
Abstract
Lung and gastrointestinal diseases often occur together, leading to more adverse health outcomes than when a disease of one of these systems occurs alone. However, the potential genetic mechanisms underlying lung-gastrointestinal comorbidities remain unclear. Here, we leverage lung and gastrointestinal trait data from individuals of European, East Asian and African ancestries, to perform a large-scale genetic cross trait analysis, followed by functional annotation and Mendelian randomization analysis to explore the genetic mechanisms involved in the development of lung-gastrointestinal comorbidities. Notably, we find significant genetic correlations between 27 trait pairs among the European population. The highest correlation is between chronic bronchitis and peptic ulcer disease. At the variant level, we identify 42 candidate pleiotropic genetic variants (3 of them previously uncharacterized) in 14 trait pairs by integrating cross-trait meta-analysis, fine-mapping and colocalization analyses. We also find 66 candidate pleiotropic genes, most of which were enriched in immune or inflammatory response-related activities. Causal inference approaches result in 4 potential lung-gastrointestinal associations. Introducing the gut microbiota as a variable establishes a relationship between the genus Parasutterella, gastro-oesophageal reflux disease and asthma. In summary, our findings highlight the genetic relationship between lung and gastrointestinal diseases, providing insights into the genetic mechanisms underlying the development of lung gastrointestinal comorbidities.
Similar content being viewed by others


Introduction
Currently, the comorbidities of lung diseases and gastrointestinal diseases have been increasingly investigated, which generally present worse health outcomes (e.g., higher morbidity and mortality) compared to a single disease alone1,2. For example, among patients with chronic obstructive pulmonary disease (COPD), those who developed inflammatory bowel disease (IBD) appeared to have a higher risk of mortality (23%) compared with those who did not3. Furthermore, among patients with asthma, those with gastro-oesophageal reflux disease (GORD) have a significantly higher risk of anxiety and depression compared to those without GORD4,5. Therefore, it is critical to understand the potential mechanisms underlying lung-gastrointestinal comorbidity.
Importantly, the gut-lung axis (GLA), as a specific axis between the gut and lung, has been reported to be responsible for functional changes in the lung and gastrointestinal tract (e.g., inflammatory cell infiltration), which may be a major cause of lung-gastrointestinal comorbidities6,7. There has been cumulative evidence to support the role of environmental factors on the GLA. For instance, cigarette smoking can promote the development of gastrointestinal diseases such as IBD and GORD in COPD patients8. The MADIET clinical trial demonstrated that higher saturated fatty acids (SFA) intake could significantly increase the risk of gastrointestinal adverse events (e.g., GORD) in patients with idiopathic pulmonary fibrosis (IPF) treated with pirfenidone9. Interestingly, previous studies have also demonstrated that genetic factors contributed to the development of GLA7, but the comprehensive genetic architecture of the GLA remains unclear.
Until now, genome-wide association studies (GWASs) have identified hundreds of genetic variants associated with lung and gastrointestinal diseases, revealing the potential genetic mechanisms that contribute to disease development. Interestingly, several studies have also reported a genetic correlation between these two diseases. For example, Lindström et al. reported a positive genetic relationship (rg = 0.27) between lung cancer (LC) and colorectal cancer (CRC) among 206,044 individuals of European (EUR) ancestry10. However, whether there is sufficient genetic overlap (e.g., shared genetic loci, susceptibility genes, or drug-targeted proteins) involved in lung-gastrointestinal comorbidities remains undetermined. Cross-trait analysis is a powerful study strategy that has been widely applied in determining the genetic basis of multiple diseases. With the gut-brain axis as an example, Gong et al.’s findings have indicated shared genetic determinants between gastrointestinal tract diseases and psychiatric disorders, highlighting biological mechanisms concerning immune responses, synaptic structure and function, and potential gut microbiome11. However, a comprehensive analysis framework for cross-trait has not been applied to improve the genetic understanding of disordered gut-lung interactions.
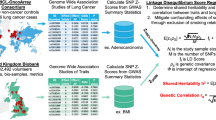
In this study, we integrated GWAS datasets for lung and gastrointestinal diseases among three populations [EUR: 180 pairs; East Asian (EAS): 84 pairs; African (AFR): 6 pairs] to perform a three-stage analysis. First, we evaluated the genome-wide and local genetic correlations between lung and gastrointestinal diseases (Fig. 1a). Second, we further applied cross-trait meta-analysis to identify pleiotropic genetic variants, followed by subsequent colocalization analysis and functional annotation (e.g., pathway-based enrichment analysis), as well as association analysis at the gene and protein levels (Fig. 1b). Third, the causal relationship was assessed using Mendelian randomisation (MR) analysis, and the role of the gut microbiome was also introduced in interpreting genetic etiology (Fig. 1c).

a Summary of the comprehensive pleiotropic analyses from different perspectives for multiple lung and gastrointestinal diseases in individuals of European, East Asian, and African ancestry. The genetic correlations at the global and local levels in each population were firstly examined. Created in BioRender. You, D. (2025) https://BioRender.com/j53h646. b Cross-trait meta-analysis and gene-based analyses were conducted to identify causal pleiotropic variants and genes. The functional annotation, TWAS, PWAS, and gene-environment interaction analyses were subsequently performed to explore biological functions. c Bi-directional MR and mediation analyses were performed to identify putative causal relationships and further clarify the shared biological mechanism between lung and gastrointestinal diseases. Created in BioRender. You, D. (2025) https://BioRender.com/j53h646. EUR: European. EAS: East Asian. AFR: African. AB: acute bronchitis. CB: chronic bronchitis. COPD: chronic obstructive pulmonary disease. FEV1: forced expiratory volume in 1 s. FVC: forced vital capacity. FEV1/FVC: FEV1/FVC ratio. PEF: peak expiratory flow. ILD: interstitial lung disease. IPF: idiopathic pulmonary fibrosis. LUAD: lung adenocarcinoma. LUSC: lung squamous cell carcinoma. SCLC: small cell lung carcinoma. LCES: lung cancer in ever smokers. LCNS: lung cancer in never smokers. Bact-pneumo: bacterial pneumoniae. Viral-pneumo: viral pneumonia. CP: colon polyp. CRC: colorectal cancer. DD: diverticular disease. GORD: gastro-oesophageal reflux disease. IBD: inflammatory bowel disease. CD: Crohn’s disease. UC: ulcerative colitis. IBS: irritable bowel syndrome. PUD: peptic ulcer disease. TWAS: transcriptome-wide association study. PWAS: proteome-wide association study. SNP: single-nucleotide polymorphism. MR: Mendelian randomisation. IV: instrumental variable.
Results
Genetic correlation between lung and gastrointestinal diseases
In the EUR population, the observed heritability derived from genome-wide single-nucleotide polymorphism (SNPs) ranged from 0.1% (viral pneumonia, liability: 1%) to 15% [forced expiratory volume in 1 s (FEV1)] for lung diseases, and from 1% [liability: 6%, peptic ulcer disease (PUD)] to 47% [liability: 24%, Crohn’s disease (CD)] for gastrointestinal diseases (Supplementary Data 1, and Supplementary Fig. S1). Similarly, among the individuals of EAS and AFR ancestry with sufficient samples, the highest heritability of lung and gastrointestinal diseases was 16% [forced vital capacity (FVC)] and 45% (liability: 17%, CD) in the EAS population; as well as 20% (FEV1) and 4% [liability: 25%, diverticular disease (DD)] in AFR population, respectively.
Subsequently, we evaluated the genetic correlations between lung and gastrointestinal diseases. At the global level, 27 significant pairs survived after Bonferroni correction in the EUR population (Fig. 2), with correlations ranging from 0.19 (P = 1.77 × 10−5, LC-GORD) to 0.65 [P = 1.02 × 10−20, chronic bronchitis (CB)-PUD], while no significant correlations were found in the EAS and AFR populations (Supplementary Data 2 and Supplementary Fig. S1). Sample overlap was considered negligible since most (76%) of the intercepts of genetic covariance across all pairs were below 0.0112.

a UpSet plot showing SNP-based heritability on the liability scale for each trait computed using LDSC (left bars, SNP-based liability heritability range: 0.02-0.21; standard error (SE) range: 0.002–0.02), global genetic correlations estimated using cross-trait LDSC (red bars, global genetic correlation range: 0.19–0.65; SE range: 0.04–0.07) and local levels computed using SUPERGNOVA [24 (17 unique) significant local genetic correlations] across 27 lung-gastrointestinal trait pairs. Lung-gastrointestinal trait pairs are indicated by blue dots. b Genome-wide map of 42 candidate pleiotropic genetic variants (dots, see also Supplementary Data 8) and the 66 corresponding candidate pleiotropic genes (see also Supplementary Data 15) for each gastrointestinal disease as a Circos plot. The region shared by multiple trait pairs is denoted by black text and a black-dotted bordered rectangle. Bonferroni correction was applied, and all reported P values were two-sided unless stated otherwise. LDSC: linkage disequilibrium score regression. CP: colon polyp. DD: diverticular disease. GORD: gastro-oesophageal reflux disease. IBS: irritable bowel syndrome. PUD: peptic ulcer disease. Bact-pneumo: bacterial pneumoniae. CB: chronic bronchitis. COPD: chronic obstructive pulmonary disease. IPF: idiopathic pulmonary fibrosis. LC: lung cancer. CRC: colorectal cancer. LUSC: lung squamous cell carcinoma. Source data were provided as a Source Data file.
Further at the local level, we identified 24 shared regions across the 27 lung-gastrointestinal trait pairs in individuals of EUR ancestry, ranging from 1 [e.g., 17q12-21.2 for asthma-colon polyp (CP)] to 5 [e.g., 1p34.3-p34.2, 1q21.3, 13q14.3, 17q21.32-q21.33 and 22q13.1-q13.2 for asthma-irritable bowel syndrome (IBS)] local regions (Supplementary Data 3, Fig. 2, Supplementary Figs. S2 and S3). Especially, three regions were found across multiple lung-gastrointestinal trait pairs, including 2q33.1-q33.3 for CB-DD and COPD-DD; 13q14.3 for asthma-IBS and COPD-IBS, and 20q13.33 for asthma-DD, asthma-GORD, COPD-DD, COPD-GORD, pneumoniae-DD and pneumoniae-GORD. In addition, for the 27 trait pairs, genetic correlations were detected in 12 functional regions, especially for the DNase I hypersensitivity site (DHS) and transcribed regions (Supplementary Data 4 and Supplementary Figs. S4 and S5).
Identification of pleiotropic SNPs using cross-trait meta-analysis
Using the genetic data of the 27 lung-gastrointestinal trait pairs with significant genetic correlations in the EUR population, we performed a cross-trait meta-analysis to identify potential pleiotropic genetic variants. As shown in Supplementary Data 5, no population stratification was observed via genomic control inflation factors.
A total of 4329 variants (144 independent variants located at 97 loci, 12 of which were not detected in previous lung or gastrointestinal disease GWASs) across 20 pairs were found at the genome-wide significance level (P ≤ 5 × 10−8), ranging from 1 (e.g., 2q33.2 for CB-DD) to 22 independent variants (e.g., 1q25.1-22q13.33 for asthma-GORD) (Supplementary Data 6 and Figure S6). Among them, the strongest pleiotropic SNP was rs921650 [17q12, P = 6.72 × 10−70 for Multi-Trait Analysis of GWAS (MTAG)], reported by previous asthma GWAS, which showed shared effects on the risk of asthma [odds ratio (OR) = 1.09, P = 3.60 × 10−79] and CP (OR = 1.03, P = 9.35 × 10−4), while obvious heterogeneity was observed (Phet = 2.04 × 10−7).
Interestingly, 30 pleiotropic loci were identified in two or more lung-gastrointestinal trait pairs; especially, 2q33.2 (asthma-IBS, CB-DD, CB-IBS, and COPD-DD), 2p16.1 (asthma-IBS, CB-IBS, COPD-DD, and COPD-IBS), 4q31.1 (asthma-DD, asthma-GORD, COPD-DD, and COPD-GORD) and 11q12.2 (asthma-CP, CB-CP, COPD-CP, and LC-CRC) were shared by four trait pairs, all of which were reported in previous GWASs.
Identification of candidate pleiotropic genetic variants
We then applied fine-mapping analysis to capture the 99% credible set at each of the 144 pleiotropic variants across 20 lung-gastrointestinal trait pairs. Totally, 609 potential causal variants were identified (Supplementary Data 7). Among these causal variants, further colocalization analysis showed that 42 independent variants were colocalized in the risk of 14 lung and gastrointestinal disease pairs [posterior probability (PP) of hypothesis H4 (PPH4) ≥ 0.5], and these 42 variants were considered candidate pleiotropic genetic variants (Supplementary Data 8 and Fig. 2). Further sensitivity analyses revealed that these candidate pleiotropic SNPs were replicated by at least one of the Genetic analysis incorporating Pleiotropy and Annotation (GPA), pleiotropic analysis under composite null hypothesis (PLACO), cross phenotype association (CPASSOC) with cross-trait statistical heterogeneity (SHet), cross phenotype meta-analysis (CPMA) and MetABF methods (Supplementary Data 9 and Supplementary Fig. S7). Additional replication analysis using independent lung-gastrointestinal GWASs also confirmed the pleiotropic effects of these 42 SNPs on lung-gastrointestinal diseases (Supplementary Fig. S8).
Notably, five loci were shared by two or more lung-gastrointestinal trait pairs, including 2q33.2, 4q24, 4q31.1, 9q33.3, and 16p11.2 (Supplementary Figs. S9, S10). Especially, both rs13135092 (4q24) and rs4837022 (9q33.3) were shared signals at two lung-gastrointestinal trait pairs (i.e., asthma-GORD and COPD-GORD).
In addition, compared to previous GWASs related to single traits, three additional loci reaching genome-wide significance were detected, including rs55673000 (17q21.1, CB-CP), rs11928675 (3p26.1, COPD-IBS), and rs28781623 (8q21.3, COPD-PUD; Supplementary Data 8). Further expression quantitative trait locus (eQTL) analysis indicated that rs55673000 may be involved in disease development by influencing the expression of multiple nearby genes (e.g., MED24), but no eQTL associations were found at the other two loci (Supplementary Data 10).
Prioritisation of pleiotropic genes and enrichment analysis
Next, we used gene-based analysis to evaluate the gene-level shared effects on the 14 lung-gastrointestinal trait pairs. At the genome-wide level, an average of 112 risk genes (PBonferroni ≤ 0.05; Supplementary Data 11) were identified, ranging from 18 (pneumoniae-CP) to 266 (asthma-IBS) genes. Subsequently, using gene set enrichment analysis (GSEA), we found that these risk genes were mainly involved in the biological process of immune or inflammatory response-related activities (Supplementary Data 12, Fig. 3, and Supplementary Fig. S11). In addition, tissue-specific enrichment analysis (TSEA) demonstrated that these genes were significantly enriched in several gastrointestinal tissues (PBonferroni ≤ 0.05; Supplementary Data 13, Fig. 3, and Supplementary Fig. S12), such as the colon, stomach, and oesophagus. Notably, cell-type specific enrichment analysis (CSEA) indicated that these risk genes were mainly related to several immune cell types (e.g., T cells), with a proportion of 61% (492 immune cells/803 total cells; Supplementary Data 14, Fig. 3, and Supplementary Fig. S13).

The heatmaps display functional annotations on the x-axis and 14 lung-gastrointestinal trait pairs on the y-axis, with significant cells filled in colour. The bars represented the number of trait pairs for each annotation item and the number of significant annotation items for each trait pair, respectively. Significantly enriched items were determined with Bonferroni-corrected P ≤ 0.05 for GSEA [normalised enrichment score > 2, GO terms and KEGG terms], TSEA, and CSEA, as well as FDR-corrected P ≤ 0.05 for TWAS and PWAS. All annotations were evaluated against a two-sided alternative hypothesis. GO and KEGG pathway enrichment analyses revealed that 24 unique biological processes related to immune or inflammatory response related activities were enriched (see also Supplementary Data 12). TSEA using the deTS method revealed 5 significantly enriched tissues (see also Supplementary Data 13). The top 5 enriched immune cell types from CSEA calculated by WebCSEA are shown (see also Supplementary Data 14). TWAS and PWAS were performed using FUSION based on 49 normal tissues from GTEx V8 and the plasma proteome built from the ARIC study, respectively (see also Supplementary Data 16-17). GO: Gene Ontology. KEGG: Kyoto Encyclopedia of Genes and Genomes. GSEA: gene set enrichment analysis. TSEA: tissue-specific enrichment analysis. CSEA: cell-type specific enrichment analysis. TWAS: transcriptome-wide association study. PWAS: proteome-wide association study. GTEx: Genotype-Tissue Expression project. CP: colon polyp. DD: diverticular disease. GORD: gastro-oesophageal reflux disease. IBS: irritable bowel syndrome. CB: chronic bronchitis. PUD: peptic ulcer disease. LUSC: lung squamous cell carcinoma. CRC: colorectal cancer. Source data were provided as a Source Data file.
In addition, compared to the genes mapped within ± 500 kb of the 42 candidate pleiotropic genetic variants, we identified 66 overlapping genes (near 28 pleiotropic SNPs) across 13 trait pairs, which were considered candidate pleiotropic genes for subsequent analysis (Supplementary Data 15, Fig. 2, and Supplementary Fig. S14).
TWAS and PWAS
Subsequently, we performed TWAS analysis to evaluate the associations between the gene expression levels of 66 pleiotropic genes in 49 normal tissues and the risk of lung-gastrointestinal diseases. Interestingly, significant associations were found for a total of 58 pleiotropic genes [false discovery rate (FDR) ≤ 0.05; Supplementary Data16, Fig. 3, and Supplementary Fig. S15]; notably, the effects of most of these genes were detected in the skin, adipose, nerve, artery, thyroid, lung, and colon tissues. Notably, decreased expression of RBM6, located near the pleiotropic variant rs13077403, could increase the risk of asthma-GORD across all tissues, with TWAS Z values ranging from − 6.57 to − 4.85.
We further applied PWAS analysis to evaluate the associations of the plasma protein levels of 66 pleiotropic genes with the risk of lung-gastrointestinal diseases. Interestingly, the expression levels of 3 proteins were associated with the risk of asthma-GORD (i.e., MANBA), COPD-DD (i.e., PPIC) and COPD-GORD (i.e., APOE) (PFDR ≤ 0.05; Supplementary Data 17 and Fig. 3).
Pleiotropic variant-exposure interactions on comorbidity risk
To explore the additional roles of candidate pleiotropic genetic variants in the development of lung-gastrointestinal diseases, we performed an interaction analysis in 377,886 individuals of EUR ancestry from the UK Biobank (UKB), to examine the interaction effect between those genetic variants and 64 common modifiable exposures on lung-gastrointestinal comorbidities (Supplementary Data 18). As expected, we found that an average of 50 exposures were associated with the risk of 14 lung-gastrointestinal disease pairs (P ≤ 0.05), especially for the positive associations of body mass index (BMI), smoking status, air pollution (e.g., nitrogen dioxide), and unfavourable mental health with the risk of all 14 lung-gastrointestinal disease pairs (Supplementary Data 19 and Supplementary Fig. S16).
Interestingly, 4 candidate pleiotropic genetic variants interacted with modifiable exposures across 4 lung-gastrointestinal disease pairs (PBonferroni ≤ 0.05), including rs2754246-unfavourable mental health (worrying too long after embarrassment) for asthma-GORD, rs71524796-dried fruit intake for COPD-CP, rs186399184-family relationship satisfaction for COPD-DD, and rs4837022-particulate matter 10 (PM10) for COPD-GORD (Supplementary Data 20).
Bi-directional MR and mediation analysis
For the 14 lung-gastrointestinal trait pairs carrying candidate pleiotropic genetic variants, we further applied bi-directional MR analysis to assess the causal associations between them (Supplementary Data 21–23, and Supplementary Fig. S17). Interestingly, we found 4 significant causal positive associations [inverse-variance weighted (IVW) estimated OR > 1, PFDR ≤ 0.05; Fig. 4], including 1 association in the lung-to-gastrointestinal direction [asthma-to-GORD: ORIVW = 1.06, PFDR = 0.034] and 3 associations in the gastrointestinal-to-lung direction [GORD-to-asthma: ORIVW = 1.25, PFDR = 5.70 × 10−4; GORD-to-CB: ORIVW = 1.99, PFDR = 5.70 × 10−11; and GORD-to-COPD: ORIVW = 1.64, PFDR = 8.46 × 10−14], notably, a bi-directional association between asthma and GORD was identified. Most associations were replicated by five alternative MR methods, and all were supported in further sensitivity analyses (Supplementary Data 21 and Supplementary Fig. S18).

a Significant bidirectional MR estimates (see also Supplementary Data 22). Data were presented as forest plots, with the OR shown as dots and the 95%CI as line segments, of the following samples per direction: asthma (n = 360,838) to GORD (n = 456,327); GORD (n = 385,276) to asthma (n = 1,376,071), CB (n = 450,422), or COPD (n = 995,917). Significance was defined as an FDR-corrected P ≤ 0.05 for IVW and a P > 0.05 for the Egger intercept, evaluated against a two-sided alternative hypothesis. Created in BioRender. You, D. (2025) https://BioRender.com/f18p045. b Mediation analysis based on IVW estimates (see also Supplementary Data 24-25). ‘Direct effect’ indicates the effect of the exposure on the outcome after adjusting for the mediator. The ‘mediation effect’ indicates the effect of the gut microbiota on the lung disease through the gastrointestinal disease, computed as the product of α (the IVW-estimated effect size of the exposure on the mediator) and β (effect size of the mediator on the outcome). The ‘mediation proportion’ was computed as the mediation effect divided by the total effect. The standard error (SE) and 95%CI of the mediation effect were calculated using the delta method. c Immune response in the pathogenesis of the Parasutterella-GORD-asthma association. Li et al. suggested that the “gut microbiota-inosine-PPARγ” axis plays an important role in the pathogenesis of chronic inflammatory disease of the intestine by regulating an inflammatory response (i.e., TNF-α and IL-6) through PPARγ signalling in colon epithelial cells, which further inhibited the immune response, leading to the increased the risk of GORD, as well as asthma14. Created in BioRender. You, D. (2025) https://BioRender.com/f18p045. All reported P values were two-sided, unless stated otherwise. MR: Mendelian randomisation. IVW: inverse variance weighted method. MR-RAPS: MR robust adjusted profile score. MR-PRESSO: MR pleiotropy residual sum and outlier. OR: odds ratio. CI: confidence interval. GORD: gastro-oesophageal reflux disease. CB: chronic bronchitis. CP: colon polyp. COPD: chronic obstructive pulmonary disease. IL: interleukin. TNF-α: tumour necrosis factor-α. PPARγ: peroxisome proliferator activated receptor γ. Source data were provided as a Source Data file.
Subsequently, by integrating the genetic data with the gut microbiota, we performed MR and mediation analysis to further assess the role of the microbiota in the above lung-gastrointestinal disease pairs. Two potential gut microbiota-to-gastrointestinal-to-lung disease pathways were identified (Supplementary Data 24, 25, and Fig. 4), including Parasutterella-GORD-asthma (mediation proportion = 38.78%) and Faecalibacterium-GORD-CB (mediation proportion = 28.63%). Importantly, the association of the Parasutterella-GORD-asthma pathway was broadly replicated in an additional Dutch Microbiome Project (DMP) cohort (Supplementary Data S23). These findings were consistent with those of previous studies in which the authors speculated that the abundance of Parasutterella was related to decreased inosine levels13, further inhibiting the immune response14, then increasing the risk of GORD15, as well as asthma16 (Fig. 4).
Discussion
In this study, we performed a large-scale cross-trait genetic analysis to assess the genetic correlation between lung and gastrointestinal diseases, as well as to identify the pleiotropic genetic variants, genes and biological functions involved in the development of this comorbidity pattern. Interestingly, we found significant global genetic correlations between 27 trait pairs, in which 24 local and 12 functional regions were identified. Furthermore, at the variant level of genome-wide significance, 42 candidate pleiotropic genetic variants were identified in 14 trait pairs, followed by 66 pleiotropic genes, most of which were enriched in the biological process of the immune or inflammatory response-related activities. Subsequent causal inference analysis demonstrated 4 potential associations, and the introduction of data from the gut microbiota further indicated a biological relationship between Parasutterella, GORD and asthma.
A total of 27 lung-gastrointestinal trait pairs with significant genetic correlations were identified, half of which were reported in previous cohorts or MR studies. For instance, we detected a high genetic correlation (rg = 0.52) for COPD-GORD, which was consistent with the findings of the studies by García Rodríguez et al. 12 and Dong et al. 13. Subsequently, using the combined approach of cross-trait meta-analysis, fine-mapping, and colocalization, we identified a total of 42 candidate pleiotropic genetic variants across 14 lung-gastrointestinal trait pairs, which further demonstrated the pleiotropy of previous GWAS SNPs. Interestingly, 3 previously unreported loci were detected among the 42 candidate pleiotropic genetic variants, including 17q21.1 for CB-CP, 3p26.1 for COPD-IBS, and 8q21.3 for COPD-PUD. Although these 3 variants were not reported in lung or gastrointestinal disease GWASs, their nearby genes have been shown to be involved in multiple biological traits, MED24 at 17q21.1 can increase the levels of various phosphatidylcholines17. GRM7, located at 3p26.1, plays a crucial role in the biological processes and development of nicotine dependence, thereby contributing to an increased risk of LC18. RUNX1T1 at 8q21.3 is involved in promoting carcinogenesis in small-cell lung carcinoma (SCLC) by inhibiting histone acetylation and upregulating E2F, while also acting as a tumour-suppressive gene in CRC by decreasing cell growth and increasing sensitivity to 5-fluorouracil19. In addition, our pathway analysis demonstrated that the genes associated with the risk of lung-gastrointestinal comorbidity were mainly enriched in immune response-related biological pathways, which was consistent with previous findings. For example, Budden et al. reported an important role of immune responses in the GLA, which are potentially mediated by the microbiota6. The epithelial surfaces of both the gastrointestinal and respiratory tracts are exposed to various microorganisms. Microbes ingested through the gastrointestinal tract can reach both sites; for instance, gut microbiota can potentially enter the lungs through aspiration6. Substantial evidence indicates that direct interactions between host epithelial and immune cells with these microorganisms and their associated cytokine responses can regulate inflammation and immune responses in distant organs such as the lung20. These findings further highlight the potential relationship between the immune response and lung-gastrointestinal comorbidities.
Given the identified genetic correlation between lung and gastrointestinal diseases, we additionally assessed the causal association between them. Notably, a total of 4 trait pairs were revealed to have positive relationships, most of which have been reported in previous observed studies. For example, a prospective follow-up study demonstrated that subjects with persistent GORD were approximately two times more likely to have asthma and other respiratory symptoms than those without GORD16. To further explore the potential risk factors involved in these 4 lung-gastrointestinal disease pairs, we performed mediation analysis to introduce the gut microbiota into these associations6, and a mechanistically distinct Parasutterella-GORD-asthma relationship was identified. Noteworthily, the potential mechanism underlying the Parasutterella-GORD-asthma association can be explained by a gut microbiota-inosine-peroxisome proliferator-activated receptor γ (PPARγ) axis, which can regulate immune response-related activities, particularly inflammation control, through PPARγ signalling in gastrointestinal epithelial cells14. In total, we speculated that the abundance of Parasutterella was related to decreased inosine levels, then upregulated the expression of proinflammatory cytokines like tumour necrosis factor α (TNF-α) and interleukin 6 (IL-6), and further inhibited the immune response, then increased the risk of GORD, as well as asthma, which could provide a molecular link between gastrointestinal bacterial metabolism, GORD, and the potential occurrence of asthma15,21. Parasutterella, a genus of Betaproteobacteria, is a member of the core fecal microbiome in the human gastrointestinal tract13. Previous studies have shown that carbohydrate intake, especially for monosaccharides, could be an energy source to promote Parasutterella growth in the gut22. Conversely, ω3 linolenic acid intake can promote anti-inflammatory effects, such as reducing pro-inflammatory cytokines like TNF-α, and support the growth of beneficial bacteria that compete with Parasutterella22. Dietary fibre and yogurt, the main sources of prebiotics and probiotics, can also promote the colonisation of beneficial bacteria (e.g., Bifidobacterium and Lactobacillus), improving the gut microbial ecosystem and enhancing beneficial physiological effects23,24. These findings suggest that dietary adjustments, such as reducing carbohydrate intake, increasing ω3 linolenic acid intake, and consuming a high-fibre diet and yogurt, combined with prebiotics and probiotics, may effectively reduce Parasutterella abundance, thereby preventing the onset of lung-gastrointestinal comorbidities.
Our study has several advantages. This is a large-scale study to systematically assess the shared genetic characteristics between multiple lung and gastrointestinal diseases. Second, several previously unreported pleiotropic variants/genes were identified, which could help explain the biological mechanisms of the genetic factors involved in lung-gastrointestinal comorbidities. In addition, some limitations should be noted. First, although we included three ancestry groups in this study, the results of EAS and AFR populations were not sufficient due to the limited sample size, which should be re-analysed in future studies. Second, more statistical analysis strategies should be applied to identify additional potential genetic correlations (e.g., local level) between lung and gastrointestinal diseases. Third, although our data were derived from various countries, that did not evenly cover all regions; therefore, additional studies in other regions are needed to validate our primary findings. Besides, the biological functions of pleiotropic genetic biomarkers should be further explored comprehensively using in vitro and in vivo experiments. Fourth, these identified biomarkers could guide potential therapeutic targets to enhance personalised medicine; therefore, the potential value of the identified biomarkers in clinical application should be evaluated in future studies. Finally, there were several issues regarding potential bias, including the misclassification and inclusion bias of study subjects with comorbidity, limited statistical power in discovering genetic evidence of several diseases with low incidence, weak instrument bias in MR analysis, and others. Although we applied several statistical analyses to correct for confounding bias, a robust study design is essential to thoroughly validate our findings.
In summary, our study systematically deciphered the shared genetic architecture between lung and gastrointestinal diseases, with the identification of previously uncharacterised pleiotropic genetic biomarkers, which may provide more insight into the genetic mechanisms of lung-gastrointestinal comorbidities.
Methods
Ethics
The research reported herein was done in compliance with all ethical requirements. Data for the present study were obtained under an approved UKB project application (ID 92675). UKB has ethics approval from the National Health Service North-West Centre Research Ethics Committee (Ref: 21/NW/0157), with informed consent obtained from all participants. The study design and conduct complied with all relevant regulations regarding the use of human study participants and was conducted in accordance with the criteria set by the Declaration of Helsinki.
Study subjects
Summary-level GWAS datasets
EUR population
Lung diseases: A total of 20 lung diseases related GWAS datasets of EUR ancestry were included in this study, including asthma (121,940 cases and 1,254,131 controls)25, bronchiectasis (2888 cases and 440,263 controls)26, bronchitis [including acute bronchitis (AB, 4483 cases and 474,818 controls), CB (10,159 cases and 440,263 controls)]26, COPD (58,559 cases and 937,358 controls)25, interstitial lung disease (ILD, 2267 cases and 467,560 controls)26, IPF (6257 cases and 947,616 controls)25, LC [29,266 cases and 56,450 controls, as well as lung adenocarcinoma (LUAD, 11,273 cases and 55,483 controls), lung squamous cell carcinoma (LUSC, 7426 cases and 55,627 controls), SCLC (2664 cases and 21,444 controls), LC in ever smokers (LCES, 23,223 cases and 16,964 controls), LC in never smokers (LCNS, 2355 cases and 7,504 controls)]27, lung function [including FEV1 (400,102 individuals), FVC (400,102 individuals), FEV1/FVC ratio (400,102 individuals), peak expiratory flow (PEF, 345,265 individuals)]28, and pneumoniae [58,174 cases and 319,103 controls, as well as bacterial pneumoniae (Bact-pneumo, 16,244 cases and 314,673 controls), viral pneumonia (Viral-pneumo, 3394 cases and 314,673 controls)]29.
Gastrointestinal diseases: Nine gastrointestinal diseases related GWAS datasets of EUR ancestry were collected, including CP (22,049 cases and 332,368 controls)26, CRC (78,473 cases and 107,143 controls)30, DD (35,617 cases and 384,914 controls)31, GORD (54,854 cases and 401,473 controls)32, IBD [12,882 cases and 21,770 controls, as well as CD (5956 cases and 14,927 controls) and UC (6968 cases and 20,464 controls)]33, IBS (53,400 cases and 433,201 controls)34, and PUD (16,666 cases and 439,661 controls)32.
EAS population
Lung diseases: A total of 12 lung diseases related GWAS datasets of EAS ancestry were included, including asthma (878 cases and 75,424 controls)35, bronchiectasis (435 cases and 75,605 controls)35, bronchitis [including AB (1765 cases and 74,675 controls) and CB (4046 cases and 74,460 controls)]35, COPD (6043 cases and 73,676 controls)35, ILD (155 cases and 75,755 controls)35, LC (1552 cases and 74,800 controls)35, LCNS (8595 cases and 8275 controls)36, lung function [including FEV1 (62,901 individuals), FVC (62,901 individuals), and PEF (62,901 individuals)]37, and pneumoniae (6903 cases and 71,487 controls)35.
Gastrointestinal diseases:Seven gastrointestinal diseases related GWAS datasets of EAS ancestry were collected, including CP (4768 cases and 166,052 controls)26, CRC (8305 cases and 159,386 controls)26, GORD (948 cases and 177,516 controls)26, IBD [14,393 cases and 15,456 controls, as well as CD (7372 cases and 15,456 controls) and UC (6862 cases and 15,456 controls)]38 and PUD (29,739 cases and 240,675 controls)39.
AFR population
Lung diseases: A total of 6 lung diseases related GWAS datasets of AFR ancestry were collected, including asthma (5051 cases and 27,607 controls)25, COPD (1978 cases and 27,704 controls)25, IPF (169 cases and 8368 controls)25 and lung function [including FEV1 (5978 individuals), FVC (5978 individuals), and PEF (5978 individuals)31].
Gastrointestinal diseases:Only a GWAS for DD (298 cases and 6338 controls)31 was included.
Detailed information for the above GWAS datasets is described in Supplementary Data 1. A more detailed description of the included GWAS datasets and quality control (QC) information can be found in the Supplementary Methods.
Individual-level data from the UKB cohort
The UKB cohort is a population-based prospective study that recruited 502,528 adults aged 40–69 years from 22 assessment centres across England, Scotland, and Wales between April 2006 and December 2010. The participants completed online and nurse-led questionnaires (e.g., sex data was acquired from central registry at recruitment) and provided biological samples. The study protocol and information about data access are available online40.
After individual-level QC, samples were removed if they were: (i) with sex discordance; (ii) identified as outliers for genotype missingness or excess heterozygosity; (iii) with genetic relatedness; (iv) from individuals other than “white British” participants of EUR ancestry; or (v) among individuals who decided not to participate in this programme, and a total of 377,886 participants remained for analysis.
Genotyping, imputation, and the QC process
Details for each GWAS dataset have been provided in Supplementary Data 1, as well as in the Supplementary Methods. We performed SNP-level QC processes in the autosome by excluding variants with (i) duplicate rsID, missing rsID and position, ambiguous strand orientation (i.e., SNPs with A/T or G/C alleles), or multi-allelic SNPs; (ii) minor allele frequency (MAF) < 1%; (iii) allele mismatches compared to the 1000 Genomes Project (1000GP); and (iv) those located within the major histocompatibility complex (MHC) region (chromosome 6: 25–34 Mb). Finally, an average of 6.67 million SNPs were retained for subsequent analysis (Supplementary Data 1). More details were presented in the Supplementary Methods.
Genetic correlation analysis
Estimation of heritability
We applied univariate linkage disequilibrium (LD) score regression (LDSC, version 1.0.1)12,41 to estimate the genome-wide SNP-based heritability (h2SNP, the proportion of phenotypic variance that can be explained by common genetic variants restricted to HapMap3, which is recognised as well imputed in most studies to minimise bias due to low imputation quality) for each trait, as well as the prevalence-corrected heritability (i.e., liability scale; Supplementary Data 1) for several diseases. Briefly, LDSC estimates heritability for a polygenic trait using the fact that SNPs in a high LD region would have higher chi-square statistics than that in a low LD region (see the Supplementary Methods).
Global-level genetic correlation analysis
To evaluate the genome-wide genetic correlation between lung and gastrointestinal diseases, we further utilised LDSC software to decipher the shared genetic architecture. Briefly, it quantifies polygenic effects by examining the relationship between LD scores and GWAS summary statistics, yielding genetic correlation from deviations in chi-square values from the null hypothesis (see Supplementary Methods). To control for multiple comparisons, we applied thresholds of a P-value ≤ 2.78 × 10−4 [0.05/180 (20 lung diseases × 9 gastrointestinal diseases)] for the EUR population, a P-value ≤ 5.95 × 10−4 [0.05/84 (12 lung diseases × 7 gastrointestinal diseases)] for the EAS population, and a P-value ≤ 0.008 [0.05/6 (6 lung diseases × 1 gastrointestinal disease)] for the AFR population to determine the significant results42,43.
Local-level genetic correlation analysis
To further estimate the pairwise local genetic correlations, we adopted SUPERGNOVA44 to investigate the genetic overlap in multiple predefined genomic regions, including 1,703 LD-independent blocks with an average size of approximately 1.6 Mb (https://bitbucket.org/nygcresearch/ldetect-data/src/master), using the 1000GP as a reference panel. A threshold of P ≤ 0.05/1703 was considered statistically significant.
In addition, partitioned LDSC was also performed to characterise the genetic overlap at the level of functional categories, recalculating 12 LD scores for the transcribed region, transcription factor binding sites (TFBS), super-enhancer, intron, DNase I digital genomic footprinting (DGF) region, DHSs, foetal DHS and histone markers H3K9ac, H3K4me1, H3K4me3, H3K27ac and repressed regions. P ≤ 0.05/12 were considered statistically significant. More details were presented in the Supplementary Methods.
Cross-trait meta-analysis
Subsequently, to identify the potential pleiotropic genetic loci associated with each pair of lung and gastrointestinal diseases, we implemented a cross-trait meta-analysis using MTAG45 options that assume equal SNP heritability for each trait (--equal-h2) and perfect genetic covariance between traits (--perfect-gencov). Briefly, MTAG applies generalised inverse-variance-weighted meta-analysis for multiple traits, and it can accommodate potential sample overlap between GWAS. We specifically focused on SNPs that demonstrated consistent directional effects (i.e., identical direction of effect for the same risk alleles) across both diseases, thereby reducing spurious associations and enhancing the biological plausibility of identified associations (i.e., shared etiological mechanisms)41,45. Genome-wide significant pleiotropic SNPs were further identified with (i) PMTAG ≤ 5 × 10−8 (i.e., SNPs associated with the cross-trait phenotypes), and (ii) Psingle-trait ≤ 0.001 for both traits. Independent loci were further extracted using PLINK 1.9 software (--clump-p1 5e-8 --clump-p2 1e-5 --clump-r2 0.001 –clump-kb 500)46. More details can be found in the Supplementary Methods.
Variant-level analysis
Fine mapping credible set analysis
Given that the significant pleiotropic variants identified in the cross-trait meta-analysis may not be causal loci, we further leveraged FM-summary47, a Bayesian fine-mapping algorithm, to calculate a credible set (99%) of causal variants in each genetic region within ± 500 kb of the pleiotropic variant. Briefly, it computes Bayes factors for different SNPs to measure the strength of evidence for causal associations. More details of this method can be found in the Supplementary Methods. A variant was considered causal if its Bayes factor-derived PP was greater than 0.05, or if it had the highest PP.
Colocalization analysis
We next carried out a colocalization analysis using the R package coloc (V5.2.2)48, a Bayesian algorithm, to investigate whether the pleiotropic signals colocalized at shared SNPs. This method provided PPs for five mutually exclusive hypotheses regarding the sharing of causal variants in a genetic region, including (i) H0: no association; (ii) H1: association with trait 1 only; (iii) H2: association with trait 2 only; (iv) H3: association with both traits but two distinct SNPs; and (v) H4: association with both traits and one shared SNP. Here, SNPs within ±500 kb of the pleiotropic variants were extracted to calculate the PPH4. We will classify a PPH4 ≥ 0.5 (suggestive evidence for colocalization) in favour of shared causal variants49,50,51. Finally, we selected the overlapping variants derived from fine-mapping and colocalization analyses as candidate pleiotropic genetic variants.
To validate the pleiotropic associations of the above candidate SNPs with lung-gastrointestinal diseases, we also performed additional cross-trait meta-analyses through GPA52, PLACO53, CPASSOC-SHet54, CPMA55, and MetABF56 methods (see the Supplementary Methods). In addition, to avoid the potential bias of sample overlapping, we also used independent GWAS summary data (lung traits, mainly from the Michigan Genomics Initiative (MGI) cohort57; gastrointestinal traits, mainly from the UKB cohort) for replication analysis.
Further functional annotation was performed using HaploReg58 (V4.2) and SNPnexus59 (V4). The variant annotations of HaploReg mainly included enhancer and promoter histone marks, identification of transcription factor-binding sites identified from ChIP-seq experiments (ENCODE Project Consortium, 2011), and altered potential regulatory motifs. Additional annotation of variants with possible clinical relevance (i.e., the potential of alterations detected in tumours to act as drivers and their possible effect on treatment response) was performed using SNPnexus. In addition, the Genotype-Tissue Expression Project (GTEx, V8)60 database was used for cis-eQTL analysis.
Gene-level analysis
Gene-based and enrichment analyses
To identify the pleiotropic effect of protein-coding genes on both traits, we further conducted gene-based analyses using summary statistics from a cross-trait meta-analysis using Multi-marker Analysis of GenoMic Annotation (MAGMA, V1.10) software61, with a Bonferroni-corrected P-value ≤ 0.05 as a threshold for deciphering pleiotropic genes11. In brief, MAGMA is an efficient tool for calculating multi-marker effects through multiple regression, accounting for LD between markers.
Subsequently, using the MAGMA-identified pleiotropic genes, we performed GSEA, TSEA, and CSEA on these genes to gain insights into the potential shared biological pathways. GSEA was completed with the R package clusterProfiler (V4.10.1) using reference gene sets derived from the Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway databases62. TSEA was performed using the R package deTS (V1.0)63 based on the GTEx project reference panel64. CSEA was conducted with the online platform WebCSEA (Web-based Cell-type Specific Enrichment Analysis of Genes) using panels of 1355 tissue-cell types from 61 different general tissues across 11 human organ systems65. We declared the significance with a Bonferroni-corrected threshold (P-value ≤ 0.05) for these 3 parallel enrichment analyses.
Transcriptome-wide association study (TWAS)
The MAGMA-identified pleiotropic genes that overlapped with the genes located within ± 500 kb of candidate pleiotropic loci were defined as candidate pleiotropic genes.
To further evaluate the risk effects of the expression of candidate pleiotropic genes on both traits, we carried out a TWAS analysis using GWAS summary statistics of the cross-trait meta-analysis by FUSION software, with the integration of the GTEx cis-eQTL dataset (V8; SNP-gene pairs within ± 500 kb distance; N = 715) of 49 normal tissues60,66,67. The TWAS Z value is calculated as formula (1).
where V is a covariance matrix across SNPs at the locus (estimated by 1000 Genomes V3 reference EUR samples), W is the weight in the imputation models trained on the GTEx V8 dataset derived from the precalculated weighted database (http://gusevlab.org/projects/fusion/#reference-functional-data), and ZMTAG is the TWAS Z value computed using the MTAG based cross-trait GWAS meta summary Z scores. We performed TWAS analysis for each lung-gastrointestinal trait pair in each tissue, as well as the combined tissues. The results were considered significant when the FDR-corrected P ≤ 0.05.
Protein-level analysis
Proteome-wide association study (PWAS)
Similarly, to expand on the biological evidence of protein abundance, we then carried out a PWAS based on GWAS summary statistics from the cross-trait meta-analysis using FUSION software, with the plasma cis-protein quantitative trait locus (cis-pQTL) dataset (SNP-gene pairs within ± 500 kb distance) of 7213 EUR American individuals from the Atherosclerosis Risk in Communities (ARIC) study (http://nilanjanchatterjeelab.org/pwas) as a reference panel (see the Supplementary Methods)66,68. The corrected P-value was calculated via the FDR method.
Gene-environment (G × E) interactions
To further explore the additional effects of candidate pleiotropic genetic variants on lung-gastrointestinal diseases, a G × E analysis was performed using genetic and phenotypic data from the UKB. A total of 64 modifiable exposures, including lifestyle factors (e.g., smoking status and second-hand smoke exposure, BMI, physical activity, alcohol drinking status and alcohol intake frequency), dietary intake (e.g., fruit and vegetables, oily fish, calcium, high-fat cheese, red meat, and processed meat), air pollutants (e.g., particulate matter air pollution, nitrogen dioxide, nitrogen oxides) and traffic-related air pollution, mood, and antibiotic/aspirin exposure69,70, were included in the analysis.
First, we evaluated the associations between 64 exposures and the risk of lung-gastrointestinal comorbidities corresponding to candidate pleiotropic genetic variants using a logistic regression model (1: comorbidity cases; 0: healthy controls). Second, significant comorbidity-related exposures were identified (P ≤ 0.05) and used in the subsequent multiply interaction analysis with candidate pleiotropic genetic variants:
where Y was the lung-gastrointestinal comorbidity, E was an exposure, G was the additively coded genotype of a pleiotropic SNP with values of 0, 1, or 2, and βInteract was the interaction coefficient measuring the G × E effects71. For each variant, significant interactions were identified using the Bonferroni-corrected method. All analyses were performed by adjusting for age, sex, assessment centre, and the first 10 principal components.
MR analyses
To further infer causal relationships between lung and gastrointestinal diseases, we conducted a bi-directional two-sample MR analysis with previously reported genome-wide significant SNPs [P ≤ 5 × 10−8; non-MHC region; derived from the GWAS Catalogue (June 30, 2023)] as instrumental variables (IVs). If the number of IVs was <10 (i.e., CB, pneumoniae, LUSC, and IBS), a suggestive threshold (P ≤ 5 × 10−6) was used. Independent variants (LD r2 < 0.001, window size = 10 Mb) with sufficient statistical power (F statistics > 10) were kept (see the Supplementary Methods). MR-pleiotropy residual sum and outlier (MR-PRESSO)72 test was further applied to exclude horizontal pleiotropic outliers (global P-value < 0.05) for subsequent MR analysis.
The primary analyses included the IVW method73, MR-Egger regression74, and Cochran’s Q heterogeneity test75. If the P-value of the heterogeneity test was > 0.05, fixed-effect based IVW results were applied; otherwise, random-effect based results were used76,77. An association was considered significant at an FDR-corrected P-value ≤ 0.05 for IVW and a P-value > 0.05 for the Egger intercept or Cochran’s Q test. In addition, several other methods, including the weighted median estimator method75, weighted mode method78, and MR robust adjusted profile score (MR-RAPS)79, were used for complementary analyses. These methods briefly employ different assumptions regarding horizontal pleiotropy, as described in the Supplementary Methods. Multiple testing correction was performed using the p.adjust function in R software with the Benjamini-Hochberg procedure.
To ensure reliable interpretation of estimates from MR, we performed several sensitivity analyses to validate the MR results80,81. First, the radial MR was utilised to flit out heterogeneity-based outliers82,83. Second, we excluded palindromic IVs (A/T, G/C), which are SNPs with identical nucleotide pairs on both the forward and reverse strands. Third, we excluded pleiotropic SNPs that were associated with potential confounding phenotypes (i.e., smoking, alcohol use, physical activity, body fat, education, and diabetes) according to the GWAS catalogue (V1.0, data download in September 2024). Finally, we conducted a leave-one-out analysis where we excluded one SNP at a time and performed IVW analysis on the remaining SNPs to evaluate the robustness of our findings.
Microbiome-level analysis
Mediation analysis
Cumulative evidence has demonstrated the potential role of the gut microbiota in the development of gastrointestinal diseases84. Therefore, based on the significant MR results of the lung-gastrointestinal diseases, we aimed to further explore the causal associations between the gut microbiota, gastrointestinal and lung diseases (i.e., GORD-asthma, GORD-CB, and GORD-COPD) using MR and mediation approaches85.
In the first MR analysis, the causal effect of the gut microbiota on gastrointestinal diseases was evaluated, of which the genetic instruments for the gut microbiota were derived from a human host-microbiome GWAS (mGWAS, 430 microbiome features from 8956 German individuals; with a P-value threshold of 1 × 10−5 and a r2 threshold of 0.001)86, following recommendations from a previous study87, with independent validation in 7738 participants of the DMP88. In the second MR analysis, we estimated the causal effect of the gut microbiota on the risk of lung disease, with the same procedure used for the first-stage MR analysis.
Subsequently, mediation analysis was used to evaluate the mediation role of gastrointestinal diseases in the association between the gut microbiota and lung diseases. The mediation proportion was calculated as the product (mediation effect) of β1 (the IVW-estimated effect size of the gut microbiota on gastrointestinal diseases) and β2 (the IVW-estimated effect size of gastrointestinal diseases on lung diseases) divided by the total effect. Besides, the standard error (SE) and 95% confidence interval (CI) of the mediation effect were calculated using the delta method89.
All analyses were conducted separately for each ancestry group using R software (V4.0.3), and a two-sided original or corrected P-value less than 0.05 indicated statistical significance.
Software used
Publicly available software (and version, where applicable) used in this paper is as follows: LDSC (V1.0.1, https://github.com/bulik/ldsc), SUPERGNOVA, MTAG, PLINK, FM-summary (https://github.com/hailianghuang/FM-summary), Coloc (V5.2.2, https://cran.r-project.org/web/packages/coloc/index.html), GPA (V1.1.0, https://github.com/dongjunchung/GPA), PLACO (V0.1.1, https://github.com/RayDebashree/PLACO), CPASSOC-Shet (http://hal.case.edu/~xxz10/zhu-web/CPASSOC/CPASSOC.zip), CPMA, MetABF (V1.0.0, https://github.com/trochet/metabf), SNPnexus (V4, https://www.snp-nexus.org), HaploReg (V4.2, https://pubs.broadinstitute.org/mammals/haploreg/haploreg.php), MAGMA (V1.10, https://cncr.nl/research/magma/), clusterProfiler (V4.10.1, http://bioconductor.org/packages/release/bioc/html/clusterProfiler.html), deTS (V1.0, https://github.com/bsml320/deTS), WebCSEA (https://bioinfo.uth.edu/webcsea/), FUSION, MR-PRESSO (V1.0, https://github.com/rondolab/MR-PRESSO), TwoSampleMR (V0.5.7, https://github.com/MRCIEU/TwoSampleMR), mr.raps (V0.2, https://github.com/qingyuanzhao/mr.raps), and RadialMR (V1.1, https://github.com/WSpiller/RadialMR/).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All analyses were based on publicly available data. The sources of GWAS summary statistics included in this study have been provided in Supplementary Data 1. Individual-level phenotype and genetic data used in this research was available from the UK Biobank resource under access application 92675. The UK Biobank data are available under restricted access for data privacy laws, access can be obtained by submitting an application to the UK Biobank Access Management System (https://ams.ukbiobank.ac.uk/ams/). Access to GWAS summary statistics from the MGI can be requested through the JIRA ticketing system by completing a data use agreement (https://precisionhealth.umich.edu/our-research/michigangenomics/). The 1000 Genomes V3 references were available from https://cncr.nl/research/magma/#reference_data. LD scores used for estimation of heritability, global-level and partitioned-level genetic correlations were available from https://console.cloud.google.com/storage/browser/broad-alkesgroup-public-requester-pays/LDSCORE. For local genetic correlations, genome partition files were available from https://bitbucket.org/nygcresearch/ldetect-data/src/master/EUR/. For MAGMA, the gene location file was available from https://ctg.cncr.nl/software/MAGMA/aux_files/NCBI37.3.zip. For TWAS, pre-computed GTEx expression weights were available from the Gusev lab at http://gusevlab.org/projects/fusion/#gtex-v8-multi-tissue-expression and http://gusevlab.org/projects/fusion/#gtex-cross-tissue-scca-expression. For PWAS, plasma proteins’ cis summary statistics of European Americans from the Atherosclerosis Risk in Communities study were available from https://jh-pwas.s3.amazonaws.com/packages/PWAS_EA.zip. For MR analysis, the sources of previously reported genome-wide significant SNPs have been provided in Supplementary Data 21. For mediation analysis, complete summary statistics of 430 microbiome features for 8,956 German individuals were available from https://www.ebi.ac.uk/gwas/publications/33462482. And, summary statistics of 7,738 participants in the Dutch Microbiome Project were available from https://www.ebi.ac.uk/gwas/publications/35115690. Source data for generating figures were provided as a Source Data file with this paper. Full data for generating Figure S6a were additionally deposited to Zenodo: https://doi.org/10.5281/zenodo.14983136. Source data are provided in this paper.
Code availability
Custom code for LDSC, SUPERGNOVA, MTAG, MAGMA, TWAS, and PWAS analysis can be found in GitHub (https://github.com/YangZhao-Lab-NJMU/shared-genetics-of-lung-gut-axis) and in Zenodo (https://doi.org/10.5281/zenodo.14983136). All software used to process the data have been referenced in the Methods section.
References
Song, X. et al. The role and mechanism of gut-lung axis mediated bidirectional communication in the occurrence and development of chronic obstructive pulmonary disease. Gut Microbes 16, 2414805 (2024).
Mackintosh, J. A. et al. In patients with idiopathic pulmonary fibrosis the presence of hiatus hernia is associated with disease progression and mortality. Eur. Respir. J. 53, 1802412 (2019).
Vutcovici, M. et al. Inflammatory bowel disease and risk of mortality in COPD. Eur. Respir. J. 47, 1357–1364 (2016).
Cheung, T. K. et al. Gastroesophageal reflux disease is associated with poor asthma control, quality of life, and psychological status in Chinese asthma patients. Chest 135, 1181–1185 (2009).
Liou, A., Grubb, J. R., Schechtman, K. B. & Hamilos, D. L. Causative and contributive factors to asthma severity and patterns of medication use in patients seeking specialized asthma care. Chest 124, 1781–1788 (2003).
Budden, K. F. et al. Emerging pathogenic links between microbiota and the gut-lung axis. Nat. Rev. Microbiol. 15, 55–63 (2017).
Hou, K. et al. Microbiota in health and diseases. Signal Transduct. Target. Ther. 7, 135 (2022).
Wang, L. et al. The bidirectional gut-lung axis in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 207, 1145–1160 (2023).
Molina-Molina, M. et al. Gastrointestinal pirfenidone adverse events in idiopathic pulmonary fibrosis depending on diet: the MADIET clinical trial. Eur. Respir. J. 62, 2300262 (2023).
Lindström, S. et al. Genome-wide analyses characterize shared heritability among cancers and identify novel cancer susceptibility regions. J. Natl. Cancer Inst. 115, 712–732 (2023).
Gong, W. et al. Role of the gut-brain axis in the shared genetic etiology between gastrointestinal tract diseases and psychiatric disorders: A genome-wide pleiotropic analysis. JAMA Psychiatry 80, 360–370 (2023).
Bulik-Sullivan, B. K. et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 47, 291–295 (2015).
Ju, T., Kong, J. Y., Stothard, P. & Willing, B. P. Defining the role of Parasutterella, a previously uncharacterized member of the core gut microbiota. Isme J. 13, 1520–1534 (2019).
Li, D. et al. Gut microbiota-derived inosine from dietary barley leaf supplementation attenuates colitis through PPARγ signaling activation. Microbiome 9, 83 (2021).
Kandulski, A. & Malfertheiner, P. Gastroesophageal reflux disease–from reflux episodes to mucosal inflammation. Nat. Rev. Gastroenterol. Hepatol. 9, 15–22 (2011).
Emilsson, Ö. I. et al. Nocturnal gastro-oesophageal reflux, asthma and symptoms of OSA: a longitudinal, general population study. Eur. Respir. J. 41, 1347–1354 (2013).
Ried, J. S. et al. Integrative genetic and metabolite profiling analysis suggests altered phosphatidylcholine metabolism in asthma. Allergy 68, 629–636 (2013).
Ji, X. et al. Identification of susceptibility pathways for the role of chromosome 15q25.1 in modifying lung cancer risk. Nat. Commun. 9, 3221 (2018).
Hu, N., Zou, L., Wang, C. & Song, G. RUNX1T1 function in cell fate. Stem Cell Res. Ther. 13, 369 (2022).
Ruff, W. E., Greiling, T. M. & Kriegel, M. A. Host-microbiota interactions in immune-mediated diseases. Nat. Rev. Microbiol. 18, 521–538 (2020).
Liaudet, L. et al. Inosine reduces systemic inflammation and improves survival in septic shock induced by cecal ligation and puncture. Am. J. Respir. Crit. Care Med. 164, 1213–1220 (2001).
Henneke, L. et al. A dietary carbohydrate - gut Parasutterella - human fatty acid biosynthesis metabolic axis in obesity and type 2 diabetes. Gut Microbes 14, 2057778 (2022).
Yang, J. J. et al. Association of dietary fiber and yogurt consumption with lung cancer risk: A pooled analysis. JAMA Oncol. 6, e194107 (2020).
Ross, F. C. et al. The interplay between diet and the gut microbiome: implications for health and disease. Nat. Rev. Microbiol. 22, 671–686 (2024).
Zhou, W. et al. Global biobank meta-analysis initiative: Powering genetic discovery across human disease. Cell Genom. 2, 100192 (2022).
Sakaue, S. et al. A cross-population atlas of genetic associations for 220 human phenotypes. Nat. Genet. 53, 1415–1424 (2021).
McKay, J. D. et al. Large-scale association analysis identifies new lung cancer susceptibility loci and heterogeneity in genetic susceptibility across histological subtypes. Nat. Genet. 49, 1126–1132 (2017).
Shrine, N. et al. New genetic signals for lung function highlight pathways and chronic obstructive pulmonary disease associations across multiple ancestries. Nat. Genet. 51, 481–493 (2019).
Kurki, M. I. et al. FinnGen provides genetic insights from a well-phenotyped isolated population. Nature 613, 508–518 (2023).
Fernandez-Rozadilla, C. et al. Deciphering colorectal cancer genetics through multi-omic analysis of 100,204 cases and 154,587 controls of European and east Asian ancestries. Nat. Genet. 55, 89–99 (2023).
Karczewski, K. et al. Pan-UK Biobank GWAS improves discovery, analysis of genetic architecture, and resolution into ancestry-enriched effects. Preprint at https://doi.org/10.1101/2024.03.13.24303864 (2024).
Wu, Y. et al. GWAS of peptic ulcer disease implicates Helicobacter pylori infection, other gastrointestinal disorders and depression. Nat. Commun. 12, 1146 (2021).
Liu, J. Z. et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet. 47, 979–986 (2015).
Eijsbouts, C. et al. Genome-wide analysis of 53,400 people with irritable bowel syndrome highlights shared genetic pathways with mood and anxiety disorders. Nat. Genet. 53, 1543–1552 (2021).
Walters, R. G. et al. Genotyping and population characteristics of the China Kadoorie Biobank. Cell Genom. 3, 100361 (2023).
Wei, X. et al. Development and evaluation of a polygenic risk score for lung cancer in never-smoking women: A large-scale prospective Chinese cohort study. Int. J. Cancer 154, 807–815 (2024).
Chen, C. Y. et al. Analysis across Taiwan Biobank, Biobank Japan, and UK Biobank identifies hundreds of novel loci for 36 quantitative traits. Cell Genom. 3, 100436 (2023).
Liu, Z. et al. Genetic architecture of the inflammatory bowel diseases across East Asian and European ancestries. Nat. Genet. 55, 796–806 (2023).
He, Y. et al. East Asian-specific and cross-ancestry genome-wide meta-analyses provide mechanistic insights into peptic ulcer disease. Nat. Genet. 55, 2129–2138 (2023).
Sudlow, C. et al. UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 12, e1001779 (2015).
Bulik-Sullivan, B. et al. An atlas of genetic correlations across human diseases and traits. Nat. Genet. 47, 1236–1241 (2015).
Jiang, X. et al. Shared heritability and functional enrichment across six solid cancers. Nat. Commun. 10, 431 (2019).
Zheng, S., Tsao, P. S. & Pan, C. Abdominal aortic aneurysm and cardiometabolic traits share strong genetic susceptibility to lipid metabolism and inflammation. Nat. Commun. 15, 5652 (2024).
Zhang, Y. et al. SUPERGNOVA: local genetic correlation analysis reveals heterogeneous etiologic sharing of complex traits. Genome Biol. 22, 262 (2021).
Turley, P. et al. Multi-trait analysis of genome-wide association summary statistics using MTAG. Nat. Genet. 50, 229–237 (2018).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Maller, J. B. et al. Bayesian refinement of association signals for 14 loci in 3 common diseases. Nat. Genet. 44, 1294–1301 (2012).
Giambartolomei, C. et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 10, e1004383 (2014).
Zhu, Z. et al. A genome-wide cross-trait analysis from UK Biobank highlights the shared genetic architecture of asthma and allergic diseases. Nat Genet 50, 857–864 (2018).
Wingo, T. S. et al. Shared mechanisms across the major psychiatric and neurodegenerative diseases. Nat. Commun. 13, 4314 (2022).
Huang, Y. et al. Deciphering genetic causes for sex differences in human health through drug metabolism and transporter genes. Nat. Commun. 14, 175 (2023).
Chung, D., Yang, C., Li, C., Gelernter, J. & Zhao, H. GPA: a statistical approach to prioritizing GWAS results by integrating pleiotropy and annotation. PLoS Genet. 10, e1004787 (2014).
Ray, D. & Chatterjee, N. A powerful method for pleiotropic analysis under composite null hypothesis identifies novel shared loci between Type 2 Diabetes and Prostate Cancer. PLoS Genet. 16, e1009218 (2020).
Zhu, X. et al. Meta-analysis of correlated traits via summary statistics from GWASs with an application in hypertension. Am. J. Hum. Genet. 96, 21–36 (2015).
Cotsapas, C. et al. Pervasive sharing of genetic effects in autoimmune disease. PLoS Genet. 7, e1002254 (2011).
Trochet, H. et al. Bayesian meta-analysis across genome-wide association studies of diverse phenotypes. Genet. Epidemiol. 43, 532–547 (2019).
Zawistowski, M. et al. The Michigan Genomics Initiative: A biobank linking genotypes and electronic clinical records in Michigan Medicine patients. Cell Genom. 3, 100257 (2023).
Ward, L. D. & Kellis, M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 40, D930–D934 (2012).
Oscanoa, J. et al. SNPnexus: a web server for functional annotation of human genome sequence variation (2020 update). Nucleic Acids Res. 48, W185–w192 (2020).
The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science. 369, 1318–1330 (2020).
de Leeuw, C. A., Mooij, J. M., Heskes, T. & Posthuma, D. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput. Biol. 11, e1004219 (2015).
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287 (2012).
Pei, G., Dai, Y., Zhao, Z. & Jia, P. deTS: tissue-specific enrichment analysis to decode tissue specificity. Bioinformatics 35, 3842–3845 (2019).
Consortium, G. T. The genotype-tissue expression (GTEx) project. Nat. Genet. 45, 580–585 (2013).
Dai, Y. et al. WebCSEA: web-based cell-type-specific enrichment analysis of genes. Nucleic Acids Res. 50, W782–w790 (2022).
Gusev, A. et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet. 48, 245–252 (2016).
Feng, H. et al. Leveraging expression from multiple tissues using sparse canonical correlation analysis and aggregate tests improves the power of transcriptome-wide association studies. PLoS Genet. 17, e1008973 (2021).
Zhang, J. et al. Plasma proteome analyses in individuals of European and African ancestry identify cis-pQTLs and models for proteome-wide association studies. Nat. Genet. 54, 593–602 (2022).
Bailie, L., Loughrey, M. B. & Coleman, H. G. Lifestyle risk factors for serrated colorectal polyps: A systematic review and meta-analysis. Gastroenterology 152, 92–104 (2017).
Huang, Y. et al. Air pollution, genetic factors, and the risk of Lung cancer: A prospective study in the UK Biobank. Am. J. Respir. Crit. Care Med. 204, 817–825 (2021).
Hunter, D. J. Gene-environment interactions in human diseases. Nat. Rev. Genet. 6, 287–298 (2005).
Verbanck, M., Chen, C. Y., Neale, B. & Do, R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat. Genet. 50, 693 (2018).
Burgess, S., Butterworth, A. & Thompson, S. G. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet. Epidemiol. 37, 658–665 (2013).
Bowden, J., Davey Smith, G. & Burgess, S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int. J. Epidemiol. 44, 512–525 (2015).
Bowden, J., Davey Smith, G., Haycock, P. C. & Burgess, S. Consistent estimation in mendelian randomization with some invalid instruments using a weighted median estimator. Genet. Epidemiol. 40, 304–314 (2016).
Jiang, R. et al. The brain structure, inflammatory, and genetic mechanisms mediate the association between physical frailty and depression. Nat. Commun. 15, 4411 (2024).
Bowden, J. et al. A framework for the investigation of pleiotropy in two-sample summary data Mendelian randomization. Stat. Med. 36, 1783–1802 (2017).
Hartwig, F. P., Davey Smith, G. & Bowden, J. Robust inference in summary data Mendelian randomization via the zero modal pleiotropy assumption. Int. J. Epidemiol. 46, 1985–1998 (2017).
Zhao, Q. Y., Wang, J. S., Hemani, G., Bowden, J. & Small, D. S. Statistical inference in two-sample summary-data mendelian randomization using robust adjusted profile score. Ann. Stat. 48, 1742–1769 (2020).
Wu, X. et al. Investigating the shared genetic architecture of uterine leiomyoma and breast cancer: A genome-wide cross-trait analysis. Am. J. Hum. Genet. 109, 1272–1285 (2022).
Xin, J. et al. Association between circulating vitamin E and ten common cancers: evidence from large-scale Mendelian randomization analysis and a longitudinal cohort study. BMC Med. 20, 168 (2022).
Bowden, J. et al. Improving the visualization, interpretation and analysis of two-sample summary data Mendelian randomization via the Radial plot and Radial regression. Int. J. Epidemiol. 47, 2100 (2018).
Stankovic, S. et al. Genetic links between ovarian ageing, cancer risk and de novo mutation rates. Nature 633, 608–614 (2024).
Richard, M. L. & Sokol, H. The gut mycobiota: insights into analysis, environmental interactions and role in gastrointestinal diseases. Nat. Rev. Gastroenterol. Hepatol. 16, 331–345 (2019).
Relton, C. L. & Davey Smith, G. Two-step epigenetic Mendelian randomization: a strategy for establishing the causal role of epigenetic processes in pathways to disease. Int. J. Epidemiol. 41, 161–176 (2012).
Rühlemann, M. C. et al. Genome-wide association study in 8,956 German individuals identifies influence of ABO histo-blood groups on gut microbiome. Nat. Genet. 53, 147–155 (2021).
Qin, Y. et al. Combined effects of host genetics and diet on human gut microbiota and incident disease in a single population cohort. Nat. Genet. 54, 134–142 (2022).
Lopera-Maya, E. A. et al. Effect of host genetics on the gut microbiome in 7,738 participants of the Dutch Microbiome Project. Nat. Genet. 54, 143–151 (2022).
Carter, A. R. et al. Understanding the consequences of education inequality on cardiovascular disease: mendelian randomisation study. Bmj 365, l1855 (2019).
Acknowledgements
We would like to thank the participants and researchers from the UKB who significantly contributed or collected data. We thank the GWAS Catalogue, the participants and the investigators of the FinnGen study, the Michigan Genomics Initiative (MGI) supported by the University of Michigan Precision Health Initiative, the global biobank meta-analysis initiative, and the National Bioscience Database Centre human database for providing GWAS summary data. We thank the following for their support for the GWAS summary data from China Kadoorie Biobank: Kadoorie Charitable Foundation (Hong Kong); Wellcome Trust; UK Medical Research Council; Cancer Research UK; British Heart Foundation; GlaxoSmithKline; National Key Research and Development Programme of China; National Natural Science Foundation of China. We thank the National Natural Science Foundation of China (82220108002 to F.C., 82173620 and 82373690 to Y.Z., and 82204156 to D.Y.), and the Priority Academic Programme Development of Jiangsu Higher Education Institution (PAPD, to Y.Z.) for funding this study. Some figures were created with BioRender.com (full licence).
Author information
Authors and Affiliations
Contributions
Y.Z. and G.J. designed the study. D.Y., Y.W. and M.L. performed the analysis. Y.Z., G.J., F.C. and D.C. supervised the study. D.Y., Y.W. and M.L. wrote the manuscript. F.S., Y.T., S.L., L.L., Z.Z., R.Z., S.S., T.L., H.X, H.M., Y.Y. and H.S. formed the core revision group. Y.Z., G.J., F.C. and D.C. provided study directions. All the authors contributed to the final revision of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Emmanuel Adewuyi, and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
You, D., Wu, Y., Lu, M. et al. A genome-wide cross-trait analysis characterizes the shared genetic architecture between lung and gastrointestinal diseases. Nat Commun 16, 3032 (2025). https://doi.org/10.1038/s41467-025-58248-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-58248-w
