
Abstract
The ether-based electrolytes are acknowledged for their compatibility with a diverse array of sodium-ion battery anodes, as well as their capability to enable efficient and reversible electrochemical reactions. However, they encounter a challenge of oxidation at high voltages. We find that a standard diglyme-based electrolyte starts to oxidize and break down at voltages exceeding 3.9 V (vs. Na+/Na). This deterioration is attributed to the nucleophilic nature of the diglyme solvent and the presence of oxygen atoms that possess two unpaired electrons. To address this issue, we incorporate foreign anions into the electrolyte system to passivate the reactive sites of terminal H on diglyme solvents, inhibiting further dehydrogenation and oxidation during battery operation. The constructed cathode electrolyte interphase, enriched with NaF and NaNxOy, substantially boosts the oxidation resistance of electrolyte to over 4.8 V (vs. Na+/Na), expanding the stability window and rendering it feasible for various high-voltage cathode materials. Our approach also ensures compatibility with either hard carbon or commercial graphite anodes, guaranteeing operation in pouch cells. This study elucidates the relationship between interfacial chemistry and oxidation tolerance at high voltages, offering an approach to the development of practical ether-based electrolytes for high-energy-density battery technologies.
Similar content being viewed by others

Introduction
Sodium-ion batteries (SIBs) are emerging as a promising sustainable energy solution, leveraging on the abundance and cost-effectiveness of sodium ions1. Unlike lithium-ion batteries (LIBs), SIBs cannot use commercial graphite negative materials due to thermodynamic instability issues of its sodium intercalated compound2, prompting the exploration of alternative negative electrode materials like hard carbon, transition metal sulfides, phosphides, and sodium metal3,4. A significant hurdle in SIBs is the compatibility of these negative electrode materials with electrolytes to ensure reversibility and cycle stability.
Unlike the poor Na+ ion storage in carbonate-based electrolyte system that is prevalent in SIBs5, commercial graphite demonstrates reversible charge storage in ether-based electrolytes through a solvent co-intercalation reaction, as illustrated in Supplementary Fig. 16. Further, ether electrolytes show good compatibility with various SIB negative electrodes, mainly owing to its capability of forming a thin and robust solid-electrolyte interphase (SEI) layer that promotes sodium ion transport and enhances electrochemical properties regarding specific capacity, rate capability, life span, and initial Coulombic efficiency (CE), as detailed in Supplementary Table 17,8. Nevertheless, the use of conventional ether-based electrolytes still struggles with high-voltage requirement of SIBs due to their ease of degradation at oxidizing potentials of positive electrodes9. A common approach to extend the electrochemical stability window is through high-concentration electrolytes, which reduce oxidative reactions by minimizing excess solvents at the electrode surface10. Yet, this strategy faces practical challenges due to increased viscosity and costs. Therefore, there is a pressing demand for the development of an economical and sustainable ether electrolyte with a normal salt concentration that can operate at high voltages11,12.
Our research is focused on the degradation mechanisms of a typical ether-based electrolyte, 1.0 M solution of sodium trifluoromethanesulfonate (NaOTf) in diethylene glycol dimethyl ether (diglyme) (denoted as G2), where we have identified the oxidation threshold at 3.9 V (vs. Na+/Na) as a pivotal cause of irreversible capacity loss. This issue stems from the C-O bonds of diglyme molecules, which are prone to nucleophilic substitution reactions. To counteract this, we added a trace amount of sodium nitrate (NaNO3, NNO) into the electrolyte, which can not only passivate the reactive sites of diglyme but also facilitate the formation of a durable cathode electrolyte interface (CEI) that is essential for long cycle life of batteries. The working mechanism of inhibiting anodic oxidation of ether electrolytes in our study diverges from that reported in lithium metal batteries13, which necessitates a significant concentration of lithium nitrate. In our sodium-based battery system, however, a mere 0.04 M concentration of NNO is sufficient to establish a CEI layer enriched with NaF and NaNxOy, which suppresses electrolyte oxidation. Validated by comprehensive theoretical and experimental research, our strategy has proven effective across various positive electrode materials and ether solvents. This highlights its potential in the advancement of high-voltage battery electrolytes.
Results
In a proof-of-concept study, we selected extensively studied G2 electrolytes to serve as the typical mode for our analysis. To evaluate high-voltage stability, we employed a P3-phase Na2/3Ni1/2Mn1/2O2 (NNM) electrode material with a particulate morphology, as shown in Supplementary Fig. 2. The Na||NNM half cell showed reversible plateau behavior with traditional carbonate electrolyte, but the cell with G2 electrolyte had a large irreversible charge capacity and a low initial CE of only 50% when charged to 4.0 V vs. Na+/Na (Fig. 1a). In contrast, a cell with a conventional carbonate electrolyte exhibited normal reversible charging and nearly 100% CE. The low CE persisted throughout the cycling process and eventually led to the premature failure of the batteries (Fig. 1b). Even at a reduced cut-off of 3.5 V, abnormal charging was seen by the 30th cycle, as depicted in Supplementary Fig. 3. We suspect that the high irreversible charge capacity is the result of severe electrolyte decomposition at the electrode-electrolyte interface, which impedes the intercalation of Na+ ions. Scanning electron microscopy (SEM) analysis of NNM particles both prior to cycling and after five cycles, as depicted in Fig. 1c, d, revealed the formation of a thick layer of byproducts on the NNM electrode surface. Notably, the presence of substantial C-containing species in Fig. 1e suggests uncontrollable parasitic reactions at the electrode-electrolyte interface, leading to uneven deterioration of the CEI layer.

a GCD profiles of Na||NNM cells using conventional carbonate and standard G2 electrolytes at a current density of 100 mA g−1. b Cycling performance of NNM positive electrodes in G2 electrolyte, within a voltage range of 2.0–4.0 V. The inset illustrates the overcharging curves during the 21st cycle. SEM images of (c) pristine and d five-cycled electrode materials. e Scanning transmission electron microscopy (STEM) images and elemental mapping of both pristine and cycled NNM particles, complemented by the elemental ratios.
Noticing the susceptibility of the G2 electrolyte with standard salt concentration to oxidation, we try to gain a more profound understanding of this phenomenon at the molecular level. The prevailing theory posits that if the Fermi level of electrode materials exceeds the highest occupied molecular orbital (HOMO) of ether during charging, the ether oxidizes preferentially14, leading to battery failure at high voltages (Fig. 2a). Density functional theory (DFT) calculations given in Fig. 2b show G2 has a higher HOMO energy than OTf−, making it prone to oxidation. However, NO3−, with much higher HOMO energy and donor number, can act as a sacrificial agent to stabilize the electrolyte15.

a Schematic representation of the cathode-driven electrolyte oxidation process in a charged state. The term LUMO refers to the lowest unoccupied molecular orbital. b Graphical depiction of the HOMO energy level and DN numbers for the G2 solvent molecule, OTf-, and NO3-. c Raman, (d) 23Na NMR, and (e) 1H NMR spectra of the G2 and R-G2 electrolytes. The insets in (c–e) display the solvation structure of CIP-OTf, CIP-NO3, and the molecular structure of the free G2 solvent, respectively. f Comparative electrostatic potential mapping and structural formulas of the predominant solvation structures. Structures dominated by NNO are highlighted within an orange dotted box.
Dominated by ion-dipole interactions between Na+, DEGDME solvent and OTf− anions, the coordinated states present as free ion (FI-OTf), contact ion pair (CIP-OTf) and aggregated ion pair (AIP-OTf), in which OTf− served as fully dissociated anion, monodentate ligand and bridged ions, respectively (Supplementary Fig. 4). The G2 electrolyte contains two characteristic peaks at 1034 and 1039 cm−1 in the Raman spectra (Supplementary Fig. 5)16,17. The former corresponds to the FI-OTf species and the latter implies the formation of the CIP-OTf. The downshift of Raman peaks signifies a transition in solvation structures to more tightly packed ion pairs18. Upon the addition of a very small amount of NNO (c.a. 0.04 M) to G2 (R-G2, reformulated G2), a noticeable decrease in the intensity of the CIP-OTf peak was observed, coinciding with the disappearance of the peak corresponding to coordinated OTf− (the bending mode of -CF3 at 760 cm−1, Fig. 2c). This indicates the dissociation of OTf− anions from the coordination structures into free ions, as illustrated by a downshift in the major solvation peak. Further analysis using nuclear magnetic resonance (NMR) spectroscopy revealed a downfield shift and broadening of the 23Na peak (Fig. 2d), indicating a de-shielding effect caused by the integrated solvation structures17. A molecular dynamics (MD) study reveals that NO3− ions are integrated into the primary solvation shell, featuring a Na+-O(NO3−) coordination distance of around 2.1 Å, as shown in Supplementary Fig. 6. Simultaneously, there is an observed reduction in the total coordination number of OTf−, indicating the release of some OTf− ions from the solvation shell. These findings are consistent with the outcomes of Raman spectroscopy and the NMR data. Overall, the electron-donating ability of NO3− weakens Na+-solvent interactions, enhancing ionic conductivity (Supplementary Fig. 7)19.
Given that the resonance of terminal H in G2 goes upfield (Fig. 2e), this feature could be the result of the shielding effect of NO3− on the nucleophilic reaction area in G2, which refers to the active hydrogen atoms20,21. Further disclosed by the DFT calculation, the electron-donating NO3− pushes the solvation structures beyond CIP, even forming the aggregated form, AIP (Fig. 2f). Furthermore, the dehydrogenation procedure serves as an associated process along with electrolyte oxidation. The active parts (H on G2, red parts) are obviously passivated in the formed CIP (CIP-NO3) and AIP (AIP-NO3) because of the participation of NO3− ions, which indicates the inhibition of the dehydrogenation and oxidation of G2 solvents during battery operation22,23.
We integrated the ether electrolyte into electrochemical cells to test its oxidation resistance. Linear sweep voltammetry (LSV) showed that the on-set anodic current of both Na||NNM and Na||stainless steel half-cells with G2 electrolyte starts from around 3.9 V vs. Na+/Na (Fig. 3a). The current of Na||NNM cells increased more rapidly, suggesting that the positive electrode surface may catalyze oxidation reactions of G2 molecules, thereby worsening electrolyte decomposition in full cells24. The enlarged detail from the LSV curves shown in Supplementary Fig. 9 displays the current peak associated with Na+ intercalation in Na||NNM cells. This finding is further supported by the galvanostatic charge-discharge (GCD) profiles of initial three cycles (Fig. 3b) and the cyclic voltammetry (CV, Supplementary Fig. 8), which showed large irreversible capacities and an average CE of 51.72% for the Na||NNM cell with G2 electrolyte. In contrast, the Na||stainless steel cell with R-G2 electrolyte demonstrated the high-voltage tolerance with an extended stable window up to 4.8 V. Meanwhile, the Na||NNM cell with R-G2 electrolyte with normal charging capacity achieved an average CE of 99.34%, indicating suppressed parasitic reactions and improved reversibility. Over the cycling process, it maintained a capacity retention of 80.5% after 100 cycles, whereas the cell with G2-electrolyte failed after 21 cycles because of the low CE triggered by severe parasitic reactions (Supplementary Fig. 10a)25.

a LSV curves comparing Na||NNM cells and Na||stainless steel cells with G2 and R-G2 electrolytes. b Charge and discharge profiles of the initial three cycles for Na||NNM cells. c 1H NMR spectra and (d) Raman spectra of cycled G2 and R-G2 electrolytes extracted from Na||NNM cells. The insets in (c) provide optical images of the separators disassembled from cells after five cycles.
We further conducted XRD analysis on the positive electrode materials after cycling. As displayed in Supplementary Fig. 11, the NNM electrode cycled with R-G2 electrolyte exhibited minimal crystallinity loss and structural degradation, suggesting that the NO3−-derived CEI layer protected both the electrolyte and the positive electrode from side reactions. The R-G2 electrolyte was also effective with other positive electrode materials like tunnel Na0.44MnO2 (NMO) and Na3V2(PO4)3 (NVP), achieving similarly improved CE (Supplementary Fig. 10b, c). For instance, Na||NNM cells with G2-electrolyte showed abnormal charge process extensions in GCD curves (Supplementary Fig. 12), indicating significant electrolyte decomposition. The deterioration was evident through the dQ/dV contour map during cycling (Supplementary Fig. 13), where redox platforms above 3.5 V faded with cycling. In contrast, half-cells with R-G2 electrolyte maintained these platforms upon long cycling period, confirming enhanced interfacial protection under high voltages.
To further assess the electrolyte stability, we analyzed cycled electrolytes from Na||NNM cells. 1H NMR showed short-chain carbon-oxide compounds corresponding to G2 solvent decomposition26, with significantly reduced intensity for the peak around 1.8 ppm with NNO additive (Fig. 3c), consistent with inhibited solvent oxidation. The separator subjected to cycling in the G2 electrolyte exhibited a darkened appearance, as shown in the inset of Fig. 3c, which is perhaps the detached CEI components or other byproducts resulting from electrolyte decomposition. In marked contrast, the R-G2 separator retained its pristine cleanliness. Raman spectra confirmed the consistent composition of R-G2 before and after cycling, with characteristic peaks attributed to C-O-C vibration of G2 molecule (850 cm−1) and -CF3 bending (756 cm−1) of OTf− anion remaining unchanged (Fig. 3d). In contrast, these peaks were nearly absent in the G2 electrolyte after only 20 cycles.
Inspired by these findings, we examined the electronic structures of specific solvation structures to explain the high-voltage endurance of R-G2 electrolytes. DFT simulations indicated that the NO3−-involved solvates, CIP-NO3 and AIP-NO3, have higher HOMO levels than the CIP-OTf of G2 electrolyte, suggesting they are more prone to decomposition (Fig. 4a). Specifically, the oxidation sites are mostly located on the NO3− of solvation clusters while the C-O bonds of the G2 solvent are thus protected. We hypothesize that NO3− decomposition promotes a protective CEI layer, thereby protecting the positive electrode material from interfacial side reactions (Supplementary Fig. 11)27. This was confirmed by the presence of N-containing species in the N 1s XPS spectra of the R-G2-derived CEI compared to the G2-derived CEI (Fig. 4b). The NaNxOy species, a good Na+ conductor28,29, likely arises from the preferential decomposition of NO3− ions. A control experiment confirmed the crucial role of CEI in protecting against oxidation of G2 solvent, as an NNM electrode pre-cycled in R-G2 with a pre-formed CEI could withstand a high potential of 4.75 V vs. Na+/Na (Fig. 4c). High-resolution transmission electron microscopy (HRTEM) revealed that the NNO-derived CEI is uniform and conformal at about 6 nm thick, while the G2-derived CEI is uneven and much thicker (~25 nm) (Fig. 4d, e)30. This thin CEI layer, further corroborated by the Mn XPS signal observed on the electrode surface (Supplementary Fig. 14), not only ensures stability but also facilitates interfacial charge transfer, as evidenced by electrochemical impedance spectroscopy (EIS) (Supplementary Fig. 15).

a LUMO and HOMO of the solvation clusters, as seen in Fig. 2f, with orbital wavefunctions in red and green. b N 1s XPS spectra of the NNM electrode cycled with R-G2-electrolyte, with Ar+ ion sputtering. c LSV measurements evaluating the stability of the CEI layer within NNM, with preconditioning in R-G2 electrolyte indicated by the blue trace and without such preconditioning by the red trace. The inset provides an enlarged view of the plot in the potential range of 2.5–4.25 V. d, e HRTEM images depicting CEI regions and (f) in-depth XPS spectra of NNM electrodes after 5 cycles in both G2 and R-G2 electrolytes.
To elucidate the CEI formation chemistry, we analyzed its components using XPS with Ar+ ion sputtering (Fig. 4f). The O 1s spectra of the G2-induced CEI showed high levels of Na2CO3/Na2CO2R, a result of uncontrolled G2 oxidation30,31. With NNO electrolyte addition, the inner layer of CEI displayed significantly reduced Na2CO3/Na2CO2R content and introduced oxidative products such as NaNxOy and RSO3Na/RSO2Na, which is also confirmed by the S 2p spectra displayed in Supplementary Fig. 1632. Free anions and solvents are absorbed into the Inner Helmholtz Plane (IHP) before cycling, decomposing to form the initial CEI. Here, NNO addition removes free solvents and releases OTf− from solvation structures, enabling OTf− decomposition in the IHP layer. This also leads to a NaF-rich composition, evident from the peak at 684.7 eV in F 1s XPS spectra33. The shift from solvent-derived to anion-derived CEI, triggered by the rearrangement of dominant solvation structures, provides an electron-insulating yet ion-conducting CEI layer between the electrolyte and electrode.
Taken together, NNO plays a dual role in CEI formation chemistry. As depicted in Fig. 5a, NO3− ions occupy in the first solvation shell, releasing some OTf− anions into free ions. These anions, occupying the IHP, decompose and form a CEI rich in NaF and RSO3Na/RSO2Na components that serve as effective electron insulators, curbing further solvent oxidation (Fig. 5b). Simultaneously, the preferential decomposition of NO3− ions acts sacrificially, integrating into the CEI as NaNxOy, and shields oxidation of the C-O bonds in G2 molecules. This synergistic action results in a thin and dense CEI layer that selectively permits Na+ transport and blocks electron transfer. In contrast, uncontrolled G2 decomposition in the absence of NNO results in a porous CEI that is prone to continuous expansion and offers minimal protection at high voltages34,35.

a Schematic illustration of electrolyte environments and CEI transformations induced by the NNO additive. b Delineation of the possible decomposition pathways for electrolyte constituents.
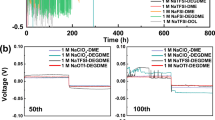
In our study, we selected the commonly used positive electrode materials like NNM, NMO, NVP, and sodium nickel iron manganese oxide (Na1.0Ni0.33Fe0.33Mn0.33O2, abbreviated as NFM), as well as graphite and HC negative electrode materials, as the representative samples. As illustrated in Fig. 6a–c and Supplementary Figs. 17 and 18, our strategy proved effective across a variety of positive and negative materials. Specifically, a battery equipped with an NNM positive electrode and a graphite negative electrode, when paired with the R-G2 electrolyte, exhibited improved cycling performance. It achieved an average CE exceeding 99%, a substantial improvement over the 50% CE observed with the G2 electrolyte alone. This enhanced cycling stability was corroborated by symmetric GCD profiles and a contour plot of dQ/dV at a 3.5 V cutoff, indicative of improved reversibility as shown in Supplementary Fig. 19.

Cycling comparison of graphite||NNM full cells at a current density of 100 mA g−1, utilizing the G2 and R-G2 electrolytes a, and G4 and R-G4 electrolytes (b), within a cut-off voltage of 4.0 V. c Cycling stability test of the HC||R-G2||NFM pouch cell at a current density of 20 mA g−1.
Our strategy was also successfully applied to the same category of TEGDME electrolyte (tetraethylene glycol dimethyl ether, denoted as G4). The graphite||NNM full battery using the reformulated R-G4 electrolyte retained 74% of its capacity after 200 cycles, as shown in Fig. 6b. This is in stark contrast to the rapid capacity decline observed with the standard G4 electrolyte. Furthermore, when using an NVP positive electrode, it maintained an impressive 83% capacity retention over 1000 cycles (Supplementary Fig. 17), coupled with a nearly perfect CE of 99.89%. This was supported by 1H NMR data (Supplementary Fig. 20), which indicated a reorganization of the solvation sheath, as evidenced by the downfield shift of integral peaks and the appearance of an additional peak in the terminal methyl groups in the G4 chains.
The incorporation of nitrate ions was found to effectively mitigate oxidation issues of ether electrolytes, thus preserving the redox stability of the electrode. The beneficial role of NO3− ions was further validated by the similar performance improvements observed with both lithium nitrate (LiNO3) and potassium nitrate (KNO3) additives, as depicted in Supplementary Fig. 21. Calculated based on the total mass of the whole battery, the HC||R-G2||NFM pouch cell shown in Fig. 6c was capable of delivering energy density of 137 Wh kg−1, while maintaining a remarkable 90% capacity retention after 100 cycles.
Discussion
Our research has made strides in addressing the issue of oxidation in ether-based electrolytes for SIBs. Through the strategic introduction of a small amount of nitrate anions (~0.04 M) into the electrolyte system, we have successfully mitigated the susceptibility of C-O bond breaking, a common cause of electrolyte degradation at high voltages. This strategic modification has catalyzed a profound change in interfacial chemistry, empowering the decomposition of OTf− and NO3− anions to form a robust cathode electrolyte interphase (CEI). The resulting CEI, enriched with NaF and NaNxOy species, has demonstrated improved high-voltage tolerance, extending the operational voltage threshold to 4.8 V vs. Na+/Na. The reformulated G2 electrolyte (R-G2) has emerged as a versatile solution, exhibiting compatibility with a wide array of high-voltage positive electrode materials. It has delivered improved Coulombic efficiency and extended cycle life, marking the advancement in the realm of SIB electrolytes. Moreover, the applicability of our approach extends beyond the G2 electrolyte, as evidenced by the enhanced cycling performance of other ether-based electrolytes, with nearly 83% capacity retention observed over 1000 cycles. Our research has not only deepened the understanding of the intricate relationship between interfacial chemistry and oxidation tolerance at high voltages but also provided a foundational blueprint for the design of high-voltage-tolerant ether electrolytes.
Methods
Materials synthesis
The P3-Na2/3Ni1/2Mn1/2O2 was prepared via a facile sol-gel method. Typically, a homogeneous acetate precursor was obtained from a 0.5 M acetate solution containing Na, Ni, and Mn ions with the proportion of 7:5:5 (Na excess), which was heated at 75 °C with vigorous stirring until a bright green sol formed and was aged at 100 °C overnight. The acetate precursor was decomposed at 500 °C in the air for 3 h, followed by the homogenized, pelletized, and annealed at 700 °C in the air for 12 h to obtain Na2/3Ni1/2Mn1/2O2 powder.
The tunnel Na0.44MnO2 was synthesized through a solid-state sintering method. NaHCO3 (99.8%, Aladdin) and Mn2O3 (99%, Aladdin) powder with the proportion of 1:1 was mixed by ball milling with rotation speed up to 500 rpm for 12 h. The mixture underwent a two-step annealing at 600 °C for 8 h and 900 °C for 10 h, respectively, to achieve Na0.44MnO2.
Cell fabrication
All the electrolyte preparation and coin cell assembly were carried out in an argon-filled glovebox with H2O and O2 levels <0.5 ppm. The modified electrolytes were prepared via direct dissolution. In electrolyte solutions, EC refers to ethylene carbonate, DEC abbreviates diethyl carbonate, and FEC stands for fluoroethylene carbonate. Typically, excessive NaNO3 (99.99%, Aladdin) was dissolved in the G2 electrolyte (99.8%, DoDoChem) in a sealed glass bottle, along with vigorous stirring at 60 °C overnight. The turbid solution was stood still for another 12 h to ensure total precipitation of undissolved NaNO3. The supernatant electrolyte was collected carefully and denoted as R-G2. R-G4 electrolyte was formulated by a similar procedure to the G4 electrolyte (99.8%, DoDoChem).
Materials characterization
The microstructure and interfacial morphology of electrodes were characterized by X-ray diffraction (XRD, Bruker-AXS D8 Advance), scanning electron microscope (SEM, Quant 250FEG), and high-resolution transmission electron microscopy (HRTEM, Tecnai G2 F30 S-TWIN). In order to obtain detailed compositions of the interfacial compound, in-depth X-ray photoelectron spectroscopy (XPS, monochromatic Al Kα source) was carried out with Ar+ ion sputtering. During our XPS etching study, the electrode was meticulously extracted from the cycled coin cell, rinsed and washed with the G2 solvent each three times to eliminate residual electrolytes, and then fully dried. This procedure was conducted in a glove box with water and oxygen levels below 0.1 ppm. The electrodes were sealed and vacuum-transferred to the XPS instrument for further analysis.
Various characterization methods were carried out to further investigate electrolyte properties during electrochemical reactions, including Raman spectroscopy (Jobin-Yvon T6400, 532 nm laser source) and nuclear magnetic resonance spectroscopy (NMR, 1H and 23Na, JEOL 600 MHz NMR spectrometer). The internal standard used in NMR experiments is tetramethylsilane and the deuterated reagents is deuterated dimethyl sulfoxide (DMSO).
Electrochemical measurement
2025-type coin cells were fabricated to test the electrochemical properties of different electrolytes. A slurry containing active materials, super P (Canrud), and poly (vinylidene fluoride) (PVDF) (Canrud) in a mass ratio of 8:1:1 was coated on Al foils and dried in a vacuum at 80 °C for 12 h, then pouched as Φ12 mm pieces. The mass loading of electrodes was controlled between 2.5 ~ 2.8 mg cm−2. The whole assembly procedure was operated in a glove box filled with high-pure Argon. The Whatman GF/F glass microfiber filters, model 1825-047 (420 μm thickness, 16 mmclateral dimension, 19 s/100 mL/in2 porosity, 0.7 μm average pore size) were used as separators in all battery systems. In half-cells, the Na foil (99.97%) was prepared directly prior to cell assembly as Φ14 mm pieces with thickness around 0.4 mm, worked as a counter electrode. Around 60 μL electrolyte were added in all cells assembling.
For full cell fabrication, the graphite and HC negative electrodes were cycled for presodiation, and the proportion of negative/positive (N/P) was set as 1.2:1. Galvanostatic charge/discharge was tested on Neware BTS-610, with constant temperature chamber of 25 °C. Pouch batteries are assembled by filling 7.0 mL R-G2 electrolyte into purchased, unfilled pouch cells. The anode consists of 9 pieces of HC weighing 6.18 g, while the cathode comprises 8 pieces of NFM with a mass of 10.679 g. The polypropylene separator has a mass of 1.02 g. Energy density is calculated based on the total mass of the unfilled pouch cell including the packaging (24.318 g). CE is calculated by dividing the discharge capacity by the charge capacity in the previous cycle. The LSV, EIS, and ionic conductivity profiles of different electrolytes were achieved using a Corrtest workstation. LSV was measured between 2 ~ 5 V, with a scanning rate of 0.5 mV s−1. EIS was conducted using galvanostatic perturbation at 10 mV amplitude in the frequency range from 105 Hz to 10−2 Hz, with 80 data points. Measurements were taken at the quasi-stationary potential after an 8-h open-circuit potential stabilization period. The ionic conductivity was measured in a 2025-type half-cell in 25 °C, in which two pieces of stainless steel worked as blocking electrodes. The ionic conductivity of standard 1.0 M LiPF6 in EC: DEC (1:1 in volume) electrolyte (8.0 mS cm−1) was tested as the control group.
Theoretical calculation
The oxidative tendency of different solvation structures was carried out using Gaussian 09 package based on density functional theory (DFT). Solvation structures were constructed through classical solvation theory and optimized via G16 to find the optimal conformation. The HOMO/LUMO calculation was carried out with a def2-TZVP basis set. According to frontier molecular orbital theory, a solvation cluster with higher HOMO may attribute to preferential oxidation compared to other components. The M06-2X was used in all calculations.
MD simulations: Two models were constructed: one comprising 25 NaOTf molecules and 175 G2 molecules with a total of 4250 atoms; the other comprising 25 NaOTf molecules, 175 G2 molecules, and one NaNO3 molecule with a total of 4255 atoms. All electrolyte models were initially equilibrated in the NPT ensemble (isobaric-isothermal ensemble) for a duration of 20 ns to maintain a pressure of 1 atm and a temperature of 298 K, utilizing time constants of 1 ps for pressure coupling and 0.1 ps for temperature coupling. Subsequently, a production run lasting for 1000 ns was performed in the NVT ensemble. Only the final segment of this run, encompassing the last 500 ns, was utilized to analyze the radial distribution function, solvation structures, and coordination numbers.
Data availability
The data that support the findings detailed in this study are available in the Article and its Supplementary Information or from the corresponding authors upon request. Source data are provided with this paper.
References
Jia, S., Kumakura, S. & McCalla, E. Unravelling air/moisture stability of cathode materials in sodium ion batteries: characterization, rational design, and perspectives. Energy Environ. Sci. 17, 4343–4389 (2024).
Liu, Z., Lu, Z., Guo, S., Yang, Q. H. & Zhou, H. Toward high performance anodes for sodium-ion batteries: from hard carbons to anode-free systems. ACS Cent. Sci. 9, 1076–1087 (2023).
Qiao, S. et al. Advanced anode materials for rechargeable sodium-ion batteries. ACS Nano 17, 11220–11252 (2023).
Zhang, F. et al. Emerging chemistry for wide-temperature sodium-ion batteries. Chem. Rev. 124, 4778–4821 (2024).
Li, Y. et al. Ether-based electrolytes for sodium ion batteries. Chem. Soc. Rev. 51, 4484–4536 (2022).
Kim, H. et al. Sodium storage behavior in natural graphite using ether‐based electrolyte systems. Adv. Funct. Mater. 25, 534–541 (2014).
Dong, R. et al. Elucidating the mechanism of fast Na storage kinetics in ether electrolytes for hard carbon anodes. Adv. Mater. 33, e2008810 (2021).
Li, K. et al. Evolution of the electrochemical interface in sodium ion batteries with ether electrolytes. Nat. Commun. 10, 725 (2019).
Zhang, J. et al. Ethers illume sodium‐based battery chemistry: uniqueness, surprise, and challenges. Adv. Energy Mater. 8, 1801361 (2018).
Liang, H. J. et al. Ether-based electrolyte chemistry towards high-voltage and long-life Na-ion full batteries. Angew. Chem. Int. Ed. 60, 26837–26846 (2021).
Liu, Q. et al. Stabilizing cathode-electrolyte interphase by localized high-concentration electrolytes for high-voltage sodium-ion batteries. Nano Energy 123, 109389 (2024).
Zheng, J. et al. Extremely stable sodium metal batteries enabled by localized high-concentration electrolytes. ACS Energy Lett. 3, 315–321 (2018).
Zhang, W. et al. Engineering a passivating electric double layer for high performance lithium metal batteries. Nat. Commun. 13, 2029 (2022).
Goodenough, J. B. & Park, K. S. The Li-ion rechargeable battery: a perspective. J. Am. Chem. Soc. 135, 1167–1176 (2013).
Liu, J. et al. Cationic covalent organic framework with ultralow HOMO energy used as scaffolds for 5.2 V solid polycarbonate rlectrolytes. Adv. Sci. 9, e2200390 (2022).
Jiang, L. et al. High-voltage aqueous Na-Ion battery enabled by inert-cation-assisted water-in-salt electrolyte. Adv. Mater. 32, e1904427 (2020).
Ma, G. et al. Reshaping the electrolyte structure and interface chemistry for stable aqueous zinc batteries. Energy Storage Mater. 47, 203–210 (2022).
Endrikat, A. et al. Electrochemical reduction mechanism of NbF5 and NbCl5 in the ionic liquid 1-butyl−1-methylpyrrolidinium trifluoromethanesulfonate. Electrochim. Acta 321, 134600 (2019).
Qin, M. et al. Dipole–dipole interactions for inhibiting solvent co-intercalation into a graphite anode to extend the horizon of electrolyte design. Energy Environ. Sci. 16, 546–556 (2023).
Ming, J. et al. Molecular-scale interfacial model for predicting electrode performance in rechargeable batteries. ACS Energy Lett. 4, 1584–1593 (2019).
Ming, J. et al. New insight on the role of electrolyte additives in rechargeable lithium ion batteries. ACS Energy Lett 4, 2613–2622 (2019).
Yan, Y. et al. Tailoring electrolyte dehydrogenation with trace additives: stabilizing the LiCoO2 cathode beyond 4.6 V. ACS Energy Lett. 7, 2677–2684 (2022).
Zhang, Y. et al. Revealing electrolyte oxidation via carbonate dehydrogenation on Ni-based oxides in Li-ion batteries by in situ Fourier transform infrared spectroscopy. Energy Environ. Sci. 13, 183–199 (2020).
Mu, L. et al. Deciphering the cathode–electrolyte interfacial chemistry in sodium layered cathode materials. Adv. Energy Mater. 8, 1801975 (2018).
Sun, Y., Shi, P., Xiang, H., Liang, X. & Yu, Y. High-safety nonaqueous electrolytes and interphases for sodium-ion batteries. Small 15, e1805479 (2019).
Huang, J. et al. Nanostructures of solid electrolyte interphases and their consequences for microsized Sn anodes in sodium ion batteries. Energy Environ. Sci. 12, 1550–1557 (2019).
Fang, W. et al. Constructing inorganic-rich solid electrolyte interphase via abundant anionic solvation sheath in commercial carbonate electrolytes. Nano Energy 104, 107881 (2022).
Li, P. et al. Nitrofullerene as an electrolyte-compatible additive for high-performance sodium metal batteries. Nano Energy 89, 106396 (2021).
Zhang, Q. et al. Tertiary‐amine based network polymer electrolyte for improving the cyclic stability of Na metal batteries with large capacity. Adv. Energy Mater. 14, 2303791 (2024).
Ba, D. et al. Robust cathode-ether electrolyte interphase on interfacial redox assembled fluorophosphate enabling high-rate and ultrastable sodium ion full cells. Nano Energy 94, 106918 (2022).
Wang, H. et al. Formation of NaF-Rich solid electrolyte interphase on Na anode through additive-induced anion-enriched structure of Na+ solvation. Angew. Chem. Int. Ed. 61, e202208506 (2022).
Zhang, J. et al. Achieving superb sodium storage performance on carbon anodes through an ether-derived solid electrolyte interphase. Energy Environ. Sci. 10, 370–376 (2017).
Jin, T. et al. High-energy aqueous sodium-ion batteries. Angew. Chem. Int. Ed. 60, 11943–11948 (2021).
Tian, Z. et al. Electrolyte solvation structure design for sodium ion batteries. Adv. Sci. 9, e2201207 (2022).
Wang, E. et al. Mitigating electron leakage of solid electrolyte interface for stable sodium-ion batteries. Angew. Chem. Int. Ed. 62, e202216354 (2023).
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Nos. 22272080 [M.Y.], 52272218 [H.X.]), Natural Science Foundation of Jiangsu Province (No. BK20200076 [M.Y.]), Jiangsu Province Carbon Peak and Neutrality Innovation Program (Industry tackling on prospect and key technology) (No. BE2022002-4 [H.X.]). We express our gratitude to Prof. Qi Liu from the City University of Hong Kong, who is now the Ph.D. supervisor of X.W., for his support throughout the revision process. We are also grateful to Mr. Zhengyang Li and Mr. Chenying Li (postgraduate students of Prof. Mei Yang.) for their assistance with figure format revisions.
Author information
Authors and Affiliations
Contributions
Mei Yang conceived and contributed the project. Xingyu Wang, Qi Fan and Ziheng Liu performed experimental tests and materials characterization. Xingyu Wang also analyzed the data and wrote the original manuscript. Xinyue Zhu, Zhiyuan Guo, and Yu Jing focused on the theoretical calculations. Yuting Chen and Liuqi Wang assisted in completing the experimental sections and data plotting and visualization. Mei Yang and Hui Xia revised the manuscript and provided funding for this work.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Xing-Long Wu, Shipeng Jia and the other, anonymous, reviewer for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, X., Fan, Q., Liu, Z. et al. Anion-mediated approach to overcome oxidation in ether electrolytes for high-voltage sodium-ion batteries. Nat Commun 16, 2536 (2025). https://doi.org/10.1038/s41467-025-57910-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-57910-7
