
Abstract
The employment of self-assembled molecular hybrid could improve buried interface in perovskite solar cells (PSCs). However, the interplay among hybrid self-assembled monolayers (SAMs) during the deposition process has not been well-studied. Herein, we study the interaction between co-adsorbents and commonly used SAM material, [4-(3,6-dimethyl-9H-carbazol-9-yl)butyl]phosphonic acid (Me-4PACz) for wide-bandgap (WBG) PSCs. It is found that the co-adsorbent, 6-aminohexane-1-sulfonic acid (SA) tends to fill the uncovered sites without interference with Me-4PACz, ensuring the formation of a dense hole selective layer. Moreover, the use of SA/Me-4PACz mixed SAMs could effectively reduce the interfacial non-radiative recombination loss, optimize the energy alignment at the buried interface and regulate the crystallization of WBG perovskite. As a result, the 1.77 eV WBG PSCs deliver a power conversion efficiency (PCE) of 20.67% (20.21% certified) and an impressive open-circuit voltage (VOC) of 1.332 V (1.313 V certified). By combining with a 1.26 eV narrow-bandgap (NBG) PSC, we further fabricate 2-terminal all-perovskite tandem solar cells (TSCs) with a PCE of 28.94% (28.78% certified) for 0.087 cm2 and 23.92% for mini-module with an aperture area of 11.3 cm2.
Similar content being viewed by others


Introduction
All-perovskite tandem solar cell (TSC), consisting of a narrow-bandgap (NBG) bottom subcell and a wide-bandgap (WBG) top subcell, has exhibited great ability to significantly minimize the optical loss and adequately harvest the photons in solar spectrum to break the Shockley-Queisser limit, giving a theoretical efficiency over 43%1,2. Taking advantages of the tremendous efforts on addressing the fast crystallization process and undesired oxidation of Sn2+ issues in NBG bottom subcells, the efficiencies of TSCs have been improved to 30.1%3. However, WBG perovskite top subcells are still facing issues of large open-circuit voltage (VOC) deficit (defined by \({E}_{{{{\rm{g}}}}}/{{{\rm{q}}}}-{V}_{{{{\rm{OC}}}}}\)) induced by the interfacial defects-mediated non-radiative charge recombination and inefficient charge extraction induced by the mismatched energy level alignment at charge transport layer/perovskite interface, limiting the further efficiency improvement of TSCs4,5.
To date, most of the interface engineering studies are mainly focus on the passivation of top surface defects for WBG perovskite films to improve the device performance of TSCs6,7. Beyond that, stabilizing the buried interface is equally important, which is, however, much more challenging. Recently, regulating the self-assembled monolayers (SAMs)/perovskite interface has been demonstrated to be an effective strategy to reduce the interfacial energy loss of single junction inverted perovskite solar cells (PSCs)8,9,10,11,12,13,14,15. However, achieving a dense coverage of SAMs on the substrate remains challenging. This is mainly due to the amphiphilic nature of self-assembled molecules, making them tend to form micellar nanoparticles in solution, thus leading to the formation of clusters and incomplete coverage of the substrate during the deposition process11,13. Thus, the deposition of compact and uniform SAMs largely contributes to the reduced VOC deficit16,17. Introducing co-adsorbents to form mixed SAMs is an effective method to achieve dense SAMs18,19. However, the interplay between the main self-assembled molecules and the co-adsorbents has not been fully studied.
Here, we studied co-adsorbents based on bifunctional alkylamine with different anchoring groups, including (6-aminohexyl)-phosphonic acid (PA), 7-aminoheptanoic acid (CA), and 6-aminohexane-1-sulfonic acid (SA), together with [4-(3,6-dimethyl-9H-carbazol-9-yl)butyl]phosphonic acid (Me-4PACz) to construct NiOx/SAMs bilayer hole selective layers for 1.77 eV WBG PSCs. We conducted molecular dynamics (MD) simulations to study the competitive adsorption behavior of co-adsorbents with Me-4PACz. It is found that PA shows an unexpected competitive deposition behavior against Me-4PACz during the co-adsorption process. Whereas the competitive adsorption capacity of CA against Me-4PACz is much weaker, and a best co-adsorption mode is achieved by using SA as co-adsorbent together with Me-4PACz. The SA/Me-4PACz co-adsorption strategy could not only reduce the interfacial non-radiative recombination loss, but also optimize the energy alignment at SAMs/perovskite interface and regulate the crystallization behavior of perovskite films. Moreover, we fabricated WBG devices with 1.77 eV, and SA co-adsorption devices achieved a champion power conversion efficiency (PCE) of 20.67% (20.21% certified), corresponding to an outstanding VOC of 1.332 V (1.313 V certified). We further attempted to integrate efficient WBG subcells to all-perovskite TSCs and obtained a champion PCE of 28.94% (28.78% certified) for 0.087 cm2 and 23.92% for mini-module with an aperture area of 11.3 cm2.
Results and discussion
The competitive adsorption of hybrid adsorbed molecules and interface properties
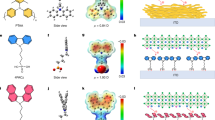
We first used density functional theory (DFT) to investigate the binding modes of the co-adsorbents PA, CA, and SA with NiOx and indium tin oxide (ITO) (Supplementary Fig. 1-2). Geometry optimizations showed that the co-adsorbents preferred lying on the NiOx surface, anchored with the acid groups rather than forming perpendicular contacts with the acid or amino terminus. The binding energies of PA, CA and SA in their most favored configurations are –3.90, –3.25 and –3.78 eV, respectively. It suggests that CA could form the weakest adsorption on NiOx while PA exhibits the strongest adsorption. To investigate interactions between the co-adsorbents and Me-4PACz during the co-adsorption process, we performed MD simulations in 1 ns at 373.15 K, using a mixture of PA/CA/SA with Me-4PACz according to the experimental molar ratio of 5:8 on a 10 nm × 10 nm NiOx surface. Supplementary Fig. 3 illustrates side views of the three substrate models established by MD simulations. The MD simulations for Me-4PACz without the co-adsorbents showed substantial agglomeration, resulting in a surface coverage of 0.68 (Fig. 1a) due to dimers formation (Supplementary Movie 1). The introduction of co-adsorbents increased the coverage of the substrates to 0.74–0.76 (Fig. 1b–d). We sampled the dimer structures of the co-adsorbents and Me-4PACz from the last MD snapshots (Supplementary Fig. 4). Both amino and acid groups of the co-adsorbents could bind with the phosphate group of Me-4PACz. The calculated binding energies indicated stronger interactions between the acid groups in the (Me-4PACz is abbreviated as Me in dimers) PA-Me (–0.61 eV), CA-Me (–0.58 eV), and SA-Me dimers (–0.98 eV) compared to the co-adsorbent dimers PA-PA (–0.40 eV), CA-CA (–0.27 eV), and SA-SA (–0.47 eV). These results suggested that the co-adsorbents tend to disperse and bind with the Me-4PACz. Moreover, we noted that the SA-Me binding energy (–0.98 eV) is larger than the Me-Me dimer (–0.75 eV), which could aid in breaking the micelles of Me-4PACz and reducing the SAMs defects.

a–d Top view of adsorption equilibrium of different types of mixed SAMs on ITO/NiOx surface simulated by molecular dynamics. Among them, purple, green, blue and pink represent Me-4PACz, PA, CA and SA molecules respectively. The NiOx surface coverage ratios are 0.68, 0.76, 0.74, and 0.75 for Me-4PACz, PA-Me-4PACz, CA-Me-4PACz, and SA-Me-4PACz hybrids. e–h XPS N 1 s spectra for control, PA, CA and SA samples, respectively. box plot of PCE (i) and VOC ( j) of 1.77 eV bandgap perovskite devices. k J-V curves of champion control, PA-, CA-, SA-treated devices at reverse scan. l J-V curves of champion SA-based device under forward and reverse scans.
The MD simulations for PA/Me-4PACz hybrid (Supplementary Movie 2) showed that PA can only bind with the neighboring Me-4PACz as the phosphate groups are strongly adsorbed on the NiOx surface, consistent with the adsorption energy calculations. Consequently, PA tended to compete with Me-4PACz for the adsorption site rather than facilitating the dispersion of Me-4PACz on NiOx surface. The CA/Me-4PACz hybrid (Supplementary Movie 3) showed rapid motions of CA on the NiOx surface through the Me-4PACz clusters, due to its relatively weak surface adsorption energy and intermolecular binding energies, potentially leading to desorption during Me-4PACz SAMs formation. The SA/Me-4PACz hybrid results (Supplementary Movie 4) indicated balanced surface and intermolecular interactions, allowing SA to first bind with the NiOx surface and then enhance Me-4PACz adsorption via the SA-Me dimers. We computed the molecular ratio between the co-adsorbents and Me-4PACz in the SAMs, defined by all molecules 5 Å above the NiOx surface. The PA:Me−4PACz, CA:Me-4PACz, and SA:Me-4PACz ratios are 0.65, 0.50, and 0.59, respectively (Supplementary Fig. 5). It suggests that SA could form a moderate mixture with Me-4PACz in the adsorbed molecular layer, while PA and CA could lead to excessive or insufficient mixing with the co-adsorbents, respectively.
Considering the incomplete coverage of NiOx nanoparticles on the ITO surface, we performed the same simulations on the ITO surface (Supplementary Fig. 6 and Supplementary Movies 5–8). In addition, we also considered the NiOx/ITO interface to study the co-adsorptions on the surface with imperfect deposition of the NiOx including exposed ITO area (Supplementary Fig. 7 and Supplementary Movies 9–12). It was shown that SA could assist Me-4PACz in filling the exposed ITO area to homogenize the adsorbed molecular layers. We also computed work functions to study how co-adsorbents could affect the hole-transportation performance of Me-4PACz (Supplementary Fig. 8). We found that the SA-Me dimer achieves better energy level alignment, which could facilitate the carrier transport and prolong the carrier lifetime.
To verify the predicted co-adsorbent interactions with Me-4PACz, we conducted X-ray photoelectron spectroscopy (XPS) measurements to study the chemical states of the co-adsorbed mixed adsorbed molecular layer. For ease of description, we denoted substrates based on Me-4PACz alone as control sample, and PA/Me-4PACz, CA/Me-4PACz, and SA/Me-4PACz substrates as PA, CA, and SA samples, respectively. For the control sample, the presence of Me-4PACz is evidenced by the appearance of the characteristic C–C peak and C–P peak in the XPS spectrum (Supplementary Fig. 9a). In the case of mixed SAMs, PA and CA are indicated by the enhanced C–P peak (Supplementary Fig. 9b) and the appeared O–C = O peak, respectively (Supplementary Fig. 9c)20. SA is denoted by the C 1 s (Supplementary Fig. 9d) and S 2p spectra (Supplementary Fig. 9e) compared with the control sample (Supplementary Fig. 9f). We further checked the N 1 s signals in XPS spectra (Fig. 1e–h), and found that PA, CA, and SA samples present both the N peak of carbazole and the N peak of amino group as compared to the control sample21. The ratio of Namino/Ncarbazole indicates the proportion of co-adsorbents in hybrid SAMs, where the PA shows the largest ratio and CA the lowest (Supplementary Fig. 10). These results are consistent with the predicted molecular ratios in the MD simulations.
To verify the predicted features of SA in enhancing the adsorption of Me-4PACz, we conducted a comparative analysis of the surface properties of ITO/NiOx, ITO/NiOx/Me-4PACz and ITO/NiOx/SA samples by analyzing the elements of O and Ni signals in XPS results. From the XPS spectra of O 1 s (Supplementary Fig. 11), notably, there is a pronounced shift in the energy bands for both ITO/NiOx/Me-4PACz and ITO/NiOx/SA samples compared to ITO/NiOx, with the latter one exhibiting more significant shifts. This indicates that SA molecules could enhance the binding capability of Me-4PACz to NiOx. Based on the XPS spectra of Ni 2p3/2 shown in Supplementary Fig. 12, we further analyzed the ratio of Ni3+/Ni2+. The ratio of Ni3+/Ni2+ in ITO/NiOx, ITO/NiOx/Me-4PACz and ITO/NiOx/SA is 0.98, 1.33 and 1.90, respectively. The increasing ratio indicates enhanced conductivity in ITO/NiOx/SA films22, which is beneficial for the hole transportation23.
To further investigate the dynamic adsorption process of mixed adsorbed molecules on the NiOx surface, we employed dynamic light scattering (DLS) to detect micelles in solutions. (Supplementary Fig. 13). The introduction of the co-adsorbents reduced the average hydrodynamic diameter of the micelles, indicating that the co-adsorbents indeed have inhibitory effects on the formation of Me-4PACz micelles13. Furthermore, we immersed the ITO/NiOx substrate in different mixed SAMs solutions and recorded the ultraviolet visible (UV-vis) absorption spectra of the samples at different time intervals (Supplementary Fig. 14). The absorbance intensity provides a reliable indication of the loading amount of Me-4PACz molecules on ITO/NiOx substrates24. It was observed that all of these mixed adsorbed molecules exhibit rapid adsorption in the first hour, after which the rate was gradually decreased due to the saturation of adsorption. Notably, CA/Me-4PACz and SA/Me-4PACz exhibited faster adsorption rates and higher loading amounts compared to Me-4PACz alone. This could be attributed to the surfactant-like behavior of CA/SA molecules, which reduce the formation of Me-4PACz micelles or clusters and increase the amount of monodispersed Me-4PACz molecules in solution that could be well adsorbed on the surface of the ITO/NiOx substrate11.
To investigate the influence of co-adsorbents on the interfacial properties of the hole transport layers (HTLs), we measured the contact angle of the mixed SAMs. As shown in Supplementary Fig. 15, the contact angles of control, PA, CA and SA substrates are 69.9°, 74.8°, 78.7°, and 80.9°, respectively. The increase of contact angles indicates that the introduction of three co-adsorbents does change the interface contact situation. We deduce that the increased contact angle enables more hydrophobic surfaces, which is mainly due to the better coverage of hybrid SAMs that reduces the exposure of highly hydrophilic groups on the ITO/NiOx surface22.
The change in interfacial properties should be correlated with the variation of surface potential. To verify this point, we also conducted Kelvin probe force microscopy (KPFM) characterizations. As shown in Supplementary Fig. 16, the introduction of co-adsorbents improves the uniformity of the surface potential, which can be ascribed to the elimination of bare NiOx or ITO regions and improved coverage by forming more compact and uniformly distributed mixed SAMs. Meanwhile, the decrease in contact potential difference (CPD) of SA sample could be beneficial for carrier extraction25.
Photovoltaic performance of WBG PSCs employing co-adsorption strategy
To study the influence of the mixed SAMs on the device performance, we fabricated WBG PSCs with configuration of ITO/NiOx/SAMs/FA0.8Cs0.2Pb(I0.6Br0.4)3/1,3-propanediamine dihydriodide (PDAI2)/C60/bathocuproine (BCP)/Ag based on the above mixed SAMs (Supplementary Fig. 17). Figure 1i-j and Supplementary Fig. 18-19 summarize the statistical photovoltaic performance of the devices. It is found that the mixed SAMs-based devices showed higher PCEs than the control case. Especially, SA-based devices gave the best PCEs. From the typical J-V curves of the devices, the improvement of PCE is mainly due to the increase in VOC and fill factor (FF) (Fig. 1k). Detailed photovoltaic parameters are shown in Supplementary Table 1. The external quantum efficiency (EQE) integrated short-circuit current density (JSC) is 17.42, 17.60, 17.64 and 18.06 mA cm−2 for the control, PA, CA and SA-based devices, respectively, which is well matched with the JSC extracted from the J-V scans (Supplementary Fig. 20). To further demonstrate that the device performance was not overestimated, we measured the steady-state output power at the maximum power point and the results are consistent with the J-V results (Supplementary Fig. 21). It should be mentioned that the SA-based device gave a champion PCE of 20.67% (20.50%) under reverse (forward) scan with a VOC of 1.332 V (1.331 V), a JSC of 18.29 mA cm−2 (18.21 mA cm−2), an FF of 84.84% (84.56%) (Fig. 1l). This PCE is the highest PCE reported for 1.77 eV WBG PSCs with an impressive VOC of 1.332 V (Supplementary Fig. 22 and Supplementary Table 2). We also sent one of the best devices to Shanghai Institute of Microsystems and Information Technology (SIMIT) for certification and achieved a certified PCE of 20.21% (Supplementary Fig. 23).
Characterization of WBG perovskite films and buried interface
To investigate the effect of interface modification on the nucleation and growth kinetics of perovskite, we used in-situ photoluminescence (PL) spectroscopy to monitor the crystallization behavior of perovskite on control and SA-based substrates during the stages of spin-coating and annealing (Supplementary Fig. 24). It was found that both samples exhibited initial fluorescence emission centered at 700 nm during the spin-coating process, but the fluorescence intensity was stronger for SA-based films (Supplementary Fig. 24a, b). This may be due to the good uniformity of the mixed adsorbed molecules and the interaction of the functional end groups of SA with perovskite precursors at the buried interface, likely providing nucleation sites for perovskite and increased the nucleation rate26. During the annealing process, we observed a sequential evolution of PL intensity, i.e., an initial rapid increase, followed by a decrease, and ultimately, a subsequent increase (Supplementary Fig. 24c-d). This observation implied a crystallization-dissolution-recrystallization dynamic occurring on the surface27. Importantly, the perovskite deposited on SA-based substrate had stronger PL intensity, which indicated a higher quality perovskite film28,29.
Although the perovskite films deposited on various mixed SAMs showed nearly identical optical absorption properties and bandgaps around 1.77 eV (Supplementary Fig. 25), the interfacial properties of the mixed SAMs substrates are considered to play an important role in the perovskite film quality and the device performance. To investigate this point, we conducted scanning electron microscopy (SEM) measurements to examine the perovskite surface morphology. As shown in Fig. 2a–d, numerous nanovoids are observed at the grain boundaries of the perovskite grown on the control substrate. Nevertheless, the voids are eliminated at the perovskite deposited on SA-based substrate. This is likely because the introduction of co-adsorbents with the amino terminal groups could interact with perovskite precursors and facilitate the heterogeneous nucleation, leading to the growth of compact perovskite films30. The cross-sectional SEM image of the perovskite deposited on SA-based substrate indicates grain penetration, which makes for longitudinal transport of carriers and diminished hysteresis caused by interfacial charge accumulation31.

a–d Top view and cross-sectional SEM images of perovskite films deposited on control, PA-, CA-, SA-based substrates, respectively. e, f The surface KPFM results and g, h AFM images of the perovskite films deposited on control and SA-based substrates, respectively. i–l PL mapping of the perovskite films deposited on control, PA-, CA-, and SA-based substrates, respectively.
To further study the effect of mixed SAMs-based substrates on the crystallinity of the deposited perovskite films, X-ray diffraction (XRD) measurements were conducted (Supplementary Fig. 26). It is found that the (100) diffraction peak of the perovskite film deposited on the SA-based substrate has the highest intensity and the lowest full width at half maximum (FWHM). These results show that the crystallinity of perovskite film deposited on SA-based substrate is significantly enhanced, which is consistent with the SEM results. These characterizations demonstrate that SA-based substrates help to fabricate perovskite films with better quality32. This point is further supported by KPFM results obtained on the surface of the perovskite films, which showed a more uniform CPD of the films deposited on the SA-based substrate compared to the control substrate (Fig. 2e, f). In addition, the atomic force microscope (AFM) results also confirm that the SA-based perovskite film showed a root-mean-square (RMS) roughness of 10.4 nm, lower than that of the control film (13.9 nm) (Fig. 2g, h). The flatter film surface of the SA sample could contribute to the reduction in leakage currents and interfacial recombination33.
To study the influence of the residual stress of the films, grazing incidence X-ray diffraction (GIXRD) was performed (Supplementary Fig. 27). As the incident angle increased from 5° to 45° with a penetration depth of 200 nm, the peak of 2θ = 32.4° in perovskite deposited on control substrate was gradually shifted to a smaller angle, which could be due to the vacancy defects at the bottom of the film. By contrast, the peak shift of perovskite deposited on SA-based substrate is almost negligible at increasing angles of incidence, which may be attributed to the amino groups introduced by SA molecules passivating the uncoordinated Pb2+ at the bottom, thereby reducing or eliminating the stress of the film. This is favourable for the performance and stability of PSCs34.
Due to the direct contact with mixed SAMs, the quality of the perovskite at the bottom interface should be affected16. We thus used a scatheless peel-off technique to obtain exposed bottom surface of perovskite deposited on various substrates for grazing-incidence wide-angle X-ray scattering (GIWAXS) and PL mapping characterization measurements to further investigate this point35. All GIWAXS scattering results showed no change in the preferential orientation of the perovskite crystals (Supplementary Fig. 28). However, the scattering intensity of the (100) peak of the perovskite film deposited on Me-4PACz was significantly lower than three mixed SAMs cases. Remarkably, the perovskite film on SA-based substrate exhibited the strongest intensity in the (100) peak, indicating the improvement of the crystal crystallinity and the reduction of amorphous phases of the perovskite films at the buried region. This is beneficial for inhibiting the migration of perovskite ions and reduce the defect density34,36. PL mapping results (Fig. 2i–l) showed that the SA-based perovskite film exhibits an improved uniformity when compared with the control one. Moreover, the SA sample showed higher PL intensity, suggesting that the non-radiative recombination at the bottom interface is suppressed. This is likely due to the improved perovskite quality and the passivation effect of the mixed SAMs37.
In addition to the quality of perovskite, the modification of ITO/NiOx with SAMs should also affect the charge transport at the buried interface. We thus further carried out ultraviolet photoelectron spectroscopy (UPS) measurement to assess the energy level arrangement of the prepared HTL. Compared to the control sample, the valence band maximum (VBM) of ITO/NiOx modified with the SA is lower (–5.04 vs –5.51 eV) (Fig. 3a and Supplementary Fig. 29), which facilitates efficient hole extraction and transport from the perovskite layer to the HTL38,39. To investigate the charge transfer at the SAMs/perovskite interface, we conducted time-resolved photoluminescence (TRPL) measurements (Fig. 3b, c). TRPL decays typically exhibit two stages, in which the rapid decay indicates the transfer of carriers from the perovskite to the charge transport layer, followed by a slower decay due to carrier recombination40. Compared with control sample, the initial charge extraction rate of SA-based sample is faster, while the charge recombination rate is significantly reduced. The carrier lifetime of perovskite deposited on SA-based substrate is 637 ns, as determined by double exponential function fitting, which is much longer than the control. The extended carrier lifetime indicates that non-radiative recombination at the interface has been suppressed, which enhances the device performance.

a Energy levels of valence band maximum (VBM) and conduction band minimum (CBM) derived from UPS spectra of the control and SA-treated substrates. b TRPL decays of perovskite films deposited on various mixed SAMs. c Differential lifetime computed from fits to the TRPL. d PLQY values and e QFLS values for WBG perovskite films deposited on glass and various HTL. f Dark J-V curves, (g) VOC-light intensity dependent, (h) the EQEEL measurements and (i) detailed VOC loss analysis of the control and SA-based WBG devices.
To further understand the device performance improvements from the perspective of device physics, we measured photoluminescence quantum yield (PLQY) of perovskite deposited on different substrates and got the corresponding quasi-Fermi level splitting (QFLS) (Fig. 3d, e). The PLQY value of perovskite film deposited on glass substrate is 4.231%, corresponding to a QFLS value of 1.388 eV. For perovskite films deposited on mixed SAMs, SA sample achieves a maximum PLQY value of 3.005%, corresponding to a high QFLS value of 1.379 eV. All of the above indicate that SA co-adsorption strategy can reduce non-radiative recombination loss at SAMs/perovskite interface15. In addition, we also performed space charge limited current (SCLC) to estimate the defect density of the perovskite films41. As shown in Supplementary Fig. 30, the defect state density decreased from 2.75 × 1016 cm−3 in the control device to 1.64 × 1016 cm−3 in the SA-based device. The reduced trap-filled limit voltage (VTFL) and decreasing defect state density of SA sample indicate an improvement of film quality. The results of transient photocurrent (TPC) decay and transient photovoltage (TPV) decay demonstrate that SA sample has more efficient hole extraction and suppressed interfacial non-radiative recombination, respectively (Supplementary Fig. 31)42. The Mott-Schottky results can reflect the built-in field of the device. The SA-based device achieves a larger built-in field, indicating a stronger driving force for carrier transport, which is consistent with the improved VOC (Supplementary Fig. 32)24. Dark J–V characteristics current measurements suggest that the SA-based device has a smaller leakage current (Fig. 3f), indicating an increase in interface recombination resistance and suppression of non-radiative charge recombination, which may be related to the improved coverage of SAMs43. We also measured the VOC-light intensity dependence of devices (Fig. 3g). Ideal factor can reflect defect-assisted recombination in device. The ideal factor value of the SA sample is closer to 1, indicating that the defect-assisted trap states at the buried interface were suppressed to improve the FF44.
External quantum efficiency electroluminescence (EQEEL) was used to further understand the origin of the improved device VOC. Under the current density equivalent to JSC under AM 1.5 G illumination, the external quantum yield values of EQEEL for the control and SA-based devices were 0.04% and 1.15%, respectively (Fig. 3h). Compared to the control device, the EQEEL of SA-based device increased by almost 28 times, which can be attributed to the reduction of non-radiative recombination loss. Furthermore, we conducted a quantitative analysis of VOC loss, as shown in Fig. 3i. The VOC loss can be divided into loss related to thermal radiation, radiative recombination, and non-radiative recombination (Supplementary Fig. 33). Using the results of EQEEL for quantitative calculation, the non-radiative recombination loss is reduced by ~90 mV, which is the main reason for the reduction of VOC loss8,45.
Motivated by the improved film quality, we directly quantify ion migration of the WBG perovskite films based on temperature-dependent conductivity measurements (Supplementary Fig. 34). It is found that the ion migration active energy (Ea) of the perovskite film deposited on SA-based substrate was more than twice that of the control sample. To evaluate the notorious photoinduced phase segregation of this mixed halide WBG perovskite, we performed the time-dependent PL measurements. As shown in Supplementary Fig. 35, the PL intensity of the control films at 700 nm becomes weak within 10 min and an additional PL peak packet appears at 760 nm, indicating the presence of light-induced phase separation and the formation of I-rich phase in perovskite film. In contrast, the SA sample exhibits higher PL intensity under illumination. And over time, the PL peak just exhibits a weak redshift, indicating suppressed phase segregation, which is favorable to the improvement of device performance and stability.
Performance and stability of all-perovskite tandems and mini-modules
To demonstrate the potential of SA co-adsorption strategy for applications in tandem devices, we further integrated the improved WBG perovskite subcells and 1.26 eV NBG (FA0.6MA0.3Cs0.1Pb0.5Sn0.5I3) subcells into 2-terminal all-perovskite TSCs (Fig. 4a). The best-performing NBG PSC exhibited a PCE of 22.55%, with a VOC of 0.868 V, a JSC of 32.56 mA cm−2 and an FF of 79.76% (Supplementary Fig. 36 and Supplementary Table 3). As shown in Fig. 4b, the VOC and PCE of SA-based tandem devices were higher than the control devices (Supplementary Table 4). According to the J-V curves (Fig. 4c), the SA-based best-performing tandem device achieves an impressive PCE of 28.94% (28.59%), with a remarkable VOC of 2.146 V (2.149 V), a JSC of 16.19 mA cm−2 (16.27 mA cm−2) and an FF of 83.29% (81.72%) under reverse (forward) scan. The corresponding steady-state PCE measured by the maximum power point tracking (MPPT) was 28.72% (Supplementary Fig. 37). The EQE-integrated JSC values for WBG and NBG subcells measured from a tandem device are 15.94 and 15.71 mA cm−2, respectively (Fig. 4d). On this basis, we sent the SA-based encapsulated TSCs to SIMIT for certification, and obtained a certified PCE of 28.78% and a VOC of 2.145 V with an aperture area of 0.087 cm2 (Supplementary Fig. 38).

a Cross-sectional SEM image of SA-based all-perovskite TSC. b Photovoltaic performance statistics of control and SA-based all-perovskite TSCs. c J-V curves and d EQE spectra of the champion SA-based all-perovskite TSC. e J-V curves of SA-based all-perovskite tandem mini-module with an aperture area of 11.3 cm2. f Long-term operational stability of the control and SA-based encapsulated tandem devices under 1 sun continuous illumination with MPPT in ambient air.
Furthermore, to evaluate the upscaling potential, we fabricated 50 SA-based all-perovskite tandem mini-modules containing five subcells (Supplementary Fig. 39). The average PCE of SA-based mini-modules was 21.82%. The J-V curves of the champion SA-based tandem mini-module measured from both reverse and forward scans were presented in Fig. 4e. As a result, the tandem mini-module delivered a champion PCE of 23.92% and a VOC of 10.10 V with an aperture area of 11.3 cm2 (Supplementary Table 5).
Finally, we investigated the operational stability of the encapsulated small-area tandem device under a continuous AM 1.5 G illumination at MPPT in ambient air (Fig. 4f). The SA-based tandem device exhibited promising operational stability and maintained 85% of its initial PCE after 500 h continuous operation. Moreover, we also performed thermal and damp-heat (85 °C and 85% relative humidity) aging tests of the tandem devices (Supplementary Fig. 40). The enhanced stability exhibited by the SA-based devices further demonstrates that effectiveness of our employed co-adsorption strategy in enhancing device performance.
In summary, we demonstrated a feasible hybrid SAMs strategy at the buried interface of perovskite films for efficient all-tandem perovskite solar cells and modules. We studied the interaction between co-adsorbents and commonly used Me-4PACz for WBG PSCs. We found that the co-adsorbent SA tends to fill the uncovered sites without interference with Me-4PACz, ensuring the formation of a dense hole selective layer. Moreover, the use of SA co-adsorption strategy could effectively reduce the interfacial non-radiative recombination loss, optimize the energy alignment at the buried interface and regulate the crystallization behavior of WBG perovskite films. As a result, the 1.77 eV WBG PSC delivered a PCE of 20.67% (20.21% certified) and an impressive VOC of 1.332 V (1.313 V certified). By combining with a 1.26 eV NBG PSC, we further fabricated 2-terminal all-perovskite TSCs with a PCE of 28.94% (28.78% certified) for 0.087 cm2 and 23.92% for mini-module of 11.3 cm2. The efficiencies of single-junction WBG devices, small-area TSCs and mini-modules are all the top-level device performance reported to date. Moreover, the fabricated TSCs maintained 85% of its initial PCE after 500 h of continuous operation at the MPPT. Our work demonstrates a promising avenue for favorable contact at the buried interface of perovskite films for efficient and stable inverted PSCs and TSCs.
Methods
Materials
All reagents were obtained from commercial vendors and used as received. N, N-dimethylformamide (DMF, 99.9%), dimethyl sulfoxide (DMSO, 99.9%), chlorobenzene (CB, 98%), isopropanol (IPA, 99.9%), ethanol (99.8%), toluene (99.9%), ethyl acetate (EA, 99.9%), potassium thiocyanate (KSCN), tin (II) fluoride (SnF2), tin (II) iodide (SnI2) and glycine hydrochloride (GlyHCl,) were obtained from Sigma-Aldrich. Me-4PACz, caesium iodide (CsI), guanidine thiocyanate (GuaSCN) and bathocuproine (BCP) were obtained from TCI chemicals. NiOx, formamidinium iodide (FAI), lead bromide (PbBr2), lead iodide (PbI2) and methylammonium iodide (MAI) were obtained from Advanced Election Technology Co., Ltd. Potassium iodide (KI) and 7-Aminoheptanoic acid (CA) were obtained from Shanghai Aladdin Biochemical Technology Co., Ltd. (6-aMinohexyl)-Phosphonic acid (PA) was obtained from Xueyanhui (Wuhan) Technology Co., Ltd. 6-Aminohexane-1-sulfonic acid (SA) was obtained from Shanghai Kaiwei Chemical Technology Co., Ltd. 1,3-propanediamine dihydriodide (PDAI2), fullerene (C60), poly (3,4-ethylenedioxythiophene)-poly (styrenesulfonate) (PEDOT:PSS) aqueous solution (Al-4083) and ethylenediammonium diiodide (EDAI2) were obtained from Xi’an Yuri Solar Co., Ltd.
Preparation of precursor solution
For the 1.77 eV FA0.8Cs0.2Pb(I0.6Br0.4)3 perovskite, 1.2 M perovskite precursor solution was prepared by mixing 0.0623 g CsI, 0.1651 g FAI, 0.2213 g PbI2 and 0.2642 g PbBr2 in 750 μL DMF and 250 μL DMSO mixed solvent. In addition, 0.0029 g KSCN were added to the precursor as additive.
For the 1.26 eV FA0.6MA0.3Cs0.1Pb0.5Sn0.5I3 perovskite, 1.9 M perovskite precursor solution was prepared by mixing 0.1960 g FAI, 0.0906 g MAI, 0.0494 g CsI, 0.4380 g PbI2 and 0.3539 g SnI2 in 750 μL DMF and 250 μL DMSO mixed solvent. In addition, 0.0149 g SnF2, 0.0046 g GuaSCN and 0.0042 g GlyHCl were added to the precursor as additives.
Device fabrication
Wide-bandgap perovskite solar cells (WBG PSCs): ITO substrates were ultrasonically cleaned by detergent, deionized water and ethanol for 15 min, respectively. Before use, the ITO substrates were treated with ultraviolet ozone for 20 min. Then the NiOx layer was fabricated by spin-coating the NiOx dispersions (5 mg mL−1 dispersed in deionized water) on ITO substrates at 3000 rpm for 25 s, and annealed at 150 °C for 10 min in ambient air. Then these substrates were transferred into a N2-filled glove box. For control devices, the Me-4PACz (0.6 mg mL−1 in ethanol) was spin-coated on NiOx at 3000 rpm for 25 s, and annealed at 100 °C for 10 min. For PA, CA, SA devices, just the composition of SAMs solution was changed to a mixture (Me-4PACz: 0.6 mg mL−1 in ethanol and PA/CA/SA: 0.2 mg mL−1 in ethanol with a molar ratio of 8:5). Then the perovskite layer was fabricated by spin-coating 60 μL precursor on SAMs at 5000 rpm for 60 s and dropping 400 μL EA on the film at 25-30 s before the end of the spin-coating. Subsequently, the film was annealed at 100 °C for 10 min. The passivation layer was fabricated by spin-coating the PDAI2 (1.5 mg mL−1 in IPA) on perovskite at 4000 rpm for 20 s, and annealed at 100 °C for 5 min. The electron transport layer (ETL) was fabricated by thermal evaporation of C60 (20 nm) at a rate of 0.1 Å s−1. Finally, BCP (5 nm) at a rate of 0.1 Å s−1, and Ag (100 nm) were thermally evaporated.
Narrow-bandgap perovskite solar cells (NBG PSCs): ITO was treated the same as WBG PSCs. The PEDOT:PSS was spin-coated on ITO substrates at 5000 rpm for 30 s, and annealed at 150 °C for 20 min in ambient air. Then these substrates were transferred into glove box filled with N2. Then the perovskite layer was fabricated by spin-coating 60 μL precursor on PEDOT:PSS with a two-step process (1000 rpm for the initial 10 s and 4000 rpm for the final 40 s) and dropping 350 μL CB on the film at 20 s before the end of the spin-coating. Subsequently, the film was annealed at 100 °C for 10 min. The passivation layer was fabricated by spin-coating the EDAI2 (0.5 mg mL−1 in 1:1 IPA:toluene) on perovskite at 4000 rpm for 20 s, and annealed at 100 °C for 5 min. Finally, C60 (20 nm), BCP (5 nm) at a rate of 0.1 Å s−1 and Ag (100 nm) were thermally evaporated.
Tandem solar cells: For the bottom subcell, the preparation process was the same as the fabrication of WBG PSCs before the thermal evaporation of BCP. Then these substrates were deposited 30 nm SnO2 at 100 °C by the atomic layer deposition (ALD). Subsequently, 1 nm Au was thermally evaporated. And next, the deposition of PEDOT:PSS, NBG perovskite, EDAI2, C60, BCP and Ag were the same as the fabrication of NBG PSCs.
Mini-modules: The mini-modules were fabricated on 5 cm × 5 cm sized glass/ITO substrates. First of all, these ITO substrates were etched by a 532 nm nanosecond laser with a power of 4 W to form P1 lines isolated subcells with a width of 6.8 mm. Then the fabrication process was the same as the small-area tandem solar cells above, before the thermal evaporation of Ag. But the accelerations during spin-coating processes needs to be changed to 1000 rpm s–1 to ensure that the solution can be evenly coated on the substrate. Then the substrates were etched by the nanosecond laser with a power of 0.5 W to form P2 lines. Next, Cu (100 nm) was deposited by thermal evaporation. Subsequently, the substrates were etched by the nanosecond laser with a power of 0.5 W to form P3 lines. Finally, the tinned copper tape was attached to both sides of the modules and encapsulated with 0.5 cm wide butyl tape.
Computational details
DFT calculations: The geometry optimizations for molecules and surface adsorption used the Vienna ab initio Simulation Package (VASP) code, employing the generalized gradient approximation (GGA) with Perdew-Burke-Ernzerh (PBE) functional for the exchange-correlation functional of electrons and the Grimme’s DFT-D3 dispersion correction46,47,48,49,50. The kinetic energy cut-off is 500 eV for the expansion of the wavefunctions. The convergence thresholds were set to 10−6 eV for energy and −0.05 eV/Å for the norm of the atomic forces. We built a NiOx slab based on the NiO (100) surface with a 5 × 5 × 2 supercell. The ITO slab was approximated with the In2O3 (111) surface in a cell of 14.48 Å × 25.10 Å × 5.51 Å. A 12 Å vacuum space was added to the top of the adsorbed molecules. Dipole corrections are applied to the z-direction for all adsorption calculations. All ionic optimizations were done at the gamma point and the bottom two atom layers were fixed during the optimizations. We applied spin-polarization with antiferromagnetic moments and DFT + U corrections (U = 8 eV and J = 0.95 eV) for Ni atom51. The adsorption energy was determined using the equation \(E={E}_{{total}}-{E}_{{slab}}-{E}_{{molecule}}\), where Etotal, Eslab and Emolecule represents the total energy of the slab with molecule, the slab and molecule, respectively. The binding energy between molecules was computed in the same way by replacing Eslab with the electronic energy of the other molecule.
The work function calculations for SAMs only included the SAM molecules to show their intrinsic capability of losing one electron. The work functions are defined by Ve – Ef, where Ve is the vacuum electrostatic potential and Ef is the Fermi level of the molecules. Because the SAM molecules are isolated by the vacuum space in the x, y, and z direction, the values of Ve are almost identical in all directions. Thus, we used the Ve in the z-direction to compute the work function. The work function calculations for NiO surface model used the same equation. The Ve was computed in the z-direction perpendicular to the surface. The Fermi level was obtained from the above spin-polarization DFT + U calculations.
MD simulation: We prepared three adsorption models: (1) NiOx surface based on a 100.47 Å × 100.47 Å × 8.37 Å NiO (100) supercell, (2) ITO surface based on the a 100.74 Å × 99.71 Å × 8.81 Å In2O3 (111) supercell, and (3) NiOx/ITO interface based on a 50.24 Å × 50.24 Å × 8.37 Å NiO (100) supercell adsorbed on 100.74 Å × 99.71 Å × 8.81 Å In2O3 (111) supercell. On top of the surface, we used the Packmol program52 to generate the SAMs layer with (1) 160 Me-4PACz molecules, (2) 160 Me-4PACz and 100 PA, (3) 160 Me-4PACz and 100 CA, and (4) 160 Me-4PACz and 100 SA, following the experimental molar ratio between Me-4PACz and co-adsorbents (8:5).
Classical molecular dynamics simulations were performed with the LAMMPS molecular dynamics simulator53. The equation of motions was integrated with a time step of 1 fs using the Velocity Verlet method54. The trajectories are propagated in a canonical ensemble (NVT) with the Nosé-Hoover thermostat55,56 with a time constant of 0.1 fs. Periodic boundary conditions were applied to the x and y directions, while the bottom surface layers were fixed. The velocities were initialized at 298.15 K and equilibrated in 0.1 ns. Then we increased the temperature to the experimental condition at 373.15 K in 0.1 ns for the production run in 1 ns. The surface adsorption analysis used our in-house code, which assigned SAMs to adjacent surfaces and computed the surface coverage by searching any atoms of SAMs above the surface within a distance threshold of 3 Å and a radial threshold of 2 Å.
The MD simulations used the GAFF2 force field for organic molecules, INTERFACE force field57 for NiO, and Buckingham potential for In2O358. The interactions between molecules and surfaces were described by combining their force field parameters using the geometric mixing rule, where the Buckingham parameters of In2O3 was fitting to suitable Lennard-Jones parameters59. The atomic charges for the GAFF2 force fields used the restrained electrostatic potential (RESP) charges60, computed at the PBE0/cc-pVDZ level, using the ORCA 5.0.4 program61.
Characterization and measurements
The J-V curves of all the PSCs were obtained using a Keithley 2400 source measurement unit and a solar simulator (Oriel, model 9119) with an AM 1.5 G spectrum. The stabilized power outputs of the devices were obtained by monitoring the stabilized current density output under MPP bias voltage (extracted from the reverse scan J-V curves). A QE system (Enlitech Co., Ltd.) was used to measured EQE spectra of single-junction solar cells in ambient. For tandem solar cells, the measurements of the WBG and NBG subcells were achieved using two highly-bright LEDs with emission peaks of 850 and 460 nm, respectively. A Lambda 950 UV-vis spectrophotometer (PerkinElmer Co., USA) was used to measure the absorption spectra and transmission spectra. XPS measurements were performed on a Thermo ESCALAB 250XI using 100 W monochromatic Al Kα X-ray source (1486.6 eV). The FTIR spectra were obtained by Nicolet iS50R FTIR spectrometer (Thermo Scientific Co., America) The DLS spectra of hybrid SAMs solution were obtained by a ZetaSizer Nano-ZS 90 (Malvern Instruments Ltd., Worcestershire, UK). KPFM and AFM measurements were obtained using a dimension icon scanning probe microscope (Asylum Research, MFP-3D-Stand Alone) in the tapping mode. The contact angles were obtained using a CA200 automatic optical contact angle measuring instrument. The SEM images were gained by a Nova NanoSEM 450 scanning electron microscope at 10 kV operating voltage. The in-situ PL measurement was achieved by laser excitation at 532 nm and the target area was fixed on the micro-spectral measurement system (ProSp-Micro40-VIS), together with spectrograph (Ocean Insight, USA). The GIWAXS measurement was carried out with the Xeuss 3.0. The GIXRD spectra was gained by a Rigaku Smartlab with Cu Kα radiation in the 2θ range of 31.5°– 33.5°. The XRD spectra was obtained using an X-ray diffractometer (PANalytical B.V. Co.) with Cu Kα radiation. The temperature-dependent conductivity was obtained by a Keithley 2400 at different temperatures. The UPS spectra was conducted on an AXIS-ULTRA DLD-600W Ultra spectrometer with a He discharge lamp, emitting ultraviolet energy at 21.22 eV. The TRPL measurements were achieved on a HORIBA Delta Flex spectrometer. The photoluminescence quantum yield (PLQY) measurements were conducted on a Quanta Master 8000 with a 532 nm laser to photoexcite the samples. And the Quasi-Fermi level splitting (QFLS) was acquired by the data of PLQY. The EQEEL spectra were measured on a Keithley 2400 Source Measurement Unit with a light emitting diode PL quantum-yield measurement system. The dark J-V curves, light intensity dependence of VOC of PSCs, SCLC of the hole-only devices, transient photovoltage decay (TPV), transient photocurrent decay (TPC) and Mott-Schottky plots were acquired using a Zennium electrochemistry workstation (Zahner, Germany).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data that support the findings of this study are available in the Supplementary Information/Source Data file (Figshare https://doi.org/10.6084/m9.figshare.28573850). Source data are provided with this paper.
References
Eperon, G. E., Hörantner, M. T. & Snaith, H. J. Metal halide perovskite tandem and multiple-junction photovoltaics. Nat. Rev. Chem. 1, 1–18 (2017).
Leijtens, T., Bush, K. A., Prasanna, R. & McGehee, M. D. Opportunities and challenges for tandem solar cells using metal halide perovskite semiconductors. Nat. Energy 3, 828–838 (2018).
Green, M. A. et al. Solar cell efficiency tables (Version 64). Prog. Photovolt. Res. Appl. 32, 425–441 (2024).
Nie, T., Fang, Z., Ren, X., Duan, Y. & Liu, S. F. Recent Advances in Wide-Bandgap Organic–Inorganic Halide Perovskite Solar Cells and Tandem Application. Nano-Micro Lett. 15, 70 (2023).
He, R. et al. Wide-bandgap organic–inorganic hybrid and all-inorganic perovskite solar cells and their application in all-perovskite tandem solar cells. Energy Environ. Sci. 14, 5723–5759 (2021).
Chen, H. et al. Regulating surface potential maximizes voltage in all-perovskite tandems. Nature 613, 676–681 (2023).
Lai, H. et al. High‐Performance Flexible All‐Perovskite Tandem Solar Cells with Reduced VOC‐Deficit in Wide‐Bandgap Subcell. Adv. Energy Mater. 12, 2202438 (2022).
Yi, Z. et al. Achieving a high open-circuit voltage of 1.339 V in 1.77 eV wide-bandgap perovskite solar cells via self-assembled monolayers. Energy Environ. Sci. 17, 202–209 (2024).
Zheng, X. et al. Co-deposition of hole-selective contact and absorber for improving the processability of perovskite solar cells. Nat. Energy 8, 462–472 (2023).
Cao, Q. et al. Co-Self-Assembled Monolayers Modified NiOx for Stable Inverted Perovskite Solar Cells. Adv. Mater. 36, 2311970 (2024).
Liu, M. et al. Compact Hole-Selective Self-Assembled Monolayers Enabled by Disassembling Micelles in Solution for Efficient Perovskite Solar Cells. Adv. Mater. 35, 2304415 (2023).
He, R. et al. Improving interface quality for 1-cm2 all-perovskite tandem solar cells. Nature 618, 80–86 (2023).
Park, S. M. et al. Low-loss contacts on textured substrates for inverted perovskite solar cells. Nature 624, 289–294 (2023).
Zhang, S. et al. Minimizing buried interfacial defects for efficient inverted perovskite solar cells. Science 380, 404–409 (2023).
Liu, S. et al. Buried interface molecular hybrid for inverted perovskite solar cells. Nature 632, 536–542 (2024).
Cui, H. et al. Lead halide coordination competition at buried interfaces for low V OC -deficits in wide-bandgap perovskite solar cells. Energy Environ. Sci. 16, 5992–6002 (2023).
Liu, L. et al. Self-Assembled Amphiphilic Monolayer for Efficient and Stable Wide-Bandgap Perovskite Solar Cells. Adv. Energy Mater. 13, 2202802 (2023).
Al-Ashouri, A. et al. Wettability Improvement of a Carbazole-Based Hole-Selective Monolayer for Reproducible Perovskite Solar Cells. ACS Energy Lett. 8, 898–900 (2023).
Deng, X. et al. Co-assembled Monolayers as Hole-Selective Contact for High-Performance Inverted Perovskite Solar Cells with Optimized Recombination Loss and Long-Term Stability. Angew. Chem. Int. Ed. 61, e202203088 (2022).
Jiang, Q. et al. Surface reaction for efficient and stable inverted perovskite solar cells. Nature 611, 278–283 (2022).
Li, L. et al. Flexible all-perovskite tandem solar cells approaching 25% efficiency with molecule-bridged hole-selective contact. Nat. Energy 7, 708–717 (2022).
Zhang, J. et al. Elimination of Interfacial Lattice Mismatch and Detrimental Reaction by Self-Assembled Layer Dual-Passivation for Efficient and Stable Inverted Perovskite Solar Cells. Adv. Energy Mater. 12, 2103674 (2022).
Wang, S. et al. Critical Role of Removing Impurities in Nickel Oxide on High-Efficiency and Long-Term Stability of Inverted Perovskite Solar Cells. Angew. Chem. 134, e202116534 (2022).
Li, E. et al. Bonding Strength Regulates Anchoring-Based Self-Assembly Monolayers for Efficient and Stable Perovskite Solar Cells. Adv. Funct. Mater. 31, 2103847 (2021).
Li, S. et al. A Generic Strategy to Stabilize Wide Bandgap Perovskites for Efficient Tandem Solar Cells. Adv. Mater. 36, 2307701 (2024).
Li, D. et al. Regulating Charge Transport Dynamics at the Buried Interface and Bulk of Perovskites by Tailored-phase Two-dimensional Crystal Seed Layer. Angew. Chem. 136, e202400708 (2024).
Szostak, R. et al. In Situ and Operando Characterizations of Metal Halide Perovskite and Solar Cells: Insights from Lab-Sized Devices to Upscaling Processes. Chem. Rev. 123, 3160–3236 (2023).
Liu, C. et al. Flexible Indoor Perovskite Solar Cells by In Situ Bottom-Up Crystallization Modulation and Interfacial Passivation. Adv. Mater. 36, 2311562 (2024).
Pei, F. et al. A binary 2D perovskite passivation for efficient and stable perovskite/silicon tandem solar cells. Nat. Commun. 15, 7024 (2024).
Dunlap-Shohl, W. A., Zhou, Y., Padture, N. P. & Mitzi, D. B. Synthetic Approaches for Halide Perovskite Thin Films. Chem. Rev. 119, 3193–3295 (2019).
Shao, S. & Loi, M. A. The Role of the Interfaces in Perovskite Solar Cells. Adv. Mater. Interfaces 7, 1901469 (2020).
Meng, X. et al. Effective Inhibition of Phase Segregation in Wide-Bandgap Perovskites with Alkali Halides Additives to Improve the Stability of Solar Cells. Sol. RRL 7, 2201099 (2023).
Wang, J. et al. Bimolecular Crystallization Modulation Boosts the Efficiency and Stability of Methylammonium-Free Tin–Lead Perovskite and All-Perovskite Tandem Solar Cells. Adv. Energy Mater. 14, 2402171 (2024).
Luo, C. et al. Engineering the buried interface in perovskite solar cells via lattice-matched electron transport layer. Nat. Photonics 17, 856–864 (2023).
Gao, Y. et al. Elimination of unstable residual lead iodide near the buried interface for the stability improvement of perovskite solar cells. Energy Environ. Sci. 16, 2295–2303 (2023).
Fei, C. et al. Lead-chelating hole-transport layers for efficient and stable perovskite minimodules. Science 380, 823–829 (2023).
An, Y. et al. Optimizing Crystallization in Wide-Bandgap Mixed Halide Perovskites for High-Efficiency Solar Cells. Adv. Mater. 36, 2306568 (2024).
Al-Ashouri, A. et al. Monolithic perovskite/silicon tandem solar cell with >29% efficiency by enhanced hole extraction. Science 370, 1300–1309 (2020).
Chen, R. et al. Reduction of bulk and surface defects in inverted methylammonium- and bromide-free formamidinium perovskite solar cells. Nat. Energy 8, 839–849 (2023).
Liu, S., Biju, V. P., Qi, Y., Chen, W. & Liu, Z. Recent progress in the development of high-efficiency inverted perovskite solar cells. NPG Asia Mater. 15, 1–28 (2023).
Chen, R. et al. Robust hole transport material with interface anchors enhances the efficiency and stability of inverted formamidinium–cesium perovskite solar cells with a certified efficiency of 22.3%. Energy Environ. Sci. 15, 2567–2580 (2022).
Sun, H. et al. Suppressed Phase Segregation with Small A-Site and Large X-Site Incorporation for Photostable Wide-Bandgap Perovskite Solar Cells. Small Methods 8, 2400067 (2024).
Lin, R. et al. Monolithic all-perovskite tandem solar cells with 24.8% efficiency exploiting comproportionation to suppress Sn(II) oxidation in precursor ink. Nat. Energy 4, 864–873 (2019).
Tan, H. et al. Efficient and stable solution-processed planar perovskite solar cells via contact passivation. Science 355, 722–726 (2017).
Peng, W. et al. Reducing nonradiative recombination in perovskite solar cells with a porous insulator contact. Science 379, 683–690 (2023).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Kresse, G. & Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 47, 558–561 (1993).
Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 27, 1787–1799 (2006).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Dudarev, S. L., Botton, G. A., Savrasov, S. Y., Humphreys, C. J. & Sutton, A. P. Electron-energy-loss spectra and the structural stability of nickel oxide: An LSDA+U study. Phys. Rev. B 57, 1505–1509 (1998).
Martínez, L., Andrade, R., Birgin, E. G. & Martínez, J. M. PACKMOL: A package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 30, 2157–2164 (2009).
Thompson, A. P. et al. LAMMPS - a flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales. Comput. Phys. Commun. 271, 108171 (2022).
Verlet, L. Computer ‘Experiments’ on Classical Fluids. I. Thermodynamical Properties of Lennard-Jones Molecules. Phys. Rev. 159, 98–103 (1967).
Nosé, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 81, 511–519 (1984).
Hoover, W. G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 31, 1695–1697 (1985).
Kanhaiya, K. et al. Accurate Force Fields for Atomistic Simulations of Oxides, Hydroxides, and Organic Hybrid Materials up to the Micrometer Scale. J. Chem. Theory Comput. 19, 8293–8322 (2023).
Walsh, A., Catlow, C. R. A., Sokol, A. A. & Woodley, S. M. Physical Properties, Intrinsic Defects, and Phase Stability of Indium Sesquioxide. Chem. Mater. 21, 4962–4969 (2009).
Lim, T.-C. Alignment of Buckingham Parameters to Generalized Lennard-Jones Potential Functions. Z. F. ür. Naturforsch. A 64, 200–204 (2009).
Bayly, C. I., Cieplak, P., Cornell, W. D. & Kollman, P. A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. Well-Behaved Electrost. Potential Based Method Using Charg. Restraints Deriving. Charg. RESP Model 97, 1026910280 (1993).
Neese, F. Software update: The ORCA program system—Version 5.0. WIREs Comput. Mol. Sci. 12, e1606 (2022).
Acknowledgements
W.C. acknowledge the financial support from the National Natural Science Foundation of China (W2412077, U20A20252), the Innovation Project of Optics Valley Laboratory (OVL2021BG008) and the Ministry of Science and Technology of China (2021YFB3800104). Z.L. acknowledge the financial support from the National Natural Science Foundation of China (52473301), the Natural Science Foundation of Hubei Province (2022CFA093), the Self-determined and Innovative Research Funds of HUST (2020kfyXJJS008) and the Young Elite Scientists Sponsorship Program by CAST. Z.Y. acknowledge the financial support from the National Natural Science Foundation of China (62205187) and the Fundamental Research Program of Shanxi Province (202103021223032). R.C. acknowledge the financial support from China Postdoctoral Science Foundation (2023M731172). Q.Z. acknowledge the financial support from China Postdoctoral Science Foundation (2024M751002, GZC20240528). J.W. acknowledge the financial support from the Fundamental Research Funds for the Central Universities, HUST (2023JYCXJJ041). The authors thank the Analytical and Testing Center of HUST for the facilities’ support of sample measurements. The authors also would like to acknowledge the Center for Computational Science and Engineering at Southern University of Science and Technology.
Author information
Authors and Affiliations
Contributions
W.C., Z.L., J.L. and C.S. conceived the project. C.S. was mainly responsible for fabrication and certification of high-performance wide-bandgap and tandem devices. J.W. was mainly responsible for optimization of narrow-bandgap and preparation of tandem devices. Q.Z. was mainly responsible for DFT calculations and module preparation. W.W. carried out MD simulation. X.L. revised the DFT calculations and the MD simulations. C.S., J.W., S.L., J.Z., Z.T. and W.L. conducted the material and device characterizations. Z.Y., S.L., H.Z, R.C. and Y.P. provided suggestions for writing the paper. Zhengjing Z., Z.C., X.Q. and Zhiguo Z. contributed materials and analysis tools. C.S., J.W., X.L., J.L., Z.L. and W.C. analysed all experimental data. And C.S., J.W., X.L., Q.Z. and W.W. prepared the paper under the supervision of J.L., Z.L. and W.C. C.S., J.W., X.L., Q.Z. and W.W. contributed equally to this work. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Shi, C., Wang, J., Lei, X. et al. Modulating competitive adsorption of hybrid self-assembled molecules for efficient wide-bandgap perovskite solar cells and tandems. Nat Commun 16, 3029 (2025). https://doi.org/10.1038/s41467-025-58111-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-58111-y
