
Abstract
Trastuzumab deruxtecan (T-DXd) is an antibody-drug conjugate (ADC) targeting HER2, exhibiting significant clinical efficacy in breast cancer (BC) with varying HER2 expression, including HER2-low and HER2-ultralow. However, the precise mechanism underlying its efficacy and the contribution of immune activation in these settings remain unclear. Here, we demonstrate that T-DXd efficacy in HER2-low and HER2-negative BC is independent of HER2 engagement and ADC internalization. Instead, its activity relies on extracellular proteases, such as cathepsin L (CTSL), within the tumor microenvironment. Irrespective of their HER2 status, tumor and stromal compartments of invasive BC abundantly express CTSL, which efficiently cleaves the specialized linker of T-DXd, facilitating payload release and inducing cytotoxicity against HER2-low/negative tumors. In HER2-positive BC, the antibody backbone of T-DXd engages Fcγ-receptors and drives antibody-dependent cellular phagocytosis (ADCP). Concurrently, its cytotoxic payload (DXd) induces immunogenic cell death, further activating myeloid cells via TLR4 and STING pathways to enhance tumor antigen presentation to CD8+ T cells. Notably, T-DXd cytotoxicity also upregulates tumor CD47 expression, dampening immune activation. Combining T-DXd with CD47 checkpoint blockade significantly enhances anti-tumor immune responses in a HER2-transgenic BC mouse model, while also inducing durable CD8+ T cell memory to prevent tumor recurrence after therapy cessation.
Similar content being viewed by others


Introduction
Antibody-drug conjugates (ADCs) are a groundbreaking therapeutic class in oncology, designed to enhance chemotherapeutic efficacy while minimizing systemic toxicity. By harnessing the specificity of antibodies, ADCs combine a monoclonal antibody (mAb) with a linker-conjugated cytotoxic payload to selectively target cancer cells in the tumor microenvironment (TME)1. While mAbs target cancer cell antigens and potentially activate anti-tumor immunity2,3,4, the role of Fcγ-receptors (FCGR) in ADC efficacy is unclear. Linkers serve as a molecular bridge, connecting the antibody to the cytotoxic payload, with various cleavage mechanisms influencing payload release5. Cytotoxic payloads exploit different tumor cell proliferative pathways, occur in different drug-to-antibody ratios for a given ADC, and likely trigger diverse types of tumor cell death6. Ultimately, ADC design aims for precise, targeted drug delivery to cancer cells to reduce systemic side effects and improve therapeutic outcomes. While there have been notable successes in the use of ADCs, the fundamental mechanisms that underlie clinical efficacy remain incompletely understood. These unknowns are perhaps best illustrated in human epidermal growth factor receptor-2 positive (HER2+) breast cancer (BC), in which a recently developed ADC, trastuzumab deruxtecan (T-DXd), dramatically outperformed the previously approved ADC, ado-trastuzumab emtansine (T-DM1), in recent clinical trials.
T-DXd has achieved remarkable success in multiple clinical trials, leading to its initial approval in 2019 for the treatment of metastatic HER2-positive BC patients7. Subsequently, T-DXd received approvals for HER2-positive gastric and non-small-cell lung cancers8,9, paving its way for a recent tissue-agnostic approval by the FDA in 2024, the first ever for an ADC10. In contrast, T-DM1 is an alternative HER2-targeting ADC first approved in 2014. Although both ADCs utilize the same mAb backbone (trastuzumab), T-DXd differs from T-DM1 in several components: T-DXd contains a specialized linker, formed by a cleavable maleimide tetrapeptide (Gly-Gly-Phe-Gly) that is designed for enzymatic cleavage by cathepsin proteases, allowing for earlier and targeted release inside tumor endosomes11. In contrast, T-DM1 utilizes a non-cleavable thioether linker (N-succinimidyl-4-(N-maleimidomethyl) cyclohexane-1-carboxylate), and can only release its payload upon antibody hydrolysis in the cellular lysosome12. A second distinct feature is the conjugated payload, as T-DXd carries DXd, a potent topoisomerase-I inhibitor13, while T-DM1 utilizes DM1, a derivative of maytansine that inhibits microtubule polymerization12. Finally, T-DXd has a drug-to-antibody ratio (DAR) of ~8 compared to ~3.5 for T-DM113. Collectively, these differences may all contribute to different preclinical/clinical efficacy and pharmacodynamics between the two ADCs.
HER2 status in BC is determined by immunohistochemistry (IHC) for HER2 protein expression and in situ hybridization (ISH) for HER2 gene amplification. In the phase-III clinical trial (DESTINY-Breast03), T-DXd significantly improved progression-free (PFS) and overall survival (OS) compared with T-DM1 in patients with metastatic HER2-positive (IHC3+ or IHC2+/ISH+) BC14. Notably, long-term survival analysis demonstrated T-DXd achieved a 36-month PFS rate of 45.7%, compared to 12.4% in the T-DM1 arm. Thus, despite their shared backbone, the differences in linker and payload underpin the enhanced and broader clinical responses of T-DXd. Furthermore, these structural variations contribute to distinct safety profiles, with T-DXd and T-DM1 exhibiting different patterns of adverse events15. Intriguingly, T-DXd demonstrated surprising efficacy in “HER2-low” (IHC1+ or IHC2+/ISH−) BC16, which led to its approval for this cancer, encompassing ~60% of all BC patients17. More recently, a phase-III DESTINY-Breast06 trial revealed that T-DXd significantly outperformed conventional chemotherapy in “HER2-ultralow” metastatic BC patients (defined as IHC0/ISH−, with incomplete and faint membrane HER2 staining in >0 but ≤10% of tumor cells), with comparable PFS observed between HER2-low and HER2-ultralow cohorts18,19. Despite these successes, many patients still experience relapse or incomplete responses to T-DXd14, suggesting that additional approaches will be needed to extend its efficacy. Importantly, while T-DXd has achieved clinical success, the exact mechanisms of action (MOA) behind its superior anti-tumor efficacy, particularly in HER2-low/ultralow BC, remain unclear and a subject of active investigation13,20,21.
In our study, we sought to address these unknowns by investigating the anti-tumor MOAs of T-DXd. Using a variety of in vitro and in vivo models and approaches, we uncovered two previously unreported MOAs that may help explain the clinical success of T-DXd. First, we confirmed the previously reported “bystander killing effect”20 of T-DXd, a mechanism widely believed to be a key factor in T-DXd’s superior clinical efficacy over T-DM1. Crucially, we discovered a different MOA, in which T-DXd can release its cytotoxic payload in an extracellular manner independent of HER2 binding, contributing to its efficacy in HER2-low and even HER2-negative tumors in vivo. This unique feature of T-DXd is attributed to its specialized linker, which we found could be cleaved by cathepsin L, in contrast to the widely reported cathepsin B (CTSB)11. This cleavage occurs both within intracellular tumor endosomes and in the extracellular environment, enabling T-DXd to exert a significant impact on non-HER2-expressing cancers and potentially explaining the observed clinical success in HER2-low/ultralow breast cancer.
In the second part of our study, we found that T-DXd’s cytotoxic payload induces immunogenic cell death (ICD) and activates the STING and TLR4 pathways. This activation stimulates macrophages and dendritic cells, which in turn enhances tumor antigen presentation and the priming of CD8+ T cells. However, this cytotoxic payload effect also upregulates tumor CD47 expression, which limits immune activation22. The combination of CD47/SIRPα checkpoint blockade therapy with T-DXd improved tumor phagocytosis and further boosted innate immune activation, resulting in a more robust anti-tumor adaptive immune response. Notably, this combination led to complete tumor regression against the majority of large tumors in an immunocompetent endogenous HER2-driven BC mouse model and prevented tumor relapse by inducing CD8+ T cell-mediated immune memory, even in the presence of ongoing HER2 oncogenic signaling.
In summary, while novel ADCs like T-DXd represent a promising avenue for future cancer therapies, many aspects of their success remain poorly understood. Our results underscore the importance of elucidating the underlying mechanisms of payload release and immune activation to fully harness ADC potential. These findings offer vital insights that may guide the rational design and development of next-generation ADCs.
Results
T-DXd cytotoxicity against HER2-low and HER2-negative tumors in vivo occurs independently of HER2-high cells in proximity
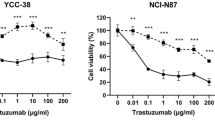
In our initial studies we assessed T-DXd targeted internalization into cancer cells, a key reported MOA for ADCs. Given the demonstrated efficacy of T-DXd in HER2-low breast cancer16, leading to its approval as the new standard-of-care for this subset of BC patients in 2022, and its unexpected efficacy in “HER2-ultralow” BC18,19, we tested cell lines with different surface HER2 levels: KPL4 and Au565 (HER2-high), CAPAN-1 (HER2-low), MDA-MB-231 (HER2-low) and MDA-MB-468 (HER2-negative), all verified by flow cytometry (Supplementary Fig. 1a). To assess internalization, we labeled T-DXd and T-DM1 with pHrodo, which fluoresces in low pH endosomal compartments (Supplementary Fig. 1b). We found that both T-DXd and T-DM1 were efficiently internalized by HER2-high cells, but only weakly by HER2-low cells, with no observable uptake by HER2-negative cells after 24 h treatment in culture (Fig. 1a, b and Supplementary Fig. 1c). Expectedly, these internalization differences correlated with their sensitivities to T-DXd cytotoxicity (Fig. 1c). Both ADCs were highly cytotoxic to HER2-high cells (Supplementary Fig. 1d, e), but not to HER2-low (Supplementary Fig. 1f, g) or HER2-negative cells (Supplementary Fig. 1h–j), indicating sufficient ADC internalization is crucial for T-DXd and T-DM1 cytotoxicity in vitro.

a T-DXd internalization in cancer lines with different HER2 expression levels. T-DXd or control antibody (rituximab) were labeled with pHrodo to assess internalization into the endosome. Endocytosis was analyzed at 24 h post treatment. Representative flow histograms are shown for each T-DXd concentration. b Summary of T-DXd internalization. c Cell lines with different HER2 expression were treated with indicated ADCs or antibodies for 4 days, and cell viability was assessed by cellular ATP quantification. d In vitro bystander killing analysis. Vybrant DiD-labeled Au565 cells and Cell-Trace Violet-labeled MDA-MB-468 cells were co-cultured (1:2 ratio) with antibodies/ADCs (100 ng/mL) for 2 and 6 days. Apoptosis was assessed by AnnexinV/PI staining. e, f Summary of direct Au565 killing and bystander MDA-MB-468 killing. Total AnnexinV+ percentages (early and late apoptosis combined) are plotted. g, h Similar co-culture experiment using labeled HER2-low CAPAN-1 and MDA-MB-468, treated with 1 µg/mL of antibodies/ADCs. Apoptosis was assessed by AnnexinV/PI staining. b, c, e–h Two-way ANOVA with Tukey’s multiple comparisons test (n = 3). i–k HER2-ADC therapeutic efficacies on various BC xenografts with different HER2 expression engrafted in mammary fat pads of SCID mice. Tumor-bearing animals were treated weekly (arrows indicated) with T-DXd or T-DM1 (10 mg/kg each) or vehicle control PBS. l Diagram of KPL4 and MDA-MB-468 dual-implantation into the right and left mammary fat pads of SCID mice. Created in BioRender. Hartman, Z. (2025) https://BioRender.com/f86p031. m, n Tumor-bearing mice in (l) were treated weekly with rituximab, T-DXd, or T-DM1 (10 mg/kg, arrows indicated). Tumor growths from each side are plotted. i–n Mixed-effects analysis (REML) with Tukey’s multiple comparisons. i, k, m, n n = 5, (j) (n = 10). All data is presented as mean ± SEM with p values. n.s. = not significant.
However, previous studies documented “bystander” killing of HER2-negative cells after co-culture with HER2-positive cells, attributed to DXd’s membrane-permeability and subsequent “payload spreading“20. To accurately study this bystander killing, we employed cell labeling with Vibrant-DiD dye for HER2-high Au565 cells and Cell-Trace Violet dye for HER2-negative MDA-MB-468 cells, enabling their identification via flow cytometry (Fig. 1d). HER2-high Au565 cells after both T-DXd and T-DM1 treatment showed significant apoptosis (AnnexinV/PI staining) within two to six days (Fig. 1e and Supplementary Fig. 2a). Interestingly, bystander apoptosis in HER2-negative cells was modestly observed at later time points on day six with T-DXd (22.5% ± 0.9 total apoptotic cells), but no differences between T-DM1 and control treatments (Fig. 1f and Supplementary Fig. 2b). This bystander effect occurred only in co-culture conditions, and not with MDA-MB-468 monocultures (Supplementary Fig. 2e), highlighting the requirement of dying HER2-positive cells for payload spreading and bystander killing by T-DXd. Notably, when HER2-high Au565 cells are replaced with HER2-low CAPAN-1 cells in this co-culture setting, we did not detect any significant apoptosis in any cell type, even with higher doses of T-DXd (Fig. 1g, h and Supplementary Fig. 2c). This highlights the lack of T-DXd internalization in HER2-low cells (Fig. 1a, b), which strongly limited both direct killing (Fig. 1g) and subsequent payload spreading for bystander killing (Fig. 1h). Collectively, these results confirm the ability for the T-DXd payload to spread, but reveal this ‘bystander’ cytotoxic MOA to have a modest impact and require a significant presence of HER2-high cells in a heterogenous tumor mixture.
Recent clinical trial results18 have shown comparable T-DXd efficacy in HER2-low and HER2-ultralow BC patients, suggesting that T-DXd may induce tumor killing in the absence of HER2-high cells within the in vivo TME. We hypothesized this anti-tumor efficacy may not depend heavily on HER2-mediated internalization of T-DXd or subsequent payload spreading, particularly in HER2-ultralow BC, which has very limited HER2 expression. Notably, a recent pharmacokinetic study using human xenografts in mice demonstrated that T-DXd effectively delivered its DXd payload to HER2-negative MDA-MB-468 tumors, achieving anti-tumor efficacy comparable to that observed in HER2-low tumor models23. To further explore these findings and test this hypothesis, we evaluated the in vivo efficacy of T-DXd against human BC xenografts with varying HER2 expression, engrafted in SCID mice. As expected, HER2-high KPL4 tumors responded robustly to both ADC treatments (Fig. 1i). Surprisingly, HER2-low MDA-MB-231 and HER2-negative MDA-MB-468 BC both exhibited significant responses to T-DXd, whereas no detectable responses occurred with T-DM1 (Fig. 1j, k). Additional experiments using HER2-negative, non-breast cancer models like K562 (chronic myelogenous leukemia) and SUDHL-10 (B cell lymphoma) also demonstrated significant T-DXd efficacy without T-DM1 effects in these models (Supplementary Fig. 4b, c). Importantly, T-DXd did not demonstrate cytotoxicity against these HER2-low or HER2-negative cell lines in vitro (Supplementary Fig. 1g–j), suggesting that the mechanism of T-DXd payload delivery and tumor killing for these HER2-low/negative tumors may differ between in vivo and in vitro settings. Moreover, there were no significant differences in sensitivity between DXd and DM1 payloads across these cell lines (Supplementary Fig. 3d–g), and HER2 expression levels in these models were confirmed by flow and IHC analysis (Supplementary Figs. 1a and 4a), thus suggesting an alternative MOA for the observed anti-tumor effect.
To assess and compare the treatment efficacy of T-DM1 and T-DXd against HER2-high and HER2-negative tumors within the same animal, we contralaterally implanted HER2-high KPL-4 cells in the right mammary fat pad and HER2-negative MDA-MB-468 cells in the left mammary fat pad (Fig. 1l) before beginning T-DM1 or T-DXd treatment. Following dual implantation, both T-DXd and T-DM1 treatment showed potent anti-tumor efficacy against HER2-high KPL4 tumors in the right fat pad (Fig. 1m). Consistent with the results seen in Fig.1k, T-DXd also exhibited significant anti-tumor activity against contralateral HER2-negative MDA-MB-468 tumors, whereas T-DM1 did not (Fig. 1n). Analysis of isolated tumor cells and tumor sections from these mice confirmed HER2 expression exclusively in KPL-4, with none observed in the HER2-negative tumor models (Supplementary Fig. 4d), affirming T-DXd efficacy is mediated by both HER2-dependent and HER2-independent mechanisms. These results indicated that T-DXd can induce cell death in HER2-negative tumors without neighboring HER2-high cells present in the same TME. These observations suggested that in vivo T-DXd efficacy against HER2-low/negative tumors may be distinct from the purported “bystander killing effect” observed in vitro, which is dependent on T-DXd uptake by HER2-high cells within close proximity (Fig. 1f).
HER2-independent DXd payload release by extracellular Cathepsin L (CTSL) and its expression in breast cancer biospecimens
Our findings indicated that T-DXd in vivo anti-tumor activity may not depend on HER2 recognition and ADC internalization. To demonstrate HER2-binding independence for T-DXd efficacy in HER2-low BC, we conducted a competition experiment using excess trastuzumab to compete against T-DXd binding in the treatment of MDA-MB-231 tumors in vivo. We found that co-administration of a fivefold excess dose of trastuzumab had no impact on T-DXd efficacy in HER2-low tumors (Fig. 2a). This suggested that in HER2-low/negative tumors, T-DXd efficacy did not rely upon HER2 binding, consistent with the lack of observable ADC internalization into these cancer cells (Fig. 1a, b).

a MDA-MB-231 tumors treated with T-DXd (4 mg/kg) were co-injected with fivefold higher dose of trastuzumab (20 mg/kg) to assess T-DXd efficacy after HER2 binding competition. Mixed-effects analysis (REML) with Tukey’s multiple comparisons (n = 10). Arrows indicate treatment administered. b Liquid chromatography-mass spectrometry (LC-MS) quantification of DXd molecules in tumors treated with T-DXd (10 mg/kg) for 3 days. DXd levels per tumor mass were quantified. One-way ANOVA with Tukey’s multiple comparisons test (n = 13). c Systemic DXd payload levels in plasma from same treated animals in (b). DXd levels (ng) per plasma volume (mL) were quantified. No statistical significance between plasma groups was observed (n = 13). d T-DXd was incubated with recombinant human CTSL or CTSB for 6 h in low pH assay buffer as described in Methods. Released DXd levels were quantified by LC-MS. e Cytotoxicity of cleaved T-DXd was assessed on MDA-MB-468 cells. f Immunohistochemistry analysis of primary core needle biopsies from BC patients. Representative HER2 expression (top row) is shown. CTSL expression and localization in the same patient’s biopsy are shown (bottom row). Positive staining in tumor beds (red arrows) and stromal compartments (black arrows) are indicated. g–i Tissue microarray comprising 321 invasive BC, 20 DCIS, and 49 normal breast samples were assessed for CTSL expression via IHC. Pixelwise H-scores were used for quantification by QuPath. One-way ANOVA with Tukey’s multiple comparisons test. g CTSL expression differences in invasive BC, DCIS, and normal breast. h CTSL expression among lymph node-negative, lymph node-positive, or metastatic BC. i CTSL expression among BC with different HER2 IHC scores.
To verify the amount of DXd payload release in vivo, we quantified DXd levels in HER2-positive KPL4 and HER2-negative MDA-MB-468 tumor tissues using liquid chromatography-tandem mass spectrometry (LC-MS). As expected, KPL4 tumors contained the highest levels of released DXd (Fig. 2b), congruent with efficient ADC binding and internalization observed in HER2-high BC cells. However, significant DXd release was also detected in MDA-MB-468 tumors, implying an alternative mechanism of extracellular DXd payload cleavage. To rule out undetectable “ultralow” HER2 expression in MDA-MB-468 tumors contributing to intracellular payload release, we repeated the analysis with engrafted HER2-null murine tumor MM3MG. T-DXd treatments showed comparable DXd concentrations in MM3MG and MDA-MB-468 tumors (Fig. 2b), demonstrating payload release independent of human HER2 expression in a HER2-negative TME. Further analysis of plasma DXd levels in treated animals showed detectable systemic concentrations without differences between engrafted tumor types and in non-tumor-bearing mice (Fig. 2c). Interestingly, median DXd concentration in MDA-MB-468 tumors was 23-fold higher than in the plasma (comparing Fig. 2b, c), suggesting tumor-specific payload release mechanisms in HER2-negative TMEs over systemic release. These findings strongly suggested a potential extracellular payload release mechanism in HER2-low/negative BC, which would account for the clinical efficacy of T-DXd observed in HER2-ultralow BC patients18,19.
T-DXd’s payload release reportedly depends on tumor-specific cathepsin proteases, such as CTSB and CTSL, which can cleave its specialized linker sequence (Gly-Gly-Phe-Gly)11. To examine this cleavage activity, we incubated T-DXd with recombinant human CTSB or CTSL under optimized buffer conditions and duration, previously verified using a fluorescent substrate cleavage assay (Supplementary Fig. 5a, b). Notably, we found that recombinant CTSL at low pH efficiently released the payload from T-DXd (Fig. 2d), with an average of 22,800 ng/mL DXd detected (~90% of input from 1 mg/mL T-DXd). However, recombinant CTSB or low pH alone did not release any significant payloads. Accordingly, CTSL-treated T-DXd promoted cytotoxicity against HER2-low/negative cells in vitro (Fig. 2e and Supplementary Fig. 5c). Neither cathepsins affected T-DM1’s cytotoxicity against HER2-low/negative BC (Supplementary Fig. 5d, e), consistent with T-DM1’s non-cleavable linker. These results establish proof of concept for extracellular payload release mediated by CTSL, promoting T-DXd cytotoxicity against HER2-low/negative BC. Notably, tumor cathepsins are often upregulated with altered localization in many cancer types, and exhibit proteolytic activity both in the endosomes and in the tumor extracellular matrix (ECM)24.
To evaluate the clinical relevance of CTSL expression in breast cancer, we conducted IHC analysis of core needle biopsies from BC patients at Duke Cancer Institute. HER2 expression and localization in tumor beds were first confirmed across biopsies with different HER2 IHC scores (Fig. 2f, left). However, CTSL staining revealed CTSL expression not only in tumor beds but also strongly in stromal compartments of all biopsies, regardless of HER2 status (Fig. 2f, right). Because T-DXd therapy has shown clinical efficacy in metastatic BC patients, we analyzed additional lesions of BC metastasis in the brain, bone, and lymph node and observed similar CTSL expression patterns (Supplementary Fig. 5f). To further validate these findings, we performed CTSL IHC on BC tissue microarrays (TMA) comprising 321 invasive breast cancers, 20 ductal carcinoma in situ (DCIS), and 49 normal breast epithelium specimens (Fig. 2g–i and Supplementary Fig. 6). Quantitative analysis of CTSL expression using Pixelwise H-scores25 revealed significant upregulation of CTSL in invasive BC and DCIS compared to normal breast tissues (Fig. 2g), consistent with a role for CTSL in cancer disease progression24,26. Notably, heterogeneous expression of CTSL was observed across invasive BC specimens, irrespective of disease stage (Fig. 2h) or HER2 IHC status (Fig. 2i). Collectively, these results suggest that in many HER2-low/ultralow BC, the high levels of extracellular CTSL in the TME may be critical in facilitating tumor-targeted drug release to elicit an anti-tumor effect.
Tumor cathepsin L mediates extracellular T-DXd payload release in the tumor microenvironment, contributing to therapeutic efficacy in HER2-low BC
To first document CTSL’s intracellular role in payload release, we treated HER2-high Au565 cells with a CTSL-specific inhibitor ((Rac)-Z-Phe-Phe-FMK), which significantly decreased T-DXd cytotoxicity (Fig. 3a), but not T-DM1 cytotoxicity (Fig. 3b). These data highlight the importance of intracellular CTSL-mediated cleavage of T-DXd in the endosomes. To more specifically confirm the role of CTSL in payload release, we generated Au565 and MDA-MB-231 lines to overexpress CTSL or knock-out CTSL expression using CRISPR lentiviral vectors (Supplementary Fig. 7a). CTSL is initially expressed as an inactive zymogen with an N-terminal propeptide chain blocking its catalytic active cleft, which can be removed by mature CTSL or other cathepsin proteases24,27. To bypass this activation requirement, we expressed the mature form of CTSL (without the propeptide). Consequently, mature CTSL overexpression increased Au565 sensitivity to T-DXd cytotoxicity by 10-fold, while CTSL-KO modestly reduced T-DXd efficacy by about 3-fold in vitro (Fig. 3c). As expected, CTSL expression or KO did not affect T-DM1 cytotoxicity in Au565 cells (Supplementary Fig. 7b). Notably, CTSL modification in MDA-MB-231 cells did not alter T-DXd sensitivity (Supplementary Fig. 7c–e), consistent with the limited ADC internalization by HER2-low BC in vitro (Fig. 1a, b).

a, b Au565 cells were assessed for T-DXd (a) or T-DM1 (b) cytotoxicity in vitro in the presence of CTSL inhibitor (3 µM Z-Phe-Phe-FMK). c Au565 cells overexpressing CTSL or lacking CTSL were assessed for T-DXd cytotoxicity in vitro. d Extracellular CTSL secretion by MDA-MB-231 lines was quantified with ELISA analysis of conditioned media. e Secreted proteins in conditioned media of indicated cell lines were concentrated and quantified for CTSL enzymatic activity in assay buffer. a–e Two-way ANOVA with Tukey’s multiple comparisons test (n = 3 per group). Nonlinear regression curve fit to calculate IC50 values. f Parental and CTSL-overexpressed MDA-MB-231 cells were implanted in SCID mice, and treated with T-DXd (5 mg/kg) or control PBS. g Control-KO and CTSL-KO MDA-MB-231 cells were implanted in SCID mice, and treated with T-DXd (10 mg/kg) or control PBS. f–h Mixed-effects analysis (REML) with Tukey’s multiple comparisons (n = 10). Arrows indicate time of treatments administered. h Tumor sizes (mm3) measured at end point for studies using MDA-MB-231 CTSL-KO lines. i Immunohistochemistry verifications of CTSL expression in parental or CTSL-modified MDA-MB-231 tumors. j LC-MS quantification of DXd in MDA-MB-231 parental (n = 15) versus CTSL-overexpressed tumors (n = 10), treated with T-DXd (10 mg/kg) for 5 days. k LC-MS quantification of DXd in MDA-MB-231 control-KO versus three different CTSL-KO pools, treated with T-DXd (10 mg/kg) for 3 days (n = 10 for all groups, except n = 6 for CTSL-KO #2). h, k One-way ANOVA with Tukey’s multiple comparisons. j Two-sided Mann–Whitney test. All data is presented as mean ± SEM with p values indicated.
CTSL is also known to be secreted and activated in the tumor microenvironment and plays a crucial role in ECM remodeling, cancer progression, invasion, and metastasis24. Given these roles and our observations, we next examined the ability of CTSL to mediate extracellular T-DXd cleavage. We first confirmed extracellular secretion of CTSL by analyzing conditioned media, observing increased CTSL in overexpressed cells, whereas CTSL-KO cells had undetectable levels (Fig. 3d). Surprisingly, we observed limited CTSL activity in conditioned media from CTSL-overexpressing MDA-MB-231 cells (Supplementary Fig. 7f), correlating with a lack of T-DXd cytotoxicity in vitro (Supplementary Fig. 7d). This suggested that secreted CTSL had limited enzymatic activity in culture media. We, therefore, isolated CTSL from their conditioned media and tested proteolytic activity using pH-buffer-adjusted conditions. Indeed, purified and pH-adjusted CTSL from conditioned media of overexpressed cells showed enhanced cleavage activities (Fig. 3e), underscoring the requirement for low pH conditions for CTSL activity, typical in the TME in vivo24.
To evaluate the in vivo role of extracellular CTSL activity on T-DXd therapeutic efficacy in HER2-low BC, we implanted parental and CTSL-overexpressing MDA-MB-231 lines in SCID mice. In contrast to a lack of cytotoxicity in vitro, CTSL-overexpressing MDA-MB-231 tumors showed significantly enhanced T-DXd efficacy in vivo over parental tumors (Fig. 3f), thus underscoring the importance of the TME in eliciting T-DXd efficacy against HER2-low BC in vivo. Conversely, CTSL knockout (KO) significantly decreased T-DXd efficacy in two out of three CTSL-KO MDA-MB-231 lines established for growth in SCID mice (Fig. 3g, h and Supplementary Fig. 7h, i). IHC analysis confirmed the overexpression and knockout of CTSL proteins within the TME (Fig. 3i). To confirm an altered T-DXd payload release by CTSL in HER2-low BC in vivo, we performed LC-MS analysis to quantify tumor DXd levels after therapy. As anticipated, CTSL-overexpressed tumors showed significantly increased DXd levels compared to parental controls (Fig. 3j). Moreover, two out of three CTSL-KO lines (CSTL-KO #1 and #2) demonstrated decreased DXd levels over the control-KO tumor (Fig. 3k), corresponding to their reduced T-DXd sensitivity. Notably, all three CTSL-KO lines had minimal CTSL protein levels, including CTSL-KO #3 (Fig. 3i and Supplementary Fig. 7g), suggesting other TME extracellular proteases may also mediate T-DXd cleavage. Collectively, these studies demonstrate a critical role for CTSL in mediating T-DXd therapeutic efficacy and payload release in HER2-low BC.
T-DXd cytotoxicity induces immunogenic tumor cell death, activating antigen presentation by myeloid immune cells
Having demonstrated differences in payload release mechanisms, we next explored the responses elicited by T-DXd and T-DM1 payload-mediated cell death. While there are many recognized types of regulated cell death, there is emerging evidence that ICD is crucial for instigating tumor antigen-specific immunity and potentially important in the success of anti-cancer therapies28,29. We, therefore, compared the ICD responses of topoisomerase inhibitor DXd and microtubule inhibitor DM1 in our HER2+ breast cancer cell lines. To examine cellular responses independent of the antibody component, we initially evaluated the cytotoxic impact of unconjugated DXd and DM1. Both payloads readily killed Au565 and KPL4 cells at low concentrations with comparable IC50s in Au565 (Supplementary Fig. 3a, b). To evaluate ICD responses, we first quantified extracellular ATP (eATP), a chemoattractant for phagocytes30. We found a significantly higher release of eATP by DXd in comparison to DM1 cytotoxicity (Fig. 4a and Supplementary Fig. 8a). Consistent results were obtained when cancer cells were treated with T-DXd versus T-DM1 (Fig. 4b and Supplementary Fig. 8b). We next examined the release of the Damage-Associated-Molecular-Pattern (DAMP) protein High Mobility Group Box 1 (HMGB1), a chromatin protein that is secreted by tumor cells undergoing ICD, acting as an alarmin to activate innate immunity31. Utilizing HMGB1 immunoassay, we observed DXd induced substantially higher secretion of HMGB1 in comparison to DM1 (Fig. 4c, d). Lastly, we examined calreticulin (CRT), typically confined to the endoplasmic reticulum, and known to translocate to the cellular surface during ICD to activate phagocytes32. Surface CRT levels on dying tumor cells were also significantly increased following DXd-mediated cytotoxicity, compared to DM1 (Fig. 4e and Supplementary Fig. 8c). Collectively, these findings suggest that the DXd payload instigates a more robust ICD signature than DM1.

a, b Au565 cells were treated with DXd, DM1, or HER2-ADCs for 3 days, and extracellular ATP was measured. c, d Au565 and KPL4 cells were treated for 3 days, and extracellular HMGB1 release was quantified by Lumit-Immunoassays normalized to cell viability. e Surface Calreticulin (CRT) on dying tumor cells was measured by flow cytometry on Au565 cells after treatment for 2 days. a–e Two-way ANOVA with Tukey’s multiple comparisons (n = 3). f–j Flow cytometry assessment of human macrophage surface expression of (f) HLA-A2, (g) HLA-DR, (h) CD80, (i) CD40, and (j) CCR7 after 2-days co-culturing with Au565 cells pre-treated with indicated antibodies or ADCs. Macrophage experiments derived from two representative PBMC donors are shown. MFI Mean Fluorescence Intensity. One-way ANOVA with Tukey’s multiple comparisons test (n = 3 per donor). k, l RNA-seq analysis of gene expression by human macrophages after one day co-culture with Au565 cells pre-treated with indicated antibodies/ADCs. Relative gene expression involved in (k) antigen presentation and (l) chemotaxis were assessed. Heat maps represent average gene counts (n = 3 per group) normalized with z-scores. All data is presented as mean ± SEM with p values.
Secretion of eATP and HMGB1 and induction of surface CRT indicates the potential for recruitment and activation of immune phagocytes like macrophages and dendritic cells (DC). Therefore, we comprehensively analyzed immune activation markers on human monocyte-derived macrophages after co-culturing with ADC-treated Au565 cells. Critically, we found that terminally differentiated macrophages, unlike proliferating cancer cells, are not susceptible to T-DXd or payload cytotoxicity in vitro (Supplementary Fig. 8d, e). However, examination of co-cultured macrophages derived from multiple PBMC donors revealed that stimulation by T-DXd-treated Au565 cells upregulated multiple antigen-presentation surface markers on the macrophages (Fig. 4f–j and Supplementary Fig. 9a), including MHC molecules (HLA-A2, HLA-DR) and co-stimulatory molecules (CD80, CD40, and CCR7). A similar activation profile was observed on co-cultured human DCs and mouse bone marrow-derived macrophages (BMDM) (Supplementary Fig. 9b, c). To assess innate immune induction more broadly, we performed RNA-seq analysis of co-cultured macrophages that revealed significant upregulation of antigen presentation genes (Fig. 4k) and chemokine genes (Fig. 4l) by both HER2 ADC treatments, with T-DXd showing a more potent effect than T-DM1, despite similar tumor cell cytotoxicity. We further found that T-DXd and T-DM1 both upregulated the expression of various inflammatory cytokines over trastuzumab, suggesting the potent impact of payload-mediated immune activation (Supplementary Fig. 9d). Overall, these results imply that T-DXd cytotoxicity of BC cells activates nearby myeloid antigen-presenting cells (APCs), potentially priming them for tumor antigen presentation, while simultaneously enhancing chemokine and cytokine secretion to impact the activation of tumor antigen-specific adaptive immunity.
T-DXd activates FCGRs and promotes antibody-dependent cellular phagocytosis, enabling tumor antigen-specific activation of CD8 T cells
We next investigated antibody-mediated immune activation by HER2-ADCs through FCGRs. Our research and that of others have illustrated trastuzumab’s capability to robustly engage with these FCGRs, facilitating tumor antibody-dependent cellular phagocytosis (ADCP) by macrophages4,33,34. Notably, mouse FCGR4, the ortholog of human activating FCGR3A, exhibits comparable affinity to the human IgG1 antibody Fc-region, enabling macrophage-mediated Fc-effector functions such as ADCP35,36,37. To determine whether T-DXd and T-DM1 could similarly activate these FCGRs, we employed an established Jurkat-FCGR NFAT-luciferase reporter assay4. Upon binding to HER2 on tumor cells, both ADCs activated human FCGR3A and mouse FCGR4, albeit slightly less than trastuzumab, but significantly more than control antibody (Fig. 5a, b). This indicates that the conjugation of payloads to trastuzumab did not significantly interfere with its FCGR engagement. Building on previous findings of trastuzumab cross-reactivity with mouse FCGR44, our data confirmed and extended such cross-reactivity to trastuzumab-based ADCs. To further investigate FCGR-mediated anti-tumor mechanisms, we established a co-culture system to assess ADCP, involving fluorescently-labeled Au565 target cells with macrophage effector cells4. Using human and mouse macrophages as effector cells, we observed that both T-DXd and T-DM1 effectively promoted tumor ADCP, comparable to trastuzumab (Fig. 5c–e). This finding indicates that the conjugated payloads of these HER2-ADCs do not impair or enhance the anti-tumor mechanism inherent to the parental antibody. Instead, they retain the ability to interact with human and mouse FCGR-effector cells, thereby facilitating ADCP.

a, b KPL4 cells treated with indicated antibodies/ADCs and co-cultured for 4 h with Jurkat effector cells expressing (a) human FCGR3A or (b) mouse FCGR4. FCGR signaling to lead to activation of NFAT-luciferase promoter-reporter in effector cells was assessed. c Representative flow graph demonstrating phagocytosis of Au565 cells by human monocyte-derived-macrophages upon treatment with indicated antibodies/ADCs (1 µg/mL) for 4 h. d, e Quantification of ADCP is mediated by (d) human macrophages or (e) mouse Bone-marrow-derived-macrophages (BMDMs). The percentage of macrophages containing tumor-label dye was plotted. f–i Tumor antigen (eGFP) presentation to Just-EGFP-Death-Inducing (JEDI) CD8+ T cells by mouse BMDM. Au565 expressing eGFP were treated overnight with indicated antibodies, ADCs, or unconjugated payloads, then co-cultured with BMDMs for two days. CD8 + JEDI T cells were added to BMDM for 3 days to allow antigen presentation, T cell activation, and proliferation. f Representative flow graphs visualizing proliferating T cells, defined as Cell-TraceLow populations. g Summary of T cell proliferation. h Representative flow graphs of JEDI T cell activation assessment by surface CD44 expression. i Summary of CD44 expression on JEDI T cells. a, b Two-way ANOVA with Tukey’s multiple comparisons test. (n = 3). d, e, g, i One-way ANOVA with Dunnett’s multiple comparisons test (n = 3). All data plotted in this figure are presented as mean ± SEM with p values.
Our observation of DXd-induced ICD and myeloid APC activation, coupled with the ability of the antibody component to elicit ADCP, suggested potential enhancement of tumor-specific adaptive T cell responses by T-DXd treatment. To formally test this hypothesis, we employed a T cell activation model using eGFP-specific JEDI (Just-eGFP-Death-Inducing) CD8+ T cells38, co-cultured with BALB/c BMDMs to allow for T cell expansion (demonstrated in pilot experiments using eGFP peptide, Supplementary Fig. 10a). In this model, HER2-positive Au565 cells expressing eGFP were co-cultured with BMDMs, along with ADCs, mAbs, or unconjugated payloads DXd and DM1. Following co-culture and activation of macrophages, JEDI T cells were added to assess tumor-eGFP antigen presentation and activation. Our experiment revealed that T-DXd treatment led to higher activation of JEDI T cells compared to T-DM1 and trastuzumab, evidenced T-cell proliferation (Fig. 5f, g) CD44 expression (Fig. 5h, i). Specifically, T-DXd treatment resulted in 10-fold higher T-cell proliferations over rituximab, and up to threefold higher over T-DM1 and trastuzumab. Similarly, T-DXd treatment resulted in 2–3-fold higher CD44 expression over rituximab, T-DM1, or trastuzumab. Importantly, while unconjugated DXd alone induced tumor ICD (Fig. 4a, c–e), its efficacy in T cell activation required the presence of an active anti-HER2 antibody Fc region, emphasizing the importance of both components of T-DXd in facilitating immune activation, tumor antigen phagocytosis and presentation to antigen-specific T cells. A graphical summary of these findings is provided in Supplementary Fig. 10b.
T-DXd mediated immune activation depends on STING/IFN-I and TLR4 signaling pathways
Having demonstrated T-DXd induction of ICD and adaptive immunity, we next investigated the innate immune pathways underlying this response. Prior research suggested the importance of the cGAS/STING pathway in topoisomerase inhibitor-mediated immune activation through type-1 interferon signaling39. However, HMGB1 release (Fig. 4c, d) mediated by DXd cytotoxicity suggested Toll-Like-Receptor-4 (TLR4) pathway activation31. Indeed, RNA-seq data from macrophage co-cultures revealed upregulation of both TLR4 pathway genes and interferon-stimulated genes in the T-DXd treated group (Fig. 6a, b).

a, b RNA-seq analysis of human macrophages co-cultured with treated Au565 cells. Relative gene expression in TLR4 signaling pathway and interferon-stimulated genes were analyzed. Heat maps show average gene counts (n = 3 per group) normalized with z-scores. c–g BMDM generated from wild-type, TLR4-mutant or STING-KO mice and co-cultured for 2 days with Au565 cells with indicated treatments. BMDM surface expression of c MHC-II, d CD40, e CD80, and f CD86 were assessed. g IL-6 secretion by BMDM assessed with ELISA. h, i Au565 cells treated with indicated agents were co-cultured with plasmacytoid dendritic cells (pDC) derived from WT or STING-KO mice. IFNα1 secretion by pDCs was assessed by ELISA. c–i Two-way ANOVA with Tukey’s multiple comparisons test was performed between indicated treatment groups or BMDM genotypes (n = 3). j–l Tumor antigen (eGFP) presentation to JEDI CD8+ T cells by wild type (WT), TLR4-mutant or STING-KO BMDM after co-culture with T-DXd treated Au565-eGFP cells. j Representative JEDI T cells proliferation plot shown for each BMDM co-cultured. k Summary of JEDI T cells proliferation (defined as Cell-TraceLow) and l activation (CD44+) after co-culture with WT, TLR4-mutant and STING-KO BMDMs. Two-way ANOVA with Šidák’s multiple comparisons test. (n = 3). All data is presented as mean ± SEM with p values. n.s. not significant.
To explore the importance of TLR4 and STING pathways in T-DXd-induced anti-tumor immune activation, we generated BMDM from C3H/HeJ mice (bearing a dominant-negative Tlr4 gene mutation)40 and STING-KO mice (with a deletion of Sting exon 3). Repeating our co-culture experiment of HER2 + BC with macrophages using TLR4-mutant and STING-KO BMDMs revealed that both pathways were necessary for full immune activation of macrophages induced by T-DXd cytotoxicity, as deficiencies in either gene prevented upregulation of MHC-II and CD40 (Fig. 6c, d). Other co-stimulatory molecules, such as CD80 and CD86, remained inducible but at significantly lower levels compared to wild-type (WT) macrophages (Fig. 6e, f). Notably, T-DXd stimulated IL-6 secretion was completely TLR4-dependent (Fig. 6g), while IFNα production by co-cultured plasmacytoid dendritic cells (pDCs) depended on STING (Fig. 6h, i), highlighting the distinct roles of these pathways.
To assess the significance of these pathways in T-DXd-promoted tumor antigen presentation to adaptive T cells, we utilized WT, TLR4-mutant, and STING-KO BMDMs as APCs in our co-culture assay with eGFP+/HER2+Au565 cells and JEDI CD8+ T cells. As expected, JEDI T cell activation after T-DXd treatment was significantly diminished in both TLR4-mutant and STING-KO APCs compared to WT APCs. Notably, JEDI T cell proliferation was strongly impaired by the knockout of either pathway in APCs (Fig. 6j, k), whereas JEDI T cell activation, measured by CD44 expression, was more TLR4-dependent (Fig. 6l). Overall, these studies reveal that ability of T-DXd induced ICD stimulates innate immune cells through both the TLR4 and STING pathways, which are essential for their ability to promote tumor-specific adaptive immunity.
T-DXd-mediated immune activation is restricted by tumor CD47 expression
Our past studies of trastuzumab revealed its dependence on ADCP for therapeutic efficacy, which can be intensified by CD47 blockade4, a finding that has since been confirmed by other researchers33,41. This led us to explore if targeting the CD47/SIRPα checkpoint could enhance T-DXd potency by augmenting ADCP to elicit innate effector function and subsequent expansion of tumor-specific CD8+ T cells. As previous studies documented increased tumor CD47 expression following radiotherapy-induced DNA damage42, we investigated if DXd cytotoxicity similarly increased tumor CD47. Interestingly, we found that DXd treatment raised surface CD47 levels in HER2 + BC cells, while DM1 decreased CD47 expression (Fig. 7a). Gene expression analysis also showed CD47 upregulation post-T-DXd cytotoxicity (Supplementary Fig. 8a), suggesting that T-DXd could trigger tumor CD47 expression, potentially hindering ADCP and tumor antigen uptake over time.

a Flow cytometry assessment of surface CD47 expression on Au565 cells after 2 days of treatment with DXd or DM1. b ADCP assessment of parental or CD47-KO KPL4 cells by human macrophages after 4 h co-culture. The percentage of macrophages containing fluorescent-labeled KPL4 was assessed by flow cytometry. c–i Human macrophages co-culture with indicated KPL4 target cells and treatments for 2 days. Macrophage surface expression of c HLA-A2, d HLA-DR, e CD80, and f CD40 were assessed by flow cytometry. Inflammatory cytokines g TNFα, h IL-6, and chemokine (i) CCL4 secreted by macrophages assessed by ELISA. n Tumor antigen (eGFP) presentation to JEDI CD8 T cells using BMDM co-cultured with treated KPL4-eGFP or KPL4-eGFP-CD47-KO cells. Representative flow graphs for JEDI T-cell proliferation assessment by Cell-Trace (j) or T-cell activation assessment by CD44 staining (l) are shown for each treatment condition. k JEDI T-cell proliferation summary and m CD44 expression summary. a–m Two-way ANOVA with Tukey’s or Šidák’s multiple comparisons test (n = 3 per group in all experiments). Data presented as mean ± SEM with p values. MFI mean fluorescence intensity.
To assess CD47 suppression of T-DXd ADCP, we generated CD47-KO HER2 + KPL4 cells (Supplementary Fig. 8b). Consistent with our previous findings with trastuzumab4, CD47-KO rendered KPL4 cells more susceptible to macrophage-mediated ADCP upon T-DXd treatment (Fig. 7b). Additionally, loss of tumor CD47 significantly boosted T-DXd-mediated immune activation, manifested by elevated expression of HLA molecules, CD80 and CD40 on co-cultured macrophages (Fig. 7c–f), and increased secretion of inflammatory cytokines and chemokines (Fig. 7g–i). Thus, these studies underscore that the conjugation of trastuzumab with DXd enables stimulation of tumor cell ADCP along with immune activation of APCs, which are both in turn enhanced by CD47 blockade.
To determine if these observations extend to stronger antigen presentation and activation of tumor-specific T cells, we repeated our JEDI T cells and BMDM co-culture experiment using KPL4 or KPL4-CD47-KO target cells expressing eGFP (Fig. 7j). Notably, the magnitude of tumor-specific JEDI T cell activation by T-DXd was modest with the treatment of parental tumor cells (Fig. 7j, top), congruent with our earlier observations (Figs. 5f–i, and 6j, k). Strikingly, we observed that T-DXd treatment of CD47-KO KPL4 resulted in six-fold higher JEDI T cell proliferation over treatment of parental tumors (29.6% vs 5.3%, Fig. 7j, k). Similarly, T-DXd-treated CD47-KO tumors significantly activated JEDI T cells as determined by CD44 expression (Fig. 7l, m). These observations collectively demonstrate that T-DXd-mediated innate and adaptive immune activation is markedly restricted by tumor CD47 expression.
CD47 blockade enhances T-DXd anti-tumor efficacy in vivo
Having demonstrated the impact of CD47 blockade on T-DXd in vitro, we next assessed the therapeutic potential of T-DXd combined with CD47 blockade in vivo. For these studies, we utilized an immune-competent HER2Δ16 transgenic mouse model previously employed in anti-HER2 mAb studies4,43. Using doxycycline chow, we induced HER2 oncogenic signaling in mammary tissues (Supplementary Fig. 12a) to establish HER2 + BC. Once tumors were palpable, mice were randomized to trastuzumab or T-DXd treatment arms. To address potential mouse anti-human-IgG1 responses as a confounding factor in this immunocompetent model, we included a non-binding human IgG1 antibody (rituximab) as a control for comparison. Initial comparisons (Supplementary Fig. 12b–e) revealed that trastuzumab treatment delayed tumor growth compared to this control, reaffirming the anti-HER2 effect of trastuzumab previously demonstrated in this model4. However, trastuzumab did not achieve complete regression in any animal. In contrast, T-DXd exhibited significantly greater efficacy, with over two-thirds of the mice showing pronounced tumor suppression and extended survival. This highlighted the potency of DXd payload cytotoxicity against an endogenous HER2-positive mammary tumor, prompting us to investigate its potential impact on anti-tumor adaptive immunity. IFNγ-ELISPOT assessment of splenic T cells revealed a modest increase of HER2-specific T cell responses in T-DXd treated mice compared to control IgG1 or trastuzumab-treated groups (Supplementary Fig. 12f). Additionally, serum analysis for anti-HER2 humoral immune responses showed significantly increased anti-HER2 mouse IgG levels in T-DXd treated mice (Supplementary Fig. 12g), indicating T-DXd’s ability to break HER2 tolerance and promote tumor-specific adaptive immunity.
We next investigated whether T-DXd anti-tumor efficacy could be enhanced by CD47 blockade. Given the strong efficacy of T-DXd in the preliminary experiments (Supplementary Fig. 12d, e), we utilized a delayed treatment strategy to treat larger tumors (tumor volume ~500 mm3), and we observed that while T-DXd monotherapy prolonged survival, it resulted in limited complete tumor regression. Congruent with our past studies using this transgenic model, we found limited efficacy by CD47 blockade therapy alone4. Intriguingly, combining T-DXd with the anti-CD47 blocking antibody significantly increased treatment efficacy, producing complete regression and survival in 60% of treated mice, in contrast to T-DXd or anti-CD47 monotherapy (Fig. 8a and Supplementary Fig. 12h–k). Combinatio`n therapy also elicited more effective endogenous humoral immune responses, evidenced by significantly enhanced anti-HER2 IgG responses in the serum (Fig. 8b). After one week of treatment (Supplementary Fig. 13a), the combination treatment significantly increased tumor phagocytosis, CD40 and MHC-II expression on tumor-associated macrophages over control IgG1 treatment, while the T-DXd monotherapy only significantly enhanced phagocytosis (Fig. 8d–f). Similar effects were seen on tumor-infiltrating DCs (Supplementary Fig. 13b). Moreover, at four weeks post-treatment, combination therapy significantly promoted CD8+ T cell infiltration into the tumor (Fig. 8g), accompanied by elevated levels of cytotoxic (CD107a+) and effector (CD44+/CD62L−) CD8+ T cells over the control groups, while suppressing levels of naive (CD44-/CD62L+) CD8+ T cells (Fig. 8h–j). Similar trends were detected in infiltrating CD4+ T cells, although these increases were not statistically significant (Supplementary Fig. 13c). Spatial analysis upon immunohistochemistry of tumor sections revealed that combination treatment resulted in significantly higher CD8+ T cells infiltration into the tumor bed, whereas T cells were restricted to the stromal compartment in control and T-DXd monotherapy groups (Fig. 8k, l and Supplementary Fig. 14a–c). These findings demonstrate the anti-tumor potential of combining T-DXd with CD47 blockade, enhancing tumor destruction and augmenting innate and adaptive immune responses, allowing for increased infiltration and activation of myeloid cells and CD8+ T cells.

a Tumor-bearing (>500 mm3) HER2-transgenic mice were treated weekly for the first four weeks with control IgG1 antibody (rituximab 10 mg/kg), T-DXd (5.4 mg/kg), anti-CD47 (see Methods) or in combination. Animal survival is plotted. Log-rank (Mantel-Cox) test between T-DXd monotherapy versus combination therapy (n = 15 per group). b Anti-tumor humoral responses in serum from mice in (a) were assessed by ELISA quantification of mouse IgG against HER2. Two-way ANOVA with Tukey’s multiple comparisons (n = 15). c–f Intratumor macrophages (live CD45+/CD11b+/Gr1−/F/480+) analysis after 1 week of treatment with control IgG1 (n = 8), T-DXd (n = 7) or T-DXd plus anti-CD47 (n = 6). c Macrophage percentage, (d) phagocytosis of GFP+ tumor cells, (e) CD40 expression and (f) MHC-II expression were assessed by flow cytometry. g–j Tumor-infiltrating CD8+ T cells (live CD45+/CD11b-/CD8+) analysis after 4 weeks of treatment with control IgG1 (n = 8), T-DXd (n = 12) or T-DXd plus anti-CD47 (n = 6). g Percentage of infiltrated CD8+ T cells. h CD107a cytotoxic marker expression. i naive CD8 T cells (CD62L+/CD44−). j Activated effector CD8 T cells (CD62L−/CD44+). k Immunohistochemistry assessment of tumor-infiltrating T cells levels and spatial location. CD8+ (brown), CD4+ (blue) and FoxP3+ (red), intratumor T cells (black arrows), T cells in stroma (green arrows) are shown for representative sections for each group. l Ratio of quantified CD8+ counts in tumor bed versus stroma in (k) (n = 5 per group). c–l One-way ANOVA with Tukey’s multiple comparisons test. m HER2-transgenic mice with moderate tumor sizes (>150 mm3) were treated weekly with T-DXd (10 mg/kg) and anti-CD47. Complete tumor regressors were randomly assigned to CD8 depletion group or control depletion group. T-DXd and anti-CD47 treatments were halted, and mouse CD8 depletion antibodies (10 mg/kg) or PBS control were administered weekly. Tumor regression after combination therapy and relapse after CD8 depletion were monitored over time. Each line represents tumor growth from one animal subject. n Percentage of tumor-free animals in control versus CD8 depleted groups in (m). Log-rank (Mantel-Cox) test (n = 12). All data is presented as mean ± SEM with p values indicated.
While the delayed kinetics of adaptive stimulation suggest that rapid tumor regressions are primarily the product of payload cytotoxicity, our observations suggested the potential of adaptive immune memory in preventing tumor relapse and being critical for long-term survival after treatment cessation. To explore this, we treated tumor-bearing transgenic mice with T-DXd + anti-CD47 for up to 5 weeks (Fig. 8m). Animals that responded with complete tumor regression were divided into control and CD8+ T cells depletion groups, then subjected to weekly injections of control or a CD8 depletion antibody (preliminary validation of CD8 depletion efficiency shown in Supplementary Fig. 15). These studies revealed CD8 depletion of complete regressors resulted in a significantly higher and faster tumor relapse rate compared to the control group (75% vs. 33% relapse rate) (Fig. 8m, n), demonstrating the impact of CD8+ T cell-mediated immune memory post cessation of T-DXd + anti-CD47 combination treatment. Taken together, with observed immune infiltration and activation in the TME, our study demonstrates a critical role for T-DXd induction of anti-tumor immunity, which can be significantly augmented by blockade of the CD47-SIRPα axis.
Discussion
Trastuzumab deruxtecan (T-DXd), also known as fam-trastuzumab deruxtecan-nxki, has emerged as a groundbreaking ADC with demonstrated clinical benefit across a variety of different HER2+ cancers, including HER2-low BC (encompassing ~60% of all BCs)16,17. Most notably, T-DXd outperformed chemotherapy in BC patient cohorts with limited detectable HER2 expression (IHC0 with incomplete and faint membrane staining in >0 but ≤10% of tumor cells), recently termed as “HER2 ultralow”. This effect was first reported in the phase-II DAISY trial19 in 2023, and later confirmed in the larger phase-III DESTINY-Breast06 trial18. Furthermore, other clinical findings revealed enduring T-DXd-mediated clinical benefit across a spectrum of solid tumors displaying HER2-high expression10, resulting in its landmark approval as the first tissue-agnostic ADC in the US. Interestingly, similar trastuzumab-based ADC (T-DM1) has not yielded similar clinical responses in past and comparison trials. Unlike T-DM1, T-DXd utilizes an enzyme-cleavable linker to connect trastuzumab with Deruxtecan (DXd), a topoisomerase-I inhibitor. This cleavable linker is thought to be active in endosomes and allows for the cleavage and diffusion of DXd to adjacent tumor cells as the purported MOA for T-DXd20. However, this MOA alone does not fully account for T-DXd’s clinical efficacy in HER2-low/ultralow BC16,19, nor the more potent impact in multiple HER2-high solid cancers in comparison to T-DM17,10. Consequently, unraveling the molecular mechanisms governing T-DXd’s efficacy in distinct HER2-expressing cancers is essential for improving its clinical performance and expanding the application of ADCs against other targets.
To understand the MOA of T-DXd, we first assessed its impact against various HER2-negative and HER2-low tumor xenograft models, as well as payload release in HER2-negative TMEs. Notably, we observed limited T-DXd internalization by HER2-low and HER2-negative BC cells, but profound anti-tumor efficacy against these cancers in vivo (Fig. 1), with detectable DXd payload release in HER2-negative tumors. These surprising findings align with a recent pharmacokinetics and pharmacodynamics study of clinical grade T-DXd in HER2-low and HER2-negative tumor xenografts23. Collectively, these observations suggested the potential importance of extracellular tumor-specific proteases, such as cysteine cathepsins, in facilitating T-DXd payload release outside of tumor cells within HER2-low/negative TMEs. Cysteine cathepsins are frequently dysregulated and exhibit heightened activity within the tumor ECM, playing a pivotal role in cancer progression, invasion, and metastasis24,27. In breast cancer patients, overexpression of CTSL and CTSB correlates with poor prognosis for relapse and OS26. Intriguingly, our investigation revealed that CTSL, but not CTSB, promotes extracellular cleavage of T-DXd, enhancing cytotoxicity on HER2-low/negative xenografts (Figs. 2–3). Critically, our analysis of BC biospecimens revealed elevated CTSL levels in both tumor and stromal compartments, with increased CTSL protein expression in invasive BC relative to benign breast epithelial tissue—regardless of HER2 status (Fig. 2). Functionally, we demonstrate that tumor CTSL expression results in extracellular CTSL secretion and proteolytic activity at low pH, consequently bolstering T-DXd efficacy and payload release in HER2-low tumors in vivo. Conversely, CTSL knockout diminishes, but does not entirely abolish, T–DXd–mediated tumor suppression and DXd release, indicating that other ECM proteases may also participate in T-DXd cleavage. Additionally, linker instability reported in other ADCs44 could lead to premature payload release. Our data suggest T-DXd’s extracellular linker sensitivity to CTSL confers a tumor-targeted drug-delivery advantage over chemotherapy, even in HER2-low and ultralow tumors, where limited ADC internalization occurs. Together, these findings provide a plausible explanation for T-DXd’s improved clinical efficacy over chemotherapy in HER2-ultralow BC cohorts from the DESTINY-Breast06 trial18. This has critical implications for clinical trials using this linker-DXd combination in other ADCs45,46, as tumor CTSL expression and other ECM proteases might be vital efficacy determinants and serve as potential predictive biomarkers or companion diagnostics.
While previous studies have documented “bystander killing” of HER2-negative cells adjacent to HER2-positive cells, attributed to DXd membrane-permeability and subsequent “payload spreading” in HER2 heterogenous tumors or co-cultures20—which we have confirmed occur at a modest albeit significant level (Fig. 1d–f) — our findings propose a distinct mechanism in HER2-low/ultralow tumors. In these instances, limited HER2 expression throughout the TME restricts T-DXd uptake and intracellular payload release, restricting conventional bystander killing. This limitation makes extracellular payload release, facilitated by proteolytic activities within the ECM, a likely dominant contributor to the efficacy of T-DXd seen in tumors with minimal HER2 expression. This mechanism may help T-DXd overcome resistance seen in other ADCs like T-DM1, which rely on high HER2 expression and intracellular ADC trafficking12. Moving forward, both ECM protease activity and HER2 expression may be necessary to fully define T-DXd’s treatment spectrum in HER2-low settings. Finally, toxicities associated with T-DXd dosing, such as interstitial lung disease and pneumonitis47, may also hinge on CTSL or other cathepsins, warranting further investigations of their expression and extracellular functions in lungs or other organs of T-DXd treated patients.
In the second part of this study, we highlight the superior immunogenic cell death (ICD) response induced by T-DXd compared to T-DM1. While T-DXd’s higher DAR may partly explain its potency, prior evidence suggests that topoisomerase inhibition can cause extensive DNA damage, eliciting potent ICD responses known to instigate tumor antigen-specific immunity and influence long-term therapeutic outcomes28,29. Specifically, our data reveal that DXd, a potent topoisomerase inhibitor, triggers the release of danger-associated-molecular patterns such as eATP and HMGB1, accompanied by increased surface expression of CRT. These molecules play pivotal roles in recruiting immune phagocytes, activating innate immunity through Toll-like receptors, and facilitating phagocytic uptake of dying tumor cells, respectively30,31,32. These responses trigger immune activation of nearby APCs, including macrophages and dendritic cells (DCs), leading to upregulation of HLA molecules, co-stimulatory markers (e.g., CD40, CD80, CD86), chemokines, and cytokines (Fig. 4). Additionally, the trastuzumab component of T-DXd binds Fcγ-receptors on effector cells, enabling ADCP of tumor antigens (Fig. 5). The DXd-mediated cytotoxicity further amplifies this immune activation through ICD. Together, these two distinct features of T-DXd significantly improved the activation and expansion of tumor antigen-specific CD8+ T cells, surpassing the effects observed with trastuzumab, T-DM1, or DXd alone, as demonstrated in our JEDI T cell co-culture studies. Mechanistically, we show that TLR4 and STING/IFN-I pathways in APCs (Fig. 6) are important mediators of T-DXd’s antitumor immune response. While the role of ICD induction in clinical settings remains to be fully determined, our study suggests this as a critical difference between T-DXd and T-DM1, warranting future study. Our findings imply that T-DXd may function like an in situ cancer vaccine, potentially explaining its broad success in various HER2-high solid tumors10.
Finally, we explored the potential of combining T-DXd therapy and CD47 checkpoint blockade. Previous studies, including ours, have elucidated the pivotal role of trastuzumab in promoting tumor ADCP, significantly enhanced by CD47 checkpoint blockade4, an effect corroborated by other researchers33,41. CD47, frequently upregulated on solid tumor cells, signals “don’t eat me” through SIRPα, inhibiting phagocytosis and potentially suppressing APC-mediated immune activation22. Notably, while DXd-induced toxicity increases surface CRT (Fig. 4e), a pro-phagocytic signal, it also upregulates CD47 (Fig. 7a and Supplementary Fig. 11a), which could counteract ADCP of dying tumor cells. Our findings align with previous reports on radiation-induced tumor CD47 expression42, suggesting potential therapeutic benefits of CD47 blockade with DNA-damaging agents such as T-DXd. Through CRISPR-engineered CD47 knockout experiments, we demonstrate robust enhancements between tumor CD47 knockout and T-DXd-mediated ADCP and macrophage immune activation, leading to robustly enhanced tumor eGFP antigen presentation and JEDI T cell activation (Fig. 7). In a HER2Δ16 transgenic BC mouse model—previously noted for its limited response to CD47 blockade alone4 —treatment with anti-CD47 plus T-DXd significantly slowed tumor progression and improved survival relative to either therapy alone (Fig. 8). This combination therapy also boosted tumor-specific humoral responses, as well as local innate and adaptive immune responses within the TME. Notably, the HER2Δ16 oncogene confers an immune-cold TME with sparse immune infiltrates48, yet we observed that T-DXd plus CD47 inhibition considerably augmented tumor-infiltrating CD8+ T cells, enhancing their infiltration into the tumor bed. Importantly, the depletion of CD8+ T cells revealed the essential role of memory CD8+ T cells in preventing cancer recurrence after the cessation of ADC treatment (Fig. 8m, n)—a phenomenon that may mirror prolonged survival in patients following transient ADC regimen. These studies suggest the potential impact of CD47/SIRPα blockade to enhance T-DXd efficacy in HER2-high BC and offer insight into strategies to limit the duration of T-DXd treatment. CD47 blockade is currently being explored in clinical trials (NCT05765851 and NCT05868226), although careful evaluation of toxicity is likely to be required for this combination, which could vary between different cancers22,47. Notably, ADCP enhancement through CD47 blockade requires sufficient expression of HER2 targets for immune recognition by Fc-effector cells22,34, suggesting alternative combination strategies would likely be needed to improve T-DXd efficacy in HER2-low/ultralow BC.
In conclusion, our study elucidates multiple pivotal anti-tumor MOAs for T-DXd that cooperate to allow for superior anti-tumor efficacy. High levels of CTSL expression and secretion in invasive BC permits extracellular proteolytic cleavage that allows for targeted DXd payload release independently of HER2 binding and internalization. Additionally, our study has revealed that the DXd payload induces robust immunogenic cell death, which activates nearby myeloid sentinel immune cells through TLR4 and STING pathways. Moreover, the antibody component of T-DXd elicits ADCP via FcγR engagement. These mechanisms cooperate to enhance immune activation and stimulate tumor-specific CD8+ T cells against HER2-high BC. Furthermore, CD47 blockade strongly enhanced ADCP and these adaptive responses, which translated into enhanced anti-tumor efficacy in an endogenous model of non-immunogenic HER2 + BC. These findings suggest that strategies to augment ADC-induced immunity may be clinically impactful, and highlight other potential immune-mediated T-DXd activities, (e.g. complement activation with pertuzumab49). Overall, our results underscore that fully capitalizing on ADCs may require a broader understanding of their payload release mechanisms and immunostimulatory capacities, alongside ongoing efforts to optimize tissue penetration and dosing strategies50.
Methods
Study approval
All animals were maintained and bred in accordance with Duke Institutional Animal Care and Use Committee–approved protocol (A198-18-08), and supervised by Division of Laboratory Animal Resources (DLAR). All studies utilizing human breast cancer biospecimens are approved by Duke Health Institutional Review Board under protocol ID 00116330.
Therapeutic antibodies, antibody-drug-conjugates, and unconjugated payloads
Clinical Grade T-DXd (Enhertu), T-DM1 (Kadcyla), trastuzumab (Herceptin) and rituximab (Rituxan) were obtained from Duke Medical Center. Unconjugated payloads DXd (HY-13631D) and DM1 (HY-19792) were purchased from MedChemExpress. Anti-CD47 antibody (clone MIAP410, #BE0283), and anti-CD8b depletion antibody (clone Lyt3.2, #BE0223) were purchased from BioXcell.
Cell lines and genetic modifications strategies
Lentiviral transductions of CRISPR-Cas9 vectors were used to knockout human CTSL (gRNA: CTTTGTGGACATCCCTAAGC) and CD47 (gRNA: ATCGAGCTAAAATATCGTGT). When indicated, a control knockout (empty gRNA) was generated in parallel. We utilized the CRISPR-Cas9 construct Lenti-CRISPR_V2 ordered from Genscript containing puromycin selection. The overexpression vector of human CTSL, human HER2, HER2Δ16-TM, or EGFP was generated by synthesizing the target genes and cloning it into pENTR1a and then using L/R close to generate expression lentiviruses (pLenti-CMV-Puro or pLenti-CMV-Hygro), and cells were selected using puromycin or hygromycin. Mature CTSL gene was generated from full-length cathepsin sequence with deletion of the N-terminal propeptide chain (amino acid 18–113).
In vitro ADC and payload cytotoxicity assays
Various cancer cell lines were incubated in 96-well culture plate (2000–5000 cells/well) containing titrated ADCs, antibodies, or unconjugated payloads DXd and DM1. Treated cells were cultured for 5 days or until control groups reached reasonable cell confluency, and cell viability was assessed by total cellular ATP quantification using CellTiter-Glo Luminescent Cell Viability Assay (PROMEGA, #G7571) following manufacturer’s protocol. When indicated, 3 µM of CTSL inhibitor Z-Phe-Phe-FMK (MCE, #HY-115733) or 3 µM of chloroquine (MCE, #HY-17589A) were added before ADC treatment.
ADC internalization and endocytosis assessment
One mg of Rituximab and T-DXd were labeled with pH-RHODO using Deep Red pHrodo Antibody Labeling Kit (Thermo, P35356) following manufacturer’s protocol. Labeled ADCs were added to various HER2-expressing cancer cells. 6 to 24 h after incubation, cells were harvested and analyzed by flow cytometry for internalization of the labeled antibody/ADC.
Bystander killing analysis
HER2-positive cell lines Au565 or CAPAN-1 were labeled with Vybrant DiD dye (5000× dilution), and HER2-negative cell line MDA-MB-468 was labeled with CellTrace-Violet (2000× dilution). Both labelings were performed with 1 million cells per mL volume at 37 °C for 20 min. Labeled cells were washed twice with PBS, and co-cultured together at a 1:2 ratio of HER2-positive cells versus HER2-negative cells (300,000 total cells in 12-well plates). Cells were treated with ADCs or antibodies (100–1000 ng/mL), and apoptosis of HER2+ and HER2- cells were analyzed on day 2 and day 6 post-treatment by AnnexinV/PI staining (Biolegend 640914). Mono-culture controls labeled Au565, CAPAN-1, and MDA-MB-468 were also performed.
Liquid chromatography–tandem-mass spectrometry (LC/MS/MS) assays for measurement of DXd
The DXd concentration in plasma and tumor tissue was measured by LC/MS/MS assays optimized for the required sensitivity/concentration range and the nature/available quantity of the sample. The pure materials, DXd (>98%) and DXd-d5 (>98.5%) were purchased from GLP-Bio and used as calibration standards and internal standards, respectively. Agilent 1100/1200 LC, AB/Sciex API-5500 QTrap MS/MS instrument, and Analyst® version 1.6.2 (AB Sciex, Ontario, Canada) were used for all measurements. A 100 µL aliquot of plasma or tumor homogenate (1 part tissue, 3 parts water) was mixed with 10 µL of working internal standard solution (20 ng/mL DXd-d5 in acetonitrile), 100 µL water, and 400 µL of chloroform. The sample was vigorously agitated in Fast Prep 120 (Thermo-Savant) at speed 4.0 for 20 s, 2 cycles. After centrifugation at 13,000 × g for 5 min, upper aqueous layer was discarded, and 200 µL of organic phase was transferred to glass tube and evaporated to dryness by gentle nitrogen stream. The residue was reconstituted with 30 µL of 50% A/50% B mobile phase mix (see below), sonicated 5 min, centrifuged, and 20 µL transferred into autosampler vial at 4 °C. 5 µL was injected into LC/MS/MS system. The LC/MS/MS conditions were as follows. Column: Eclipse Plus C18, 4.6 × 50 mm, 1.8 μm, at 40 °C. Mobile phase A: 0.1%, formic acid in water, 2% acetonitrile; mobile phase B: acetonitrile; flow: 1 mL/min; elution gradient (linear): 0–1 min 10–90% B, 1–1.5 min 90% B, 1.5–1.7 min 90–10% B. The analyte and internal standard were measured in positive ion mode. The following MS/MS transitions of the respective [M + H]+ ions were used. DXd: m/z 494/375, DXd-D5 (int.std.): m/z 499/380. Calibration standards in 0–2 ng/mL range were prepared in pooled drug-free plasma or 0–30 ng/mL in drug-free tumor homogenate. The lower limit of quantification for plasma was 0.016 ng/mL, and for tumor tissue 0.05 ng/g wet tissue.
Cathepsin L and B activity assay and assessment
All reagents were purchased from R&D System. Recombinant human CTSL (#952-CY) and CTSB (#953-CY) were dissolved in assay buffers specific to their pH requirement. CTSL assay buffer: 50 mM MES, 5 mM DTT, 1 mM EDTA, and 0.005% Brij-35, pH 6.0. CTSB assay buffer: 25 mM MES, 5 mM DTT, pH 5.0. A fluorogenic peptide substrate, Z-Leu-Arg-AMC (#ES008), was used for both enzymes. CTSL or CTSB (1 µg/mL) were incubated with 100 µM of substrate for 15 to 60 min in a 96-well Black Maxisorp plate, and cleavage activity was measured on fluorescent plate reader (Ex/Em 380/460 nm). For cleavage of HER2-ADC, CTSL or CTSB (1 µg/mL) were incubated with 2 mg/mL of T-DXd or T-DM1 for 6 h. Cleaved ADCs were added to MDA-MB-468 or MDA-MB-231 cells to access cytotoxicity of released payloads. To assess CTSL activity in conditioned media of CTSL-overexpressing cells, supernatants were filtered through 45 µM membrane and concentrated by 20-fold using Spin-X UF concentrator (Corning 431483). The concentrated proteins were suspended in a CTSL assay buffer (5 µg/mL), and the CTSL cleavage activity of the substrate was measured. When indicated, 12 µM of CTSL inhibitor Z-Phe-Phe-FMK (#HY-115733, MCE) were added to demonstrate exclusive CTSL-mediated activity.
Extracellular cathepsin L assessment by ELISA
Extracellular Cathepsin L levels from MDA-MB-231 and Au565 cells were quantified using Human Cathepsin L DuoSet ELISA (DY952, R&D Systems). Parental, CTSL-overexpressed or knockout cells were seeded (50,000 per well in 96-well plate), and cultured for 1–4 days. Conditioned media were harvested and diluted in assay diluent, and ELISA was performed according to the manufacturer’s protocol.
Western blot
Three million cells of each cell line of interest were lysed in 300 µL of RIPA buffer (Sigma, R0278) containing 1× Protease Inhibitor cocktail (Cell Signaling, #5871). 30 µL of lysate were incubated with 10 µL of 4× loading buffer (LI-COR, 928-40004) and heated at 95 °C for 10 min. Denatured protein samples were run on 10% PROTEAN TGX gels (Bio-Rad, #4568033). Proteins were then transferred to the PVDF membrane for 1 hour at 4 °C, following standard protocol. Membranes were probed with the following primary antibodies (from Cell Signaling): anti-Cathepsin L (clone E3R3P, # 55914S), anti-HER2 N-terminal domain (clone D8F12, # 4290S), and anti-HER2 C-terminal domain (clone 29D8, #2165S), β-actin (clone 8H10D10, #3700S). All primary antibodies were diluted 2000-fold in Odyssey PBS Blocking Buffer +0.2% Tween 20 and incubated with membrane overnight at 4 °C. Secondary antibodies IRDye 680RD Donkey anti-Mouse IgG (926-68072), and IRDye 800CW Goat anti-Rabbit IgG (926-32211) were from purchased from LI-COR, and diluted 10,000-fold dilution in Odyssey PBS Blocking Buffer +0.2% Tween 20 + 0.01% SDS and incubated with the membrane for 1 hour at room temperature. After washing, membranes were allowed to air dry and imaged on the LI-COR CXL machine at the highest resolution.
Breast cancer biospecimen and tissue microarray analysis
Data of breast cancer patients receiving T-DXd (ENHERTU) therapy at Duke Cancer Institute were identified and extracted using the Duke Enterprise Data Unified Content Explorer (DEDUCE), and approved by Duke Health Institutional Review Board under protocol ID 00116330. Core needle biopsies sections from these patients were then requested from the Duke BioRepository and Precision Pathology Center. Breast Cancer TMA was purchased from the Cooperative Human Tissue Network (CHTN). Four blocks of TMAs (case sets 4, 6, 18, and 19) were used from the Cancer Diagnosis Program (CDP) BC Progression Tissue (https://chtn.sites.virginia.edu/cdp-progression-info). Primary BC core needle biopsies and TMAs were assessed for CTSL expression via IHC analysis using CTSL antibody clone E3R3P (1:500 dilution). High-resolution images were acquired using the Cytation7 machine. QuPath software was used for Pixelwise quantification of CTSL DAB staining intensity and H-score calculation following the published method, using the uploaded script: https://shorturl.at/0o0bH.
Immunogenic cell death assessment
KPL4 or Au565 cells were treated with titrated doses of HER2 ADCs or unconjugated payloads DXd and DM1 for 3 days in white opaque-walled 96-well plates. Extracellular ATP in conditioned media was measured using RealTime-Glo™ Extracellular ATP Assay (Promega GA5010), with luminescence read 3 h after adding the eATP reagent substrate. HMGB1 release was measured using the Lumit® HMGB1 Human/Mouse Immunoassay (Promega W6110) per manufacturer’s protocol: cells were treated for 3 days, followed by the addition of anti-HMGB1 antibody mixtures for 90 min, a 5-min incubation with Lumit Detection reagent substrate, and luminescence measurement. HMGB1 levels were normalized to cell viability using CellTiter-Glo after the Lumit HMGB1 Immunoassay. Surface CRT was assessed by flow cytometry using anti-CRT antibody (Abcam, ab209577) compared to rat IgG2a-PE isotype control. Au565 cells were treated with titrated doses of DXd or DM1 for 2 days for surface CRT assessment on dying tumor cells.
FCGR activation assay
Jurkat cells expressing human FCGR3A or mouse FCGR4 with NFAT-Luciferase reporter were generated with lentiviral transduction and selected with puromycin, as validated in our previous publications. For the assay, HER2 + KPL4 cells were first plated and treated with serially diluted doses (1 ug/mL to 1 ng/mL) of T-DXd, T-DM1, trastuzumab or rituximab for 1 hour. Jurkat-FCGR-NFAT-LUC effector cells were added and co-cultured for 4 h. FCGR signaling activation was assessed by luciferase activity quantification.
Primary human and mouse macrophages or dendritic cell generation
Human PBMCs (HLA-A2+) were obtained from HemaCare (https://www.criver.com/products-services/cell-sourcing, product code: PB001F-2). PBMCs from three different donors were used for experiments (Sample IDs: W313717041653; W313717042818; W313718047140). Human macrophages were generated from 50 million PBMCs in 10 cm2 dish differentiated with 50 ng/mL human M-CSF (Peprotech, 300-25) for 10 days. Similarly, human dendritic cells were generated with 50 ng/mL GM-CSF (Peprotech 300-03) and 50 ng/mL IL-4 (Peprotech 200-04) for 10 days. Mouse BMDMs were generated from isolated bone marrows in the mouse tibia, differentiated for 10 days with 50 ng/mL mouse MCSF (Peprotech 315-02). 50 million bone marrow cells were plated in 10 cm2 dish with corresponding cytokines on day 0. Unattached cells in the supernatant were removed, and fresh media + cytokines were supplemented on day 3 and day 6.
In vitro macrophage and dendritic cells activation analysis by flow cytometry and RNA-Seq
HER2 + BC cell lines KPL4 or Au565 (250,000 cells) were pre-treated with 1 µg/mL of T-DXd, T-DM1, trastuzumab, or rituximab for one day. Primary human and mouse macrophages or DCs (250,000 cells) were then added to the pre-treated dying tumor cells, and co-cultured for 2 days. When indicated, 50 nM unconjugated DXd payload was used as pre-treatment. When indicated, CD47-KO KPL4 were used as target tumor cells. On the day of analysis, macrophages/DCs were harvested using a cell scraper after 30 min incubation with Accutase. Antigen-presentation and activation surface markers were analyzed by flow cytometry. FcR blockers (Biolegend) were used before staining with flow antibodies. The following human panel was used: HLA-DR FITC, CD80 PE, CCR7 PE-TR, CD40 PE-Cy7, CD45 PacBlue, HLA-A2 APC. The following mouse panel was used: CD40 FITC, CD80 PE, MHC-II PerCP-Cy5.5, MHC-I PE-Cy7, CD45 BV605, CD83 APC, CD86 APC-Cy7. Representative FACS gating strategies can be found in Supplementary Fig. 16a. Unstained flow controls for each marker were performed for gating purposes, but not subtracted from the presented MFI data. Activated macrophages from one day-culture experiment were used for RNA-seq analysis. Tumor cells were removed from co-culture by brief trypsin treatment. Total RNA from macrophages was harvested using RNeasy plus mini kit (Qiagen, 74134). RNA quality and concentration were determined using an Agilent 2100 Bioanalyzer. Whole transcriptome mRNA sequencing was performed by Novogene on an Illumina NovaSeq 6000. Read alignment, quality control, and gene count analysis were performed by Novogene using their standard pipeline. Gene expression visualizations were performed using GENAVI with z-score normalization.
In vitro ADCP assays
Primary human and mouse macrophages were used for ADCP assays on day 10 of differentiation. HER2 + BC cell lines (KPL4 or Au565) were labeled with Brilliant Violet 450 (BD 562158). Macrophages (250,000 cells) and labeled target cells (250,000 cells) were incubated with (1 µg/mL) rituximab, trastuzumab, T-DXd or T-DM1, or (50 nM) DXd in 12-well plates for 4 h or overnight at 37 °C. After co-culture, phagocytosis of labeled tumor cells by macrophages (CD45+) was analyzed by flow cytometry.
JEDI T cell co-culture and activation
HER2 + BC lines (KPL4 or Au565) were engineered to express eGFP via lentiviral transduction and puromycin selection. These HER2+ eGFP+ BC cells were used as target cells. 250,000 target cells were pre-treated with 1 µg/mL of T-DXd, T-DM1, trastuzumab, or rituximab, or 50 nM of DXd or DM1 for one day. Fully differentiated BMDMs derived from Balb/c mice were then co-cultured with the pre-treated target tumor cells for two days to allow phagocytoses and immune activation. Tumor cells were removed with brief trypsin treatment and fresh T cell media (RPMI, 50 µM 2-Mercaptoethanol, 100 IU/mL murine IL-2) were added to macrophages. Whole splenocytes were harvested from JEDI mice, and CD8+ T cells were isolated (STEMCELL 19853), labeled with Cell-Trace Violet, and added (100,000 cells) to each well with macrophages. A positive control for T cell activation using 1 µg/mL of eGFP peptide (HYLSTQSAL) was included. Three days after co-culture, T cells were analyzed by flow cytometry for proliferation and activation using the following antibody panel: CD8 PE-Cy7, CD44 PerCP-Cy5.5, CD45 BV605, Live/Dead Far-Red. Representative FACS gating strategies can be found in Supplementary Fig. 16b.
pDC isolation and IFNα assessment
Single-cell splenocyte suspensions from WT or STING-KO mice were isolated, and pDCs were enriched using EasySep Mouse Plasmacytoid DC Isolation Kit (STEMCELL, #19764). Enriched pDCs were 80% double positive in pDC markers PDCA1 and CD11c. Au565 cells were pre-treated with ADCs (1 µg/mL) or DXd (50 nM) for two days, and enriched pDCs were added onto treated Au565 cells at 2:1 ratio, and co-cultured for 2 days. Conditioned media were analyzed for IFNα1 levels by ELISA (Biolegend, #447904).
Mouse strains
All mice experiments were performed on female mice, which is required for relevance in breast cancer studies. SCID-beige (C.B-Igh-1b/GbmsTac-Prkdcscid-Lystbg N7; Taconic Biosciences) mice between the ages of 6 and 10 weeks old were used for all human cell line xenograft experiments. The HER2Δ16 transgenic model was generated by crossing MMTV-rtTA strain (a kind gift by Dr. Lewis Chodosh, UPenn, Philadelphia, USA) with TetO-HER2d16-IRES-EGFP strain (a kind gift by Dr. William Muller, McGill University, Montreal, Canada). 6-week-old female mice were put on a doxycycline diet and enrolled for experiments when they develop palpable breast tumors (usually in 4–6 weeks post dox diet). JEDI mice (STOCK Ptprca TcrbLn1Bdb TcraLn1Bdb H2d/J, Strain #:028062), TLR4-mutant mice (C3H-HeJ, Strain #:000659), STING-KO mice (BALB/c-Sting1em3Vnce/J, Strain #:036638), and BALB/cJ wild-type controls (Strain #:000651) were purchased from The Jackson Laboratory. All experiments were performed with a minimum of n = 5 per treatment group.
Orthotopic implanted xenograft models and therapeutic ADC/antibody treatments
KPL4, MDA-MB-231, and MDA-MB-468 cells were implanted into mammary fat pads (1 × 106 to 5 × 106 cells in 50% Matrigel/PBS) of SCID-beige mice. K562 and SUDHL-1O cells were implanted into mice flank subcutaneously (1 × 106 cells in 50% Matrigel/PBS). Tumor growth was measured with caliper-based tumor volume measurement (length × width × width/2) over time. For therapeutic treatments, animals were randomized and were administered weekly with T-DXd, T-DM1, or trastuzumab (10 mg/kg intraperitoneally) at the start of indicated time points post tumor implantation. For MDA-MB-231 CTSL-KO cells, three individual CTSL-KO tumors successfully established to grow in vivo were isolated and used for further studies. Stable knockout of CTSL in these three KO lines was validated by western blot and IHC. A control-KO tumor line was isolated and established in parallel for comparison.
Transgenic HER2Δ16 mouse model and therapeutic antibody treatments
The HER2Δ16 transgenic mouse model was generated by crossing two strains of mice, TetO-HER2Δ16-IRES-EGFP and MMTV-rtTA. This system was described previously, but utilizes a TET-ON system (with MTV-rtTA) to drive expression of HER2Δ16 to generate HER2 + BC. For experiments, one-month-old mice were put on Doxycycline diet (200 mg/kg, Bio-Serv) to induce spontaneous HER2-driven breast cancer. Individual animals were randomly enrolled into a treatment group when palpable breast tumors were detected in any of the eight mammary fat pads at a predetermined tumor size (criteria indicated for each experiment in figure legend). Control antibody rituximab (10 mg/kg), trastuzumab (10 mg/kg), or T-DXd (10 mg/kg or 5.4 mg/kg) were treated intraperitoneally weekly when animals were enrolled in experiment group, whereas anti-CD47 (MIAP410) was treated weekly with a priming dose of 5 mg/kg for the first two weeks, followed by 15 mg/kg per week. Treatments were administered for up to four weeks. Animals were terminated once their total body tumor burden volume reached >1500 mm3, unless tumor ulceration necessitated earlier termination. For CD8 depletion, animals were treated intraperitoneally weekly with anti-CD8b (Lyt3.2, 10 mg/kg).
Flow Cytometry analysis of tumor-infiltrating immune cells
When tumor growth reached humane endpoint size (>2000 mm3), whole tumors from mice were harvested and chopped into <1 mm piece, and incubated for 1 hour in digestion buffer (DMEM + 100 µg/mL collagenase + 0.2 U/mL DNAse + 1 µg/mL hyaluronidase). Single-cell suspensions were spun down through a 70 µm filter and washed with media. Approximately 5–10 million cells were used for staining and flow cytometry analysis on LSRII machine. FcR blockers (Biolegend 101319) were used before staining with flow antibodies. The following panel of myeloid cell markers (Biolegend) were used: CD45 PE-TR, CD11b APC-Cy7, Gr-1 PerCP-Cy5.5, F4/80 PE-Cy7, CD11c PacBlue, CD40 PE, CD86 BV605, MHC-II APC, and viability dye (Aqua, Invitrogen). Tumor-associated macrophages (TAM) were identified by live F4/80 + Gr1- CD11b+CD45+ gating. DCs were identified as live CD11c+CD11b+Gr1- CD45+. In vivo tumor phagocytosis was identified by eGFP+ macrophages or DCs. The following panel T cell markers (Biolegend) were used: CD45 BV605, CD11b PE-Cy7, CD3-APC, CD8 APC-CY7, CD4 PE-TR, CD44 PerCP-CY5.5, CD62L-PacBlue, CD107a-PE and viability dye (Aqua, Invitrogen). Tumor-infiltrating T cells were identified by CD8 and CD4 gating in the live CD45+/CD11b−/CD3+ population. CD44/CD62L double gating was used to identify effector and naive T cells. CD107a was used to identify cytotoxic T cells. Representative FACS gating strategies can be found in Supplementary Fig. 16c, d.
Immunohistochemistry analysis of tumor-infiltrated T cells
Tumor tissues were harvested and fixed in 10% formalin solution for 2 days. Tumors were paraffin-embedded and sectioned. Antigen unmasking was performed with antigen retrieval buffer A. Endogenous peroxidases/phosphatases were quenched with BLOXALL blocking solution (Vector), and tissues were blocked with Animal-Free Blocker R.T.U. (Vector). Sections were probed with primary antibodies overnight at 4 °C. Primary antibodies were from Cell Signaling and used at the following dilutions: CD8 (D4W2Z, 1:300), CD4 (D7D2Z, 1:200), FoxP3 (FJK-16S, 1:100) and Cathepsin L (E3R3P, 1:500). After probing, sections were washed with PBS, and incubated with the appropriate ImmPRESS polymer detection reagent (Vector) for 30 min at room temperature. Visualization was performed by incubation with 3,3′-diaminobenzidine (DAB) (Vector), ImmPACT Vector Red (Vector), or a Green HRP staining kit (Novus), For triple IHC, a second round of retrieval was performed with R-Buffer A after developing the first round of HRP and AP stains. Tissues were counterstained with Gill No.3 Hematoxylin (Sigma-Aldrich), coverslipped, and imaged on Cytation7 machine at ×20 objective. Quantification of intratumor versus stromal CD8+ T cells was conducted using ImageJ. Six representative fields were measured for each tumor section. Stromal areas were manually drawn in each field, and stromal area (mm2) was calculated with ImageJ. Tumor bed area was calculated by subtracting “stromal area” from “total area measured” in the field. CD8+ cells (brown) were independently counted in stromal vs tumor regions using Cell Counter plug-in feature of ImageJ.
ELSIPOT assay
Splenocytes from transgenic HER2Δ16 mice experiments were isolated into single cell suspensions, red blood cells lysed (RBC lysis buffer, Thermo), and live cells counted. 500,000 live splenocytes were stimulated with 1 µg/mL HER2 peptide pool or control HIV/Gag peptide pool on ELISPOT plates coated with anti-IFNγ antibody (AN-18), incubated for 24 h. IFN-γ spots were developed using biotinylated IFNγ antibody (R4-6A2), developed with Streptavidin-HRP and AEC substrate. Spots were counted using Cytation 7.
Serum ELISA analysis of anti-HER2 IgG
Blood was drawn from euthanized animals by heart puncture and serum on the top layer was isolated by centrifugation 10 min 15,000 RCF. Serum were diluted 50-fold, followed by twofold serial dilutions, and analyzed by ELISA for anti-HER2 mouse IgG levels. 50 ng/mL HER2 protein (Sinobiological, 10004-H08H) in PBS was used to coat ELISA plates overnight, and HRP conjugated anti-mouse-IgG antibodies were used for detection.
Statistical analysis
Statistics were performed using Prism 9 (GraphPad Software). All numerical data are expressed as the mean ± SEM. All data were assumed a normal distribution. One-way and two-way analysis of variance tests with Tukey’s multiple comparisons tests were performed to determine differences in multi-groups experiments. Tumor growth differences were analyzed with a mixed-effects model with Geisser-Greenhouse correction and Tukey’s multiple comparisons test. Log-rank (Mantel–Cox) test was used for animal survival analysis. P < 0.05 was considered statistically significant. All data plotted are presented as mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001, ****p < 0.0001.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Values for all data points in graphs and uncropped blots are reported in the Source Data File. RNA sequencing data have been deposited in the Sequence Read Archive database under accession code PRJNA1144387, [https://www.ncbi.nlm.nih.gov/bioproject/1144387]. LC-MS for DXd analysis has been deposited at Duke University’s Research Data Repository, accessible via the https://doi.org/10.7924/r4862qh18. Source data are provided with this paper.
References
Dumontet, C., Reichert, J. M., Senter, P. D., Lambert, J. M. & Beck, A. Antibody-drug conjugates come of age in oncology. Nat. Rev. Drug Discov. 22, 641–661 (2023).
Bournazos, S., Gupta, A. & Ravetch, J. V. The role of IgG Fc receptors in antibody-dependent enhancement. Nat. Rev. Immunol. 20, 633–643 (2020).
Tsao, L. C., Force, J. & Hartman, Z. C. Mechanisms of therapeutic antitumor monoclonal antibodies. Cancer Res. 81, 4641–4651 (2021).
Tsao, L. C. et al. CD47 blockade augmentation of trastuzumab antitumor efficacy dependent on antibody-dependent cellular phagocytosis. JCI Insight 4, https://doi.org/10.1172/jci.insight.131882 (2019).
Sheyi, R., de la Torre, B. G. & Albericio, F. Linkers: an assurance for controlled delivery of antibody-drug conjugate. Pharmaceutics 14, https://doi.org/10.3390/pharmaceutics14020396 (2022).
Fu, Z., Li, S., Han, S., Shi, C. & Zhang, Y. Antibody drug conjugate: the “biological missile” for targeted cancer therapy. Signal Transduct. Target Ther. 7, 93 (2022).
Hurvitz, S. A. et al. Trastuzumab deruxtecan versus trastuzumab emtansine in patients with HER2-positive metastatic breast cancer: updated results from DESTINY-Breast03, a randomised, open-label, phase 3 trial. Lancet 401, 105–117 (2023).
Shitara, K. et al. Trastuzumab deruxtecan in previously treated HER2-positive gastric cancer. N. Engl. J. Med. 382, 2419–2430 (2020).
Li, B. T. et al. Trastuzumab deruxtecan in HER2-mutant non-small-cell lung cancer. N. Engl. J. Med. 386, 241–251 (2022).
Meric-Bernstam, F. et al. Efficacy and safety of trastuzumab deruxtecan in patients with HER2-expressing solid tumors: primary results from the DESTINY-PanTumor02 phase II trial. J. Clin. Oncol. 42, 47–58 (2024).
Shiose, Y., Ochi, Y., Kuga, H., Yamashita, F. & Hashida, M. Relationship between drug release of DE-310, macromolecular prodrug of DX-8951f, and cathepsins activity in several tumors. Biol. Pharm. Bull. 30, 2365–2370 (2007).
Hunter, F. W. et al. Mechanisms of resistance to trastuzumab emtansine (T-DM1) in HER2-positive breast cancer. Br. J. Cancer 122, 603–612 (2020).
Ogitani, Y. et al. DS-8201a, a novel HER2-targeting ADC with a novel DNA topoisomerase I inhibitor, demonstrates a promising antitumor efficacy with differentiation from T-DM1. Clin. Cancer Res. 22, 5097–5108 (2016).
Cortés, J. et al. Trastuzumab deruxtecan versus trastuzumab emtansine in HER2-positive metastatic breast cancer: long-term survival analysis of the DESTINY-Breast03 trial. Nat. Med. 30, 2208–2215 (2024).
Liu, F. et al. Adverse event profile differences between trastuzumab emtansine and trastuzumab deruxtecan: a real-world, pharmacovigilance study. J. Cancer 14, 3275–3284 (2023).
Modi, S. et al. Trastuzumab deruxtecan in previously treated HER2-low advanced breast cancer. N. Engl. J. Med. 387, 9–20 (2022).
Holthuis, E. I. et al. Real-world data of HER2-low metastatic breast cancer: a population based cohort study. Breast 66, 278–284 (2022).
Bardia, A. et al. Trastuzumab deruxtecan after endocrine therapy in metastatic breast cancer. N. Engl. J. Med. https://doi.org/10.1056/NEJMoa2407086 (2024).
Mosele, F. et al. Trastuzumab deruxtecan in metastatic breast cancer with variable HER2 expression: the phase 2 DAISY trial. Nat. Med. 29, 2110–2120 (2023).
Ogitani, Y., Hagihara, K., Oitate, M., Naito, H. & Agatsuma, T. Bystander killing effect of DS-8201a, a novel anti-human epidermal growth factor receptor 2 antibody-drug conjugate, in tumors with human epidermal growth factor receptor 2 heterogeneity. Cancer Sci. 107, 1039–1046 (2016).
Shitara, K. et al. Discovery and development of trastuzumab deruxtecan and safety management for patients with HER2-positive gastric cancer. Gastric Cancer 24, 780–789 (2021).
Maute, R., Xu, J. & Weissman, I. L. CD47-SIRPα-targeted therapeutics: status and prospects. Immunooncol. Technol. 13, 100070 (2022).
Vasalou, C. et al. Quantitative evaluation of trastuzumab deruxtecan pharmacokinetics and pharmacodynamics in mouse models of varying degrees of HER2 expression. CPT Pharmacomet. Syst. Pharm. 13, 994–1005 (2024).
Olson, O. C. & Joyce, J. A. Cysteine cathepsin proteases: regulators of cancer progression and therapeutic response. Nat. Rev. Cancer 15, 712–729 (2015).
Ram, S. et al. Pixelwise H-score: A novel digital image analysis-based metric to quantify membrane biomarker expression from immunohistochemistry images. PLoS One 16, e0245638 (2021).
Foekens, J. A. et al. Prognostic significance of cathepsins B and L in primary human breast cancer. J. Clin. Oncol. 16, 1013–1021 (1998).
Yadati, T., Houben, T., Bitorina, A. & Shiri-Sverdlov, R. The ins and outs of cathepsins: physiological function and role in disease management. Cells 9, https://doi.org/10.3390/cells9071679 (2020).
Kroemer, G., Galluzzi, L., Kepp, O. & Zitvogel, L. Immunogenic cell death in cancer therapy. Annu. Rev. Immunol. 31, 51–72 (2013).
Fucikova, J. et al. Detection of immunogenic cell death and its relevance for cancer therapy. Cell Death Dis. 11, 1013 (2020).
Elliott, M. R. et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature 461, 282–286 (2009).
Apetoh, L. et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat. Med. 13, 1050–1059 (2007).
Obeid, M. et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 13, 54–61 (2007).
Upton, R. et al. Combining CD47 blockade with trastuzumab eliminates HER2-positive breast cancer cells and overcomes trastuzumab tolerance. Proc. Natl. Acad. Sci. USA 118, https://doi.org/10.1073/pnas.2026849118 (2021).
Van Wagoner, C. M. et al. Antibody-mediated phagocytosis in cancer immunotherapy. Immunol. Rev. 319, 128–141 (2023).
Nimmerjahn, F. et al. FcγRIV deletion reveals its central role for IgG2a and IgG2b activity in vivo. Proc. Natl Acad. Sci. USA 107, 19396–19401 (2010).
Nimmerjahn, F. & Ravetch, J. V. Fcgamma receptors as regulators of immune responses. Nat. Rev. Immunol. 8, 34–47 (2008).
Dekkers, G. et al. Affinity of human IgG subclasses to mouse Fc gamma receptors. MAbs 9, 767–773 (2017).
Agudo, J. et al. GFP-specific CD8 T cells enable targeted cell depletion and visualization of T-cell interactions. Nat. Biotechnol. 33, 1287–1292 (2015).
Wang, Z. et al. cGAS/STING axis mediates a topoisomerase II inhibitor-induced tumor immunogenicity. J. Clin. Invest. 129, 4850–4862 (2019).
Poltorak, A. et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 282, 2085–2088 (1998).
Candas-Green, D. et al. Dual blockade of CD47 and HER2 eliminates radioresistant breast cancer cells. Nat. Commun. 11, 4591 (2020).
Ghantous, L. et al. The DNA damage response pathway regulates the expression of the immune checkpoint CD47. Commun. Biol. 6, 245 (2023).
Turpin, J. et al. The ErbB2ΔEx16 splice variant is a major oncogenic driver in breast cancer that promotes a pro-metastatic tumor microenvironment. Oncogene 35, 6053–6064 (2016).
Su, D. & Zhang, D. Linker design impacts antibody-drug conjugate pharmacokinetics and efficacy via modulating the stability and payload release efficiency. Front Pharm. 12, 687926 (2021).
Lisberg, A. et al. Datopotamab deruxtecan in locally advanced/metastatic urothelial cancer: preliminary results from the phase 1 TROPION-PanTumor01 study. J. Clin. Oncol. 42, 603–603 (2024).
Bardia, A. et al. Datopotamab deruxtecan versus chemotherapy in previously treated inoperable/metastatic hormone receptor-positive human epidermal growth factor receptor 2-negative breast cancer: primary results from TROPION-breast01. J. Clin. Oncol. Jco2400920, https://doi.org/10.1200/jco.24.00920 (2024).
Dowling, G. P. et al. Efficacy and safety of trastuzumab deruxtecan in breast cancer: a systematic review and meta-analysis. Clin. Breast Cancer 23, 847–855.e842 (2023).
Attalla, S. S. et al. HER2Δ16 engages ENPP1 to promote an immune-cold microenvironment in breast cancer. Cancer Immunol. Res. 11, 1184–1202 (2023).
Tsao, L. C. et al. Trastuzumab/pertuzumab combination therapy stimulates antitumor responses through complement-dependent cytotoxicity and phagocytosis. JCI Insight 7, https://doi.org/10.1172/jci.insight.155636 (2022).
Chen, P., Bordeau, B. M., Zhang, Y. & Balthasar, J. P. Transient inhibition of trastuzumab-tumor binding to overcome the “binding-site barrier” and improve the efficacy of a trastuzumab-gelonin immunotoxin. Mol. Cancer Ther. 21, 1573–1582 (2022).
Acknowledgements
This research was supported by grants from the National Institutes of Health (NIH) (1 R01CA238217-01A1/02S1 to ZCH), Susan G Komen for the Cure (OG22873853) and the Department of Defense (DOD) (W81XWH-16-1-0354/ BC191055 to ZCH, W81XWH2120031/ BC201085 to HKL HT9425-24-1-0041/BC230508 to ZCH). IS acknowledges NCI Cancer Center Support Grant P30CA014236, for the partial support of LC/MS/MS analysis.
Author information
Authors and Affiliations
Contributions
L.C.T. conceptualized the study, devised, performed, and directed all animal and cell culture experiments, analyzed data, compiled the figures, and wrote the manuscript. Z.C.H. provided supervision and support for the study, contributed to study conceptualization, and edited the manuscript. H.K.L. provided clinical perspectives and discussions, and provided support for the study. M.M. provided clinical-grade therapeutics from Duke University Cancer Center Pharmacy for use in this study and also provided clinical perspectives and discussions. J.W., S.S., X.M., and J.R. assisted with cell culture and animal experiments. J.P.W. provided support for lentivirus production. B.L. and G.L. supported cloning for gene expression and CRISPR constructs. X.Y. and C.X.L. provided support in animal experiments and maintenance. T.W. provided support for flow cytometry. J.W. charted and extracted T-DXd-treated patients at Duke Cancer Institute using DEDUCE, and requested clinical specimens/biopsies for CTSL/HER2 assessment. T.N.T. provided support in IHC analysis and JEDI T-cell experimental model and edited the manuscript. J.M. assisted with IHC staining. IS supervised and performed LC-MS analysis with technical support from P.F. We especially thank the members of the Duke Surgery Center for Applied Therapeutics for technical assistance, thoughtful discussion, and insight into this project.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Tsao, LC., Wang, J.S., Ma, X. et al. Effective extracellular payload release and immunomodulatory interactions govern the therapeutic effect of trastuzumab deruxtecan (T-DXd). Nat Commun 16, 3167 (2025). https://doi.org/10.1038/s41467-025-58266-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-58266-8
This article is cited by
-
SC134-deruxtecan, a fucosyl-GM1 targeting ADC for small cell lung cancer therapy
Journal of Translational Medicine (2025)
-
Antibody–bottlebrush prodrug conjugates for targeted cancer therapy
Nature Biotechnology (2025)
-
Unveiling the molecular and immunological drivers of antibody–drug conjugates in cancer treatment
Nature Reviews Cancer (2025)
-
Antibody–drug conjugates in breast cancer: mechanisms of resistance and future therapeutic perspectives
npj Breast Cancer (2025)
-
Antibody drug conjugates in metastatic brain tumors: current landscape, therapeutic potential and challenges
Journal of Neuro-Oncology (2025)
