
Abstract
The industrial implementation of coupled electrochemical hydrogen production systems necessitates high power density and high product selectivity for economic viability and safety. However, for organic nucleophiles (e.g., methanol, urea, and amine) electrooxidation in the anode, most catalytic materials undergo unavoidable reconstruction to generate high-valent metal sites under harsh operation conditions, resulting in competition with oxygen evolution reaction. Here, we present unique Ni(II) sites in Prussian blue analogue (NiFe-sc-PBA) that serve as stable, efficient and selective active sites for ethylene glycol (EG) electrooxidation to formic acid, particularly at ampere-level current densities. Our in situ/operando characterizations demonstrate the robustness of Ni(II) sites during EG electrooxidation. Molecular dynamics simulations further illustrate that EG molecule tends to accumulate on the NiFe-sc-PBA surface, preventing hydroxyl-induced reconstruction in alkaline solutions. The stable Ni(II) sites in NiFe-sc-PBA anodes exhibit efficient and selective EG electrooxidation performance in a coupled electrochemical hydrogen production flow cell, producing high-value formic acid compared to traditional alkaline water splitting. The coupled system can continuously operate at stepwise ampere-level current densities (switchable 1.0 or 1.5 A cm−2) for over 500 hours without performance degradation.
Similar content being viewed by others
Introduction
Water electrolysis driven by renewable energy sources can yield high-purity green hydrogen, offering a sustainable alternative to the conventional steam methane reforming process1,2,3. Nevertheless, the high theoretical potential (1.23 V versus reversible hydrogen electrode, vs. RHE; all potentials are vs. RHE if not mentioned) of the sluggish oxygen evolution reaction (OER) demands substantial power input and poses an explosion risk due to hydrogen-oxygen mixing in industrial settings4,5. To mitigate these issues, alternative oxidations of small molecules have been developed6, such as hydrazine7, urea8,9, certain alcohols10,11,12 and aldehydes13. Notably, the alcohol oxidation reaction (AOR)14,15 has attracted considerable attention as an alternative anode reaction, prized for its higher product value and lower theoretical potential (0.003 V for glycerol16, 0.103 V for methanol (MeOH)17 and 0.57 V for ethylene glycol (EG)18) with no vapor products. Annually, the production of polyethylene terephthalate (PET) reaches approximately 70 million tons annually14,19; however, the disposal of waste PET through incineration or landfilling contributes significantly to environmental degradation20. Alternatively, wasted PET can be recycled and dissolved out the EG monomer with strong alkali, and then upgraded via electrochemical EG oxidation reaction (EGOR)21,22, generating products of formic acid (FA)18,23, glycolic acid (GA)24, oxalic acid (OA)25, carbonate, etc., of which FA is considered a high-value product essential for advancing the cradle-to-gate approach toward carbon neutralization26.
Recently, there has been considerable progress in developing efficient and durable electrocatalysts for EGOR, including transition metal phosphides (CoNi0.25P15, Co-Ni2P27, CoNi0.2P-uNS28), selenides (Ni2Se18, NiMn-MOF-Se14), sulfides (Co, Cl-NiS29) targeted at producing FA, alongside noble metal-based materials optimized for GA generation24,30. For FA-selective electrocatalysts, most of these transition metal-based materials undergo structural reconfiguration at high potentials, forming metal (oxy)hydroxides in a manner akin to the OER process14,15. These high-valence metal sites within the (oxy)hydroxides can reach considerable FEFA in the specific conditions, however, the elevated reaction rates (especially at ampere-level current densities) facilitate the adsorption and desorption of oxygen-relative intermediates31, leading to unavoidable OER competition. In this regard, it is imperative to engineer structurally stable and highly efficient electrocatalytic materials that promote selective and rapid EGOR without undergoing reconfiguration to form OER-active species.
In comparing the electrolytes used for EGOR and OER, the alternative anode reactions differentiate in the reactants deriving from the substrate rather than water alone. This complexity is further compounded by the absorption and desorption processes of the substrate, making the interaction between the substrate and the anode material a critical factor in both the catalytic activity and the structural evolution of the electrocatalyst32,33. Generally, two predominant types of interactions between the substrate and catalyst have been identified: substrate-promoted catalyst structural transformation34,35,36 and substrate-stabilized catalyst structure9,11. On the one hand, our group34 has reported a “reactant-induced activation strategy” for NiCoBx, which enhances the formation of Co(III) active sites crucial for the oxidation of 5-hydroxymethylfurfural (HMF); Bedford et al.35 have proposed that HMF reactant enables reciprocal reconfiguration of metal sites, and that high-valence (+3 and +4) sites accelerate the reaction kinetics and enable highly selective generation of 2,5-furandicarboxylic acid (FDCA). On the other hand, in studies regarding substrate-stabilized catalyst structure11, unexpectedly, Qiao et al.9 discovered that Prussian blue analogues (PBA) facilitate urea oxidation on low-valence nickel sites, effectively preventing catalyst reconstruction over short periods. In previous studies on AORs, systematic elucidation of the interaction between EG and the anode material remains sparse, and, to our knowledge, no electrocatalyst has yet demonstrated stability without structural reconstruction, particularly under ampere-level current densities6. PBAs with open backbone and stable coordination structure have been widely studied as anode materials, especially for OER. However, the interaction between alcohol substrates and cyano-ligand structures has not been systematically investigated in AORs, and the unique metal-cyanine ligands would possibly maintain its original motif due to strong metal-CN-metal bonding.
Herein, we have successfully demonstrated stable Ni(II) sites in Prussian blue analogue (NiFe-sc-PBA) for selective and fast EGOR, achieving low applied potentials of 1.484 and 1.625 V to reach current densities (j) of 500 and 1000 mA cm−2,respectively, with a peak 98.5% Faradaic efficiency of formic acid (FEFA) and robust stability at 1.60 V over 50 h (FEFA > 90%). In situ/operando structural characterizations have confirmed the stability of Ni(II) sites during EG electrooxidation. Additionally, molecular dynamics simulations further illustrate that EG molecules preferentially accumulate on the NiFe-sc-PBA surface, inhibiting hydroxyl-induced reconstruction. Moreover, when integrated into a coupled electrochemical hydrogen production system, this NiFe-sc-PBA anode not only reduces cell voltage energy consumption by 450 and 530 mV at current densities of 1.0 and 1.5 A cm−2 compared to a conventional alkaline water splitting system, but also facilitates the production of high value-added products, enhancing techno-economic efficiency. Impressively, the coupled system can operate continuously at stepwise ampere-level current densities for more than 500 h without performance degradation. Our work underscores the importance of understanding the interaction between substrates and stable Ni(II) active sites in EGOR process, providing a roadmap for future development of high-performance electrocatalysts for the electrooxidation of organic nucleophiles.
Results and discussion
Synthesis and structural characterization of Ni(II) sites in NiFe-sc-PBA
The NiFe-sc-PBA was synthesized directly on nickel foam (NF) via one-step hydrothermal method, using sodium citrate as a reducing agent to modulate the valence of Fe ions in the ligand center to form structurally well-defined NiFe-sc-PBA. Upon synthesis, the blank NF changed from its original silvery-white to green (Supplementary Fig. 1), representing the successful formation of NiFe-sc-PBA on NF. The catalyst crystallizes in the cubic Fm̅3m space group (Supplementary Fig. 2). To avoid the influence of strong peaks from NF, we performed an X-ray diffraction (XRD) pattern using the surplus NiFe-sc-PBA in the hydrothermal autoclave. As shown in Supplementary Fig. 3, a series of diffraction peaks at 17.6°, 25.0°, 36.6°, 40.0°, etc. match the (200), (220), (400), (420), etc. crystallographic planes of K2Ni[Fe(CN)6] (JCPDS No. 20-0915), respectively. We studied the chemical states and coordination structure of NiFe-sc-PBA using X-ray absorption near-edge spectroscopy (XANES). The first derivative normalized XANES spectra for Ni K-edge are shown in Supplementary Fig. 4a, and the topical peaks of NiFe-sc-PBA are essentially identical to NiO and NiOOH. To be specific, the absorption edge of NiFe-sc-PBA is located between NiO and NiOOH, indicating that the bulk average valence states of Ni species are below +3. Extended X-ray absorption fine structure (EXAFS) results of the investigation into the coordination environment. As shown in Supplementary Fig. 4b, the Ni R space of NiFe-sc-PBA exhibited prominent peaks at around 1.56 and 2.63 Å, attributed to the Ni-N/O and Ni-C scattering paths37,38. Correspondingly, the valence of Fe was +3 (Supplementary Fig. 5), for the absorption edge located between FeO and Fe2O3. The peaks at around 1.41 and 2.42 Å are attributed to Fe-C/O and Fe-N scattering paths37.
Scanning electron microscope (SEM) images (Supplementary Fig. 6) demonstrated the microstructure of the catalysts. NiFe-sc-PBA exhibited a classical structure of nanocubes, growing abundantly and uniformly on the NF. There was a certain height distribution and some pore structure (Supplementary Fig. 6b), which also increase the overall specific surface area of the catalyst. High-resolution transmission electron microscopy (HRTEM) images of NiFe-sc-PBA (Supplementary Fig. 7a) showed a clear indication of a well-defined nanocube structure. As shown in the inset of Supplementary Fig. 7b, NiFe-sc-PBA shows the characteristic spacings of 0.249 nm for the (400) lattice planes of K2[NiFe(CN)6]. In order to further clarify the elemental distribution, we performed energy dispersive X-ray spectroscopy (EDS) detection, and the elements of Ni, Fe, K, C, and N showed a homogeneous distribution (Supplementary Fig. 8).
Evidence of low-valence Ni(II) sites for motivating EGOR
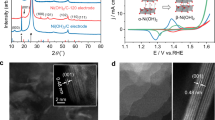
Figure 1a shows the schematic of the catalytic behavior for the NiFe-sc-PBA in the EGOR and OER processes. For EGOR, ethylene glycol accumulates on the NiFe-sc-PBA surface for protection, preventing the further oxidation of the NiFe-sc-PBA, and enabling the oxidation of ethylene glycol to formic acid at low-valence Ni(II) sites. For OER, NiFe-sc-PBA is oxidized to NiOOH under the attack of OH- without ethylene glycol addition, while simultaneously causing the leaching of ferrocyanide. We conducted EGOR or OER at 1.6 V for different time to investigate structural evolution for NiFe-sc-PBA. From the corresponding SEM images (Fig. 1b, c and Supplementary Fig. 9), it can be observed that the morphology of NiFe-sc-PBA remained unchanged after EGOR, presenting the standard nanocube morphology. In contrast, after OER (Fig. 1d and Supplementary Fig. 10), the catalyst underwent comprehensive reconstruction into a sheet-like structure. Moreover, the cracking of the catalyst layer on NF after OER also indicated the reconstruction of NiFe-sc-PBA (Supplementary Fig. 10). The crystalline structures of NiFe-sc-PBA did not undergo any change after EGOR (Fig. 1e). In contrast, all the diffraction peaks disappeared after OER, suggesting that NiFe-sc-PBA underwent reconstruction and an amorphous or poorly crystallized structure was generated.

a Schematic illustration of the protective role of EG during EGOR and the structure evolution of NiFe-sc-PBA during OER. SEM images of NiFe-sc-PBA for pristine (b), EGOR after 2 h (c) and OER after 2 hours (d) corresponding to catalytic behaviors shown in (a). e The XRD patterns of NiFe-sc-PBA for pristine, post-EGOR and post-OER samples. The High-resolution XPS spectra of NiFe-sc-PBA for pristine, post-EGOR and post-OER samples in f Ni 2p and g Fe 2p regions, the gray circle lines are for the original spectrum. Source data are provided as a Source Data file.
To illustrate the chemical environment after EGOR and OER, NiFe-sc-PBA was investigated by high-resolution X-ray photoelectron spectroscopy (XPS). Peaks at 288.1 and 286 eV are attributed to C=N-O and C≡N (Supplementary Fig. 11a) for NiFe-sc-PBA and Post EGOR; however, after OER, peaks appearing at 288.8 and 285.6 eV are attributed to O-C=O and C-OH. Peaks at 402.5, 400.1 and 398 eV are attributed to NO3-, N-H, and C≡N (Supplementary Fig. 11b)39. A significant increase in the intensity of the N-H peak after OER corresponded to a decrease in the intensity of the C≡N peak. The combination of C 1s and N 1s spectra can illustrate the retention of C≡N after EGOR in NiFe-sc-PBA. Whereas, after OER, C≡N was oxidized as well as dissolved, O-C=O and C-OH appeared40. The increase in the strength of N-H could be attributed to the partial oxidative bond breaking of C≡N. For NiFe-sc-PBA, peaks at 874.6 and 857.3 eV are assigned to 2p1/2 and 2p3/2 states of Ni3+, peaks at 872.8 and 855.5 eV are assigned to 2p1/2 and 2p3/2 states of Ni2+, respectively, with two satellite peaks (Fig. 1f)41. After EGOR, peaks of Ni2+ underwent a weak negative shift (~0.1 eV) but the peaks of Ni3+ were significantly negatively shifted (~0.4 eV). For OER, the main peaks of Ni were significantly positively shifted (Supplementary Table 1), demonstrating the surface Ni species are strongly oxidized. Importantly, the ratio of Ni2+/Ni3+ increased after EGOR and decreased after OER (Supplementary Table 1), which indicates that the overall Ni sites might experience a reduction process because of EG, suggesting the role of EG in protecting the catalyst from oxidation. However, without EG during OER, the catalyst suffers from a strong oxidation, which results in an increase in the valence of Ni. It is noteworthy that the Fe 2p orbital was particularly weak after OER (Fig. 1g). The low Fe content after OER indicated the Fe leaching from NiFe-sc-PBA during OER process.
After OER, the electrolyte turned light yellow similar to ferricyanide. The leaching of Fe was further detected by ultraviolet-visible (UV-vis) spectroscopy (Supplementary Fig. 12). The absorption of the electrolytes after OER was identical to that of standard [Fe(CN)6]4-, with little time-dependent variation in peak intensities. This suggests that the reconstruction of NiFe-sc-PBA proceeds rapidly during OER, leading to the rapid release of ferrocyanide ions.
We performed cyclic voltammetry scans at 0.1 mV s−1 to avoid the “pseudo-inductive” currents (Supplementary Fig. 13)8,9. During the OER process, NiFe-sc-PBA began to show a current rise at 1.34 V, exhibiting a broad oxidation peak, indicating the obvious oxidation from Ni(II) to Ni(III). This oxidation peak range includes both the oxidation current and the inductive current. In contrast, during the EGOR process, NiFe-sc-PBA showed an onset potential around 1.31 V, which is significantly lower than the oxidation potential of Ni(II) to Ni(III). In the reported Ni2+/Ni3+ redox process18,29, the oxidation of ethylene glycol occurred during the reduction from Ni3+ to Ni2+, thus the onset potential must be higher than or equal to the oxidation potential of Ni(II). However, the onset point of the EGOR process for NiFe-sc-PBA is clearly earlier than the oxidation potential of Ni(II), indicating that the EGOR process of NiFe-sc-PBA did not rely on the oxidation from Ni(II) to Ni(III) nor is it a Ni2+/Ni3+ redox process.
In situ Raman spectroscopy was conducted to detect structural evolution behaviors during EGOR and OER processes, with the device schematic diagram shown in Supplementary Fig. 14. In Raman spectra, two strong peaks at 2104 and 2141.8 cm−1 are characteristic of cyanide stretching, coexisting with the peak at 251 cm−1 belonging to the Ni-N stretching vibration and the peak at 510 cm−1 belonging to the Fe-C stretching vibration (Supplementary Fig. 15)9. Vitally, in the EGOR process (Fig. 2a and Supplementary Fig. 15a), the Raman spectra are similar to those at the pristine and the open current potential (OCP) at various potentials. The intensity of twin peaks of cyanide is strengthened with increasing potentials because of the self-adaption in the electrolyte. As a counterpart, in the OER process (Fig. 2b and Supplementary Fig. 15b), the NiOOH double peaks at 482 and 561 cm−1 appear after 1.40 V as the twin peaks of cyanide get smaller9,31. It indicates the different catalytic behaviors in the EGOR and OER processes. NiFe-sc-PBA would not undergo reconstruction during EGOR and the Ni sites would not be oxidized to high valence; however, during OER, NiFe-sc-PBA would undergo reconstruction to NiOOH after 1.40 V, which is above the potential of Ni2+ to Ni3+ (NiOOH) oxidation (~1.36 V). Time-dependent Raman spectra shared the same conclusion, as EGOR proceeded, faint peaks were observed at 528 cm−1 of the adsorption of OH* after 600 seconds (Supplementary Fig. 16). Because this is the accumulation of the important intermediate, absorbed OH*, for EGOR42.

a In situ Raman spectra of NiFe-sc-PBA in a 3 M KOH with 1 M EG and b 3 M KOH. Bode plots of NiFe-sc-PBA in c 3 M KOH with 1 M EG and d 3 M KOH at various potentials. e Ni K-edge XANES analysis of NiFe-sc-PBA before and after reaction (inset: the enlargement of NiFe-sc-PBA and Post EGOR samples). f Fourier transformation curves of EXAFS spectra at the Ni K-edge in the R-space for NiFe-sc-PBA before and after EGOR and OER process (without phase correction), showing the robustness of Ni-N and Ni-C bonds post EGOR. Source data are provided as a Source Data file.
In order to elucidate the preservation of the active site structure, we employed operando electrochemical impedance spectroscopy (EIS) (Fig. 2c, d) to study the electrical behavior of NiFe-sc-PBA during the reaction process. The detailed and typical explanation of Bode phase plots is presented below Supplementary Fig. 1734,43. For EGOR (Fig. 2c), after 1.30 V, the phase degree in the low-frequency region rapidly decreased, indicating the rapid adsorption and desorption of intermediates, which suggests that the oxidation of EGOR can occur rapidly at lower potential regions. It is noteworthy that a new phase degree peak (~101 Hz) appeared in the mid-frequency region at potentials below 1.55 V, attributed to the adsorption of EG*, and this peak disappeared as the potential increased, indicating the rapid desorption of EG* at higher potentials. Most importantly, the shoulder position of the high-frequency region remained at a certain level as the potential increased, indicating that NiFe-sc-PBA was not fully oxidized during the EGOR process. Conversely, for OER (Fig. 2d), after exceeding 1.30 V, the shoulder position of the high-frequency region disappeared, indicating the oxidation of NiFe-sc-PBA to NiOOH, and the phase degree in the low-frequency region only began to decrease after 1.55 V, indicating that the kinetics of OER remained slow and the intrinsic activity was poor after the oxidation of NiFe-sc-PBA. Taking these pieces of evidence together, it is convincing that the reconstructed structure is NiOOH (the amorphous nickel (oxy)hydroxides). We further investigated the transformation of the electronic and geometric structures post EGOR using XANES and EXAFS. Both Ni and Fe K-edge XANES spectra of the spent NiFe-sc-PBA of EGOR remarkably shifted toward lower-energy region (Fig. 2e and Supplementary Fig. 18a), corresponding well to the above characterizations. The EXAFS spectra also verified the phenomenon that NiFe-sc-PBA kept its structure during EGOR (Fig. 2f). All the above characterizations demonstrate the existence of an interaction between ethylene glycol and NiFe-sc-PBA, which prevents NiFe-sc-PBA restructuring into NiOOH. Throughout the catalytic process, the nickel active sites exist in a low-valence state, avoiding competition between EGOR and OER. For comparation, we successfully synthesised sheet-like Ni(OH)2 (Supplementary Figs. 19, 20) and CoFe-sc-PBA (Supplementary Figs. 21, 22) to ecludidate the unique Ni(II) active sites in NiFe-sc-PBA. As shown in Supplementary Fig. 23, the peak at 449 cm−1 was attributed to the Ni2+-OH bond, while the broad peak ranging from 983 to 1087 cm−1 was attributed to the adsorption of OH- by Ni(OH)2 in the electrolyte42. With increasing potential during the EGOR process, distinct double peaks at 454 and 544 cm−1, corresponding to NiOOH, can be clearly observed. After the reaction, Ni(OH)2 still retained its layered structure, but some fragmentation occurred (Supplementary Fig. 24), which is due to the reconstruction process from Ni(OH)2 to NiOOH under alkaline conditions. This process can be well optimized, but it inevitably leads to misalignment and fragmentation during the recovery of the layered structure. Consistent with extensive research, the Ni2+/Ni3+ redox process indeed occurred on Ni(OH)2 during alcohols oxidation. In addition, to delve deeper into different active sites and their interaction between the substrate and material itself. As illustrated in Supplementary Fig. 25, with increasing potential, the peaks at 2089.7 and 2130.2 cm−1 associated with the cyano groups in CoFe-sc-PBA diminished and ultimately vanished above 1.45 V, coinciding with the emergence of double peaks at 449.5 and 556.7 cm−1 attributed to high-valent cobalt in CoO244. This observation suggests that CoFe-sc-PBA underwent leaching of ferrous cyanide and subsequent reconstruction. After EGOR, CoFe-sc-PBA exhibited significant reconstruction, resulting in a flaky appearance of the surface material (Supplementary Fig. 26). Thus, in bimetallic PBA sites with cyclic units of A-NC-B-CN-A, metal A (e.g., Ni, Co) usually serves as active site and metal B (e.g., Fe, Co) usually plays a structural role39,44,45. Our study indicates that only Ni active sites in PBA would maintain their low valence during EGOR, while Co active sites in PBA coupled with a Co3+/Co4+ redox process and traditional Ni-based materials coupled with a Ni2+/Ni3+ redox process.
Theoretical studies of catalyst/substrate interface and intrinsic catalytic bahavior of Ni(II)
The species distribution on PBA surface with/without EG molecules was simulated by molecular dynamics (MD) simulations (simulated configurations are given in Supplementary Fig. 27). For EGOR system, EG molecules would enrich on the NiFe-sc-PBA surface, remove OH- from the surface, and prevent the oxidative attack of OH-, as shown in Fig. 3a. On the contrary, OH- species were enriched on the NiFe-sc-PBA surface in the water system, promoting the OER process (Fig. 3b). The species number on NiFe-sc-PBA surface with/without EG molecules was counted in Fig. 3c, d to quantify the results of MD simulations. In the EG system, the number of OH- near the NiFe-sc-PBA surface was lower than that far away from the surface, while H2O molecules exhibited the opposite trend (Supplementary Fig. 28). In addition, EG molecules tended to accumulate on the NiFe-sc-PBA surface as shown in Supplementary Fig. 29. It was indicated that EG molecules would attract H2O molecules to the NiFe-sc-PBA surface and prevent the adsorption of OH-, which would prevent the occurrence of OER. For the OER system, OH- would prefer to adsorb on the NiFe-sc-PBA surface, and boost the water oxidation process. To further analyze the role of EG molecules, the hydrogen bond number on NiFe-sc-PBA surface in the system with/without EG molecules was counted in Fig. 3e. It was found that the hydrogen bond number near the NiFe-sc-PBA surface in the EG system was more than that in the water system, because the EG molecules enriched on the surface formed more hydrogen bonds than the water molecules (Fig. 3f). This result also confirms that EG molecules can form a rich hydrogen bond network with H2O molecules and prevent the adsorption of OH- on the PBA surface.

a MD simulation: final simulated snapshot of system a with EG and b without EG after 2 ns. The mass density distribution along the Z axis of c OH- and d H2O and EG and on K2Ni[Fe (CN)6] surface in the system with/without EG. e The H-bond number on K2Ni[Fe (CN)6] surface of the EG and water system, inserted schematic for counting the number of hydrogen bonds. f The average number of hydrogen bonds formed by each molecule on K2Ni[Fe(CN)6] surface of the EG system, inserted schematic diagram of hydrogen bonds formed by EG molecules as well as water molecules. Source data are provided as a Source Data file. The MD simulations are provided as a Supplementary Data file.
We employed differential charge density analysis and Bader charge analysis to calculate the charge transfer between Fe and Ni. As illustrated in Supplementary Fig. 30, the brown Fe atoms exhibited a loss of electrons, whereas the Ni atoms displayed an acquisition of electrons, as evidenced by the planar surface of the differential charge. According to the Bader charge analysis, each Ni atom receives 0.15 |e| of electricity from the Fe atoms. Consequently, Fe may provide electrons to the Ni, thereby maintaining Ni in a low-valent state and potentially impeding its oxidation. Typically, the alkali metal ions (e.g., K+) do not determine the reconstruction behavior of PBAs45,46, but our theoretical studies indicate that it has influence on the stability of Ni(II). As shown in Supplementary Fig. 31, the projected density of states was calculated for structure with and without potassium ions. With the doping of potassium ions, the peak at the Fermi energy level in the 3d orbitals of Fe is shifted toward the low-energy region, indicating that the 3d orbitals of Fe lose electrons. The 3d orbitals of Ni, on the other hand, showed more peaks at the Fermi surface and the peaks of the original peaks are higher, indicating that the 3d orbitals of nickel have gained electrons. It means that the introduction of potassium will help transfer the electrons from Fe to Ni, possibly making Ni a lower valence state and more stable.
Drawing from previously reported article as well as the corresponding experiments, we selected two pathways for ethanoic acid and glyoxal for density functional theory (DFT) calculation. The distinction between the two pathways lies in whether the glycolaldehyde is oxidized to glyoxal or glycolic acid. The Fe and Ni sites on the PBA (200) surface were chosen as catalytic sites and compared with trivalent Ni sites on the NiOOH (001) surface. As shown in Supplementary Fig. 32, the oxidation of EG to formic acid at all sites is exothermic. For Ni@PBA as well as Fe@PBA, the potential-determining step (PDS) in both sites is the oxidation of *CH2OHCH2OH to *CHOCH2OH, with Gibbs free energy changes (ΔG) of 0.17 eV and 0.47 eV, respectively. During the oxidation of *CHOCH2OH on both sites, the ΔG of the *COOHCH2OH pathway decreases more and the ΔG of the next step is also decreasing, which suggests that EGOR of PBA surface is dominated by the generation of ethanoic acid, which is consistent with the experiment. For Ni@NiOOH, the ΔG of the PDS of the ethanoic acid and glyoxal paths are 0.60 eV and 0.44 eV, respectively. To sum up, the EGOR on the Ni@PBA has the lowest PDS (0.17 eV), indicating the *COOHCH2OH path on this site is the most favorable for EGOR. In addition, performed in in situ attenuated total reflection infrared absorption spectroscopy (in situ ATR-IRAS) shown in Supplementary Fig. 33, to identify the reaction intermediates. After increasing the potential to 1.35 V, a peak corresponding to glycolic acid appeared around 1464 cm−1 18. This suggests that NiFe-sc-PBA may follow a reaction pathway involving glycolic acid as an intermediate during the EGOR process.
Electrocatalytic performance evaluations for EGOR
Electrochemical measurements towards EGOR and OER were performed in a three-electrode system with an H-type cell. In spite of NiFe-sc-PBA, Ni(OH)2 has also been studied because it would be readily reconfiged into NiOOH during the reaction47. To better illustrate the role of ethylene glycol, we used 3 M KOH to create a harsh and strong oxidative environment. As shown in the linear scanning voltammetry (LSV) curves (Fig. 4a and Supplementary Fig. 34), NiFe-sc-PBA boasted a superior EGOR performance, showing obviously higher current densities than its OER and NiO’s EGOR. Only 1.484 and 1.625 V were required to attain 500 and 1000 mA cm−2 for EGOR on NiFe-sc-PBA, respectively. The excellent activity was further confirmed by the Tafel slope analysis (Supplementary Fig. 35), and the Tafel slope of NiFe-sc-PBA (73.3 mV dec−1) is much lower than that of Ni(OH)2 (125.1 mV dec−1), suggesting that NiFe-sc-PBA has a faster reaction kinetics for EGOR. In addition, electrochemical active area (ECSA) was further evaluated via analyzing double-layer capacitance (Cdl) of the materials (Supplementary Figs. 36, 37), showing that the electrochemically active area of NiFe-sc-PBA (Cdl = 1.13 mF cm−2) is larger than that of Ni(OH)2 (Cdl = 0.72 mF cm−2).

a LSV curves of NiFe-sc-PBA in 3 M KOH with 1 M EG and 3 M KOH, and Ni(OH)2 control at a scan rate of 1 mV/s, iRreal compensation for NiFe-sc-PBA EGOR and iRconstant compensation for Ni(OH)2 EGOR (0.8 Ω) and NiFe-sc-PBA OER (1.3 Ω). b FEs of NiFe-sc-PBA for FA and OA production at different applied potentials in 3 M KOH with 0.3 M EG. The error bars represent the standard deviation of three independent measurements. c Chronoamperometric (i-t curve) profiles of NiFe-sc-PBA in H-type cell at 1.60 V without iR compensation. Source data are provided as a Source Data file.
Apart from electrochemical characterization, the Faradaic efficiency (FE) is a significant indicator to illustrate the oxidative attribution and products of EGOR. The FE values were obtained by high performance liquid chromatography (HPLC). Supplementary Fig. 38 shows the calibration curves of HPLC with excellent correspondence between peak area and solution concentration, all the R2 ≥ 0.985. We performed product analysis at different potential with 0.3 M EG (Fig. 4b), and FA was the main product of EGOR accompanied by OA as a byproduct. At a low potential of 1.40 V, the low FEFA (~86.2%) and a certain amount of OA might be attributed to the low energy supply, and the OA cleavage step could not be carried out fast enough leading to the accumulation of OA. However, it was then solved after increasing the applied potential. Overall, FEFA was high in the wide potential region, reaching 98.6% ± 2.5% at 1.45 V and 94.6% ± 3.1% at 1.70 V with 0.3 M EG. When the EG concentartion was increased to 1 M (Supplementary Fig. 39), the FEFA reached a higher level, achieving FEFA of 98.42% ± 0.01% at 1.55 V. For comparison, NiFe-sc-PBA was applied at 1.60 V for 1 h in 3 M KOH to be completely reconstructed to NiOOH, after which product detection was performed (Supplementary Fig. 40). When the potential and correspond current density increased, the FEFA dropped to 74.7% ± 4.9% at 1.65 V showing the severe competition between EGOR and OER. Not surprisingly, the FEFA of Ni(OH)2 exhibited an overall decreasing trend, reaching approximately 80% at 1.7 V (Supplementary Fig. 41). And this decline was accompanied by significant competition from OER or carbon loss phenomena, resulting in a total liquid-phase FE of less than 100%. Because NiOOH operates as an active species for both EGOR and OER, it is undoubtful for them to compete for the active sites.
To further validate the protective effect of EG as well as to explore the applicability of NiFe-sc-PBA under industrial conditions, the alkali concentration was further increased on purpose (Supplementary Fig. 42). The performances were boosted as the KOH concentration increased and the curves followed the same trend as the original curve (Fig. 4a). After EGOR for 2 h at 1.60 V, the peaks of Ni2+ were retained to a large extent in XPS spectra (Supplementary Fig. 43), and the ratios of Ni2+/Ni3+ were maintained at a high level (Supplementary Table 1). The results indicate that ethylene glycol remains effective in inhibiting catalyst oxidation under high alkaline concentration and more oxidative environment. In contrast, the LSV curves of Ni(OH)2 exhibited a peak range that indicates a pronounced exacerbation of the reconstruction of Ni(OH)2 with increasing alkali concentration (Supplementary Fig. 44a). The emergence of high-valent nickel sites (NiOOH) due to this reconstruction also functioned as active sites for OER, resulting in considerable competition with EGOR processes. Moreover, as the alkaline concentration increases, after a 2-hour reaction at 1.6 V, as shown in Supplementary Fig. 44b, the main peaks of nickel were positively shifted, signifying a notable increase in the oxidation state of nickel within Ni(OH)2. Importantly, EG concentration exerts a significant influence on the stability of Ni(II) sites. Consequently, we adjusted the EG concentration (from 0.1 to 100 mM) in 3 M KOH and conducted a single LSV test for each electrolyte (Supplementary Fig. 45). At low EG concentrations (0.1 mM EG), a distinct oxidation peak was observed (from 1.355 V to 1.422 V). However, as the concentration increased, the electrooxidation of EG became dominant, and the current-rising position of the current signal progressively shifted to lower potentials, indicating that the material did not undergo oxidation. Meanwhile, at lower EG concentrations (less than 5 mM), a disturbance in the current signal was noted around 1.56 V. This could be attributed to the low concentration of EG affecting its diffusion in the solution, or it may indicate a competitive phenomenon related to OER occurring at these lower EG concentrations. Following the single LSV tests, NiFe-sc-PBA underwent completely reconstruction with 0.1 mM EG (Supplementary Fig. 46a), similar to OER. However, with 0.2 mM and 0.5 mM EG (Supplementary Fig. 46b, c), it can be clearly seen that only the surface layer of NiFe-sc-PBA underwent reconstruction, while the sub-surface crystal structure remained well-defined. As the EG concentration continued to increase, NiFe-sc-PBA maintained its pristine structure without reconstruction. A trace amount of ethylene glycol is sufficient to protect NiFe-sc-PBA from reconstruction, demonstrating the reliability of the catalyst/substrate interaction. Other organic nucleophiles, such as glycerol, exhibited a similar effect in preventing reconstruction of NiFe-sc-PBA. It showed superior performance in GOR (Supplementary Fig. 47a), and after 2-hour treatment at 1.6 V, the peak areas of Ni2+ had a great advantage over that of Ni3+ (Supplementary Fig. 47b). Correspondingly, the microstructure of NiFe-sc-PBA was well preserved (Supplementary Fig. 48). From the above results, we propose that other organic nucleophiles, such as glycerol, possess properties similar to those of EG. Furthermore, NiFe-sc-PBA maintained a j of around 400 mA cm−2 at 1.60 V with high FEFA (>90%), indicating the high operation stability of NiFe-sc-PBA for EG oxidation (Fig. 4c). There were short-term increases in current density which might be attributed to the self-adaption of the catalyst-substrate environment after refreshing the electrolyte. After stability test, inductively coupled plasma optical emission spectroscopy (ICP-OES) analysis was performed to detect component leaching (Supplementary Table 2). It showed that the ionic concentration was negligibly low, illustrating the super stable structure of NiFe-sc-PBA without structure reconstruction or component leaching during EGOR process. NiFe-sc-PBA possesses significant advantages over similar alternative anode reaction (Supplementary Table 3) and possesses high intrinsic activity and stability, showing promising potentials for practical applications in three-electrode coupled electrochemical hydrogen production system.
Coupled hydrogen production in EGOR//HER flow cell system
To further evaluate the practical coupled hydrogen production performance, we assembled an EGOR//HER system in the membrane electrode assembly (MEA) shown in Supplementary Figs. 49, 50. To ensure the durability of the commercial alkaline membrane, we conducted the MEA system in 1 M KOH with/without 1 M EG. All MEA data were not iR compensated. With EG supplied in the electrolyte, the cell voltage was significantly decreased (Fig. 5a). The cell voltages to obtain at 0.5, 1.0 and 1.5 A cm−2 are 1.86, 2.02 and 2.15 V for EGOR//HER system, compared to 2.17, 2.47 and 2.68 V for OER//HER system, which indicates the energy saving capability of EGOR//HER system. It also means that the coupled system can reduce the electricity input of ~1.29 kWh/m3 of H2 at 1.5 A cm−2.

a MEA cell voltage of NiFe-sc-PBA//Pt/C at different current densities without iR compensation in 1 M KOH with or without 1 M EG. Faradaic efficiencies and formate productivities of EGOR//HER electrolyzer device b at different current densities and c at 1.5 A cm−2 for different time. The error bars represent the standard deviation of three independent measurements. d Stability test of EGOR//HER electrolyzer device at step current densities of 1.5 A cm−2 and 1.0 A cm−2 without iR compensation. Rotation rate: 15 rpm; electrolyte: 1 M KOH with 1 M EG and operation at room temperature. e XRD pattern of anodic product (KDF), inserted digital image of KDF. Source data are provided as a Source Data file.
As shown in Fig. 5b, the anode NiFe-sc-PBA exhibited high Faradaic efficiency (FEFA > 90%) in the wide current density regions from 0.2 to 1.8 A cm−2. The formate productivity linearly increased with the increase in current densities, reaching 20.3 mmol cm−2 h−1 at 1.8 A cm−2. Moreover, at a current density of 1.5 A cm−2, FEFA did not significantly decrease as operating time increased and remained above 92% (Fig. 5c). It is acknowledge that temperature not only affects the performance of (MEA but also influences its product selectivity. At 50 °C, only 1.88 V and 2.03 V were required to achieve current densities of 1 A cm−2 and 2 A cm−2, respectively (Supplementary Fig. 51). Furthermore, as illustrated in Supplementary Fig. 52, within the current density range of 200 to 2000 mA cm−2, the NiFe-sc-PBA catalyst consistently maintained a high FEFA, with the lowest value approaching 90% Unlike the H-type cell, the flowing electrolyte resulted in product glycolic acid (GA) in a low Faradaic efficiency. Overall, the protective effect of EG is still applicable in MEA for NiFe-sc-PBA Thus, the high FEFA and corresponding no oxygen produced would avoid the risk of H2/O2 mixing explosion in industrial applications. The flow electrolyte system can overcome the mass transfer limitations of traditional H-cell or single-cell, making the stability within the MEA a crucial indicator for the industrial applicability of the catalyst.
In addition, we conducted long-term stability tests to validate the practical feasibility of this system. Impressively, the EGOR MEA system operated stably at low cell voltages of ~2.13 and ~2.02 V at the step j of 1.5 and 1.0 A cm−2 for over 500 h (Fig. 5d), surpassing all previously reported alcohol oxidation coupled systems (Supplementary Table 4). Moreover, changing the current density at 100 h and 200 h did not affect the stability, demonstrating the excellent real-time response capability and outstanding stability of this coupled hydrogen production system. During the tests, we observed a slight orange color of the electrolytes and conducted UV-vis spectroscopy (Supplementary Fig. 53), which revealed distinct peaks at 280 cm−1 attributed to glyoxal with the FE being less than 0.5%. After EGOR, there were no changes in the crystallographic parameters of NiFe-sc-PBA (Supplementary Fig. 54), although the intensity of the peak decreased due to the compression when assembling MEA and the dropping of NiFe-sc-PBA caused by the flow of electrolyte, as well as a decrease in the crystallinity. In addition, SEM images showed a standard nanocube morphology after EGOR (Supplementary Fig. 55). After conducting 100 h operation in MEA, part of the electrolyte was conducted simple acidification, rotary evaporation, precipitation and filtration to obtain a large quantity of the product potassium diformate (KDF), which is the condensation of two potassium formate molecules. XRD patterns indicated that the separated crystals were pure KDF (Fig. 5e). The stability comparison with different applied current densities and conduction time demonstrates the advantage and privilege of our coupled hydrogen production MEA in energy-saving and H2 producing, which will drive the deployment of coupled hydrogen production devices to industrial applications. The detailed parameters are displayed in Supplementary Table 4. In parallel, we also assembled a conventional water splitting system that could be operated for more than 40 h at 400 mA cm−2 with a relatively high cell voltage (Supplementary Fig. 56). In order to better evaluate the coupled EGOR//HER hydrogen system, we performed a simplified techno-economic analysis (TEA) based on some models currently reported34,48,49. As shown in Supplementary Fig. 57, our coupled system demonstrates significantly improved profitability compared to traditional water electrolysis devices, requiring only a power supply cost of less than $0.269/kWh to achieve profitability, while the OER//HER system shows almost no profitability. The high operating current density, selectivity and stability all demonstrate the extraordinary potential of EGOR//HER coupled hydrogen production system using our NiFe-sc-PBA as anode material for industrial applications. However, to achieve large-scale production, several issues still need to be addressed, including the durability of the membrane, performance degradation due to the decrease in substrate concentration, product separation, the reduction of side reaction and so on. All the detailed calculations indicate that Ni(II) can be stabilized by the Fe and K ions and protected by EG during EGOR process.
Discussion
In summary, we have demonstrated the unique efficacy of Ni(II) sites within a Prussian blue analogue for efficient and durable alcohol electrooxidation at ampere-level current densities. In situ and operando structural characterizations reveal the structural stability of Ni(II) sites during the EGOR process; additionally, molecular dynamics simulations highlight the interaction at the catalyst/substrate interface with EG molecules densely populating the catalyst surface, preventing its reconstruction in harsh alkaline electrolytes. In a practical coupled hydrogen production system, the assembled EGOR//HER MEA electrolyzer can save power supply of ~1.29 kWh/m3 H2 at 1.5 A cm−2, along with high-value formic acid production. The coupled MEA system maintains stable operation at ampere-level current densities for over 500 h without performance degradation. This work provides a promising understanding of the catalyst/substrate interface interaction and offers valuable insights for further exploration of efficient and stable electrocatalytic materials for organic nucleophiles electrooxidation.
Method
Materials
Analytical grade potassium ferricyanide (K3Fe(CN)6, 99.5%), potassium hydroxide (KOH, 85%) was purchased from Shanghai Lingfeng Chemical Reagent. Sodium citrate (C6H5O7Na3, 98%) and ethylene glycol (>99.5%, GC) were purchased from Shanghai Macklin Biochemical Co., Ltd. Analytical grade nickel (II) chloride hexahydrate (NiCl2·6H2O, 98%), cobalt (II) chloride hexahydrate (CoCl2·6H2O, 98%) ammonium fluoride (NH4F, 96%), nickel (II) nitrate hexahydrate (Ni(NO3)2·6H2O, 98%) and urea were purchased from Sinopharm Chemical Reagent Co., Ltd. Polyvinylpyrrolidone (PVP, M.W. 58000) was purchased from Thermo Fisher Scientific. The TGP-H-060 carbon paper was provided by Toray. Nafion solution (5 wt%) was purchased from Sigma-Aldrich. 40% Pt/C was purchased from Shanghai Hesen Electronics Co., Ltd. All chemicals were used as received without further purification and all water used for synthesis and analysis was purified to >18.2 MΩ cm by Millipore system. Nickel foam (thickness: 1.6 mm, bulk density: 0.23 g·cm−3) was purchased from Heze Tianyu Technology Development Co., Ltd.
Synthesis of NiFe-sc-PBA
All the chemicals used in the experiment were of analytical grade and used with no further purification. Prior to the synthesis, nickel foam (NF) was cut into 4 * 3.5 cm2 and then ultrasonically cleaned in acetone to clean the surface, 3 M hydrochloric acid to remove the nickel oxide layer and ethanol until the final ethanol solution appears colorless and transparent without light green Ni2+ ions. NiFe-sc-PBA supported on NF was synthesized by a simple one-step hydrothermal method. Specifically, 0.3568 g NiCl2·6H2O, 0.6176 g C6H5O7Na3 and 1.16 g PVP were dissolved in 40 mL DI water and stirred until a homogeneous light green solution A was formed; 0.1646 g K3Fe(CN)6 was dissolved in 30 mL DI water as solution B. Under vigorous stirring, solution A was all added dropwise to solution B to obtain solution C. Then solution C was transferred into a 100 mL Teflon-lined stainless-steel autoclave with a piece of the pre-treated NF put into. The hydrothermal reaction was conducted at 80 °C for 27 h. After the reaction was completed and cooling down naturally, NiFe-sc-PBA grown on NF was washed with DI water and ethanol for 3 times and then put into a vacuum oven at 60 °C for overnight drying. Considering certain characterization, the powder in the solution was centrifuged, washed and vacuum dried overnight.
Synthesis of CoFe-sc-PBA
CoFe-sc-PBA was synthesized using a similar method to NiFe-sc-PBA, simply replacing an equimolar amount of NiCl2·6H2O with CoCl2·6H2O.
Synthesis of Ni(OH)2
Ni(OH)2/NF was synthesized by a typical one-step hydrothermal method. Specifically, 0.5934 g Ni(NO3)2·6H2O, 0.1860 g NH4F and 0.6 g urea were dissolved in 35 mL DI water and the solution was transferred into a 50 mL Teflon-lined stainless-steel autoclave with a piece of the pre-treated NF (2 cm * 3 cm) put into. The hydrothermal reaction was conducted at 120 °C for 6 hours. After that, the Ni(OH)2/NF was washed as the same as PBAs above.
Preparation of Pt/C electrodes
For the preparation of Pt/C on GDL, 20 mg 40% Pt/C and 100 μL Nafion solution were dispersed in DI water and ethanol (1:1). Then the ink was sonicated for 30 min. After the ink became homogenous, it was sprayed onto GDL on a 60 °C heating table. Importantly, the GDL was weighed before and after spraying to obtain the Pt/C load.
Characterization
The characterizations have partly illustrated in our previous work.[34] To be specific, XRD (D8 Advance ECO) measurements were conducted to verify the crystal structure of the samples. The morphology of the samples was characterized by SEM (Hitachi S4800, Thermo Fisher Helios G4 UC). TEM characterization was performed on a ThermoFisher Talos F200X under 200 kV. High angle annular dark field (HAADF)-STEM images were recorded using a convergence semi angle of 11 mrad, and inner- and outer collection angles of 59 and 200 mrad, respectively. Energy dispersive X-ray spectroscopy (EDS) was carried out using 4 in-column Super-X detectors. Chemical states of the elements were examined by (XPS, Kratos Axis Ultra DLD). ICP-OES was carried out on Agilent ICP-MS 7800 to determine the Ni and Fe concentration. UV-vis was performed on *UV-2450/2550 with spectral wavelength from 220 to 1400 nm.
XAFS measurements in the transmission mode were conducted at on the Ni, Fe K-edge at beamline BL14W1 of the Shanghai Synchrotron Radiation Facility. The electron beam energy was set at 3.5 GeV, with a stored current of 230 mA (top-up mode). A 38-pole wiggler, featuring a maximum magnetic field strength of 1.2 T, was employed in the straight section of the storage ring. XAFS data acquisition utilized a fixed-exit double-crystal Si (111) monochromator. The raw data were subsequently analyzed using the IFEFFIT software package, following standard data analysis protocols.
Raman spectroscopy was performed using a Leica DMLM microscope (Renishaw) equipped with the 514 nm excitation laser. In situ Raman measurements were performed on the LabRAM HR spectrometer, employing the same 514 nm excitation laser and a 50× microscope objective with a numerical aperture of 0.5 and a working distance of 10.6 mm. Prior to the experiments, calibration was executed using the peak at 520 cm−1 from a silicon wafer standard. NiFe-sc-PBA served as the working electrodes, while a platinum wire and Ag/AgCl electrode were used as the counter electrode and reference electrode, respectively. During the in situ experiment, the electrodes were immersed in the appropriate electrolyte. For each specific potential, a stabilization period of 200 seconds was allowed to achieve a steady-state electrode condition before the acquisition of Raman signals.
Electrochemical measurements
The electrochemical data collected for EGOR and OER were conducted in a three-electrode system within an H-type electrochemical cell (50 mL), employing either the XP20 from Ivium or the CHI 760E electrochemical workstation. The catalytic materials by geometric surface areas of 0.5 cm2 or 1 cm2, served as the working electrode, while a Hg/HgO electrode and a platinum mesh were designated as the reference and counter electrodes, respectively. All electrolytes were stored in polytetrafluoroethylene containers at ambient temperature. Calibration treatment was carried out in a hydrogen-saturated KOH electrolyte (1.0 M), with platinum mesh as the counter electrode. CV curves were conducted at the scan rate of 1 mV s−1, and the average of the two potentials at which the current surpassed zero was considered the thermodynamic potential for hydrogen electrode reactions (0.957 V in 3.0 M KOH).[34] All potentials were referenced to RHE by the following calculation: ERHE = EHg/HgO + 0.957 V. The LSV curves were collected at a scan rate of 1 mV s−1 within the potential range of 0.20 V to 0.80 V (vs. Hg/HgO). All LSV data were corrected with 85% iR compensation (where Rreal for real-time compensation, Rconstant for manual compensation measured through CHI 760). Durability assessments were carried out at 1.60 V vs. RHE without iR compensation. EIS measurements were conducted across a frequency spectrum of 105 Hz to 10−2 Hz, utilizing an amplitude of 5 mV. Cdl of various samples were determined via CV in a potential region devoid of Faradaic processes to calculate the ECSA. EGOR and OER tests were conducted in 3 M KOH with and without the inclusion of 1 M EG.
Alkaline anion-exchange membrane water electrolysis test
An AEMWE system was fabricated, featuring two titanium end plates with an active surface area of 1.2 cm2. The anode catalyst utilized was NiFe-sc-PBA/NF, while a commercial 40% Pt/C-coated GDL served as the cathode. The loading of Pt/C was maintained at 1 mg cm−2, with the anode and cathode layers sandwiched between a Sustainion X37-FA anion-exchange membrane. The electrolytes were delivered into the system via peristaltic pumps at 15 rpm. For EGOR//HER system, the anode was supplied with an electrolyte of 1 M KOH containing 1 M EG and the cathode was supplied with 1 M KOH. In conventional OER//HER system both the anode and cathode were pumped with 1 M KOH. Stability tests were performed at 1.5 A cm−2 and 1.0 A cm−2.
Product quantification
Organic acid products were qualified and quantified by high performance liquid chromatography (HPLC, Shimazu LC-40D), with a UV-Vis detector set at 210 nm. A 7.7 ×300 mm Angilent Hi-Plex H, 8 um column was used. HPLC eluent consisted of 5 mM sulfuric acid (H2SO4) solution and was performed at a flow rate of 0.6 mL/min over 20 min. The HPLC samples were prepared by neutralizing and diluting 80 μL electrolyte with 60 μL H2SO4 aqueous solution and H2O to 1 mL. The Faradaic efficiency for the chronoamperometry and chronopotentiometry test in H-type cell and MEA was obtained by collecting electrolyte either during the reaction or when the reaction was finished.
The following equations were used to calculate the FE of oxidation products and FA productivity:
where n is the number of electron transfer for each product formation, n = 3 for formic acid, n = 4 for glycolic acid and n = 8 for oxalic acid; F for the Faraday constant (96485 C mol−1).
The electricity consumption per m3 of H2 produced (W, kWh/m3 H2) was calculated as:
where n is the number of electrons transferred for each product molecule (n = 2), U is the applied voltage and Vm is the molar volume of gas at normal temperature and pressure (22.4 mol L–1).
Computational details
The MD simulations were implemented using GROMACS simulation package50. The SPC/E model51 was chosen for water molecules. Force field parameters for ions were taken from the EG in CHARMM36 using SwissParam 2023 web-based tool52 and all system expect EG molecule were solved by CHARMM36 force field53, respectively. Van der Waals interactions were treated with a cut-off method using a cut-off radius of 2 nm. Electrostatic interactions were handled using the particle mesh Ewald (PME) method with a cut-off radius of 2 nm. The size of the electrode is 4.08 nm × 4.08 nm, and the spacing between the two electrode surfaces is 10 nm, packing a solution with 3 M KOH. For system with EG, EG was added to a concentration of 1 M. The electrodes are fixed in the x, y, and z directions, with an additional 2 nm vacuum layer. Before simulation, the inner surfaces of the positive and negative electrodes were fixed charges of ±0.1 C/m2. All systems were first exposed to energy minimization. After that, the simulations were performed in the NVT ensemble at 300 K for 20 ns, with the Berendsen thermostat (the relaxation constant used was 1 ps), using a timestep of 1 fs. Then, VMD software54 was used to obtain the snapshots. The molecular dynamics trajectories of the optimized computational models in this work are provided in GRO format (Supplementary Data 1).
All the density functional theory (DFT) calculations were based on the projector augmented ware pseudopotential method (PAW)55,56 by Vienna ab initio Simulation Package (VASP)57. And the exchange and correlation energies between nucleus and electron were established by the generalized gradient approximation (GGA) with Perdew-Burke-Ernzerhof functional (PBE)58. The kinetic energy cutoff of plane-wave basis set was set to 520 eV. And the DFT-D359 method was adopted to correct the dispersion between the molecules and substrate model as well as the spin polarization was considered simultaneously. In the process of optimizing the structures, the convergence conditions of energy and force were set to a value of less than 1 × 10−5 eV/atom and 0.02 eV/Å, respectively. The atomic coordinates of the optimized computational models in this work are provided in CIF format (Supplementary Data 1).
The calculation module of PBA (200) surface, which contains 84 atoms, was placed in a lattice consisting of a = b = 10.2 Å and c = 25.2 Å. And this slab module contains both Ni@PBA and Fe@PBA catalytic sites. A vacuum layer of 15 Å to prevent the interaction along the Z direction was set in two slab modules. And calculation module of the β-NiOOH (001) surface contains 142 atoms and was placed in a 10.3 Å × 11.9 Å × 22.6 Å lattice. A 3 × 3 × 1 Γ-centered k-point was employed for structure optimization of all modules. To calculate the Bader charge analysis, differential charge density analysis and density of states, the convergence of energy was set to a value of less than 1 × 10−6 eV/atom. Furthermore, a Γ-centered k-point of 5 × 5 × 1 was employed.
The Gibbs free energy(G) was calculated as following formula:
where E and EZPE represent the total energy and the zero-point energy calculated by DFT, Cp denotes the heat capacity, T is temperature equal to 298.15 K, and S is entropy. The computational hydrogen electrode (CHE)60 model was used to treat the free energy of proton-coupled electron transfer.
Data availability
Source Data and Supplementary Data are provided with this paper. Source data are provided with this paper.
References
Li, A. et al. Atomically dispersed hexavalent iridium oxide from MnO2 reduction for oxygen evolution catalysis. Science 384, 666–670 (2024).
Chong, L. et al. La- and Mn-doped cobalt spinel oxygen evolution catalyst for proton exchange membrane electrolysis. Science 380, 609–616 (2023).
Wang, X. et al. Pivotal role of reversible NiO6 geometric conversion in oxygen evolution. Nature 611, 702–708 (2022).
Xie, H. et al. A membrane-based seawater electrolyser for hydrogen generation. Nature 612, 673–678 (2022).
Ze, H. et al. In situ probing the structure change and interaction of interfacial water and hydroxyl intermediates on Ni(OH)2 surface over water splitting. J. Am. Chem. Soc. 146, 12538–12546 (2024).
Tian, C. et al. Progress and roadmap for electro-privileged transformations of bio-derived molecules. Nat. Catal. 7, 350–360 (2024).
Zhu, Y. et al. Constructing Ru-O-TM bridge in NiFe-LDH enables high current hydrazine-assisted H2 production. Adv. Mater. 36, 2401694 (2024).
Yan, Y. et al. Nonredox trivalent nickel catalyzing nucleophilic electrooxidation of organics. Nat. Commun. 14, 7987 (2023).
Geng, S. K. et al. Nickel ferrocyanide as a high-performance urea oxidation electrocatalyst. Nat. Energy 6, 904–912 (2021).
Yan, Y., Zhong, J., Wang, R., Yan, S. & Zou, Z. Trivalent nickel-catalyzing electroconversion of alcohols to carboxylic acids. J. Am. Chem. Soc. 146, 4814–4821 (2024).
Li, Y., Wei, X., Han, S., Chen, L. & Shi, J. MnO2 electrocatalysts coordinating alcohol oxidation for ultra‐durable hydrogen and chemical productions in acidic solutions. Angew. Chem. Int. Ed. 60, 21464–21472 (2021).
Liu, F. et al. Concerted and selective electrooxidation of polyethylene-terephthalate-derived alcohol to glycolic acid at an industry-level current density over a Pd−Ni(OH)2 catalyst. Angew. Chem. Int. Ed. 62, e202300094 (2023).
Sun, Y. et al. Highly Selective electrocatalytic oxidation of amines to nitriles assisted by water oxidation on metal-doped α-Ni(OH)2. J. Am. Chem. Soc. 144, 15185–15192 (2022).
Qi, J. et al. Energy-saving and product-oriented hydrogen peroxide electrosynthesis enabled by electrochemistry pairing and product engineering. Nat. Commun. 14, 6263 (2023).
Zhou, H. et al. Electrocatalytic upcycling of polyethylene terephthalate to commodity chemicals and H2 fuel. Nat. Commun. 12, 4679 (2021).
Fang, Y. et al. Sulfur-doped manganese-cobalt hydroxide with promoted surface reconstruction for glycerol electrooxidation assisted hydrogen production. Nano Energy 127, 109754 (2024).
Qi, Y. et al. Insights into the activity of nickel boride/nickel heterostructures for efficient methanol electrooxidation. Nat. Commun. 13, 4602 (2022).
Li, J. et al. Selective ethylene glycol oxidation to formate on nickel selenide with simultaneous evolution of hydrogen. Adv. Sci. 10, 2300841 (2023).
Chen, Z. et al. Biodegradation of highly crystallized poly(ethylene terephthalate) through cell surface codisplay of bacterial PETase and hydrophobin. Nat. Commun. 13, 7138 (2022).
Lee, K., Jing, Y., Wang, Y. & Yan, N. A unified view on catalytic conversion of biomass and waste plastics. Nat. Rev. Chem. 6, 635–652 (2022).
Song, M. et al. Corrosion engineering of part-per-million single atom Pt1/Ni(OH)2 electrocatalyst for PET upcycling at ampere-level current density. Adv. Mater. 36, 2403234 (2024).
Shi, R. et al. Electrocatalytic reforming of waste plastics into high value-added chemicals and hydrogen fuel. Chem. Commun. 57, 12595–12598 (2021).
Liu, K. et al. Selective electrocatalytic reforming of PET-derived ethylene glycol to formate with a Faraday efficiency of 93.2% at industrial-level current densities. Chem. Eng. J. 473, 145292 (2023).
Shi, K., Si, D., Teng, X., Chen, L. & Shi, J. Pd/NiMoO4/NF electrocatalysts for the efficient and ultra-stable synthesis and electrolyte-assisted extraction of glycolate. Nat. Commun. 15, 2899 (2024).
Qin, Y. et al. Extraordinary p–d Hybridization Interaction in Heterostructural Pd-PdSe Nanosheets Boosts C−C Bond Cleavage of Ethylene Glycol Electrooxidation. Angew. Chem. Int. Ed. 61, e202200899 (2022).
Verma, S., Lu, S. & Kenis, P. J. A. Co-electrolysis of CO2 and glycerol as a pathway to carbon chemicals with improved technoeconomics due to low electricity consumption. Nat. Energy 4, 466–474 (2019).
Li, Y. et al. Coupling of PET waste electroreforming with green hydrogen generation using bifunctional catalyst. Sustain. Energy Fuels 6, 4916–4924 (2022).
Wang, X. H. et al. Ultra-thin CoNi0.2P nanosheets for plastics and biomass participated hybrid water electrolysis. Chem. Eng. J. 465, 142938 (2023).
Chen, Z. et al. Dual-doped nickel sulfide for electro-upgrading polyethylene terephthalate into valuable chemicals and hydrogen fuel. Nano Micro Lett. 15, 210 (2023).
Yan, Y. et al. Electrocatalytic Upcycling of biomass and plastic wastes to biodegradable polymer monomers and hydrogen fuel at high current densities. J. Am. Chem. Soc. 145, 6144–6155 (2023).
Cai, M. et al. Formation and stabilization of NiOOH by introducing α-FeOOH in LDH: Composite electrocatalyst for oxygen evolution and urea oxidation reactions. Adv. Mater. 35, 2209338 (2023).
Zhang, R. et al. Tracking the role of defect types in Co3O4 structural evolution and active motifs during oxygen evolution reaction. J. Am. Chem. Soc. 145, 2271–2281 (2023).
Ge, R. et al. Selective electrooxidation of biomass-derived alcohols to aldehydes in a neutral medium: promoted water dissociation over a nickel-oxide-supported ruthenium single-atom catalyst. Angew. Chem. Int. Ed. 61, e202200211 (2022).
Xu, H. G. et al. Reactant-induced activation over amorphous metal-metalloid electrocatalysts for HMF electrooxidation. Chem 10, 2147–2169 (2024).
Yang, Y. et al. Defect-Promoted Ni-Based Layer Double hydroxides with enhanced deprotonation capability for efficient biomass electrooxidation. Adv. Mater. 35, 2305573 (2023).
He, Z. et al. Promoting biomass electrooxidation via modulating proton and oxygen anion deintercalation in hydroxide. Nat. Commun. 13, 3777 (2022).
Zhou, J. et al. In situ exploring of the origin of the enhanced oxygen evolution reaction efficiency of metal(Co/Fe)–organic framework catalysts via postprocessing. ACS Catal. 12, 3138–3148 (2022).
Xiao, Y. et al. Prussian blue analogues for sodium-ion battery cathodes: A review of mechanistic insights, current challenges, and future pathways. Small 20, 2401957 (2024).
Yu, Z. Y. et al. Unconventional CN vacancies suppress iron-leaching in Prussian blue analogue pre-catalyst for boosted oxygen evolution catalysis. Nat. Commun. 10, 2799 (2019).
Wang, S. et al. Enhancing oxygen evolution reaction performance in Prussian blue analogues: Triple-play of metal exsolution, hollow interiors, and anionic regulation. Adv. Mater. 35, 2304494 (2023).
Zhang, Y., Jin, F., Liu, H. & Jin, Z. In-situ XPS monitoring bimetallic Prussian blue analogs and metal oxides heterojunction for photocatalytic hydrogen production. Fuel 361, 130655 (2024).
Lee, S., Chu, Y. C., Bai, L., Chen, H. M. & Hu, X. Operando identification of a side-on nickel superoxide intermediate and the mechanism of oxygen evolution on nickel oxyhydroxide. Chem. Catal. 3, 100475 (2023).
Zhao, J., Cano, M., Giner-Casares, J. J., Luque, R. & Xu, G. Electroanalytical methods and their hyphenated techniques for novel ion battery anode research. Energy Environ. Sci. 13, 2618–2656 (2020).
Xu, H. G. et al. Oxygen plasma triggered Co−O−Fe motif in Prussian blue analogue for efficient and robust alkaline water oxidation. Angew. Chem. Int. Ed. 64, e202415423 (2024).
Lai, C. et al. 3D spatial combination of CN vacancy-mediated NiFe-PBA with N-doped carbon nanofibers network toward free-standing bifunctional electrode for Zn–air batteries. Adv. Sci. 9, 2105925 (2022).
Zhao, Y. et al. Operando-reconstructed polyatomic ion layers boost the activity and stability of industrial current–density water splitting. Sci. Bullet. 69, 3384–3394 (2024).
Chen, W. et al. Activity origins and design principles of nickel-based catalysts for nucleophile electrooxidation. Chem 6, 2974–2993 (2020).
Michaelides, A. et al. Identification of general linear relationships between activation energies and enthalpy changes for dissociation reactions at surfaces. J. Am. Chem. Soc. 125, 3704–3705 (2003).
Liu, S. Q. et al. A coupled electrocatalytic system with reduced energy input for CO2 reduction and biomass valorization. Energy Environ. Sci. 16, 5305–5314 (2023).
Abraham, M. J. et al. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1-2, 19–25 (2015).
Berendsen, H. J. C., Grigera, J. R. & Straatsma, T. P. The missing term in effective pair potentials. J. Phys., Chem. 91, 6269–6271 (1987).
Bugnon, M. et al. SwissParam 2023: A modern web-based tool for efficient small molecule parametrization. J. Chem. Inf. Model. 63, 6469–6475 (2023).
Lee, J. et al. CHARMM-GUI input generator for NAMD, Gromacs, Amber, Openmm, and CHARMM/OpenMM simulations using the CHARMM36 additive force field. Biophys. J. 110, 641a (2016).
Humphrey, W., Dalke, A. & Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph 14, 33–38 (1996).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953 (1994).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758 (1999).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169 (1996).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865 (1996).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Peterson, A. A., Abild-Pedersen, F., Studt, F., Rossmeisl, J. & Nørskov, J. K. How copper catalyzes the electroreduction of carbon dioxide into hydrocarbon fuels. Energy Environ. Sci. 3, 1311–1315 (2010).
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (22239001, 22379043, 51920105003 and 22309053), the Shanghai Pilot Program for Basic Research (22TQ1400100-12), the Science and Technology Commission of Shanghai Municipality (23520710700, 21DZ1207101 and 22ZR1415700), and the Fundamental Research Funds for the Central Universities. The authors also thank the Frontiers Science Center for Materiobiology and Dynamic Chemistry, the crew of the BL14W1 beamline at the Shanghai Synchrotron Radiation Facility (SSRF) for their constructive assistance with the XAFS measurements and data analyses.
Author information
Authors and Affiliations
Contributions
H.G.Y. and P.F.L. directed the research. J.K.L. performed the experiments and data analyses. C.L. guided the MD simulations. M.K. and K.H. performed the computational simulations. H.G.X., Y.X.W., X.Y.Z., H.F. and S.R.F. participated in the characterization studies. Y.Z. (Yan Zhu) assisted with the SEM tests and data analyses. Y.Z. (Yi Zhou) provided assistance with the HPLC characterization. All the authors participated in writing and editing the manuscript, and contributed their efforts to the discussion.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Yong Chen, Tiancheng Mu and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, J.K., Kang, M., Huang, K. et al. Stable Ni(II) sites in Prussian blue analogue for selective, ampere-level ethylene glycol electrooxidation. Nat Commun 16, 3458 (2025). https://doi.org/10.1038/s41467-025-58203-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-58203-9
