
Abstract
Maintaining genomic integrity and faithful transmission of genetic information is essential for the survival and proliferation of cells and organisms. DNA damage, which threatens the integrity of the genome, is rapidly sensed and repaired by mechanisms collectively known as the DNA damage response. The RNA demethylase FTO has been implicated in this process; however, the underlying mechanism by which FTO regulates DNA repair remains unclear. Here, we use an unbiased quantitative proteomic approach to identify the proximal interactome of endogenous FTO protein. Our results demonstrate a direct interaction with the DNA damage sensor protein PARP1, which dissociates upon ultraviolet stimulation. FTO inhibits PARP1 catalytic activity and controls its clustering in the nucleolus. Loss of FTO enhances PARP1 enzymatic activity and the rate of PARP1 recruitment to DNA damage sites, accelerating DNA repair and promoting cell survival. Interestingly, FTO regulates PARP1 function and DNA damage response independent of its catalytic activity. We conclude that FTO is an endogenous negative regulator of PARP1 and the DNA damage response in cells beyond its role as an RNA demethylase.
Similar content being viewed by others


Introduction
Human cells experience a high frequency of DNA lesions induced by endogenous and exogenous insults that must be rapidly repaired to avoid the accumulation of catastrophic mutations and genomic instability, which can lead to cell death or the development of diseases such as cancer1,2. To ensure genomic integrity, cells have evolved versatile mechanisms, collectively known as the DNA damage response (DDR), that sense and repair DNA lesions. The recruitment of poly(ADP-ribose) polymerase 1 (PARP1) to DNA damage sites represents one of the earliest events in multiple DNA repair pathways, including single-strand break repair, base excision repair, nucleotide excision repair and double-strand break repair through homologous recombination or non-homologous end-joining pathways (NHEJ)3,4. It is well established that active PARP1 catalyzes the post-translational attachment of poly(ADP-ribose) (PAR) onto itself and essential DNA repair proteins, recruiting them to the damaged sites. However, the mechanisms and factors that restrict PARP1 activation in undamaged conditions remain poorly understood.
Fat mass and obesity-associated (FTO) protein is an Fe2+/α-ketoglutarate-dependent dioxygenase primarily known to demethylate multiple RNA species at the N6 (m6A) or N1 (m1A) position of adenosine and the N6, 2’-O-dimethyladenosine (m6Am)5,6,7. FTO expression is misregulated in many types of cancers8. Upregulation of FTO, which leads to downregulation of m6A marks in its target mRNAs such as RARA, MYC, ASB2, PARP1 and CEBPA, contributes to tumorigenesis by promoting cancer stem-cell renewal and differentiation, cell growth, migration, and invasion in most cancer types8,9,10,11,12. Small molecule inhibitors targeting FTO are therefore of clinical importance for the treatment of acute myeloid leukemia, glioblastoma, breast and pancreatic cancers13,14,15. However, FTO can also exhibit a tumor suppressor function in lung, breast, liver and cervical cancers by modulating gene expression in an m6A-dependent manner8,16,17. Thus, a better understanding of the cellular function of FTO in DDR may provide mechanistic insight into the contribution of FTO in maintaining genomic stability in cells.
N6-methyladenosine (m6A) signaling has recently emerged as a critical regulator of the DDR18,19,20,21. It is the most prevalent post-transcriptional RNA modification in eukaryotes, which is deposited co-transcriptionally by the m6A writer complex containing the catalytically active methyltransferase-like 3 (METTL3) and reversed by the m6A RNA demethylases, FTO or AlkB homolog 5 (ALKBH5)6,22,23,24,25. Upon ultraviolet (UV) light exposure, METTL3 is recruited to the DNA damage site and increases the m6A level in a PARP1-dependent manner, which through an unknown mechanism then facilitates the recruitment of polymerase κ (Polκ) for efficient DNA repair18. Although the role of FTO in the DDR pathway has recently been indicated in a few studies26,27,28, the exact mechanisms and functions of FTO in these processes are unclear.
Results
FTO loss promotes UV-induced DNA repair
To determine the role of FTO in the UV-induced DDR, we quantified the levels of cyclobutene pyrimidine dimers (CPDs), the dominant DNA lesions caused by UV exposure29, in two independent lines of FTO knockout (KO) HeLa cells (Supplementary Fig. 1a)30. Our results revealed that FTO KO cells exhibited significant reductions in CPD levels (Fig. 1a, b). Notably, there were no significant differences in the level of CPDs formed shortly after UV exposure between wild-type (WT) and FTO KO cells (Supplementary Fig. 1b–e), indicating that loss of FTO promotes CPD removal in HeLa cells. Next, we examined the phosphorylation of the histone variant H2A.X at Ser-139 (γH2A.X), a marker of DNA double-strand breaks31. Consistent with our CPD data, we observed lower UV-induced γH2A.X in FTO KO cells than in WT HeLa cells by immunostaining (Fig. 1c, d) and western blot assays (Fig. 1e, f). The reductions in DNA damage markers in FTO KO HeLa cells are associated with a more robust cell survival after 5–25 J UV irradiation than WT cells (Fig. 1g, h). To validate our findings, we generated FTO KO in two distinct cell lines, namely U2OS (one clone) and HCT116 (two clones), and determined the impact of FTO depletion on DDR. Corroborating the findings in HeLa cells, our results showed that FTO KO U2OS and HCT116 cells had reduced CPD and γH2A.X levels following UV treatment, resulting in higher percentages of cell survival after UV irradiation (Supplementary Fig. 2). Collectively, these findings demonstrate that FTO suppresses the repair of UV-induced DNA lesions and cellular resistance to UV.

a Dot-blot detection of the CPDs in genomic DNA of HeLa cells upon UVC irradiation. Methylene blue showed the amount of DNA loaded onto the membrane. b Quantification of CPD levels. Data were normalized to the non-irradiated controls (N = 9 per group) and analyzed using two-way ANOVA with Dunnett’s multiple comparison test (*WT vs KO.1 and #WT vs KO.2). c WT or FTO KO HeLa cells were pre-treated with BrdU, irradiated with 100 J of UVC, and allowed to recover for 30 min and 6 h. Cells were fixed and stained with the DNA double-strand break marker γH2A.X (green) and DAPI (blue). Scale bar, 20 μm. d Quantification of γH2A.X levels. Data were normalized to the WT group (N = 4 biological replicates; WT-30 min = 176 cells; KO.1-30 min = 301 cells; KO.2–30 min = 291 cells; WT-6 h = 198 cells; KO.1–6 h = 266 cells; KO.2-6 h = 272 cells) and analyzed using one-way ANOVA with Dunnett’s multiple comparison test. e Same experiment as c, but cells were lyzed and subjected to western blotting analysis with antibodies against γH2A.X, FTO and α-tubulin. f Quantification of γH2A.X levels after data were normalized to the 6 h—WT group (N = 5 per group). Data were analyzed using two-way ANOVA with Sidak’s multiple comparison tests. g WT or FTO KO HeLa cells were subjected to UVC irradiation at the indicated dosages. Colonies of surviving cells were stained with crystal violet 14 days post-UVC exposure. h The percentage of cell survival was determined by quantifying the number of colonies and normalizing them to the non-irradiated control groups (N = 4–7 per group). Data were analyzed using two-way ANOVA with Tukey’s multiple comparison test (*WT vs KO.1 and #WT vs KO.2). All data are represented as mean ± SEM. Source data are provided as a Source Data file.
Identification of endogenous FTO proximal proteome
To determine the underlying mechanisms of FTO function in DDR, we performed an unbiased screen to identify FTO proximal interactome in HeLa cells by combining the biotinylation by antibody recognition (BAR) labeling technique with stable isotope labeling with amino acids in cell culture (SILAC)-based mass spectrometry (MS) analysis (Supplementary Fig. 3a, b)32. This method does not require overexpression of FTO protein tagged with a biotin ligase or peroxidase, which enabled us to capture the endogenous FTO proximal interactome recognized only by specific antibodies against FTO. In WT cells, FTO BAR generated significantly stronger biotin labeling as revealed by immunostaining with Alexa-488-conjugated streptavidin than that observed in FTO KO HeLa cells (Supplementary Fig. 3c). Moreover, only low to negligible levels of biotin labeling could be detected in conditions where no primary antibodies, biotin phenol or hydrogen peroxide (H2O2) were added to the reactions (Supplementary Fig. 3c), validating the specificity of the assay. The normalized MS data revealed 243 proteins in the FTO proximal proteome (adjusted P-value < 0.05), which had significantly higher levels of biotinylation in WT than FTO KO cells (Fig. 2a, b and Supplementary Data 1). Many of these proteins function in mRNA splicing (DDX17, SFPQ, SRSF6, PUF60 and WTAP) and the spliceosome complex (snRNP70, PRPFs and HNRNPs) (Fig. 2c and Supplementary Data 2), consistent with the established role of FTO in these processes33,34,35. In addition, we also identified proteins involved in the ribonucleoprotein biogenesis complex in the FTO proximal proteome (multiple members of the DDX, RPL and RPS family of proteins), indicating a cellular function of FTO in ribosome biogenesis. We also identified several FTO interactors that had been reported in previous proteomic studies27,36, including DDX17, KIFC1, HNRNPH1 and SFPQ (Supplementary Fig. 4a), confirming the ability of our assay to isolate known interacting proteins.

a Clustering and heat-map diagram of mean z-scored biotinylated protein abundances that were identified from the FTO BAR SILAC-MS analyses from 3 independent experiments. b A ranking plot of FTO proximal interacting proteins based on the mean fold-change of biotinylated protein abundance in WT compared to FTO KO HeLa cells. c Gene ontology (GO) enrichment analyses of the FTO proximal interactome. d Chord diagram displaying the relationship between a subset of FTO proximal interacting proteins and selected cellular functions associated with GO terms enriched in the FTO proximal proteome. GO enrichment analysis was performed using the R-package clusterProfiler55 utilizing over-representation analysis based on Fisher’s exact test (one-sided). e Interactions with proteins involved in nuclear division, DNA repair, DNA replication, telomere maintenance and chromosome organization were mapped in a STRING network using Cytoscape with a 0.7 confidence score cut-off.
Despite being a critical component in RNA processing, we found many FTO proximal interacting proteins are involved in DNA replication, telomere maintenance, DNA repair, chromosomal organization and nuclear division, suggesting a prominent role of FTO in regulating the stability and integrity of the nuclear genome in cells (Fig. 2d, e). Many of these FTO proximity interactors can function in multiple cellular pathways that maintain genomic integrity, such as members of the MCM replicative helicases, XRCC5 (Ku80), PRKDC (DNA-PK), ERCC4 and PARP1. Although FTO itself was not identified in the list of 585 PARP1 proximal-interacting proteins37, we found approximately 37% of FTO proximity interactors (89 out of 243 proteins) in the PARP1-proximal proteome (Supplementary Fig. 4b). Many of them are nuclear proteins with nucleic acid binding and helicase activities, indicating the functional interaction between these two proteins.
FTO interacts with PARP1 and regulates its clustering in cells
To validate the interaction between FTO and PARP1, we performed BAR assays with anti-FTO antibodies followed by western blotting. We could pull down biotinylated PARP1 and its interacting proteins that form the core component of the NHEJ machinery, DNA-PK and Ku80 in WT but not in FTO KO cells (Fig. 3a, b)38,39. Proximity-based ligation assays (PLA) confirmed the interaction between endogenous FTO and PARP1. FTO-PARP1 PLA puncta were observed in the nuclei of WT cells but were significantly reduced in two independent lines of FTO KO cells (Fig. 3c, d). Furthermore, in vitro binding assays revealed that recombinant Flag-PARP1 protein only co-precipitated on a nickel column in the presence of His-FTO (Fig. 3e, f), demonstrating that PARP1 can directly interact with FTO.

a Validation of FTO proximal interaction using the BAR assay and western blotting. b Quantification of (a). Data were normalized to the non-irradiated controls using two-tailed unpaired t-tests (N = 3 per group). c PLA signals (gray) between endogenous FTO and PARP1 in nuclei (DAPI, blue). Scale bar, 20 μm. d PLA puncta per nucleus were quantified and analyzed using one-way ANOVA with Tukey’s multiple comparison test (WT, N = 508 cells; KO.1, N = 183 cells; KO.2, N = 346 cells). e Recombinant Flag-PARP1 interacts with His-FTO proteins. f Quantification of PARP1 binding. Data were normalized to the no His-FTO group (N = 5) and analyzed using a two-tailed paired t-test. g FTO-PARP1 PLA puncta (gray) in the nucleus (blue) of control and UVC irradiated cells. Scale bar, 20 μm. h PLA puncta per nucleus were quantified and analyzed using a two-tailed Mann-Whitney test (Ctrl, N = 315 cells; UV, N = 299 cells). i Representative structured illumination microscopy (SIM) images of HeLa cells immunostained for FTO (green) and PARP1 (magenta). Scale bars, 10 μm and 1 μm (inset). j Frequency distribution of the nearest distance between FTO to PARP1. Data were binned at 0.05 μm (Ctrl, N = 18 cells; UV, N = 20 cells) and analyzed using a two-tailed Mann-Whitney test. k Representative SIM images of WT and FTO KO HeLa cells. Scale bars, 10 μm and 1 μm (inset). l, m The number of PARP1 clusters per nucleus (l, N = 21–27 cells per group) and the ratio of the individual PARP1 cluster to the nucleus area (m, N = 60–133 clusters per group) were quantified and analyzed using one-way ANOVA with Dunnett’s multiple comparison test. n Relative frequency distribution of PARP1 volume (N = 80–202 particles per group, minimal size of 0.4 μm3) was analyzed using a two-tailed Kolmogorov-Smirnov test. All data are represented as mean ± SEM, except for the violin, and box and whiskers plots, which showed the median and quartile values. Source data are provided as a Source Data file.
UV exposure in cells could modulate the strength of the interaction between FTO and PARP1. Interestingly, the FTO-BAR assays revealed a significant reduction in biotinylated PARP1, Ku80 and DNA-PK 4 min post-UV irradiation (Fig. 3a, b). We also observed a similar decrease in FTO-PARP1 PLA puncta in cells (Fig. 3g, h), suggesting that UV exposure weakens the interaction between FTO and PARP1. The reduced FTO-PARP1 interaction was not due to changes in PARP1 expression because FTO knockout or overexpression did not affect the abundance of PARP1 protein (Supplementary Fig. 5a, b). Furthermore, there were no significant changes in the levels of PARP1 mRNA in FTO KO compared to wild-type HeLa cells (Supplementary Fig. 5c). Next, we determined the localization of FTO and PARP1 using super-resolved structured illumination microscopy. As expected, high-intensity clusters of PARP1 molecules were localized within the nucleoli of HeLa cells (Fig. 3i). Conversely, FTO had a more even distribution across the nucleus although a higher level of labeling could be observed close to PARP1 within the nucleoli (Fig. 3i). Consistent with the results obtained with the BAR and PLA assays, we found that the nearest distance between FTO and PARP1 molecules increased post-UV treatment, further confirming the weakening of the FTO-PARP1 interaction in cells (Fig. 3i, j and Supplementary Fig. 6a). Based on the high-resolution images, we also observed a dispersal of the high-intensity PARP1 clusters that localized to the nucleoli following UV treatment (Fig. 3i, k). This was evidenced by the reduced number of high-intensity PARP1 clusters as well as the wider area occupied by the dispersed PARP1 clusters in response to UV treatment (Fig. 3k–m). This phenomenon was accompanied by a significant decrease in the volume of individual PARP1 puncta and an alteration in their three-dimensional shape (Fig. 3n and Supplementary Fig. 6b). Strikingly, the loss of FTO expression led to PARP1 dispersal into smaller clusters that occupied larger surface areas (Fig. 3k–n). UV exposure did not induce further dispersal of PARP1 molecules in FTO KO cells (Fig. 3k–n). Moreover, UV treatment only had minor effects on the volume and three-dimensional shape of FTO puncta (Supplementary Fig. 6c, d). Collectively, these data indicate that FTO plays a structural role in maintaining PARP1 clusters in cells.
FTO negatively regulates PARP1 function
Our results predicted that UV-induced dissociation of the FTO-PARP1 interaction allows PARP1 to disperse and be efficiently recruited to DNA damage sites. To test this hypothesis, we directly visualized the recruitment of GFP-PARP1 to the sites of DNA lesions elicited by UVA micro-irradiation in live cells. UV micro-irradiation caused rapid association of GFP-PARP1 to the DNA damage sites with a time constant (τ) of ~8.1 s (Fig. 4a–d). By contrast, HeLa cells lacking FTO exhibited a significantly faster rate of GFP-PARP1 recruitment (τ ~ 5.8 s) in two independent KO lines without affecting the amount of GFP-PARP1 bound to damaged DNA (Fig. 4a–e). Although overexpression of myc-FTO did not slow the recruitment rate, the abundance of GFP-PARP1 at sites of DNA damage was dramatically reduced (Supplementary Fig. 7a–d).

a Live imaging of WT or FTO HeLa KO cells expressing GFP-PARP1 following UVA micro-irradiation. Scale bar, 5 μm. b, c Average (b) and normalized (c) traces showing relative changes in GFP-PARP1 fluorescence intensity at the site of DNA damage over time. Solid and dashed lines indicate means and SEM, respectively. d, e Quantification of the time-constant (τ) of fluorescence recovery after photobleaching (d) and the peak fluorescent intensity (e) of GFP-PARP1 at DNA damage sites (WT, N = 24; KO.1, N = 19; KO.2, N = 17). Data were analyzed using one-way ANOVA with Dunnett’s multiple comparison test. f UVC-induced global PARylation in WT or FTO KO HeLa cells. g Quantification of total PAR levels. Data were normalized to the 1 min timepoint of the WT group (N = 7 per group) and analyzed using two-way ANOVA with Holm-Šídák’s multiple comparison test (*WT vs KO.1 and # WT vs KO.2). h Recombinant Flag-PARP1 (indicated by the asterisk) was incubated with or without NAD + , Olaparib and His-FTO for 30 min at 30 °C. i Quantification of the levels of PARP1 auto-PARylation (arrowhead in h). Data were normalized to the no FTO group (N = 5 per group) and analyzed using a two-tailed paired t-test. j UVC-induced PARP1 auto-PARylation in WT or FTO KO HeLa cells. The high molecular weight smear of PARylated PARP1 was eliminated in the Olaparib-treated cells. k Quantification of the levels of PARP1 auto-PARylation in cells. Data were normalized to the 1 min timepoint of the WT group (N = 7 per group) and analyzed using two-way ANOVA with Holm-Šídák’s multiple comparison test (*WT vs KO.1 and # WT vs KO.2). All data are represented as mean ± SEM. Source data are provided as a Source Data file.
The binding of PARP1 to DNA strand breaks results in a conformational change that dramatically enhances its enzymatic activity and catalyzes rapid poly(ADP)ribosylation (PARylation) of itself (auto-PARylation) and of many other nuclear proteins40,41. Consistent with the faster recruitment of PARP1 to the UV damage sites, we found that UV-induced global protein PARylation was markedly upregulated in FTO KO cells (Fig. 4f, g). On the contrary, overexpression of myc-FTO significantly suppressed PARP1 enzymatic activity following UV treatment (Supplementary Fig. 7e, f). Changes in the kinetics of PARP1 relocation to DNA strand breaks, which are associated with its polymerase activity, also prompted us to test the possibility that FTO directly affects the enzymatic activity of PARP1. Indeed, we observed that His-FTO inhibited PARP1 activity by limiting the length of the ADP-ribose polymer formed in vitro (Fig. 4h, i). Moreover, significant increases in PARP1 auto-PARylation could be observed in FTO KO cells (Fig. 4j, k), underscoring the role of FTO as an endogenous negative regulator of PARP1 function in cells.
To establish a direct involvement of FTO in DDR through PARP1 interaction, we utilized AlphaFold to predict the binding interface between FTO and PARP142. AlphaFold generated several models with different interfaces; all showed relatively low confidence scores. However, this may be expected as there is currently no experimentally determined structure of full-length PARP1. Despite this, several models predicted the N-terminal domain of FTO to be involved in the interaction with PARP1 (Fig. 5a and Supplementary Fig. 8a). Based on this prediction, we produced recombinant His-FTO protein with a deletion in the first 28 amino acids at the N-terminal region (FTO-Δ28). In vitro binding assays revealed a significant reduction in the binding of recombinant Flag-PARP1 protein to His-FTO-Δ28 compared to full-length His-FTO protein (Fig. 5b, c), demonstrating that FTO directly interacts with PARP1 in part through its N-terminal helix. Next, we transfected full-length myc-FTO or myc-FTO-Δ28 into FTO KO HeLa cells and performed PLA assays to determine their proximal interactions with endogenous PARP1. We found that myc-FTO-Δ28 formed significantly less PLA puncta than full-length myc-FTO, which could be further reduced when the cells were exposed to UV irradiation (Fig. 5d, e). Consequently, re-expression of myc-FTO-Δ28 could only partially rescue dispersed PARP1 clusters, while full-length myc-FTO WT completely restored high-intensity PARP1 clusters in the FTO KO cells (Fig. 5f, g).

a A schematic diagram of the domain structure of FTO depicting binding sites for Fe2+ and PARP1. b Recombinant Flag-PARP1 proteins have reduced binding to His-FTO-Δ28 (28 amino acid deletion at the N-terminal) compared to full-length (FL) His-FTO. c Quantification of PARP1 binding. Data were normalized to the His-FTO-FL group (N = 7) and analyzed using a two-tailed paired t-test. d FTO KO HeLa cells expressing myc-FTO (WT, Δ28 or DD) were irradiated with 100 J UVC and recovered for 4 min. PLA signals (gray) between endogenous PARP1 and myc-FTO were present within nuclei (marked by dashed lines that outline the DAPI stain). Transfected cells were identified by the presence of myc staining (green). Scale bar, 20 μm. e Quantification of the number of PLA puncta per nucleus. (WT-Ctrl, N = 55 cells; WT-UV, N = 51 cells; Δ28-Ctrl, N = 48 cells; Δ28-UV = 29 cells; DD-Ctrl = 43 cells; DD-UV = 35 cells). Data were analyzed using one-way ANOVA with Tukey’s multiple comparison test. f Representative Airyscan2 confocal images of FTO KO HeLa cells overexpressing myc-FTO (WT, Δ28 or DD) that were irradiated with 100 J UVC or not, and immunostained for myc (green), PARP1 (magenta) and DAPI (blue). Scale bars, 10 μm and 1 μm (inset). g Quantification of the number of PARP1 clusters per nucleus area (KO-Ctrl, N = 80 clusters; KO-UV, N = 64 clusters; WT-Ctrl, N = 64 clusters; WT-UV, N = 39 clusters; Δ28-Ctrl, N = 52 clusters; Δ28-UV = 36 clusters; DD-Ctrl = 59 clusters; DD-UV = 43 clusters). Data were analyzed using one-way ANOVA with Tukey’s multiple comparison test. All data are represented as mean ± SEM. Source data are provided as a Source Data file.
Using a molecular replacement strategy, we re-introduced myc-FTO (WT or Δ28) in FTO KO HeLa cells and determined their effects on PARP1 functions in DDR. Firstly, we found that the expression of myc-FTO WT not only normalized the rate of GFP-PARP1 association to lesioned DNA but also reduced the amount of GFP-PARP1 recruitment (Fig. 6a–e), mimicking the effect of FTO overexpression in cells (Supplementary Fig. 7a–d). In contrast, the effects of myc-FTO-Δ28 expression on the rate and extent of GFP-PARP1 recruitment to the DNA damage sites were indistinguishable from those in FTO KO cells (Fig. 6a–e). Secondly, UV-induced global protein PARylation was markedly downregulated in FTO KO cells that expressed myc-FTO WT, but was only partially rescued by myc-FTO-Δ28 (Fig. 6f, g). Consistent with these findings, re-expression of myc-FTO WT significantly reduced the rate of DNA repair (measured by the levels of CPD, Fig. 6h, i) and resulted in poor cell survival compared to FTO KO cells (Fig. 6j). On the other hand, the expression of myc-FTO-Δ28, which significantly reduced the interaction with PARP1, only exhibited partial effects (Fig. 6h–j). These data demonstrate the requirement of the FTO N-terminal helix in interacting with and regulating PARP1 functions in cells.

a Live imaging of FTO HeLa KO cells expressing GFP-PARP1 and myc-FTO (WT, Δ28 or DD) following UVA micro-irradiation. Scale bar, 5 μm. b and c Average (b) and normalized (c) traces of GFP-PARP1 fluorescence intensity changes over time. Solid and dashed lines indicate means and SEM, respectively. d and e Quantification of the peak fluorescent intensity (d) and the time-constant (τ) of fluorescence recovery after photobleaching (e) of GFP-PARP1 at DNA damage sites (KO, N = 11 cells; WT, N = 13; Δ28, N = 12; DD, N = 11). Data were analyzed using one-way ANOVA with Tukey’s multiple comparison test. f UVC-induced PARylation in FTO KO HeLa cells were transduced with or without lentiviral particles expressing myc-FTO (WT, Δ28 or DD). Total proteins were visualized by Revert Protein Stains on the PVDF membrane. g Quantification of total PAR levels. Data were normalized to the 1 min timepoint of the KO group (KO, N = 6; WT, N = 7; Δ28, N = 5; DD, N = 6) and analyzed using two-way ANOVA with Holm-Šídák’s multiple comparison test. h Dot-blot detection of the CPDs in genomic DNA of FTO KO HeLa cells expressing myc-FTO (WT, Δ28 or DD). Methylene blue showed the amount of DNA loaded onto the membrane. i Quantification of CPD levels. Data were normalized to the 1 min timepoint of the KO group (KO, N = 8; WT, N = 8; Δ28, N = 8; DD, N = 9) and analyzed using two-way ANOVA with Tukey’s multiple comparison test. j The percentage of cell survival was determined by quantifying the number of colonies and normalizing them to the non-irradiated control groups (KO, N = 9; WT, N = 8; Δ28, N = 9; DD, N = 8). Data were analyzed using two-way ANOVA with Tukey’s multiple comparison test. All data are represented as mean ± SEM. Source data are provided as a Source Data file.
However, it is important to note that FTO contains two nuclear localization signals (NLS), one at the N-terminus (residues 1–31) and another at the C-terminus43. Due to the deletion of the N-terminal NLS, myc-FTO-Δ28 became partially mislocalized in the cytoplasm (Fig. 5d, f and Supplementary Fig. 8b, c), which may also explain why this FTO deletion mutant failed to rescue the phenotypes associated with FTO KO cells. Regardless of the exact underlying mechanisms, FTO-Δ28 represents a loss-of-function mutant that allows us to demonstrate the functional importance of direct FTO-PARP1 interaction and/or FTO nuclear localization in DDR.
FTO regulates PARP1 function independent of its RNA demethylase activity
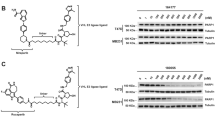
Given the emerging role of m6A signaling in DDR18,19,20,21, we next investigated the involvement of FTO RNA demethylase activity in regulating PARP1 function and the DNA repair pathway using two complementary approaches. First, we used a small molecule FTO inhibitor FB23-2 (ref. 14) and confirmed that 72 h treatment with this compound resulted in a significant accumulation of m6A levels in HeLa cells (Supplementary Fig. 9a, b). Interestingly, we found that pharmacological inhibition of FTO demethylase activity with 10 μM FB23-2 did not significantly affect the UV-induced GFP-PARP1 recruitment in cells (Supplementary Fig. 9c–f), PARP1 catalytic activity (Supplementary Fig. 9g, h) or the interaction between FTO and PARP1 in control or UV-treated cells (Supplementary Fig. 9i, j). Furthermore, we also found that FB23-2 treatment did not change the levels of CPD formation or cell survival following UV exposure (Supplementary Fig. 9k–m).
To corroborate these findings, we performed molecular replacement analyses of FTO KO cells overexpressing a catalytically inactive FTO H231A/D233A (DD) mutant44, which abrogates its ability to bind Fe2+ (Fig. 5a). Myc-FTO-DD mutant displayed the expected nuclear localization and interacted with PARP1 to a level comparable to wild-type FTO (Fig. 5d, e). The re-introduction of myc-FTO-DD also effectively restored high-intensity PARP1 clusters in the nucleoli of FTO KO cells (Fig. 5f, g). Like my-FTO-WT, overexpression of myc-FTO-DD suppressed the recruitment of GFP-PARP1 to the DNA damage sites (Fig. 6a–e), reduced the levels of UV-induced global protein PARylation (Fig. 6f, g) and the rate of DNA repair (Fig. 6h, i), resulting in poorer cell survival compared to FTO KO cells (Fig. 6j). Overall, these results provide strong evidence supporting the notion that the role of FTO as a negative regulator of PARP1 function in UV-induced DDR is independent of its m6A-demethylase activity.
FTO is a substrate of PARP1
Considering the close interaction between FTO and PARP1, we also examined whether FTO is a direct target of PARP1, revealing that FTO could indeed be PARylated by PARP1 in vitro (Fig. 7a). Due to its proximity to PARP1, a low FTO PARylation could be detected in cells under basal conditions (Fig. 7b, c). Consistent with the activation of PARP1 catalytic activity, FTO became hyper-PARylated 1 min post-UV exposure. However, as the activity of PARP1 waned, the FTO PARylation was significantly down-regulated at 4 min post-UV exposure and returned to baseline at 10 min. Interestingly, the level of PARP1 auto-PARylation showed an inverse correlation with the extent of FTO-PARP1 interaction in cells measured by two independent assays, namely PLA (Fig. 7d, e) and BAR followed by western blotting analysis (Fig. 7f, g). Moreover, the time course of FTO PARylation also inversely correlated with the dispersal of PARP1 clusters in cells following UV treatment (Supplementary Fig. 10a–c). These data suggest a role for protein PARylation in regulating the interaction between these two proteins. To address this possibility, we performed an in vitro binding assay and found that the addition of the PARP1 substrate, nicotinamide adenine nucleotide (NAD + ), dramatically enhanced the interaction between FTO and PARP1 (Fig. 7h). In contrast, when auto-PARylation of PARP1 occurred before His-FTO was added, no augmentation of FTO-PARP1 binding was observed. The dynamic interaction between FTO and PARP1 was not affected by inhibition of FTO demethylase activity (Fig. 5d, e). Based on these results, we propose a model in which a low level of FTO PARylation supports the interaction between FTO and PARP1 under basal conditions. As the activity of PARP1 increases following UV exposure, its auto-PARylation creates steric hindrance and charge repulsion, which subsequently promotes its dissociation from FTO (Fig. 7i).

a Recombinant His-FTO proteins are PARylated in vitro. b Endogenous FTO protein is PARylated in response to UVC irradiation in HeLa cells. c Quantification of FTO PARylation levels. Data were normalized and compared to non-irradiated control groups using a two-tailed Mann-Whitney test (0 min, N = 7; 1 min, N = 5; 4 min, N = 7; 10 min, N = 6). d Time course of FTO-PARP1 proximal interaction in WT HeLa cells upon UVC irradiation. Scale bar, 20 μm. e Quantification of the number of PLA puncta per nucleus (Ctrl, N = 194 cells; UV-1 min, N = 154 cells; UV-4 min, N = 153 cells; UV-10 min, N = 93 cells). Data were analyzed using one-way ANOVA with Tukey’s multiple comparison test. f Time course of FTO-PARP1 proximal interaction determined by BAR assays. g Quantification of biotinylated PARP1. Data were normalized and compared to non-irradiated control groups using a two-tailed Mann-Whitney test (0 min, N = 4; 1 min, N = 3; 4 min, N = 4; 10 min, N = 3). All data represent mean ± SEM. h Recombinant Flag-PARP1 was incubated with a combination of NAD+, Olaparib or His-FTO for 30 min at 30 °C, followed by Ni2+-Sepharose pull-down. In condition a, Olaparib was added at the end of 30 min incubation to stop the PARylation reaction. In condition b, His-FTO was added after the PARylation reaction. Representative blots from 4 independent experiments are shown. i A schematic diagram of the working model. We propose that FTO maintains the clustering PARP1 and dampens its catalytic activity in the nucleolus. Upon UV exposure, PARP1 dissociates from FTO, allowing it to be recruited to the sites of DNA damage. Loss of FTO expression enhances PARP1 enzymatic activity and the rate of PARP1 recruitment to DNA damage sites, thereby accelerating DNA repair and promoting cell survival. Created in BioRender. Anggono, V. (2025) https://BioRender.com/r35n837. Source data are provided as a Source Data file.
Discussion
In this study, we used quantitative MS to map the proximal interactome of endogenous FTO and identified a functional interaction with the DNA damage sensor PARP1. Interestingly, this interaction was not observed in previous studies that rely on overexpressing the FTO-BioID2 construct or chemical cross-linking approaches27,36. Furthermore, there are minimal overlaps with those reported in those studies27,36 or the BioGRID database45. Strikingly, approximately one-third of FTO interacting proteins were also reported in a recent PARP1 proximal interactome study37, suggesting that they share common cellular functions, which include DNA repair, ribosome biogenesis, RNA splicing and chromatin remodeling.
Recent studies have implicated FTO in the DDR pathway induced by various stressors12,26,27,28. Consistent with our findings that the loss of FTO promotes DNA repair in 3 different cell lines (HeLa, U2OS and HCT116), epidermis-specific deletion of FTO also prevents arsenic- and UVB-induced skin tumorigenesis in mice28. In contrast, some studies have reported that FTO is required to protect cells from DNA-damaging agents such as 2-hydroxyurea, H2O2, cisplatin or UVC irradiation12,26,27. Generally, these studies indicate that FTO functions as an RNA demethylase to control the expression of genes involved in the DNA repair pathway, including PARP1 (ref. 12). However, we did not observe any significant changes in the expression of PARP1 protein or PARP1 mRNA in FTO KO HeLa cells. More importantly, pharmacological inhibition of FTO demethylase activity with FB23-2 or overexpression of the catalytic-inactive FTO-DD mutant did not have any significant effect on UV-induced DNA repair and cell survival, suggesting that FTO has an RNA demethylase-independent function in the DDR pathway.
Here we demonstrate that FTO regulates the DDR by physically interacting with PARP1 through the N-terminal helix and regulating its function. Firstly, FTO acts as a scaffolding molecule to cluster inactive PARP1 inside the nucleolus. Upon UV exposure, PARP1 rapidly dissociates from FTO and is recruited to the sites of DNA lesions to promote DNA repair. In FTO KO HeLa cells, PARP1 molecules appear more dispersed and are recruited at a faster rate to DNA damage sites. Secondly, FTO inhibits PARP1 enzymatic activity both in vitro and in vivo. The apparent increases in the levels of global protein PARylation and PARP1 auto-PARylation coincide with more efficient DNA repair in FTO KO cells, likely by promoting the recruitment of DNA repair proteins and facilitating the release of PARP1 from damaged DNA. Moreover, FTO overexpression suppresses the recruitment of PARP1 and decreases the level of UV-induced protein PARylation. Furthermore, we showed that the N-terminal deletion of FTO significantly reduces PARP1 binding and can only partially regulate PARP1 function in cells. These lines of evidence directly support the role of FTO as an endogenous suppressor of the DDR by negatively regulating PARP1 function.
FTO is a direct target of PARP1 and is PARylated in cells. Mechanistically, the interaction between FTO and PARP1 relies on the basal level of FTO PARylation. The activation of PARP1 triggers auto-PARylation, which may create a steric hindrance and force its dissociation from FTO. The high level of FTO PARylation observed shortly after UV exposure could also contribute to its dissociation not only with PARP1 but also with other repair proteins, including DNA-PK and Ku80. Interestingly, the interaction of FTO and PARP1 is largely independent of the former’s RNA demethylase function. Therefore, our study highlights an important mechanism of FTO protein in the DDR beyond its role as an RNA demethylase. Given that METTL3 recruitment to DNA lesions is dependent on PARP1 function, our findings are consistent with the apparent increase in the level of m6A observed in FTO KO cells following UV irradiation18. However, based on our working model, we propose that the increase in m6A level is due to enhanced PARP1 function, thus facilitating the more efficient recruitment of METTL3 to sites of DNA damage instead of the loss of RNA demethylase function in FTO KO cells per se.
The enzymatic activity of PARP1 can be positively modulated by several interacting proteins, including the mammalian sirtuin SIRT6 (ref. 46), the DNA repair protein XPC47, the microtubule-associated protein TPX2 (ref. 37), and the histone PARylation factor HPF1 (ref. 48). HPF1 robustly interacts with PARP1 and completes the enzymatic active site, which in turn determines the specificity of PARP1 towards serine-linked PARylation48,49,50. Interestingly, none of these PARP1 positive modulators was identified in the FTO proximal proteome, further supporting the role of FTO as a unique suppressor of PARP1 activity in cells. One possible mechanism of action may involve the ability of FTO to bind NADP+ and NAD+ (ref. 51), which limits the availability of PARP1 substrate. FTO could therefore be responsible for preventing hyperactivation of PARP1 which could be detrimental to cells due to excessive consumption of NAD+ substrate, leading to energy collapse and cell death52.
In conclusion, our study uncovers an unexpected role of FTO in regulating multiple aspects of PARP1 function, which are not affected by the inhibition of its RNA demethylase activity. We suggest that FTO acts as a scaffolding molecule that is required for the clustering of PARP1 in the nucleolus. The dissociation of FTO regulates the dynamic dispersal of nucleolar PARP1 upon UV-induced DNA damage, suggesting a regulatory mechanism that controls the recruitment of PARP1 to the sites of DNA lesions. Our work also highlights that FTO negatively modulates PARP1 catalytic activity, suggesting the potential clinical use of FTO antisense oligonucleotide to enhance PARP1 sensitivity and increase the efficacy of PARP1 inhibitors for the treatment of many cancer types in human.
Methods
Generation of FTO KO cell lines
HeLa (CCL-2), U2OS (HTB-96) and HCT116 (CCL-247) cells were obtained from ATCC. FTO KO cell lines were generated using the CRISPR/Cas-9 system as previously described30. Three single-guide RNAs (sgRNAs) were designed to target exon 1 of the FTO gene (sgRNA#1 – CACCGAAGCGCACCCCGACTGCCG, sgRNA#2 – CACCGAGCTTCGCGCTCTCGTTCCT and sgRNA#3 – CACCGCTCTCGTTCCTCGGCAGTCG) and cloned into pSpCas9(BB)-2A-GFP vector (a gift from Feng Zhang, Addgene plasmid #48138)53. After transfection, GFP-positive cells were isolated by fluorescence-activated cell sorting and grown as single clones. FTO KO cells were screened and confirmed by genomic DNA sequencing and western blot analysis. The resulting cell lines were HeLa FTO KO.1 (sgRNA#1), FTO KO.2 (sgRNA#2), U2OS FTO KO (sgRNA#1), HCT116 FTO KO.1 (sgRNA#1) and HCT116 KO.3 (sgRNA#3). Most experiments were performed with FTO KO.2 HeLa cells, unless otherwise stated.
Antibodies
The following antibodies were obtained commercially: mouse anti-β-actin (clone C4, Santa Cruz Biotechnology, sc-47778, WB 1:5000), mouse anti-CPD (clone TDM-2, Cosmo Bio USA, CAC-NM-DND-001, WB 1:1000), rabbit anti-DNA PK (Cell Signaling Technology, #4602, WB 1:1000), rabbit anti-FLAG (clone D6W5B, Cell Signaling Technology, #14793, WB 1:1000), rabbit anti-FTO (clone EPR6894, Abcam, ab126605, WB 1:1000, IF 1:250, BAR 1:2000, PLA 1:250), rabbit anti-phospho-Ser-139 Histone H2A.X (clone 20E3, Cell Signaling Technology, #9718, WB 1:1000, IF 1:500), rabbit anti-Ku80 (clone C48E7, Cell Signaling Technology, #2180, WB 1:1000), rabbit anti-m6A (Abcam, ab151230, WB 1:1000), rabbit anti-myc (clone A-14, Santa Cruz Biotechnology, sc-789, IF 1:500, PLA 1:500), mouse anti-Myc (clone 9E10, Santa Cruz Biotechnology, sc-40, WB 1:500), mouse anti-PARP1 (clone A6.4.12, Bio-Rad, MCA1522G, WB 1:1000, IF 1:250, PLA 1:250), mouse anti-PAR (clone 10HA, Trevigen, 4335-MC-100, WB 1:1000), mouse anti-PAR (10H, Merck, MABC547, WB 1:1000) and mouse anti-α-tubulin (clone DM1A, Cell Signaling Technology, #3873, WB 1:10,000). Alexa-conjugated secondary antibodies or streptavidin were purchased from Thermo Scientific. Horseradish peroxidase (HRP)-conjugated secondary antibodies were obtained from GE Healthcare.
DNA plasmids
FTO cDNA was isolated from the human cDNA library by standard PCR with the following primers: 5′- TTGTCGACCAGCATGAAGCGCACCCCGACT-3′ (sense) and 5′- TTGCGGCCGCCTAGGGTTTTGCTTCCAGAA-3′ (anti-sense), and cloned into the pRK5-myc vector or FUW-myc lentiviral vector using the SalI and NotI restriction sites. FTO-Δ28 and FTO-DD (H231A/D233A) were generated using a standard and an overlapping PCR protocol, respectively.
Cell culture
All cell lines were grown in Dulbecco’s modified Eagle’s medium (DMEM) with 4.5 g/L glucose supplemented with 10% fetal bovine serum (FBS), 50 U/mL penicillin and 50 mg/mL streptomycin in a humidified 5% CO2 incubator at 37 °C. Cells were transfected using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol and processed the next day or transduced with lentiviral particles. For SILAC labeling, HeLa cells were cultured in DMEM (Silantes) containing unlabeled or labeled 2H4-lysine (Lys-4) and 13C6-arginine (Arg-6) for 8 passages. Labeling efficiency reached ~96% after five passages. To induce DNA damage, cells were pre-treated with 10 μM bromodeoxyuridine (BrdU, Sigma) overnight before UV irradiation exposure. UVC irradiation (254 nm) was performed in a Stratalinker 2400 (Stratagene) or the CL-508 UV cross-linker (UVITec) at the indicated dose (0–100 J/m2). UVA micro-irradiation (405 nm) was performed on an LSM710 confocal microscope (Zeiss). The PARP1 inhibitor Olaparib and FTO blocker FB23-2 were purchased from Selleck. They were dissolved in DMSO and used at the indicated concentrations.
Lentiviral packaging
Lentiviral particles encoding myc-FTO (WT, Δ28 or DD) were generated in HEK293T cells (ATCC, CRL-3216) that had been transfected with 10 μg of FUW-myc-FTO plasmid, 5 μg each of pMD2.G envelope plasmid, pRSV-Rev encoding plasmid and pMDLg/pRRE packaging constructs via the calcium-phosphate precipitation method. Lentivirus-containing supernatant was collected 48 h after transfection and passed through a 0.45 μm cellulose acetate low protein binding membrane. Lentiviral particles were pelleted by ultracentrifugation at 106,559 g at 4 °C for 2 h, resuspended in OptiMEM medium (Invitrogen), snap-frozen in liquid nitrogen and stored at -80 °C.
Cyclobutene pyrimidine dimer (CPD) dot blot assay
Genomic DNAs were extracted from HeLa cells using the DNAzol reagent (Invitrogen) according to the manufacturer’s instructions. DNAs (500 ng) were spotted onto Hybond-N+ nylon membranes (GE Healthcare) and air-dried for 10 min. The membrane was stained with 0.04% methylene blue solution (in sodium acetate pH 5.2) for 5 min to visualize the loading of total RNA on a ChemiDoc XRS+ gel imaging system (Bio-Rad). After extensive washing with methanol, membranes were blocked in 5% skim milk (in PBS containing 0.1% Tween-20) for 1 h and incubated with anti-CPD antibodies (Cosmo Bio USA) overnight at 4 °C. Membranes were washed the next day and incubated with HRP-secondary antibody for 1 h at room temperature. They were then washed extensively and developed using the enhanced chemiluminescence (ECL) method (PerkinElmer). Images were acquired with an Odyssey Fc imaging system (LiCOR) and quantified using ImageStudio software.
γH2A.X immunostaining
Cells were fixed with Parafix solution (4% paraformaldehyde, 4% sucrose in PBS) for 10 min at room temperature, followed by a 2 min methanol fixation at -20 °C. They were then permeabilized with 0.025% Triton X-100 in PBS for 10 min, washed and blocked with 10% normal goat serum (NGS) in PBS for 1 h. After incubation with anti-phospho-Ser-139 H2A.X antibodies (Cell Signaling Technologies) overnight at 4 °C, cells were washed five times in PBS and incubated with Alexa-conjugated secondary antibodies, washed and stained with DAPI in PBS. Images were taken with Zeiss Axio Imager fluorescence microscope and analyzed using ImageJ software (National Institutes of Health).
Colony formation assay
Cells were seeded at low density (500 cells per 6 cm dish) a day prior to UV treatments at the indicated dose, then returned to culture for 14 days prior to being fixed and stained with crystal violet solution (0.05% w/v crystal violet, 1% v/v formaldehyde, 1% v/v methanol in PBS) for 20 min. Tissue culture plates were washed extensively and air-dried.
Biotinylation by antibody recognition (BAR) assay
BAR assays were performed according to a previously published protocol with minor changes32. Briefly, cells were fixed, permeabilized and oxidized with 0.5% H2O2. They were then blocked in 3% bovine serum albumin (BSA) in PBS containing 0.1% Tween-20 (PBS-T) for 1 h at room temperature and incubated with anti-FTO antibodies (Abcam; diluted in 3% BSA containing PBS-T) overnight at 4 °C. After extensive washes with PBS-T, samples were incubated with HRP-conjugated anti-rabbit antibodies (in 3% BSA containing PBS-T) for 2 h at room temperature. Samples were washed three times with PBS and incubated with 500 mM of biotin tyramide (Iris Biotech) in PBS for 30 min. The biotin labeling reaction was induced by adding 1 mM H2O2 for 1 min and stopped by adding 500 mM of sodium ascorbate to the reaction. After washing with PBS-T, cells were processed for immunostaining or affinity-purification for biochemical analyses.
For immunostaining, biotinylated proteins and FTO immunostaining were detected with Alexa-488-conjugated streptavidin and Alexa-568-conjugated anti-rabbit antibodies, respectively, followed by analysis using a Zeiss Axio Imager epifluorescence microscope.
For biochemical analysis, cells were lyzed with ice-cold RIPA buffer (50 mM Tris, 100 mM NaCl, 2 mM EDTA, 2 mM EGTA, 10 mM Na-pyrophosphate, 50 mM NaF, 1% Triton X-100, 0.5% Na-deoxycholate and 0.1% SDS) supplemented with Complete-EDTA free protease inhibitor cocktails (Roche). Cell lysates were sonicated, boiled for 30 min and centrifuged at 20,627 g for 20 min at 4 °C. Cleared lysates were then incubated with Dynabeads M-280 streptavidin magnetic beads (Invitrogen) or neutravidin agarose resin (Thermo Scientific), followed by mass spectrometry or western blot analyses, respectively.
Mass spectrometry
Immunoprecipitated samples were separated by SDS-PAGE stained with Coomassie Brilliant Blue R-250. Each sample was excised into 3 equal fractions and cut into 0.5-1 mm gel cubes. Proteins were equilibrated in 100 µL of 100 mM ammonium bicarbonate (pH 8.0), destained in 50 mM ammonium bicarbonate/50% acetonitrile (pH 8.0), and dehydrated in 100% acetonitrile. The gel pieces were dried and incubated with 10 mM 1,4-dithiothreitol (Roche) for 30 min at 55 °C and then alkylated in 20 mM iodoacetamide (Sigma) for 30 min at room temperature. After washing, trypsin (12.5 ng/µL, Promega) was added to the gel pieces and they were incubated overnight at 37 °C. Following overnight digestion, tryptic peptides were extracted with 50% acetonitrile/2% formic acid and dried under vacuum centrifugation. They were resuspended in 0.1% formic acid for desalting using pre-equilibrated C18 OMIX tips (Agilent). Bound peptides were eluted with 50% acetonitrile/0.1% formic acid, dried under vacuum centrifugation, and stored at -80 °C until analysis. The peptides were resuspended in 0.1% formic acid, bath sonicated for 15 min and centrifuged at 14,000 g for 15 min to remove any insoluble debris. The clarified peptides were transferred into glass vials for liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis.
The tryptic peptides were separated on an Ultimate 3000 nano-LC (Thermo Scientific) fitted with the Acclaim PepMap C18 reversed-phase column (15 cm length, 75 μm diameter and 2 μm particle size, Thermo Fisher Scientific) and eluted with a linear gradient from 2 to 80% acetonitrile containing 0.1% formic acid) for 60 min. Eluted peptides were subsequently ionized into a Q Exactive Plus mass spectrometer (Thermo Scientific). Precursor ions were selected for MS/MS fragmentation using a data-dependent “Top 10” method operating in Fourier transform (FT) acquisition mode with higher-energy C-trap dissociation (HCD) fragmentation. FT-MS analysis on the Q Exactive Plus was carried out at 70,000 resolution with an automated gain control (AGC) target of 5 × 105 ions in full MS. MS/MS scans were carried out at 17,500 resolution with an AGC target of 1 × 105 ions. Maximum injection times were set to 50 and 110 ms, respectively. The charge exclusion was set to unassigned and 1+ charged state with a dynamic exclusion of 20 s. The ion selection threshold for triggering MS/MS fragmentation was set to 44,000 counts, and an isolation width of 1.5 m/z was used to perform HCD fragmentation with a normalized collision energy of 27.
Peptide identification and bioinformatic analyses
Raw spectra files were processed using the Proteome Discoverer software 2.2 (Thermo Scientific) incorporating the Sequest search algorithm. Peptide identifications were determined using a 20-ppm precursor ion tolerance and a 0.1-Da MS/MS fragment ion tolerance for FT-MS and HCD fragmentation. Carbamidomethylation modification ( + 57.021 Da) of cysteines was considered a static modification while oxidation ( + 15.995 Da) of methionine, 13C615N2 ( + 8.014 Da) and 2H4 ( + 4.025 Da) of lysine, 13C615N4 ( + 10.008 Da) and 13C6 ( + 6.020 Da) of arginine, biotin ( + 226.078 Da) of lysine and acetyl ( + 42.011 Da) modification on N-terminal residues were set as variable modifications allowing for maximum two missed cleavages. The data were processed through Percolator to estimate false discovery rates (FDRs). Protein identifications were validated using a q-value of 0.01. PSMs and peptides were validated using a strict FDR for PSMs of 0.01 and 0.05 for a relaxed FDR, respectively. Peptides shorter than 6 amino acids were filtered out. Peptide-spectrum matches (PSMs) were chromatographically aligned for each input file in a sample set with a mass tolerance of 10 ppm and a maximum retention time (RT) shift of 10 min. Peptide groups used for protein quantification were analyzed using the default parameters which set a peptide as unique if it was included in only one protein group. The quantification was processed using unique and razor peptides (peptides shared among multiple proteins group or proteins) with the precursor abundance based on the intensity. Protein abundance was calculated as a sum of the individual peptide group abundances and the ratio was based on a pairwise ratio using a geometric median of the peptide group ratios. A one-way ANOVA was used for the hypothesis test, using the background population of ratios for all peptides and proteins to determine whether any given single peptide or protein was significantly changing relative to that background.
Differential enrichment protein analysis was performed with the R-package DEP54. Only proteins detected in at least 2 out of 3 replicates with an adjusted P-value < 0.05 were considered significantly enriched. Gene ontology (GO) enrichment analysis was performed using the R-package clusterProfiler55. The protein-protein interaction (PPI) network of selected GO terms was generated with STRING (minimum required interaction score > 0.7) and visualized with Cytoscape56,57.
Proximity ligation assay (PLA)
Monolayers of WT and FTO KO HeLa cells were fixed with Parafix solution for 10 min at room temperature, followed by a further 2 min fixation with 100% methanol at -20 °C. Cells were then permeabilized with 0.25% Triton X-100/PBS for 10 min and blocked in 10% normal goat serum for 1 h, after which they were incubated with rabbit anti-FTO (Abcam), rabbit anti-myc (Santa Cruz Biotechnology) and mouse anti-PARP1 (Bio-Rad) antibodies at 4 °C overnight. PLA was carried out with the Duolink® kit (Sigma) according to the manufacturer’s protocol. Briefly, cells were incubated with mouse PLUS and rabbit MINUS Duolink PLA probes at 37 °C for 1 h, ligated at 37 °C for 30 min and amplified (Texas Red) at 37 °C for 100 min. Nuclei were visualized with DAPI staining. Images were acquired on a Zeiss LSM510 confocal microscope with a 63× oil-immersion objective (1.4 N.A.). PLA puncta number and fluorescence intensities within cell nuclei were quantified using the BioVoxxel Toolbox (Speckle Inspector) plugin in ImageJ software (NIH). Nuclei were set as big objects with a minimum object size of 3000 pixels and the PLA fluorescence was set as small speckles with a minimum speckle size of 7 pixels.
Structured illumination and confocal microscopy
Cells were fixed, blocked, permeabilized and stained with rabbit anti-FTO and mouse anti-PARP1 antibodies. FTO and PARP1 were visualized by staining with Alexa-488-conjugated anti-rabbit and Alexa-568-conjugated anti-mouse secondary antibodies, respectively. Nuclei were visualized with DAPI staining. Coverslips were mounted on a glass slide with Vectashield (Vector Laboratories). Images were acquired on a Zeiss ELYRA inverted structured illumination microscope equipped with a 63x Plan-Apochromat oil-immersion objective (1.4 N.A.). Raw SIM files were manually processed with the Zeiss Zen microscopy software using the structured illumination function (noise filter at -5.0 to -6.0 and SR frequency weighting at 1.0), followed by spatial misalignment adjustment (channel alignment method) with calibrated polystyrene beads coated with multiple fluorophores. In some experiments, images were acquired on a Zeiss LSM 980 NLO Airyscan 2 confocal microscope built around an Axio Observer 7 body and equipped with a 63x Plan-Apochromat oil-immersion objective (1.4 N.A.), Airyscan 2 super-resolution detector, a 34-channel spectral PMT (photomultiplier tube) array, two internal GaAsP PMTs, a transmission PMT, and controlled by Zeiss Zen Blue software. Raw images were processed on the Zeiss Zen Blue software using the Airyscan joint deconvolution, and the number of iterations was set at 8.
The rendering of FTO and PARP1 surfaces in the nucleus was created using the surface tool (smooth surface detail set at 0.02 mm) in the Imaris image analysis software (Oxford Instruments), which were subsequently used to calculate the volume (μm3), sphericity and nearest distance (μm) of FTO and PARP1 particles. Nuclear PARP1 clusters and area analyses were performed using the ImageJ software. The minimum cluster radius of PARP1 was thresholded at 3.0 pixels. The number of PARP1 clusters per nucleus was manually counted. The area of the nucleus and each PARP1 cluster was determined by tracing the region of interest (ROI).
Laser micro-irradiation and GFP-PARP1 recruitment assay
A plasmid expressing GFP-PARP1 was generated by subcloning PARP1 cDNA from the pCMV-PARP1-3x-Flag construct (a gift from Thomas Muir, Addgene plasmid #111575)58 into the pEGFP-C3 vector (Clontech). Cells were transfected with the pEGFP-PARP1 construct and treated with 10 μM BrdU in DMEM without phenol red but containing 10% FBS and 1% penicillin and streptomycin overnight. The recruitment of GFP-PARP1 was imaged at 5 Hz on a Zeiss LSM710 confocal microscope. DNA damage was induced by irradiating a 10 pixel-width of the nucleus with a 405 nm laser (line scan mode). The laser was set with a line attenuator transmission of 92%, 1 iteration and a pixel dwell time of 177.32 µs. ROIs were drawn to the DNA damage site to measure the GFP fluorescence over time using the ImageJ software. DNA damage-induced changes in fluorescence were calculated by normalizing the background subtracted values to the baseline fluorescence prior to UVA micro-irradiation (ΔF/Fo). The recruitment time constant (τ) was calculated by fitting the fluorescence recovery after micro-irradiation to a single exponential function.
Cellular PARylation assay
Cells were lyzed in 1% SDS (in PBS) and diluted in 10 volumes with ice-cold cell lysis buffer (1% Triton X-100, 1 mM EDTA, 1 mM EGTA, 50 mM NaF, 5 mM Na-pyrophosphate in PBS) supplemented with Complete EDTA-free protease inhibitor cocktails (Roche). Cell lysates were sonicated and centrifuged at 20,627 g for 20 min at 4 °C. Global PARylation of cellular proteins were detected by SDS-PAGE and western blotting with anti-PAR antibodies (Trevigen or Merck). α-tubulin or β-actin blots were used as loading controls. To detect PARP1 auto-PARylation, total cell lysates were probed with anti-PARP1 antibodies (BioRad) to reveal the slower migrating bands, which were not present in Olaparib-treated cells (negative control). To detect PARylation of endogenous FTO protein, cell lysates were subjected to immunoprecipitation with anti-FTO antibodies (Abcam) that were coupled to protein A Sepharose beads at 4 °C overnight. Bound proteins were washed and eluted with 2X SDS sample buffer for 10 min at 95 °C. Eluates were resolved on SDS-PAGE and blotted with anti-PAR and anti-FTO antibodies.
In vitro binding assay
The full-length FTO open-reading frame was subcloned into the pRSFDuet1 bacterial expression vector. FTO construct lacking the first 28 amino acids (Δ28) was also subcloned into the same bacterial expression vector. The expression and purification of recombinant His-FTO proteins (full-length or Δ28) in Escherichia coli BL21(DE3) cells were performed according to a previously published method59. For the in vitro binding assay, 5 μg of purified His-FTO recombinant proteins were pre-coupled to 30 μL of Ni Sepharose beads (Cytiva, pre-blocked with 1.5% BSA in PBS-T) in binding buffer (0.5% Triton X-100, 0.5 mM EDTA, 0.5 mM EGTA, 25 mM NaF, 2.5 mM Na-pyrophosphate, 10 mM Na phosphate, 0.25 M NaCl, 10 mM imidazole in 0.5× PBS, PH 7.4) for 1 h at room temperature. The beads were washed twice in the binding buffer before incubation with 1 μg of recombinant Flag-PARP1 proteins (Abcam) for 1 h at room temperature. The beads were then washed four times and eluted by boiling them in 2× SDS sample buffer for 10 min at 95 °C. Eluates were resolved on SDS-PAGE, and either blotted with anti-Flag and anti-FTO antibodies or stained with Coomassie Brilliant Blue R-250.
In vitro PARylation assay
The in vitro PARylation assay was performed by adding 1 μg of recombinant Flag-PARP1 protein in 50 μL of reaction buffer (20 mM Tris-Cl, 100 mM NaCl, 10 mM MgCl2, 10 mM ZnCl2, 10% glycerol, 300 mM NAD + , pH 8.0). The addition of 10 μM of PARP1 inhibitor Olaparib or the omission of NAD+ in a reaction served as negative controls. Recombinant His-FTO recombinant proteins (5 μg) were added to the reaction mix as specified. The reactions were performed at 30 °C for 30 min. PARP1 PARylation was analyzed by western blotting using anti-Flag antibodies (Cell Signalling Technology). To detect FTO PARylation, the reaction containing His-FTO was incubated with Ni Sepharose beads, washed and eluted in 2X SDS sample buffer for 10 min at 95 °C. FTO PARylation was analyzed by western blotting using anti-PAR antibodies.
Western blotting
Samples were loaded in 7.5 or 10% SDS-PAGE gels and separated at 110 V for 1-2 h. Proteins were then transferred to a PVDF membrane at 100 V for 2 h. Membranes were blocked in 5% skim milk (in TBS containing 0.1% Tween-20, TBS-T) for 1 h and washed in TBS-T three times at 5 min intervals prior to overnight incubation with primary antibodies at 4 °C. Membranes were washed in 1% milk/TBS-T five times and incubated with HRP-conjugated secondary antibodies (GE Healthcare, 1:10,000) for 1 h at room temperature. They were then washed extensively and developed using the enhanced chemiluminescence method (PerkinElmer). In some experiments, the total protein load was visualized by Revert Protein Stains (LiCOR) according to the manufacturer’s instructions. Images were acquired with a LiCOR imaging system and quantified with the ImageStudio software. Uncropped blots are provided in the source data file.
m6A dot blot
Total RNA was extracted using Trizol (Invitrogen) and normalized to the same concentration. RNA was denatured at 95 °C for 3 min and then chilled on ice. An equal volume of RNA was spotted on a Hybond-N+ nylon membrane (GE Healthcare), air-dried and UV crosslinked. The membrane was stained with 0.04% methylene blue solution (in sodium acetate pH 5.2) for 5 min to visualize the loading of total RNA on a ChemiDoc XRS+ gel imaging system (Bio-Rad). After extensive washing with methanol, the membrane was blocked with 5% skim milk in PBS containing 0.1% Tween, incubated with anti-m6A antibody (Abcam) overnight at 4 °C, followed by incubation with HRP-conjugated secondary antibodies (GE Healthcare, 1:10,000) and detection with the enhanced chemiluminescence method (PerkinElmer).
Quantitative RT-PCR
Reverse transcription of total RNA from wild-type or FTO KO HeLa cells was performed using SuperScript IV (Invitrogen) according to the manufacturer’s instructions. Quantitative real-time PCR with Rotor-Gene Q real-time cycler (Qiagen) was performed using SYBR-green amplification (Takara Bio). PARP1 and ACTB transcript levels were measured using the following primers: PARP1 FP 5′-GGGCAAGCACAGTGTCAAAG-3’ and PARP1 RP 5′-CCAGATGAAATCCCGGTCCC-3′, ACTB FP 5′-GCAGGAGTATGACGAGTCCGGC-3′ and ACTB RP 5′-GTAACAACGCATCTCATATTTG-3′, respectively. PARP1 expression was calculated using the standard comparative Ct method after normalizing to the housekeeping gene ACTB.
Statistical analysis
The sample size (N) reported in figure legends represents individual cells or wells generated from at least three independent experiments, unless otherwise stated. Statistical analysis was performed in Graph Pad Prism 9.0 using one-way or two-way analysis of variance (ANOVA). A two-tailed student’s t-test was employed to compare between two groups. All data are reported as mean ± standard error of the mean (SEM), unless stated otherwise.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE60 partner repository with the dataset identifier PXD041638. Source data are provided with this paper.
References
Tubbs, A. & Nussenzweig, A. Endogenous DNA damage as a source of genomic instability in cancer. Cell 168, 644–656 (2017).
Negrini, S., Gorgoulis, V. G. & Halazonetis, T. D. Genomic instability-an evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 11, 220–228 (2010).
Ray Chaudhuri, A. & Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 18, 610–621 (2017).
Huang, D. & Kraus, W. L. The expanding universe of PARP1-mediated molecular and therapeutic mechanisms. Mol. Cell 82, 2315–2334 (2022).
Wei, J. et al. Differential m(6)A, m(6)Am, and m(1)A demethylation mediated by FTO in the cell nucleus and cytoplasm. Mol. Cell 71, 973–985 (2018).
Jia, G. et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 7, 885–887 (2011).
Mauer, J. et al. Reversible methylation of m(6)Am in the 5’ cap controls mRNA stability. Nature 541, 371–375 (2017).
Li, Y., Su, R., Deng, X., Chen, Y. & Chen, J. FTO in cancer: functions, molecular mechanisms, and therapeutic implications. Trends Cancer 8, 598–614 (2022).
Li, Z. et al. FTO plays an oncogenic role in acute myeloid leukemia as a N(6)-methyladenosine RNA demethylase. Cancer Cell 31, 127–141 (2017).
Su, R. et al. R-2HG exhibits anti-tumor activity by targeting FTO/m(6)A/MYC/CEBPA signaling. Cell 172, 90–105.e123 (2018).
Liu, W. et al. FTO regulates the DNA damage response via effects on cell-cycle progression. Mutat. Res. Genet Toxicol. Environ. Mutagen 887, 503608 (2023).
Wang, J. et al. FTO promotes colorectal cancer progression and chemotherapy resistance via demethylating G6PD/PARP1. Clin. Transl. Med. 12, e772 (2022).
Su, R. et al. Targeting FTO suppresses cancer stem cell maintenance and immune evasion. Cancer Cell 38, 79–96.e11 (2020).
Huang, Y. et al. Small-molecule targeting of oncogenic FTO demethylase in acute myeloid leukemia. Cancer Cell 35, 677–691 (2019).
Huff, S., Tiwari, S. K., Gonzalez, G. M., Wang, Y. & Rana, T. M. m(6)A-RNA demethylase FTO inhibitors impair self-renewal in glioblastoma stem cells. ACS Chem. Biol. 16, 324–333 (2021).
Huang, H. et al. FTO-dependent N(6)-methyladenosine modifications inhibit ovarian cancer stem cell self-renewal by blocking cAMP signaling. Cancer Res. 80, 3200–3214 (2020).
Yang, X. et al. WNT/beta-catenin-suppressed FTO expression increases m(6)A of c-Myc mRNA to promote tumor cell glycolysis and tumorigenesis. Cell Death Dis. 12, 462 (2021).
Xiang, Y. et al. RNA m(6)A methylation regulates the ultraviolet-induced DNA damage response. Nature 543, 573–576 (2017).
Zhang, C. et al. METTL3 and N6-methyladenosine promote homologous recombination-mediated repair of DSBs by modulating DNA-RNA hybrid accumulation. Mol. Cell 79, 425–442 (2020).
Yang, Z. et al. METTL14 facilitates global genome repair and suppresses skin tumorigenesis. Proc. Natl. Acad. Sci. USA 118, e2025948118 (2021).
Li, E. et al. METTL3 promotes homologous recombination repair and modulates chemotherapeutic response in breast cancer by regulating the EGF/RAD51 axis. Elife 11, e75231 (2022).
Murakami, S. & Jaffrey, S. R. Hidden codes in mRNA: Control of gene expression by m6A. Mol. Cell 82, 2236–2251 (2022).
Shi, H., Wei, J. & He, C. Where, when, and how: Context-dependent functions of RNA methylation writers, readers, and erasers. Mol. Cell 74, 640–650 (2019).
Zheng, G. et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol. Cell 49, 18–29 (2013).
Liu, J. et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 10, 93–95 (2014).
Zhang, Q. et al. The RNA demethylase FTO is required for maintenance of bone mass and functions to protect osteoblasts from genotoxic damage. Proc. Natl. Acad. Sci. USA 116, 17980–17989 (2019).
Covelo-Molares, H. et al. The comprehensive interactomes of human adenosine RNA methyltransferases and demethylases reveal distinct functional and regulatory features. Nucleic Acids Res. 49, 10895–10910 (2021).
Cui, Y. H. et al. Autophagy of the m(6)A mRNA demethylase FTO is impaired by low-level arsenic exposure to promote tumorigenesis. Nat. Commun. 12, 2183 (2021).
Mitchell, D. L. The relative cytotoxicity of (6-4) photoproducts and cyclobutane dimers in mammalian cells. Photochem. Photobiol. 48, 51–57 (1988).
Zhu, T., Yong, X. L. H., Xia, D., Widagdo, J. & Anggono, V. Ubiquitination regulates the proteasomal degradation and nuclear translocation of the fat mass and obesity-associated (FTO) protein. J. Mol. Biol. 430, 363–371 (2018).
Mah, L. J., El-Osta, A. & Karagiannis, T. C. gammaH2AX: a sensitive molecular marker of DNA damage and repair. Leukemia 24, 679–686 (2010).
Bar, D. Z. et al. Biotinylation by antibody recognition-a method for proximity labeling. Nat. Methods 15, 127–133 (2018).
Zhao, X. et al. FTO-dependent demethylation of N6-methyladenosine regulates mRNA splicing and is required for adipogenesis. Cell Res. 24, 1403–1419 (2014).
Bartosovic, M. et al. N6-methyladenosine demethylase FTO targets pre-mRNAs and regulates alternative splicing and 3’-end processing. Nucleic Acids Res. 45, 11356–11370 (2017).
Mauer, J. et al. FTO controls reversible m(6)Am RNA methylation during snRNA biogenesis. Nat. Chem. Biol. 15, 340–347 (2019).
Song, H. et al. SFPQ Is an FTO-binding protein that facilitates the demethylation substrate preference. Cell Chem. Biol. 27, 283–291 (2020).
Mosler, T. et al. PARP1 proximity proteomics reveals interaction partners at stressed replication forks. Nucleic Acids Res. 50, 11600–11618 (2022).
Yang, G. et al. Super-resolution imaging identifies PARP1 and the Ku complex acting as DNA double-strand break sensors. Nucleic Acids Res. 46, 3446–3457 (2018).
Spagnolo, L., Barbeau, J., Curtin, N. J., Morris, E. P. & Pearl, L. H. Visualization of a DNA-PK/PARP1 complex. Nucleic Acids Res 40, 4168–4177 (2012).
Dawicki-McKenna, J. M. et al. PARP-1 activation requires local unfolding of an autoinhibitory domain. Mol. Cell 60, 755–768 (2015).
Langelier, M. F., Eisemann, T., Riccio, A. A. & Pascal, J. M. PARP family enzymes: regulation and catalysis of the poly(ADP-ribose) posttranslational modification. Curr. Opin. Struct. Biol. 53, 187–198 (2018).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Aas, A. et al. Nucleocytoplasmic shuttling of FTO does not affect starvation-induced autophagy. PLoS One 12, e0168182 (2017).
Han, Z. et al. Crystal structure of the FTO protein reveals basis for its substrate specificity. Nature 464, 1205–1209 (2010).
Oughtred, R. et al. The BioGRID database: a comprehensive biomedical resource of curated protein, genetic, and chemical interactions. Protein Sci. 30, 187–200 (2021).
Mao, Z. et al. SIRT6 promotes DNA repair under stress by activating PARP1. Science 332, 1443–1446 (2011).
Blessing, C. et al. XPC-PARP complexes engage the chromatin remodeler ALC1 to catalyze global genome DNA damage repair. Nat. Commun. 13, 4762 (2022).
Gibbs-Seymour, I., Fontana, P., Rack, J. G. M. & Ahel, I. HPF1/C4orf27 is a PARP-1-interacting protein that regulates PARP-1 ADP-ribosylation activity. Mol. Cell 62, 432–442 (2016).
Bonfiglio, J. J. et al. Serine ADP-ribosylation depends on HPF1. Mol. Cell 65, 932–940 e936 (2017).
Suskiewicz, M. J. et al. HPF1 completes the PARP active site for DNA damage-induced ADP-ribosylation. Nature 579, 598–602 (2020).
Wang, L. et al. NADP modulates RNA m(6)A methylation and adipogenesis via enhancing FTO activity. Nat. Chem. Biol. 16, 1394–1402 (2020).
Fouquerel, E. & Sobol, R. W. ARTD1 (PARP1) activation and NAD(+) in DNA repair and cell death. DNA Repair (Amst.) 23, 27–32 (2014).
Ran, F. A. et al. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308 (2013).
Zhang, X. et al. Proteome-wide identification of ubiquitin interactions using UbIA-MS. Nat. Protoc. 13, 530–550 (2018).
Wu, T. et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innov. (Camb.) 2, 100141 (2021).
Szklarczyk, D. et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47, D607–D613 (2019).
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003).
Liszczak, G., Diehl, K. L., Dann, G. P. & Muir, T. W. Acetylation blocks DNA damage-induced chromatin ADP-ribosylation. Nat. Chem. Biol. 14, 837–840 (2018).
Huang, J. et al. Structural basis of the zinc-induced cytoplasmic aggregation of the RNA-binding protein SFPQ. Nucleic Acids Res. 48, 3356–3365 (2020).
Perez-Riverol, Y. et al. The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 50, D543–D552 (2022).
Acknowledgements
We thank R. Amor and A. Gaudin for their support with microscopy; V. Nink for assisting with the flow cytometry; and R. Tweedale for comments on the manuscript. This work was partly supported by Australian Medical Research Future Fund (Clem Jones Centre for Ageing Dementia Research Flagship Project Grant to V.A.). V.A. holds an Australian Research Council (ARC) Future Fellowship (FT220100485). J.W. was supported by an ARC Discovery Early Career Researcher Award (DE170100112), ARC Discovery Project Grant (DP230102041), a University of Queensland (UQ) Amplify Award and the Peter Hilton Alzheimer’s Fund. J.Z.A.T. was supported by a UQ Researcher Retention (RSA2) Fellowship. T.Z., L.R., S.S.D. and H.H. were supported by UQ Research Training Scholarships. Imaging was performed at the Queensland Brain Institute’s Advanced Microscopy Facility, supported by the Australian Government through ARC LIEF grant LE130100078.
Author information
Authors and Affiliations
Contributions
Conceptualization: T.Z., J.W., and V.A. Methodology: T.Z., J.Z.A.T., L.Z., H.H., S.S.D., M.J.K.J., M.L., A.L., J.W., and V.A. Investigation: T.Z., J.Z.A.T., L.Z., F.C., P.P., M.L., A.L., J.W. and V.A. Visualization: T.Z., J.Z.A.T., L.Z., H.H., M.L., J.W., and V.A. Funding acquisition: J.W. and V.A. Project administration: J.W. and V.A. Supervision: J.W. and V.A. Writing—original draft: T.Z., J.W. and V.A. Writing—review & editing: T.Z., J.Z.A.T., L.Z., P.P, M.J.K.J., M.L., A.L., J.W., and V.A.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhu, T., Tan, J.Z.A., Zhang, L. et al. FTO suppresses DNA repair by inhibiting PARP1. Nat Commun 16, 2925 (2025). https://doi.org/10.1038/s41467-025-58309-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-58309-0
