
Abstract
Catalytic methods to couple alkynes and alkenes are highly valuable in synthetic chemistry. The cobalt-catalyzed intermolecular reductive coupling of alkenes and alkynes is particularly attractive due to the unique reactivity and cost-effectiveness of cobalt catalysts. However, the enantioselective transformations of this kind are less developed. The limited successful enantioselective examples are restricted to the use of electronically biased activated olefins as the coupling partners. Herein, we report an asymmetric desymmetric reductive coupling of electronically unbiased succinimide-containing cyclobutenes with alkynes to synthesize enantioenriched, synthetically important vinyl cyclobutanes via photoredox and cobalt dual catalysis. Excellent enantioselectivities, good diastereoselectivities and regioselectivities are obtained. Preliminary mechanistic studies suggest that Hantzsch ester is a better reducing reagent when used in combination with Et3N. Density functional theory calculations reveal that the reaction proceeds more likely through a Co(III)-H migratory insertion mechanism.
Similar content being viewed by others
Introduction
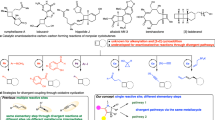
Transition metal-catalyzed intermolecular reductive coupling of alkenes and alkynes, which resemble two readily available and stable feedstocks, is an efficient and powerful strategy for rapidly constructing structurally diverse carbon–carbon bonds1,2,3,4,5,6,7. The low-valent cobalt-catalyzed reactions of this kind are particularly attractive due to the cost-effectiveness and low toxicity of cobalt catalysts8. Pioneering work reported by Cheng employed stoichiometric amounts of Zn as the terminal reductant and electronically biased acrylates as the coupling partners, with monoenes exclusively formed through protonation of the Co–C intermediate, rather than β-hydride elimination, which typically results in 1,3- or 1,4-dienes9. Subsequent advancements in this area have led to a series of remarkable achievements and new reaction modes, significantly broadening the scope of this transformation10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26. However, cobalt-catalyzed intermolecular coupling reactions of alkynes with electronically unbiased unactivated alkenes are sporadically investigated, and the 1,4-dienes are commonly formed through β-hydride elimination of the cobaltacyclopentene intermediate (Fig. 1a)27,28,29. Recently, the thriving development of metallaphotoredox catalysis30,31,32,33,34,35,36,37 and electrochemical catalysis38 has led to significant advancements, stoichiometric amounts of terminal metal reductants (e.g., Zn, Mn) are not required and mild reaction conditions are allowed. Despite these important advances, the cobalt-catalyzed enantioselective reductive coupling reactions of alkenes and alkynes are still less developed. In 2011, Cheng achieved the asymmetric reductive coupling of alkynes with cyclic enones using stoichiometric Zn as the terminal reductant14. In 2022, Xia developed an enantio-, regio- and stereoselective reductive coupling reaction of alkynes and alkenyl nitriles via photoredox cobalt dual catalysis (Fig. 1b)39. Nevertheless, alkenes in these successful enantioselective examples are restricted to the electronically biased activated ones. To the best of our knowledge, an efficient cobalt-catalyzed enantioselective reductive coupling of electronically unbiased unactivated alkenes and alkynes remains unexplored, probably due to the challenge of completing β-hydride elimination of the cobaltacyclopentene intermediate to afford 1, 4-dienes27,28.

a Cobalt-catalyzed reductive coupling of alkynes and alkenes. b Asymmetric cobalt-catalyzed reductive coupling of alkynes and alkenes. c photoredox cobalt-catalyzed asymmetric desymmetric reductive coupling of alkynes with electronically unbiased unactivated cyclobutenes (this work).
Multi-substituted enantioenriched vinyl cyclobutanes are useful building blocks in organic synthesis and are frequently found in natural products, bioactive compounds and pharmaceutical agents40,41,42. Therefore, the development of an efficient and general strategy for their rapid preparation is in high demand. Cyclobutenes are versatile building blocks43,44,45,46,47,48,49,50 for the synthesis of chiral multi-substituted vinyl cyclobutanes. For example, in 2021, Hall reported a copper-catalyzed enantioselective protocol using cyclobutene 1-carboxyester to synthesize cis-β-boronyl cyclobutylcarboxyesters, which can be subsequently exclusively transformed into trans-β-vinyl cyclobutylcarboxylesters through photoredox and nickel dual catalysis51. More recently, Lu showcased a palladium-catalyzed enantioselective 1,3-difunctionalization of cyclobutenes with aryl iodine and alkenyl boric acid, resulting in chiral vinyl cyclobutanes with a quaternary carbon center52. During the revision of this manuscript, Meng reported photoredox/cobalt-catalyzed enantioselective reductive coupling of 1,1-disubstituted allenes and cyclobutenes, cyclobutanes containing tetrasubstituted alkenes were successfully prepared53.
Herein, we report a photoredox (S,S)-Ph-BPE/Co(II) catalyzed asymmetric desymmetric reductive coupling reaction of electronically unbiased unactivated succinimide-containing cyclobutenes with alkynes under photoredox mild conditions to prepare a variety of chiral vinyl cyclobutanes with high regio-, enantioselectivity, and high efficiency (Fig. 1c). Challenges in this reaction include: (1) the competing β-hydride elimination and the direct reductive elimination of the cobaltacyclopentene intermediate would lead to the formation of complex mixtures22,27,54,55; (2) potential cobalt-catalyzed alkyne trimerization would result in aromatic by-products56; (3) the cyclobutene is known to be prone to be activated under light irradiation, which might lead to the radical involved side products formation57,58; and (4) in the case of unsymmetric alkynes, the simultaneous control of both regioselectivity and enantioselectivity would be challenging33.
Results
Reaction development
We commenced our study by employing succinimide-containing cyclobutene (1a) and 3-hexyne (2a) as the model substrates. The optimal conditions to afford the desired chiral vinyl cyclobutane (3a) were determined to be the use of the Co[(S,S)-Ph-BPE]Cl2 as the catalyst, 3DPA2FBN as the photocatalyst, the combination of triethyl amine (Et3N) and Hantzsch Ester (HE) as the proton sources and reducing agents, CH3CN as the solvent under the irradiation of blue LEDs at room temperature for 48 h, resulting in a 76% isolated yield with >20/1 dr, >20/1 E/Z ratio, and 98% ee (Table 1, entry 1). The potential undesired dienes and the tricyclic products were not detected. A detailed comparison of the changes in each reaction parameter is summarized in Table 1 (see Supplementary Tables S1–S6 for more details). The chiral diphosphine ligand plays a crucial role in maintaining both the enantioselectivity and yield. Other commonly used bidentate chiral phosphine ligands, including (R)-CyBINAP(L1), (R)-H8-BINAP (L2), (R)-MeO-BIPHEP (L3), (R)-(S)-Ph2PF-PPh2 (L4), and (S,S)-BDPP (L6) resulted in inferior results (entries 2–6). Further control experiments confirmed the indispensable roles of the chiral diphosphine ligand, cobalt catalyst (entries 7, 8), photocatalyst (entry 9), Et3N (entry 10), and light (entry 12), as the reaction completely shut down in their absence. Interestingly, the desired product was obtained in a 16% NMR yield in the absence of HE (entry 11), implying that Et3N can be employed as both reductant and proton source.
Substrate scope
With the optimal conditions in hand, the scope of the reaction was next systematically investigated (Fig. 2). A wide range of succinimide-containing cyclobutenes bearing various alkyl or aryl groups on the nitrogen atom were found to be well tolerated. In most cases (except 3h, 3j, 3s), the desired products were obtained with excellent diastereoselectivities (>20/1 dr), E/Z ratios (>20/1), and enantioselectivities (>90% ee) (3a–3t). Notably, cyclobutene containing an allyl group, which could serve as a potential handle for further manipulations, reacted well (3b). The benzyl group on the nitrogen atom was also tolerated (3c). A wide range of functionalized phenyl groups, regardless of their electronic nature, substitution patterns, and steric properties, were found to be compatible (3e–3r). More importantly, a number of functional groups commonly found in laboratories, including aryl fluoride (3e, 3f), chloride (3g), bromide (3h), trifluoromethyl (3i), trifluoromethoxy (3j), ester (3k), ketone (3l), methoxyl (3n) and amine (3o) remained intact. Furthermore, the substitution pattern of the phenyl showed a negligible effect on the overall performance of the reaction (3p–3r). Unfortunately, when a 2-naphthyl was introduced to the nitrogen atom in the cyclobutene, the ee dropped significantly (3s), the exact reason is not clear at the current stage. Finally, the heteroaryl pyridine containing cyclobutene reacted smoothly, resulting in the formation of the desired product in good yield and excellent stereocontrol (3t). Unfortunately, substrates with a methyl group (1b), a sterically hindered t-butyl group (1c), or a phenyl group bearing a para-nitro on the nitrogen were found to be unproductive. The succinimide moiety was found crucial for the reactivity, as no desired products were observed in its absence (1v–1x). The scope with regard to internal alkynes was next examined. Both symmetrical and unsymmetrical alkynes were suitable substrates, affording the desired product in high yields and excellent ees (3u–3am). Besides the ethyl-substituted symmetrical

aUnless otherwise stated, all reactions were performed on a 0.15 mmol scale. Dr, rr and E/Z ratios were determined by crude 1H NMR analysis. For unsymmetrical alkynes, unless otherwise noted, >20/1 rrs were observed. The yields were isolated yields. Ee values were determined by chiral HPLC. For details, see Supplementary Information section 6. bDr could not be determined by crude 1H NMR, and the minor diastereomer was not observed after column purification. cThe minor Z isomer was not observed after column purification. dCo[(S,S)-Ph-BPE]Cl2 (20 mol%) was used. eAcr Mes+ClO4- (3.0 mol%) was used instead of 3DPA2FBN. f4CzTPN-(8tBu) (3.0 mol%) was used instead of 3DPA2FBN. g4DPAIPN (3.0 mol%) was used instead of 3DPA2FBN. h(R)-H8-BINAP (L2, 10 mol%) was used instead of (S,S)-Ph-BPE, b/l was determined by crude 1H NMR analysis.
alkynes, the carbon chain extended n-butyl (3u) and aromatic phenyl (3v) substituted ones are well tolerated. For the unsymmetrical alkynes, dialkyl alkynes were initially tested and found to work smoothly, although the regioselectivities were not high (3w, 3x). A variety of substituted phenylacetylenes were then examined and shown to be good substrates (3y–3am). Phenyl with different substitution patterns and substituted groups showed no significant impact on both the yield and the stereocontrol (3y–3ag). Several functionalities, including aryl fluoride (3z, 3aa), chloride (3ab), trifluoromethyl (3ac), methoxyl (3ae), cyano (3af), and ketal (3ag) remained intact. Phenylacetylenes with longer carbon chains (3ah) and ether (3ai, 3aj), ester (3ak), alkyl chloride (3al), and cyclopropyl (3am) functionalized alkyls were found to be well compatible, though in some cases higher loading of catalyst is required. Finally, terminal alkyl alkyne delivered the branched olefin as the major product with a high level of ee (3an). It was found that sterically hindered groups, such as TMS (2h) and t-butyl (2j) substituted alkynes were not reactive. Furthermore, alkyl bromide (2l), nitro (2v), aromatic cyano (2x), free hydroxyl (2m, 2ac), 2-ethynyl naphthalene (2ab) and aryl-terminal alkynes (2af–2ah) containing alkynes were not tolerated.
Configuration determination and product derivatization
The absolute configuration of the desired products was assigned as 1R, 5S, 6R, and the olefin was in the E configuration, both of which were unambiguously determined by X-ray single-crystal diffraction of 3f. The regioselectivity, favoring the formation of the new C(sp²)–C(sp³) bond at the alkyl-substituted position, and the E configuration of the olefin in the unsymmetric alkyne cases were also unambiguously determined by X-ray single-crystal diffraction of 3y (Fig. 3a).

a X-ray structures of 3f and 3y. b scale-up reactions. c derivatizations (See supplementary information section 7).
To further demonstrate the practicability of this reaction, scale-up reactions and product transformations were conducted (Fig. 3b). Treatment of 1.5 mmol of both cyclobutene 1a or 1 d with 2a under the optimal conditions, the desired products were obtained with slightly decreased yields and enantioselectivities (93% ee for 3a, 95% ee for 3c). The desenly functionalized enantioenriched products were amendable to further transformations with excellent retention of the chirality (Fig. 3c). The olefin in 3c can be cleaved by exposure to ozone, resulting in the formation of a ketone with a 72% yield. Treating 3a with m-CPBA afforded the multi-substituted epoxide 5 in a quantitative yield, albeit the diastereoselectivity was not well-controlled. Due to the significance of chiral multi-substituted cyclobutanes in organic synthesis, it is valuable to further transform the succinimide moiety into other useful functional groups. Treating 3a with LiAlH4 in THF yielded tertiary amine 6 with a yield of 87%. Upon sequential treatment 3c with NaBH4, Et3N/HOAc, and LiAlH4, diol 7 was obtained in a total 66% yield.
Preliminary mechanistic studies & DFT calculations
To determine whether the isolated isomers in the unsymmetrical phenylacetylene derivatives are diastereomers or olefin Z, E isomers, several control experiments were conducted. First, both pure 3v and 3v′ were subjected to ozonation, only ketone 8 with single diastereomer was observed (Fig. 4a), suggesting that 3v and 3v′ are most likely olefin Z and E isomers. Moreover, upon treatment of both pure 3v and 3v′ under the standard reaction conditions and in the absence of a cobalt catalyst, Et3N, and HE, otherwise identical standard conditions (Fig. 4b), both isomers were detected with ratios close to that observed in the coupling reaction, implying that the Z, E isomers were generated through photo-induced olefin isomerization59. Further optimization of the photocatalysts revealed that excellent Z/E ratios can be obtained by either using Acr Mes+ClO4−, 4CzTPN-8tBu or 4DPAIPN (Fig. 4c).

a Olefin ozonation. b Control experiments. c Photocatalysts evaluation.
The mechanism for the reductive coupling of cyclobutenes and internal alkynes was next explored. First, the cobalt complex used in the reaction was unambiguously confirmed by X-ray crystallography (Fig. 5a). Second, the roles of Et3N and HE were investigated. Both can serve as reductants and proton sources, as reported in the literature60,61. However, in our reaction, satisfactory results are only achieved by using them in combination (see Table 1, entries 10 and 11). Fluorescence quenching experiments of both HE and Et3N at the optimal concentrations were conducted to reveal some clues (Fig. 5b). The emission intensity of the excited 3DPA2FBN* was dramatically diminished in the presence of the supernatant of the optimal quantity of HE in the optimal volume of acetonitrile (the solubility of HE in acetonitrile is poor, leaving a large quantity in a solid state). In sharp contrast, the emission intensity of the excited 3DPA2FBN* was only slightly diminished in the presence of Et3N. These results suggest that single-electron transfer (SET) between the excited 3DPA2FBN* and HE is faster in the photoredox catalytic cycle. Together with the observation that no product was detected in the absence of HE, a semi-stoichiometric amount of Et3N is optimal (see Table 1). The major terminal reductant is likely the HE or its related HEH•, and Et3N probably reacts as a proton shuttle to facilitate the protonation of the cobalt–carbon species. It is worth noting that substrate 1a also exhibits emission intensity, which was dramatically diminished in the presence of HE or Et3N, Thus, HE and Et3N might help circumvent the formation of the olefin-photo-activated radical involved by-products (see Supplementary Figs. S2–S5 for more details).

a X-ray structure of Co[(S,S)-Ph-BPE]Cl2. b Fluorescence quenching of Et3N and HE. c Plausible catalytic cycles. d Density functional theory (DFT) studies. The energies are in kcal/mol.
Based on the literature30,34,39,62,63,64 and our experiments, we proposed two plausible catalytic cycles (Fig. 5c). In the left catalytic cycle, The excited photocatalyst 3DPA2FBN* is first reductively quenched by HE or HEH•, generating 3DPA2FBN•− and HE•+ or HEH+. The former species then mediates the reduction of LnCo(II) to LnCo(I) with concomitant regeneration of the ground state of 3DPA2FBN to close the photoredox catalytic cycle. HE•+ or HEH+ engages in a rapid proton transfer with Et3N, resulting in the formation of Et3N·H+. The LnCo(I) species undergoes protonation to form the LnCo(III)-H species, which inserts into cyclobutene 1a, leading to the formation of intermediate I. The insertion of intermediate I into alkyne 2a generates the alkenyl cobalt species II, which upon reduction and protonation, releases the desired product and regenerates the LnCo(II) species. The LnCo(II) species is then further reduced by the reduced form of the photocatalyst to close the cobalt catalytic cycle. In the right pathway, oxidative cyclometallation of 1a, 2a, and the low-valent LnCo(I) leads to the formation of the cobaltacyclopentene intermediate IV. The intermediate IV is reduced by the reduced form of the photocatalyst to intermediate V. This intermediate undergoes double protonation by Et3N∙H+ to release both the desired product 3a and LnCo(II) species. Preliminary density functional theory (DFT) calculations were performed to distinguish between the two proposed catalytic cycles. DFT results suggest that both the Co(III)-H insertion and oxidative cyclometalation prefer to occur on the triplet free energy profile. In the key Co(III)-H migratory insertion step, the triplet transition state 3TS-1 exhibits a lower energy barrier (ΔG‡ = 13.8 kcal/mol) compared to the singlet structure (ΔG‡ = 16.8 kcal/mol; Fig. 5d). During the oxidative cyclometalation step, the triplet transition state 3TS-2 demonstrates a substantially lower energy barrier (ΔG‡ = 23.4 kcal/mol) than the singlet structure (ΔG‡ = 51.3 kcal/mol; Fig. 5d). The comparison between 3TS-1 and 3TS-2 indicates that the Co(III)-H migratory insertion mechanism is more likely involved in this reaction, which is consistent with previous report35.
Discussion
In conclusion, we have developed an enantioselective desymmetric reductive coupling reaction of alkynes with electronically unbiased succinimide-containing cyclobutenes to access the important chiral multi-substituted vinyl cyclobutanes via photoredox cobalt dual catalysis. High yields (up to 96%), good regio- (up to >20/1 rr), and excellent enantioselectivities (up to 99.7% ee) were obtained. The reaction operates under mild conditions and without the requirement for additional stoichiometric heterogeneous metals as the reducing agents, thereby, broad functional group tolerance was achieved. Preliminary mechanistic studies reveal that HE is a better-reducing reagent when used in combination with Et3N and the photo-activation of cyclobutene can be prevented by HE or Et3N in this reaction. DFT calculations reveal that the reaction prefers the Co(III)-H migratory insertion mechanism. These findings should be useful in future reaction design.
Methods
General procedure for the enantioselective reductive coupling of alkynes with succinimide-containing cyclobutenes
An oven-dried 8.0 mL screw-cap vial equipped with a magnetic stir bar was transferred into a nitrogen-filled glove box. Co[(S,S)-Ph-BPE]Cl2 (9.6 mg, 10 mol%), 1 (0.15 mmol, 1.0 equiv), PC (3.0 mol%), Hantzsch Ester (38.0 mg, 0.15 mmol, 1.0 equiv) and anhydrous CH3CN (0.5 mL) were added to the vial. Then, the internal alkyne (2, 0.45 mmol, 3.0 equiv) and Et3N (7.6 mg, 0.075 mmol, 0.5 equiv) were subsequently added to the above reaction mixture. The vial was sealed before being transferred out of the glove box. The reaction mixture was stirred at room temperature and irradiated with blue LEDs (21 W, wavelength: 448–452 nm) for 48 h. Upon completion of the reaction, the reaction mixture was concentrated under reduced pressure to give the crude product. If applied, 1,1,2,2-tetrachloroethane (~0.10 mmol) was added as the internal standard for 1H NMR analysis of the NMR yield. Further purification by chromatography on silica gel afforded the desired product 3.
Data availability
Crystallographic data for the structures reported in this Article have been deposited at the Cambridge Crystallographic Data Centre, under deposition numbers CCDC 2392153 ([Co(S,S)-Ph-BPE]Cl2), 2369036 (3f) and 2369037 (3y). Copies of the data can be obtained free of charge via https://www.ccdc.cam.ac.uk/structures/. Supplementary Information and chemical compound information are available in the online version of the paper. For NMR analysis and HPLC traces of the compounds in this article, see Supplementary Information. Correspondence and requests for materials should be addressed to Jun Zhu. Source data are provided with this paper. All data are available from the corresponding author upon request. Source data are provided with this paper.
References
Jeganmohan, M. & Cheng, C. Cobalt‐ and nickel‐catalyzed regio‐ and stereoselective reductive coupling of alkynes, allenes, and alkenes with alkenes. Chem. Eur. J. 14, 10876–10886 (2008).
Reichard, H. A. & Micalizio, G. C. A site‐ and stereoselective intermolecular alkene–alkyne coupling. Process. Angew. Chem. Int. Ed. 46, 1440–1443 (2007).
Mannathan, S., Jeganmohan, M. & Cheng, C. Nickel‐catalyzed borylative coupling of alkynes, enones, and bis(pinacolato)diboron as a route to substituted alkenyl boronates. Angew. Chem. Int. Ed. 48, 2192–2195 (2009).
Trost, B. M. & Cregg, J. J. Ruthenium-catalyzed alkene–alkyne coupling of disubstituted olefins: application to the stereoselective synthesis of trisubstituted enecarbamates. J. Am. Chem. Soc. 137, 620–623 (2015).
Trost, B. M., Koester, D. C. & Herron, A. N. Stereocontrolled synthesis of vinyl boronates and vinyl silanes via atom‐economical ruthenium‐catalyzed alkene–alkyne coupling. Angew. Chem. Int. Ed. 54, 15863–15866 (2015).
O’Rourke, N. F. & Micalizio, G. C. Cyclopropenes in metallacycle-mediated cross-coupling with alkynes: convergent synthesis of highly substituted vinylcyclopropanes. Org. Lett. 18, 1250–1253 (2016).
Gutiérrez-González, A., Marcos-Atanes, D., Cool, L. G., López, F. & Mascareñas, J. L. Ruthenium-catalyzed intermolecular alkene–alkyne couplings in biologically relevant media. Chem. Sci. 14, 6408–6413 (2023).
Tohidi, M. M., Paymard, B., Vasquez-García, S. R. & Fernández-Quiroz, D. Recent progress in applications of cobalt catalysts in organic reactions. Tetrahedron 136, 133352 (2023).
Wang, C.-C., Lin, P.-S. & Cheng, C.-H. Cobalt-catalyzed highly regio- and stereoselective intermolecular reductive coupling of alkynes with conjugated alkenes. J. Am. Chem. Soc. 124, 9696–9697 (2002).
Gandeepan, P. & Cheng, C.-H. Cobalt catalysis involving π components in organic synthesis. Acc. Chem. Res. 48, 1194–1206 (2015).
Wu, M.-S., Rayabarapu, D. K. & Cheng, C.-H. Cobalt-catalyzed cyclotrimerization of diynes with norbornenes in one efficient step. Tetrahedron 60, 10005–10009 (2004).
Chang, H.-T., Jayanth, T. T., Wang, C.-C. & Cheng, C.-H. Cobalt-catalyzed reductive coupling of activated alkenes with alkynes. J. Am. Chem. Soc. 129, 12032–12041 (2007).
Mannathan, S. & Cheng, C.-H. Cobalt-catalyzed regio- and stereoselective intermolecular enyne coupling: an efficient route to 1,3-diene derivatives. Chem. Commun. 46, 1923–1925 (2010).
Wei, C.-H., Mannathan, S. & Cheng, C.-H. Enantioselective synthesis of β-substituted cyclic ketones via cobalt-catalyzed asymmetric reductive coupling of alkynes with alkenes. J. Am. Chem. Soc. 133, 6942–6944 (2011).
Mannathan, S. & Cheng, C. Synthesis of trans‐disubstituted alkenes by cobalt‐catalyzed reductive coupling of terminal alkynes with activated alkenes. Chem. Eur. J. 18, 11771–11777 (2012).
Santhoshkumar, R. Manna than, S. & Cheng, C.-H. Cobalt-catalyzed hydroarylative cyclization of 1,6-enynes with aromatic ketones and esters via C–H activation. Org. Lett. 16, 4208–4211 (2014).
Santhoshkumar, R., Mannathan, S. & Cheng, C.-H. Ligand-controlled divergent C—H functionalization of aldehydes with enynes by cobalt catalysts. J. Am. Chem. Soc. 137, 16116–16120 (2015).
Wang, G. et al. Cobalt-catalyzed ligand-controlled divergent regioselective reactions of 1,6-enynes with thiols. Organometallics 39, 2037–2042 (2020).
Whyte, A., Bajohr, J., Torelli, A. & Lautens, M. Enantioselective cobalt‐catalyzed intermolecular hydroacylation of 1,6‐enynes. Angew. Chem. Int. Ed. 59, 16409–16413 (2020).
Whyte, A. et al. Cobalt-catalyzed enantioselective hydroarylation of 1,6-enynes. J. Am. Chem. Soc. 142, 9510–9517 (2020).
Herbort, J. H., Lalisse, R. F., Hadad, C. M. & RajanBabu, T. V. Cationic Co(I) catalysts for regiodivergent hydroalkenylation of 1,6-enynes: an uncommon cis-β-C–H activation leads to Z-selective coupling of acrylates. ACS Catal. 11, 9605–9617 (2021).
Pagar, V. V. & RajanBabu, T. V. Tandem catalysis for asymmetric coupling of ethylene and enynes to functionalized cyclobutanes. Science 361, 68–72 (2018).
Ghosh, K. K. & RajanBabu, T. V. Ligand effects in carboxylic ester- and aldehyde-assisted β-C–H activation in regiodivergent and enantioselective cycloisomerization–hydroalkenylation and cycloisomerization–hydroarylation, and [2 + 2 + 2]-cycloadditions of 1,6-enynes. J. Am. Chem. Soc. 146, 18753–18770 (2024).
Wang, H., Jie, X., Chong, Q. & Meng, F. Pathway-divergent coupling of 1,3-enynes with acrylates through cascade cobalt catalysis. Nat. Commun. 15, 3427 (2024).
Wei, C., Mannathan, S. & Cheng, C. Regio‐ and enantioselective cobalt‐catalyzed reductive [3+2] cycloaddition reaction of alkynes with cyclic enones: a route to bicyclic tertiary alcohols. Angew. Chem. Int. Ed. 124, 10744–10747 (2012).
Fujihara, T. et al. Cobalt‐catalyzed reductive coupling of alkynes and acrylates bearing a leaving group: construction of cyclobutene rings. Asian J. Org. Chem. 7, 2456–2458 (2018).
Hilt, G. & Treutwein, J. Cobalt‐catalyzed alder–ene reaction. Angew. Chem. Int. Ed. 46, 8500–8502 (2007).
Hilt, G., Paul, A. & Treutwein, J. Cobalt catalysis at the crossroads: cobalt-catalyzed Alder−Ene Reaction versus [2 + 2] cycloaddition. Org. Lett. 12, 1536–1539 (2010).
Hilt, G., Erver, F. & Harms, K. Regioselective cobalt-catalyzed alder-ene reaction toward silicon- and boron-functionalized building blocks. Org. Lett. 13, 304–307 (2011).
Rai, P., Maji, K. & Maji, B. Photoredox/cobalt dual catalysis for visible-light-mediated alkene–alkyne coupling. Org. Lett. 21, 3755–3759 (2019).
González, M. J. & Breit, B. Visible‐light‐driven intermolecular reductive ene–yne coupling by iridium/cobalt dual catalysis for C(sp3)−C(sp2) bond formation. Chem. Eur. J. 25, 15746–15750 (2019).
Li, Y.-L., Zhang, S.-Q., Chen, J. & Xia, J.-B. Highly regio- and enantioselective reductive coupling of alkynes and aldehydes via photoredox cobalt dual catalysis. J. Am. Chem. Soc. 143, 7306–7313 (2021).
Zhang, S.-Q. et al. Regio- and stereoselective divergent cross-coupling of alkynes and disubstituted alkenes via photoredox cobalt dual catalysis. Org. Chem. Front. 10, 6070–6080 (2023).
Gu, Z., Li, W., Li, Y., Cui, K. & Xia, J.-B. Selective reductive coupling of vinyl azaarenes and alkynes via photoredox cobalt dual. Catal. Angew. Chem. Int. Ed. 62, e202213281 (2023).
He, X.-K. et al. Desymmetrization–addition reaction of cyclopropenes to imines via synergistic photoredox and cobalt catalysis. J. Am. Chem. Soc. 146, 18892–18898 (2024).
Ren, X.-Y. et al. Photoredox cobalt-catalyzed stereodivergent synthesis of 1,4-dienes. Chin. J. Catal. 61, 291–300 (2024).
Nakamura, K., Nishigaki, H. & Sato, Y. Dual photoredox/cobalt-catalyzed reductive cyclization of alkynals. ACS Catal. 14, 3369–3375 (2024).
Gao, S., Liu, C., Yang, J. & Zhang, J. Cobalt-catalyzed electrochemical reductive coupling of alkynes and alkenes. Chin. J. Org. Chem. 43, 1559 (2023).
Cui, K., Li, Y.-L., Li, G. & Xia, J.-B. Regio- and stereoselective reductive coupling of alkynes and crotononitrile. J. Am. Chem. Soc. 144, 23001–23009 (2022).
Tumlinson, J. H. et al. Sex pheromones produced by male boll weevil: isolation, identification, and synthesis. Science 166, 1010–1012 (1969).
Fujiwara, Y. et al. Two new alkaloids, pipercyclobutanamides A and B, from Piper nigrum. Tetrahedron Lett. 42, 2497–2499 (2001).
Hwang, S. et al. Structure revision and the biosynthetic pathway of tripartilactam. J. Nat. Prod. 83, 578–583 (2020).
Chen, Y.-J., Hu, T.-J., Feng, C.-G. & Lin, G.-Q. Synthesis of chiral cyclobutanes via rhodium/diene-catalyzed asymmetric 1,4-addition: a dramatic ligand effect on the diastereoselectivity. Chem. Commun. 51, 8773–8776 (2015).
Guisán‐Ceinos, M., Parra, A., Martín‐Heras, V. & Tortosa, M. Enantioselective synthesis of cyclobutylboronates via a copper‐catalyzed desymmetrization approach. Angew. Chem. Int. Ed. 55, 6969–6972 (2016).
Nóvoa, L., Trulli, L., Parra, A. & Tortosa, M. Stereoselective diboration of spirocyclobutenes: a platform for the synthesis of spirocycles with orthogonal exit vectors. Angew. Chem. Int. Ed. 60, 11763–11768 (2021).
Goetzke, F. W., Hell, A. M. L., Van Dijk, L. & Fletcher, S. P. A catalytic asymmetric cross-coupling approach to the synthesis of cyclobutanes. Nat. Chem. 13, 880–886 (2021).
Nóvoa, L., Trulli, L., Fernández, I., Parra, A. & Tortosa, M. Regioselective monoborylation of spirocyclobutenes. Org. Lett. 23, 7434–7438 (2021).
Goetzke, F. W., Sidera, M. & Fletcher, S. P. Catalytic asymmetric hydrometallation of cyclobutenes with salicylaldehydes. Chem. Sci. 13, 236–240 (2022).
Liang, Z. et al. Cobalt-catalyzed diastereo- and enantioselective carbon–carbon bond forming reactions of cyclobutenes. J. Am. Chem. Soc. 145, 3588–3598 (2023).
Yuan, F. et al. Diversified synthesis of chiral fluorinated cyclobutane derivatives enabled by regio‐ and enantioselective hydroboration. Angew. Chem. Int. Ed. 63, e202401451 (2024).
Nguyen, K. et al. Catalytic enantioselective synthesis of a cis-β-boronyl cyclobutylcarboxyester scaffold and its highly diastereoselective nickel/photoredox dual-catalyzed Csp3–Csp2 cross-coupling to access elusive trans-β-aryl/heteroaryl cyclobutylcarboxyesters. ACS Catal. 11, 404–413 (2021).
Wang, Z., Zhu, J., Wang, M. & Lu, P. Palladium-catalyzed divergent enantioselective functionalization of cyclobutenes. J. Am. Chem. Soc. 146, 12691–12701 (2024).
Wu, Q., Zhang, Z., Chong, Q. & Meng, F. Photoredox/cobalt‐catalyzed chemo‐, regio‐, diastereo‐ and enantioselective reductive coupling of 1,1‐disubstituted allenes and cyclobutenes. Angew. Chem. Int. Ed. 64 e202416524 (2025).
Chao, K. C., Rayabarapu, D. K., Wang, C.-C. & Cheng, C.-H. Cross [2 + 2] cycloaddition of bicyclic alkenes with alkynes mediated by cobalt complexes: a facile synthesis of cyclobutene derivatives. J. Org. Chem. 66, 8804–8810 (2001).
Parsutkar, M. M., Pagar, V. V. & RajanBabu, T. V. Catalytic enantioselective synthesis of cyclobutenes from alkynes and alkenyl derivatives. J. Am. Chem. Soc. 141, 15367–15377 (2019).
Kumon, T., Yamada, S., Agou, T., Kubota, T. & Konno, T. Highly regioselective cobalt-catalyzed [2+2+2] cycloaddition of fluorine-containing internal alkynes to construct various fluoroalkylated benzene derivatives. J. Fluor. Chem. 213, 11–17 (2018).
Du, J. & Yoon, T. P. Crossed intermolecular [2+2] cycloadditions of acyclic enones via visible light photocatalysis. J. Am. Chem. Soc. 131, 14604–14605 (2009).
Miller, Z. D., Lee, B. J. & Yoon, T. P. Enantioselective crossed photocycloadditions of styrenic olefins by lewis acid catalyzed triplet sensitization. Angew. Chem. Int. Ed. 56, 11891–11895 (2017).
Li, H. et al. Selective synthesis of Z ‐cinnamyl ethers and cinnamyl alcohols through visible light‐promoted photocatalytic E to Z isomerization. Chem. Asian J. 15, 555–559 (2020).
Constantin, T. et al. Aminoalkyl radicals as halogen-atom transfer agents for activation of alkyl and aryl halides. Science 367, 1021–1026 (2020).
Kong, M. et al. Radical cross coupling and enantioselective protonation through asymmetric photoredox catalysis. Adv. Sci. 11, 2307773 (2024).
Sun, M., Chen, J.-F., Chen, S. & Li, C. Construction of vicinal quaternary carbon centers via cobalt-catalyzed asymmetric reverse prenylation. Org. Lett. 21, 1278–1282 (2019).
Ghorai, S., Chirke, S. S., Xu, W.-B., Chen, J.-F. & Li, C. Cobalt-catalyzed regio- and enantioselective allylic amination. J. Am. Chem. Soc. 141, 11430–11434 (2019).
Wang, L., Wang, L., Li, M., Chong, Q. & Meng, F. Cobalt-catalyzed diastereo- and enantioselective reductive allyl additions to aldehydes with allylic alcohol derivatives via allyl radical intermediates. J. Am. Chem. Soc. 143, 12755–12765 (2021).
Acknowledgements
We are grateful for the financial support from NSFC 22101214 (J.Z.), 22201222 (X.Q.) and 22374110 (Z.W.) the National Key R&D Program of China (2023YFA1508600, X.Q.), and the startup from Wuhan University for financial support. We acknowledge the Core Research Facilities of CCMS (WHU) for access to analytic equipments. We thank Profs. Q. Zhou, W. -B. Liu, and H. Lv at Wuhan University for the sharing of the basic instruments and chemicals. X.Q. acknowledges the supercomputing system in the Supercomputing Center of Wuhan University.
Author information
Authors and Affiliations
Contributions
T.Z. and J.Z. conceived and designed the experiments. T.Z., Y.L., L.W. and Q.H. performed all the experiments and analyzed all the data. Y.H., J.C., and X.Q. performed the DFT calculation and analyzed the computational data. Y.-Y.L., Z.W. performed the optical focusing inductive electrospray (iESI) HRMS and analyzed the data. T.Z. and J.Z. wrote the manuscript with contributions from all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Kento Nakamura and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zeng, T., He, Y., Li, Y. et al. Photoredox cobalt-catalyzed asymmetric desymmetric reductive coupling of cyclobutenes with alkynes. Nat Commun 16, 3102 (2025). https://doi.org/10.1038/s41467-025-58315-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-58315-2
