
Abstract
Organic electrode materials with bipolar-redox activity are a promising candidate for high-energy aluminum-ion batteries (AIBs), but face the capacity ceiling due to limited active sites and low electron transfer number. To universally address this issue, seeking for a kind of multisite bipolar organic material to achieve multielectron transfer is a prerequisite but challenging. Herein, we develop a 12-electron transfer tetraalkynylporphyrin macrocycle positive electrode with two p-type amine (‒NH‒) motifs, two n-type imine (C = N) motifs and four n-type alkynyl (C ≡ C) motifs. The bipolar 18π-electron porphyrin macrocycle can alternately bind and release AlCl4− anions at ‒NH‒ sites and AlCl2+ cations at C = N sites (oxidized from 18π to 16π or reduced from 18π to 20π), achieving four electrons transfer. Furthermore, each terminal C ≡ C site can also coordinate with two AlCl2+ cations, thereby delivering eight electrons. The designed aluminum-organic battery achieves a high capacity of up to 347 mAh g−1 (3-6 times that of conventional graphite positive electrode, 60-120 mAh g−1) and a high specific energy of 312 Wh kg−1 (up to 150% compared to cells with graphite as positive electrode) based on the mass of positive electrode materials.
Similar content being viewed by others


Introduction
Rechargeable aluminum-ion batteries (AIBs) are a promising high-energy electrochemical energy storage device due to the high volumetric capacity (8046 mAh cm−3) and gravimetric capacity (2980 mAh g−1) of Al metal negative electrode1. Unfortunately, AIBs are facing the challenges of low actual capacity and insufficient cycle life due to the lack of suitable positive electrode materials for storing large-sized Al-complex ions. Despite great efforts have been devoted to various inorganic positive electrode materials, such as carbon materials2,3,4, transition metal chalcogenides5,6,7, and chalcogens8, the electrochemical performance is still unsatisfactory. For example, graphitic positive electrode materials show broad application prospects due to their high operating voltage (>2.0 V) and impressively long cycle life (thousands of cycles). However, the intercalation mechanism of monovalent AlCl4− anion leads to a low specific capacity (<120 mAh g−1)3, which restricts their specific energy. In contrast, based on the multielectron conversion reaction mechanism, transition metal chalcogenides and chalcogens offer high initial capacity but suffer from severe capacity deterioration and short cycle life (< 1000 cycles) because of their large volume change and dissolution of active species6,8. Till now, it remains a critical challenge to develop high-energy and long-life positive electrode materials for AIBs.
Emerging organic materials have been promoted as a new solution for high-energy AIBs due to their diverse redox activity, flexible molecular designability as well as unique charge storage mechanism. According to the stored charge species, organic electrode materials can be categorized into n-type (coordinated with cations), p-type (coordinated with anions), and bipolar-type (coordinated with cations and anions alternately). Carbonyl compounds are the most widely studied n-type organic positive electrodes in AIBs, such as quinones9,10, imides11, and ketones12, which undergo reduction reactions first and coordinate with monovalent AlCl2+ or divalent AlCl2+ cations through the enolization of carbonyl groups (C = O). Furthermore, some organics with nitrogen-containing groups, including imines (C = N)13, nitriles (C ≡ N)14, and azo compounds (N = N)15, can also generate anionic radicals that coordinate with AlCl2+ cations. These n-type organic positive electrodes typically exhibit a high specific capacity and good redox reversibility but suffer from poor cycling stability due to substantial dissolution in acid ionic liquid electrolytes. In contrast, p-type organics generally undergo oxidation reactions first and coordinate with AlCl4− anions via charge delocalization along their molecular skeletons, such as polypyrenes16 and aromatic amines17. However, given the limited active sites and single-electron transfer, these p-type organic positive electrodes only offer finite specific capacity. Innovatively, some bipolar-type organics, exhibiting both n-type and p-type redox characteristics, have been explored for the alternate storage of dual-ions (AlCl4− anions and AlCl2+ cations) to obtain a higher capacity18,19. Nevertheless, the low electron transfer number per active unit in most organic materials leads to a low ceiling of theoretical capacity (<200 mAh g−1), especially for AIBs using large-sized Al-complex ions as charge carriers.
Porphyrins, a bipolar-redox 18π-electron conjugated macrocyclic compound capable of multielectron transfer, show great competitiveness in electrochemical energy storage20,21. Benefitting from the small LUMO-HOMO gap, porphyrins can not only be oxidized by donating π-electrons (from 18π to 16π) but also reduced by accepting π-electrons (from 18π to 20π) at their 18π-electron macrocycles. However, the high molecular weight of porphyrins confines their specific capacity, which must be improved through ingenious molecular design. Fortunately, the electrochemical properties of porphyrins can be easily regulated by connecting different functional groups at their meso- or β- sites. Some recent works have shown the great energy storage potential of porphyrin-based electrode materials for AIBs. Han et al.22 reported a stable 5,10,15,20-tetraphenylporphyrin (H2TPP) positive electrode material with a specific capacity of 101 mAh g−1. The electrochemical mechanism involves the reversible coordination/dissociation reactions between two imine (C = N) nitrogen atoms and AlCl2+ cations. The redox activity of H2TPP can be regulated by introducing electron-withdrawing or electron-donating functional groups, which can also serve as active sites to store more AlCl2+ cations23. In another work, Chowdhury et al.24 introduced four active diphenylamine groups into H2TPP to store AlCl4− anions, enhancing the rate capability to 83 mAh g−1 at 1 A g−1. Differently, the nitrogen atoms in the porphyrin macrocycle can be protonated due to the acidity of the electrolyte and serve as the storage sites for AlCl4− anions. Nevertheless, owing to the unipolar-redox activity and single-electron-transfer groups, the insufficient electron transfer number per porphyrin unit (no more than 6 electrons) restricts the specific capacity (< 180 mAh g−1). Notably, the redox activity of the porphyrin macrocycle may be related to the Lewis basicity of imine (C = N) groups and acidity of amine (‒NH‒) groups22,23,24,25. Functional groups with strong electron-withdrawing or electron-donating effects may cause low basicity (or acidity) of nitrogen atoms, reducing their ability to coordinate with cations (or anions). This, in turn, results in weak bipolar-redox activity or even a unipolar redox mechanism. Encouragingly, the alkynyl group (C ≡ C–H) can induce a weak electron-withdrawing effect due to its moderate electronegativity, which provides an opportunity for designing porphyrins with appropriate basicity (or acidity) to achieve bipolar redox. More importantly, the unsaturated C ≡ C bond can also serve as the active site to accept two electrons and transform into a C = C bond, enabling dual-electron transfer26. In addition, studies have shown that the porphyrins containing terminal alkynyl groups can be self-polymerized by oxidative coupling reaction27,28, which is conducive to better cycling stability. These potential advantages, including bipolar redox, multielectron transfer, and self-polymerization, make alkynyl-porphyrins a highly desirable organic positive electrode material for AIBs.
Herein, we demonstrate 12-electron bipolar-redox chemistry of tetraalkynylporphyrin (H2TEPP) macrocycle positive electrode for high-energy aluminum-organic batteries (Fig. 1). The bipolar-redox activity of 18π-electron macrocycle enables alternate storage of dual carriers (AlCl4− anions and AlCl2+ cations) at two p-type amine (‒NH‒) sites and two n-type imine (C = N) sites during charge/discharge processes, thereby contributing to four electrons transfer. Impressively, four n-type alkynyl (C ≡ C) sites can also coordinate with more AlCl2+ cations during the discharge process due to their modest electronegativity and affinity to cationic carriers, achieving eight electron transfers. Both experimental results and theoretical calculations confirm the 12-electron transfer charge storage mechanism. As a result, the Al | |H2TEPP battery achieves a high capacity of 347 mAh g−1 and a high specific energy of 312 Wh kg−1, which is up to 150% compared to cells with graphite as the positive electrode. Moreover, the H2TEPP positive electrode also exhibits good cyclability of 87% capacity retention after 6500 cycles due to the in situ electropolymerization of terminal alkynyl groups. This work heralds a promising strategy for designing multielectron-transfer bipolar-redox organic positive electrode materials to realize high-energy AIBs.

The 12-electron bipolar-redox charge storage mechanism of H2TEPP positive electrode in AIBs.
Results
Molecular design and structural characterizations
A high capacity is highly desirable to achieve the high specific energy of batteries. The specific capacity of organic molecules mainly depends on the electron transfer number per weight. To achieve maximum electron transfer, we select three bipolar-redox porphyrin molecules 5,10,15,20-tetraphenylporphyrin (H2TPP), 5,10,15,20-tetra(4-pyridyl) porphyrin (H2TPyP) and 5,10,15,20-tetraalkynylporphyrin (H2TEPP) (Fig. 2a and Supplementary Fig. 1) as positive electrode materials for AIBs. Each porphyrin molecule consists of an 18π-electron macrocycle (containing two imine (C = N) groups and two amine (–NH–) groups) and four functional phenyl substituent groups. Density functional theory (DFT) calculations is performed to study their electronic properties. We first calculate the molecular electrostatic potential (MESP) to predict the active sites for nucleophilic and electrophilic reactions (Fig. 2a)29. Generally, the blue areas present negative electrostatic potential, acting as electrophilic sites that tend to bind with positive charge carriers, while the red regions manifest positive electrostatic potential, serving as nucleophilic sites that attract negative charge carriers. In the MESP maps, it can be seen that the surface minimum value points (cyan balls) of three porphyrins are mainly located on the imine (C = N) groups at the center of the 18π-electron macrocycle, while the maximum value points (orange balls) are distributed around the amine (–NH–) groups. The calculated surface maximum and minimum values of MESP are shown in Supplementary Table 1. Compared with the unfunctionalized H2TPP, the pyridine (C = N) groups of H2TPyP and alkynyl (C ≡ C) groups of H2TEPP exhibit significant negative electrostatic potential, which means that these functional groups may provide extra active sites for electrophilic reaction. The introduction of these potential active groups can significantly increase the electron transfer number of per porphyrin unit, contributing to higher capacity. Notably, the strongly electronegative pyridine groups significantly reduce the basicity of imine groups in the macrocycle, which may lead to weak electrophilic activity, while the moderately electronegative alkynyl groups with weak electron-withdrawing effect induce appropriate basicity of imine groups and acidity of amine groups. Furthermore, we calculate the energies of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) of three porphyrins (Fig. 2b). The frontier electrons of all porphyrin molecules are mainly distributed on the central macrocycle, which means that the electronic structure change preferentially occurs at the 18π-electron macrocycle during redox process. Impressively, H2TEPP shows a smaller LUMO-HOMO gap (2.631 eV) than H2TPP (2.671 eV) and H2TPyP (2.722 eV), indicating its higher electronic conductivity. In addition, the localized orbital locator-π (LOL-π) color-filled maps of three porphyrins show efficient π-electron delocalization throughout the 18π-electron macrocycle (Fig. 2c and Supplementary Fig. 2), which ensures fast electron transfer. Considering the significant advantage of alkynyl groups in facilitating electron transfer, H2TEPP is selected as the preferred positive electrode material for AIBs.

a Molecular structures and MESP distributions of three porphyrins (cyan balls: minimum value points, orange balls: maximum value points). b Calculated LUMO-HOMO energy levels and energy gaps (∆E). The green and blue isosurfaces represent positive and negative orbital phases, respectively. c LOL-π map of H2TEPP. d Scatter plot of IRI vs. sign(λ2)ρ of H2TEPP. e Isosurface map of IRI for the intermolecular π-π interaction of H2TEPP. f Comparison of specific capacity with increased electron transfer number. g Contour plots of specific energy compared with reported organic and graphite positive electrode materials in AIBs.
To reveal the potential multielectron transfer capability, the interaction region indicator (IRI) method is applied to unveil the intermolecular interaction of H2TEPP. The detected green spikes in the sign(λ2)ρ from − 0.02 to 0.00 arb. units (Fig. 2d) and green isosurfaces (Fig. 2e) indicate the existence of π-π interaction between H2TEPP molecules. Such π-π interaction in the out-of-plane direction between H2TEPP molecules makes it easy to donate or accept electrons, ensuring efficient charge transfer, multielectron redox activity, and high accessibility of the active sites. Generally, aromatic molecules exhibit prominent structural stability due to the π-electron delocalization effect. The iso-chemical shielding surface (ICSS) method30 is used to reveal the aromaticity of H2TEPP. Protruding green magnetic shielding isosurface in the perpendicular direction are observed in the Z-component of ICSS (ICSSZZ) for the macrocycle plane (Supplementary Fig. 3a), surrounded by a circular blue deshielding isosurface. Positive ICSS(0)ZZ (31.7 ppm) and ICSS(1)ZZ (34.1 ppm) values indicate a high level of π-aromaticity and in-plane π-electron delocalization within the 18π-electron macrocycle. In addition, the ICSSZZ slice planes (pink to red area) exhibit a clear magnetic shielding effect and smooth isosurface variation in the inner area of the macrocycle (Supplementary Fig. 3b). Such strong aromaticity along the whole molecular skeleton guarantees high structure stability, allowing for the injection and release of multiple electrons due the redox process25.
The molecular structures of the three porphyrins are further characterized by Fourier transform infrared spectroscopy (FTIR) (Supplementary Fig. 4). The bands at about 1600 cm−1 of H2TPP and H2TEPP are associated with the C = C stretch in benzene groups31, while the band at 1593 cm−1 of H2TPyP is assigned to the C = C stretch in pyridine groups32. For H2TEPP, the peaks at 1349 and 1473 cm−1 correspond to the C‒N and C = N vibrations in the 18π-electron macrocycle, respectively33,34. The peaks at 2108 and 3290 cm−1 are assigned to the C ≡ C and C ≡ C‒H vibrations of alkynyl groups, respectively31. X-ray diffraction (XRD) patterns show obvious crystal characteristics of the three porphyrins (Supplementary Fig. 5). Scanning electron microscopy (SEM) images show that all porphyrins present irregular micron block morphology (Supplementary Fig. 6). Thermogravimetric analysis (TGA) reveals high thermal stability of H2TEPP with an onset decomposition temperature of up to 400 °C under N2 atmosphere (Supplementary Fig. 7).
As analyzed above, the capacity of porphyrins can be improved by introducing more active sites to increase the electron transfer number. Figure 2f shows the relationship between the specific capacity and electron transfer number of three porphyrins. Compared with H2TPP, which can only transfer four electrons, H2TPyP and H2TEPP can theoretically transfer up to 8 or 12 electrons due to the introduction of four single-electron-accepting pyridine (C = N) sites and four dual-electron-accepting alkynyl (C ≡ C) sites. According to the results of the electrochemical test, the specific capacities of H2TPP, H2TPyP, and H2TEPP are 118, 251, and 347 mAh g−1, respectively. With the electron transfer number increasing from 4 to 12, the specific capacity of H2TEPP is increased by 3 times. Benefitting from the high specific capacity, the H2TEPP positive electrode presents a competitive specific energy over 300 Wh kg−1 compared with the recently reported organic positive electrode materials in AIBs, including n-type, p-type, and bipolar-type organics (Fig. 2g and Supplementary Table 2). This result represents an apparent advantage of multielectron-transfer organic materials, which break through the specific energy limitation of conventional graphite positive electrode materials (100–200 Wh kg−1).
Electrochemical performance of Al-porphyrin batteries
The electrochemical performances of Al-porphyrin batteries are evaluated using Al foil as the negative electrode, three porphyrins as the positive electrode, and aluminum chloride/1-ethyl-3-methylimidazolium chloride (AlCl3/[EMIm]Cl) ionic liquid as the electrolyte. Galvanostatic charge/discharge tests are performed in a voltage range of 0.1–2.2 V. Despite a larger molecular weight than H2TPP and H2TPyP, the H2TEPP delivers a high initial discharge capacity of 347 mAh g−1 at 0.2 A g−1 (Fig. 3a and Supplementary Fig. 8), which means that H2TEPP may offer a higher electron transfer number. The irreversible charge/discharge capacities and large overpotentials in the first few cycles are mainly attributed to the unique self-polymerization process of porphyrins28,35. This irreversibility is frequently observed in porphyrin-based electrode materials, especially at low current rates, which may also be related to the formation of solid electrolyte interphase (SEI)24. Notably, the charge/discharge curves of the three porphyrin electrodes gradually tend to be stable after activation (Supplementary Fig. 9). Compared with H2TPP and H2TPyP, H2TEPP delivers a higher average discharge voltage of 1.3 V with a lower overpotential of 0.32 V. Even after 400 cycles, H2TEPP still maintains a high reversible capacity of 200 mAh g−1 with good cycling stability, which is much higher than H2TPP (83 mAh g−1) and H2TPyP (64 mAh g−1) (Fig. 3b). The capacity experiences a decay in the first few cycles, mainly owing to the inevitable initial dissolution of porphyrin molecules in the electrolyte (Supplementary Fig. 10). However, compared with the obvious dissolution of H2TPP and H2TPyP, H2TEPP shows a lighter dissolution phenomenon.

a Galvanostatic charge/discharge curves of H2TEPP positive electrode at 0.2 A g−1. b Comparison of cycling performance of H2TEPP, H2TPP, and H2TPyP positive electrodes at 0.2 A g−1. c The dQ/dV plots of the first three discharge curves of H2TEPP. d, e Rate performance of H2TEPP at different current rates from 0.1 to 5 A g−1. f Comparison of the specific energy and specific power of H2TEPP with recently reported organic and graphitic positive electrode materials in AIBs. The source of the literature data shown in this figure can be found in Supplementary Information, Table 2. g Long-term cycling stability of H2TEPP at 2 A g−1. h Performance comparison of H2TEPP and other porphyrin-based positive electrode materials in AIBs.
To investigate the electrochemical behavior of H2TEPP during the first few cycles, cyclic voltammetry (CV) test is performed at 0.1 mV s−1 (Supplementary Fig. 11). In the first anodic scan, three distinct irreversible oxidative peaks appear at 1.93, 2.01 and 2.14 V, suggesting an intense oxidation reaction of H2TEPP, which can be attributed to the insertion of Al-complex anions and irreversible oxidation self-polymerization of alkynyl groups (as discussed below). In subsequent cycles, the peak at 2.01 V disappears, and the peaks at 1.93/2.01 V gradually decrease, showing enhanced redox reversibility. Impressively, the cathodic scan displays multiple reductive peaks at 1.36, 1.57, 1.70, 1.84, and 1.98 V, indicating a multiple-electron redox process, which contributes to a high specific capacity. The dQ/dV curves of H2TEPP in the first three cycles manifest three potential peaks, indicating a three-step discharge process (Fig. 3c). The weakened reductive peak (1.36 V) and potential peaks (b and c) indicate that there may be incomplete coordination of H2TEPP with Al-complex ions as the cycle progresses.
To further explore the irreversible oxidation reaction in the first cycle, in-situ FTIR is performed to analyze the H2TEPP electrode (Supplementary Fig. 12a). The vibration of C ≡ C–H group at 3264 cm−1 significantly weakens after fully charged to 2.2 V in the first charge process, but does not recover in subsequent discharge and recharge processes, indicating that the terminal alkynyl groups are in-situ self-polymerized under the electric field28,34. The weak residual absorption signal after fully recharging may be due to incomplete polymerization of alkynyls. Furthermore, XRD patterns show that H2TEPP changes from pristine crystalline characteristic to amorphous polymer characteristic after the first charge process (Supplementary Fig. 12b). SEM is used to observe the morphology change of H2TEPP before and after cycling (Supplementary Fig. 13). The pristine electrode shows willow leaf-like morphology of H2TEPP with a size of ~ 5 × 1 μm. After the first cycle, these micro-leaves are interconnected and transformed into large microsheets. After 400 cycles, the large microsheets are further connected into a whole without an obvious dissolution phenomenon. The above results indicate that the porphyrins containing terminal alkynyl groups might be in-situ electrochemically polymerized into conjugated polymers during the first cycle (Supplementary Fig. 14)27. Such in-situ electropolymerization behavior of H2TEPP guarantees its long cycling stability.
The changes in electronic and ionic conductivity of H2TEPP are investigated by electrochemical impedance spectroscopy (EIS) analysis (Supplementary Fig. 15). The corresponding distribution of relaxation times (DRT) suggests that both charge transfer and ion diffusion resistances significantly decrease after the first cycle. Additionally, DFT calculations are performed to study the electronic structure properties of H2TEPP after polymerization. Compared with the monomer, the H2TEPP dimer shows a smaller LUMO-HOMO gap (Supplementary Fig. 16), indicating an enhanced electronic conductivity. The MESP of H2TEPP dimer shows that both central nitrogen atoms and terminal alkynyls still maintain obvious negative electrostatic potential (Supplementary Fig. 17). In fact, the alkynyl groups have special affinity to metal cations given their modest electronegativity, which can provide additional active sites for ion storage36. These results suggest that electropolymerization can effectively improve the electronic and ionic conductivity of organic materials, which allows for better rate performance. Furthermore, the binding energy (∆E) of H2TEPP upon self-polymerization is also calculated (Supplementary Fig. 18 and Table 4). The positive ∆E value indicates that the oxidation polymerization of alkynyl groups (C ≡ C–H) is an endothermic process, meaning that the electropolymerization occurs in the charging process, which may lead to a large reaction energy barrier and overpotential in the initial cycle.
To verify the influence of electronic conductivity on cycling stability, we have carried out a controlled experiment by changing the amount of conductive additive in the H2TEPP electrode (Supplementary Fig. 19). The capacity contribution of conductive carbon is neglectable. With the additive increasing from 30 wt.% (6:3:1) to 40 wt.% (5:4:1), the reversible capacity of H2TEPP increases from 234 mAh g−1 to 259 mAh g−1 at 0.2 A g−1 with a similar cycling stability. However, when the additive decreases to 20 wt.% (7:2:1), H2TEPP electrode shows an apparently lower capacity of 171 mAh g−1 and an inferior cycling stability. This may be because of the decrease in conductivity that hinders the electropolymerization process of H2TEPP in the first cycle. Considering the specific energy of the whole electrode, the H2TEPP electrode containing 30 wt.% conductive carbon is preferred.
The rate performance of porphyrin electrodes after activation is tested at different current rates from 0.1 to 5 A g−1. H2TEPP delivers reversible capacities of 269, 221, 180, 152, 127 and 99 mAh g−1 at 0.1, 0.2, 0.5, 1, 2 and 5 A g−1 (Fig. 3d). The discharge capacity recovers to 263 mAh g−1 when the current rate returns to 0.1 A g−1, maintaining 98% of the initial capacity, which is better than H2TPP and H2TPyP (Fig. 3e and Supplementary Fig. 20). Benefitting from the multi-electron transfer and good rate performance, H2TEPP shows competitive comprehensive performance compared with previously reported organic positive electrode materials in AIBs (Fig. 3f and Supplementary Table 2). The long cycling performance of H2TEPP is also evaluated at 2 A g−1. Even after 6500 cycles, H2TEPP still maintains a high reversible capacity of 127 mAh g−1 with a capacity retention of 87% (Fig. 3g), demonstrating long-term cycling stability. With the increase of cycle number, the charge-discharge profiles gradually tend to be stable (Supplementary Fig. 21). FTIR spectra show that H2TEPP still maintains the main vibration bands of 18π-electron macrocycle after thousands of cycles (Supplementary Fig. 22), indicating its high structural stability, which benefits from the aromaticity and flexibility of porphyrin macrocycle. Furthermore, H2TEPP can effectively store Al-complex ions even after 6000 cycles because in-situ electropolymerization inhibits the initial dissolution (Supplementary Fig. 23). Consequently, compared with the reported porphyrin-based positive electrode materials in AIBs, H2TEPP shows significant advantages, especially in terms of electron transfer number, specific capacity, specific energy and cycle life (Fig. 3h). In fact, porphyrin-based redox-active materials have great application potential in energy storage due to their natural availability, chemical versatility and environmental friendliness20,21.
To elucidate the charge storage mechanism of H2TEPP, CV measurements are performed at various scan rates from 0.5 to 10 mV s−1 to analyze the capacity contribution of faradaic and non-faradaic processes (Supplementary Fig. 24a). The faradaic process involves ion diffusion and charge transfer in the redox reaction process, while the non-faradaic process relates to the capacitive ion adsorption in the electric double layer12. The capacity contributions from these two processes can be identified by fitting the relationship between peak current (i) and scan rate (v) according to the following Equation:
where the b value can be determined by the slope of (log v)-(log i) plot. The b = 0.5 corresponds to a diffusion-controlled process, whereas b = 1 indicates a capacitive-controlled process. The calculated b value of the oxidation (peak 1) and reduction (peaks 2 and 3) are 0.73, 0.72, and 0.78, respectively (Supplementary Fig. 24b), indicating that the capacity is contributed by both ion diffusion and capacitance.
To further explore the ion diffusion kinetics of H2TEPP, the galvanostatic intermittent titration technique (GITT) is carried out after activation at 0.1 A g−1 (Supplementary Fig. 25). During the charging process, the Al-complex ions (SOC below 50%) in H2TEPP show relatively fast kinetics with a diffusion coefficient (D) on the order of 10−8–10−9 cm2 s−1, while with the progress of charging (SOC 50–100%), the D value gradually decreases to 10−9–10−11 cm2 s−1. This process may be caused by the insertion/extraction of large-sized Al-complex anions, such as AlCl4−. In the discharge process, the Al-complex ions extract (insert) quickly from (into) H2TEPP with D ≈ 10−8–10−9 cm2 s−1 (SOC 100–50%), and the subsequent Al-complex ions slightly slower with D ≈ 10−9–10−10 cm2 s−1 (SOC below 50%). The magnitude of these diffusion coefficients indicates fast diffusion of the ions within the H2TEPP positive electrode, which is in a similar range of AlCl4− ion in graphite (10−9–10−11 cm2 s−1)37. Molecular dynamics (MD) simulations are further carried out to evaluate the diffusion behavior of Al-complex anion (AlCl4−) and cation (AlCl2+) in H2TEPP (Supplementary Fig. 26). Considering the self-polymerization characteristic of H2TEPP, the periodic structure model of H2TEPP polymer is established in the simulation box. The simulation snapshots show that AlCl4− anions tend to migrate along the pore channels of the H2TEPP polymer, while AlCl2+ cations tend to migrate along the interlayer channels. Meanwhile, a quantitative characterization of AlCl4− and AlCl2+ diffusivity in the transport process is simulated by the mean square displacement (MSD) (Supplementary Fig. 27). Both AlCl4− and AlCl2+ display fast ion diffusion with a diffusion coefficient on the order of 10−9 cm2 s−1, which are comparable to the order of magnitudes of experimental results.
Redox reaction mechanism of H2TEPP
To clarify the redox reaction mechanism of H2TEPP, ex-situ Raman spectroscopy, in-situ FTIR, and ex-situ X-ray photoelectron spectroscopy (XPS) are performed on the electrodes at different charge and discharge states. Raman spectroscopy is conducted to monitor the structural evolution of H2TEPP at the selected states in Fig. 4a. The assignment of different bands of pristine H2TEPP positive electrode is shown in Supplementary Fig. 28 and Table 333,38,39. As shown in Fig. 4a, all peaks are broadened after charging and discharging compared with the pristine H2TEPP, which can be due to the interaction of Al-complex anions and cations with H2TEPP that changes the symmetrical structure40. The CIm = N peak at 1468 cm−1 occurs redshift to 1450 cm−1 during the charge process and then occurs blueshift to 1469 cm−1 during the discharge process. The blueshift is caused by the increased force constants of CIm = N bonds due to the coordination with Al-complex cations. The decreased peak intensity after fully charged to 2.2 V may be ascribed to the insertion of large-sized Al-complex anions that distort the planar structure of the porphyrin macrocycle. Furthermore, the C = C peak at 1556 cm−1 first decreases and then increases during the charging process, and the discharge process also shows the same change tendency. Such reversible change is related to the structural rearrangement of C = C bonds caused by the redox reaction of 18π-electron porphyrin macrocycle. Similar to the CIm = N peak, the C ≡ C peak at 1496 cm−1 also exhibits reversible red shift and blue shift during charge and discharge processes. Notably, it can be observed that the peak intensity of C ≡ C increases after charging and decreases after discharging. On the contrary, the new characteristic peak of C = C (sp2 C) at 1585 cm−1 decreases during the charging process and increases during the discharge process. The increased intensity of C = C peak after discharging can be attributed to the reduction of C ≡ C bonds to C = C bonds26, which forms the hybrid sp2 C. This process may be accompanied by the coordination of Al-complex cations because of the electrophilic activity of the C ≡ C group and its special affinity for cationic charge carriers36. In fact, this process is similar to the overlithiation reaction of the unsaturated C ≡ C bond of alkynyl-based electrode materials in lithium-ion batteries26,41. Notably, the reduction of the C ≡ C group usually involves only dual-electron transfer (from C ≡ C to C = C rather than C‒C bond) because of the strong steric-hindrance and inductive effect of the coordination intermediate26. This means that the four C ≡ C groups can accept up to 8 electrons during the discharge process, which contributes to a high specific capacity.

a Ex-situ Raman spectra of H2TEPP at different charge/discharge states. b In situ FTIR spectra of H2TEPP during the second cycle. c–e Ex situ XPS spectra of (c) N 1 s, (d) Al 2p, (e) Cl 2p. f The proposed 12-electron bipolar-redox reaction mechanism of H2TEPP.
To further clarify the redox reaction mechanism, in-situ FTIR characterization is carried out to unveil the structural evolution of H2TEPP (Fig. 4b). The peak signals of ‒NH‒ (790 cm−1)25, C = N (1490 cm−1) and C ≡ C (2108 cm−1) groups34 decrease in the discharge process and increase in the recharge process, indicating their strong redox activity. It is noted that two new signals appear at 743 and 832 cm−1 after fully discharged, which can be attributed to the formation of N‒Al bonds42,43 due to the synergetic coordination of pyrrole N atoms (‒NH‒) and imine N atoms (C = N) with Al-complex cations. More importantly, the peak signal at 1570 cm−1 assigned to C = C bonds is strengthened during the discharge process, meanwhile, a strong response signal at 1164 cm−1 emerges due to the formation of coordination intermediates (Al‒C = C‒Al) of C ≡ C groups and Al-complex cations25. The Raman and FTIR spectra results indicate that both ‒NH‒/C = N and C ≡ C groups are likely to be the active sites for storing Al-complex ions.
The charge storage mechanism of H2TEPP is further identified by XPS. Generally, the soluble halides of Al element undergo asymmetric cleavage in chloroaluminate electrolyte to generate AlCl4− anions and AlCl2+ cations9, which can serve as active charge carriers for p-type and n-type redox reactions in organic electrodes. The pristine N1s XPS spectrum (Fig. 4c) shows two characteristic peaks with binding energies at 398.0 and 400.2 eV, which are assigned to the ‒NH‒ and C = N groups of H2TEPP, respectively44. After fully charged to 2.2 V, a new peak appears at a higher binding energy of 401.6 eV, which is attributed to the generation of =NH+‒ cationic radicals due to the p-type oxidation reaction of porphyrin macrocycle from 18π to 16π25. This allows the AlCl4− anions in the electrolyte to insert into H2TEPP and interact with the N atoms of ‒NH‒ groups. After fully discharged to 0.1 V, the =NH+‒ peak disappears, and an intense peak signal is detected at 402.1 eV, which can be assigned to the formation of N‒Al bonds. This means that the AlCl2+ cation can also insert into H2TEPP to coordinate with the N atoms due to the n-type reduction reaction of porphyrin macrocycle from 18π to 20π accompanied by the transformation from C = N to C‒N18, which is verified by the decrease of C = N peak. Moreover, compared with the pristine Al 2p and Cl 2p spectra, both charged-state and discharged-state spectra show obvious Al peaks and Al‒Cl peaks (Fig. 4d, e), further confirming the insertion of Al-complex ions. Notably, the intensity of the Al‒Cl signal at the discharged state is remarkably higher than that at the charged state, suggesting the insertion of more AlCl2+ cations due to the reduction reaction of extra C ≡ C groups. In addition, the energy-dispersive spectroscopy (EDS) spectra show that the Cl/Al atomic ratio is 3.76 after fully charging to 2.2 V, indicating that the coordinated anionic carriers are AlCl4− (Supplementary Fig. 29). After fully discharging to 0.1 V, the content of Al and Cl elements significantly increase, while the Cl/Al atomic ratio decreases to 2.10, indicating that abundant AlCl2− cations are inserted into the H2TEPP positive electrode (Supplementary Fig. 30). These results indicate that both Al-complex anions and cations participate in the redox reactions. As a result, a 12-electron bipolar-redox reaction mechanism is proposed, as shown in Fig. 4f. Due to the bipolar-redox activity of the 18π-electron macrocycle, AlCl4− anions and AlCl2+ cations can be alternately inserted into/extracted from H2TEPP during charge/discharge processes. Furthermore, four active C ≡ C groups can also coordinate with more AlCl2+ cations, and the whole process can transfer up to 12 electrons.
To further confirm the alternate storage mechanism of AlCl4− anions and AlCl2+ cations, we perform time-of-flight secondary ion mass spectrometry (TOF-SIMS) on fully charged/discharged H2TEPP electrodes. The chemical composition of the electrode’s surface and interior can be accurately determined by bombarding the electrode with a primary bismuth (Bi) ion beam to produce secondary ions (Fig. 5a). According to the flight time of different secondary ions from the electrode to the detector, the composition and content of these ions can be obtained. The 2D sputtering images show four secondary ions (Al−, Cl−, AlCl2− and AlCl4−) on the electrode surface (Supplementary Fig. 31). These secondary ion fragments mainly originate from the Al-complex anions/cations inserted in the electrode. The 3D rendering images further display the distribution of different secondary ions inside the electrode (Fig. 5b). At the charge-2.2 V state, the intense signals of Cl− and AlCl4− components can be observed, indicating that the AlCl4− anion is inserted into H2TEPP after fully charged. At the discharge-0.1 V state, the weakened signal of AlCl4− suggests that the AlCl4− anion is extracted from H2TEPP after fully discharged. Notably, the intensity of Al−, Cl−, and AlCl2− components at discharge-0.1 V state is significantly higher than that at the charge-2.2 V state, which indicates the insertion of abundant AlCl2+ cations after full discharge. Furthermore, the depth profiles show the intensity distribution of different secondary ions in depth at charge-2.2 V and discharge-0.1 V states (Fig. 5c). As shown in the magnified views of the depth profiles (Fig. 5d, e), the sputter amount of AlCl4− at charge-2.2 V state is more than that at discharge-0.1 V state, while the sputter amount of Al−, Cl−, and AlCl2− at charge-2.2 V state is significantly less than that at discharge-0.1 V state. These results further demonstrate the bipolar-redox reaction mechanism, which involves the alternate storage of AlCl4− anion and AlCl2+ cation. Interestingly, with the increase of sputtering depth in the range of 0–25 μm, the intensity of all secondary ions increases first and then tends to be stable in both charge and discharge processes. It is worth noting that strong AlCl4− and AlCl2− signals are still observed even at a sputtering depth of 25–50 μm, indicating the diffusion of active ions into the interior of the electrode. The high accessibility of multiple active sites in H2TEPP promotes the storage of Al-complex anions/cations, thus contributing to a high capacity.

a Schematic diagram of TOF-SIMS. b 3D sputtering images of H2TEPP electrode at charge-2.2 V and discharge-0.1 V states. c The depth profiles of different secondary ions at charge-2.2 V and discharge-0.1 V states. d, e Comparison of the magnified depth profiles in different ranges of (d) 0–25 and (e) 25–50 μm sputtering depth. The data are smoothed.
Theoretical calculations
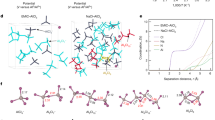
DFT calculations are performed to further reveal the charge storage mechanism of H2TEPP. We first calculate the MESP distribution to determine the active sites (Fig. 6a). The oxidation species H2TEPP2+ displays intensely positive charge characteristics throughout the molecule, especially at the N atoms region of ‒NH‒ groups, which can serve as nucleophilic sites to bind with anions. In contrast, the reduction species H2TEPP2− and H2TEPP10− exhibit distinctly negative charge characteristics at the regions of central C = N groups and terminal C ≡ C groups, which are electrophilic sites that bind with cations. The MESP results theoretically evidence that the N atoms at the center of the macrocycle and C ≡ C groups are the active sites. Anisotropy of the induced current density (AICD) and nucleus-independent chemical shift (NICS) calculations are conducted to evaluate the π-aromaticity and molecular stability of H2TEEP during the redox process (Fig. 6b–e)45. In AICD plots of neutral H2TEPP (18π), the clockwise diamagnetic continuous current flows over the whole molecular backbones, indicating a global aromatic characteristic. The corresponding NICS(1)ZZ map displays significant negative values. In contrast, the oxidation-state H2TEPP2+ (16π) and reduction-state H2TEPP2− (20π) show obvious antiaromatic characteristics on the porphyrin macrocycle, as suggested by the anticlockwise current in AICD plots and the positive NICS(1)ZZ values. However, owing to the conjugation effect46, they can be well stabilized by the four aromatic benzene rings during the redox process. Even after accepting 10 electrons to H2TEPP10−, the porphyrin macrocycle still maintains its antiaromaticity of the 20π-electron configuration. Such aromatic-antiaromatic transformation follows Huckel’s rule and shows significant π-electron delocalization, suggesting that both the oxidation and reduction states are thermodynamically stable. To further clarify the structure stability of H2TEPP at different redox states, we calculate the LUMO-HOMO energy levels of H2TEPP at different redox states to illustrate the position of external electrons and qualitative information of their electronic properties (Supplementary Fig. 32). The frontier electrons of H2TEPP are mainly distributed on the porphyrin macrocycle, and are well positioned and retained in the molecular structure of all redox states, indicating that all the redox states are stable. This means that the H2TEPP can carry 1–2 positive charges or 1–10 negative charges. In addition, the LUMO-HOMO gaps of different redox states are smaller than that of the neutral state, indicating faster electron transfer, which facilitates the interaction between H2TEPP and anions (cations) during charging (discharging).

a MESP distributions of H2TEPP2+, H2TEPP2−, H2TEPP10−. b–e AICD plots and NICS(1)ZZ maps of (b) neutral H2TEPP (18π), (c) oxidation-state H2TEPP2+ (16π), (d) reduction-state H2TEPP2− (20π), and (e) reduction-state H2TEPP10− (20π). The red arrows in AICD plots refer to the direction of the induced current. f Calculated ΔG values of H2TEPP during charge/discharge processes and optimized geometries of H2TEPP and its coordination compounds (H2TEPP-2AlCl4−, H2TEPP-2AlCl2+, H2TEPP-10AlCl2+). g–i Scatter plots and gradient isosurfaces of IGMH of (g) H2TEPP-2AlCl4− (State I), (h) H2TEPP-2AlCl2+ (State II), and (i) H2TEPP-10AlCl2+ (State III). The blue region represents notable attraction, the green region represents weak interaction, and the red region represents notable repulsion.
To confirm the 12-electron bipolar-redox mechanism of H2TEPP, we calculate the binding energies (∆E) between H2TEPP with different ions in the electrolyte (Supplementary Fig. 33). The positive ∆E values indicate the interaction between H2TEPP and anions during the charge process47, while the negative ∆E values suggest a spontaneous reaction between H2TEPP and cations during the discharge process. Compared with large-sized Al2Cl7− anion, the binding of AlCl4− to H2TEPP is more favorable because of the lower binding energy, while the more negative binding energy indicates that H2TEPP prefers to bind with AlCl2+ cation rather than EMI+. In this case, all possible configurations of H2TEPP upon binding with AlCl4− anions or AlCl2+ cations are considered and optimized to obtain the most stable structures (Supplementary Fig. 34). The calculated total energies and corresponding binding energies of H2TEPP-mAlCl4− (m = 1, 2) and H2TEPP-nAlCl2+ (n = 1, 2, 3, 4, 5, 6, 7, 8, 9, 10) are shown in Supplementary Table 5. Compared with H2TEPP-AlCl2+_b (−6.65 eV), the more negative ∆E value of H2TEPP-AlCl2+_a (− 8.43 eV) indicates a preferential binding of AlCl2+ cation to the central N atoms in H2TEPP, which is attributed to the stronger electrophilic activity of C = N groups than C ≡ C groups. Notably, when C ≡ C groups bind with AlCl2+ cations, the ∆E value of the second AlCl2+ is significantly lower than that of the first AlCl2+, suggesting that the C ≡ C groups may prefer to bind with two AlCl2+ to form a more stable structure. In addition, all ∆E values of H2TEPP and AlCl2+ at each reaction step are negative, indicating that H2TEPP can bind with up to 10 AlCl2+.
The Gibbs free energy changes (∆G) of H2TEPP upon binding with AlCl4− anions and AlCl2+ cations are also calculated based on the most stable coordination configurations (Supplementary Fig. 35 and Table 6). In the charge process, the positive ΔG indicates the reaction of H2TEPP with AlCl4− anion is an endothermic process48. The increased ∆G (ΔG1 < ΔG2) means that the two-step single-electron reactions of H2TEPP with AlCl4− can occur successively. In the discharge process, the negative ΔG suggests the reaction of H2TEPP with AlCl2+ cations can take place spontaneously. The ΔG decreases step by step in the whole discharge process, indicating that the H2TEPP can stably bind with 10 AlCl2+ cations in thermodynamics. Similar to the charge process, the increased ∆G (ΔG1 < ΔG2) means that the two-step single-electron reactions of H2TEPP with AlCl2+ can also occur successively. Considering the fast electron transfer ability of the 18π-electron macrocycle, a dual-electron redox reaction may occur during the charge/discharge process. However, the ΔG of each step from H2TEPP to H2TEPP-10AlCl2+ (from ΔG1 to ΔG10) does not increase continuously, indicating that the discharge process may not be successive ten-step single-electron reactions, but two-step successive dual-electron and 8-electron reactions. Notably, the sum of the ΔG values of two-step reactions for each C ≡ C group is nearly equal, indicating that four alkynyl sites can coordinate with 8 AlCl2+ cations simultaneously. The more negative value of ΔG1 + ΔG2 suggests a preferential coordination of porphyrin N atoms with AlCl2+. Therefore, a three-step 12-electrons-transfer reaction process is proposed (Fig. 6f). Furthermore, the calculated LUMO-HOMO levels show that the energy gaps of all H2TEPP complexes are smaller than the fundamental gap of H2TEPP (Supplementary Fig. 36), which further demonstrates the thermodynamically favorable reaction between H2TEPP and AlCl4− anions (or AlCl2+ cations).
We further perform an independent gradient model based on Hirshfeld partition (IGMH) analysis to reveal the interaction between H2TEPP and AlCl4−/AlCl2+ carriers (Fig. 6g–i)49. In the charge complex H2TEPP-2AlCl4− (state I), the detected green isosurfaces and the corresponding green spike in the sign(λ2)ρ from − 0.02 to 0.00 arb. units indicate a weak interaction between H2TEPP and AlCl4− anions. In the discharged complexes H2TEPP-2AlCl2+ (state II) and H2TEPP-10AlCl2+ (state III), bule isosurfaces and bule spikes in the sign(λ2)ρ from −0.05 to − 0.02 arb. units are observed, suggesting the strong N-Al and C-Al coordination interaction between H2TEPP and AlCl2+ cations. Moreover, the charge transfer between H2TEPP and AlCl4−/AlCl2+ carriers is also revealed by the electron density difference (EDD) plots (Supplementary Fig. 37). When combined with AlCl4− anions, there is an electron depletion region (blue) on the porphyrin macrocycle, indicating that the H2TEPP participate in the electron-donating process. When combined with AlCl2+ cations, the significant electron accumulation regions (red) are observed around center N atoms and terminal C ≡ C groups, suggesting the strong charge transfer from AlCl2+ carriers to H2TEPP and their strong coordination interactions to form stable configurations. In addition, the LOL-π method is used to reveal the π-electron delocalization behavior of H2TEPP during charge/discharge processes (Supplementary Fig. 38). In pristine H2TEPP, the continuous LOL-π isosurfaces throughout the 18π-electron macrocycle shows high conjugation and π-electron delocalization. After binding with two AlCl4− anions, H2TEPP displays a slight break in π-electron isosurfaces, which could be attributed to the distortion of the molecular structure caused by large-sized anions. Impressively, H2TEPP exhibits favorable π-electron isosurfaces even after binding with ten AlCl2+ cations, indicating high structural stability.
As a result, a 12-electron bipolar-redox mechanism has been demonstrated for H2TEPP (Supplementary Fig. 39), which involves the alternate storage of dual charge carriers (AlCl4− anion and AlCl2+ cation). In the first charge process, H2TEPP loses two π-electrons from 18π to 16π configuration and coordinates with two AlCl4− anions. During the discharge process, H2TEPP first accepts two π-electrons from 16π to 18π accompanied by the release of two AlCl4− anions simultaneously. Due to the bipolar-redox activity, a reduction reaction from 18π to 20π enables two AlCl2+ cations to insert into H2TEPP. In the subsequent discharge process, four terminal C ≡ C sites can store up to eight AlCl2+ cations, and each site coordinates with two AlCl2+. The whole charge/ discharge process involves 12 electron transfer and three-step redox reactions. Such alternate storage of opposite charges and multielectron-redox reaction make the utmost of active sites in H2TEPP, remarkably improving the electrochemical activity and storage capacity (Supplementary Fig. 40). Besides, compared with conventional single-ion storage mode, the unique dual-ion alternate storage mode is more likely to accelerate ion transport kinetics and contribute to better rate performance25,50.
Discussion
In summary, we have demonstrated a 12-electron transfer bipolar-redox tetraalkynylporphyrin organic positive electrode to realize high-energy AIBs. The bipolar 18π-electron porphyrin macrocycle achieves a 4-electron redox reaction to alternately bind and release dual charge carriers (AlCl4− anions and AlCl2+ cations) during charge/discharge processes, thus doubling the capacity of the unipolar organic positive electrode. Simultaneously, four terminal alkynyl (C ≡ C) electrophilic sites undergo 8-electron redox to reversibly coordinate/dissociate more AlCl2+ cations with 2-electron transfer for each active site. Consequently, the constructed aluminum-organic battery delivers a high capacity of up to 347 mAh g−1 (3–6 times that of the graphite positive electrode, 60-120 mAh g−1) and a high specific energy of 312 Wh kg−1 (up to 150% compared to cells with graphite as positive electrode) based on the mass of positive electrode materials. The multielectron-transfer molecular design strategy can be extended to other bipolar organic electrode materials for achieving high-capacity and high-energy metal-organic batteries.
Methods
Materials
5,10,15,20-tetrakis(4-ethynylphenyl) porphyrin (H2TEPP, 97%) was purchased from Aladdin (Aladdin, Shanghai, China). 5,10,15,20-tetraphenyl porphyrin (H2TPP, 97%), 5,10,15,20-tetra(4-pyridyl) porphyrin (H2TPyP, 97%), aluminum chloride anhydrous (AlCl3, 99.9%), 1-ethyl-3-methylimidazolium chloride ([EMIm]Cl, 98.0%), N-methyl-2-pyrrolidinone (NMP, 99.5%) and acetonitrile (99.5%) were obtained from Macklin (Shanghai, China). All chemicals and reagents were purchased and used without further purification. Aluminum foil (50 μm thickness, 99.99%), and tantalum foil (10 μm thickness, 99.95%) were purchased from Qing Yuan metal co. LTD (China). Molybdenum mesh (20 μm thickness, 100 mesh, 150 μm pore size, 99.99%) was purchased from Guan Tai Metal Co. LTD (China). All metal materials were used without any chemical treatments prior to electrode fabrication.
Material characterizations
Scanning electron microscopy (SEM, Zeiss Gemini 300) with energy-dispersive spectroscopy (EDS) was used to characterize the micromorphology and elements of samples. Fourier transform infrared (FTIR) spectra were conducted on Thermo Fisher Scientific Nicolet iS20 spectrometer from 400 to 4000 cm−1. In-situ electrochemical FTIR spectra were performed on a Thermo IS50 spectrometer from 650 to 4000 cm−1. The X-ray diffraction (XRD) patterns were collected using a Rigaku D/max-RB X-ray diffractometer with Cu Kα radiation (λ = 1.5406 Å). Thermogravimetric analysis (TGA) was carried out on a Rigaku TG/DTA8122 thermal analyzer at a heating rate of 10 °C min−1 under N2 flow. Raman spectra were collected from a HORIBA LabRAM/HR800 confocal Raman microscope with a laser wavelength of 532 nm to investigate the structural evolution of organic molecules. X-ray photoelectron spectroscopy (XPS) tests were performed on a Thermo Scientific K-Alpha spectrometer to analyze the chemical states of the electrodes. Time-of-flight secondary ion mass spectrometry (TOF-SIMS) depth profiling and analysis were carried out on a TOF secondary ion mass spectrometer (TOF-SIMS ion tof Gmhb 5) using a primary bismuth ion source (negative ion mode).
All electrode samples for ex-situ characterizations were disassembled from the Swagelok cells at different charged/discharged states in an argon-filled glove box (O2 < 0.1 ppm, H2O < 0.1 ppm). The electrodes were washed three times with anhydrous acetonitrile to remove the electrolyte, followed by drying under vacuum for 2 h. The electrodes for ex-situ XPS and TOF-SIMS characterizations were transferred from the glove box to the test chamber with a vacuum transfer vessel to avoid exposure to oxygen and moisture. The electrodes for ex-situ Raman, FTIR, and SEM-EDS characterizations were separately sealed in the glove box and stored in a vacuum chamber, and the packages were opened during testing.
Cell preparation and electrochemical measurements
The electrochemical performances were measured using pouch cells or Swagelok cells. To prepare the organic positive electrode, active material (H2TEPP, H2TPP and H2TPyP), acetylene black, and polyvinylidene fluoride (PVDF) binder were mixed in NMP solvent by a weight ratio of 6:3:1. At room temperature and air atmosphere, the mixture was mixed on an automatic mixer for 12 h to form a homogeneous slurry, which is then cast onto a tantalum foil current collector using a wet film coater. The electrode was dried overnight at 60 °C in a vacuum drying oven, and the mass loading of active material is ca. 1.0 mg cm−2. To prepare the ionic liquid electrolyte, in an argon-filled glove box (O2 < 0.1 ppm, H2O < 0.1 ppm), AlCl3 was slowly added to [EMIm]Cl in a molar ratio of 1.3:1 (AlCl3:[EMIm]Cl) in a glass bottle. The electrolyte was then stirred using a stir bar at room temperature for 12 h. The pouch cells were assembled with an organic positive electrode (1.5 × 1.5 cm), a modified glass fiber separator (Whatman GF/A, 2.5 × 2.5 cm), an aluminum foil negative electrode (2 × 2 cm), and 350 μL AlCl3/[EMIm]Cl electrolyte in the argon-filled glove box. The Swagelok cells were assembled with an organic positive electrode (12 mm diameter), a modified glass fiber separator (Whatman GF/A, 14 mm diameter), an aluminum foil negative electrode (14 mm diameter), and 100 μL AlCl3/[EMIm]Cl electrolyte in the argon-filled glove box. The Swagelok cells using polytetrafluoroethylene shells (14 mm inner diameter), and molybdenum rods (14 mm diameter) as the current collector. Galvanostatic charge-discharge measurements were conducted using pouch cells on the multichannel battery testing system (Neware BTS-53) in a voltage range of 0.1‒2.2 V at 25 °C and air environment. Cyclic voltammetry (CV) was performed on the EC-Lab electrochemical workstation in a range of 0.1‒2.3 V at different scan rates from 0.1 to 10.0 mV s−1. Electrochemical impedance spectroscopy (EIS) measurements were carried out on an EC-Lab electrochemical workstation in a frequency range of 100 kHz to 0.01 Hz with a perturbation amplitude of 5 mV under the open circuit potential. Galvanostatic intermittent titration technique (GITT) measurement was performed with a galvanostatic pulse current of 0.1 A g−1 for 6 min, followed by an open-circuit relaxation of 1 h. The CV, EIS, and GITT measurements were performed using Swagelok cells to study the in-situ electropolymerization behavior and electrochemical kinetics of porphyrins.
For the in situ electrochemical FTIR spectra test, the H2TEPP positive electrode was prepared using the same method with molybdenum mesh as the current collector. The weight ratio of H2TEPP, acetylene black, and PVDF binder is 6:3:1, and the mass loading of active material is ca. 1.0 mg cm−2. The Al | |H2TEPP cell was assembled with an H2TEPP positive electrode (12 mm diameter), a glass fiber separator (Whatman GF/A, 18 mm diameter), an aluminum foil negative electrode (14 mm diameter), and 150 μL AlCl3/[EMIm]Cl electrolyte in the argon-filled glove box. The charge-discharge test was performed in a voltage range of 0.1‒2.2 V at 0.1 A g−1.
Density functional theory calculations
All DFT calculations were performed using the Gaussian 16 software package51. The geometry structures of all porphyrin molecules, ions, and coordination compounds were fully optimized at the level of B3LYP52/def2-SVP53 with Grimme’s DFT-D3(BJ) empirical dispersion correction54 to describe the weak interaction. The vibration frequency calculations of the optimized structures were performed at the same level to confirm that the optimized geometry is the local minimum point on the potential energy surface (no imaginary frequencies) and obtain the thermal correction of Gibbs free energy. The implicit solvation model based on density-generic ionic liquid (SMD-GIL)55 is used to consider the solvation effect of AlCl3/[EMIm]Cl ionic liquid electrolyte for geometry optimization and frequency calculations. The average ionic liquid solvent parameters of SMD-GIL are shown in Supplementary Table 7.
The precise single point energy (E) of the optimized structures was further calculated at a higher level of M06-2X56/def2-TZVP53, and the solvation effect was also considered by SMD-GIL. The binding energies (∆E) between H2TEPP and different ions in the electrolyte are calculated using the formula:
where \({E}_{(x){ion}}\), \({E}_{(x-1){ion}}\) and \({E}_{{ion}}\) are the single point energy for H2TEPP with x ions, H2TEPP with x − 1 ions, and the ion, respectively; x is the number of ions.
The Gibbs free energy (G) was calculated using the Shermo program57 based on the precise single point energy, the thermal correction value of Gibbs free energy, and the zero point energy (ZPE) correction factor. According to the ZPVE15/10 database58, the ZPE correction factor of the B3LYP/def2-SVP level was fitted as 0.9826. The Gibbs free energy change (∆G) of H2TEPP in each step of the reaction process can be calculated using the formula:
where \({G}_{f}\), \({G}_{i}\), and \({G}_{{ion}}\) are the Gibbs free energy for the final and initial state of H2TEPP, and for the Al-complex ions (AlCl4−or AlCl2+), respectively; n is the number of Al-complex ions.
The calculated results of molecular electrostatic potential (MESP), molecular orbital (MO) composition, localized orbital locator-π (LOL-π), iso-chemical shielding surface (ICSS), nucleus-independent chemical shift (NICS), interaction region indicator (IRI), independent gradient model based on Hirshfeld partition (IGMH) and electron density difference (EDD) were performed via Multiwfn 3.8 program59. The isosurface maps were visualized by Visual Molecule Dynamics (VMD) 1.9.3 software60. The anisotropy of the induced current density (AICD) were calculated by the AICD program61.
Molecular dynamics simulations
Molecular dynamics (MD) simulations were conducted using the GROMACS software62 to elucidate the diffusion behaviors of AlCl2+ cations and AlCl4− anions within the H2TEPP matrix. The UFF force field63 was employed for these simulations. Force field parameters for the AlCl2+ cation and AlCl4− anion were derived using the Seminario method64 in conjunction with the RESP charge model. The periodic structure of the H2TEPP polymer was parameterized using the UFF force field. To accurately model the Coulombic interactions between H2TEPP and Al-complex ions, the H2TEPP polymer structure was refined using the CP2K quantum chemistry software package65. Subsequently, the REPEAT atomic charges66 of H2TEPP polymer at varying charge states were calculated to serve as input parameters for the MD simulations.
The initial simulation boxes were populated AlCl2+ cations and AlCl4− anions, ensuring that the overall net charge of each system was neutral. The simulation box contained 160 repeated H2TEPP units, with a charge state of 50% for both AlCl2+ and AlCl4−, resulting in 160 AlCl4− and 960 AlCl2+ ions, respectively. The systems were subjected to energy minimization followed by a thermal annealing process from 0 K to 400 K within 1 ns (time step: 1 ps) to achieve equilibrium67. The systems were then equilibrated at 298.15 K using a velocity-rescale thermostat68 (relaxation time constant is 1 ps) and maintained at 1.01325 × 105 Pa with a Berendsen’s barostat (isothermal compressibility constant is 4.5 × 105). Considering the periodicity of the systems, periodic boundary conditions were imposed in all dimensions. Particle-mesh Ewald (PME) method was used to calculate the electrostatic interactions and van der Waals forces, and the cut-off distance was set to 15 Å. Finally, a 20 ns MD simulation was conducted under the NVT ensemble, recording the trajectory snapshots every 1 ps. Subsequent statistical analysis was performed on the trajectory data using GROMACS tools. The simulation box visualization was accomplished using the VMD program.
The translation diffusion coefficient was calculated by linear fitting of the mean square displacement (MSD) of the center of mass of a molecule:
where τ is the lag time between the two positions. The coefficient before the MSD takes the value of 1/6 for a three-dimensional system.
Evaluation of the specific energy
The specific energy of aluminum batteries can be calculated using the following equation:
where E is the specific energy (Wh kg−1), V is the average output voltage (V) of the battery, C is the capacity (mAh), and m is the mass (mg) of the active material. Therefore, the specific energy of the Al | |H2TEPP battery based on the active material (H2TEPP) reaches 312 Wh kg−1, this value is higher than those reported in most literature for organic and graphitic positive electrode materials in AIBs.
Data availability
All relevant data supporting the findings of this study are available in this article and Supplementary Information. The data are available from the corresponding author on request. Source data are provided in this paper.
References
Tu, J. et al. Nonaqueous rechargeable aluminum batteries: Progresses, challenges, and perspectives. Chem. Rev. 121, 4903–4961 (2021).
Lin, M.-C. et al. An ultrafast rechargeable aluminium-ion battery. Nature 520, 324–328 (2015).
Wang, D.-Y. et al. Advanced rechargeable aluminium ion battery with a high-quality natural graphite cathode. Nat. Commun. 8, 14283 (2017).
Chen, H. et al. Ultrafast all-climate aluminum–graphene battery with quarter-million cycle life. Sci. Adv. 3, 2375–2548 (2017).
Wang, S. et al. High-performance aluminum-ion battery with CuS@C microsphere composite cathode. ACS Nano 11, 469–477 (2017).
Cai, T. et al. Stable CoSe2/carbon nanodice@reduced graphene oxide composites for high-performance rechargeable aluminum-ion batteries. Energy Environ. Sci. 11, 2341–2347 (2018).
Yu, Z. et al. Construction of double reaction zones for long-life quasi-solid aluminum-ion batteries by realizing maximum electron transfer. Nat. Commun. 14, 5596 (2023).
He, S. M. et al. Rechargeable Al-chalcogen batteries: Status, challenges, and perspectives. Adv. Energy Mater. 11, 2100769 (2021).
Kim, D. J. et al. Rechargeable aluminium organic batteries. Nat. Energy 4, 51–59 (2019).
Bitenc, J. et al. Concept and electrochemical mechanism of an Al metal anode–organic cathode battery. Energy Stor. Mater. 24, 379–383 (2020).
Zhou, J. W., Yu, X. Z., Zhou, J. & Lu, B. G. Polyimide/metal-organic framework hybrid for high performance Al-Organic battery. Energy Stor. Mater. 31, 58–63 (2020).
Yoo, D.-J., Heeney, M., Glöcklhofer, F. & Choi, J. W. Tetradiketone macrocycle for divalent aluminium ion batteries. Nat. Commun. 12, 2386 (2021).
Mao, M. et al. A pyrazine-based polymer for fast-charge batteries. Angew. Chem. Int. Ed. 58, 17820–17826 (2019).
Guo, F. et al. Active cyano groups to coordinate AlCl2+ cation for rechargeable aluminum batteries. Energy Stor. Mater. 33, 250–257 (2020).
Qin, K. et al. Synergy of carbonyl and azo chemistries for wide-temperature-range rechargeable aluminum organic batteries. Nano Energy 101, 107554 (2022).
Walter, M., Kravchyk, K. V., Böfer, C., Widmer, R. & Kovalenko, M. V. Polypyrenes as high-performance cathode materials for aluminum batteries. Adv. Mater. 30, 1705644 (2018).
Wang, G. et al. An efficient rechargeable aluminium–amine battery working under quaternization chemistry. Angew. Chem. Int. Ed. 61, e202116194 (2022).
Guo, Y., Wang, W., Lei, H., Wang, M. & Jiao, S. Alternate storage of opposite charges in multisites for high-energy-density Al–MOF batteries. Adv. Mater. 34, 2110109 (2022).
Liu, Y. et al. Redox-bipolar polyimide two-dimensional covalent organic framework cathodes for durable aluminium batteries. Angew. Chem. Int. Ed. 62, e202306091 (2023).
Zhao-Karger, Z. et al. New organic electrode materials for ultrafast electrochemical energy storage. Adv. Mater. 31, 1806599 (2019).
Min Park, J., Lee, J. H. & Jang, W.-D. Applications of porphyrins in emerging energy conversion technologies. Coord. Chem. Rev. 407, 213157 (2020).
Han, X. et al. Stable high-capacity organic aluminum-porphyrin batteries. Adv. Energy Mater. 11, 2101446 (2021).
Jiao, S. et al. Functional group-driven competing mechanism in electrochemical reaction and adsorption/desorption processes toward high-capacity aluminum-porphyrin batteries. Angew. Chem. Int. Ed. 63, e202410110 (2024).
Chowdhury, S. et al. π-Conjugated metal free porphyrin as organic cathode for aluminum batteries. Batteries Supercaps 7, e202300285 (2024).
Song, Z. et al. Multielectron redox-bipolar tetranitroporphyrin macrocycle cathode for high-performance zinc-organic batteries. Angew. Chem. Int. Ed. 63, e202401049 (2024).
Cao, Y. et al. Alkynyl boosted high-performance lithium storage and mechanism in covalent phenanthroline framework. Angew. Chem. Int. Ed. 62, e202302143 (2023).
Wu, K., Guo, J. & Wang, C. Gelation of metalloporphyrin-based conjugated microporous polymers by oxidative homocoupling of terminal alkynes. Chem. Mater. 26, 6241–6250 (2014).
Gao, P. et al. A porphyrin complex as a self-conditioned electrode material for high-performance energy storage. Angew. Chem. Int. Ed. 56, 10341–10346 (2017).
Liu, L. et al. Molecular electrostatic potential: A new tool to predict the lithiation process of organic battery materials. J. Phys. Chem. Lett. 9, 3573–3579 (2018).
Liu, Z., Lu, T. & Chen, Q. An sp-hybridized all-carboatomic ring, cyclo[18]carbon: Bonding character, electron delocalization, and aromaticity. Carbon 165, 468–475 (2020).
Wang, Z. et al. Nanoporous porphyrin polymers for gas storage and separation. Macromolecules 45, 7413–7419 (2012).
Li, D., Swanson, B. I., Robinson, J. M. & Hoffbauer, M. A. Porphyrin based self-assembled monolayer thin films: Synthesis and characterization. J. Am. Chem. Soc. 115, 6975–6980 (1993).
Feng, X. et al. Stabilization of organic cathodes by a temperature-induced effect enabling higher energy and excellent cyclability. ACS Appl. Mater. Interfaces 13, 7178–7187 (2021).
Feng, X. et al. A bipolar organic molecule toward a universal pseudocapacitive cathode for stable dual ion charge storage. Energy Stor. Mater. 42, 454–463 (2021).
Wu, X. et al. Porphyrin-thiophene based conjugated polymer cathode with high capacity for lithium-organic batteries. Angew. Chem. Int. Ed. 63, e202317135 (2024).
Wang, N. et al. Graphdiyne-based materials: Preparation and application for electrochemical energy storage. Adv. Mater. 31, 1803202 (2019).
Han, D. et al. Initial electrode kinetics of anion intercalation and de-intercalation in nonaqueous Al-graphite batteries. Chin. J. Chem. 39, 157–164 (2021).
Huang, H., Li, F., Zhang, Y. & Chen, Y. Two-dimensional graphdiyne analogue Co-coordinated porphyrin covalent organic framework nanosheets as a stable electrocatalyst for the oxygen evolution reaction. J. Mater. Chem. A 7, 5575–5582 (2019).
Wang, J. et al. Porphyrin conjugated polymer grafted onto BiVO4 nanosheets for efficient Z-scheme overall water splitting via cascade charge transfer and single-atom catalytic sites. Adv. Energy Mater. 11, 2003575 (2021).
Wu, X. et al. Thiophene functionalized porphyrin complexes as novel bipolar organic cathodes with high energy density and long cycle life. Energy Stor. Mater. 46, 252–258 (2022).
Shea, J. J. & Luo, C. Organic electrode materials for metal ion batteries. ACS Appl. Mater. Interfaces 12, 5361–5380 (2020).
Mori, Y. & Sugahara, Y. One-pot synthesis of soluble precursors possessing both Al–N and B–N backbones and their pyrolysis. Bull. Chem. Soc. Jpn. 79, 1681–1687 (2006).
Meng, F., Yu, G. & Huang, B. Polymer-supported zirconocene catalyst for ethylene polymerization. J. Polym. Sci. Part A Polym. Chem. 37, 37–46 (1999).
Zhang, L., Wang, K., Qian, X., Liu, H. & Shi, Z. Porous conjugated polymer nanotip arrays for highly stable field emitter. ACS Appl. Mater. Interfaces 5, 2761–2766 (2013).
Ju, C.-W. et al. Modular synthesis of pentagonal and hexagonal ring-fused NBN-phenalenes leading to an excited-state aromatization-induced structural planarization molecular library. J. Am. Chem. Soc. 143, 5903–5916 (2021).
Tie, Z. et al. A symmetric all-organic proton battery in mild electrolyte. Angew. Chem. Int. Ed. 61, e202115180 (2022).
Jiang, Q. et al. A redox-active 2D metal-organic framework for efficient lithium storage with extraordinary high capacity. Angew. Chem. Int. Ed. 59, 5273–5277 (2020).
Yan, L. et al. Towards high-performance aqueous zinc batteries via a semi-conductive bipolar-type polymer cathode. Angew. Chem. Int. Ed. 61, e202211107 (2022).
Lu, T. & Chen, Q. Independent gradient model based on Hirshfeld partition: A new method for visual study of interactions in chemical systems. J. Comput. Chem. 43, 539–555 (2022).
Sun, W. et al. Ion co-storage in porous organic frameworks through on-site Coulomb interactions for high energy and power density batteries. Angew. Chem. Int. Ed. 62, e202300158 (2023).
Frisch, M. J. et al. Gaussian 16 Rev. C.01. (2016).
Stephens, P. J., Devlin, F. J., Chabalowski, C. F. & Frisch, M. J. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 98, 11623–11627 (1994).
Weigend, F. & Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 7, 3297–3305 (2005).
Goerigk, L. & Grimme, S. Efficient and accurate double-hybrid-meta-GGA density functionals—Evaluation with the extended GMTKN30 database for general main group thermochemistry, kinetics, and noncovalent interactions. J. Chem. Theory Comput. 7, 291–309 (2011).
Bernales, V. S., Marenich, A. V., Contreras, R., Cramer, C. J. & Truhlar, D. G. Quantum mechanical continuum solvation models for ionic liquids. J. Phys. Chem. B 116, 9122–9129 (2012).
Zhao, Y. & Truhlar, D. G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 120, 215–241 (2008).
Lu, T. & Chen, Q. Shermo: A general code for calculating molecular thermochemistry properties. Comput. Theor. Chem. 1200, 113249 (2021).
Alecu, I. M., Zheng, J., Zhao, Y. & Truhlar, D. G. Computational thermochemistry: Scale factor databases and scale factors for vibrational frequencies obtained from electronic model chemistries. J. Chem. Theory Comput. 6, 2872–2887 (2010).
Lu, T. & Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 33, 580–592 (2012).
Humphrey, W., Dalke, A. & Schulten, K. V. M. D. Visual molecular dynamics. J. Mol. Graph. 14, 33–38 (1996).
Geuenich, D., Hess, K., Köhler, F. & Herges, R. Anisotropy of the induced current density (ACID), a general method to quantify and visualize electronic delocalization. Chem. Rev. 105, 3758–3772 (2005).
Abraham, M. J. et al. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1-2, 19–25 (2015).
Rappe, A. K., Casewit, C. J., Colwell, K. S., Goddard, W. A. III & Skiff, W. M. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J. Am. Chem. Soc. 114, 10024–10035 (1992).
Allen, A. E. A., Payne, M. C. & Cole, D. J. Harmonic force constants for molecular mechanics force fields via hessian matrix projection. J. Chem. Theory Comput. 14, 274–281 (2018).
Kühne, T. D. et al. CP2K: An electronic structure and molecular dynamics software package-Quickstep: Efficient and accurate electronic structure calculations. J. Chem. Phys. 152, 194103 (2020).
Gabrieli, A., Sant, M., Demontis, P. & Suffritti, G. B. Partial charges in periodic systems: Improving electrostatic potential (ESP) fitting via total dipole fluctuations and multiframe approaches. J. Chem. Theory Comput. 11, 3829–3843 (2015).
Li, N. et al. Customization nanoscale interfacial solvation structure for low-temperature lithium metal batteries. Energy Environ. Sci. 17, 5468–5479 (2024).
Bussi, G., Donadio, D. & Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 014101 (2007).
Acknowledgements
This work was financially supported by the Beijing Natural Science Foundation (No. L243019), National Natural Science Foundation of China (Nos. 52374298 and 52474318), Natural Science Foundation of Chongqing (No. CSTB2023NSCQ-MSX0662), Beijing Nova Program (No. Z211100002121082) and Interdisciplinary Research Project for Young Teachers of USTB (Fundamental Research Funds for the Central Universities, No. FRF-IDRY-GD23-005). The authors also thank the Xiaomi Foundation for the financial support of the Xiaomi Young Scholar Program.
Author information
Authors and Affiliations
Contributions
S.J., Y.Z., and W.W. conceived the concept. S.J., W.W., and Y.Z. designed the experiments. Y.G. carried out the experiments and simulations. Y. G. and W. W. co-wrote the manuscript. K.G., X.C., Z.H., M.W., and W.S. participated in characterizations and results analysis. All authors discussed the results.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Guo, Y., Wang, W., Guo, K. et al. A bipolar-redox tetraalkynylporphyrin macrocycle positive electrode with 12-electrons-transfer for high-energy aluminum-organic batteries. Nat Commun 16, 2794 (2025). https://doi.org/10.1038/s41467-025-58126-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-58126-5
