
Abstract
Endochondral ossification generates most of the load-bearing bones, recapitulating it in human cells remains a challenge. Here, we report generation of SOX9+ sclerotomal progenitors (scl-progenitors), a mesenchymal precursor at the pre-condensation stage, from human pluripotent stem cells and development of osteochondral induction methods for these cells. Upon lineage-specific induction, SOX9+ scl-progenitors have not only generated articular cartilage but have also undergone spontaneous condensation, cartilaginous anlagen formation, chondrocyte hypertrophy, vascular invasion, and finally bone formation with stroma, thereby recapitulating key stages during endochondral ossification. Moreover, self-organized growth plate-like structures have also been induced using SOX9+ scl-progenitor-derived fusion constructs with chondro- and osteo-spheroids, exhibiting molecular and cellular similarities to the primary growth plates. Furthermore, we have identified ITGA9 as a specific surface marker for reporter-independent isolation of SOX9+ scl-progenitors and established a culture system to support their expansion. Our work highlights SOX9+ scl-progenitors as a promising tool for modeling human skeletal development and bone/cartilage bioengineering.
Similar content being viewed by others

Introduction
The skeletal system, consisting of cartilage and bones, provides essential physical support and protection for the human body, as well as attachment sites for muscles, tendons, and ligaments that support locomotion1. Skeletal disorders, such as knee osteoarthritis, affected more than 16% of the adult population worldwide2, with limited clinical treatment options and unsatisfactory outcomes3,4,5. Genetic variants for many human bone development disorders, including scoliosis6, idiopathic bone cavities7, and juvenile osteoporosis8, remain largely unknown. Establishing appropriate models to recapitulate human skeletal development, particularly endochondral ossification, is critical for uncovering the molecular mechanism underlying these genetic variants.
Endochondral ossification is a conserved mechanism in mammals that generates most of the load-bearing bones. It begins with the condensation of mesenchymal progenitors, followed by anlagen formation, chondrocyte hypertrophy, vascular invasion, and ultimately bone formation with marrow cavities and hematopoiesis-supporting stroma1. Notably, the formation of growth plates, characterized by columns of proliferative chondrocytes, is a key feature that mediates polarized bone growth and serves as a major residence for skeletal stem cells9,10. Traditionally, the molecular mechanisms governing endochondral ossification have been studied primarily in mouse models, which have helped characterize key signaling pathways such as BMPs11,12, FGFs13, Hedgehogs14, and Wnt15. However, the initiation, regulation, and progression of human endochondral ossification, as well as the formation of distinct osteochondral zones, remain elusive due to two major challenges: the limitations of current cell sources and the lack of efficient differentiation methods to faithfully model the process.
To model human skeletal development, various stem cell sources have been explored, including mesenchymal stem cells (MSCs) from different tissues, with or without biomaterials16,17,18,19,20, and human pluripotent stem cell (hPSC)-derived cells21,22,23,24,25, achieving varying levels of success. MSCs-based approaches suffered from unclear developmental lineages, cellular heterogeneity, and individual variations26,27. For hPSC-based studies, much of the focus has been on generating articular chondrocytes for cartilage regeneration using hPSC-derived paraxial mesodermal progenitors and their progenies22,28, ectomesodermal neural crest stem cells23,29,30, and more recently limb bud-like cells24. Meanwhile, it was also attempted to model skeletal development starting from the hypertrophic cartilage stage22,25,31, where cells were typically first differentiated to chondroprogenitors and then to hypertrophic chondrocytes for bone formation. However, forced chondrocyte hypertrophy by hormones bypassed important steps such as spontaneous mesenchymal condensation and cartilaginous anlagen formation, deviating from the natural course of endochondral ossification. There was also a lack of evidence for the formation of bone marrow cavities with stroma and distinct osteochondral zones. These findings suggested that the osteogenic differentiation of artificially induced hypertrophic chondrocytes might still differ significantly from their in vivo counterparts. Most importantly, none of the previous studies were able to induce the formation of polarized growth plate-like structures that could mediate longitudinal bone growth, a critical feature of long bones. Therefore, it is imperative to identify a more suitable, developmentally relevant stem cell source and develop accompanying differentiation methods in order to comprehensively model the natural course of human skeletal development.
Developmentally, the human skeleton is derived from three lineages: paraxial mesoderm, lateral plate mesoderm, and neural crest32. The paraxial mesoderm arises from the primitive streak and gives rise to the axial skeleton through somite and sclerotome33. The sclerotome further develops into the vertebrae and associated ribs34, tendons35,36, and other tissues32. Notably, the induction of sclerotome from hPSCs has been established recently37,38,39, and direct transplantation of hPSC-derived sclerotomal cells in vivo could spontaneously generate bone-like tissues40. These findings suggested that hPSC-derived sclerotomal cells might be a promising stem cell source. However, as these cells contained a heterogeneous cell population that formed non-osteochondral tissues upon direct transplantation39,41, and proper differentiation methods to promote the osteochondral fate in these cells are still lacking, more thorough characterization is warranted to dissect and identify the key population within sclerotomal cells with true osteochondral differentiation capability and develop suitable differentiation strategies.
In this study, we characterized hPSC-derived sclerotomal cells and identified SOX9+ sclerotomal progenitors (scl-progenitors) as the key cell group with osteochondral differentiation capability. We then developed a differentiation strategy to derive these progenitors from hPSCs with an efficiency of 99.21% ± 0.24% on average across multiple cell lines. By development of lineage-specific induction methods, SOX9+ scl-progenitors were differentiated into either chondroprogenitors for cartilage regeneration or osteogenesis-committed early chondrocytes capable of recapitulating key steps of the endochondral ossification process, including condensation, anlagen formation, chondrocyte hypertrophy, vascular invasion, and bone formation with marrow cavities and stroma. Furthermore, chondro- and osteo-spheroids derived from SOX9+ scl-progenitors self-organized into polarized and elongating growth plate-like structures, exhibiting molecular and cellular similarities to in vivo growth plates. Additionally, ITGA9 was identified as a specific marker for reporter-independent isolation of SOX9+ scl-progenitors and an in vitro culture system was established to to support their continuous expansion. Our findings present an integrated hPSC-based toolkit for modeling human skeletal development and bioengineering cartilage and bone tissues.
Results
Characterization and efficient derivation of SOX9 + sclerotomal progenitors from hPSCs
Sox9 is a critical determining factor during the early development of the mammalian skeleton, with its expression detected in all the condensed mesenchyme and osteochondral progenitor cells42,43. Lineage tracing and in vivo functional studies also have highlighted that Sox9 is a key regulator during both the condensation stage and the downstream osteochondral specification for sclerotome44,45. Thereby, the expression of Sox9 might be an important marker of mesenchymal progenitors at the pre-condensation stage, and we hypothesized that SOX9+, but not the SOX9- subpopulation of hPSC-derived sclerotomal cells would represent sclerotomal progenitors with true osteochondral differentiation capability. To test the hypothesis, a SOX9-IRES-tdTomato reporter knockin cell line was constructed using CRISPR/Cas9 with previously established UC01 hiPSCs (human induced pluripotent stem cells)46 as the parental cells. An IRES-tdTomato cassette was inserted into the SOX9 locus through non-homologous end joining (NHEJ)-mediated gene knockin (Supplementary Fig. 1A). The edited cells were characterized to confirm the correct insertion of the reporter cassette (Supplementary Fig. 1B–D) and the maintenance of pluripotency markers (Supplementary Fig. 1E-F). Hereafter, this cell line would be referred to as SOX9-tdTomato hPSCs.
To differentiate SOX9-tdTomato hPSCs towards sclerotomal lineage, a previously reported traditional method was adopted, where cells were differentiated through primitive streak (PS)38, presomitic mesoderm (PSM)47,48, somite specification (SM)37, and then sclerotome (SCL)37,38,39,49,50 (Supplementary Fig. 2A-B). RT-qPCR confirmed the sequential induction of stage-specific markers during the differentiation (Supplementary Fig. 2C). After six days, SOX9+ cells were observed both by immunostaining (Supplementary Fig. 2D) and flow cytometry analysis (Supplementary Fig. 2E). As a portion of the cells were found negative for SOX9 and TWIST expression (Supplementary Fig. 2D-E), SOX9+ and SOX9- cells were sorted by FACS for further characterization. RT-qPCR confirmed that sclerotome-specific genes were only enriched in the SOX9+ population (Supplementary Fig. 2F). The presence of about 20% SOX9- cells indicated that the traditional sclerotomal differentiation was still heterogeneous (Supplementary Fig. 2D-E), consistent with other reported observations41. To investigate if SOX9+ cells were the key cell population that retained osteochondral differentiation capability, they were FACS sorted and subjected to in vitro osteochondral induction. For chondrogenic differentiation, pelleted SOX9+ scl-progenitors were treated with a low dose of BMP2 together with TGFβ3 and bFGF in a modified serum-free medium (chondrogenic induction medium, CI medium), while for osteogenic differentiation, the classic serum-containing medium51 was adopted (Supplementary Fig. 2G). Indeed, upon induction, SOX9+ cells, but not SOX9- ones, efficiently formed Alcian blue/COL II/Toluidine blue-positive but COL I/X negative cartilaginous spheroids (Supplementary Fig. 2H-I) and calcified Alizarin red-positive regions (Supplementary Fig. 2J), supporting that these cells were the osteochondral progenitors. Hereafter, they were referred to as SOX9+ sclerotomal progenitors (SOX9+ scl-progenitors).
As the differentiation efficiency for SOX9+ scl-progenitors was only around 80% using the traditional method and heterogeneous cells could also be observed from the culture (Supplementary Fig. 2D-E), which generated unwanted non-osteochondral tissues upon direct transplantation39,41, we sought to eliminate the heterogeneity and improve the differentiation. Interestingly, it was previously reported that chondrogenic sclerotomal cells could be induced directly by SHH treatment in chicken PSM explants without going through somite specification44 and the initial cell density strongly affected differentiation efficiency as mesenchymal cells tend to become over-confluent during differentiation24,38. Therefore, we investigated whether the strategy could be adopted in human cell differentiation to bypass the somite stage (somite-skipping), and directly induce the sclerotomal fate from the Day 2-replated human PSM cells by activating SHH signaling with smoothened agonist (SAG) while inhibiting BMP and WNT pathways with LDN193189 and XAV939 to suppress the dermomyotome fate (Fig. 1A). Indeed, improved morphological changes were observed during the stepwise induction, with uniform-look mesenchymal cells shown on day 4 already (Supplementary Fig. 3A), while traditional sclerotome differentiation typically took at least 6 days (Supplementary Fig. 2A-B). RT-qPCR confirmed the induced expression of stage-specific markers in each population, with OCT4 and NANOG as pluripotency markers52, BRACHYURY (T) and MIXL1 as the markers of PS53,54, MSGN1 and TBX6 as the markers of PSM55,56, and PAX1, NKX3-2, TWIST1 and SOX9 as the markers of sclerotome37,44,57 (Fig. 1B and Supplementary Fig. 3B). Single-cell RNA-seq was also used to analyze the differentiated cells at each stage and the four cell groups were separated in the t-SNE plot (Supplementary Fig. 3C). Pseudotime analysis and marker gene expression dynamics all confirmed the intended differentiation trajectory and induction of respective markers at each stage (Supplementary Fig. 3D-E). Expressions of SOX9 and TWIST1 were further confirmed by immunostaining (Fig. 1C) and western blotting (Fig. 1D). The percentage of SOX9+ cells increased from an average of 82.01% to 99.30% (Fig. 1E-F). To test batch-to-batch variations of the optimized method, samples from multiple independent experiments were analyzed. The optimized method exhibited higher consistency compared with the traditional one (Fig. 1F and Supplementary Fig. 4A). Similar results were also obtained using other established hPSC lines from different sources, including hPSC lines (H1 and H9) and hiPSCs (IMR904 and NC3-1) (Supplementary Fig. 4B-C), supporting the general applicability of the optimized method. To investigate whether osteochondral potentials were still maintained in the SOX9+ scl-progenitors derived from the optimized method, cells were subjected to the same in vitro differentiation protocols as specified previously (Supplementary Fig. 2G). Alcian blue, COL II, Toluidine blue, and Safranin O-positive but COL I/X-negative chondral spheroids, as well as Alizarin red-rich regions, were readily induced from the SOX9+ scl-progenitors (Supplementary Fig. 5A-B). Direct transplantation of SOX9+ scl-progenitors in vivo also formed cartilaginous and bone-like tissues (Supplementary Fig. 5C), supporting that the SOX9+ scl-progenitors differentiated from the optimized method still retained the osteochondral differentiation capability.

A The strategy for stepwise induction of SOX9+ sclerotomal progenitors (scl-progenitors) (top), mimicking the developmental process (bottom). B RT-qPCR confirmed sclerotome-specific gene expression in SOX9+ Scl-progenitors. Error bars: mean ± sd), n = 6 samples. ACTB (beta-actin) was used as the housekeeping control. ND: not detected in 40 cycles. C SOX9 and TWIST1 immunostaining in SOX9+ scl-progenitors at day 4. Nuclear staining: DAPI. Scale bars: 200 μm. D Western blotting on day-4 SOX9+ scl-progenitors confirmed expression of SOX9 and TWIST1. E Flow cytometry analysis revealed near uniform derivation of SOX9+ scl-progenitors. Traditional and optimized methods were compared. Undifferentiated SOX9-tdTomato hPSCs served the negative control. F The multi-batch analysis confirmed the consistent performance of the optimized differentiation method. Independent batches were analyzed by flow cytometry. The box (extending from 25th to 75th percentiles with median in the middle) and whiskers (minima to maxima) represent data from independent batches of differentiation (n = 9 for optimized method; n = 10 for traditional method). Statistics: Student’s t-test (two-tailed), by SPSS v26.0. ***p < 0.001 (p = 0.0002). G Experimental strategy for expanding SOX9+ scl-progenitors in vitro. H Continuous expansion of SOX9+ scl-progenitors for ~2 months in the defined medium. I SOX9+ scl-progenitors exhibited further maturation during in vitro expansion. RT-qPCR analysis showed increased expression of sclerotome-specific genes in the expanded cells. Error bars: mean ± sd), n = 6 samples. N.D., not detected in 40 cycles. Statistics: Student t-test (two-tailed), by SPSS v26.0. *p < 0.05 (p = 0.0127), **p < 0.01 (p = 0.0048 for PAX1 and p = 0.0024 for TWIST1); ****p < 0.0001 (p = 0.000038). J Immunostaining of SOX9 and TWIST1 confirmed the maintenance of cell identity in expanded scl-progenitors. Nuclear staining: DAPI. Scale bars: 200μm. K, L Flow cytometry analysis confirmed the maintenance of SOX9+ scl-progenitor’s identity at different passages. K The percentage of SOX9+ cells at different passages. L Repeatability test of three independent samples of expanded scl-progenitors at different passages by flow cytometry. M The expansion medium was specific for supporting SOX9+ scl-progenitors. SOX9+ scl-progenitor and SOX9- cells were sorted and compared in the expansion medium for multiple passages. Source data are provided as a Source Data file.
We next sought to investigate whether an in vitro expansion culture system could be established for SOX9+ scl-progenitors to exploit their potential in skeletal modeling and bone/cartilage regeneration. To develop such an expansion system, the following rationales were considered: (i) SHH signaling alone was sufficient for the specification of the sclerotomal fate from presomitic mesoderm (PSM) or somite (SM) cells58,59, indicating continuous stimulation with SHH activator might be a prerequisite for the maintenance of SOX9+ scl-progenitors; (ii) sclerotomal cells were induced to chondrogenesis by BMPs during development44,60, so BMP inhibition by LDN193189 might be required to prevent cells from differentiation; (iii) supplements promoting cell viability, such as ROCK inhibitors61, KSR62, and bovine serum albumin (BSA)63, along with factors promoting cell proliferation, such as bFGF22, might be needed as candidates for long-term cell expansion. We thus tested different combinations of pathway-modulating molecules in a basal medium containing SAG and LDN193189 (BMP inhibitor) and discovered that simultaneous SHH activation and BMP inhibition, supplemented with ROCK inhibitor, bFGF, KSR and BSA were essential for supporting the continuous expansion of SOX9+ scl-progenitors (Fig. 1G and Supplementary Fig. 6A–C). With such a culture system, SOX9+ scl-progenitors could be maintained for at least 2 months (~15 passages), which was about a 15,000-fold increase from the start (from initial 2x105 cells to 3x109 cells after 15 passages), with a population doubling time of 3.882 days (Fig. 1H and Supplementary Fig. 6D). RT-qPCR and western blotting further confirmed the maintenance of SOX9+ scl-progenitor’s identity, where sclerotome-specific markers were highly enriched in the expanded cells (Fig. 1I and Supplementary Fig. 6E). Interestingly, the expression levels of sclerotome-specific markers in the expanded SOX9+ scl-progenitors were even higher than the freshly differentiated ones. This suggested that there might be a maturation process during expansion, which was not uncommon in hPSC-derived functional cells such as neural progenitor cells64, cardiomyocytes65, and more recently, the limb-bud-like mesenchymal cells24. Besides RT-qPCR and western blotting, immunostaining also revealed that almost all the expanded cells were SOX9 and TWIST positive (Fig. 1J), which was further supported by flow cytometry analysis, where tdTomato-positive cells were over 99.0% with consistent minimal batch-to-bath variations across different passages (Fig. 1K-L). Moreover, when both sorted SOX9+ scl-progenitors and SOX9- cells (prepared by the traditional method in Supplementary Fig. 2A) were examined in the expansion culture, only the SOX9+ scl-progenitors could be expanded, supporting the expansion culture was specific for SOX9+ scl-progenitors (Fig. 1M).
Together, these data indicated that SOX9+ cells within the traditional sclerotomal differentiation were the key cell population with osteochondral differentiation capability. We have established an efficient differentiation strategy to derive SOX9+ scl-progenitors and they could be further propagated in scale using the expansion culture in vitro.
SOX9 + scl-progenitors could faithfully model key steps of endochondral ossification
Endochondral ossification is a critical process that forms most of the human skeleton during development. In previous studies, cells were typically centrifuged together to mimic the condensation and then artificially induced to hypertrophic chondrocytes in vitro using hormones before transplantation for further ossification in vivo. One such study first generated chondrocytes (FGF2 for 4-6 weeks) and then induced hypertrophy with T3 (triiodothyronine) for 10-21 days prior to transplantation25. An alternative strategy used high-density micromass culture to induce chondrocyte differentiation with TGFβ3 for 10 days, followed by BMP4 treatment for 4-12 weeks to induce chondrocyte hypertrophy22. As both strategies utilized hypertrophic chondrocytes for bone formation in vivo, the pre-hypertrophic phases (or chondrocytic phases) of endochondral ossification process12,66 could not be recapitulated under such conditions, such as spontaneous condensation, differentiation into cartilaginous analgen and the induction of growth plates with proliferating chondrocyte columns where skeletal stem cells reside9,10. In addition, the extremely long treatment process made it difficult to maintain experimental consistency. Therefore, we investigated whether a more robust osteogenic differentiation protocol could be developed to recapitulate the endochondral ossification process including the chondrocytic phases.
We reasoned that, as sclerotome forms the axial skeleton during development33, SOX9+ scl-progenitors, if indeed resembling their counterparts in vivo, should be at the pre-condensation stage. Upon proper induction, they should have the ability to spontaneously condense, differentiate and form cartilaginous analgen, and undergo endochondral ossification. A micromass culture system was then developed based on the following considerations: (i) sclerotomal cells were driven by BMP signals to induce chondrogenic anlagen formation in vivo, at which stage the early chondrocytes emerged44, suggesting that high-dose of BMPs might be required for sclerotomal specification towards the osteogenic fate; (ii) the proximal-distal (bone-cartilage) patterning of ribs was partially determined by SHH gradient67, indicating SHH activation might help to prevent chondrocytes from adopting the permanent cartilage fate. Therefore, a micromass culture with high-dose BMP2 (100 ng/ml) in combination with SHH stimulation by SAG was tested to induce mesenchyme condensation and differentiation of osteogenesis-committed early chondrocyte in SOX9+ scl-progenitors (Fig. 2A).

A Experimental strategy to recapitulate endochondral ossification using SOX9+ scl-progenitors. B 4-week grafts showed hypertrophic chondrocytes and initial calcification. i Graft overview with calcified hypertrophic region outlined (black dotted line). ii human origin confirmed by anti-KU80. iii-iv cartilaginous tissues visualized by Safranin O and COL II staining. v hypertrophy indicated by COL X immunostaining. vi calcified region marked by Alizarin red. Scale bars: 1 mm. C RT-qPCR indicated that the day-12 early chondrocytes lacked COL10A1 expression. Error bars: mean ± sd (n = 6 samples). Statistics: Student’s t-test (two-tailed), one-way ANOVA (SPSS v26.0). NS, not significant (p = 0.5715). ****p < 0.0001 (p = 0.00000000020). D 8-week grafts showed vascular invasion and multiple ossification centers. i blood vessel network on graft surface. ii-iii anti-KU80 confirmed human origin. iv multiple calcified regions and ossification centers revealed by Alizarin red. v Safranin O staining revealed multi-directional growth-plate-like tissues surrounding ossification centers. Scale bars: 1 mm (i, ii and iv), 500 μm (iii, v). E Representative growth plate-like tissues: anti-COL II (proliferating zone), anti-COL X (hypertrophic zone). Scale bars: 200 μm. F Columnar chondrocytes in growth plate-like tissues (8-week grafts). RZ: resting zone; PZ: proliferating zone; HZ: hypertrophic zone. Scale bars: 100 μm. G Masson’s trichrome staining revealed Bone-like tissues (8-week grafts). Scale bars: 200 μm. H Complete bone with a medullary cavity at 16 weeks. i bone formation and vascularization. ii-iv H&E staining showed bone marrow cavity formation with adipocytes, stromal, and hematopoietic cells. TB, trabecular bone. v-vi anti-COL I and anti-human VIM confirmed bone formation and its human origin. Arrowheads, COL I, and human VIM colocalization. vii-ix adipocytes were stained by Oil red and anti-SCD1. Anti-KU80 confirmed the human origin of some adipocytes. x host hematopoietic cells stained by anti-mouse CD45. Scale bars: 1 mm (i), 500 μm (ii, v), 50 μm (iii, vii-x), 20 μm (iv, vi). I Vascular architecture and mesenchymal stroma in 16-week grafts. i anti-COL I, and anti-EMCN revealed trabecular bone intertwined by sinusoidal vessels. ii host mesenchymal stromal cells verified by anti-LEPR. iii-iv niche cytokines were stained by anti-CXCL12 and anti-SCF. v EMCN+vascular endothelial cells enfolded by KU80+ human cells. Scale bars: 100 μm. Source data are provided as a Source Data file.
Indeed, when SOX9+ scl-progenitors were induced with such a method, a gradual increase of condensation marker CDH2 (N-CAD)68,69 was observed (Supplementary Fig. 7A). CDH2 expression reached the peak and started to drop at day 12, while SDC3, a marker of condensation termination and boundary setting70, was induced, alongside early chondrocyte markers ACAN, COL9A1, and EPYC (Supplementary Fig. 7A). These results were further confirmed by fluorescent microscopy, where SOX9+ scl-progenitors showed spontaneous condensation during the differentiation, accompanied by cell contraction (Supplementary Fig. 7B-C), initial compartmentalized expression of tdTomato, N-CAD, and COL II, and later the formation of cartilaginous ACAN+ cells (Supplementary Fig. 7C-D). To compare it with the reference micromass-based method, which induced hypertrophic chondrocytes from hPSC-derived paraxial mesoderm (theoretically covering the early chondrocyte phase)22, SOX9+ scl-progenitors were induced using both procedures and analyzed at day 12 post differentiation (Supplementary Fig. 8A-B). Although the reference procedure also induced the expression of the condensation markers (CDH2 and SDC3) and chondrogenic initiation genes (SOX5, SOX6 and ACAN)44,71, much lower expression levels of COL9A1 and EPYC were observed by day 12 (Supplementary Fig. 8C). These results suggested that the current method was more efficient in inducing early chondrocytes from SOX9+ scl-progenitors within the same time frame. Notably, human bone marrow-derived mesenchymal stem cells (BMSCs, characterized as CD90+CD73+CD105+CD45-CD14-CD34-CD19-HLA-DR- in Supplementary Fig. 8D) did not respond well to this induction method, with only a slight increase in CDH2 and SDC3 expression and no induction of chondrogenic initial genes (COL2A1, SOX5, SOX6 and ACAN) and early chondrocyte markers (COL9A1 or EPYC) (Supplementary Fig. 8E-G), indicating the method was specific for SOX9+ scl-progenitors only.
To evaluate whether the early chondrocytes could serve as the cartilaginous anlagen template and spontaneously form hypertrophic chondrocytes and bones in vivo, we injected the micromass-derived cells subcutaneously in NOD-SCID mice and harvested the samples at various time points (4, 8 and 16 weeks) for analysis (Fig. 2A). At 4 weeks, most of the transplants, whose human origin was confirmed by KU80 staining (Fig. 2B, i-ii), remained cartilaginous, as shown by positive staining of COL II and Safranin O (Fig. 2B, iii-iv). Hypertrophy and calcification had already started in some areas, as indicated by COL X and Alizarin Red staining, respectively (Fig. 2B, v-vi). RT-qPCR confirmed the induction of COL10A1 expression in the 4-week transplants, but not in early chondrocytes or SOX9+ scl-progenitors (Fig. 2C), supporting the non-hypertrophic nature of the micromass-derived cells, which was distinct from the previous methods where cells were directly induced to hypertrophic chondrocytes prior to transplantation. At 8 weeks, the vascular invasion was evident, and multiple regions appeared to be calcified (Alizarin red positive), while Safranin O staining revealed that a substantial portion of the transplant remained cartilaginous (Fig. 2D). Immunostaining of COL II and X indicated that these regions contained chondrocytes aligned in columns, undergoing gradual hypertrophy (Fig. 2E).
Interestingly, closer examination of the regions using Masson’s trichrome staining revealed multiple growth plate-like structures (Fig. 2F), where proliferating chondrocytes in columns were visible between the resting and hypertrophic zones. This suggested that the formation of growth plate-like structures might be a cell-autonomous process. In some areas, more matured bone-like structures were observed with Masson’s trichrome staining (Fig. 2G). Moreover, as recent reports suggested that growth plates provided the major residence for skeletal stem cells9,10, we investigated whether such stem cells could be detected in the transplants. Indeed, when the same gating strategies suggested by the original studies10,72 were applied (Supplementary Fig. 9A), FACS analysis of the SOX9+ scl-progenitor-derived human mini-bones at 8 weeks confirmed the presence of human skeletal stem cells as non-hematopoietic (CD235a-, CD45-), non-endothelial (TIE2-, CD31-), and PDPN+CD146-CD73+CD164+ (Supplementary Fig. 9B), which could be further verified by immunostaining (Supplementary Fig. 9C).
Most importantly, at 16 weeks, the transplants further matured, where cortical and trabecular bones with a clear presence of bone marrow cavities, hematopoietic cells, and adipose tissues could be observed (Fig. 2H, i-iv). Co-immunostaining of human VIMENTIN and COL I confirmed that the bone-like tissues were indeed derived from the transplanted cells (Fig. 2H, v-vi). Oil Red, SCD173, and human KU80 staining confirmed the adipocytes were also derived, at least partially, from the transplanted cells (Fig. 2H, vii-ix), while mouse CD45 staining indicated hematopoietic cells were from the host (Fig. 2H, x). Immunostaining of bone marrow markers further revealed that the formation of supportive niches for hematopoiesis within the grafts (Fig. 2I), with COL I and EMCN to mark trabecular bone intertwined by sinusoidal vessels74, LEPR to mark recruited host stromal cells75, and CXCL12 and SCF to mark cytokines secreted by the stromal cells75,76. Interestingly, some EMCN+ vascular endothelial cells were enfolded by cells of human origin (KU80+), suggesting that part of the stroma might be directly derived from transplanted cells.
Together, these data suggested that by establishing a micromass-based induction method, hPSC-derived SOX9+ scl-progenitors could undergo spontaneous condensation to form early chondrocytes. These osteogenesis-committed chondrocytes could further differentiate into hypertrophic chondrocytes and induced vascular invasion and ossification upon in vivo transplantation, forming growth plate-like structures and bone marrow stroma, thus faithfully modeling the key steps of endochondral ossification of human bone development.
Generating polarized growth plate-like structures by SOX9 + scl-progenitors-derived osteochondral fusions
During the chondrocytic phase of endochondral ossification, one of the key features is the formation of the growth plate, where proliferating chondrocytes are aligned and organized into columns, and then gradually mature, transit to hypertrophic chondrocytes, and cause longitudinal growth by growing away from the ossification center66. The localization of the growth plate is largely dictated by local paracrine regulators, such as parathyroid hormone-related protein (PTHrP) and Indian hedgehog (IHH), secreted by the resting zone chondrocytes and the ossification center, respectively77,78. How growth plates in the human skeleton are formed remains elusive due to the lack of a model that can recapitulate the process.
From our data mentioned above, when micromass-derived osteogenesis-committed early chondrocytes were subcutaneously injected in single-cell suspensions, there were multiple random ossification centers and at the interface between the ossification centers and non-calcified cartilaginous tissues, growth plate-like structures could be observed (Fig. 2D-F). We hypothesized that, if a fusion construct could be generated with one end resistant to hypertrophy and the other prone to hypertrophy and ossification, it might be possible to induce the formation of growth plate-like structures in a more defined and polarized manner at the osteochondral interface.
To ensure the generation of a single center with synchronized ossification, the micromass-derived early chondrocytes were centrifuged and cultured in CI medium for condensation as spheroids (hereafter, named osteo-spheroids), and then transplanted both subcutaneously and under kidney capsule to evaluate the ossification process (Fig. 3A). Under the kidney capsule, the osteo-spheroid underwent synchronized hypertrophy and ossification within one week (Supplementary Fig. 10A), while the subcutaneous transplant remained cartilaginous at such time point (Supplementary Fig. 10B), suggesting that the kidney capsule might be the more suitable transplantation site for fast ossification. Within 4 weeks, the osteo-spheroids became hypertrophic and were completely calcified, as revealed by positive staining of Alizarin red and COL X/II staining (Fig. 3B), indicating the generation of a single ossification center.

A Experimental procedure to construct osteogenic spheroids. The osteogenesis-committed early chondrocytes (micromass-derived) were centrifuged for spheroid formation followed by being cultured in CI medium for 7 days to further condense. B The osteo-spheroids formed calcified tissues at 4 weeks under kidney capsule, as revealed by Alizarin red, COL II, and COL X staining. Scale bars: 500 μm (before transplantation), 100 μm (post transplantation). C Experimental procedure to construct chondrogenic spheroids. The SOX9+ scl-progenitors were centrifuged for spheroid formation followed by being cultured in CI medium for chondroprogenitor (Cp) differentiation. D Representative images of the repaired proximal tibial plateau compared to the sham control. Scale bars: 500 μm. E Chondroprogenitors (Cps, day 4) could efficiently generate articular cartilage in vivo. H&E: visualization of tissue morphology. Toluidine blue staining, Alcian blue, and Safranin O were used to detect cartilaginous extracellular matrix (ECM). COL II and ACAN: chondrocyte markers. Human vimentin (VIM): human origin marker. PRG4 marked the superficial layer of the articular cartilage. No typical cartilage tissues were formed in the sham control. For each group, n = 3 mice. Scale bars: 100 μm. F No hypertrophy or ossification was detected in Cp-derived cartilaginous tissues. COL X: hypertrophic chondrocyte marker. COL I: the marker for fibrous and bone tissues. The mouse femur was used as the positive control. Scale bars: 100 μm. G Transplanted Cps did not contribute to the subchondral bone. COL I: the marker for fibrous and bone tissues. human VIMENTIN: the marker for transplanted Cps. Scale bars: 200μm. (H) The chondro-spheroids alone only formed cartilaginous tissues under kidney capsule. Samples were analyzed at 4 weeks post-transplantation. The histological evaluations were conducted the same as (B). Scale bars: 500 μm (upper), 100 μm (lower).
Next, we sought to generate a hypertrophy-resistant chondrogenic center using our modified chondrogenic induction method (Supplementary Fig. 2G and Fig. 3C). SOX9+ scl-progenitors were cultured as spheroids and differentiated into chondroprogenitors (Cps) in a CI medium. RT-qPCR revealed that Cp-markers were induced as early as day 4 of differentiation, including SOX9, COL2A1, SOX5, SOX6, COL9A1, and ACAN44,71 (Supplementary Fig. 11A). Fluorescence detection and immunostaining of these Cp spheroids further confirmed that the cells highly expressed SOX9 and COL II, but not COL I/X (Supplementary Fig. 11B). These results were also compared with the previously reported methods22,25,41 and consistently showed higher induction of Cp markers (Supplementary Fig. 12A–C), indicating that the optimized protocol was more efficient in inducing Cp differentiation. In addition, BMSCs also did not achieve comparable Cp marker expression levels under such conditions, (Supplementary Fig. 12D–F), suggesting that the induction was cell-specific and more suitable for the development-relevant SOX9+ scl-progenitors.
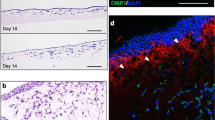
To evaluate the chondrogenic potential and resistance to hypertrophy of differentiated Cps, injury sites were created in the proximal medial tibial plateau, and Cp cells were transplanted to repair the cartilage. At 8 weeks, the defects were efficiently and consistently repaired by the transplanted Cps, whereas the sham control group failed to do so (Fig. 3D-E and Supplementary Fig. 13). The cartilaginous nature of the regenerated tissue was further confirmed by the negative immunostaining of COL I and X (Fig. 3F). Moreover, co-staining of the repaired site with COL I and human VIMENTIN indicated that transplanted Cps did not participate in the subchondral bone formation (Fig. 3G). Besides the proximal medial tibial plateau, the SOX9+ scl-progenitor-derived Cps could also repair the articular cartilage in the trochlear groove of the distal femur79 within 4 weeks and maintained their cartilage fate for at least 12 weeks (Supplementary Fig. 14). No tendency of hypertrophy and ossification was observed in the repaired tissues (Supplementary Fig. 14D and H), and the transplanted cells did not form any subchondral bone (Supplementary Fig. 15). When the transplanted cells were live traced for 8 weeks with AkaLuc, a reporter capable of tracing cells in single-cell resolution80, they showed no abnormal distribution or growth in the host animals (Supplementary Fig. 16). Together, these data indicated that the differentiated chondroprogenitors could maintain their chondrogenic potential in vivo and were resistant to ossification.
We next sought to evaluate the formation of chondrogenic center under kidney capsule. Chondro-spheroids were generated using Cps differentiated for 14 days, as RT-qPCR revealed that the induction reached a plateau at day 14, with sharp upregulation of both early (COL2A1, ACAN) and late (EPYC, COMP) chondrocyte markers, which were also confirmed by immunostaining (Supplementary Fig. 17). When the chondro-spheroids were transplanted under the kidney capsule for 4 weeks, they remained cartilaginous, as indicated by positive staining of COL II but negative staining for Alizarin red and COL X (Fig. 3H), which were in sharp contrast with the calcified osteo-spheroids-derived tissues (Fig. 3B).
To generate the fusion construct, the osteogenesis-committed early chondrocytes and day-14 chondroprogenitors were sequentially centrifuged together at the ratio of 1:1 followed by 7-day culture in CI medium before further transplantation under the kidney capsule (Fig. 4A). Interestingly, within 4 weeks, growth plate-like structures with columnar chondrocytes were detected at the interface between chondrogenic and osteogenic regions (Fig. 4B). The human origin of these structures was also confirmed by KU80 staining. More importantly, the orientation of the columnar chondrocytes was longitudinal towards the ossification center consisting of calcified hypertrophic chondrocytes, as revealed by staining with Alizarin red, Masson’s trichrome, and Safranin O (Fig. 4B). The growth plate-like structures were consistently present only in the fusion constructs with defined proliferative chondrocyte zones, but not in chondro- or osteo-spheroids alone (Fig. 4C). Moreover, immunostaining of regional markers, COL II (a marker for epiphyseal chondrocytes), IHH (a marker for pre-hypertrophic chondrocyte), COL X (a marker for hypertrophic chondrocyte), SP7 (a marker for osteoblast/progenitor cells) and EMCN (a marker for endothelial cells) further supported that the cellular organization of the growth plate-like structures closely resembled the primary mouse growth plates at PN9 (Fig. 4D-E).

A The experimental procedure to construct osteochondral fusion using SOX9+ scl-progenitor-derived osteo-spheroids (containing osteogenesis-committed early chondrocytes) and chondro-spheroids (containing day-14 chondroprogenitors. Scale bars: 500 μm. B The osteochondral fusions generated polarized growth plate-like tissues with unidirectional proliferating chondrocyte columns. Alizarin red: calcified region. Masson’s trichrome and Safranin O showed the overall morphology and the growth plate-like regions. The dotted line: proliferating zone. KU80 marked human cells. RZ, resting zone. PZ, proliferating zone. HZ, hypertrophic zone. Scale bars: 100μm. C Consistent formation of polarized growth plate-like tissues in the osteochondral fusion grafts. The width of the proliferating zones was measured and compared with transplants with chondro- and osteo-spheroids as controls. Error bars (mean ± sd) represented data from five independent transplants (two sections per transplant). Scale bars: 100 μm. Statistics: Student’s t-test (two-tailed), by SPSS v26.0. **p < 0.01 (p = 0.0011), ****p < 0.0001 (p = 0.00000025). D, E Similar expression of regional markers between the osteochondral fusion (E) with primary growth plate tissues from mouse femur at PN9 (D). COL II, pan-chondrocyte marker. IHH, prehypertrophic chondrocyte marker. COLX, hypertrophic chondrocyte marker. SP7, osteoblast, and bone progenitor marker. EMCN, endothelial marker. Scale bars: 100 μm. Source data are provided as a Source Data file.
Together, these results indicated that polarized growth plate-like structures could be generated at the defined regions by creating a chondrogenic and osteogenic interface using SOX9+ scl-progenitors. These structures expressed regional markers similar to primary growth plates.
Functional and cellular characterization of induced growth plate-like structures
Next, we sought to investigate if the growth plate-like tissues were capable of mediating longitudinal bone growth, a critical functional feature of primary growth plates. Immunostaining indicated that proliferative chondrocytes (PCNA+COL2A1+ flattened) indeed resided in the proliferating zone of the osteochondral fusion, similar to the primary mouse growth plate at PN9 (Fig. 5A). When analyzed weekly over a 4-week period, the fusion constructs gradually increased in size, with the average length increasing from 0.55 mm to 2.43 mm and width from 0.53 mm to 1.98 mm (Fig. 5B and C). Growth in width appeared to slow over time, while growth in length became more evident. The aspect ratio (i.e., length/width) increased from approximately 1.0 (round-shaped) prior to transplantation to an average of 1.4 at 4 weeks, indicating polarized and longitudinal elongation (Fig. 5D). Linear regression analysis showed a growth rate of 0.61 mm/week, approximately half the growth rate of reported embryonic long bones81,82. As the primary long bones typically have two growth plate regions, one on each end, the growth rate of the our fusion constructs was thus similar to the primary ones. Furthermore, H&E staining indicated that the enlargement was largely due to an increased spongiosa compartment, supporting the possible contribution from the growth plate-like structures (Fig. 5E). Additionally, the proliferating zone (flattened cells stained by COL II) appeared as early as the 1-week time point and was itself elongating, supplying chondrocyte pools for maturation (Fig. 5F-G). Therefore, these results supported that the growth plate-like structures were indeed functional and could mediate longitudinal bone growth.

A Anti-COL II and anti-PCNA confirmed proliferative cells in growth plate-like structures of osteochondral fusions, resembling PN9 mouse femur. Scale bars: 100 μm. B–E Osteochondral fusions mediated longitudinal bone growth. Grafts were harvested weekly for 4 weeks for analysis. B graft overview. Scale bar: 500 μm. C tissue growth quantification. Each dot represents a single fusion graft (n = 6 for week-0, n = 3 for each time point). D increasing aspect ratios indicated longitudinal elongation. Error bars: mean ± sd. Statistics: Student t-test (two-tailed). p = 0.0190 (week-4 vs. week-2); p = 0.0031 (week-4 vs. week-1); p = 0.00000081 (week-4 vs. week-0). E H&E staining. Scale bar: 200 μm. F, G Anti-COL II confirmed PZ (proliferative chondrocyte zone) elongation. F PZs marked by dotted lines (flattened cells stained by anti-COL II). Nuclear staining: DAPI. Scale bars: 200 μm. G quantitative analysis of PZ elongation. Data collected from independent samples (n = 6 for week-0, n = 3 for each time point). Statistics: Student t-test (two-tailed), *p < 0.05, ***p < 0.001, ****p < 0.0001. p = 0.0337 (week-4 vs. week-3); p = 0.0007 (week-4 vs. week-2); p = 0.000085 (week-4 vs. week-1); p = 0.0000010 (week-4 vs. week-0). H, I Mechanical testing by nanoindentation. H 4-week grafts coronally cryosectioned, fixed, and polished with indents made on resting/proliferative zone (R/PZ), hypertrophic zone (HZ), and spongiosa. I elastic modulus, and hardness calculated using the Oliver-Pharr method. Error bars: mean ± sd, n = 6 indents for each zone. Statistics: Student t-test (two-tailed) and One-way ANOVA, **p < 0.01, ****p < 0.0001. **p = 0.0061, ****p = 0.0000045 (Young’s modulus); **p = 0.0023, ****p = 0.000059 (hardness). J ScRNA-seq identified human skeletal and host cells in 4-week grafts. EpiphyChon, epiphyseal chondrocytes. ProlifChon, proliferative chondrocytes. PrehyperChon, prehypertrophic chondrocytes. HyperChon, hypertrophic chondrocytes. OB, osteoblast/progenitor cells. PeriChon, perichondral cells. EndoCell, endothelial cells. HematoCell, hematopoietic cells. StromCell, stromal cells. K Marker gene expression in each cluster by dot plot. (L-O) Integration analysis with human embryonic long bones (PCW8.0). L UMAP visualization of integrated dataset. M overlaps between osteochondral fusions and embryonic long bones across cell types. N similar gene expression profiles between fusion grafts and embryonic long bones. O comparable expression of endochondral ossification lineage-specific genes. Source data are provided as a Source Data file.
Besides the elongation, we also measured the mechanical properties of the constructed osteochondral fusions. The 4-week osteochondral fusions were analyzed using nanoindentation, with elastic modulus and hardness determined via the Oliver-Pharr method83 (Fig. 5H). The mechanical properties of the osteochondral fusions displayed zonal variations, with the average modulus increasing approximately 160-fold (from 26.1 MPa to 4.16 GPa) and hardness rising about 240-fold (from 2.15 MPa to 0.51 GPa) from growth plate cartilage to bone, indicating tissue maturation (Fig. 5I). Within the growth plate cartilage, hypertrophic chondrocytes were observed with lower modulus and hardness compared to the resting and proliferating chondrocytes, consistent with previous reports in bovines and rabbits84,85. Spongiosa bone displayed the highest rigidity, with its average modulus (4.16 GPa) and hardness (0.51 GPa) resembling human fetal long bones86 (Fig. 5I).
To further characterize the osteochondral fusions at the single-cell level, we conducted scRNA-seq on the 4-week grafts using the 10x Genomics platform. From the dataset, cells of both human and mouse origins were detected (Fig. 5J). The human fraction contained the full spectrum of cells during growth-plate maturation (Fig. 5K and Supplementary Fig. 18A), including epiphyseal chondrocytes (COL2A1+IHH-COLX-SP7-)87 with resting-zone cells (UCMA+ IGF2+ MKI67- TOP2A- PCNA-)88, proliferating chondrocytes (MKI67+ TOP2A+ PCNA+)89, prehypertrophic chondrocytes (IHH+ ALPL+ COLXlow)77, hypertrophic chondrocytes (IHH+ COLXhigh VEGFA+ MMP13+)90,91, and osteoblasts/progenitor cells (SP7+ DLX5+ SPP1+ BGLAP2+ IBSP+)92,93,94,95. These observations were in sharp contrast to the hPSC-derived hyaline chondrocytes generated in vitro, which was compared as a negative control dataset41 and showed minimal clustering with our fusion-derived cells (Supplementary Fig. 18B). Additionally, the referenced hPSC-derived hyaline chondrocytes expressed lower levels of pan-chondrocyte (COL2A1, COL9A3, COMP, MATN4)41 and proliferating genes (PCNA and TOP2A) compared to our fusion-derived matured chondrocytes. No expression of resting (UCMA), prehypertrophic (IHH, ALPL), hypertrophic (COL10A1, VEGFA), and osteoprogenitor/osteoblast (SP7) markers were observed in the reference dataset either, supporting that the detected endochondral ossification-related genes in our dataset were specific (Supplementary Fig. 18C). Meanwhile, host-derived Emcn+ endothelial cells and hematopoietic cells were also detected (Fig. 5K and Supplementary Fig. 18A), indicating vascular invasion into the spongiosa. Additionally, our scRNA-seq data was also compared with that from primary human embryonic long bones containing growth plate structures (PCW8.0)96. Integrated analysis revealed general overlaps across clusters of the growth-plate lineage between the two datasets (Fig. 5L-M), with similarities in both the overall (Fig. 5N) and selected stage-specific (Fig. 5O) gene expressions for each cell cluster.
Collectively, these results indicated that the osteochondral fusions could induce the formation of growth plate-like structures, which exhibited functional and cellular similarities to in vivo primary growth plates and could mediate longitudinal bone growth.
ITGA9 enabled reporter-independent isolation of SOX9 + scl-progenitors
Although SOX9+ scl-progenitors exhibited strong osteochondrogenic differentiation capability, the current enrichment strategy still relied on the expression of the tdTomato reporter, which hindered their potential use in translational studies. To establish a reporter-independent isolation method for these cells, transcriptome analysis comparing both SOX9+ scl-progenitors vs SOX9- cells or hPSCs, was conducted. The cells were derived from the traditional method (Supplementary Fig. 2A) as the improved strategy did not yield enough SOX9- cells for comparison.
From the transcriptome analysis, candidate surface markers enriched in SOX9+ scl-progenitors were identified (Figs. 6A, S19A) and their expressions were validated by RT-qPCR (Fig. 6B). By ranking gene fold changes and TPMs (Transcripts per Million) in SOX9+ scl-progenitors, ITGA9 was identified as one of the most significantly enriched candidates (Supplementary Fig. 19B-C). Itga9 expression in mouse primary sclerotome was further confirmed by re-analyzing previously published scRNA-seq data97 (Supplementary Fig. 20A-B). Immunostaining on the E10.5 mouse embryo also verified its enrichment in the sclerotomal region (Sox9+Pax9+Nestin-) (Supplementary Fig. 20C-D), consistent with the FACS analysis data showing that Itga9+ cells of the E10.5 trunk region were also Sox9+ (Supplementary Fig. 20E). These results indicated that ITGA9 might be a specific marker for SOX9+ scl-progenitors.

A RNA-seq analysis revealed multiple surface markers enriched in SOX9+ scl-progenitors. Transcriptomic analysis was done by comparing SOX9+ scl-progenitors vs SOX9- cells sorted from Day-6 sclerotomal cells from the traditional method (n = 3 for each group). Statistics: Exact test based on the negative binomial distribution (two-sided) by edgeR. B Confirmation of differential gene expression between SOX9+ scl-progenitors and SOX9- cells by RT-qPCR. For RT-qPCR, error bars (mean ± sd) represented data from six biological replicates; for RNA-seq, error bars (mean ± sd) represented data from three biological replicates. C Confirmation of SOX9 and ITGA9 co-expression in differentiated sclerotomal cells by flow cytometry. Isotype antibody served as the control. The proportion of SOX9+ cells among the ITGA9+ population was shown on the right, which was gated according to undifferentiated hPSCs. D Confirmation of SOX9 and ITGA9 co-expression by immunostaining. ITGA9+ cells were sorted and stained with anti-SOX9. Scale bars: 50 μm. E Sclerotome-specific genes were highly enriched in ITGA9+ cells. Error bars:mean ± sd, n = 6 samples. F ITGA9+ cells retained osteochondral competence. Cell differentiation was induced the same as in Supplementary Fig. 2G. Scale bars: 200 μm (Alizarin red); 100 μm (spheroids). G Negative immunostaining of hypertrophic and osteogenic markers indicated no ossification. COL X: hypertrophic chondrocyte marker. COL I: the marker for fibrous and bone tissues. The mouse femur was used as the positive control. Scale bars: 100 μm. H, I Sclerotome-specific genes were also enriched in ITGA9+ cells derived by the current method. Error bars:mean ± sd, n = 6 samples. ACTB served as the housekeeping control. Statistics: Student t-test (two-tailed), by SPSS v26.0. ***p < 0.001, ****p < 0.0001. p = 0.0000021 (ITGA9); p = 0.00000045 (SOX9); p = 0.0000086 (PAX1); p = 0.0002 (PAX9); p = 0.0000084 (TWIST1); p = 0.0000062 (NKX3-2). Source data are provided as a Source Data file.
Next, Cell sorting using the anti-human ITGA9 antibody confirmed that SOX9+ scl-progenitors were indeed enriched in the ITGA9+ fraction (Fig. 6C), consistent with SOX9 immunostaining (Fig. 6D) and RT-qPCR analysis of sclerotome-specific markers (Fig. 6E). To confirm that ITGA9+ cells were indeed scl-progenitors with robust osteochondral differentiation capability, ITGA9+ cells were subject to in-vitro differentiation as mentioned previously (Supplementary Fig. 2G). Compared to ITGA9- cells, only ITGA9+ cells exhibited strong osteochondrogenic competence (Fig. 6F) and showed no expression of COL I/X (Fig. 6G). Since these ITGA9+ cells were derived using the traditional method, we also confirmed these observations in the optimized differentiation method. Under such conditions, ITGA9+ cells were also present and enriched with sclerotome-specific marker expressions (Fig. 6H-I). Together, these data indicated that ITGA9 was a specific surface marker for SOX9+ scl-progenitors and enabled reporter-independent isolation of these cells.
Discussion
In this study, we characterized and identified SOX9+ scl-progenitors as the key population in the hPSC-derived sclerotomal cells with strong osteochondral potential. To resolve the cell heterogeneity issue of the traditional method, an improved stepwise induction protocol was then developed, which could achieve an average of 99.21% ± 0.24% efficiency in deriving SOX9+ scl-progenitors from multiple hPSC lines. A differentiation toolkit was then established for inducing these cells further into either chondroprogenitors that could generate articular cartilage or osteogenesis-committed early chondrocytes that faithfully model key steps of endochondral ossification, including spontaneous condensation, anlagen formation, chondrocyte hypertrophy, vascular invasion, and bone formation. Notably, when osteo- and chondro-spheroids derived from these progenitors were fused, they self-organized into elongating growth plate-like structures that exhibited molecular and cellular similarities to primary growth plates. Additionally, we identified ITGA9 as a specific marker for isolating SOX9+ progenitors and established a culture system that supported their continuous expansion in vitro. Our findings highlight SOX9+ scl-progenitors as a valuable cell source for modeling human skeletal development and bioengineering of cartilage and bones (Fig. 7).

Illustrative summary highlighting key features of this study.
Mammalian skeletal development and homeostasis can be divided into two phases: the developmental phase, where bones are formed through mesenchymal progenitors following developmental processes, and the postnatal homeostatic phase, where osteoblasts and osteoclasts coordinate to maintain normal bone metabolism and remodeling1. While substantial research has been done in studying the function of resident skeletal stem cells, osteoblasts, and osteoclasts in bone remodeling and repair1,98,99, less is known on early bone development. Especially, the molecular mechanisms on how endochondral ossification is initiated and growth plate regions are organized and regulated during human long bone development are still unclear. Genetic mutations that affect growth plate formation have been found to cause pathological conditions in patients, including Hutchinson-Gilford progeria syndrome (HGPS) caused by mutant Lamin A (LMNA)100,101, Achondrogenesis Type IB caused by mutant SLC26A2 (or DTDST)102, and Hereditary multiple osteochondromas (HMO)103, a rare condition causing benign bone tumors to grow along the growth plates. Besides these serious diseases, short stature is another growth plate-related condition, and the genetic variants that caused most of the cases of short stature were still unknown (thus often defined as Idiopathic Short Stature, ISS), which makes it a challenge for clinical diagnosis and effective management104. Given the difficulty of recapitulating many of these pathological conditions in mouse models and the limitations of other human PSC-based strategies in modeling key steps of early endochondral ossification, our method for generating SOX9+ sclerotomal progenitors, along with the accompanying osteochondral differentiation as well as growth-plates induction strategies, may offer a promising tool for studying early human skeletal development. In particular, the basic biological mechanisms underlying key factors such as self-condensation, chondrogenic induction, cellular elements involved in growth plate organization, and genetic mutations associated with osteochondral disorders could be systematically investigated.
In addition to skeletal modeling by constructing osteochondral fusions, the SOX9+ scl-progenitors-derived chondroprogenitors reported here were also demonstrated having ability to repair focal cartilage injuries in NOD/SCID mice, suggesting their potential for applications in cartilage regenerative medicine. Compared to autologous chondrocyte implantation (ACI), SOX9+ scl-progenitors-derived Cps could avoid potential surgery-caused donor-site morbidity and may be better suited for elderly patients when autologous tissues were also affected by ageing-caused degeneration. Additionally, hPSC-derived chondrocytes resembled a juvenile state, demonstrating higher proliferative potential and resistance to inflammatory cytokines compared to adult chondrocytes105. However, further investigation on SOX9+ scl-progenitors-derived Cps and their tissue regeneration potential within the inflammatory microenvironment as well as treatment safety is essential to determine their applicability in treating cartilage defects in OA patients.
As a critical regulator during early skeletal development, including the pre-condensation (the mesenchyme) and downstream lineage-specific progenitor stages, Sox9 was also used previously by other studies as marker of chondrogenic progenitors. For example, it was reported that hPSCs could acquire an ectomesenchymal fate by TGFβ inhibitor (SB431542) treatment followed by further propagation in a medium containing FGF2 and SB431542. This strategy could obtain expandable CD271+CD73+ cells, where ~20% of them were SOX9- positive30. In another study, the hPSCs were first induced to a mesodermal fate (expressing TCF15 and MEOX1), followed by cell expansion to obtain the SOX9+ subpopulation28. Although those cells were shown to be chondrogenic upon further lineage induction, their developmental relevance was not clear. Both of these cells showed SOX9 expression as well as some mesenchymal cell markers (e.g., CD271 and CD73), but the heterogeneity was clearly observed. Therefore, further research is required to compare the different hPSC-derived progenitors and the in vivo counterparts in terms of their molecular similarities and functional differences to better define their developmental relationships.
Additionally, the identification of ITGA9, encoding integrin alpha-9, as a specific marker for SOX9+ scl-progenitors has provided reporter-free approaches to isolate sclerotomal cells from differentiated hPSCs. In situ immunostaining revealed co-expression of Sox9 and Itga9 in the sclerotome, highlighting intriguing aspects of sclerotome development. Previous studies have shown that integrin alpha-9 has been associated with processes such as cell migration, invasion, and epithelial-mesenchymal transition (EMT) in tumor cells106. During sclerotome specification, the epithelial somite undergoes EMT, transitioning from a compact, adhesive state to a motile mesenchymal state. This transition is critical for generating a migratory cell population capable of positioning around midline axial structures to form the axial column107,108. Itga9 may contribute to this process by promoting EMT, facilitating cell migration, and regulating proliferation. Interestingly, from published data on chromatin immunoprecipitation of SOX9 protein, it had direct binding on known Itga9 promoter or enhancer regions in multiple samples, including mouse chondrocytes109 as well as both mouse and bovine fetal testis110. Similarly, ITGA9 expression might also be directly regulated by SOX9 during sclerotome specification, which would require further investigation.
It is also important to note that under physiological conditions, sclerotomal progenitors give rise to the axial skeleton, not the appendicular skeleton. Therefore, it remains to be determined whether this strategy can be used to generate appendicular growth plates and articular cartilage. Although the elongation rates seemed comparable to primary ones, the actual size was still smaller than human fetal long bones. The initial cell number, the host environment (mouse physiology), as well as the transplantation site, may influence the size and growth of the transplanted tissues. Nanoindentation indicated that the hardness of the bone obtained in the study was comparable to that during the embryonic stage. However, further optimization is still needed to improve the construction of osteochondral fusions in order to better mimic the architecture of primary long bones and eventually use it as a robust preclinical drug evaluation platform. Moreover, the current approach still depends on in vivo transplantation to enable proper vascular invasion and bone marrow formation, emphasizing the need for continued efforts to fully replicate these processes in vitro.
Methods
Cell culture
All experiments involving human pluripotent stem cells were reviewed and approved by the Institutional Animal Care and Use Committee at the College of Life Sciences, Sichuan University. H1 hPSCs, H9 hPSCs, NC3-1 hiPSCs, IMR904 hiPSCs (gifts from Prof. Yan Liu at Nanjing Medical University), UC01 hiPSCs (a gift from Prof. Jinglei Cai from GIBH-CAS), SOX9-tdTomato UC01 hiPSC and AkaLuc-BsdR-SOX9-tdTomato UC01 hiPSCs (constructed using UC01 hiPSCs) were routinely propagated in Pluripotency Growth Master 1 (PGM1, Cellapy, Cat# CA1007500) medium on cell culture plates coated with Matrigel (Corning, Cat# 354230 or MeisenCTCC Cat#MS0101ZY). The working solution of Matrigel (MatriWS) was prepared using DMEM/F12 (Gibco, Cat# C11330500) at 1 mg/12 ml, and the coating was conducted at 37 °C for at least 1 h. All the hPSC/iPSC lines were maintained carefully to avoid any spontaneous differentiation. Cell splitting was carried out at 1:5 with 0.5 mM EDTA (ThermoFisher, Cat# AM9260G) in PBS (Gibco, Cat# C10010500BT) when the confluency reached 80%. The cells should be lifted from plates as fine clumps, and the ROCK inhibitor was excluded during routine culture unless otherwise mentioned. Human bone marrow stem cells (hBMSCs, OriCell, Cat# HUXMA-01001) were cultured in MEM alpha (Gibco, Cat# 12571063) supplemented with 20% v/v lot-selected non-heat inactivated fetal bovine serum (FBS) (Hyclone, Cat# SH30084.03) and 1% v/v penicillin/streptomycin (pen-strep, Gibco, Cat#15140-122). The cells were passaged with 0.25% trypsin-EDTA (Thermofisher, Cat# 25200056) when the confluency reached ~70% and replated at the ratio of 1:5.
UC01 (SOX9-tdTomato) hiPSC line construction
Knock-in was achieved by non-homologous mediated end joining using the CRISPR/Cas9 system with guide RNAs (gRNAs) targeting the 3’ untranslated region (UTR) of SOX9 (Supplementary Fig. 1A). Four gRNAs were designed using the online tool CRISPR111, with PAM sequences located within 100 base pairs (bp) downstream from the stop codon of SOX9. The targeting vector containing the IRES-tdTomato-PGK-PuroR cassette was synthesized by BGI Group, Beijing. The gRNAs were annealed and subcloned into both the PX330 vector harboring the Cas9-expressing cassette and the targeting vector. To generate the UC01 (SOX9-tdTomato) hiPSC line, parental UC01 hiPSCs were seeded sparsely (~40% confluency) as single cells on MatriWS-coated 12-well plates. The next day, the cells were co-transfected with 800 ng of PX330-SOX9 gRNA and 500 ng of IRES-tdTomato-PGK-PuroR targeting vector by Lipofectamine Stem Transfection Reagent (ThermoFisher, Cat# STEM00015) using manufacturer’s protocol. Drug selection was carried out using puromycin for 10 days till the confluency reached 80%. The concentration of puromycin increased gradually from 0.2 to 0.5 μg/ml and was kept constant at 0.5 μg/ml for subsequent colony screening. To pick correct colonies, the transfection pools were dissociated into single cells by Accutase (Innovative Cell Technologies, Cat#AT-104) and seeded at a density of 6–10 cells/well in MatriWS-coated 96-well plates and cultured with PGM1 supplemented with 0.5 μg/ml puromycin and 10 μM ROCK inhibitor, Y27632 (MCE, Cat#HY-10071). This density would result in ~15% single colonies in each plate, which were propagated further till sufficient cells could be harvested for genotyping. To verify correct integration, both 5’ and 3’ ends were amplified by PCR and were subject to Sanger sequencing. gRNAs used are listed in Supplementary Data 1.
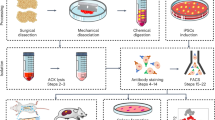
Generation of SOX9 + sclerotomal progenitors (scl-progenitors)
Before differentiation, routinely cultured hPSCs were re-split to another MatriWS-coated plate. To control the starting cell density, the cells were detached from culture plates by 0.5 mM EDTA as fine clumps when the confluency reached 80% and split at 1:5. Once plating, vigorous shaking should be done several times to make sure the clumps were evenly distributed across the culture plates. The next day, fine and separated colonies should be present on the culture plates and any signs of over-confluency represented by large-sized integrated multi-colonies should be avoided to maintain differentiation efficiency.
All differentiation was conducted in the two-dimensional serum-free condition using a modified chemically defined medium with insulin (CDMi)37. To prepare CDMi, IMDM (Gibco, Cat# 31980-097) and F12 (Gibco, Cat# 31765-092) were mixed at 1:1 supplemented with 1% v/v concentrated lipids (Gibco, Cat# 11905-031), 450 μM monothioglycerol (Sigma, Cat# M6145), 0.7 μg/mL insulin (Solarbio, Cat# I8830), 15 μg/ml transferrin (Sigma, Cat# T0665), 1% v/v pen-strep, and 1 mg/mL polyvinyl alcohol (Sigma, Cat# P8136-250G). The polyvinyl alcohol was first solved at 25 mg/ml in ultrapure water (UP H2O) by heating to 85°C and then cooled on ice before being added to the medium.
Our method to generate SOX9+ scl-progenitors was optimized based on the traditional one37,38,39,41 as illustrated in Supplementary Fig. 2A. To start traditional differentiation, day-0 hPSCs were gently washed with the wash medium (formulated as 3 mg/ml bovine serum albumin (BSA) in DMEM/F12) and were induced to primitive streak (PS) in CDMi containing Activin A (30 ng/ml, Solarbio, Cat#P00101), CHIR99021 (7 μM, MCE, Cat#HY-10182) and bFGF (20 ng/ml, Peprotech, Cat#100-18B) for 24 h; the next day, day-1 PS was washed with wash medium and differentiated to presomitic mesoderm (PSM) in CDMi containing SB431542 (20 μM, MCE, Cat#HY-10431), CHIR99021 (3μΜ), LDN193189 (125 nM, MCE, Cat#HY-12071A), and bFGF (40 ng/ml) for another 24 h; subsequently, the day-2 PSM was differentiated to somite (SM) in CDMi containing XAV939 (0.5 μM) and PD0325901 (1 μM, MCE, Cat#HY-10254) for 24 h; the following day, the SM was washed with wash medium before induced to sclerotome by CDMi containing SAG (200 nM) and LDN193189 (600 nM) for 72 h with medium changed every day. The final SOX9+ scl-progenitors were obtained by fluorescence-activated cell sorting (FACS).
Our optimized method was established inspired by the chicken PSM explant culture experiments44,59, where the PSM is induced to a sclerotomal fate directly without somite specification (somite-skipping). To start optimized differentiation, the induction of PS and PSM was kept the same as the traditional method38. Subsequently, the day-2 PSM was dissociated to single cells by TrypLE (Gibco, Cat#12605028) and four-fold volume of wash medium was used to stop digestion. The cells were then pelleted by centrifugation at 300 × g for 3 min and resuspended for further wash and cell counting. Next, cell suspension was re-collected by centrifugation again at 300 g for 3 min and resuspended in CDMi supplemented with SAG (200 nM, MCE, Cat#HY-12848), LDN193189 (600 nM), XAV939 (0.5 μM, MCE, Cat#HY-15147), and Y27632 (10 μM) for the following SOX9+ scl-progenitor differentiation. The cells were seeded onto another MatriWS-coated plate at the density of 1 × 105 cells/cm2. Also, vigorous shaking should be done several times immediately after plating to make sure the cells are evenly distributed across the culture plates. The cells were cultured for 24 h where the medium was replaced with CDMi without Y27632 for another 24 h of induction. (Note: the plating density of PSM could be variable for different hPSC lines. Cell death may occur when cells are plated too sparsely, while overcrowding may cause unintended and nonuniform differentiation. Optimization should be done when changing the lines).
Quantitative PCR
Total RNAs were extracted from cells using TRI Reagent (Sigma-Aldrich, Cat# T9424) and then reverse-transcribed to cDNA with the PrimeScript RT reagent kit with gDNA Eraser (TAKARA, Cat# RR047A) according to the manufacturer’s protocol. The quantitative PCR was performed using a CFX384 Optics Module (Bio-Rad, USA) with AceQ Universal SYBR qPCR Master Mix (Vazyme, Cat# Q511-02), and the relative expression levels were calculated with the comparative threshold cycle (∆∆CT) method. Primers are listed in Supplementary Data 1.
Immunocytochemical staining
Adherent cells cultured on a cover glass or confocal dish were fixed with 4% paraformaldehyde (PFA) for 8 min at room temperature (RT) before being washed twice with PBS. Permeabilization was conducted for intramembranous antigen with 0.3% PBST (v/v Triton X-100 in PBS), followed by blocking with blocking buffer (5% bovine serum albumin (BSA, Sigma, Cat# A3311-10G) in PBS) at RT for 1 h. Subsequently, the cells were incubated with primary antibodies diluted in blocking buffer overnight at 4 °C and then were washed three times with PBS. Next, the cells were stained with secondary antibodies in the dark at RT for 1 h and were washed again with PBS. For cells cultured on cover glass, the samples were mounted using the mounting medium with 4,6-diamidino-2-phenylindole (DAPI) (ZSJB-BIO, Cat# ZLI-9557) for nuclear staining. For cells cultured on the confocal dish, the nuclei were stained by Hoechst 33342 (ThermoFisher, Cat#H3570). Immunofluorescent microscopy was conducted with Leica TCS SP5II and Olympus VS200. Antibodies used are listed in Supplementary Data 2.
Western blotting
Proteins of the day-0 undifferentiated SOX9-tdTomato hPSCs, day-4 SOX9+ scl-progenitors, and expanded scl-progenitors were extracted with RIPA lysis buffer (Cat# 89901, Thermo Fisher) and were quantified using a Pierce™ BCA Protein Assay Kits (Cat# 23227, Thermo Fisher) under manufacturer’s instructions. 10ug proteins were separated by electrophoresis using SDS–PAGE followed by being transferred to 0.45 μm PVDF membranes (Cat# 88518, Thermo Fisher). After blocking with 5% (w/v) skim milk in PBST (0.02% (v/v) Tween 20 in PBS), the membranes were incubated with Anti-SOX9 and TWIST1 (Cat# 25465-1-AP, Proteintech) overnight at 4 °C. After incubation with horseradish peroxidase (HRP)-conjugated antibodies, the membranes were reacted with ECL Western Blotting Substrate (Cat# KF8005, Affinity) to detect signals using the Bio-Rad Gel Doc XR+ Documentation System. Antibodies used are listed in Supplementary Data 2.
Flow cytometry
For analysis or sorting SOX9+ cells using the SOX9-tdTomato hPSCs, the cells were first dissociated into single cells by Accutase and were washed once with 5% BSA. The cells were then resuspended in PBS and were filtered with a 70-μm strainer for downstream experiments. Day-0 undifferentiated SOX9-tdTomato hPSCs were used as the negative control.
For intracellular or cell-surface protein staining, cells were dissociated into single cells by Accutase and were counted with Cell Counter (ThermoFisher). 1 × 106 cells were used for a single test. Permeabilization was performed using 0.3% PBST for intracellular antigens. The cells were then incubated with 100 μl of primary antibodies in 5% BSA at RT for 40 minutes and were then washed twice with 5% BSA. The cells were subsequently stained with 100 μl of secondary antibodies (1:400) in 5% BSA at RT for 30 min protected from light and washed twice with 5% BSA. Samples stained with isotype IgG were used as the negative control.
For detection of the secreted proteins, including COL II, COL X, COL I, etc., the cells were first treated with 10 μg/ml of brefeldin A supplemented in the culture medium for 24 h to block protein secretion from the endoplasmic reticulum. For xenografts transplanted in NOD/SCID mice, 500 μl of 0.5 mg/ml brefeldin A in PBS was injected intravenously into the host mice 6 h before tissue harvesting. The samples were then dissociated into single cells and were subject to routine intracellular protein staining protocol as mentioned above. All the analysis and sorting were done by BD Fortessa and Aria III (BD Biosciences, USA). Antibodies used are listed in Supplementary Data 2.
Generation of chondroprogenitors (Cps) and cartilaginous/chondral spheroids from SOX9+ scl-progenitors
Adherent cells were first dissociated into single cells by TrypLE, and sorted cells were collected by centrifugation at 300 × g for 3 min. After being washed once with wash medium, 1.5 × 105 cells were collected in the U-shaped ultra-low attachment 96-well plate, suspended in CDMi supplemented with 200 nM SAG, 600 nM LDN193189, and 10 μM Y27632 followed by centrifugation at 300 × g for 6 min to form spheroids overnight. After being washed once with wash medium, the spheroids were cultured by chondrocyte induction (CI) medium formulated as DMEM supplemented with 0.1 μM dexamethasone (DEX) (MCE, Cat#HY-14648), 50 μM ascorbic acid (Sigma, Cat#A8960), 50 μg/ml L-proline (Sigma-Aldrich, Cat#P0380), 20 ng/ml TGFβ3 (Peprotech, Cat#100-36E), 20 ng/ml ΒMP2 (Peprotech, Cat#120-02), 20 ng/ml bFGF, 1% v/v ITS + 1 (Sigma-Aldrich, Cat#12521-5 ML), 1% v/v sodium pyruvate (Gibco, Cat#11360070), and 1% v/v pen-strep. For articular cartilage repair assays (Fig. 3D-G and Supplementary Figs. 13–16), the cell induction lasts for 4 days to obtain day-4 Cps; for construction of osteochondral fusions, the induction lasts for 14 days to generate day-14 Cps (Figs. 3H, 4–5 and Supplementary Fig. 17); and for evaluation of chondrogenic competence by in vitro differentiation, the induction should be 42 days to generate cartilaginous /chondral spheroids (Fig. 6F-G and Supplementary Fig. 2G-I and 5A).
Osteogenic induction in vitro for Alizarin red staining
Osteogenic induction was conducted to evaluate the osteogenic competence of SOX9+ scl-progenitors, expanded scl-progenitors, SOX9- cells, ITGA9+ and ITGA9- cells. Adherent cells were dissociated into single cells by TrypLE, and sorted cells were collected by centrifugation at 300 g for 3 min. After being washed once with wash medium, 1 × 105 cells were seeded on a 96-well plate and cultured with α-MEM supplemented with 10% v/v FBS, β-glycerophosphate (10 mM, Millipore, Cat#35675), DEX (0.1 μM), ascorbic acid (50 μg/ml) and 1% v/v pen-strep for 30 days.
Histological analysis for in vitro osteochondrogenic assays
Day-42 chondroprogenitors were fixed with 4% PFA overnight at 4 °C. After being washed twice in PBS, samples were embedded in paraffin. Sections were made at 6 μm using paraffin microtome (Leica RM2245) and were stained by Alcian blue, Safranin O, Toluidine blue O, COL II, COL I, COL X after rehydration in xylene and ethanol. For Alcian blue staining, 5% w/v Alcian blue 8GX (Sigma, Cat#A5268-100G) stock solution was first prepared in PBS and brought to 1% w/v working solution using 0.1 N HCl. The sections were treated with 0.1 N HCl for 2 min, followed by staining with Alcian blue working solution at RT for 30 min. After being washed in 0.1 N HCl for 2 min, the sections were rinsed in ultrapure water to remove any acidic content for subsequent nuclei staining by hematoxylin. For Safranin O staining, nuclei staining was first conducted with Wiegert’s hematoxylin (Solarbio, Cat#G1371). The sections were then stained with 0.1% w/v Fast Green FCF (Sigma-Aldrich, Cat#F7252-100G, in UP H2O) for 90 s, followed by rinsing in 1% glacial acetic acid for 90 s. Subsequently, the sections were stained with 5% Safranin O (Sigma, Cat#S2255-100G, in UP H2O) for 1 min. After rapid dehydration in 95% ethanol and 100% ethanol for 1 min each, the sections were cleared in xylene and mounted with neutral resin. The Toluidine blue O staining was conducted using the manufacturer’s protocol (Solarbio, Cat# G2543). For Col II staining, antigen retrieval was first carried out using pepsin (Sigma-Aldrich, Cat#R2283) for 30 min at 37 °C. The sections were then treated with 5% w/v BSA (Sigma-Aldrich, Cat#A9418-100G) and incubated with Col II antibody overnight at 4 °C. After being washed in PBS three times, the subsequent secondary antibody staining and DAB reaction were conducted using the Universal IHC Staining Kit (ZSJB-BIO, Cat# PV6000) according to the manufacturer’s protocols. The nuclei were stained with hematoxylin. The same procedures were used for COL I and COL X staining except that the heat-induced antigen retrieval was conducted using citrate buffer (pH 6.0). Antibodies used are listed in Supplementary Data 2.
Day 30 osteogenic cells were fixed with 4% PFA at RT for 8 min and washed twice with PBS. The fixed cells were then stained by 2% w/v Alizarin red (Sigma-Aldrich, A5533) at RT for 2 min, followed by washing three times with PBS.
Comparative evaluation assays for chondroprogenitor generation
Current protocol vs. reference methods
For the current protocol, the chondroprogenitors were generated using SOX9+ scl-progenitors by centrifugation as mentioned above, and were cultured in the CI medium for 4 days. For reference method I41, the SOX9+ scl-progenitors were induced to chondroprogenitors in CDMi containing 20 ng/ml BMP4 for indicated days. For reference method II22, 2 × 105 SOX9+ scl-progenitors were seeded on MatriWS-coated plate suspended in 20 μl base chondrogenic medium (DMEM supplemented with 1% v/v ITS, 50 μg/ml ascorbic acid, 40 μg/ml L-proline, 0.1 μM DEX) supplemented with 20 ng/ml bFGF to adhere at 37 °C for 30 min–1 h. The micromass was then covered by the base chondrogenic medium supplemented with TGFβ3 (10 ng/ml) and was maintained in the medium for indicated days. For reference method IΙΙ25, the chondroprogenitors were generated using SOX9+ scl-progenitors by centrifugation as mentioned above. The spheroids were cultured in APEL2 (Stem cell technology, Cat#05270) supplemented with 5% PFHM II (ThermoFisher, Cat# 12040077), 20 ng/ml bFGF, and 1% Pen-strep for indicated days. All differentiation was conducted in the same time frame (4 days) for comparison by RT-qPCR.
hBMSCs differentiation
The hBMSCs (human bone marrow mesenchymal stem cells) were dissociated into single cells using TrypLE and were differentiated into chondroprogenitors using the current protocol as mentioned above. The hBMSCs-derived chondroprogenitors were harvested on day 4 for analysis by RT-qPCR.
Animal use
All animal experiments were approved by the Institutional Animal Care and Use Committee at the College of Life Sciences, Sichuan University (Protocol numbers: SCU2003013 and SCU2203010). For all animal experiments, 8-week-old male NOD/SCID mice (NOD/ShiLtJGpt-Prkdcem26Cd52/Gpt, #T001492, GemPharmatech Co., Ltd, Jiangsu) were used. The animals were maintained under standardized conditions with the temperature (18-23 °C), humidity (50% ± 10%), and light (12 h light/dark cycle) controlled, in individually ventilated cages always with companion mice, and had free access to food and water.
Articular cartilage repair assays
The NOD/SCID mice were anesthetized with avertin (1.25% w/v 2,2,2-tribromoethanol) at 20 μl/g (body weight). The working solution of avertin was prepared by mixing 1.25 g of 2,2,2-tribromoethanol (Sigma-Aldrich, Cat#T48402) with 2.5 ml of 2-methyl-2-butanol (Sigma-Aldrich, Cat#721123) and dissolving in 97.5 ml of UP H2O. Subsequently, the skin of implantation sites was shaved and sterilized under standard procedures. A 3 mm incision was made medial to the left knee, and the femoral condyle was exposed by lateral dislocation of the patella. A 0.5 mm wide and 1 mm depth focal defect was then made on the articular surface of the distal femur or the medial tibial plateau to reach the underlying subchondral bone. Subsequently, 2–3 day-4 chondroprogenitors were implanted in the defect site and were secured by the blood from the subchondral area. After the patella was repositioned, the muscle and skin were closed with 8-0 and 6-0 prolene suture, respectively. The mice with defects but no cell implantation were prepared as sham controls.
Transplantation of SOX9 + scl-progenitors under renal capsule
Day-4 SOX9+ scl-progenitors were dissociated to single cells by TrypLE and 5.0 × 105 cells were suspended in CDMi supplemented with SAG (200μΜ), LDN193189 (600nΜ) and Y27632 (10 μM) followed by centrifugation at 300 × g for 6 min in the U-shaped ultra-low attachment 96-well plate for spheroid formation overnight. Before transplantation, the spheroids were embedded in 10 μl of Collagen I-Matrigel mixture (1:1) at 37 °C for 5 min for solidification. The Collagen I solution (2.4 mg/ml) was prepared by adding 1 ml of MEM (10x, Gibco, Cat# 11430030) to 8 ml of Type I Collagen stock (3 mg/ml, Bovine, Advanced Biomatrix, Cat# 5005). After pH adjustment to 7.0 by 1 M NaOH, the volume was brought to 10 ml by DMEM (Gibco, Cat# 11965092).
For transplantation, 8-week NOD/SCID mice were anesthetized, and the skin was disinfected as mentioned above. The spheroids were then transplanted under the kidney capsule, and the grafts were collected 8 weeks post-transplantation for histological evaluations.
Derivation of osteogenesis-committed early chondrocytes in micromass culture
6 μl of MatriWS was first added on the central region of a 24-well plate for pre-coating at 37 °C for at least 1 h. 2 ×105 SOX9+ scl-progenitors resuspended in 5 μl of CDMi containing SAG (200 nM), LDN193189 (600 nM), and Y27632 (10 μM) were seeded on the coated region and were cultured for 30 min at 37 °C to adhere. Subsequently, an adequate volume of medium mentioned above was added to cover the cells. The next day, the medium was changed to the induction medium formulated as CDMi containing SAG (200nΜ), ΒΜP2 (100 ng/ml), and 1% v/v knockout serum replacement (KSR, Gibco, Cat#10828082) after washing with the wash medium. The micromass was then cultured for 12 days with the induction medium refreshed daily.
Subcutaneous injection of micromass-derived early chondrocytes
Each micromass was dissociated into single cells by Accutase and was suspended in 70 μl of Matrigel. The skin was then tented using the tip of the insulin syringe (BD, Cat#324903), and the cells were injected into the subcutaneous site left to the midline of mice.
Histological section, evaluation, and immunostaining
For the frozen section, all the samples were fixed with 4% PFA overnight and were dehydrated in 30% sucrose solution until sunk. The knee joints were decalcified in 19% w/v EDTA (in PBS, pH = 7.0) at RT for 24 h before dehydration. The dehydrated tissues were then embedded with OCT (optimal cutting temperature, SAKURA, Cat#4583) and placed at −80 °C for 24 h. All the samples were sectioned at 6μm thickness with the microtome cryostat (Leica M1950). Tissue sections were stored at −20 °C before histological analysis.
For the paraffin section, the samples were fixed with 4% PFA overnight and were dehydrated in a serial gradient of ethanol from 50% to 100%. Also, the knee joints were decalcified before dehydration as mentioned above. After tissue clearing in xylene, the samples were embedded in paraffin overnight.
For the histological staining, sections were re-hydrated and stained with H&E, Masson’s trichrome (Solarbio, Cat# G1346), and Oil red (Solarbio, Cat# G1260) according to the manufacturer’s protocol. Safranin O, Toluidine blue, and Alcian blue staining were conducted as mentioned before.
For the RNAscope assay, the Probe-Mm-Pax9-C2 (ACDbio, Cat# 454321-C2) and Opal 690 Reagent (Asbio, Cat# ASOP690) were used following the manufacturer’s protocols. All slides were examined under Olympus VS200 after nuclear staining by DAPI.
For the immunofluorescent staining, antigen retrieval was conducted after re-hydration, if necessary, followed by a double rinse in PBS to remove the retrieval reagent. For intracellular antigens, the sections were permeabilized with 0.3% Triton X-100 for 8 min. Then, the sections were treated with blocking solution (5% BSA in PBS) at RT for 1 h and incubated overnight at 4 °C with primary antibodies diluted in blocking solution. Secondary antibodies were stained at RT for 1 hr protected from light and then washed three times with PBS before being mounted with DAPI. All the sections were examined under Leica TCS SP5II and Olympus VS200. Antibodies used are listed in Supplementary Data 2.
For the immunostaining assays in the main figures, FACS analysis was also performed to further confirm the presence of key cell types (Supplementary Fig. 21). Representative images of negative control (isotype IgG) were shown in Supplementary Fig. 22.
Comparative evaluation assays for osteogenesis-committed early chondrocyte generation
Current protocol vs the reference method
Osteogenesis-committed early chondrocyte induction using the current protocol has been described above. For the reference method22, 2 × 105 SOX9+ scl-progenitors were seeded on MatriWS-coated plate in 20 μl base chondrogenic medium (DMEM supplemented with 1% v/v ITS, 50 μg/ml ascorbic acid, 40 μg/ml L-proline, 0.1 μM DEX) supplemented with 20 ng/ml bFGF to adhere at 37 °C for 30 min–1 h. The micromass was then covered by base chondrogenic medium supplemented with TGFβ3 (10 ng/μl) only and was maintained in the medium for 10 days at which time the TGFβ3 was replaced by 50 ng/ml BMP4 for the rest of the experiments (2 days). All the differentiation was conducted in the same time frame (12 days) for comparison by RT-qPCR.
hBMSCs differentiation by the current protocol
The hBMSCs were differentiated by micromass using the current protocol following the same procedure. The medium was refreshed every day. The samples were harvested on day 12 for analysis by RT-qPCR.
Characterization of skeletal stem cells in micromass-derived tissues
The micromass was generated from SOX9+ scl-progenitors as mentioned above followed by subcutaneous injection to NOD/SCID mice. The grafts were collected 8 weeks post-transplantation for either FACS analysis or immunostaining. For FACS analysis, the grafts were dissociated to single cells by Collagenase I and Collagenase II mixture and were stained by indicated reagent and antibodies (DAPI, CD235a, CD31, TIE2, CD45, PDPN, CD146, CD73, CD164). The gating strategy was carefully defined using Fluorescence Minus One (FMO) control. Antibodies used are listed in Supplementary Data 2.
Tissue clearing for the imaging of immunostained micromass
The osteogenesis-committed early chondrocytes were generated using SOX9+ scl-progenitor on the confocal dish as mentioned above, and the immunostaining was conducted with routine protocols. After nuclear staining by Hoechst 33342, the cells were dehydrated in the increasing gradient of 30%, 50%, 80%, and 100% ethanol for 5 min each. After an extra 10 min dehydration in 100% ethanol, the cells were cleared with a clearing solution containing 80% v/v ethyl cinnamate (ECi) (Sigma, Cat# 112372) and 20% v/v polyethylene glycol (Sigma, Cat# 447943) at RT for 10 min. For longer preservation, the samples were immersed in the clearing solution at 4 °C until imaging with the lid sealed by paraffin film. The imaging was conducted with Olympus SpinSR10.
Construction of osteochondral fusions
The osteochondral fusions were constructed by the integration of chondroprogenitors and osteogenesis-committed early chondrocytes. The chondroprogenitors were first generated by pelleting 1 × 105 SOX9+ scl-progenitors suspended in CI medium supplemented with Y27632 (10 μM) in Ultra-low attachment 96-well plate and were cultured in CI medium for 14 days for chondrogenic differentiation and maturation. The osteogenesis-committed early chondrocytes were prepared as mentioned above in micromass (12 days). For reconstitution, the osteogenesis-committed early chondrocytes were first dissociated to single cells by Accutase and resuspended using CI medium supplemented with 10 μM of Y27632 as 70 μl/micromass. Each micromass suspension (70 μl) was then placed into the low attachment plate for another round of centrifugation at 300 × g for 6 min. The day-14 chondroprogenitors were then fused with the pelleted osteogenesis-committed early chondrocytes at 1:1 by centrifugation at 300 × g for 6 min. After being cultured overnight, the medium was changed to CI medium without Y27632. The fusions were cultured in vitro for 7 days in CI medium with the medium changed every 2 days before transplanted under the renal capsule of NOD/SCID mice. The osteogenesis-committed early chondrocyte alone (osteo-spheroids) and chondroprogenitor alone (chondro-spheroids), were prepared as control and were cultured in the CI medium for the same duration (7 days) followed by transplantation.
Bulk-population RNA sequencing for cell-surface marker screening
For bulk-population RNA-seq analysis, day-0 hPSCs were differentiated to sclerotome using the traditional 6-day protocol (Supplementary Fig. 2A), and total RNAs were extracted respectively from undifferentiated human PSCs, sorted SOX9+ scl-progenitors and SOX9- cells using TRI Reagent. Library preparation and sequencing were performed by Novogene Inc., Beijing. Raw data in FASTQ format were obtained using the CASAVA 1.8.4 pipeline and were filtered to remove the adapters and paired reads with N.A. bases of more than 10%. The clean reads were then mapped to hg19 using the HISAT2 software (version 2.2.1). Read counts for each gene were based on sense-strand data obtained using the featureCounts (version 2.0.1) software from the Subread package. Transcripts per million (TPM) was used for the normalization of gene expression levels accounting for both gene length and sample variations112. Differential gene expression analysis was conducted using edgeR113 comparing the SOX9+ scl-progenitors vs SOX9- cells and SOX9+ scl-progenitors vs hPSCs. For cell-surface marker screening, the candidate list was obtained by setting the cutoff line as log2(fold change)>1 and adjusted p-value < 0.05. Genes with TPM < 50 in the SOX9+ population and genes with TPM > 50 in SOX9- cells were excluded. Moreover, genes with TPM > 10 in hPSCs were further discarded to remove any genes enriched in undifferentiated cells. Bulk-population RNA-seq data have been submitted to the GEO database (GSE222109).
Single-cell RNA sequencing
Derivation of the SOX9+ scl-progenitors(cell hashing-based scRNA-seq): Cell hashing using TotalSeqTM reagents enabled to run simultaneously multiple samples in a single lane. Undifferentiated UC01 hiPSCs, PS, PSM, and SOX9+ scl-progenitors derived by the current method (Fig. 1A) were prepared as mentioned above and were dissociated into single cells using Accutase. After being washed once with 5% BSA (in PBS), the cells were filtered with a 40-μm strainer followed by cell counting. Then indicated number of cells was incubated with TotalSeqTM antibodies using the manufacturer’s protocol. The undifferentiationUC01, PS, PSM, and SOX9+ scl-progenitors were labeled by TotalSeq™-A0251 anti-human Hashtag 1 (Biolegend, Cat# 394601), TotalSeq™-A0252 anti-human Hashtag 2 (Biolegend, Cat# 394603), TotalSeq™-A0253 anti-human Hashtag 3 (Biolegend, Cat# 394605) and TotalSeq™-A0254 anti-human Hashtag 4 (Biolegend, Cat# 394607), respectively.
After washing and resuspending in 0.4% BSA (in PBS), an equal number of cells from each stage were combined for cell viability assessment using Countess II Automated Cell Counter (Thermo Fisher, Cat# AMQAX1000). Library preparation and sequencing were performed by Singleron Biotechnologies Inc, Nanjing using GEXSCOPE® Single Cell RNA Library Kit for library construction.
Generation of the osteochondral fusions(10x Genomics-based scRNA-seq): 4-week osteochondral fusion grafts were prepared for single-cell RNA sequencing where three transplants were pooled as a single sample to obtain enough cells. The host tissue (from kidney) was thoroughly removed to prevent contamination. The grafts were then put into 0.4 WÃ1/4nsch units/ml of LiberaseTM (Roche, Cat# 5401127001) digestion medium and were cut into small pieces followed by incubation at 37 °C for 15 min with shaking (150 rpm). The supernatant was then collected into the wash buffer composed of DMEM/F12, 10% FBS, 1% pen-strep after filtered by 70 μm cell strainers. These steps were repeated for several times until no large fragments could be found. After centrifuged at 300 × g for 5 min, the cells were treated with hemolysis reagents (Cat# C3702-500ml, Beyotime) for 10 min at RT. Then the cells were centrifuged again at 300 × g for 5 min and were resuspended in the wash buffer for another filtering using 40 μm strainers.
Library preparation was conducted using Chromium Single Cell 3’ Library & Single Cell 3ʹ v3 Gel Beads under manufacture’s instruction by Shanghai OE Biotech. Co., Ltd., and sequencing was performed with Illumina Novaseq 6000.
Processing of scRNA-seq raw data
Cell hashing-based scRNA-seq: The CeleScope software was obtained from Git Hub (https://github.com/singleron-RD/CeleScope). Alignment, filtering, barcode counting, and UMI counting were performed with the CeleScope pipeline to generate a feature-barcode matrix and determine clusters. scRNA-seq data have been submitted to the GEO database (GSE232109).
10x Genomics-based scRNA-seq: The 10x Genomics data were processed using Cell Ranger (ver. 7.1.0) for barcode counting and UMI counting. The human and mouse reference genomes (Human (GRCh38) and mouse (GRCm39) reference-20240A) were used for sequence alignment. ScRNA-seq data have been submitted to the GEO database (GSE275943).
Downstream analysis of the scRNA-seq data
Cell hashing-based scRNA-seq: quality control, t-SNE visualization of cell clustering, reconstruction of differentiation trajectories, and gene expression dynamics profiling: The feature-barcode matrix was processed using R and Seurat v4.2.0 package. For the hashtag library, signal-to-noise ratio (SNR) was defined as the ratio of the highest and the second-highest UMI count value of hashtags for a certain cell. Cells that have low UMI counts (highest UMI count less than 200) and low SNR (SNR less than 20) were filtered out as multiplets and undetermined cells. The cells with mitochondrial gene content of more than 10% were further removed as low-quality cells. Subsequently, we filtered out cells that have UMI counts (number of counts, nCounts) and expressing genes (number of features, nFeatures) under the 10th percentile among the total cells. The gene expression levels for each cell were normalized by the sequencing depth, multiplying a default size factor of 10,000, and then log-transformed. To reduce variance introduced by cell cycle stages, Seurat’s function “ScaleData” was used to regress out the effect with cell cycle markers extracted from the Seurat database “cc.genes”114. Next, dimensionality reduction and visualization were performed using Seurat’s function “RunTSNE” where the cells were colored by hashtags in a tSNE (t-Distributed Stochastic Neighbor Embedding) plot.
For reconstruction of differentiation trajectories, datasets of cells passed the quality control steps including UMI and SNR threshold for hashtag library, mitochondrial gene filtering, nCount, and nFeature threshold as mentioned above were converted into a Monocle object using Monocle 2. To cluster and order the cells in an unsupervised approach, the genes used were selected based on the level of expression and variability across cells by using the “dispersionTable” function to calculate the average expression and dispersion values of each gene. Genes with a mean expression higher than 1 were selected and the trajectories were visualized using the Monocle function “plot_cell_trajectory”. Subsequently, the gene expression dynamics across the trajectories were plotted using the “plot_genes_in_pseudotime” function to show the correct induction of stage-specific genes during differentiation.
10x Genomics-based scRNA-seq: quality control, UMAP visualization of cell clustering and integration of datasets: The feature-barcode matrix was processed using Seurat v4.3.0 package. Quality control was performed before subsequent downstream analysis. Cells with expressing genes (number of features, nFeatures) less than 200 and more than 7500, and with UMI counts (number of counts, nCounts) less than 50,000 were removed. Cells with mitochondrial gene content of more than 10% were further discarded as low-quality cells. The gene expression levels for each cell were normalized by the sequencing depth, multiplying a default size factor of 10,000, and then log-transformed. Next, dimensionality reduction and clustering were performed using “RunPCA”, “FindNeighbors” and “FindClusters” and were visualized in a UMAP (Uniform Manifold Approximation and Projection) plot by “RunUMAP”.
For comparison of our osteochondral fusion data to the published PW8.0 embryonic long bone data (GSE143753), the cells carrying mouse genes were first filtered by removing the cells with murine gene content more than 0.2%. The datasets were then integrated using “FindIntegrationAnchors” and “IntegrateData”. The integrated data were then normalized using “ScaleData” followed by dimensionality reduction and clustering using “RunPCA”, “FindNeighbors” and “FindClusters” and were then visualized by “RunUMAP” with cells colored by clusters.
Re-clustering of mouse organogenesis atlas
To test if Itga9 was expressed in primary sclerotome during mouse development, single-cell RNA sequencing data on mouse embryonic organogenesis was re-analyzed by dissecting the sclerotome populations present in the E10.5 embryo from the reference atlas97 and re-clustered using non-linear dimensionality reduction algorithm to visualize clusters on T-distributed Stochastic Neighbor Embedding (t-SNE) plot. The sclerotome population was annotated using differentially expressed genes.
Expansion culture of SOX9 + scl-progenitors
SOX9+ scl-progenitors were dissociated to single cells with Accutase. After being washed once with wash medium, cells were seeded on MatriWS-coated plates at 1 × 105 cells/cm2 and cultured with CDMi supplemented with SAG (200 nM), LDN19389 (600 nM), Y27632 (10 μM), bFGF (10 ng/ml), 1% v/v KSR, 5 mg/ml BSA for 3–5 days before the next splitting. The medium was refreshed every day. (Note: the optimal concentration of LDN193189 varied for different hPSC lines. Cell death may occur when the concentration was too high. Optimization should be done when changing the lines).
Mechanical assessment of osteochondral fusions by nanoindentation
To identify zonal mechanical properties of our polarized growth plate-like structures, 4-week osteochondral fusions were collected for mechanical testing by Nanoindenter (Bruker, Hysitron TI-950). The tissues were stored at −80 °C until use and were cryo-sectioned coronally to reflect a flat plane for indentation. The samples were fixed in epoxy resin, polished with a soft cloth, and measured with a 10 μm-diameter diamond, Berkovich tip (Bruker, Cat# TI0039; Young’s modulus, 1140 Gpa; Poisson’s ratio, 0.07). Indentations were made on resting/proliferative zone (R/PZ), hypertrophic zone (HZ), and the spongiosa bone using peak forces of 100 μN for R/PZ and HZ, and 10 mN for spongiosa bone, respectively. For R/PZ and HZ, the loading rate was keeping constant at 20 μN/s for 5 s, followed by 2 s of loading and 5 s of unloading. For spongiosa bone, the loading rate was keeping constant at 2 mN/s for 5 s, followed by 2 s of loading and 5 s of unloading. The Young’s modulus and hardness of the detected regions were calculated with the Poisson’s ratio of 0.3 following the Oliver-Pharr method83.
Bioluminescence imaging
AkaLuc-BsdR UC01 (SOX9-tdTomato) hPSCs was constructed by co-transfection of piggyBac transposase-expressing vector (pBase) and the donor vector harboring EF1-AkaLuc and PGK-EM7-BsdR cassette between transposon-specific inverted terminal repeats (ITR) to the parental UC01 (SOX9-tdTomato hPSC) cells. Articular cartilage repair assays were conducted using the AkaLuc-BsdR cell-derived chondroprogenitors. Upon imaging, the mice we
Statistics and reproducibility
Statistical analysis was performed with SPSS 26.0 software. All data were expressed as mean ± standard deviation (SD). The statistical method used for each dataset was described in the figure legend respectively. p < 0.05 was deemed statistically significant. Representative images of SOX9+ scl-progenitor characterization, expansion, surface marker identification, and in vitro differentiation were obtained from at least three independent experiments. Staining assays for all tissue transplants, including cartilage repair and transplantation of cells under the subcutaneous site or renal capsule, were performed on samples from three or more independent procedures. RT-qPCR experiments were conducted on six biologically independent samples. And in other figures, the experiments were performed on three or more independent differentiations.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All RNA sequencing data collected in this study have been deposited in the NCBI Gene Expression Omnibus (GEO) (accession number GSE222109 https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE222109; GSE232109 https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE232109; GSE275943 https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE275943). We have also acquired published RNA-seq datasets from previous studies, including human embryonic long bones (PCW8.0, GSE143753), mouse organogenesis atlas (GSE119945 [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE119945]) and hPSC-derived chondrocytes (GSE160787 [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE160787]). All other data generated in this study are provided in the Supplementary Information and Source Data files. Source data are provided with this paper.
References
Salhotra, A., Shah, H. N., Levi, B. & Longaker, M. T. Mechanisms of bone development and repair. Nat. Rev. Mol. Cell Biol. 21, 696–711 (2020).
Cui, A. et al. Global, regional prevalence, incidence and risk factors of knee osteoarthritis in population-based studies. EClinicalMedicine 29–30, 100587 (2020).
Makris, E. A., Gomoll, A. H., Malizos, K. N., Hu, J. C. & Athanasiou, K. A. Repair and tissue engineering techniques for articular cartilage. Nat. Rev. Rheumatol. 11, 21–34 (2015).
Latourte, A., Kloppenburg, M. & Richette, P. Emerging pharmaceutical therapies for osteoarthritis. Nat. Rev. Rheumatol. 16, 673–688 (2020).
Katz, J. N., Arant, K. R. & Loeser, R. F. Diagnosis and treatment of hip and knee osteoarthritis: a review. JAMA 325, 568–578 (2021).
Janicki, J. A. & Alman, B. Scoliosis: Review of diagnosis and treatment. Paediatr. Child Health 12, 771–776 (2007).
Chrcanovic, B. R. & Gomez, R. S. Idiopathic bone cavity of the jaws: an updated analysis of the cases reported in the literature. Int. J. Oral. Maxillofac. Surg. 48, 886–894 (2019).
Ciancia, S. et al. Osteoporosis in children and adolescents: when to suspect and how to diagnose it. Eur. J. Pediatr. 181, 2549–2561 (2022).
Chan, C. K. et al. Identification and specification of the mouse skeletal stem cell. Cell 160, 285–298 (2015).
Chan, C. K. F. et al. Identification of the human skeletal stem cell. Cell 175, 43–56.e21 (2018).
Lim, J. et al. BMP–Smad4 signaling is required for precartilaginous mesenchymal condensation independent of Sox9 in the mouse. Dev. Biol. 400, 132–138 (2015).
Salazar, V. S., Gamer, L. W. & Rosen, V. BMP signalling in skeletal development, disease and repair. Nat. Rev. Endocrinol. 12, 203–221 (2016).
Ornitz, D. M. & Marie, P. J. Fibroblast growth factor signaling in skeletal development and disease. Genes Dev. 29, 1463–1486 (2015).
Alman, B. A. The role of hedgehog signalling in skeletal health and disease. Nat. Rev. Rheumatol. 11, 552–560 (2015).
Teufel, S. & Hartmann, C. in Current Topics in Developmental Biology Vol. 133 (ed Bjorn R. Olsen) 235-279 (Academic Press, 2019).
Gawlitta, D. et al. Modulating endochondral ossification of multipotent stromal cells for bone regeneration. Tissue Eng. Part B Rev. 16, 385–395 (2010).
Brown, C. et al. Mesenchymal stem cells: cell therapy and regeneration potential. J. Tissue Eng. Regen. Med. 13, 1738–1755 (2019).
Whelan, I. T. et al. A microphysiological model of bone development and regeneration. Biofabrication 15 https://doi.org/10.1088/1758-5090/acd6be (2023).
Bernero, M., Zauchner, D., Müller, R. & Qin, X. H. Interpenetrating network hydrogels for studying the role of matrix viscoelasticity in 3D osteocyte morphogenesis. Biomater. Sci. 12, 919–932 (2024).
Gehre, C. et al. Guiding bone cell network formation in 3D via photosensitized two-photon ablation. Acta Biomater. 174, 141–152 (2024).
Oldershaw, R. A. et al. Directed differentiation of human embryonic stem cells toward chondrocytes. Nat. Biotechnol. 28, 1187–1194 (2010).
Craft, A. M. et al. Generation of articular chondrocytes from human pluripotent stem cells. Nat. Biotechnol. 33, 638–645 (2015).
Lee, M. S. et al. Comparative evaluation of isogenic mesodermal and ectomesodermal chondrocytes from human iPSCs for cartilage regeneration. Sci. Adv. 7 https://doi.org/10.1126/sciadv.abf0907 (2021).
Yamada, D. et al. Induction and expansion of human PRRX1(+) limb-bud-like mesenchymal cells from pluripotent stem cells. Nat. Biomed. Eng. 5, 926–940 (2021).
Lamandé, S. R. et al. Modeling human skeletal development using human pluripotent stem cells. Proc. Natl Acad. Sci. USA 120, e2211510120 (2023).
Galderisi, U., Peluso, G. & Di Bernardo, G. Clinical trials based on mesenchymal stromal cells are exponentially increasing: Where are we in recent years? Stem Cell Rev. Rep. 18, 23–36 (2022).
Zhou, T. et al. Challenges and advances in clinical applications of mesenchymal stromal cells. J. Hematol. Oncol. 14, 24 (2021).
Pothiawala, A. et al. GDF5+ chondroprogenitors derived from human pluripotent stem cells preferentially form permanent chondrocytes. Development 149 https://doi.org/10.1242/dev.196220 (2022).
Lee, G. et al. Isolation and directed differentiation of neural crest stem cells derived from human embryonic stem cells. Nat. Biotechnol. 25, 1468–1475 (2007).
Umeda, K. et al. Long-term expandable SOX9+ chondrogenic ectomesenchymal cells from human pluripotent stem cells. Stem Cell Rep. 4, 712–726 (2015).
Pretemer, Y. et al. Differentiation of hypertrophic chondrocytes from human iPSCs for the in vitro modeling of chondrodysplasias. Stem Cell Rep. 16, 610–625 (2021).
Tani, S., Chung, U. I., Ohba, S. & Hojo, H. Understanding paraxial mesoderm development and sclerotome specification for skeletal repair. Exp. Mol. Med. 52, 1166–1177 (2020).
Williams, S., Alkhatib, B. & Serra, R. Development of the axial skeleton and intervertebral disc. Curr. Top. Dev. Biol. 133, 49–90 (2019).
Scaal, M. Development of the amniote ventrolateral body wall. Dev. Dyn. 250, 39–59 (2021).
Berendsen, A. D. & Olsen, B. R. Bone development. Bone 80, 14–18 (2015).
Nakajima, T. et al. Grafting of iPS cell-derived tenocytes promotes motor function recovery after Achilles tendon rupture. Nat. Commun. 12, 5012 (2021).
Matsuda, M. et al. Recapitulating the human segmentation clock with pluripotent stem cells. Nature 580, 124–129 (2020).
Loh, K. M. et al. Mapping the pairwise choices leading from pluripotency to human bone, heart, and other mesoderm cell types. Cell 166, 451–467 (2016).
Adkar, S. S. et al. Step-wise chondrogenesis of human induced pluripotent stem cells and purification via a reporter allele generated by CRISPR-Cas9 genome editing. Stem Cells 37, 65–76 (2019).
Tani, S. et al. Stem cell-based modeling and single-cell multiomics reveal gene-regulatory mechanisms underlying human skeletal development. Cell Rep. 42, 112276 (2023).
Wu, C. L. et al. Single cell transcriptomic analysis of human pluripotent stem cell chondrogenesis. Nat. Commun. 12, 362 (2021).
Wright, E. et al. The Sry-related gene Sox9 is expressed during chondrogenesis in mouse embryos. Nat. Genet 9, 15–20 (1995).
Mori-Akiyama, Y., Akiyama, H., Rowitch, D. H. & de Crombrugghe, B. Sox9 is required for determination of the chondrogenic cell lineage in the cranial neural crest. Proc. Natl. Acad. Sci. USA 100, 9360–9365 (2003).
Zeng, L., Kempf, H., Murtaugh, L. C., Sato, M. E. & Lassar, A. B. Shh establishes an Nkx3.2/Sox9 autoregulatory loop that is maintained by BMP signals to induce somitic chondrogenesis. Genes Dev. 16, 1990–2005 (2002).
Akiyama, H., Chaboissier, M. C., Martin, J. F., Schedl, A. & de Crombrugghe, B. The transcription factor Sox9 has essential roles in successive steps of the chondrocyte differentiation pathway and is required for expression of Sox5 and Sox6. Genes Dev. 16, 2813–2828 (2002).
Zhou, T. et al. Generation of induced pluripotent stem cells from urine. J. Am. Soc. Nephrol. 22, 1221–1228 (2011).
Umeda, K. et al. Human chondrogenic paraxial mesoderm, directed specification and prospective isolation from pluripotent stem cells. Sci. Rep. 2, 455 (2012).
Cheung, C., Bernardo, A. S., Trotter, M. W., Pedersen, R. A. & Sinha, S. Generation of human vascular smooth muscle subtypes provides insight into embryological origin-dependent disease susceptibility. Nat. Biotechnol. 30, 165–173 (2012).
Xi, H. et al. In vivo human somitogenesis guides somite development from hPSCs. Cell Rep. 18, 1573–1585 (2017).
Zhao, J. et al. Small molecule-directed specification of sclerotome-like chondroprogenitors and induction of a somitic chondrogenesis program from embryonic stem cells. Development 141, 3848–3858 (2014).
Pittenger, M. F. et al. Multilineage potential of adult human mesenchymal stem cells. Science 284, 143–147 (1999).
Adewumi, O. et al. Characterization of human embryonic stem cell lines by the International Stem Cell Initiative. Nat. Biotechnol. 25, 803–816 (2007).
Pearce, J. J. H. & Evans, M. J. Mml, a mouse Mix-like gene expressed in the primitive streak. Mechan. Dev. 87, 189–192 (1999).
Wilson, V. & Beddington, R. Expression of T protein in the primitive streak is necessary and sufficient for posterior mesoderm movement and somite differentiation. Dev. Biol. 192, 45–58 (1997).
Chalamalasetty, R. B. et al. Mesogenin 1 is a master regulator of paraxial presomitic mesoderm differentiation. Development 141, 4285–4297 (2014).
Chapman, D. L., Agulnik, I., Hancock, S., Silver, L. M. & Papaioannou, V. E. Tbx6, a mouse T-Box gene implicated in paraxial mesoderm formation at gastrulation. Dev. Biol. 180, 534–542 (1996).
Della Gaspera, B. et al. Lineage tracing of sclerotome cells in amphibian reveals that multipotent somitic cells originate from lateral somitic frontier. Dev. Biol. 453, 11–18 (2019).
Fan, C. M. & Tessier-Lavigne, M. Patterning of mammalian somites by surface ectoderm and notochord: evidence for sclerotome induction by a hedgehog homolog. Cell 79, 1175–1186 (1994).
Murtaugh, L. C., Chyung, J. H. & Lassar, A. B. Sonic hedgehog promotes somitic chondrogenesis by altering the cellular response to BMP signaling. Genes Dev. 13, 225–237 (1999).
Yoon, B. S. et al. Bmpr1a and Bmpr1b have overlapping functions and are essential for chondrogenesis in vivo. Proc. Natl. Acad. Sci. USA 102, 5062–5067 (2005).
Beers, J. et al. Passaging and colony expansion of human pluripotent stem cells by enzyme-free dissociation in chemically defined culture conditions. Nat. Protoc. 7, 2029–2040 (2012).
Hayashi, K., Ohta, H., Kurimoto, K., Aramaki, S. & Saitou, M. Reconstitution of the mouse germ cell specification pathway in culture by pluripotent stem cells. Cell 146, 519–532 (2011).
De Castro, M., Orive, G., Gascón, A. R., Hernandez, R. M. & Pedraz, J. L. Evaluation of human serum albumin as a substitute of foetal bovine serum for cell culture. Int. J. Pharm. 310, 8–14 (2006).
Cheng, L. et al. Generation of neural progenitor cells by chemical cocktails and hypoxia. Cell Res. 24, 665–679 (2014).
Buikema, J. W. et al. Wnt activation and reduced cell-cell contact synergistically induce massive expansion of functional human iPSC-derived cardiomyocytes. Cell Stem Cell 27, 50–63.e55 (2020).
Kovacs, C. S. et al. The role of biomineralization in disorders of skeletal development and tooth formation. Nat. Rev. Endocrinol. 17, 336–349 (2021).
Fogel, J. L., Lakeland, D. L., Mah, I. K. & Mariani, F. V. A minimally sufficient model for rib proximal-distal patterning based on genetic analysis and agent-based simulations. Elife 6 https://doi.org/10.7554/eLife.29144 (2017).
Woods, A., Wang, G., Dupuis, H., Shao, Z. & Beier, F. Rac1 signaling stimulates N-cadherin expression, mesenchymal condensation, and chondrogenesis. J. Biol. Chem. 282, 23500–23508 (2007).
Chimal-Monroy, J. & Díaz de León, L. Expression of N-cadherin, N-CAM, fibronectin and tenascin is stimulated by TGF-beta1, beta2, beta3 and beta5 during the formation of precartilage condensations. Int. J. Dev. Biol. 43, 59–67 (1999).
Bhumiratana, S. et al. Large, stratified, and mechanically functional human cartilage grown in vitro by mesenchymal condensation. Proc. Natl. Acad. Sci. USA 111, 6940–6945 (2014).
Lefebvre, V., Behringer, R. R. & de Crombrugghe, B. L-Sox5, Sox6 and Sox9 control essential steps of the chondrocyte differentiation pathway. Osteoarthr. Cartil. 9, S69–S75 (2001).
Hoover, M. Y. et al. Purification and functional characterization of novel human skeletal stem cell lineages. Nat. Protoc. 18, 2256–2282 (2023).
Ntambi, J. M. et al. Loss of stearoyl-CoA desaturase-1 function protects mice against adiposity. Proc. Natl. Acad. Sci. USA 99, 11482–11486 (2002).
Kim, Y. W. et al. Integration of single-cell transcriptomes and biological function reveals distinct behavioral patterns in bone marrow endothelium. Nat. Commun. 13, 7235 (2022).
Ding, L., Saunders, T. L., Enikolopov, G. & Morrison, S. J. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature 481, 457–462 (2012).
Sugiyama, T., Kohara, H., Noda, M. & Nagasawa, T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity 25, 977–988 (2006).
St-Jacques, B., Hammerschmidt, M. & McMahon, A. P. Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev. 13, 2072–2086 (1999).
Chung, U. I., Lanske, B., Lee, K., Li, E. & Kronenberg, H. The parathyroid hormone/parathyroid hormone-related peptide receptor coordinates endochondral bone development by directly controlling chondrocyte differentiation. Proc. Natl. Acad. Sci. USA 95, 13030–13035 (1998).
Fitzgerald, J. et al. Evidence for articular cartilage regeneration in MRL/MpJ mice. Osteoarthr. Cartil. 16, 1319–1326 (2008).
Iwano, S. et al. Single-cell bioluminescence imaging of deep tissue in freely moving animals. Science 359, 935–939 (2018).
Wiśniewski, M., Baumgart, M., Grzonkowska, M., Szpinda, M. & Pawlak-Osińska, K. Quantitative anatomy of the ulna’s shaft primary ossification center in the human fetus. Surg. Radio. Anat. 41, 431–439 (2019).
Suzuki, Y. et al. Morphogenesis of the femur at different stages of normal human development. PLoS One 14, e0221569 (2019).
Oliver, W. C. & Pharr, G. M. An improved technique for determining hardness and elastic modulus using load and displacement sensing indentation experiments. J. Mater. Res. 7, 1564–1583 (1992).
Eckstein, K. N. et al. The heterogeneous mechanical properties of adolescent growth plate cartilage: a study in rabbit. J. Mech. Behav. Biomed. Mater. 128, 105102 (2022).
Fischenich, K. M., Schneider, S. E., Neu, C. P., Payne, K. A. & Ferguson, V. L. Material properties and strain distribution patterns of bovine growth plate cartilage vary with anatomic location and depth. J. Biomech. 134, 111013 (2022).
Su, X. W., Feng, Q. L., Cui, F. Z. & Zhu, X. D. Microstructure and micromechanical properties of the mid-diaphyses of human fetal femurs. Connect. Tissue Res. 36, 271–286 (1997).
Zhao, Q., Eberspaecher, H., Lefebvre, V. & De Crombrugghe, B. Parallel expression of Sox9 and Col2a1 in cells undergoing chondrogenesis. Dev. Dyn. 209, 377–386 (1997).
Zhang, B. et al. A human embryonic limb cell atlas resolved in space and time. Nature https://doi.org/10.1038/s41586-023-06806-x (2023).
Chen, Y. et al. A high-resolution route map reveals distinct stages of chondrocyte dedifferentiation for cartilage regeneration. Bone Res. 10, 38 (2022).
D’Angelo, M. et al. MMP-13 is induced during chondrocyte hypertrophy. J. Cell Biochem. 77, 678–693 (2000).
Gerber, H. P. et al. VEGF couples hypertrophic cartilage remodeling, ossification and angiogenesis during endochondral bone formation. Nat. Med. 5, 623–628 (1999).
Nakashima, K. et al. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell 108, 17–29 (2002).
Kawane, T. et al. Dlx5 and mef2 regulate a novel runx2 enhancer for osteoblast-specific expression. J. Bone Min. Res. 29, 1960–1969 (2014).
Pan, L. C. & Price, P. A. The propeptide of rat bone gamma-carboxyglutamic acid protein shares homology with other vitamin K-dependent protein precursors. Proc. Natl. Acad. Sci. USA 82, 6109–6113 (1985).
Hojo, H. & Ohba, S. Gene regulatory landscape in osteoblast differentiation. Bone 137, 115458 (2020).
He, J. et al. Dissecting human embryonic skeletal stem cell ontogeny by single-cell transcriptomic and functional analyses. Cell Res. 31, 742–757 (2021).
Cao, J. et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature 566, 496–502 (2019).
Dirckx, N., Moorer, M. C., Clemens, T. L. & Riddle, R. C. The role of osteoblasts in energy homeostasis. Nat. Rev. Endocrinol. 15, 651–665 (2019).
Sims, N. A. & Martin, T. J. Osteoclasts provide coupling signals to osteoblast lineage cells through multiple mechanisms. Annu. Rev. Physiol. 82, 507–529 (2020).
Gordon, C. M. et al. Hutchinson-Gilford progeria is a skeletal dysplasia. J. Bone Min. Res. 26, 1670–1679 (2011).
Gordon, L. B. et al. Disease progression in Hutchinson-Gilford progeria syndrome: impact on growth and development. Pediatrics 120, 824–833 (2007).
Superti-Furga, A. et al. Achondrogenesis type IB is caused by mutations in the diastrophic dysplasia sulphate transporter gene. Nat. Genet. 12, 100–102 (1996).
Pacifici, M. Hereditary multiple exostoses: new insights into pathogenesis, clinical complications, and potential treatments. Curr. Osteoporos. Rep. 15, 142–152 (2017).
Inzaghi, E., Reiter, E. & Cianfarani, S. The challenge of defining and investigating the causes of idiopathic short stature and finding an effective therapy. Horm. Res. Paediatr. 92, 71–83 (2019).
Lee, J., Smeriglio, P., Chu, C. R. & Bhutani, N. Human iPSC-derived chondrocytes mimic juvenile chondrocyte function for the dual advantage of increased proliferation and resistance to IL-1β. Stem Cell Res. Ther. 8, 244 (2017).
Mostovich, L. A. et al. Integrin alpha9 (ITGA9) expression and epigenetic silencing in human breast tumors. Cell Adh. Migr. 5, 395–401 (2011).
Smith, C. A. & Tuan, R. S. Functional involvement of Pax-1 in somite development: somite dysmorphogenesis in chick embryos treated with Pax-1 paired-box antisense oligodeoxynucleotide. Teratology 52, 333–345 (1995).
Šošić, D., Brand-Saberi, B., Schmidt, C., Christ, B. & Olson, E. N. Regulation of paraxis expression and somite formation by ectoderm- and neural tube-derived signals. Dev. Biol. 185, 229–243 (1997).
Ohba, S., He, X., Hojo, H. & McMahon, A. P. Distinct transcriptional programs underlie Sox9 regulation of the mammalian chondrocyte. Cell Rep. 12, 229–243 (2015).
Rahmoun, M. et al. In mammalian foetal testes, SOX9 regulates expression of its target genes by binding to genomic regions with conserved signatures. Nucleic Acids Res. 45, 7191–7211 (2017).
Concordet, J. P. & Haeussler, M. CRISPOR: intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res. 46, W242–w245 (2018).
Wagner, G. P., Kin, K. & Lynch, V. J. Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theory Biosci. 131, 281–285 (2012).
Anders, S. et al. Count-based differential expression analysis of RNA sequencing data using R and Bioconductor. Nat. Protoc. 8, 1765–1786 (2013).
Tirosh, I. et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196 (2016).
Acknowledgements
We would like to thank the Core Facilities in the College of Life Sciences for their technical assistance. Profs. Jinglei Cai from GIBH-CAS and Yan Liu from Nanjing Medical University for their kind gifts of hPSC cell lines. Xiaoyu Li and Xiong Lin from the College of Polymer Science & Engineering for their assistance on atomic force microscopy. Profs. Rui Yue of Tongji University and Lijian Hui of CAS Shanghai for critical reading of the manuscript. This work was supported by the National Key Research and Development Program of China (2022YFA1104401, 2021YFA1100601), the National Natural Science Foundation of China (32471507, 3247120327, 32071455, 32271295), Sichuan University (020SCUNL109), West China School of Stomatology Sichuan University (RD-03-202106), the Major Science and Technology Projects in Yunnan Province (202302AA310038), and the Fundamental Research Funds for the Central Universities (SCU2019D013).
Author information
Authors and Affiliations
Contributions
Z.L., Y.Y., and J.X. conceived the study. J.X., Y.L., K.X., Yuqing W., C.S., and Y.D. performed most of the wet experiments. Hanyi C., J.X., and Haoyang C. performed the bioinformatic analysis. R.M, G.Y., Yifu W., Q.Z. and Y.Z. provided experimental assistance. Z.L. and W.G. oversaw the collection of results and data interpretation. Z.L. and J.X. wrote the manuscript. All authors have seen and approved the final version of the paper.
Corresponding authors
Ethics declarations
Competing interests
Patents for isolation, characterization, and expansion of SOX9+ Scl-progenitors have been filed (application nos. 202211228107.2, 202211220041.2 and 202310160109.0). For all patents, the patent applicant: Sichuan University; name of inventors: Zhonghan Li and Jingfei Xiong; status of application: 202211228107.2 and 202310160109.0 have been granted. All other authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Hongwei Ouyang, and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.”
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xiong, J., Ma, R., Xie, K. et al. Recapitulation of endochondral ossification by hPSC-derived SOX9+ sclerotomal progenitors. Nat Commun 16, 2781 (2025). https://doi.org/10.1038/s41467-025-58122-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-58122-9
