygen in argon (or nitrogen), respectively, as the attempts above clearly indicate a moderate oxidizing environment is required. As shown in Fig.1c, the three products are all dominated by the target O3-Na[Li1/3Mn2/3]O2. The diffraction signals arisen from the honeycomb superstructure is also clearly visible (in the 2θ range of 20°–30°), in excellent agreement with the literature24. The products synthesized with 1% O2 and 2% O2 only contain trace amounts of Li2MnO3 impurities, while 5% O2 sample contains a small amount of a P3-NLMO phase. In-depth explorations of the reaction mechanisms are required to understand the critical role of oxygen content during the synthesis.
Operando studies of O3-Na[Li1/3Mn2/3]O2 synthesis during temperature ramping
We combine three operando techniques, including XRD, thermal gravimetric analysis (TGA) and gas chromatography (GC) to track the solid and the gaseous components evolution during the synthesis process. The operando GC is developed in-house, and is able to accurately quantify the oxygen content in the gas flow with a temporal resolution of ~2 min (Supplementary Fig. 2; Methods). Overall, the combined operando results (Fig. 2) of the synthesis conducted under 2% O2 (in argon or nitrogen) show that the reaction is highly complex featured by a series of structural changes, companied by distinct oxygen release and uptake. For clarity, we divide the temperature ramping section into three reaction phases (Fig. 2a), primarily based on whether it is oxygen releasing or uptake, and discuss in details below.

a The temperature profile, b operando XRD, c operando GC and d TGA results during the temperature ramping of the O3-Na[Li1/3Mn2/3]O2 synthesis under 2% O2. e A summary of the reaction processes and intermediate compounds during the temperature ramping. Source data are provided as a Source Data file.
Phase I: room temperature (RT) to ~290 °C; oxygen release. The diffraction signals from the precursors, i.e., Na2O2, Li2O and MnO2 are clearly observed at the beginning of the ramping process (Fig. 2, Supplementary Fig. 3). When the temperature reaches ~160 °C, a P3-NaxLiyMn1-yO2 phase is formed but quickly disappears as the temperature continues to increase, forming α-Na1-δMnO2 (O'3-NMO, Supplementary Fig. 3). Simultaneously, a large amount of oxygen is released during this phase, as evidenced by operando GC results (Fig. 2c) and the drastic weight loss in the TGA (Fig. 2d). Note that the amount of released oxygen and the weight loss is significantly higher that of the Na2O2 disproportionation reaction alone, as supported by both the GC (theoretical: 2500 µmol; experimental: 3341 µmol) and TGA (theoretical weight loss: 7.84%; experimental: 10.03%) results (Supplementary Fig. 4). The extra gas release and weight loss can be attributed to the oxygen loss from the reaction between NaOx/LiOx and MnO2 because lower Mn valence compounds such as α-Na1-δMnO2 is formed. This is particularly important as it indicates the nominal oxidation state of manganese is now lower than +IV by the end of phase I, and in order to return back +IV as in the O3-Na[Li1/3Mn2/3]O2, an oxidative environment is required for the rest of the calcination process.
Phase II: ~290 °C to ~550 °C; oxygen uptake. As the temperature increases, the amount of α-Na1-δMnO2 decreases as evidenced by the reduced intensities of the diffraction signals. Note that α-Na1-δMnO2 is still present at these temperatures as confirmed by ex situ high-resolution synchrotron X-ray diffraction results (Supplementary Fig. 5). Concurrently, x-Na3MnO4 forms, which denotes one or multiple polymorphs of Na3MnO4 that are structurally unknown as it cannot be indexed by major structural databases (see Supplementary Note 1 and Supplementary Figs. 6–8 for a detailed discussion of this assignment). Upon further increasing the temperature beyond 450 °C, x-Na3MnO4 rapidly transforms to γ-Na3MnO4 (Fig. 2b, Supplementary Fig. 5). Lithium manganese compounds (Spinel-like) are observed at the early stage of II (Supplementary Fig. 5). During phase II, obvious oxygen uptake is observed (Fig. 2c) and the weight of the solid gradually increases (Fig. 2d), in agreement with the presence of more oxidized Mn compounds as compared to those at the end of phase I. Note that one discrepancy occurs at ~390 °C, i.e., an oxygen uptake signal is detected while no obvious mass increase is observed, which we attribute to a H2O-release reaction (Supplementary Fig. 4). This issue does not affect the qualitative trend obtained by the operando GC and we performed additional experiments using anhydrous sodium hydroxide instead of highly hydroscopic Na2O2 and the results confirm that the nominal manganese valence initially decreases (phase I) but increases to above +IV after the oxygen-consuming phase II (Supplementary Fig. 9).
Phase III: ~550 °C to 700 °C; oxygen release. During this phase, γ-Na3MnO4, α-Na1-δMnO2 and Li2MnO3 are identified as the key intermediates that further react to form the target product O3-Na[Li1/3Mn2/3]O2 (Fig. 2b, Supplementary Note 2 and Supplementary Figs. 10–11), based on the operando PXRD and ex situ synchrotron PXRD. Therefore, assuming α-Na1-δMnO2 is stoichiometric (i.e., δ = 0), the reaction (reaction 2) toward O3-Na[Li1/3Mn2/3]O2 can be described as follows:
Based on this mechanism, no change in the solid mass nor oxygen release or uptake is expected during this phase. However, TGA and operando GC results reveal that this reaction is oxygen releasing as evidenced by a slight decrease in the sample mass (91.77% to 91.56%; Fig. 2d) and an increase in the cumulative amount of oxygen released (1946.8 µmol to 2151.5 µmol; Supplementary Fig. 4a). This discrepancy can be explained by the excessive formation of Na3MnO4, which is balanced by presence of Na-deficient α-Na1-δMnO2 to maintain the overall Na:Mn ratio, thereby raising the nominal oxidation state of manganese to higher than that +IV (Supplementary Fig. 9). Based on this understanding, we propose the following generalized reaction (reaction 3) which accounts for this variation:
Note that the values of y and δ are interrelated by the total amount of sodium, and they further determine the nominal Mn valence of the mixture of α-Na1-δMnO2, Li2MnO3 and Na3MnO4.
Collectively, the results from operando XRD, GC and TGA clearly show that the reactions occur during the synthesis of O3-Na[Li1/3Mn2/3]O2 are highly complex, featured by the formation of multiple Na-Mn-O and Li-Mn-O intermediate phases as well as distinct oxygen uptake and release, demonstrating that this process is sensitive to the atmospheric condition, i.e., the oxygen chemical potential (Fig. 2e). Notably, our results unambiguously explain why calcinations conducted using conventional conditions, no matter inert atmosphere such as argon or nitrogen, air, or pure oxygen, lead to various impurities in the final product: (1) for an inert environment, the manganese is reduced at low temperature (phase I) and cannot be oxidized back to +IV with low oxygen chemical potential at higher temperature, resulting in low-valence impurities such as α-Na1-δMnO2; (2) for strong oxidizing environments such as air and pure oxygen, high valence impurities such as Na3MnO4 exhibit preferential formation thermodynamically.
Operando studies of Na-Mn-O and Li-Mn-O ternary systems during ramping
To further understand the independent phase evolution upon heating in Na-Mn-O and Li-Mn-O ternary systems and the interplay between them, operando experiments were also conducted using the same precursor sets: Na2O2 + MnO2, or Li2O + MnO2, respectively, under the same atmosphere of 2% O2 in argon. The molar ratio of the reactants is maintained the same as that in the NLMO synthesis, i.e., Na2O2:MnO2 = 3:4 and Li2O:MnO2 = 1:4, respectively. For the Na-Mn-O system, the operando XRD results (Fig. 3a, b) show that multiple phase changes occur, leading to the formation of α-Na1-δMnO2 and then Na3MnO4 as the temperature increases to 700 °C, which is in close similarities to the observation in the NLMO synthesis (Supplementary Fig. 12), suggesting that the reaction occur during the NLMO synthesis is predominantly governed by Na-Mn-O. The unidentified species visible in phase II is also presented upon heating the ternary Na-Mn-O system, indicating that these are some unknown Na-Mn-O materials. For the Li-Mn-O system, spinel LixMnOy also starts to form above ~300 °C and persists till 700 °C (a minor amount of Mn2O3 is produced between ~500 °C and 650 °C; Fig. 3d, e), which is different from the Na-Li-Mn-O quaternary system where Li2MnO3 is formed at high temperatures. We propose that the formation of various Na-Mn-O species with different Na:Mn stoichiometry results in a dynamic Li:Mn ratio, thus continuously alters the reaction thermodynamics for the Li-Mn-O ternary system along the calcination process, leading to the formation of different Li-Mn-O compounds.

a The temperature profile, b operando XRD and c operando GC results of the Na-Mn-O system during the temperature ramping process. d The temperature profile, e operando XRD and f operando GC results of the Li-Mn-O system during the temperature ramping process. The calcination for both systems were carried out under a composition fixed atmosphere containing 2% O2. Source data are provided as a Source Data file.
Such intricate interplays also strongly impact the oxygen uptake/release behavior, which exhibits a stark contrast between the Na-Li-Mn-O quaternary system and the two ternary systems (Fig. 3c, f). For the Na-Mn-O system, similar oxygen releasing processes associated with peroxide disproportionation and formation of α-Na1-δMnO2 are observed at the beginning of the calcination. However, unlike the presence of phase II (~280 °C to ~550 °C) for the NLMO during which a significant amount of oxygen is consumed, minimum oxygen uptake is observed during the ramping processes for both of the two ternary systems. Based on the fact that strong α-Na1-δMnO2 diffraction signals persist in the Na-Mn-O system (Fig. 3b) while their intensity drops significantly in the Na-Li-Mn-O system (Fig. 2b) after the first oxygen-uptake spike at 390 °C in NLMO (Fig. 2c), we argue that this oxygen-uptake process is closely related to the oxidation of α-Na1-δMnO2 to a new phase with a higher Mn valence, which is a crucial step to form O3-Na[Li1/3Mn2/3]O2 in the Na-Li-Mn-O system. These results show that the reaction thermodynamics of the two ternary systems, Na-Mn-O and Li-Mn-O, strongly affect each other, leading to the dynamic change in the oxygen release and uptake behavior, which plays an essential role in the phase evolution to form O3-Na[Li1/3Mn2/3]O2.
Impact of oxygen content and dynamic controlled atmosphere
To unravel the impact of oxygen chemical potential on the reaction thermodynamics and kinetics, we track the environmental oxygen content evolution (using operando GC) upon calcination under three different atmosphere conditions, i.e., 1%, 2% and 5% oxygen in Ar/N2, respectively. All the three reactions exhibit a similar qualitative and quantitative evolution till ~400 °C, by which oxygen release associated with the disproportionation of peroxide and the formation of reduced manganese compounds (phase I) occur followed by a subsequent oxygen uptake (Fig. 4a, Supplementary Fig. 13). The variation in the synthesis atmosphere primarily leads to different oxygen uptake/release behavior from ~400 °C and onwards where a higher O2 environment exhibits more oxygen uptake during the remaining phase II and less oxygen release during phase III. Thus, a constant oxidizing environment throughout the calcination process can result in over-oxidation of Mn-containing species, preventing the formation of a phase-pure O3-Na[Li1/3Mn2/3]O2.

a Cumulative oxygen release using composition fixed atmosphere containing 1%, 2% and 5% O2, respectively. b TGA result of the synthesis using dynamic controlled atmosphere approach, which includes a 2% O2 flow during the ramping process and a pure nitrogen flow during the temperature hold. Bragg peak (Na3MnO4) evolution during the calcination using composition fixed atmosphere (2% O2) (c) and DCA approach (d). e XRD results of O3-NLMO synthesized via dynamically controlled atmosphere containing 2%, 5% and 25% O2 for the ramping process, respectively. Source data are provided as a Source Data file.
Based on the generalized reaction (3), we further investigate the thermodynamic influence of the oxygen partial pressure through theoretical calculations of the reaction energy (Supplementary Table 1). The results (Supplementary Note 3 and Supplementary Fig. 14) clearly indicate that decreasing the oxygen partial pressure lowers the reaction energy, thereby favoring the formation of O3-NLMO. In addition, a less Na-deficient α-Na1-δMnO2 requires a significantly lower oxygen partial pressure to achieve the same reaction energy when comparing Na0.83MnO2 to Na0.75MnO2. Guided by these theoretical insights, a less oxidizing or inert environment is recommended for phase III during the ramping and the subsequent high temperature holding at 700 °C in order to alter the reaction thermodynamics, favoring the formation of phase-pure O3-NLMO. We therefore adopt a DCA approach which contains oxygen for the ramping process but switches to inert atmosphere such as argon or nitrogen at high temperatures. Indeed, the TGA result clearly shows a sudden mass drop (~0.4%; Fig. 4b) when the atmosphere is switched to nitrogen (controlled by built-in mass flow controllers), which is not observed when a constant 2% O2 flow is supplied (Fig. 2a). This mass loss is attributed to oxygen release as a result of the reduction of high valence Mn impurities such as Na3MnO4 (Fig. 4c, d). For reaction with fixed environmental oxygen content, only slight decrease in intensity (Fig. 4c), suggesting that majority of Na3MnO4 impurity remains unreacted. In contrast, a rapid conversion of Na3MnO4 impurity at high temperature can be identified after switching the atmosphere from 2% O2 to pure nitrogen in the DCA synthesis as suggested by the attenuation of corresponding Bragg peaks (Fig. 4d). Note that distinct peak shifts to lower angles were observed in both Fig. 4c, d during the temperature ramping (Supplementary Fig. 15), which are attributed to the thermal expansion of Na3MnO4. Moreover, we also applied other oxygen content during the ramping, 5% and 21% to test the generality of our method. The 21% is selected to simulate the air condition. Note that using fixed (in composition) gas flow (Fig. 1c), the product contains impurities such as P3-NLMO. By dynamically switching to N2 at high temperature, the formation of O3-Na[Li1/3Mn2/3]O2 can be clearly promoted as a result of Mn reduction in an inert atmosphere as shown in Fig. 4e. Only a minimum amount of Li2MnO3 and Na3MnO4 remains in the DCA-5% and DCA-21% O2 samples, which may be eliminated by extending the duration of temperature holding.
O3-type titanium-substituted NaLi1/3Mn2/3-xTixO2 cathode materials
The DCA approach we proposed by systematic tracking of the phase, weight, and gas environment evolution upon solid-state synthesis can be highly expandable. As a demonstration, three O3-type titanium-substituted NaLi1/3Mn2/3-xTixO2 (x = 1/18, 1/9 and 1/6) oxygen redox cathode materials were synthesized using the same method. The ramping process during the calcination was carried out under 2% O2 environment whereas the constant-temperature and cooling steps are under pure argon, i.e., DCA-2% O2. Similar to the synthesis of NLMO, the DCA approach suppresses the formation of Li2MnO3 and Na3MnO4 impurities (Supplementary Fig. 16), as confirmed by the XRD results (Fig. 5a for NLMTO-1/6; NLMTO-1/18 and NLMTO-1/9 are shown in Supplementary Fig. 11). The lattice parameters for the NLMTOs (based on the C2/m structural model) are summarized in Fig. 5b (more details in Supplementary Fig. 17 and Supplementary Table 2; Supplementary Fig. 18 and Supplementary Table 3 for refinement results based on the \(R\bar{3}m\) structure). Notably, the a, b and c lattice parameters gradually increase as the Ti4+ content increases, which is ascribed to its larger ionic radius (0.605 Å) compared to that of the Mn4+ (0.53 Å), indicating successfully incorporation of Ti into the structure (stoichiometries confirmed by inductively coupled plasma-optical emission spectroscopy; Supplementary Table 4).

a XRD pattern and Rietveld refinement of the pristine NLMTO-1/6. b Lattice parameters of NaLi1/3Mn2/3O2 and NaLi1/3Mn2/3-xTixO2 (x = 1/18, 1/9 and 1/6) from Rietveld refinements. Structural model, \(C2/m\). c SEM-EDX elemental mapping of the pristine NLMTO-1/6. Scale bar, 1 µm. d HAADF-STEM image of the pristine NLMTO-1/6. Scale bar, 1 nm. Red and blue rectangles illustrate monoclinic unit cells viewed from the [100] and [110] zone axes, respectively. Yellow, green, purple circles represent sodium, lithium, and transition metals (manganese and titanium), respectively. e Voltage profiles of NLMO and NLMTOs during the first cycle at a rate of C/20 between 1.5 and 4.5 V (i.e., 9.5 mA/g; 1 C = 190 mA/g). Source data are provided as a Source Data file.
The as-synthesized NLMTO-1/6 has a plate-like morphology with a particle size of ~2 µm as shown in the scanning electron microscope (SEM) images (Supplementary Fig. 19), and the energy-dispersive spectroscopy (EDS) mappings confirm that Na, Mn, Ti, and O are uniformly distributed in the particle (Fig. 5c). The aberration-corrected high-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) image (Fig. 5d) reveals the typical layered structure with dumbbell-like features arisen from the honeycomb ordering46. The compound also presents various stacking faults, which can be attributed to rotational47 and/or transitional stacking faults (Supplementary Note 4, Supplementary Figs. 20, 21). The degree of such stacking faults is estimated to be approximately 18% using FAULTS (Supplementary Table 5, Supplementary Fig. 22), which propagate along the c axis (Fig. 5d, Supplementary Fig. 22). The distances between adjacent TM layers are measured to range from 5.39 Å to 5.56 Å (Supplementary Fig. 23), which is close to value determined from lattice parameters from the refinement (cM×sin(βM), 5.31 Å; Fig. 5b). The discrepancy is attributed to sample preparation, including possible lattice strain from grinding and de-sodiation from sonication in ethanol, as well as potential measurement errors and lattice distortion due to electron beam irradiation. The line-scan electron energy loss spectroscopy (EELS) measurement along the transition metal layer confirms the presence of Ti and Mn in the TM layer and the Mn L-edge X-ray absorption spectra confirm the Mn in NLMTO-1/6 is close to Mn(IV) state (Supplementary Fig. 24).
The electrochemical performance of NLMO and NaLi1/3Mn2/3-xTixO2 (x = 1/18, 1/9 and 1/6) synthesized using DCA-2% O2 approach was investigated in sodium half cells and the corresponding first-cycle voltage profiles of the four materials are shown in Fig. 5e. All four charging curves display a prolonged plateau at approximately 3.6 V, succeeded by a sloping region, while the discharging curves exhibit a slope shape. Interestingly, the titanium substitution demonstrates a noticeable impact on the charge and discharge capacities: a minor substitution of 1/18 initially enhances the capacity, whereas further increases to 1/9 and 1/6 result in lower capacities. The NLMTO-1/18 sample exhibits the highest charge and discharge capacities of 271.8 mAh/g and 204.3 mAh/g, respectively. Upon increasing the rate to 0.2 C (38 mA g-1), the discharge capacities for NLMO, NLMTO-1/18, NLMTO-1/9, and NLMTO-1/6 are 160.1 mAh g-1, 159.9 mAh g-1, 138.0 mAh g-1, and 102.4 mAh g-1, respectively, during the first 0.2 C (38 mA g-1)cycles (Supplementary Fig. 25), further demonstrating that excessive cooperation of Ti leads to lower reversible capacities. These results indicate that higher levels of Ti substitution lead to lower reversible capacities, which can be attributed to the electrochemically inert nature of Ti4+. However, the Ti-substituted samples exhibit a significantly improved capacity retention over long-term aging compared to the NLMO cathode. Among the Ti-substituted samples, NLMTO-1/18 demonstrated the best overall performance, delivering a similar capacity at 0.2 C (38 mA g-1) compared to the unsubstituted NLMO while significantly improving capacity retention. This improvement is tentatively attributed to an enhanced reversibility of sodiation due to suppressed parasitic reactions related to oxygen redox as well as Jahn-Teller distortion of manganese, as seen in Ti-substituted P2-type anionic redox cathode materials48. Nonetheless, further comprehensive investigation into the electrochemical performance and structural evolution during sodiation and de-sodiation is required, although it lies beyond the scope of this study.
Discussion
In this work, we have decisively unraveled the intricate reaction mechanism underlying O3-NLMO synthesis and extended this understanding to the successful creation of a series of novel Ti-substituted O3-NLMTOs. Through fine adjustment of oxygen content in the calcination atmosphere, we have demonstrated the feasibility of synthesizing O3-NLMO under low oxygen environments. Employing a combination of operando characterization techniques, including operando XRD, GC and TGA, we have shown a remarkably complex reaction scheme characterized by multiple intermediates and distinct oxygen dynamics of release and uptake. These findings, distinct from those of Na-Mn-O and Li-Mn-O ternary systems, underscore the pivotal role of oxygen in governing the thermodynamics and kinetics of quaternary compound synthesis. Importantly, we have pioneered a DCA approach to manipulate reaction dynamics, favoring the formation of O3-type oxide cathode materials, and have successfully synthesized three NLMTOs which all have high reversible capacities exceeding 190 mAh g-1.
Our work creates a versatile synthesis approach for high-capacity oxide cathode materials by advancing conventional synthesis conditions, thereby propelling advancements in sodium-ion battery research and development. Furthermore, the methodology we have developed for characterizing solid-state synthesis, which tracks the evolution of both solid and gaseous components concurrently, is indispensable for comprehensively unraveling reaction mechanisms.
Methods
Theoretical calculations
First-principles calculations were conducted on various Li-substituted Na2MnO3 structures using the PWmat code49,50 to ascertain the mixing enthalpy in each instance. The generalized gradient approximation (GGA) with Hubbard-type U correction and NCPP-SG15-PBE Pseudopotential were employed, significantly mitigating calculation errors. The Monkhorst–Pack method with 6 × 3 × 6 meshes was utilized for sampling the k-point in the reciprocal space. The conjugate gradient optimization method was employed with convergence criteria of 0.01 eV Å-1 for atomic forces and 0.01 eV N-1 per atom for lattice stresses. The plane wave cutoff energy for wavefunction (ECUT) and the cutoff energies for the soft charge density and the potential (ECUT2) were established at 70 Ryd (1 Ryd = 13.6057 eV) and 280 Ryd. This configuration ensures optimal lattice refinement and adequate relaxation of the calculated structures, given that relaxation calculations necessitate the inclusion of atomic forces. For the optimization of phases with different Na:Li ratios, Na2MnO3 (C2/m space group) was used as the initial base structure and all the Na were replaced by randomly distributed Li and Na while the Mn sites were fixed. The various ordered phases were obtained by excluding duplicates based on their symmetry. The enthalpies of the mixtures, characterized by various substitution structures, cannot be directly derived from simulation calculations. Instead, they must be obtained through the following equation:
where E denotes the total energy of the system. By comparing the magnitudes of this thermodynamic quantity, it is possible to gauge the thermodynamic stability of the structure and find out the most stable phase in different substitution cases through the comparison of their magnitudes.
Reaction energies of NLMO (Na3LiMn2O6) formation at different oxygen partial pressure were calculated based on the formation energies of each reactant and product. The method for calculating the formation energies is as follows: The system energies of Li2MnO3, Na3MnO4, NaxMnO2 (x = 5/6, 3/4) and Na3LiMn2O6 are calculated by using the RELAX mode in PWmat code49,50, with the same parameters used for calculating the mixture enthalpy. The GGA + U function and the NCPP-SG15-PBE pseudopotential were used. The conjugate gradient optimization method was employed with convergence criteria of 0.01 eV Å-1 for atomic forces and 0.01 eV N-1 per atom for lattice stresses, and the plane wave cutoff energies and soft charge density and potential cutoff energies are established at 70 Ryd and 280 Ryd. The system energy of NaxMnO2 is calculated by calculating all possible Na/vacancy orderings with the GGA + U function and selecting the lowest energy structure for consideration. Then, the formation energy of each structure can be obtained by subtracting the total energy of each atom from the calculated system energy. The contribution from the oxygen partial pressure is accounted for by computing the oxygen chemical potential, \({\mu }_{{{\rm{O}}}_{2}}\), based on the equation below10,51:
where \({p}_{0}\) is the reference pressure, \({p}_{{{\rm{O}}}_{2}}\) is the oxygen partial pressure and \({k}_{{\rm{B}}}\) is the Boltzmann constant. \({h}_{{{\rm{O}}}_{2}}\) and \({s}_{{{\rm{O}}}_{2}}\) represent the enthalpy and entropy oxygen gas per molecule, respectively.
Material synthesis
The materials were synthesized through conventional solid-state methods. For O3-NaLi1/3Mn2/3O2 (denoted as O3-NLMO), stoichiometric amounts of Na2O2 (Sigma-Aldrich, ≥95%), Li2O (Sinopharm Chemical Reagent Co., Ltd. ≥99.99%), MnO2 (Aladdin, ≥99%) precursors were first mixed in an agate mortar by hand thoroughly in an argon glovebox. The Na2O2 was received in the form of granular beads (~2 mm in diameter), which were crushed, ground and sieved (300 mesh) to reduce the particle size, facilitating better contact with other precursors. Note these processes involving handling Na2O2 often led to a decrease in the moisture level in the argon glovebox (<0.1 ppm H2O), indicating its high hygroscopicity. The MnO2 was dried in a vacuum oven at 120 °C for 12 h. The mixture was calcined at 700 °C for 8 h with a temperature ramping and cooling rate of 1 °C/min using a variety of atmosphere conditions (including pure argon/nitrogen and oxygen; 1%, 2%, 5% and 21% oxygen in argon/nitrogen). For NaLi1/3Mn2/3-xTixO2 (denoted as O3-NLMTO), stoichiometric precursors of Na2O2 (Sigma-Aldrich, ≥95%), Li2O (Sinopharm Chemical Reagent Co., Ltd. ≥99%), MnO2 (Aladdin, ≥99%), TiO2 (Aladdin, ≥99.5%, 60 nanometer, rutile) were thoroughly mixed in an agate mortar by hand in an argon glovebox, followed by calcination at 800 °C for 8 h and cooled to room temperature (1 °C/min for heating and cooling) in a 2% oxygen atmosphere (98% nitrogen). Following the heat treatment, the material was promptly placed inside a glovebox filled with argon (O2 < 0.2 ppm, H2O < 0.1 ppm) to avoid any exposure to moisture.
Materials characterizations
Powder X-ray diffraction (PXRD)
Ex-situ lab-based PXRD measurements were performed on a Bruker D8 Advance diffractometer equipped with a Cu Kα X-ray source in Bragg-Brentano geometry. The measurements were recorded from 10° to 80° in the 2θ scan range with a step size of 0.02° and an exposure time of 0.8 s for each step. The current and voltage were set to 40 mA and 40 kV, respectively. Synchrotron PXRD was conducted at the BL17UM beamline using a large area detector (EIGER2-X-16M). The X-ray wavelength was λ ≈ 0.6209 and 0.4838 Å. Samples were prepared by sealing the powder with Kapton tape inside an argon-filled glovebox to avoid air exposure. Rietveld refinements were performed using the TOPAS software (version 5)31,35,52. The FAULTS program was utilized to simulate the stacking faults in the original NLMTO structure (Supplementary Table S3). The intensities of ex situ diffraction patterns have been normalized so that the highest intensity is set to 1.
Operando XRD was carried out on the same diffractometer equipped with Anton Paar XRK900 reactor chamber. The powder precursor mixture was prepared using the aforementioned method, and subsequently was pressed into pellets with a diameter of 10 mm under pressure of ~380 MPa. For the operando characterization of the Na-Mn-O system, Na2O2 and MnO2 in a molar ratio of 3:4 was mixed by hand and pressed into pellets under pressure of ~380 MPa. The same precursor preparation method was employed for the operando synthesis of Li-Mn-O, using Li2O and MnO2 in a molar ratio of 1:4. Each sample loading was carried out inside a nitrogen-filled glove bag to prevent exposure to air. The reactor was purged using a 2% O2 in N2 flow at a rate of 50 sccm for 2 h before the calcination began. The sample was then heated from room temperature to 700 °C at a rate of 1 °C/min and maintained at 700 °C for 8 h under the 2% O2 in N2 flow (50 sccm), which scanned from 10° to 60° in the 2θ scan range with a step size of 0.02° and an exposure time of 0.6 s for each step. Each scan takes ~25 min and the interval between the end of a scan and the beginning of the next one is ~1 min.
Operando gas chromatography (GC)
The home-built operando GC setup is mainly composed of, in sequence, a set of mass flow controllers (MFCs, Shaanxi Yidu Intelligence Technology Co., Ltd.), a tube furnace (TF55035C-1, Thermo Fisher), a de-moisturizing column filled with silica gel and molecular sieves (4 Å) and a gas chromagraph equipped with a thermal conductivity detector (TCD) (9790Plus, Fuli Instruments). The column used was molecular sieve 5 Å, 60–80 mesh, 2 m × 1/8 inch. The GC is also equipped with a solenoid 6-way valve, thus allowing continuous sample injection and therefore operando measurement of the composition of the gas coming out from the tube furnace.
During the measurements, the column oven and TCD detector temperatures of the GC were set at 60 °C and 120 °C, respectively. Helium (Shanghai YouJiaLi Liquid Helium Co., Ltd. ≥99.999%) was used as carrier gas with a flow rate of 20 mL/min. Each scan takes ~2 min and the interval between the end of a scan and the beginning of the next one is ~0.2 min. The temporal resolution is sufficient for this work considering the temperature ramping section is typically ~11 h (1 °C/min rate). The instrument calibration was done by measuring gas flows containing 1%, 1.2%, 1.5%, 2%, and 5% oxygen in nitrogen, respectively, and an excellent linear correlation (R2 = 0.9999) was obtained between the integral of the GC signal and the oxygen content (Supplementary Fig. 1). This calibration curve was used for processing the data from subsequent operando measurements. The response time (the time delay between the change in oxygen concentration in the reactor and the sampling in the GC) was estimated to be ~15 min, which is acceptable as the calcination experiment is significantly longer (Supplementary Fig. 1).
In a typically experiment, 5 mmol Na2O2 (or 10 mmol NaOH), 1.67 mmol Li2O and 6.67 mmol MnO2, targeting 10 mmol O3-NLMO, was mixed and loaded into a quartz tube (20 mm in diameter, 500 mm in length) in an argon glovebox. The quartz tube was sealed and transferred out from the glovebox, and then mounted onto the tube furnace with gas inlet and outlet connections. Subsequently, the whole setup was purged with the reaction gas at a flow rate of 100 sccm for 2 h prior to the calcination process. The reaction was conducted under the same gas environment and the same flow rate. The sample was heated from room temperature to 700 °C at a rate of 1 °C/min and maintained at 700 °C for 8 h.
The operando GC measurement gives a series of oxygen content values (in volume/molar percentage) as a function of time, denoted as \({C}_{{{\rm{O}}}_{2}}\left(t\right)\) (%). The incoming gas has a total volume flow rate of 100 sccm, which is equivalent to a molar flow rate (\({q}_{n}^{*}\)) of 681.6 µmol/s at 298 K assuming the ideal gas condition. The following equation was used to calculate the net change in the molar flow rate of oxygen \(\Delta {q}_{n,{{\rm{O}}}_{2}}\left(t\right)\) at a given time t:
where \({C}_{{{\rm{O}}}_{2}}^{0}\) (%) denotes the molar percentage of the oxygen in the gas flow going into the reactor. A positive \(\Delta {q}_{n,{{\rm{O}}}_{2}}\left(t\right)\) (µmol/s) value represents an increase in the oxygen flow rate, i.e., an oxygen-release reaction occurs in the reactor. The cumulative net change of oxygen was obtained by integrating the \(\Delta {q}_{n,{{\rm{O}}}_{2}}\left(t\right){\rm{vs}}{t}\) curve. The cumulative net change of oxygen (excluding the amount associated with the disproportionation of Na2O2) was further used to calculate the nominal Mn valance based on the assumption that Mn is the only element changes oxidation state during the reaction.
Thermogravimetry analysis
A TG 209 F3 Tarsus® instrument from NETZSCH was used for all the TGA experiments. The sample was prepared in an argon glovebox and then loaded into the TG (typically ~30 mg) using a nitrogen-filled glove bag (relative humidity ≤5%) to minimize moisture contamination from air. The powder samples were heated from room temperature to 700 °C with a 1 °C/min ramp-up rate under specific oxygen concentration atmosphere at a total flow rate of 100 sccm. It is worth noting that this instrument is configured so that the protection gas for the balance chamber (nitrogen) will enter the reaction chamber before venting. Therefore, the protection gas flow should be taken into account when setting up measurements targeting desired oxygen content for the reaction. All the gas supplies are controlled by built-in high precision mass flow controllers.
Operando mass spectrometry
The configuration of the operando mass spectrometry setup is similar to that of the operando GC with the only exception that a quadrupole mass spectrometer (HPR-20 EGA, Hiden) is used instead of a GC. The sample preparation was carried under the same routine as the operando GC experiments as introduced above. Gaseous species of N2, O2, H2O, and CO2 are monitored using the mass spectrometer, with ion peak channels set at 28, 32, 18, and 44, respectively. The setup was kept running for 10 h prior to the beginning of the reaction to achieve a stable signal baseline. During the temperature ramp, the powder sample was heated from room temperature to 700 °C at a rate of 1 °C/min using a 50 sccm flow of 2% O2/98% N2 gas until reaching 700 °C. The QGA software was used for quantitative analysis of the obtained results. Note that the mass spectrometer has not been calibrated so the results can only be used as a qualitative indicator.
Inductively coupled plasma-optical emission spectroscopy (ICP-OES)
The chemical composition of the obtained O3-NLMO and O3-NLMTO materials was analyzed by inductively coupled plasma-optical emission spectroscopy (ICP-OES iCAP 7400). Sample were typically prepared by dissolving 20 mg of the cathode powder in 2 mL of aqua regia overnight. The solution was then filtered through nylon filter membrane and transferred to a 50 mL volumetric bottle.
Electron microscopy
The morphology and elemental distribution of the as-synthesized materials was examined by scanning electron microscopy (SEM, JSM-IT500HR/LA, JEOL) in conjunction with an energy dispersive X-ray spectroscopy (EDS, JEOL). For transmission electron microscopy (TEM) measurements, the samples were prepared by crushing the crystals with an agate mortar and pestle and dispersed in alcohol by ultrasound. Multiple drops of suspension were deposited onto a copper-micro mesh grid. After drying, HAADF–STEM images and electron energy loss spectroscopy (EELS) were acquired on a probe aberration-corrected JEOL JEM-ARM300F transmission electron microscope operated at 300 kV.
X-ray absorption spectroscopy (XAS)
Soft XAS experiments were conducted at the beamline 02B02 on the SiP·ME2 platform of the Shanghai Synchrotron Radiation Facility (SSRF)53. The Mn L-edge spectra were acquired using the total electron yield (TEY) method and the beam size was configured to 150 µm × 50 µm at the sample location. The measurements were performed at ambient temperature under ultra-high vacuum (below 1 × 10-9 Torr). All the spectra have been energy calibrated using the result of the reference sample MnO and normalized using the ATHENA software package54. Mn K-edge XAS experiments were conducted at the BL16U1 beamline at SSRF. Data were acquired in transmission mode at room temperature. XAS data were processed and analyzed using the Demeter software package54.
Vibrating Sample Magnetometer (VSM)
Magnetic susceptibility measurements were carried out by VersaLab using a multi-function vibrating sample magnetometer (produced by Quantum Design), in the temperature range of 50–350 K and a magnetic field of 100 Oe. The sample was Ar protected to reacting with air. The results were fitted using the following equation based on a modified form of Curie-Weiss law to account for the temperature independent contribution to susceptibility: to fit the results, calculating the effective magnetic moment of the material:
where T is the temperature, \(\chi\) is the magnetic susceptibility, \({\chi }_{0}\) is a magnetic factor independent of temperature, \({\theta }_{{\rm{CW}}}\) represents the Curie–Weiss temperature, C is the Curie constant, \({\mu }_{{\rm{eff}}}\) is the effective moment and \({\mu }_{{\rm{B}}}\) is the Bohr magneton.
Batteries and electrochemistry
Self-standing cathode electrodes containing 75% (weight percentage) active material, 15% Super P conductive carbon and 10% polytetrafluoroethylene binder (PTFE, beads, Sigma-Aldrich) were prepared using the dry electrode processing method in a glovebox filled with argon (O2 < 0.2 ppm, H2O < 0.1 ppm)55. Briefly, the as-synthesized cathode powder and Super P were hand-mixed in a mortar for a few minutes, followed by adding PTFE and repeated pressing and rolling using a stainless-steel rod (316 L; diameter: 10 mm) and hand pressure until a self-standing film was obtained. 10 mm disk electrodes with a typical mass loading of ~8.6 mg cm−2 and a thickness of around 40 µm were punched out for cell assembly. The self-standing electrodes were assembled into 2032-coin cells (316 L, Canrd) with one piece of glass fiber (GF/B, Whatman) as the separator, and sodium metal (99.8%, Sigma-Aldrich) as the anode. All sodium metal electrodes were rolled by hand and stored in the glove box for less than one month and brushed with a toothbrush to remove any possible oxide layer on the surface before use. The sodium metal anode was typically 240 µm in thickness (~25 mg cm-2). The electrolyte used was 1 M NaClO4 in ethylene carbonate/ethyl methyl carbonate 3/7 (EC/EMC) (Canrd) with 5% fluoroethylene carbonate (FEC, HSC Corporation; volume percentage). The electrolyte and its additives are commercial products that are stirred with teflon magnetons in a plastic bottle before assembling the battery. Constant-current cycling was performed on the LAND battery test system (LANHE CT3002A, Wuhan LAND Electronics Co., Ltd.) between 1.5 and 4.5 V with two initial formation cycles at a rate of 0.05 C (9.5 mA g-1), followed by aging cycles at a rate of 0.2 C (38 mA g-1). All cells were kept at 25 ± 1 °C in an ambient chamber (LBI-300HT, Shanghai Longyue Instrument Equipment Co., Ltd.) during the cycling. All battery testing experiments were conducted using at least 2–3 parallel cells. The specific capacity was obtained by normalizing the cell capacity by the mass of the active material.
Data availability
All data that support the findings in this study are available within this article and its Supplementary Information. Source data are provided with this paper. Any additional relevant data are available from the corresponding authors upon request. Source data are provided with this paper.
References
Yuan, Y. et al. Zirconium nitride catalysts surpass platinum for oxygen reduction. Nat. Mater. 19, 282–286 (2020).
Han, L. et al. A single-atom library for guided monometallic and concentration-complex multimetallic designs. Nat. Mater. 21, 681–688 (2022).
Mehmood, A. et al. High loading of single atomic iron sites in Fe–NC oxygen reduction catalysts for proton exchange membrane fuel cells. Nat. Catal. 5, 311–323 (2022).
Zhu, W. et al. Ultrafast non-equilibrium synthesis of cathode materials for Li-Ion batteries. Adv. Mater. 35, 2208974 (2023).
Chen, S. et al. Ultrafast sintering for ceramic-based all-solid-state lithium-metal batteries. Adv. Mater. 34, 2200430 (2022).
Zuo, D. X. et al. Ultrafast synthesis of nasicon solid electrolytes for sodium-metal batteries. Adv. Energy Mater. 13, 2301540 (2023).
Mesnier, A. & Manthiram, A. Heuristics for molten-salt synthesis of single-crystalline ultrahigh-nickel layered oxide cathodes. ACS Appl. Mater. Interfaces 15, 12895–12907 (2023).
Yoon, M. et al. Eutectic salt-assisted planetary centrifugal deagglomeration for single-crystalline cathode synthesis. Nat. Energy 8, 482–491 (2023).
Park, H. et al. In situ multiscale probing of the synthesis of a Ni-rich layered oxide cathode reveals reaction heterogeneity driven by competing kinetic pathways. Nat. Chem. 14, 614–622 (2022).
Bianchini, M. et al. The interplay between thermodynamics and kinetics in the solid-state synthesis of layered oxides. Nat. Mater. 19, 1088–1095 (2020).
Myung, S.-T. et al. Nickel-Rich layered cathode materials for automotive lithium-ion batteries: achievements and perspectives. ACS Energy Lett 2, 196–223 (2017).
Li, W., Erickson, E. M. & Manthiram, A. High-nickel layered oxide cathodes for lithium-based automotive batteries. Nat. Energy 5, 26–34 (2020).
Noh, H.-J., Youn, S., Yoon, C. S. & Sun, Y.-K. Comparison of the structural and electrochemical properties of layered Li[NixCoyMnz]O2 (x = 1/3, 0.5, 0.6, 0.7, 0.8 and 0.85) cathode material for lithium-ion batteries. J. Power Sour. 233, 121–130 (2013).
Xie, Q., Li, W. D. & Manthiram, A. A Mg-Doped high-nickel layered oxide cathode enabling safer, high-energy-density Li-Ion batteries. Chem. Mater. 31, 938–946 (2019).
Li, H. et al. Synthesis of single crystal LiNi0.88Co0.09Al0.03O2 with a two-step lithiation method. J. Electrochem. Soc. 166, A1956–A1963 (2019).
Ying, B. et al. Monitoring the formation of Nickel-Poor and Nickel-Rich oxide cathode materials for Lithium-Ion batteries with synchrotron radiation. Chem. Mater. 35, 1514–1526 (2023).
Mesnier, A. & Manthiram, A. Synthesis of LiNiO2 at moderate oxygen pressure and long-term cyclability in Lithium-Ion full cells. ACS Appl. Mater. Interfaces 12, 52826–52835 (2020).
Kim, U.-H. et al. Heuristic solution for achieving long-term cycle stability for Ni-rich layered cathodes at full depth of discharge. Nat. Energy 5, 860–869 (2020).
Lei, Y. C., Li, X., Liu, L. & Ceder, G. Synthesis and stoichiometry of different layered sodium cobalt oxides. Chem. Mater. 26, 5288–5296 (2014).
Szymanski, N. J. et al. An autonomous laboratory for the accelerated synthesis of novel materials. Nature 624, 86–91 (2023).
Zhao, C. et al. Rational design of layered oxide materials for sodium-ion batteries. Science 370, 708–711 (2020).
Stein, A., Keller, S. W. & Mallouk, T. E. Turning down the heat: design and mechanism in solid-state synthesis. Science 259, 1558–1564 (1993).
Aykol, M., Dwaraknath, S. S., Sun, W. & Persson, K. A. Thermodynamic limit for synthesis of metastable inorganic materials. Sci. Adv. 4, eaaq0148 (2018).
Wang, Q. et al. Unlocking anionic redox activity in O3-type sodium 3d layered oxides via Li substitution. Nat. Mater. 20, 353–361 (2021).
Wu, Q. et al. Anionic redox chemistry for sodium-ion batteries: mechanisms, advances, and challenges. Energy Fuels 36, 8081–8095 (2022).
Xu, H., Guo, S. & Zhou, H. Review on anionic redox in sodium-ion batteries. J. Mater. Chem. A 7, 23662–23678 (2019).
Clément, R. J. et al. Structurally stable Mg-doped P2-Na2/3Mn1−yMgyO2 sodium-ion battery cathodes with high rate performance: insights from electrochemical, NMR and diffraction studies. Energy Environ. Sci. 9, 3240–3251 (2016).
Maitra, U. et al. Oxygen redox chemistry without excess alkali-metal ions in Na2/3[Mg0.28Mn0.72]O2. Nat. Chem. 10, 288–295 (2018).
House, R. et al. Superstructure control of first-cycle voltage hysteresis in oxygen-redox cathodes. Nature 577, 502–508 (2020).
Boivin, E. et al. Bulk O2 formation and Mg displacement explain O-redox in Na0.67Mn0.72Mg0.28O2. Joule 5, 1267–1280 (2021).
Boivin, E., House, R. A., Marie, J. J. & Bruce, P. G. Controlling iron versus oxygen redox in the layered cathode Na0.67Fe0.5Mn0.5O2: mitigating voltage and capacity fade by Mg substitution. Adv. Energy Mater. 12, 2200702 (2022).
House, R. A. et al. Delocalized electron holes on oxygen in a battery cathode. Nat. Energy 8, 351–360 (2023).
Ma, C. et al. Exploring oxygen activity in the high energy P2-Type Na0.78Ni0.23Mn0.69O2 cathode material for Na-Ion batteries. J. Am. Chem. Soc. 139, 4835–4845 (2017).
Bassey, E. N. et al. Superstructure and correlated Na+ hopping in a layered Mg-Substituted sodium manganate battery cathode are driven by local electroneutrality. Chem. Mater. 35, 10564–10583 (2023).
Yabuuchi, N. et al. New O2/P2‐type Li‐Excess layered manganese oxides as promising multi‐functional electrode materials for rechargeable Li/Na batteries. Adv. Energy Mater. 4, 1301453 (2014).
Yabuuchi, N. et al. A new electrode material for rechargeable sodium batteries: P2-type Na2/3[Mg0.28Mn0.72]O2 with anomalously high reversible capacity. J. Mater. Chem. A 2, 16851–16855 (2014).
Zhao, W., Tsuchiya, Y. & Yabuuchi, N. Influence of synthesis conditions on electrochemical properties of P2‐Type Na2/3Fe2/3Mn1/3O2 for rechargeable Na batteries. Small Methods 3, 1800032 (2019).
Rong, X. et al. Anionic redox reaction-induced high-capacity and low-strain cathode with suppressed phase transition. Joule 3, 503–517 (2019).
Kehua, D. et al. High reversibility of lattice oxygen redox quantified by direct bulk probes of both anionic and cationic redox reactions. Joule 3, 518–541 (2019).
Vergnet, J., Saubanère, M., Doublet, M.-L. & Tarascon, J.-M. The structural stability of P2-Layered Na-based electrodes during anionic redox. Joule 4, 420–434 (2020).
Gao, A. et al. Topologically protected oxygen redox in a layered manganese oxide cathode for sustainable batteries. Nat. Sustain. 5, 214–224 (2022).
Zhao, C. et al. Coexistence of (O2)n− and trapped molecular O2 as the oxidized species in P2-Type Sodium 3d layered oxide and stable interface enabled by highly fluorinated electrolyte. J. Am. Chem. Soc. 143, 18652–18664 (2021).
Liu, H. et al. Inconsistency between superstructure stability and long-term cyclability of oxygen redox in Na layered oxides. Energy Environ. Sci. 17, 668–679 (2024).
Wang, X. T. et al. Achieving a high-performance sodium-ion pouch cell by regulating intergrowth structures in a layered oxide cathode with anionic redox. Nat. Energy 9, 184–196 (2024).
Kim, D., Cho, M. & Cho, K. Rational design of Na(Li1/3Mn2/3)O2 operated by anionic redox reactions for advanced sodium-ion batteries. Adv. Mater. 29, 1701788 (2017).
Jarvis, K. A., Deng, Z., Allard, L. F., Manthiram, A. & Ferreira, P. J. Atomic structure of a Lithium-Rich layered oxide material for lithium-ion batteries: evidence of a solid solution. Chem. Mater. 23, 3614–3621 (2011).
Eum, D. et al. Electrochemomechanical failure in layered oxide cathodes caused by rotational stacking faults. Nat. Mater. 23, 1093–1099 (2024).
Cao, X. et al. Restraining oxygen loss and suppressing structural distortion in a newly Ti-Substituted layered oxide P2-Na0.66Li0.22Ti0.15Mn0.63O2. ACS Energy Lett. 4, 2409–2417 (2019).
Jia, W. et al. The analysis of a plane wave pseudopotential density functional theory code on a GPU machine. Comput. Phys. Commun. 184, 9–18 (2013).
Jia, W. et al. Fast plane wave density functional theory molecular dynamics calculations on multi-GPU machines. J. Comput. Phys. 251, 102–115 (2013).
Ong, S. P., Wang, L., Kang, B. & Ceder, G. Li−Fe−P−O2 phase diagram from first principles calculations. Chem. Mater. 20, 1798–1807 (2008).
House, R. A. et al. What triggers oxygen loss in oxygen redox cathode materials? Chem. Mater. 31, 3293–3300 (2019).
Ren, G. et al. Photon-in/photon-out endstation for studies of energy materials at beamline 02B02 of Shanghai Synchrotron Radiation Facility. Chin. Phys. B 29, 016101 (2020).
Ravel, B. & Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron. Radiat. 12, 537–541 (2005).
Li, L., Tan, G., Tao, J., Lun, Z. & Xu, C. Benchmarking the performance of moisture-sensitive battery materials: the importance of the electrode preparation method. ACS Appl. Energy Mater. 6, 6883–6889 (2023).
Acknowledgements
This work is supported by the National Natural Science Foundation of China (Grant No. 52302286, 11227902) and the ShanghaiTech University Start-up Fund. Z.L. acknowledges funding support from the National Natural Science Foundation of China; the Institute of Weiqiao-UCAS Science and Technology; the Fundamental Research Funds for the Central Universities. J.W. acknowledges support from the National Natural Science Foundation of China (Grant No. 52302264). The authors are grateful to X. Guan and C. Xie for discussions on the operando GC experiments and L. Du for assisting the operando MS experiments. Theoretical calculations were supported by HPC platform of ShanghaiTech University. The authors thank the support from Analytical Instrumentation Center (contract no. SPST-AIC10112914), SPST, ShanghaiTechUniversity. The authors thank beamline 02B02 of the Shanghai Synchrotron Radiation Facility for providing the beamtime. The synchrotron experiment was carried out with the support of Shanghai Synchrotron Radiation Facility, beamlines BL17UM (proposal 2023-SSRF-PT-505347, 2024-SSRF-JJ-507636), BL16U1 (proposal 2024-SSRF-JJ-509632) and BL02B02 (proposal 2023-SSRF-PT-505346).
Author information
Authors and Affiliations
Contributions
C.X. conceived the idea. C.X., Z.L., and J.W. supervised the project. Y.Q., J.T., and Q.L. performed operando and ex situ PXRD and analysed data with assistance from F.L. W.W. and Y.W. assisted performing synchrotron X-ray diffraction. D.W., N.Y., and Y.Y. assisted the operando PXRD measurements. Y.Q. performed operando GC, TGA, SEM, XAS, electrochemistry experiments and analysed results with assistance from G.T. and N.Z. Q.L. performed VSM measurement, FAULTS analysis and theoretical calculations under guidance of J.W. J.T. performed operando MS and analysed data. P.Y. and K.C. conducted (S)TEM experiments and analysed data. C.X., Y.Q., and Q.L. wrote the manuscript with input from all co-authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Yu Qiao, and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Qiu, Y., Liu, Q., Tao, J. et al. Enabling the synthesis of O3-type sodium anion-redox cathodes via atmosphere modulation. Nat Commun 16, 2343 (2025). https://doi.org/10.1038/s41467-025-57665-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-57665-1
This article is cited by
-
Effect of Sodiation Temperature on the Electrochemical and Structural Properties of NaNi0.85Fe0.10Mn0.05O2 Cathodes for Sodium-Ion Batteries
Electronic Materials Letters (2026)
