
Abstract
Nucleolytic resection of DNA ends is critical for homologous recombination, but its mechanism is not fully understood, particularly in mammalian meiosis. Here we examine roles of the conserved MRN complex (MRE11, RAD50, and NBS1) through genome-wide analysis of meiotic resection during spermatogenesis in mice with various MRN mutations, including several that cause chromosomal instability in humans. Meiotic DSBs form at elevated levels but remain unresected if Mre11 is conditionally deleted, thus MRN is required for both resection initiation and regulation of DSB numbers. Resection lengths are reduced to varying degrees in MRN hypomorphs or if MRE11 nuclease activity is attenuated in a conditional nuclease-dead Mre11 model. These findings unexpectedly establish that MRN is needed for longer-range extension of resection beyond that carried out by the orthologous proteins in budding yeast meiosis. Finally, resection defects are additively worsened by combining MRN and Exo1 mutations, and mice that are unable to initiate resection or have greatly curtailed resection lengths experience catastrophic spermatogenic failure. Our results elucidate MRN roles in meiotic DSB end processing and establish the importance of resection for mammalian meiosis.
Similar content being viewed by others

Introduction
Homologous recombination during meiosis initiates with DNA double-strand breaks (DSBs) made by SPO11 protein, which remains covalently bound to DNA 5’ ends after strand breakage1,2. These DSBs must then be nucleolytically processed (resected) to generate the single-stranded DNA (ssDNA) needed for homology search and strand exchange (Fig. 1a)3. Despite its central role in recombination, meiotic DSB resection remains poorly understood, particularly in mammals.

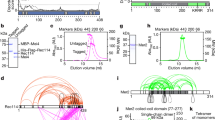
a Early meiotic recombination steps and sequencing strategy. i, SPO11 (magenta ellipses) cuts DNA via a covalent protein–DNA intermediate that is nicked (arrowheads) to give an entry point(s) for exonuclease(s). Bi-directional resection releases SPO11-oligo complexes and exposes ssDNA tails. ii, Two-step resection model: MRE11 hands off to other exonucleases. iii, Sequencing adapters are ligated to duplex ends after removal of ssDNA. Adapted from Fig. 1A of ref. 34 under a CC-BY 4.0 license (https://creativecommons.org/licenses/by/4.0/). b Conditional deletion of Mre11. c Reduced MRN protein in immunoblots of whole testis lysates from 5–7 wk old Mre11-cKO animals. Numbers below the MRE11 blot indicate amounts of a dilution series of wild-type (wt) lysate or band intensities for mutants relative to wild type, normalized to loading control (tubulin). d Seminiferous tubule sections at 5–7 wk stained for MRE11 (brown). In wild type, all cells except elongated spermatids stained positive for MRE11. In Mre11-cKO, tubules were categorized based on presence of MRE11-positive spermatocytes. Arrowheads, examples of MRE11-positive spermatogonia (red) or spermatocytes with (purple) or without (green) MRE11 staining. e Quantification of tubule classes in (d). Number of animals tested: n = 1 for wild type and n = 2 for Mre11-cKO (mean ± range). f Strand-specific S1-seq [reads per million mapped reads (RPM)] at a representative DSB hotspot. Signals from three (wild type) or two (Spo11–/– and Mre11-cKO) biological replicate libraries were averaged and plotted after smoothing (151-bp Hann window). Choice of ages is explained in Methods. SPO11-oligo sequencing throughout is from our previous study37. The y-axis baseline for each plot is 0. g Stereotyped distribution of resection endpoints around DSB hotspots. Heatmaps (data in 40-bp bins) show strand-specific reads around SPO11-dependent DSB hotspots from wild type (n = 13,960 from our previous study37). Each hotspot is shown as a horizontal line, strongest at the top. Sequencing signals were locally normalized by dividing by the total signal in a 4001-bp window around each hotspot’s center. Each hotspot thus has a total value of 1, to facilitate comparisons of spatial patterns between hotspots of different strengths. Arrowheads, the position of subsidiary peaks. Source data are provided as a Source data file.
The most detailed understanding of meiotic resection is currently for budding yeast, which uses a two-step DSB-processing mechanism (Fig. 1a)3. In the first step, the conserved Mre11-Rad50-Xrs2 (MRX) complex together with Sae2 (homologous to MRE11-RAD50-NBS1 (MRN) and CtIP in mouse) nicks the Spo11-bound strands using the endonuclease activity of Mre11 and degrades ssDNA towards the DSB using the 3’→5’ exonuclease activity of Mre11. We will refer to this as resection initiation. In the second step, the more processive 5’→3’ exonuclease activity of Exo1 further degrades ssDNA away from the DSB. This step has been referred to as long-range resection4, although it should be noted that this is considerably shorter than the long-range resection that can occur in non-meiotic contexts3.
The combined action of these nucleases generates long 3’-terminal ssDNA and releases Spo11 still covalently bound to short oligonucleotides (oligos)2,4,5,6,7,8,9,10,11,12,13. The average resection length in wild-type yeast is ~800 nt and this is reduced by more than half (to ~375 nt) in exo1 nuclease-deficient mutants4,12. This residual meiotic resection in exo1 mutants is similar in length to MRX/Sae2-dependent short-range resection in vegetative cells3.
It is comparatively elusive how resection is executed in mammalian meiocytes. Similar to yeast, mouse DSB processing generates long 3’-terminal ssDNA (averaging ~1100 nt) and releases covalent SPO11-oligo complexes8,14,15. Unlike yeast, however, net resection length is decreased by only ~10% in mouse Exo1 mutants, suggesting that EXO1 plays only a modest role in resection or is redundant with another nuclease(s)14,15.
The contribution of MRN to resection has been difficult to address, in part because of experimental challenges due to the embryonic lethality of knockout mouse models lacking MRN subunits16,17,18. Conditional deletion of Nbs1 in germ cells resulted in reduced numbers of foci of RAD51 and other recombination proteins; this was interpreted to reflect a role for MRN in DSB processing, but resection per se was not directly assessed19. More recently, conditional Rad50 deletion was found to cause an increased occurrence of unresected DSBs and to reduce resection tract lengths20. However, how MRN promotes resection remains unclear. Finally, it also remains unclear how resection length ties in with successful execution of meiotic recombination and thus with fertility.
We address these questions here through genome-wide analysis of meiotic DSB resection in mouse spermatocytes conditionally depleted of MRE11 or carrying various MRN mutations, including several that model hypomorphic genetic variants found in human chromosomal instability syndromes21,22,23,24,25,26,27,28. Our findings indicate that MRN is essential for resection initiation in mammals, but, surprisingly, it is also needed to generate most of the length of resection tracts that the yeast paradigm would instead ascribe to (Mre11-independent) long-range resection. Our findings further reveal how resection defects alter the binding of recombination factors to ssDNA and impinge on faithful execution of meiosis.
Results
MRE11 is required to initiate resection
To circumvent the lethality of an Mre11 null mutation17, we used a conditional deletion strategy combining a floxed Mre11 allele29 with Ngn3-Cre30,31,32 (Fig. 1b). Ngn3-Cre is expressed in spermatogonia during the mitotic divisions prior to meiotic entry, but not in spermatogonial stem cells (SSCs, Supplementary Fig. 1a)31,33. We will refer to Mre11flox/– Ngn3-Cre+ as Mre11-cKO (for conditional knockout), Mre11flox/+ Ngn3-Cre– or Mre11flox/flox Ngn3-Cre– as phenotypic wild type (wt), and Mre11flox/+ Ngn3-Cre+ and Mre11flox/– Ngn3-Cre– as cHet and Het, respectively.
MRE11 protein was reduced by about 20 fold in immunoblots of whole testis extracts from young adult Mre11-cKO males (5 wk old, Fig. 1c). We used immunohistochemistry on seminiferous tubule sections to determine which cell types retained MRE11 protein (Fig. 1d, e). Virtually all cells were positive for MRE11 in wild type, including the layers of spermatogenic cells and Sertoli cells within the tubules as well as interstitial somatic cells.
In Mre11-cKO mice, most tubules contained a basal layer(s) of spermatogonia and spermatocytes but lacked later-stage germ cells, and 16.1% of tubules lacked spermatocytes entirely (detailed further below) (Fig. 1d, e). Spermatogonia and interstitial cells remained uniformly MRE11-positive as expected, but spermatocytes, when present, either lacked detectable MRE11 or were a mix of MRE11-positive and -negative in the same tubule. These findings indicate that the Mre11-flox allele is recombined efficiently but incompletely by Ngn3-Cre expression in spermatogonia, resulting in a substantial population of spermatocytes entering meiosis with no or low MRE11 protein. As described before29, RAD50 and NBS1 levels were also decreased when MRE11 was depleted (Fig. 1c).
We visualized resection endpoints genome-wide using S1-sequencing (S1-seq), in which genomic DNA is digested with the ssDNA-specific nuclease S1 and the blunted DNA ends are captured and sequenced (Fig. 1a)4,15,34. For some experiments, we used a combination of exonuclease VII and exonuclease T from Escherichia coli instead of nuclease S1 (Exo7/T-seq; based on END-seq14,35,36).
Most DSBs are formed in hotspots whose locations can be identified by sequencing of SPO11 oligos37,38 (Fig. 1f, top). S1-seq and Exo7/T-seq yield Spo11-dependent reads distributed ~0.3–2 kb away from hotspot centers with defined polarity: top-strand reads from rightward-moving resection tracts and bottom-strand reads from leftward tracts15 (Fig. 1a, f, g and Supplementary Fig. 1b, c). Both methods also generate reads close to hotspot centers (Fig. 1f, g and Supplementary Fig. 1b, c). This central signal is thought to arise from recombination intermediates, possibly D loops14,15 (Supplementary Fig. 1d). Importantly, both S1-seq and Exo7/T-seq can also detect DSBs that have not been resected at all (Supplementary Fig. 1e)4,14,15,39,40.
S1-seq and Exo7/T-seq profiles at hotspots were dramatically altered in young adult Mre11-cKO mice (Fig. 1f, g and Supplementary Fig. 1b, c). Virtually all signals accumulated at hotspot centers in a pattern strikingly similar to SPO11 oligos in wild type, with a strong central cluster and weaker flanking clusters to either side (Fig. 1f, g and Supplementary Fig. 1b, c). We conclude that DSBs remain unresected in the absence of MRE11. Our observations may also indicate that MRE11 is dispensable for DSB formation, but other interpretations are possible (Discussion).
MRE11 controls DSB numbers and restrains double cutting
To summarize global patterns and to facilitate comparisons between genotypes, we plotted genome-wide profiles around DSB hotspots by co-orienting and averaging the sequencing signal from top and bottom strand reads (Fig. 2a). Compared to wild type, Mre11-cKO mice had elevated read counts at hotspots, with an increase well beyond the sample-to-sample variation in wild type (Fig. 2a, b). In response to DSBs, MRN complexes activate ataxia-telangiectasia mutated (ATM) protein kinase (Tel1 in budding yeast)41,42,43. During meiosis, ATM/Tel1 regulates the formation of DSBs via a negative feedback mechanism (reviewed in44) (Fig. 2c). We therefore surmised that the increased sequencing read count in Mre11-cKO mice reflects, at least in part, an increase in the number of DSBs per cell because of a loss of MRN-dependent ATM activation.

a Genome-average S1-seq and Exo7/T-seq at hotspots in 5 wk old Mre11-cKO animals. Bottom-strand reads were flipped and combined with top-strand reads, averaged, and smoothed (151-bp Hann window; average of two biological replicates). S1-seq in 14.5-dpp wild type is shown on the same scale (average of three biological replicates). b S1-seq signal for Mre11-cKO (n = 2) and wild type (n = 10 for juvenile, 1 for adult). Each point is the area under the genome-average curve (as in panel a) from –1500 to +2500 bp relative to the hotspot center for an individual replicate. The P value is from a Student’s t test (two-sided). c Left, ATM activation by MRE11 and subsequent negative feedback regulation of SPO11. Right, absence of resection and increase in SPO11 double cuts in the absence of MRE11. d Offset top- and bottom-strand reads at hotspot centers in Mre11-cKO, consistent with double cutting. The plot shows the smoothed genome-average strand-specific profile close to hotspot centers (51-bp Hann window) with the SPO11-oligo profile for comparison. e Increased SPO11-oligo complexes with altered electrophoretic mobility in Mre11-cKO. The autoradiograph shows SPO11-oligo complexes immunoprecipitated from testis extracts from 5–6 wk old mice, radiolabeled with terminal transferase and [α-32P]-dCTP, and separated by SDS-PAGE. The signal intensity of the region above the non-specific band, normalized to wild type, is indicated below. Asterisk: non-specific labeling. f SPO11 oligo lengths in Mre11-cKO. SPO11 oligos were purified from two mice of each genotype by immunoprecipitation and protease digestion, then radiolabeled with terminal transferase and [α-32P]-GTP and separated by denaturing urea PAGE. Note that SPO11 oligos appear slightly larger than their true lengths because of nucleotide(s) added by terminal transferase and residual amino acid(s) not removed by protease. Both samples were run on the same gel, but the intensity of the Mre11-cKO signal was increased approximately threefold to facilitate comparison. Asterisks: non-specific species; nt: markers in nucleotides. Lane traces (background-subtracted and normalized to the total lane signal) are shown to the right. Expected migration positions of species arising from MRE11-dependent (canonical) resection and from SPO11 double cutting are indicated. Source data are provided as a Source data file.
If so, we anticipated that Mre11-cKO mutants would also display another hallmark of ATM deficiency, namely, a high frequency of double cutting in which multiple SPO11 complexes cut the same DNA molecule in close proximity45,46,47. In support, the top-strand signal of the central S1-seq peak was shifted to the right of the SPO11-oligo peak, and the bottom-strand signal was shifted to the left (Fig. 2d). This pattern is distinct from the central signal derived from recombination intermediates in wild type (Supplementary Fig. 2a), but is as expected if multiple DSBs are formed on the same DNA molecules within hotspots because only the outermost DSB ends will be represented in the final sequencing library (Figs. 2c and Supplementary Fig. 1e). Double cutting may also explain why, unlike for SPO11 oligos in wild type, the subsidiary peak to the right of the central peak is stronger on average than the left-side subsidiary peak for top-strand reads, and vice versa for bottom-strand reads (Figs. 1g and 2d).
We further tested for double cutting by radiolabeling the 3′ ends of SPO11-oligo complexes immunoprecipitated from whole-testis extracts. This assay does not detect SPO11 if it is still bound to the high molecular weight DNA of unresected DSBs, but double cutting in budding yeast can generate Spo11-oligo complexes when Mre11 nuclease activity is compromised by rad50S or sae2∆ mutations45,47 (Fig. 2c). We indeed observed SPO11-oligo complexes in Mre11-cKO mice, and these were at levels more than ten-fold greater on a per-testis basis than in wild-type or heterozygote controls (Fig. 2e and Supplementary Fig. 2b), comparable to Atm−/− mice48.
The major band of SPO11-oligo complexes from Mre11-cKO had slower mobility on SDS-PAGE than controls (Figs. 2e and Supplementary Fig. 2b). We therefore determined the size distribution of radiolabeled SPO11 oligos on denaturing PAGE (Fig. 2f). The SPO11 oligos in wild type that arise from canonical resection initiation have a bimodal size distribution, with most falling in the ranges of ~15–27 or ~31–35 nucleotides (nt) long8,37,48,49. By contrast, SPO11 oligos that arise from double cutting are at least ~33 nt and range to much larger sizes, with a pronounced ~10-nt periodicity in lengths that is a diagnostic feature of SPO11 double cuts thought to reflect geometric constraints on the orientation of adjacent SPO11 dimers relative to the DNA45,47,50. Because Atm−/− spermatocytes retain the ability to initiate resection14,15, they have a mix of SPO11 oligos arising from both resection and SPO11 double cutting45,48 (Fig. 2f).
Similar to SPO11 oligos purified from Atm−/− mice compared side by side, those from Mre11-cKO mice displayed a major species migrating slightly slower than the 34-nt marker plus a ladder of slower migrating bands with ~10-nt periodicity. Interestingly, however, compared with Atm−/−, the Mre11-cKO sample displayed substantially less labeled material migrating faster than the 34-nt marker (Fig. 2f). These findings together are consistent with the interpretation that most if not all of the SPO11-oligo complexes in Mre11-cKO spermatocytes arise from double cuts.
We also examined resection patterns in juvenile (14.5 dpp) Mre11-cKO mice. Immunoblotting of whole-testis extracts showed that MRE11 protein was reduced, but to a lesser degree than in adults ( ~ twofold lower than controls; Supplementary Fig. 2c). Similar to adults, these mice showed a greatly elevated central S1-seq signal at hotspots that had the spatial patterns diagnostic of unresected DSBs and double cutting (Supplementary Figs. 2d–f). Unlike adults, however, juveniles also had a substantial fraction of resected DSBs, although with a resection length distribution that was markedly shorter (modal length 897 nt) than in wild type (modal length 1015 nt) (Supplementary Fig. 2f). It is formally possible that spermatocytes in the initial wave of meiosis in juveniles have an additional (MRE11-independent) resection initiation mechanism that is not available in adults. We consider it more likely, however, that the weaker phenotype in juveniles reflects differences in the timing of Cre-mediated excision relative to DSB formation and/or differences in the kinetics of MRE11 protein rundown after excision.
Spermatogenesis failure in Mre11-deficient mice
Adult Mre11-cKO mice had significantly smaller testes than littermate wild-type or heterozygous controls (Fig. 3a, b). In seminiferous tubule sections, layers of spermatogonia and spermatocytes were present but postmeiotic cells were largely absent, sections of epididymis appeared empty, and epididymal sperm counts were greatly reduced (Fig. 3a, c). All Mre11-cKO tubules contained one or more layers of cells that stained positive for the mouse VASA homolog DDX4 (Supplementary Fig. 3a), which is expressed from spermatogonia to the round spermatid stage51. Tubules with apoptotic cells were also increased in frequency (Supplementary Figs. 3b, c). These Mre11-cKO phenotypes remained consistent to at least 16 wk of age (the oldest tested) (Supplementary Fig. 3d). These results, together with MRE11 staining (Fig. 1d), support the interpretation that Mre11-cKO mice stably maintain an MRE11-positive SSC population that can initiate multiple waves of spermatogenesis, but most spermatocytes in each wave are then eliminated by apoptosis. We conclude that MRE11 protein is essential for male fertility in mice, as are NBS1 and RAD5019,20.

a Bouin’s fixed testis and epididymis sections stained with hematoxylin and eosin (H&E). Sg: spermatogonia, Sc: spermatocyte, rSt: round spermatid, eSt: elongated spermatid. b Ratios of testis weight (mg per pair) to body weight (g). Each point represents one animal; error bars indicate mean ± SD. The results of Student’s t tests (two-sided) are shown; ns, not significant (P > 0.05). c Sperm counts (mean ± SD; P value from Student’s t test (two-sided)). d Representative spreads of wild-type or Mre11-cKO spermatocytes showing normal SYCP3 staining at the indicated stages or abnormal (“Zygonema-like”) cells with thickened and/or tangled axes. e Representative RPA2 staining on spermatocyte chromosome spreads. Examples are shown of Mre11-cKO cells with either low or high RPA2 focus counts. f Spermatocyte stages based on SYCP3 staining (L: leptonema, Z: zygonema, Z-like: zygonema-like, P: pachynema, D: diplonema). Error bars indicate mean ± SD from three mice of each genotype. g RPA2 focus numbers. The horizontal lines represent medians for the indicated number of cells counted from 2 mice of each genotype. The stage of each cell was determined from the SYCP3 signal. The results of two-tailed Mann-Whitney U tests are shown. Source data are provided as a Source data file.
To monitor chromosome dynamics and progression through prophase I, we immunostained spermatocyte chromosome spreads for the axis protein SYCP3, with or without costaining for the RPA2 subunit of the ssDNA-binding protein RPA. In wild type, SYCP3 forms short lines in leptonema as axes begin to form; the axes elongate and synapse with homologous partners to form stretches of synaptonemal complex (SC) in zygonema; SC spans the length of each pair of autosomes in pachynema; and then SC disassembles in diplonema (Figs. 3d and Supplementary Fig. 3e). RPA2 forms large numbers of foci on resected DSBs in leptonema and zygonema, then these foci decrease in number over the course of pachynema as DSB repair is completed (Fig. 3e)52.
Spermatocytes in Mre11-cKO mice deviated substantially from the normal patterns. We observed cells with short SYCP3 axes and no SC, similar to normal leptonema but representing a higher fraction of total spermatocytes than in wild type (Fig. 3d, f and Supplementary Fig. 3e). Most cells had very few RPA2 foci (Fig. 3e, g). We also observed cells with axes of various lengths along with SC, but most of these differed from normal zygotene cells in having only short stretches of staining for SC central region component SYCP1 and/or tangles of axes (Supplementary Fig. 3e). These “zygotene-like” cells were the most abundant class in Mre11-cKO (Fig. 3f) and most were again highly depleted for RPA2 foci compared to wild type (Fig. 3e, g). The deficit of RPA2 foci corroborates the resection defect seen in S1-seq. These results also show, not surprisingly, that unresected DSBs cannot support homologous pairing and synapsis.
In addition to these aberrant cells, there were also morphologically normal pachytene and diplotene cells, but much fewer than in wild type (Figs. 3d, f and Supplementary Fig. 3e). RPA2 focus numbers in the pachytene cells were indistinguishable from wild type (Fig. 3g). We also observed a small fraction of leptotene and zygotene cells that had RPA2 focus numbers in the normal range for these stages (Fig. 3e, g). We infer that these more normal-looking cells correspond to the MRE11-positive subpopulation of spermatocytes (Fig. 1d), perhaps cells that escaped Cre-mediated excision. If so, these escapers appear to be too small a fraction of the total leptotene and zygotene cells (Fig. 3f) to contribute an appreciable resection signal in population-average sequencing maps (Figs. 1f, g, and 2a).
Nuclease-dead MRE11 reveals an unexpected role in extending resection lengths
To test whether MRE11 nuclease activity is required for meiotic resection, we examined mice with the invariant active-site residue His-129 substituted to asparagine29. Human MRE11 with this mutation assembles MRN complexes and activates ATM, but lacks nuclease activity in vitro53,54. Mre11-H129N is cell-lethal in chicken and human cells55 and is embryonically lethal in mice29, but the phenotype in mammalian meiosis has not been addressed. To circumvent the lethality, we generated Mre11H129N/flox mice carrying Ngn3-Cre (hereafter Mre11-cHN) (Fig. 4a). Total MRE11 protein levels were normal in testis extracts from Mre11-cHN mice (Fig. 4b), consistent with the previous demonstration that the mutant mouse protein is stable and assembles MRN complexes normally29.

a Conditional Mre11-H129N strategy combining Ngn3-Cre with compound heterozygous floxed and missense Mre11 exon 5 alleles. b Normal MRE11 protein levels of whole testis lysates from 5–7 wk old Mre11-cHN mice. Here, the phenotypically wild-type control (wt) was Mre11wt/flox Ngn3-Cre– and the heterozygous HN strain (HN-Het) was Mre11H129N/flox Ngn3-Cre–. c Exo7/T-seq at the hotspot shown in Fig. 1f. d Genome-average Exo7/T-seq at hotspots (5-wk animals). Top and bottom strand reads were co-oriented, averaged, and smoothed (151-bp Hann window), then internally normalized by setting the height of the resection peak to 1. Normalization facilitates comparison of shapes of resection profiles separate from sample-to-sample variation in read frequencies34. Non-normalized plots are in Supplementary Fig. 4c. n = 2 for wt and Mre11-cHN, n = 1 for HN-Het. e MRE11 accumulation at hotspots in Mre11-cHN. MRE11 ChIP-seq signal was averaged around hotspots for Mre11-cHN (n = 3), wt (n = 2), and Atm–/– (n = 1). f, g Fewer SPO11-oligo complexes in Mre11-cHN. Autoradiograph of radiolabeled complexes immunoprecipitated from testis extracts (f; signal intensity excluding the non-specific band, relative to wild type, indicated below). Lane profiles (g) depicting fast and slow migrating species as a fraction of total signal are shown. Dashed lines represent the boundary of each species. Asterisk, non-specific labeling. h The bar graph summarizing results from two biological replicates. i Reduced testis sizes (mean ± SD). Results of Student’s t tests (two-sided) are shown. j Seminiferous tubule and epididymis sections (7 wk old; cell type labels as in Fig. 3a). k, l Increased spermatocyte apoptosis in Mre11-cHN mice (7 wk). TUNEL staining (k) and quantification (l) are shown. Error bars in (l) indicate mean and range. Wild type reproduced from Supplementary Fig. 3c. m Sperm counts (mean ± SD; P value from Student’s t test (two-sided)). Control combines wt, HN-het, and cHet. n Mre11-cHN spermatocyte spreads showing either normally synapsed autosomes and normal γH2AX-positive sex body (left) or pachytene-like cells with unpaired or tangled autosomes (middle and right; arrowheads, γH2AX-positive domains). Numbers indicate pachytene(-like) cells observed and SYCP3-positive cells counted. o Spermatocyte stages (mean ± SD). Source data are provided as a Source data file.
Because the equivalent budding and fission yeast mutants form meiotic DSBs but cannot resect them7,56, we expected that Mre11-cHN mice would accumulate only unresected DSBs if the wild-type MRE11 protein expressed from the floxed allele were sufficiently depleted (Supplementary Fig. 4a, left). On the other hand, if some wild-type MRE11 protein persisted long enough to support resection initiation at a subset of DSBs in each cell, or at all DSBs in a subset of escapee cells, we instead expected to observe that some or all of the DSBs would be fully resected (Supplementary Fig. 4a, right). The latter prediction was premised on the idea that the long-range nuclease(s) should be able to complete resection normally as long as MRE11 endonuclease had incised the DNA. Similar logic would apply to interpretation of the resection that occurs in juvenile Mre11-cKO mice.
Surprisingly, however, Exo7/T-seq patterns in young adult mice matched neither of these predictions when considering either individual hotspots (Figs. 4c and Supplementary Fig. 4b) or genome-average profiles (Fig. 4d and Supplementary Fig. 4c). We note two main findings. First, there was a modest increase in the relative amount of the sequencing signal at hotspot centers (Fig. 4d), suggesting that some unresected DSBs are present. Consistent with this interpretation, we also observed a slight increase in the sequencing signal at hotspot centers on the nonhomologous parts of the X and Y chromosomes (Supplementary Fig. 4d). Central S1-seq or Exo7/T-seq signal from recombination intermediates does not appear at X and Y hotspots in wild type, but it can be detected when there are unresected DSBs, such as in Atm–/– mutants14,15 or Mre11-cKO (Supplementary Fig. 4e).
To further test for unresected DSBs, we performed chromatin immunoprecipitation (ChIP-seq) for MRE11, which was previously demonstrated to accumulate at hotspots when unresected DSBs are present in Atm–/– mutants, but not in wild type where all DSBs are resected14. Indeed, we observed a clear MRE11 ChIP-seq signal at hotspots in Mre11-cHN mice, albeit substantially less than in Atm–/– (Figs. 4e and Supplementary Fig. 4f, g). Taken together, these results suggest that a small fraction of DSBs remains unresected in Mre11-cHN mice. If so, however, this resection initiation defect is very weak compared to Atm–/– and especially Mre11-cKO, perhaps reflecting insufficient elimination of wild-type MRE11 protein in this conditional model (see Discussion).
The second and more surprising finding was that resection tracts were substantially shorter than normal in Mre11-cHN, averaging only 672 nt (Fig. 4d). This defect was reproducible and stable, remaining the same with increasing age up to 11 wk (the oldest age tested; Supplementary Fig. 4h). We also observed a slight resection defect in mice heterozygous for Mre11-H129N (Mre11H129N/wt with Ngn3-Cre). Resection tracts in these mice averaged 1040 nt (94% of wild type) (Fig. 4d), suggesting a weak dominant-negative effect of MRE11-H129N protein. Observing shorter average resection lengths suggests that MRE11 nuclease activity contributes not just to resection initiation, but also to the extension of resection tracts beyond the lengths seen for MRX-dependent short-range resection in yeast.
Unlike in Atm–/– and Mre11-cKO, the amount of SPO11-oligo complexes was not increased in Mre11-cHN (Figs. 4f and Supplementary Fig. 4i), consistent with MRE11-HN protein being competent to activate ATM/Tel1 in both yeast and mouse29,54,57,58. Instead, the yield on a per-testis basis was reduced by about half compared to wild type. The total Exo7/T-seq signal recovered at hotspots was also diminished in Mre11-cHN (Supplementary Fig. 4j). These decreases may reflect reduced DSB frequency and/or loss of signal because of spermatocyte apoptosis (described below). We also observed a reproducible shift in the migration of SPO11-oligo complexes on SDS-PAGE gels, with faster migrating species appearing to be more depleted than slower migrating ones (Fig. 4g, h). We posit that the presence of catalytically inactive MRE11 protein sometimes occludes potential nicking sites closer to SPO11, biasing the population of SPO11 oligos generated by the residual wild-type MRE11 protein to larger sizes than normal.
We were unable to assess a role for CtIP—a cofactor needed for MRN nuclease activity3—because conditional deletion of Ctip eliminated spermatogonia prior to meiotic entry (Supplementary Figs. 4k–n). We also tested Rad50S/S mice but found normal resection (Supplementary Fig. 4o), likely because this Rad50-K22M mutation was based on a relatively weak yeast allele because mimics of stronger alleles are cell-lethal23. Thus, available mouse mutants are unable to definitively test the requirement for MRN nuclease in resection initiation.
Spermatogenesis defects in mice with nuclease-dead MRE11
Mre11-cHN mice had pronounced defects in meiotic progression. Their testes were considerably smaller than those of control littermates (Fig. 4i); seminiferous tubules contained spermatogonia and early stage spermatocytes but were depleted of spermatids and had an increased frequency of spermatocyte apoptosis (Fig. 4j–l); and epididymal sperm counts were greatly diminished (Fig. 4j, m). The rare residual sperm detected may have arisen from escapers in which Cre-mediated excision failed and/or from a subset of cells in which wild-type MRE11 protein persisted long enough after excision to support successful meiosis.
When prophase I stages were evaluated by staining spreads for SYCP3 and phosphorylated H2AX (γH2AX), we observed a subset of pachytene-like cells with unpaired or tangled chromosomes (Fig. 4n, o), indicative of sporadic synaptic failure. We also observed an increase in the proportions of leptotene and zygotene cells relative to later stages (pachytene, pachytene-like, and diplotene) (Fig. 4o). This increase may reflect a delay in completing synapsis, preferential apoptotic elimination of cells in later prophase I, or both.
Reduced resection lengths in MRN hypomorphic mutants
We further tested for MRN roles in meiotic resection using mice homozygous for hypomorphic mutations modeled on human disease alleles59 (Fig. 5a). Mre11-ATLD1 is a nonsense mutation near the 3′ end of the coding sequence, recapitulating a mutation found in ataxia telangiectasia-like disorder21. Nbs1∆B, which models Nijmegen breakage syndrome, produces an NBS1 protein lacking the N-terminal FHA and BRCT domains26. Mre11-ATLD1 and Nbs1∆B cause intra-S and G2/M checkpoint defects, DNA damage sensitivity, chromosomal instability, and reduced ATM activity in cultured cells and they cause subfertility in mice, particularly in females21,26,60. The mutations also reduce protein stability in cells21,26 and MRN nuclease activity in vitro61, but they have not previously been linked to overt resection defects.

a Schematic of MRN hypomorphs. MRE11 R632* (R632 → stop) mimics human R633* identified in ATLD patients21. Nbs1∆B produces an 80 kDa protein (red box), mimicking the 657del5 allele found in 95% of NBS patients26. b S1-seq at the hotspot from Fig. 1f, from 14.5-dpp mice. Wild type is reproduced from Fig. 1f. Two biological replicates were averaged for each of the other genotypes. Scale bars, 0.005 RPM. c Heatmaps (data in 40-bp bins) of S1-seq reads around DSB hotspots. d Genome-average S1-seq at hotspots from 14.5-dpp mice. Wild type is reproduced from Fig. 2a. Data are smoothed with a 151-bp Hann window and normalized as described in Fig. 4d. e–h Minimal if any changes in RAD51 and DMC1 focus numbers during meiosis in Rad50-L1237F or Rad50-D69Y mice. Representative spreads stained for SYCP3 and DMC1 (e) and RAD51 (g) along with quantification (f, h) are shown. Each dot in the graphs is the focus count from a single nucleus; error bars indicate means ± SD. Results of unpaired t tests (two-sided) are shown for zygonema; differences at the other stages were not statistically significant (P > 0.05). Source data are provided as a Source data file.
We also examined homozygous Rad50-D69Y and Rad50-L1237F mutants, which model recurrent mutations in human cancer affecting residues in the Walker A and B ATPase motifs, respectively62,63 (Fig. 5a). Studies in S. cerevisiae and cultured mouse cells indicated that the human RAD50-L1240F mutant (L1237F in mouse) is a separation-of-function allele that is largely proficient in DNA repair, but defective in the activation of Tel1/ATM62. Similar outcomes were observed for modeling of human RAD50-D69Y in S. cerevisiae63.
Rad50-L1237F and Rad50-D69Y mice were derived by CRISPR-mediated mutagenesis. MEFs from Rad50-D69Y mice displayed reduced ionizing radiation (IR)-provoked phosphorylation of KAP1 (an ATM target64) (Supplementary Fig. 5a); elevated mitotic indices after IR, consistent with defects in the ATM-dependent G2/M checkpoint (Supplementary Fig. 5b); and hypersensitivity to camptothecin when ATR was inhibited, as expected if compromised ATM activation cannot compensate for loss of ATR activity (Supplementary Fig. 5c). These findings confirm the separation of function predicted from prior studies in S. cerevisiae63, and verify that Rad50-D69Y, like Rad50-1237F62, is defective for ATM activation.
S1-seq maps from juvenile males demonstrated resection defects in all of these MRN mutants, demonstrating a striking allelic series (Fig. 5b–d). These findings reinforce the conclusion that the MRN complex must be fully functional to support normal resection lengths, i.e., that MRN roles in DSB processing are not limited just to resection initiation close (within a few hundred bp) to the DSB. There was only a slight increase in central S1-seq signal at hotspot centers on the sex chromosomes in Nbs1∆B, suggestive of a small number of unresected DSBs, but little if any evidence for this in the other mutants (Supplementary Fig. 5d).
Spermatogenesis in Nbs1∆B and Mre11-ATLD1 mutant males was previously shown to be nearly normal, with only mildly delayed temporal progression and persistent RAD51 localization that was attributed, at least in part, to reduced functionality of these MRN hypomorphs in meiotic DSB repair21,60. The substantially shorter resection lengths we now observe in these mutants may contribute to this apparent DSB repair delay. Spermatogenesis also appeared to be largely normal in Rad50-L1237F and -D69Y mutants. Testis sizes and sperm counts were indistinguishable from littermate controls (Supplementary Fig. 5e–g); the frequency of spermatocyte apoptosis was not significantly increased (Supplementary Fig. 5h); the distribution of prophase I stages was not substantially altered (Supplementary Fig. 5i); and the appearance and disappearance of foci of strand exchange proteins occurred normally, with the exception that both mutants showed a statistically significant but quantitatively small reduction in the number of DMC1 foci at zygonema (Fig. 5e–h). These findings show that meiotic recombination is robust in the face of even substantial deviations from normal resection lengths (e.g., up to the ~47% reduction in Mre11-ATLD1).
Side-by-side spatial organization of RAD51 and DMC1 is maintained in Nbs1ΔB mice
We tested whether the shortened resection lengths in Nbs1∆B mice altered the occupancy and spatial disposition of ssDNA binding proteins RPA, DMC1, and RAD51 by performing ChIP followed by ssDNA sequencing (SSDS)65,66. In wild type, DMC1 tends to bind closer to the broken end, RAD51 binds nearer to the ssDNA/dsDNA boundary, and RPA can be found essentially anywhere along the ssDNA65 (Fig. 6a). The SSDS signal for all three proteins was more compressed toward hotspot centers in the Nbs1∆B mutant, as expected from the decreased length of ssDNA available, but the spatial pattern of DSB-proximal DMC1 and DSB-distal RAD51 was maintained (Fig. 6b).

a, b Genome-wide profiles of DMC1, RAD51, and RPA2 ChIP followed by SSDS in wild type (a) or Nbs1∆B (b). In the graphs above, top- and bottom-strand reads around hotspots were co-oriented, combined and averaged. These maps represent sequencing coverage rather than endpoint counts reported for S1- or Exo7/T-seq. Data are smoothed with a 151-bp Hann window. An estimated background was removed by subtracting the value of signal 2.5 kb away from the hotspot center, then profiles were normalized by setting the area under each curve (from –1.0 to 2.5 kb) to 1. Heatmaps below show SSDS signals in 40-bp bins, locally normalized by dividing each signal by the total signal in a 4001-bp window around each hotspot’s center. Each hotspot thus has a total value of 1, so that spatial patterns can be compared between hotspots of different strengths. c Representative SIM images of spread spermatocytes from 14.5-dpp mice. Insets show zoomed views of the regions indicated by dashed boxes. d DMC1 to RAD51 distances for axis-associated co-foci. Tukey box plots are shown for DMC1 foci that were ≤ 450 nm from an axis and ≤ 320 nm from the nearest RAD51 focus. The plus signs indicate means; horizontal lines are medians; box edges are interquartile range; whiskers indicate the most extreme data points which are ≤ 1.5 times the interquartile range from the box; individual points are outliers. P value is from a t test (two-sided). The number of images and foci included in the analysis was summarized in Supplementary Table 1. Source data are provided as a Source data file.
Previous studies in yeast and mice showed that foci of RAD51 and DMC1 often appear side by side65,67,68. We tested using structured illumination microscopy (SIM) whether this organization at individual DSB sites was retained in Nbs1∆B mice, comparing homozygous mutants with heterozygous littermate controls (which have nearly normal resection lengths (Supplementary Fig. 6a)). Similar to previous results65, a majority of the foci of both DMC1 (93.8%) and RAD51 (79.4%) were located close to chromosome axes in Nbs1∆B/+ heterozygotes (Figs. 6c and Supplementary Fig. 6b and Supplementary Table 1). Moreover, the foci often occurred together in pairs: 80.7% of axis-associated DMC1 foci and 77.4% of axis-associated RAD51 foci were within 320 nm of the other protein (Fig. 6c and Supplementary Figs. 6c and Supplementary Table 1). The high degrees of both axis association and colocalization were retained in Nbs1∆B/∆B homozygotes (94.7% axis association for DMC1 and 86.3% for RAD51; 84.8% colocalization for DMC1 foci and 82.3% for RAD51) (Fig. 6c and Supplementary Fig. 6b, c and Supplementary Table 1).
DMC1 and RAD51 foci were nearly always offset from one another, with a distance in the controls of 103.3 ± 67.5 nm (mean ± SD) between DMC1 foci and their RAD51 mates (Fig. 6d and Supplementary Table 1). This distance was decreased significantly in Nbs1∆B mutants, but with a small effect size (96.8 nm ± 66.5).
Two conclusions emerge. First, the decreased distance between DMC1 and RAD51 foci when resection lengths are reduced supports the interpretation that paired foci represent binding of both proteins to the same ssDNA tail and not to separate DSB ends, similar to the situation inferred from yeast and mouse studies67,68. Second, the effect of the Nbs1∆B mutation on interfocus distances (less than 7% decrease on average) was much smaller than the effect on both resection length (43% decrease, Fig. 5d) and ChIP profiles (Fig. 6b). This difference between cytology and population-average molecular assays shows that ssDNA length is not the most important determinant of the three-dimensional spatial organization of DSB repair foci. This conclusion in turn fits with the idea that individual DMC1 and RAD51 filaments are fairly short, estimated at ~100 nt in yeast and ~400 nt in mouse67,68. Because filaments are only long enough to occupy a small fraction of the available ssDNA, even a substantial reduction in resection lengths should not cause a proportionate decrease in the size of cytologically observable foci.
Additive contributions of EXO1 and the NBS1 N terminus to resection length
In vitro, yeast MRX promotes Exo1 activity by providing nicks for Exo1 to act on and by fostering Exo1 binding to DNA ends69,70,71,72. To query the interplay between MRN and EXO1 in meiotic resection, we generated double mutant mice homozygous for both Nbs1∆B and Exo1 mutations. An Exo1 null is synthetically lethal with Nbs1∆B73, so we used the nuclease-deficient Exo1-D173A (DA) allele74, reasoning that the milder somatic phenotype of Exo1DA75 might allow us to obtain double mutants. Indeed, Nbs1∆B Exo1DA doubly homozygous mice were viable, albeit recovered at a sub-mendelian frequency (Supplementary Fig. 7a) and exhibiting lower body weights (Supplementary Fig. 7b).
Nbs1∆B Exo1DA double mutants showed a 111-nt decrease in the average resection length compared to Nbs1∆B alone (Figs. 7a, b and Supplementary Fig. 7c). Since this is similar to the 126-nt average decrease in Exo1DA compared to wild type, EXO1 appears to work additively with MRN. We also conclude that the Nbs1∆B resection defect is not attributable to a defect in EXO1 activity.

a S1-seq at the hotspot from Fig. 1f. Because of the smaller size of Nbs1∆B Exo1DA pups, we collected samples at 16.5 dpp for this experiment. Wild type (14.5 dpp) is reproduced from Fig. 1f; two biological replicates were averaged for each of the other genotypes. Scale bars, 0.005 RPM. b Genome-average S1-seq patterns at hotspots, smoothed with a 151-bp Hann window. Wild type and Nbs1∆B (14.5 dpp) are reproduced from Fig. 5d. Mean resection lengths are indicated. c Reduced central S1-seq signal (presumed recombination intermediates) in Nbs1∆B mutants. The plot shows the relative amount of central signal calculated as the area under the curve (AUC) for the central peak (–300 to +100 bp relative to hotspot centers) divided by the AUC for the resection peak ( + 100 to +2500 bp). For wild type, in each of three independent time courses, samples were collected from a single litter daily at approximately the same time each day. d Homolog pairing and/or synapsis defects. Representative spreads of Nbs1∆B Exo1DA spermatocytes are shown with either normal-appearing SYCP3 and γH2AX staining or with unsynapsed or tangled chromosomes within the γH2AX-stained domain (yellow arrowheads). Blue arrowheads point to examples of γH2AX flares on synapsed autosomes. e Spermatocyte stages based on SYCP3 and γH2AX staining. Error bars indicate mean ± SD of the indicated number of animals. f Percentage of pachytene or pachytene-like cells with no, 1–3, or > 3 γH2AX flares. Error bars indicate mean ± SD of the indicated number of animals. g Frequencies of tubules with the indicated number of TUNEL-positive cells. Error bars indicate mean ± range for the indicated number of animals. h Reduced testis size in Nbs1∆B Exo1DA double mutants (5–11 wk old). Error bars indicate mean ± SD. P values are from Student’s t tests (two-sided). i Sperm counts (mean ± SD). Results of Student’s t tests (two-sided) are shown. Source data are provided as a Source data file.
Relative to the S1-seq signal from resection endpoints, the central signal from recombination intermediates appeared lower in Nbs1∆B Exo1DA than in wild type or the single mutants (Fig. 7b). To evaluate this, we measured total read count (area under the curve) separately for the central peak and for the resection endpoint peak and calculated the ratio between these values (Fig. 7c). In wild type, the central peak was reproducibly ~40% of the level of the resected DSBs from 13.5 to 16.5 dpp. This ratio was lower at 12.5 dpp ( ~ 33%) in two of three experiments (Fig. 7c). Since this earlier age is likely to be more enriched for earlier prophase I spermatocytes, the lower relative amount of central signal may reflect timing differences between DSB formation (appearance of the resected DSB signal) and formation of the recombination intermediates detected by S1-seq.
In Nbs1∆B mice, the relative amount of central signal was substantially lower at 14.5 dpp (27.5% of the DSB signal; mean of n = 2) and it increased but remained lower than wild type at 16.5 dpp (34.7%; Fig. 7c). At 14.5 dpp, Mre11-ATLD1 exhibited a reduction similar to Nbs1∆B, but Rad50-L1237F and -D69Y did not (Supplementary Fig. 7d). The central signal at 16.5 dpp was even lower in Nbs1∆B Exo1DA (28.8%) but the Exo1DA mutation by itself had no effect. These decreased yields of the central signal in Mre11-ATLD1, Nbs1∆B, and Nbs1∆B Exo1DA may indicate that fewer of the relevant recombination intermediates are formed, that they turn over more rapidly, and/or that they exhibit structural features that make them more difficult to detect by S1-seq. Regardless of the source of the changes, however, it appears that Mre11-ATLD1 and Nbs1∆B mutants experience alterations in interhomolog recombination, with at least the Nbs1∆B mutant exacerbated when combined with Exo1DA.
Reinforcing this view, Nbs1∆B Exo1DA double mutants displayed an elevated frequency of pachytene-like cells with synaptic failures (tangled and/or unpaired chromosomes) and a higher proportion of zygotene cells than normal, possibly indicative of delays in completing synapsis (Fig. 7d, e). Pachytene and pachytene-like cells from the double mutant also showed an increased frequency of γH2AX flares on synapsed chromosomes, indicative of delays in DSB repair (Fig. 7d, f). Seminiferous tubule sections showed few if any elongating spermatids in Nbs1∆B Exo1DA double mutants (Supplementary Fig. 7e) and an elevated frequency of apoptotic spermatocytes (Fig. 7g and Supplementary Fig. 7f). As a consequence of this germ cell loss, the mice had substantially reduced testis sizes (Fig. 7h) and were essentially devoid of epididymal sperm (Fig. 7i and Supplementary Fig. 7e). Single Nbs1∆B or Exo1DA mutants showed few of these defects, consistent with previous studies60,75, although apoptotic tubules were observed at modestly elevated frequencies and testis sizes were reduced (Fig. 7g, h).
Discussion
Unlike the paradigmatic budding yeast pathway for meiotic DSB resection4,12, mice rely less on EXO114,15 and we now show that multiple MRN mutations confer drastic resection defects (see also20). Thus, while the canonical two-step handoff from MRN(X) to EXO1 occurs in both yeast and mammals, we find that mouse MRN is essential not just for resection initiation, but also unexpectedly for most of the normal length of resection as well. Since Exo1DA combined additively with MRN deficiency, EXO1 appears to “polish” resection tracts that are generated primarily through MRN.
ATM deficiency decreases resection lengths for a subset of DSBs14,15. Thus, because the MRN hypomorphs compromise ATM activation21,26,76, their resection defects could be an indirect consequence of diminished ATM signaling. However, Mre11-cHN mice appear to have normal ATM activity29,54,57,58 (Fig. 4f), suggesting that their defective resection is tied more directly to attenuated MRN activity per se. By extension, the resection defects in MRN hypomorphic adults and Mre11-cKO juveniles may be explained in a similar manner. We cannot exclude that reduced ATM activity also contributes, but Mre11-ATLD1 and Nbs1∆B mutants only modestly attenuate meiotic ATM activation as judged by their retention of most ATM-dependent DSB control77, so this explanation would require that resection is particularly sensitive to small decreases in ATM activation. Moreover, the resection-promoting function of ATM itself remains unknown, so the opposite causality is also plausible: i.e., resection defects in Atm mutants might hypothetically be an indirect consequence of losing ATM-dependent regulation of MRN.
We consider two plausible ways in which MRN might promote the extension of resection tracts aside from ATM activation. First, it might directly resect DNA via iterative nicking that spreads progressively from the DSB end. Although yeast MRX and human MRN preferentially cleave the 5’-terminal DNA strand ~15–45 nt away from a blocked DNA end in vitro78,79,80,81, MRN can both oligomerize and diffuse on DNA82,83. Moreover, MRX can resect unrepairable DSBs in vivo more than one kilobase in yeast lacking long-range resection enzymes84, and iterative nicking was proposed to explain both short-range resection in vegetative yeast cells85 and the ~300 nt average resection in meiotic cells lacking Exo1 activity4. If this interpretation is correct, our results indicate that the iterative action of MRN can extend resection up to ~1500 nt in mice when the nuclease activity of EXO1 is compromised.
A second, non-exclusive possibility is that MRN recruits or activates another nuclease(s). MRN enhances DNA2 nuclease in vitro69,86,87,88,89, although it remains unclear if the same is true in vivo. CtIP also promotes DNA2 activity both in vitro and in vivo90,91. Budding yeast Dna2 is unlikely to be important for meiotic resection because its partner helicase Sgs1 is dispensable4,11,12. However, the possibly mammal-specific interaction between CtIP and DNA290,91 may make DNA2 a candidate for long-range resection in mouse meiosis15.
MRN involvement in resection initiation was not surprising. However, the specific genetic dependencies are intriguing. For example, we were surprised to observe only minor apparent defects in resection initiation in Mre11-cHN mice. It is formally possible that MRE11 nuclease is dispensable for clipping SPO11 from DSBs even though MRE11 protein itself is required. However, we consider it more likely that this result instead reflects continuing presence of sufficient wild-type MRE11. For example, dimerization with continuously expressed MRE11-H129N might slow the degradation of wild-type protein compared to what happens in Mre11-cKO. In this scenario, mutant/wild-type heterodimers are still active for strand cleavage since they retain a functional nuclease active site. Alternatively, MRE11-H129N might amplify activity of a small amount of residual wild-type protein in a structural role via heterodimer formation or oligomerization on DNA. It is also possible that binding of inactive MRN complexes to DSBs allows another nuclease(s) to be recruited to initiate resection in an unconventional manner.
Interestingly, the meiotic progression defects of the Mre11-cHN mutant were considerably worse than those in Mre11-ATLD1 and Nbs1∆B despite Mre11-cHN having a longer average resection tract length. It is possible that the meiotic progression defects in Mre11-cHN reflect an as yet unknown function for MRE11 nuclease activity in downstream steps of recombination. However, it is also possible that the progression defects are entirely consequences of defects in resection initiation and/or DSB formation. For example, the reduced SPO11-oligo complexes and hotspot-associated sequencing signal may indicate that there are fewer processed DSBs available to support recombination and chromosome pairing. If so, such a reduction in processed DSBs would also explain the apparent delay during leptonema and zygonema and sporadic synapsis failure (just a few chromosomes per cell). Indeed, we previously showed that reducing DSBs to about half the normal level gives a meiotic progression defect that is quite similar to the one we document here for Mre11-cHN92.
The Mre11-cKO mutant is the first example in mammals of a yeast rad50S-like meiotic phenotype, namely, accumulation of completely unresected DSBs7,93,94,95. This finding definitively establishes that MRN is essential for resection initiation. The elevated DSB sequencing signal and double cutting in this mutant also confirms and extends the previous conclusion, based on modest elevation of SPO11-oligo complexes in Mre11-ATLD1 and Nbs1∆B, that MRN promotes ATM activation in the context of controlling SPO11 activity77. While this manuscript was in preparation, conditional deletion of Rad50 (with Stra8-Cre) was also found to cause an increased frequency of unresected DSBs, but to a much more modest extent than we observed with Mre11-cKO and without an apparent increase in DSB levels or evidence of double-cutting20. Because resection was not directly assessed for Nbs1 conditional deletion (with Vasa-Cre), it is unclear whether NBS1 is required for resection initiation19. The differences between the conditional mutants may reflect differences in timing and efficiency of gene deletion and protein disappearance rather than different requirements for the individual MRN subunits.
Our findings may also indicate that mouse MRE11 is dispensable for DSB formation, unlike in S. cerevisiae and Caenorhabditis elegans, but similar to Schizosaccharomyces pombe, Drosophila melanogaster, and Arabidopsis thaliana [reviewed in ref. 96]. However, this interpretation should be viewed cautiously because Mre11-cKO spermatocytes may retain trace amounts of MRE11 protein that are sufficient for DSB formation but not resection. In this vein, we note that Mre11 and Ctip null mutations are both cell-lethal, whereas conditional deletion of Mre11 but not of Ctip was tolerated by spermatogonia and premeiotic spermatocytes. This is consistent with both proteins being essential for mitotic germ cell divisions, but MRE11 protein persisting longer than CtIP after Ngn3-Cre-mediated excision.
In addition to defining the genetic control of DSB resection, our findings offer insight into the minimal resection length needed for successful downstream steps. The extensive pairing and synapsis defects in Mre11-cKO mice met the expectation that recombination cannot proceed without ssDNA generated by resection. However, our findings showed that meiosis is remarkably robust as long as some resection occurs. Recombination, chromosome pairing, synapsis, and chromosome segregation all occurred efficiently with slightly more than half the normal average resection length (Mre11-ATLD1). Severe problems were seen only in the Nbs1∆B Exo1DA double mutant, with its only modest further decrement in resection length. But even in this mutant, although most cells failed to complete meiosis, most chromosomes paired and synapsed well, indicating that most DSBs were successfully engaging in recombination.
It is unclear why recombination begins to fail in the Nbs1∆B Exo1DA double mutant but not appreciably in the Nbs1∆B or Mre11-ATLD1 single mutants. The minimal length of ssDNA needed to support homology searching and strand exchange in mammalian meiosis is unknown, but vegetative yeast cells can execute recombination with less than 40 bp of homology, and normal recombination efficiency appears to need only ~100–250 bp97,98,99,100,101. Moreover, RAD51 and DMC1 filaments are thought to occupy only a few hundred nucleotides67,68. Although the distribution of resection tract lengths is highly stereotyped across all mouse hotspots, there is substantial variation from DSB to DSB, and perhaps between the two sides of a single DSB. It has been suggested for yeast that normal resection lengths ensure that all DSBs are resected enough to support efficient, accurate recombination12. It is therefore possible that Nbs1∆B Exo1DA double mutants become vulnerable to recombination defects because a large enough subset of their DSBs fall below the minimum length needed for strand exchange per se or for proper choice of homologous recombination partner102.
Methods
Mice
Experiments conformed to the US Office of Laboratory Animal Welfare regulatory standards and were approved by the Memorial Sloan Kettering Cancer Center Institutional Animal Care and Use Committee (Protocol number 01-03-007). Mice were housed in solid-bottom, polysulfone, individually ventilated cages (IVCs) (Thoren Caging Systems, Hazelton, PA) on autoclaved aspen-chip bedding (PWI Industries Canada, Quebec, Canada); γ-irradiated feed (LabDiet 50531, PMI, St Louis, MO) and acidified reverse osmosis water (pH 2.5 to 2.8) provided ad libitum. The cages also contained Nestlets®, EnviroDri®, and/or EnviroPaks® as environmental enrichment. The IVC system was ventilated at approximately 30 air changes hourly. HEPA-filtered room air was supplied to each cage and the rack effluent was exhausted directly into the building’s exhaust system. Cages were changed weekly in either a HEPA-filtered vertical flow change station or a Class 2 Type A biological safety cabinet. The animal holding room was maintained at 21.5 ± 1 °C, relative humidity between 30% and 70%, and a 12:12 hour light:dark photoperiod. Mice were euthanized by CO2 asphyxiation prior to tissue harvest. Previously described Mre11-flox and Mre11-H129N29 and Ctip-flox103,104 alleles were crossed with Ngn3-Cre30 to create male germline-specific mutations. No randomization or blinding was performed.
To generate Mre11-cKO experimental mice, Mre11flox/flox (or Mre11wt/flox based on availability) mice were crossed with Mre11+/– Ngn3-Cre+ mice. The Mre11-deletion allele (Mre11–) was derived by Cre-mediated recombination of the Mre11-flox allele. To generate Mre11-cHN experimental mice, Mre11H129N/+ Ngn3-Cre+ mice were crossed with Mre11flox/flox mice. To generate Ctip-cKO mice, Ctipflox/flox mice were crossed with Ctip+/– Ngn3-Cre+ mice. The Ctip deletion allele (Ctip–) was derived by Cre-mediated recombination of Ctip-flox.
Although Ngn3-Cre is also expressed in embryonic pancreas and endocrine cells30, we did not observe any gross defects of mutant animals created with Ngn3-Cre up through the ages of testis sample collection. Previously described Atm105 Spo11106, Rad50S23, Mre11-ATLD121, Nbs1∆B26, and Exo1DA74 mutations were maintained on a congenic C57BL/6 J strain background. Nbs1∆B Exo1DA animals were generated by crossing Nbs1∆B mice with Exo1DA mice. To enhance the survival rate of double homozygous male mice for experimental purposes, female pups were euthanized by hypothermia between 4–6 dpp in accordance with the approved animal protocol.
To generate targeted Rad50 mutations, a guide RNA cassette with sequence (5’-TACATTTGTTCATGATCCCA(AGG)) and single-stranded donor DNA (5’- TTATAAAAATTTAATTCTTAAAATACAAAACTTTACAGACACCATTACCTTGGGATaATGAACAAATGTATTTCCTTTGGTTCCAGGAGGGAAATCTCCAGTACAA) harboring desired missense mutations for D69 > Y, or a guide RNA cassette with sequence (5’-TCTCGGTCGAGATTTGTTGT(CGG)) and single-stranded donor DNA (5’- GGAAACCTTCTGTCTGAACTGTGGCATCCTTGCCTTGGATGAGCCGACAACAAATtttGACCGAGAAAACATTGAGTCTCTTGCACATGCTTTGGTTGAGTAAGTA) harboring desired missense mutations for L1237 > F were microinjected into pronuclei of zygotes. Founder mice were crossed to C57BL/6 J mice purchased from Jackson Laboratories to obtain germline transmission, then heterozygous animals were backcrossed to C57BL/6 J for at least three generations.
S1-seq and Exo7/T-seq
Choice of sequencing method and animal ages
S1-seq was originally applied to juvenile testis samples because these samples lack postmeiotic cells and are thus enriched for spermatocytes that contain DSBs and recombination intermediates15. We continued to use juvenile samples for most experiments here, but found that Cre-mediated excision of the Mre11-flox construct was more penetrant in adult samples. Initial attempts to perform S1-seq on adult samples gave poor signal strength from SPO11-generated DSBs at hotspots because of the presence of S1-sensitive triplex secondary structures in the DNA107. We therefore initially used Exo7/T-seq for analysis of resection in Mre11-cKO and Mre11-cHN mice, because exonuclease VII and exonuclease T require DNA ends and thus do not give sequencing reads from many of the non-DSB structures that S1-seq can pick up. We later found that increasing the pH during the nuclease S1 digestion step greatly improved the specificity for SPO11-initiated events by avoiding the formation of triplex structures34, so we repeated the analysis of Mre11-cKO using S1-seq on adult samples. For the wild-type time courses (Fig. 7c), each time point was processed up to the agarose plug step, then S1-seq maps were prepared in a single batch after collecting the last time point.
Library preparation and sequencing
Libraries were prepared as described elsewhere34, based on S1-seq15 and END-seq14,108. Briefly, testes from mice at the indicated ages were decapsulated and digested with collagenase type IV (Worthington) and Dispase II (Sigma). The resulting seminiferous tubule preparations were then further treated with TrypLE Express enzyme (GIBCO) and DNase I (Sigma) for 15 min at 35 °C. TrypLE enzyme was inactivated with 5% FBS and tubules were further dissociated by repeated pipetting. Cells were then passed through a 70-μm cell strainer (BD Falcon).
One to two million cells from juvenile testes or two to three million cells from adult testes were embedded in plugs of 1% low-melting-point agarose (Lonza). After a brief incubation at 4 °C until the agarose became solid, plugs were incubated with proteinase K (Roche) in lysis buffer (0.5 M EDTA at pH 8.0, 1% N-lauroylsarcosine sodium salt) at 50 °C over two nights. Plugs were washed five times for 20 min with TE (10 mM Tris-HCl at pH 7.5, 1 mM EDTA at pH 8.0), and then incubated with 100 μg/ml RNase A (Thermo) for 3 hr at 37 °C. Plugs were then washed five times with TE and stored in TE at 4 °C until usage.
All library types were prepared in the same way aside from the ssDNA digestion step. For S1-seq, plugs were equilibrated with S1 buffer (50 mM sodium acetate, 280 mM NaCl, 4.5 mM ZnSO4, pH 4.7 at 25 °C), then treated with 9 U of nuclease S1 (Promega) for 20 min at 37 °C. For Exo7/T-seq, plugs were instead equilibrated with exonuclease VII buffer (50 mM Tris-HCl, 50 mM sodium phosphate, 8 mM EDTA, 10 mM 2-mercaptoethanol, pH 8 at 25 °C), then treated with 50 U of exonuclease VII (NEB) for 60 min at 37 °C. Plugs were then equilibrated in NEBuffer 4 and treated with 75 U of exonuclease T (NEB) for 90 min at 24 °C.
Plugs were next equilibrated with T4 polymerase buffer (T4 ligase buffer (NEB) supplemented with 100 μg/ml BSA and 100 μM dNTPs (Roche)) then incubated with 30 U T4 DNA polymerase (NEB) at 12 °C for 30 min. Plugs were then washed in TE and equilibrated in T4 ligase buffer on ice. Biotinylated P5 adapters were ligated to the blunted ends with 2000 U T4 DNA Ligase (NEB) at 16 °C for 20 hr. After ligation, plugs were soaked in TE overnight at 4 °C to diffuse excess unligated adapters out of the plugs. The plugs were then equilibrated in 1× β-agarase I buffer (10 mM Bis-Tris-HCl, 1 mM EDTA, pH 6.5), incubated at 70 °C for 15 min to melt the agarose with brief vortexing every 5 min, mixed well, cooled to 42 °C, and digested with 2 μl of β-agarase I (NEB) at 42 °C for 90 min. DNA was sheared to fragment sizes ranging 200–500 bp with a Covaris system (E220 Focused-ultrasonicator, microtube-500) using the following parameters: delay 300 s then three cycles of [peak power 175 W, duty factor 20%, cycles/burst 200, duration 30 s, and delay 90 s]. The DNA was precipitated with ethanol, then dissolved in TE. Unligated adapters were removed with SPRIselect beads (Beckman Colter). Fragments containing the biotinylated adapter were further purified with Dynabeads M-280 streptavidin (Thermo) and DNA ends were repaired using the End-it DNA End-repair kit (Lucigen). P7 adapters were ligated to DNA fragments, then PCR was done directly on the bead-immobilized fragments. PCR products were purified with AMPure XP beads (Beckman Colter) to remove primer dimers and unligated adapters.
After Qubit (Thermo) quantification and quality control by Agilent BioAnalyzer, libraries were pooled and sequenced on the Illumina NextSeq, HiSeq, or NovaSeq platforms equipped with HiSeq Control Software (version 2.2.68) or Real Time Analysis (version 3.1 or 4.1) in the Integrated Genomics Operation (IGO) at Memorial Sloan Kettering Cancer Center. A spike-in of bacteriophage ΦX174 DNA was added to increase diversity when necessary and for quality control purposes. We obtained paired-end reads of 50 bp.
Preprocessing and mapping
Base calls were performed using bcl-convert software v.3.9.3 to 3.10.5 or bcl2fastq software v.2.20. Reads were trimmed and filtered by Trim Galore version 0.6.6 with the arguments --paired --length 15 (http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/). Sequence reads were mapped onto the mouse reference genome (mm10) by bowtie2 version 2.3.5.1109 with the arguments -N 1 -X 1000. Duplicated reads were removed by Picard (https://broadinstitute.github.io/picard/). Uniquely and properly mapped reads (MAPQ ≥ 20) were extracted by samtools with the argument -q 20 (http://www.htslib.org/). Reads were assigned to the nucleotide immediately next to the biotinylated adapter. Mapping statistics are in Supplementary Data 1.
Bioinformatic analysis and plotting
Maps were analyzed using R version 4.0.3 to 4.3.2 (http://www.r-project.org) and R studio (version2023.06.1 + 524). To generate average profiles around individual hotspots (e.g., Fig. 1f), RPM values from biological replicates were averaged before smoothing and plotting. For genome-average profiles around hotspots (e.g., Fig. 2a), bottom-strand reads (in RPM) were co-oriented with top-strand reads by flipping the coordinates relative to the hotspot midpoint, then the combined top- and bottom-strand reads were averaged across all hotspots before smoothing and plotting. For genotypes in which resection was initiated (i.e., all genotypes except Mre11-cKO), we further normalized the genome average profiles as follows. First, an estimated background was removed by subtracting from all values the value of signal 2500 bp away from the hotspot center. The signal was then normalized to the peak height of resection endpoints, i.e, the maximum value among positions from 100 to 2,500 bp. Negative values were set as zero for plotting purposes. Where appropriate, data were smoothed with a Hann function.
To calculate the mean resection length, S1-seq signal was averaged across hotspots and an estimated background was removed by subtracting from all values the value of signal 2.5 kb away from the hot spot center. The signal close to and further away from the hot spot center was excluded by setting values of positions <100 bp and >2.5 kb to zero. Fractions of total signal were calculated every 100 bp and the mean resection length was calculated. The modal resection length in Supplementary Fig. 2f was determined by the peak position of the genome-average profile between 100 bp and 2500 bp from hotspot centers.
The color scale of heatmaps were defined after local normalization of sequencing signal at each hotspot. Signals in 40-bp bins were divided by the total signal in a 4,001-bp window around each hotspot center, so that each row has a total value of 1 regardless of the strengths of hotspots and color reflects the local spatial pattern. Normalized signals were classified into 10 groups by deciles and color-coded.
We provide analysis of variation between biological replicate maps in a separate study34. As shown there, the spatial patterns of resection and the central signal as well as the ratio of central signal to resection signal are highly reproducible between replicates (coefficient of variation of <1.4% for average resection length and < 6.8% for central:resection ratio from n = 10 wild-type S1-seq replicates). This reproducibility arises in part because these are internally controlled comparisons within a genomic map, so they are well-suited for RPM-based analysis. The absolute RPM values around hotspots are also reproducible, but somewhat less so (coefficient of variation of 26.9%) because of differences in signal:noise ratio between sequencing libraries (i.e., differences in the amount of SPO11-independent background). Because of the latter variability, we often ignore the absolute value of the RPM map, and instead focus on spatial patterns. Exceptions are cases where the difference in absolute RPM exceeds the normal range of sample-to-sample variation, and where we have clear evidence of reproducibility of differences. For example, the Mre11-cKO mutant has such a large increase in the amount of signal within hotspots that we can confidently make conclusions based on the comparison of total RPM values with wild type.
MRE11 ChIP-seq
MRE11 ChIP was performed following the previously published DISCOVER-seq protocol110. In brief, fresh testes of 5–7 wk old wild-type, Atm–/–, or Mre11-cHN mice were dissociated into single cells and resuspended in DMEM. PFA (16% stock, Pierce) was added to a final concentration of 1% and incubated for 15 min with gentle agitation, then the crosslinking was stopped by adding 2.5 M glycine to a final concentration of 125 mM, followed by 3 min incubation on ice. The nuclei were prepared as described110 and up to 10 million nuclei were resuspended in 500 μl lysis buffer (0.5% N-lauroylsarcosine, 0.1% sodium deoxycholate, 10 mM Tris-HCl pH 8.0, 100 mM NaCl, 1 mM EDTA) supplemented with protease inhibitor (Roche), and sonicated to fragment sizes ranging 200–300 bp with a Covaris system (E220 Focused-ultrasonicator, microtube-500) using the following parameters: delay 300 s then six cycles of [peak power 75 W, duty factor 10%, cycles/burst 200, duration 150 s, and delay 60 s]. Sheared nuclei were centrifuged at 20,000 g for 10 min at 4 °C to remove debris and Triton-X (1% (vol/vol) final concentration) and lysis buffer were added to make the total volume 3 ml.
For each immunoprecipitation, 10 μl of anti-MRE11 (Novus Biologicals NB100-142) was pre-mixed with 100 μl protein A Dynabeads (Thermo), then added to chromatin lysates and incubated overnight at 4 °C with end-over-end rotation. Immunoprecipitated DNA was eluted and crosslinks were reversed as described110. ChIP sequencing libraries were prepared from cleaned DNA as described for SSDS libraries111 without the boiling step after the end-repair and dA-tailing steps. After ligation with sequencing adapters, the library was amplified by 15 cycles of PCR. The amplified library was purified and sequenced on the Illumina NovaSeq platform equipped with Real Time Analysis (version 3.1 or 4.1) in the IGO. We obtained paired-end reads of 100 bp.
Base calls were performed using bcl-convert software v.3.9.3 to 3.10.5 or bcl2fastq software v.2.20. Sequencing reads were processed and mapped as described for resection libraries and uniquely and properly mapped reads were counted, and maps were further analyzed using R version 4.0.3 to 4.3.2 (http://www.r-project.org) and R studio (version2023.06.1 + 524). Mapping statistics are in Supplementary Data 1.
Single-stranded DNA sequencing (SSDS) library preparation and data analysis
ChIP followed by SSDS was performed as previously described65,111. In brief, a decapsulated, fresh or fresh-frozen testis of 8-12 wk old wild-type or Nbs1∆B mice was placed in room temperature 1% PFA (16% stock, Pierce) in PBS and incubated for 15 min with gentle agitation. The crosslinking was stopped by adding 2.5 M glycine to a final concentration of 125 mM, followed by 5 min incubation at room temperature. Fixed cells were immediately homogenized in a Dounce homogenizer with 20 strokes of the tight-fitting pestle, then passed through a 70 μm cell strainer. The nuclei were prepared as described111, resuspended in 900 μl shearing buffer (0.1% SDS, 10 mM Tris-HCl pH 8.0, 1 mM EDTA) supplemented with protease inhibitor (Roche), split into two ∼500 μl aliquots and sonicated to fragment sizes ranging 500–1000 bp with a Covaris system (E220 Focused-ultrasonicator, microtube-500) using the following parameters: delay 300 s then three cycles of [peak power 75 W, duty factor 10%, cycles/burst 200, duration 150 s, and delay 60 s]. Sheared nuclei were centrifuged at 12,000 g for 10 min at 4 °C to remove debris. The sonicated chromatin was then transferred to a 3 ml Slide-A-Lyzer G2 dialysis cassette (Pierce) and dialyzed with ChIP buffer (16.7 mM Tris-HCl, pH 8.0, 167 mM NaCl, 1.2 mM EDTA, 0.01% SDS, 1.1% Triton X-100) for 5 hr at 4 °C with constant but slow stirring. For each immunoprecipitation, 20 μl of anti-DMC1 (Abcam, ab11054), 10 μl of anti-RAD51 (Novus Biologicals, NB100-148), or 10 μl of anti-RPA2 (Abcam, ab76420) was added to chromatin lysates and incubated overnight at 4 °C with end-over-end rotation.
As described previously111, protein G or A Dynabeads (Thermo) were added to the chromatin and antibody mixture and incubated 2 hr at 4 °C, then immunoprecipitated DNA was washed and eluted. To reverse DNA-protein crosslinks, 12 μl of 5 M NaCl was added to 300 μl of eluted sample and incubated overnight at 65 °C. DNA was cleaned with MinElute PCR Cleanup Kit (Qiagen) after proteinase K (Thermo) treatment. Following end repair and dA-tailing, DNA was incubated at 95 °C for 3 min and cooled to room temperature to enrich for single-stranded DNA. After ligation with sequencing adapters, SSDS library was amplified with 12 cycles of PCR. The amplified library was purified and sequenced on the Illumina NovaSeq platform equipped with Real Time Analysis (version 3.1 or 4.1) in the IGO. We obtained paired-end reads of 100 bp.
Base calls were performed using bcl-convert software v.3.9.3 to 3.10.5 or bcl2fastq software v.2.20. We then ran a previously described bioinformatic pipeline on the resulting reads for identification of single-stranded sequences111. Sequence reads at SPO11-dependent hotspots were analyzed using R version 4.0.3 to 4.3.2 (http://www.r-project.org) and R studio (version2023.06.1 + 524).
Sperm counts
Sperm isolation was performed as described112. Briefly, both caudal and caput epididymis were dissected from > 5 wk old mice, with all excess fat and tissue trimmed away, and placed on top of filter mesh of 5 ml tubes with cell-strainer caps (BD) filled completely with PBS. After 5 min, the cap was carefully lifted out of the PBS to break the fluid surface tension, releasing plumes of sperm into the tube. This was repeated until sperm were no longer released after lifting out the cap. The cap (and tissue) was removed, and the tube sealed with parafilm. The sperm were pelleted in a swinging bucket rotor for 2 min at 4,000 g. The supernatant was aspirated down to ∼1 ml, and then gently resuspended by quick vortex pulses and incubated for 1 min at 60 °C. Inactivated sperm were mixed with Trypan Bule solution and placed on a hematocytometer for counting.
Spermatocyte chromosome spreads and immunostaining
Spreads were prepared as described with minor modifications113. When preparing a sequencing library and spreads from testes collected at the same time, testis cells were dissociated following the procedures in library preparation until passage through the 70 µm cell strainer (see above), then continued as described below. Briefly, decapsulated testis samples were placed in 2 ml TIM buffer (104 mM NaCl, 45 mM KCl, 1.2 mM MgSO4, 0.6 mM KH2PO4, 0.1% glucose, 6 mM sodium lactate, 1 mM sodium pyruvate, pH 7.3), with 200 µl collagenase (20 mg/ml), then shaken at 350 rpm for 15 min at 32 °C. After washing, tubules were resuspended in 2 ml TIM buffer with 200 µl trypsin solution (7 mg/ml) and 20 µl DNase I (400 µg/ml) and shaken for 15 min at 350 rpm at 32 °C. To stop trypsin digestion, 500 µl of FBS was added. Resuspended cells were filtered through a 70 µm cell strainer and TIM buffer was added to 15 ml. After three washes with DNase I, cells were resuspended in 10 ml of TIM buffer and distributed in 1 ml aliquots to Eppendorf tubes, centrifuged, and the supernatant was removed. The cell pellet was resuspended in 80 µl pre-warmed 0.1 M sucrose and incubated for 10 min. From the cell suspension, 40 µl was added to a Superfrost glass slide covered with 130 µl 1% PFA (with 0.1% Triton X-100, pH 9.2) in a humid chamber. Slides were kept in the closed humid chamber for at least 2 hr at 4 °C, then air dried and rinsed with 0.4% Photo-Flo (Kodak). After air drying, slides were wrapped in aluminum foil and stored at -80 °C.
For immunofluorescence, slides were blocked for 10 min with dilution buffer (0.2% BSA, 0.2% fish skin gelatin, 0.05% Tween-20 in 1× PBS) at room temperature. Using a humid chamber, slides were incubated with 100 µl primary antibody solution overnight at 4 °C. After washing, secondary antibody was added for 1 hr at 37 °C. After washing, slides were mounted with Vectashield containing DAPI (H-1000, Vector Laboratories). Primary and secondary antibodies are listed in Supplementary Data 2.
Histology
Testes and epididymides dissected from adult mice were fixed in Bouin’s fixative for 4 to 5 hr at room temperature, or in 4% PFA overnight at 4 °C. Bouin’s fixed testes were washed in 15 ml milli-Q water on a horizontal shaker for 1 hr at room temperature, followed by five 1-hr washes in 15 ml of 70% ethanol on a roller at 4 °C. PFA-fixed tissues were washed four times for 5 min in 15 ml milli-Q water at room temperature. Fixed tissues were stored in 70% ethanol before embedding in paraffin and sectioning at 5 μm. The tissue sections were deparaffinized with EZPrep buffer (Ventana Medical Systems). Antigen retrieval was performed with CC1 buffer (Ventana Medical Systems). Sections were blocked for 30 min with Background Buster solution (Innovex), and then avidin-biotin blocked for 8 min (Ventana Medical Systems). Hematoxylin and eosin (H&E) staining and immunohistochemical TUNEL assays were performed by the MSK Molecular Cytology Core Facility using the Autostainer XL (Leica Microsystems, Wetzlar, Germany) automated stainer for H&E or PAS with hematoxylin counterstain, and using the Discovery XT processor (Ventana Medical Systems, Oro Valley, Arizona) for TUNEL. Testis sections were incubated with anti-DDX4 or anti-MRE11 for 5 hr, followed by 60 min incubation with biotinylated goat anti-rabbit (Vector Labs) at 1:200 dilution (Supplementary Data 2). Detection was performed with DAB detection kit (Ventana Medical Systems) according to manufacturer’s instructions. Slides were counterstained with hematoxylin and coverslips were mounted with Permount (Fisher Scientific).
Image acquisition and analysis
Images of spread spermatocytes were acquired on a Zeiss Axio Observer Z1 Marianas Workstation, equipped with an ORCA-Flash 4.0 camera, illuminated by an X-Cite 120 PC-Q light source, with 100× 1.4 NA oil immersion objective. Marianas Slidebook (Intelligent Imaging Innovations, Denver Colorado; version 5.0) software was used for acquisition. Whole histology slides were scanned and digitized with the Panoramic Flash Slide Scanner (3DHistech, Budapest, Hungary; version 3.2.0) with a 20× 0.8 NA objective (Carl Zeiss, Jena, Germany) and processed in CaseViewer (version 2.4). High-resolution histology images were acquired with a Zeiss Axio Imager (version 3.2) microscope using a 63× 1.4 NA oil immersion objective (Carl Zeiss, Jena, Germany). All images were processed in Fiji (version 1.54 g)114. To quantify foci of RPA, DMC1, or RAD51, staging of spermatocytes was assessed by SYCP3 staining (% of axes synapsed) and only foci colocalizing with chromosome axis (SYCP3-signal) were manually counted. Statistical analysis was done with Graphpad Prism (version 9 or 10).
Structured illumination microscopy
Spermatocytes from juvenile Nbs1∆B homozygous and heterozygous littermates (13–15 dpp) were prepared for surface spreading and subsequent immunofluorescence as described previously65,115. Briefly, testes were dissected and placed in 1× PBS, pH 7.4 at room temperature before removal of the tunica albuginea. Seminiferous tubules were incubated in hypotonic extraction buffer (30 mM Tris-HCl pH 8.2, 50 mM sucrose, 17 mM trisodium citrate dihydrate, 5 mM EDTA, 2.5 mM DTT, 1 mM PMSF) for 20 min at room temperature. A homogeneous cell suspension was made in 100 mM sucrose and fixed on slides covered with a 1% PFA solution (pH 9.2) containing 0.15% Triton X-100 for at least 3 hr in a humid chamber. Slides were either used for immunofluorescence staining immediately or stored at –80 °C. For immunofluorescence, slides were blocked for 30 min at room temperature in PBST (1x phosphate-buffered saline (PBS) containing 0.1% Triton X-100) containing 3% nonfat milk. Slides were incubated with primary antibodies overnight in a humid chamber at 37 °C. Slides were washed 3 × 10 min in PBST, then incubated with secondary antibody for 1 hr at 37 °C in a humid chamber. Both primary and secondary antibodies were diluted in PBST containing 3% nonfat milk. After secondary antibody incubation, slides were washed 3 × 10 min in the dark with PBST and the slides were mounted with VECTASHIELD mounting medium containing DAPI (H-1000, Vector Laboratories).
Structured illumination microscopy (3D-SIM) was performed at the Bio-Imaging Resource Center in Rockefeller University using an OMX Blaze 3D-SIM super-resolution microscope (Applied Precision), equipped with 405 nm, 488 nm and 568 nm lasers, and 100× 1.40 NA UPLSAPO oil objective (Olympus). Image stacks of several μm thickness were taken with 0.125 μm optical section spacing and were reconstructed in Deltavision softWoRx 7.0.0 software with a Wiener filter of 0.002 using wavelength specific experimentally determined OTF functions. Maximum intensity projection images were acquired in Deltavision softWoRx 7.0.0 software. Data presented in this study were pooled from two independent staining experiments. To ensure correct alignment of fluorescent channels in all SIM experiments, multi-color fluorescent beads (TetraSpeck microspheres, 100 nm, Life Technologies, T7279) were imaged and used to correct shifts from passage of different fluorescence wave lengths through the optical path, as previously described65. SIM image analyzes were as previously described65.
Immunoblotting
About one million dissociated testis cells were boiled with NuPAGE LDS Sample Buffer (Life Technologies) and separated on 3%–8% Tris-acetate NuPAGE precast gels (Life Technologies) at 150 V for 70 min. Proteins were transferred to polyvinylidene difluoride (PVDF) membranes by wet transfer method in Tris-glycine-20% methanol, at 120 V for 40 min at 4 °C. Membranes were blocked with 5% non-fat milk in 1 × Tris buffered saline, 0.1% Tween (TBS-T) for 30 min at room temperature on an orbital shaker. Blocked membranes were incubated with primary antibodies overnight at 4 °C. Membranes were washed with TBS-T for 30 min at room temperature, then incubated with HRP-conjugated secondary antibodies for 1 hr at room temperature. Membranes were washed with TBS-T for 30 min and the signal was developed by ECL Prime (GE Healthcare) and scanned with the Image Lab (Bio-Rad; version 6.1.0 build 7). Primary and secondary antibodies are listed in Supplementary Data 2.
SPO11-oligo complexes and denaturing PAGE
Because MRE11 deficiency appears to reduce or eliminate ATM activation in response to meiotic DSBs, we considered the Atm–/– mutant to be the most appropriate comparison for analysis of the sizes of SPO11 oligos in Mre11-cKO. Moreover, the greatly elevated numbers of SPO11-oligo complexes in Atm–/–48 and Mre11-cKO (this study) allow us to measure SPO11 oligo sizes using samples from just two mice per genotype. By contrast, analysis of wild type requires many more mice48. To minimize unnecessary use of animals, we therefore decided to forego repeating a wild-type control (which has been shown previously8,48,49) in this study.
We purified SPO11-oligo complexes from testes of adults ( > 5 wk old) as previously described113. Briefly, testes were decapsulated, flash frozen in liquid nitrogen and stored at –80 °C. Testes were homogenized and extracts were cleared by ultracentrifugation. SPO11 was then immunoprecipitated with mouse monoclonal anti-SPO11-antibody 180 and protein A-agarose beads. SPO11 was eluted in SDS-containing buffer then subjected to a second round of immunoprecipitation with the same antibody. The material from the second immunoprecipitation was radiolabeled with terminal deoxynucleotidyl transferase and [α-32P]-CTP then eluted with Laemmli sample buffer and separated by SDS-PAGE. Proteins were transferred to PVDF membrane. Alternatively, the size distribution of SPO11 oligos purified from two adult Atm-KO or Mre11-cKO mice was determined by protease digestion followed by radiolabeling with [α-32P]-GTP followed by electrophoresis on a 15% denaturing polyacrylamide gel, which was then dried on filter paper as previously described116. Radio-labeled species were detected with Typhoon phosphor imager (version 4.0.0.4) after 48 hr exposure with Fuji phosphor screens, then quantified in Fiji (version 1.54 g)114.
Cell culture
Mouse embryonic fibroblasts (MEFs) were generated from E13.5 un-sexed mouse embryos and cultured as described23. Mitotic index was quantified using early passage primary MEFs by measuring Ser10 phosphorylation of histone H3 by flow cytometry 1 hr after 3 Gy of IR exposure. Cells were treated with 10 μM of ATM inhibitor (KU55933, 11855, Sigma) 30 min before IR exposure as an ATM deficient control. Two independent MEF cell lines for either wild type or Rad50-D69Y were tested. For the colony formation assay, cells were immortalized as described23, then plated with different doses of CPT for 24 hr and washed and cultured in drug-free media. For ATR inhibition, cells were treated with 50 nM of ATR inhibitor (VE822, S7102, Selleckchem) 2 hr before CPT treatment. After 10 days of growth, colonies were visualized by crystal violet stain (0.5% crystal violet, 25% methanol).
Statistics and reproducibility
For immunoblotting and histology, experiments were performed with two independent biological replicates unless otherwise indicated. We selected and included representative images of immunoblots and histology sections in this paper. For labeling of SPO11-oligo complexes, experiments were performed twice independently, and both results were included either in main figures or in Supplementary Figs. The separation of SPO11 oligos by the denaturing PAGE was performed once. For immunostaining and SIM experiments, multiple images from two independent animal samples were captured and used for analysis. The exact number of cells in each quantification was indicated in figure legends.
When experiments were performed with two biological replicates, the quantification was plotted with mean ± range. In cases with more than three data points, error bars indicated the standard deviation.
Final figures were assembled using Adobe illustrator 2023 or 2024.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Raw and processed sequencing S1-seq, Exo7/T-seq, MRE11-ChIP, and SSDS data have been deposited in the Gene Expression Omnibus (GEO) repository under accession numbers GSE266258, GSE266151, and GSE266271. We used additional S1-seq and Exo7/T-seq data from GEO accession numbers GSE265863 and GSE22945034,117. We used SPO11-oligo data from GEO accession numbers GSE8468937 and Atm–/– MRE11 ChIP-seq data from GEO accession GSE13891514. Code used for read processing and mapping is available online at https://github.com/yamadas2/mouse-S1seq, which also includes scripts for generating mean profiles and heatmaps around SPO11-oligo hotspot centers, and scatter plots to check correlation with SPO11-oligo maps and reproducibility between maps as previously described34. The mouse genome assembly mm10 (also known as GRCm38) is available at https://www.ncbi.nlm.nih.gov/datasets/genome/GCF_000001635.20/. Source data are provided with this paper.
References
Bergerat, A. et al. An atypical topoisomerase II from Archaea with implications for meiotic recombination. Nature 386, 414–417 (1997).
Keeney, S., Giroux, C. N. & Kleckner, N. Meiosis-specific DNA double-strand breaks are catalyzed by Spo11, a member of a widely conserved protein family. Cell 88, 375–384 (1997).
Cejka, P. & Symington, L. S. DNA end resection: mechanism and control. Annu Rev. Genet 55, 285–307 (2021).
Mimitou, E. P., Yamada, S. & Keeney, S. A global view of meiotic double-strand break end resection. Science 355, 40–45 (2017).
Hartsuiker, E. et al. Ctp1CtIP and Rad32Mre11 nuclease activity are required for Rec12Spo11 removal, but Rec12Spo11 removal is dispensable for other MRN-dependent meiotic functions. Mol. Cell Biol. 29, 1671–1681 (2009).
Milman, N., Higuchi, E. & Smith, G. R. Meiotic DNA double-strand break repair requires two nucleases, MRN and Ctp1, to produce a single size class of Rec12 (Spo11)-oligonucleotide complexes. Mol. Cell Biol. 29, 5998–6005 (2009).
Moreau, S., Ferguson, J. R. & Symington, L. S. The nuclease activity of Mre11 is required for meiosis but not for mating type switching, end joining, or telomere maintenance. Mol. Cell Biol. 19, 556–566 (1999).
Neale, M. J., Pan, J. & Keeney, S. Endonucleolytic processing of covalent protein-linked DNA double-strand breaks. Nature 436, 1053–1057 (2005).
Cannavo, E. & Cejka, P. Sae2 promotes dsDNA endonuclease activity within Mre11-Rad50-Xrs2 to resect DNA breaks. Nature 514, 122–125 (2014).
Garcia, V., Phelps, S. E., Gray, S. & Neale, M. J. Bidirectional resection of DNA double-strand breaks by Mre11 and Exo1. Nature 479, 241–244 (2011).
Keelagher, R. E., Cotton, V. E., Goldman, A. S. & Borts, R. H. Separable roles for exonuclease I in meiotic DNA double-strand break repair. DNA Repair (Amst.) 10, 126–137 (2011).
Zakharyevich, K. et al. Temporally and biochemically distinct activities of Exo1 during meiosis: double-strand break resection and resolution of double Holliday junctions. Mol. Cell 40, 1001–1015 (2010).
Deshpande, R. A., Lee, J. H., Arora, S. & Paull, T. T. Nbs1 converts the human Mre11/Rad50 nuclease complex into an endo/exonuclease machine specific for protein-DNA Adducts. Mol. Cell 64, 593–606 (2016).
Paiano, J. et al. ATM and PRDM9 regulate SPO11-bound recombination intermediates during meiosis. Nat. Commun. 11, 857 (2020).
Yamada, S. et al. Molecular structures and mechanisms of DNA break processing in mouse meiosis. Genes Dev. 34, 806–818 (2020).
Luo, G. et al. Disruption of mRad50 causes embryonic stem cell lethality, abnormal embryonic development, and sensitivity to ionizing radiation. Proc. Natl Acad. Sci. USA 96, 7376–7381 (1999).
Xiao, Y. & Weaver, D. T. Conditional gene targeted deletion by Cre recombinase demonstrates the requirement for the double-strand break repair Mre11 protein in murine embryonic stem cells. Nucleic Acids Res 25, 2985–2991 (1997).
Zhu, J., Petersen, S., Tessarollo, L. & Nussenzweig, A. Targeted disruption of the Nijmegen breakage syndrome gene NBS1 leads to early embryonic lethality in mice. Curr. Biol. 11, 105–109 (2001).
Zhang, B., Tang, Z., Li, L. & Lu, L. Y. NBS1 is required for SPO11-linked DNA double-strand break repair in male meiosis. Cell Death Differ. 27, 2176–2190 (2020).
Liu, Y., Lin, Z., Yan, J., Zhang, X., and Tong, M. H. A Rad50-null mutation in mouse germ cells causes reduced DSB formation, abnormal DSB end resection and complete loss of germ cells. Development 151, https://doi.org/10.1242/dev.202312 (2024).
Theunissen, J. W. et al. Checkpoint failure and chromosomal instability without lymphomagenesis in Mre11(ATLD1/ATLD1) mice. Mol. Cell 12, 1511–1523 (2003).
Yu, Z. et al. The MRE11 GAR motif regulates DNA double-strand break processing and ATR activation. Cell Res 22, 305–320 (2012).
Bender, C. F. et al. Cancer predisposition and hematopoietic failure in Rad50(S/S) mice. Genes Dev. 16, 2237–2251 (2002).
Roset, R. et al. The Rad50 hook domain regulates DNA damage signaling and tumorigenesis. Genes Dev. 28, 451–462 (2014).
Kang, J., Bronson, R. T. & Xu, Y. Targeted disruption of NBS1 reveals its roles in mouse development and DNA repair. EMBO J. 21, 1447–1455 (2002).
Williams, B. R. et al. A murine model of Nijmegen breakage syndrome. Curr. Biol. 12, 648–653 (2002).
Difilippantonio, S. et al. Role of Nbs1 in the activation of the ATM kinase revealed in humanized mouse models. Nat. Cell Biol. 7, 675–685 (2005).
Stracker, T. H., Morales, M., Couto, S. S., Hussein, H. & Petrini, J. H. The carboxy terminus of NBS1 is required for induction of apoptosis by the MRE11 complex. Nature 447, 218–221 (2007).
Buis, J. et al. Mre11 nuclease activity has essential roles in DNA repair and genomic stability distinct from ATM activation. Cell 135, 85–96 (2008).
Schonhoff, S. E., Giel-Moloney, M. & Leiter, A. B. Neurogenin 3-expressing progenitor cells in the gastrointestinal tract differentiate into both endocrine and non-endocrine cell types. Dev. Biol. 270, 443–454 (2004).
Yoshida, S. et al. Neurogenin3 delineates the earliest stages of spermatogenesis in the mouse testis. Dev. Biol. 269, 447–458 (2004).
Zheng, K. & Wang, P. J. Blockade of pachytene piRNA biogenesis reveals a novel requirement for maintaining post-meiotic germline genome integrity. PLoS Genet 8, e1003038 (2012).
Yoshida, S. et al. The first round of mouse spermatogenesis is a distinctive program that lacks the self-renewing spermatogonia stage. Development 133, 1495–1505 (2006).
Kim, S., Yamada, S., Maekawa, K., and Keeney, S. Optimized methods for mapping DNA double-strand-break ends and resection tracts and application to meiotic recombination in mouse spermatocytes. bioRxiv, https://doi.org/10.1101/2024.08.10.606181 (2024).
Canela, A. et al. Genome organization drives chromosome fragility. Cell 170, 507–521.e518 (2017).
Canela, A. et al. DNA breaks and end resection measured genome-wide by end sequencing. Mol. Cell 63, 898–911 (2016).
Lange, J. et al. The landscape of mouse meiotic double-strand break formation, processing, and repair. Cell 167, 695–708.e616 (2016).
Smagulova, F. et al. Genome-wide analysis reveals novel molecular features of mouse recombination hotspots. Nature 472, 375–378 (2011).
Claeys Bouuaert, C. et al. Structural and functional characterization of the Spo11 core complex. Nat. Struct. Mol. Biol. 28, 92–102 (2021).
Mimitou, E. P. & Keeney, S. S1-seq assay for mapping processed DNA ends. Methods Enzymol. 601, 309–330 (2018).
Stewart, G. S. et al. The DNA double-strand break repair gene hMRE11 is mutated in individuals with an ataxia-telangiectasia-like disorder. Cell 99, 577–587 (1999).
Taylor, A. M., Groom, A. & Byrd, P. J. Ataxia-telangiectasia-like disorder (ATLD)-its clinical presentation and molecular basis. DNA Repair (Amst.) 3, 1219–1225 (2004).
Uziel, T. et al. Requirement of the MRN complex for ATM activation by DNA damage. EMBO J. 22, 5612–5621 (2003).
Lukaszewicz, A., Lange, J., Keeney, S. & Jasin, M. Control of meiotic double-strand-break formation by ATM: local and global views. Cell Cycle 17, 1155–1172 (2018).
Johnson, D. et al. Concerted cutting by Spo11 illuminates meiotic DNA break mechanics. Nature 594, 572–576 (2021).
Lukaszewicz, A., Lange, J., Keeney, S. & Jasin, M. De novo deletions and duplications at recombination hotspots in mouse germlines. Cell 184, 5970–5984.e5918 (2021).
Prieler, S. et al. Spo11 generates gaps through concerted cuts at sites of topological stress. Nature 594, 577–582 (2021).
Lange, J. et al. ATM controls meiotic double-strand-break formation. Nature 479, 237–240 (2011).
Pan, J. & Keeney, S. Detection of SPO11-oligonucleotide complexes from mouse testes. Methods Mol. Biol. 557, 197–207 (2009).
Yu, Y. et al. Cryo-EM structures of the Spo11 core complex bound to DNA. Nat. Struct. Mol. Bio. 32, 113–124 (2025).
Toyooka, Y. et al. Expression and intracellular localization of mouse Vasa-homologue protein during germ cell development. Mech. Dev. 93, 139–149 (2000).
Moens, P. B. et al. The time course and chromosomal localization of recombination-related proteins at meiosis in the mouse are compatible with models that can resolve the early DNA-DNA interactions without reciprocal recombination. J. Cell Sci. 115, 1611–1622 (2002).
Arthur, L. M. et al. Structural and functional analysis of Mre11-3. Nucleic Acids Res 32, 1886–1893 (2004).
Lee, J. H. et al. Ataxia telangiectasia-mutated (ATM) kinase activity is regulated by ATP-driven conformational changes in the Mre11/Rad50/Nbs1 (MRN) complex. J. Biol. Chem. 288, 12840–12851 (2013).
Hoa, N. N. et al. Mre11 is essential for the removal of lethal topoisomerase 2 covalent cleavage complexes. Mol. Cell 64, 580–592 (2016).
Farah, J. A., Cromie, G., Steiner, W. W. & Smith, G. R. A novel recombination pathway initiated by the Mre11/Rad50/Nbs1 complex eliminates palindromes during meiosis in Schizosaccharomyces pombe. Genetics 169, 1261–1274 (2005).
Limbo, O., Porter-Goff, M. E., Rhind, N. & Russell, P. Mre11 nuclease activity and Ctp1 regulate Chk1 activation by Rad3ATR and Tel1ATM checkpoint kinases at double-strand breaks. Mol. Cell Biol. 31, 573–583 (2011).
Clerici, M., Mantiero, D., Lucchini, G. & Longhese, M. P. The Saccharomyces cerevisiae Sae2 protein negatively regulates DNA damage checkpoint signalling. EMBO Rep. 7, 212–218 (2006).
Stracker, T. H. & Petrini, J. H. The MRE11 complex: starting from the ends. Nat. Rev. Mol. Cell Biol. 12, 90–103 (2011). nrm3047 [pii].
Cherry, S. M. et al. The Mre11 complex influences DNA repair, synapsis, and crossing over in murine meiosis. Curr. Biol. 17, 373–378 (2007).
Lee, J. H. et al. Regulation of Mre11/Rad50 by Nbs1: effects on nucleotide-dependent DNA binding and association with ataxia-telangiectasia-like disorder mutant complexes. J. Biol. Chem. 278, 45171–45181 (2003).
Al-Ahmadie, H. et al. Synthetic lethality in ATM-deficient RAD50-mutant tumors underlies outlier response to cancer therapy. Cancer Discov. 4, 1014–1021 (2014).
Hohl, M. et al. Modeling cancer genomic data in yeast reveals selection against ATM function during tumorigenesis. PLoS Genet 16, e1008422 (2020).
Ziv, Y. et al. Chromatin relaxation in response to DNA double-strand breaks is modulated by a novel ATM- and KAP-1 dependent pathway. Nat. Cell Biol. 8, 870–876 (2006).
Hinch, A. G. et al. The Configuration of RPA, RAD51, and DMC1 binding in meiosis reveals the nature of critical recombination intermediates. Mol. Cell 79, 689–701.e610 (2020).
Khil, P. P., Smagulova, F., Brick, K. M., Camerini-Otero, R. D. & Petukhova, G. V. Sensitive mapping of recombination hotspots using sequencing-based detection of ssDNA. Genome Res 22, 957–965 (2012).
Brown, M. S., Grubb, J., Zhang, A., Rust, M. J. & Bishop, D. K. Small Rad51 and Dmc1 complexes often co-occupy both ends of a meiotic DNA double strand break. PLoS Genet 11, e1005653 (2015).
Slotman, J. A. et al. Super-resolution imaging of RAD51 and DMC1 in DNA repair foci reveals dynamic distribution patterns in meiotic prophase. PLoS Genet 16, e1008595 (2020).
Nicolette, M. L. et al. Mre11-Rad50-Xrs2 and Sae2 promote 5’ strand resection of DNA double-strand breaks. Nat. Struct. Mol. Biol. 17, 1478–1485 (2010).
Wang, W. et al. A DNA nick at Ku-blocked double-strand break ends serves as an entry site for exonuclease 1 (Exo1) or Sgs1-Dna2 in long-range DNA end resection. J. Biol. Chem. 293, 17061–17069 (2018).
Gobbini, E., et al. The MRX complex regulates Exo1 resection activity by altering DNA end structure. EMBO J 37. https://doi.org/10.15252/embj.201798588 (2018).
Shim, E. Y. et al. Saccharomyces cerevisiae Mre11/Rad50/Xrs2 and Ku proteins regulate association of Exo1 and Dna2 with DNA breaks. EMBO J. 29, 3370–3380 (2010).
Rein, K. et al. EXO1 is critical for embryogenesis and the DNA damage response in mice with a hypomorphic Nbs1 allele. Nucleic Acids Res 43, 7371–7387 (2015).
Zhao, X., Zhang, Y., Wilkins, K., Edelmann, W. & Usdin, K. MutLgamma promotes repeat expansion in a Fragile X mouse model while EXO1 is protective. PLoS Genet 14, e1007719 (2018).
Wang, S. et al. Role of EXO1 nuclease activity in genome maintenance, the immune response and tumor suppression in Exo1D173A mice. Nucleic Acids Res 50, 8093–8106 (2022).
Morales, M. et al. The Rad50S allele promotes ATM-dependent DNA damage responses and suppresses ATM deficiency: implications for the Mre11 complex as a DNA damage sensor. Genes Dev. 19, 3043–3054 (2005).
Pacheco, S. et al. The ATM signaling cascade promotes recombination-dependent pachytene arrest in mouse spermatocytes. PLoS Genet 11, e1005017 (2015).
Anand, R., Ranjha, L., Cannavo, E. & Cejka, P. Phosphorylated CtIP Functions as a Co-factor of the MRE11-RAD50-NBS1 Endonuclease in DNA End Resection. Mol. Cell 64, 940–950 (2016).
Deshpande, R. A. et al. DNA-dependent protein kinase promotes DNA end processing by MRN and CtIP. Sci. Adv. 6, eaay0922 (2020).
Reginato, G., Cannavo, E. & Cejka, P. Physiological protein blocks direct the Mre11-Rad50-Xrs2 and Sae2 nuclease complex to initiate DNA end resection. Genes Dev. 31, 2325–2330 (2017).
Sartori, A. A. et al. Human CtIP promotes DNA end resection. Nature 450, 509–514 (2007).
Myler, L. R. et al. Single-molecule imaging reveals how Mre11-Rad50-Nbs1 initiates DNA break repair. Mol. Cell 67, 891–898.e894 (2017).
Kissling, V. M. et al. Mre11-Rad50 oligomerization promotes DNA double-strand break repair. Nat. Commun. 13, 2374 (2022).
Gnugge, R., Reginato, G., Cejka, P. & Symington, L. S. Sequence and chromatin features guide DNA double-strand break resection initiation. Mol. Cell 83, 1237–1250.e1215 (2023).
Cannavo, E., Reginato, G. & Cejka, P. Stepwise 5’ DNA end-specific resection of DNA breaks by the Mre11-Rad50-Xrs2 and Sae2 nuclease ensemble. Proc. Natl Acad. Sci. USA 116, 5505–5513 (2019).
Cannavo, E., Cejka, P. & Kowalczykowski, S. C. Relationship of DNA degradation by Saccharomyces cerevisiae exonuclease 1 and its stimulation by RPA and Mre11-Rad50-Xrs2 to DNA end resection. Proc. Natl Acad. Sci. USA 110, E1661–E1668 (2013).
Cejka, P. et al. DNA end resection by Dna2-Sgs1-RPA and its stimulation by Top3-Rmi1 and Mre11-Rad50-Xrs2. Nature 467, 112–116 (2010).
Nimonkar, A. V. et al. BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev. 25, 350–362 (2011).
Niu, H. et al. Mechanism of the ATP-dependent DNA end-resection machinery from Saccharomyces cerevisiae. Nature 467, 108–111 (2010).
Ceppi, I. et al. PLK1 regulates CtIP and DNA2 interplay in long-range DNA end resection. Genes Dev. 37, 119–135 (2023).
Ceppi, I. et al. CtIP promotes the motor activity of DNA2 to accelerate long-range DNA end resection. Proc. Natl Acad. Sci. USA 117, 8859–8869 (2020).
Kauppi, L. et al. Numerical constraints and feedback control of double-strand breaks in mouse meiosis. Genes Dev. 27, 873–886 (2013).
Alani, E., Padmore, R. & Kleckner, N. Analysis of wild-type and rad50 mutants of yeast suggests an intimate relationship between meiotic chromosome synapsis and recombination. Cell 61, 419–436 (1990).
McKee, A. H. & Kleckner, N. A general method for identifying recessive diploid-specific mutations in Saccharomyces cerevisiae, its application to the isolation of mutants blocked at intermediate stages of meiotic prophase and characterization of a new gene SAE2. Genetics 146, 797–816 (1997).
Prinz, S., Amon, A. & Klein, F. Isolation of COM1, a new gene required to complete meiotic double-strand break-induced recombination in Saccharomyces cerevisiae. Genetics 146, 781–795 (1997).
Lam, I. & Keeney, S. Mechanism and regulation of meiotic recombination initiation. Cold Spring Harb. Perspect. Biol. 7, a016634 (2014).
Hua, S. B., Qiu, M., Chan, E., Zhu, L. & Luo, Y. Minimum length of sequence homology required for in vivo cloning by homologous recombination in yeast. Plasmid 38, 91–96 (1997).
Inbar, O., Liefshitz, B., Bitan, G. & Kupiec, M. The relationship between homology length and crossing over during the repair of a broken chromosome. J. Biol. Chem. 275, 30833–30838 (2000).
Ira, G. & Haber, J. E. Characterization of RAD51-independent break-induced replication that acts preferentially with short homologous sequences. Mol. Cell Biol. 22, 6384–6392 (2002).
Jinks-Robertson, S., Michelitch, M. & Ramcharan, S. Substrate length requirements for efficient mitotic recombination in Saccharomyces cerevisiae. Mol. Cell Biol. 13, 3937–3950 (1993).
Manivasakam, P., Weber, S. C., McElver, J. & Schiestl, R. H. Micro-homology mediated PCR targeting in Saccharomyces cerevisiae. Nucleic Acids Res 23, 2799–2800 (1995).
Huang, P. C. et al. Meiotic DNA break resection and recombination rely on chromatin remodeler Fun30. EMBO J 44, 200–224 (2025).
Bothmer, A. et al. Mechanism of DNA resection during intrachromosomal recombination and immunoglobulin class switching. J. Exp. Med 210, 115–123 (2013).
Reczek, C. R. et al. The DNA resection protein CtIP promotes mammary tumorigenesis. Oncotarget 7, 32172–32183 (2016).
Barlow, C. et al. Atm-deficient mice: a paradigm of ataxia telangiectasia. Cell 86, 159–171 (1996).
Baudat, F., Manova, K., Yuen, J. P., Jasin, M. & Keeney, S. Chromosome synapsis defects and sexually dimorphic meiotic progression in mice lacking Spo11. Mol. Cell 6, 989–998 (2000).
Maekawa, K., Yamada, S., Sharma, R., Chaudhuri, J. & Keeney, S. Triple-helix potential of the mouse genome. Proc. Natl Acad. Sci. USA 119, e2203967119 (2022).
Canela, A. et al. Topoisomerase ii-induced chromosome breakage and translocation is determined by chromosome architecture and transcriptional activity. Mol. Cell 75, 252–266.e258 (2019).
Langmead, B., Trapnell, C., Pop, M. & Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009).
Wienert, B., Wyman, S. K., Yeh, C. D., Conklin, B. R. & Corn, J. E. CRISPR off-target detection with DISCOVER-seq. Nat. Protoc. 15, 1775–1799 (2020).
Brick, K., Pratto, F., Sun, C. Y., Camerini-Otero, R. D. & Petukhova, G. Analysis of meiotic double-strand break initiation in mammals. Methods Enzymol. 601, 391–418 (2018).
Peterson, S. E., Keeney, S. & Jasin, M. Mechanistic insight into crossing over during mouse meiosis. Mol. Cell 78, 1252–1263.e1253 (2020).
Boekhout, M. et al. REC114 partner ANKRD31 controls number, timing, and location of meiotic DNA breaks. Mol. Cell 74, 1053–1068.e1058 (2019).
Schindelin, J. et al. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012).
Peters, A. H., Plug, A. W., van Vugt, M. J. & de Boer, P. A drying-down technique for the spreading of mammalian meiocytes from the male and female germline. Chromosome Res 5, 66–68 (1997).
Lam, I., Mohibullah, N. & Keeney, S. Sequencing Spo11 oligonucleotides for mapping meiotic DNA double-strand breaks in yeast. Methods Mol. Biol. 1471, 51–98 (2017).
Xu, J., Li, T., Kim, S., Boekhout, M. & Keeney, S. Essential roles of the ANKRD31-REC114 interaction in meiotic recombination and mouse spermatogenesis. Proc. Natl Acad. Sci. USA 120, e2310951120 (2023).
Acknowledgements
This article is subject to the Open Access to Publications policy of the Howard Hughes Medical Institute (HHMI). HHMI lab heads have previously granted a nonexclusive CC BY 4.0 license to the public and a sublicensable license to HHMI in their research articles. Pursuant to those licenses, the author-accepted manuscript of this article can be made freely available under a CC BY 4.0 license immediately upon publication. We thank W. Edelmann (Albert Einstein College of Medicine, New York, NY), R. Baer (Columbia University, New York, NY), Francesca Cole (MD Anderson Cancer Center, Houston, TX) and D. O. Ferguson (University of Michigan School of Medicine, Ann Arbor, MI) for generously sharing mouse lines. We thank A. Lukaszewicz (Univ. Michigan) and M. Jasin (MSK) for discussion. We thank the MSK Molecular Cytology core facility (N. Fan and M. Pulina) for histology; the MSK Integrated Genomics Operation (IGO) for sequencing; and the MSK Mouse Genetics Colony Management Group for assistance with mouse husbandry. MSK core facilities are supported by National Cancer Institute cancer center support grant P30 CA08748. The IGO was further funded by the Cycle for Survival and the Marie-Josée and Henry R. Kravis Center for Molecular Oncology. We thank M. Arter, E. Suranyi, H. Murakami, A. Shabro, V. Macera, and other members of the Keeney laboratory for discussions and experimental advice. We thank Cristina Madrid-Sandín (Universitat Autònoma de Barcelona) for assistance in generating one of the RPA2 SSDS maps from wild type. This work was supported by NIH grants R35 GM118092 (to S. Keeney), R01 GM59413 (to J.H.J.P.), R35 GM136278 (to J.H.J.P.), and the Brain Pool Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (2022H1D3A2A01096332 to S. Kim).
Author information
Authors and Affiliations
Contributions
S. Keeney and S. Kim conceived the project. S. Kim, S.Y., T.L., J.H.K., M.M., J.X., and S. Keeney designed experiments. Specific experiments were performed as follows: Fig. 3e–g, J.X.; Fig. 5e-h and Supplementary Fig. 5e–i, M.M.; SIM and analysis (Fig. 6c, d and Supplementary Fig. 6b, c), T.L.; Supplementary Fig. 5a–c, J.H.K. All other experiments were performed by S. Kim. DNA sequencing data were analyzed by S. Kim and S.Y. S. Keeney contributed to analysis of all data. Some experimental animals were provided by C.C., D.Y.E., and L.F. S. Keeney, S. Kim, and J.H.J.P. supervised the research and secured funding. S. Kim and S. Keeney wrote the paper. All authors edited the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Akira Shinohara and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kim, S., Yamada, S., Li, T. et al. Mouse MRE11-RAD50-NBS1 is needed to start and extend meiotic DNA end resection. Nat Commun 16, 3613 (2025). https://doi.org/10.1038/s41467-025-57928-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-57928-x
This article is cited by
-
EXO1 promotes the meiotic MLH1-MLH3 endonuclease through conserved interactions with MLH1, MSH4 and DNA
Nature Communications (2025)
