
Abstract
The quality and magnitude of the immune and inflammatory responses determine the clinical outcome of Leishmania infection, and contribute to the efficacy of antileishmanial treatments. However, the precise immune mechanisms involved in healing or in the chronic immunopathology of human cutaneous leishmaniasis (CL) are not well understood. Through sequential transcriptomic profiling of blood monocytes, neutrophils, and eosinophils over the course of systemic treatment with meglumine antimoniate, we revealed that a heightened and sustained Type-I interferon response signature is a hallmark of treatment failure (TF) in CL patients infected with Leishmania (Viannia) panamensis and L.V. braziliensis. The transcriptomes of pre-treatment, mid-treatment and end-of-treatment samples were interrogated to identify predictive and prognostic biomarkers of TF. A composite score derived from the expression of 11 differentially expressed genes (common between monocytes, neutrophils and eosinophils) is predictive of TF. Similarly, machine learning models constructed using data from pre-treatment as well as post-treatment samples, accurately classify treatment outcome into cure and TF. Results from this study instigate the evaluation of Type-I interferon responses as immunological targets for host-directed therapies for the treatment of CL, and highlight the feasibility of using transcriptional signatures as predictive biomarkers of outcome for therapeutic decision making.
Similar content being viewed by others


Introduction
Vector-borne infectious diseases cause more than 700,000 human deaths each year1. Among the most impactful are those caused by viruses and intracellular protozoan parasites, including malaria, visceral and cutaneous leishmaniasis (CL), Chagas disease, and dengue fever, which disproportionately affect the poor, and perpetuate the cycle of poverty and disease2. CL annually affects about one million people in 92 countries3. In the absence of vaccines and effective vector management, control of CL relies on treatment. However, the use and efficacy of most antileishmanials is limited by parenteral delivery, high levels of toxicity including hepato- and cardiotoxicity4, and increasing rates of treatment failure.
Despite the World Health Organization (WHO)/Pan American Health Organization (PAHO) guidelines favoring local treatments like thermotherapy or intralesional pentavalent antimony (SbV) for uncomplicated CL5, the vast majority of patients in endemic regions across the American continent are still treated with systemic therapies. This is due to the misalignment of local policies and global health recommendations, and the limited access to technologies like thermotherapy. Furthermore, a high proportion of patients (as much as 60%)6 are not eligible for local therapies, as they do not meet the clinical criteria for use, which include lesions in anatomical areas other than the face or near the joints, lesions < 3 cm in diameter, and patients presenting with a single lesion5. Therefore, systemic antimonials remain the treatment of choice; a clinical practice reinforced by their lower cost and higher availability. This translates into a challenge for patients inhabiting rural and dispersed communities in most endemic areas, where access to medical care is limited. Therefore, adherence to treatment and clinical follow-up are compromised, contributing to higher rates of treatment failure (TF). Consequently, there is an urgent need for predictive biomarkers of treatment outcomes for these vulnerable patient populations, but most importantly, for better therapeutic interventions.
Clinical resolution of localized CL is accompanied by a reduction in parasite burden and yet, parasite elimination per se is not the sole determinant of healing. Parasite persistence has been documented following therapeutically achieved cures in more than 40% of patients infected with Leishmania Viannia species in Central and South America7,8,9. Pathogenesis of dermal leishmaniasis (mucocutaneous and CL) is mediated by the immune and inflammatory responses; thus, resolution of the disease and control of the infection are intimately linked to the host response10. Recent studies from our group and others on localized CL infections caused by L. Viannia species are beginning to unravel the role of innate and adaptive immune components in TF upon treatment with antimonial drugs and miltefosine11,12,13,14. We have shown that lesions of TF patients are characterized by sustained local inflammation mediated by heightened expression of pro-inflammatory chemokines and cytokines, predominantly associated with activation and migration of monocytes, polymorphonuclear cells, and TH1 CD4+ T cells13. In addition, an environment of high cytolytic activity mediated by CD8+ T cells and subsequent induction of inflammation mediated by IL1β has been shown in lesion biopsies of TF patients infected with L.V. braziliensis15. Interestingly, the expression of inflammatory genes in peripheral blood mononuclear cells (PBMCs) of CL patients is modulated by in vivo exposure to antimonial drugs11, as early as 30 minutes after intramuscular drug delivery. The dynamics of expression of some of those genes, including ones involved in monocyte and neutrophil chemotaxis and activation (CCL2, CXCL2, CXCL3, CXCL8), were related to the pharmacokinetics (PK) of plasma antimony concentrations. These observations suggest that drug-dependent modulation of inflammation, especially the immunomodulation of innate responses, is a central component of healing in CL.
We hypothesized that sustained and deregulated inflammatory gene expression in innate cells leads to their continuous activation and recruitment to ulcerated cutaneous lesions, promoting immunopathology and leading to TF in patients with localized CL. Here, we present a longitudinal study of the transcriptomes of peripheral blood monocytes (Mo), neutrophils (Nφ), and eosinophils (Eφ) of CL patients obtained over the course of antileishmanial chemotherapy with meglumine antimoniate (Glucantime – GLUC). The profiles from each of the innate immune cell types interrogated here yielded clear signatures that distinguished TF patients from those who cured. Most prominent, and shared between the three cell types, was an enhanced Type I interferon (IFN) gene expression profile, which comprised the hallmark signature of innate cells in patients with TF. Characterization of the innate immune response during the course of treatment allowed the identification of host-specific prognostic signatures of the therapeutic response.
Results
Study participants and samples
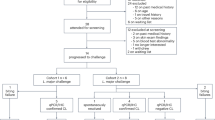
Forty adult CL patients participated in this study, and were recruited in our outpatient clinics, one in the city of Tumaco (South Pacific coast of Colombia), and another in the urban center of Cali. One patient was excluded due to progression to mucosal leishmaniasis, in accordance with the clinical protocol designed for this study (Fig. 1A). Of the remaining participants, 35 completed clinical follow-up. One participant did not donate blood or tissue samples and was therefore not included in the transcriptomic analyses. Most of the 34 enrolled participants were males of Afrocolombian descent, presented with ulcerated skin lesions, and most were infected with L.V. panamensis, the remaining with L. V. braziliensis. Five patients were treated with miltefosine (MLF), all cured. Twenty-nine patients were treated with GLUC, 19 cured, and 10 experienced TF. Due to the small sample size of MLF-treated patients and the absence of TF in this group, only samples from GLUC-treated patients (29 in total) were carried forward for transcriptomic analyses (Fig. 1B). Adherence to treatment in GLUC-treated patients was > 95% (measured as the proportion of ampoules administered from those prescribed). Significant differences were noted in the age and ethnicity of patients who cured vs. patients with TF in the aggregated data of patients recruited in Cali and Tumaco (Supplementary Table 1a). These two variables (age and ethnicity) were collinear (Supplementary Fig. 1A, B), and the magnitude of their effect on the outcome of treatment was marginal (Supplementary Fig. 1C, D). No further evidence of the influence of any of these variables on the outcome of treatment was identified in a multivariable logistic regression analysis (Supplementary Fig. 1E for the full cohort, or Supplementary Fig. 1F for patients recruited in Tumaco only).

Flow chart (A) showing the recruitment of participants in the study ending with a total of 34 enrolled participants. Study design (B) detailing participant enrollment, treatment, sample collection and analysis, and determination of treatment outcome. Created in BioRender. Gomez, M. (2025) https://BioRender.com/g83q151.
Leishmania species, drug resistance, and virulence of the infecting strain may contribute to TF. In L. V. braziliensis and L. V. guyanenesis infections, virulence has been associated with the presence of the Leishmania RNA virus (LRV)16,17. To assess the possible impact of these variables, we evaluated the Leishmania isolates from study participants for susceptibility to pentavalent antimony and the presence of LRV. Leishmania strains were isolated from 24 patients treated with GLUC (14 cures and 10 TF); isolates from the remaining 5 could not be propagated. The predominant species isolated was L.V. panamensis (n = 19), followed by L.V. braziliensis (n = 4) and L.V. guyanensis (n = 1). No statistical difference was observed between the Leishmania species and the outcome of treatment in this patient cohort (Supplementary Table 1). As intracellular amastigotes, 10 strains were susceptible to GLUC, 12 were resistant (of which 5 were isolated from patients who were cured, and 7 from TF), and data was unavailable for 2 (Supplementary Fig. 2A). No statistically significant difference was found (Fisher´s P = 0.23). All strains evaluated were negative for LRV.
To explore the possible contribution of genomic/transcriptomic differences of the parasite to the outcome of treatment, Leishmania transcriptomes were analyzed from the dual RNA-Seq data collected from lesion biopsies, Mo, Nφ, and Eφ samples from CL patients before and over the course of treatment. Only samples that passed a threshold of single mapped parasite reads ≥ 30000, and ≥ 3000 observed genes were used for the sufficient representation of the parasite genome, resulting in a total of 15 samples analyzed (corresponding to 8 biopsy samples, 1 Eφ, 1 Mo, and 5 Nφ samples from 11 patients; 6 cures and 5 with TF) (Supplementary Data 1). No significant difference in the parasite transcriptome profiles could be discerned by PCA using either uncorrected (Supplementary Fig. 2B) or surrogate variable analysis (SVA) adjusted data (Supplementary Fig. 2C). Collectively, LRV content, drug susceptibility testing, Leishmania species typing, and transcriptomic analyzes from infected human samples rule out a significant contribution of the parasite to the outcome of treatment in this cohort of patients.
Global assessment of samples and human transcriptomic profiles
Lesion biopsy samples were collected before initiation of treatment, and peripheral blood samples obtained pre-treatment (Pre-Tx), mid-way through treatment (Mid-Tx, day 8), and at the end of treatment (End-Tx, day 20). Mo, Nφ, and Eφ were isolated from all blood samples. The cell populations were evaluated by flow cytometry and light microscopy, and purity confirmed > 95% (Supplementary Fig. 3). cDNA libraries were constructed and sequenced from a total of 186 samples (Supplementary Data 1 and Fig. 2A). Following low coverage filtering (Supplementary Fig. 4), two samples were removed. We used principal component analysis (PCA) and a correlation heatmap to visualize the relationship between samples (Fig. 2B, C). The PCA resulted in the expected grouping by cell type (Fig. 2B). A similar clustering was observed in the hierarchical clustering analysis (Fig. 2C). These data support the quality, reproducibility, and specificity of transcriptomes derived from the isolated blood cells from our patient cohort.

RNA-seq was carried out on biopsies collected pre-treatment and on 3 types of leukocytes (Mo, Nφ, and Eφ) collected pre-treatment (Pre-Tx), mid-treatment (Mid-Tx), and at the end of treatment (End-Tx). C = cures, F = treatment failure (A). A principal component analysis (PCA) plot (B) and heatmap of hierarchical clustering analysis using pairwise correlations (C) are shown for all samples. The analyses were performed using all annotated protein-coding genes following filtering for low counts, CPM, quantile normalization, and log2 transformation. In the PCA plot, the first two principal components are shown on the X and Y-axes, respectively, with the proportion of total variance attributable to that PC indicated. Each sample is represented as a single point with color indicating cell type and shape indicating treatment outcome. Colors along the top of the heatmap indicate the cell type. Colors within the inset of the heatmap represent correlation values. The green line represents the frequency of correlation values.
The use of two clinics for patient recruitment (one in Tumaco and another in Cali) necessitated the evaluation of the data to account for possible clinic-associated batch effects. When all samples were colored by clinic on the same PCA plot, no grouping by clinic was discernible within cell types (Supplementary Fig. 5A – all samples); however, a grouping of samples by cell type revealed a significant amount of variance which separated Tumaco from Cali, confirming the presence of a batch effect attributable to the clinic (Supplementary Fig. 5B–D). Similar analyses were performed evaluating the effect of ethnicity (which was also cross-correlated with age, without finding any substantial contribution of this variable to the overall variability of the transcriptomic data (Supplementary Fig. 5F–H). To highlight the variability between clinics, we modeled the ‘clinic’ variable in SVA and used the surrogate variables-modified counts to generate the PCA plots, further supporting the hypothesis of a strong batch effect associated with the clinic (Fig. 3A–D).

Principal Component Analysis (PCA) plots of the global gene expression profiles of all samples combined (A) and individual blood cells (eosinophils, monocytes, and neutrophils) and lesion biopsies collected from study participants (B–E) at two different clinics (one in Tumaco and another in Cali), and three visits (pre-treatment and twice during the course of treatment). Here, and based on the results shown in Supplementary Fig. 5, we modeled the ‘clinic’ variable in SVA and used the surrogate variables-modified counts to generate the PCA plots. The analyses were performed using all annotated protein-coding genes following filtering for low counts, CPM normalization, and log2 transformation. In the PCA plots, the first two principal components are shown on the X and Y-axes, respectively, with the proportion of total variance attributable to that PC indicated. Each sample is represented as a single point with color indicating either the clinic (A), or clinic, treatment and outcome (B–E). The inner and outer colored ellipses represent the 90% and 95% confidence intervals, respectively. These are only shown for classes containing more than 3 samples.
The relative contribution of metadata factors to the transcriptional profile was further examined via a series of principal component (PC) and surrogate variable (SV) loading analyses. This was performed by taking the first 5 PCs of the normalized (CPM and filtered) data and collecting their F-statistics and associated P-values with respect to a series of empirically observed important metadata factors: clinic, donor, and visit number. PCs and factors with high F-statistics and low P-values were deemed noteworthy. The normalized data was then passed to SVA with cure/TF status as the variable of interest, and the F and P-values were collected. We found that the most prominent SV in the expression data was ‘clinic’ (SV4 and SV5), followed by modest F-statistic/P-values for ‘donor/participant’ (SV4) and, to a much lesser extent, ‘visit number’ (SV1 and SV2). The same values were then collected from the SVA-adjusted transcriptional profile (Supplementary Data 2). Indeed, the effect of ‘clinic’ increased significantly relative to the other two factors (as shown by higher F and P-values in the Post-SVA PC1), indicating a persistent batch effect associated with the clinic.
Based on the significant batch effect introduced by the patient recruitment clinic and the skewed representation of cured patients recruited in Cali (9 of 10 CL patients recruited in this site cured), we excluded all samples obtained from patients in the Cali clinical site from the initial transcriptomic analyses and biomarker discovery. Therefore, the transcriptomic variance associated with therapeutic outcome was analyzed on SVA-adjusted data sets from samples derived from patients recruited in Tumaco. After subsetting to include only samples collected from Tumaco and carrying out similar loadings analyses as described above (Supplementary Data 2), the effect of ‘visit’ continued as minor, showing that the expression changes in the samples are modest between samples collected Pre-Tx, Mid-Tx or at End-Tx. The largest effect remained attributable to the ‘donor’.
Transcriptomes from lesion biopsies obtained pre-treatment corroborate heightened tissue cytolytic activity in treatment failure patients
A recent comparative transcriptome analysis of Pre-Tx lesion biopsies from CL patients infected with L.V. braziliensis showed a gene signature of heightened cytolytic activity in lesions from TF patients, compared to those who cured15. To explore the congruity of those findings with infections with other L. Viannia species (L.V. panamensis), we examined CL lesion transcriptomes in our study cohort. Overall, the transcriptomic profiles of lesion biopsies from patients who cured were indistinguishable from those of patients with TF (Supplementary Fig. 5E), even following SVA (Fig. 3E). As expected, only few genes (n = 28) were differentially expressed (DE) (P < 0.05; |log2FC | ≥ 1), 17 of which were up-regulated and 11 down-regulated in patients with TF, compared to cures (Supplementary Data 3a). Notably, among up-regulated genes, a signature of increased cytolytic activity (GZMB, NCR1, SH2D1B, PRF1, KLRC1, GNLY, FGFBP2, KIR2DL4, CCL3 and CCL4) was found in tissue samples from TF patients (Supplementary Data 3b), consistent with previous findings from TF patients infected with L.V. braziliensis15. The minimal difference in the global transcriptomes of skin lesions from cured and TF patients was expected since bulk transcriptomes from complex multicellular tissues are often skewed to reflect the most abundant or transcriptionally active cells within the sample. Nevertheless, the fact that a clear transcriptomic signature of enhanced cytolytic activity was detected in TF suggests that systemic differences leading to the activation and/or recruitment of cytotoxic Natural Killer (NK) and CD8+ T cells could be contributing to this enhanced inflammatory state.
Transcriptomic profiles of innate immune cells do not change over the course of treatment
Our understanding of the participation of innate immune responses in the outcome of antileishmanial therapy is almost exclusively limited to the role of macrophages as primary host cells for the parasite. However, mounting evidence shows that other innate cells, including Nφ, and more recently Eφ and NK cells, participate in the inflammatory responses that contribute to CL immunopathology, and thus their functions are relevant to the outcome of treatment18.
Among the hallmarks of innate immune cell functions are the velocity and robustness of their elicited responses, which in turn require tight mechanisms of control to avoid host injury. To explore the dynamics of these responses and their participation in therapeutic responsiveness, we analyzed samples collected from CL patients Pre-Tx, Mid-Tx, and at End-Tx. PCA plots of the transcriptomes of individual cell types did not reveal any significant clustering of samples based on visit (i.e., over the course of treatment) (Fig. 4A–C). Furthermore, a correlation analysis of DE genes (DEGs) derived from SVA-adjusted data using two different models, one which explicitly included visits in the DE model and the other which did not, showed strong and significant correlations for the log2FC values in Mo, Eφ, and Nφ (r > 0.9), as well as for the respective P-values (ρ > 0.82) (Fig. 4D–F). These data rule out a substantial effect of including visits in the DE model for cure vs. TF. Based on the above, we opted to group transcriptome samples from all time points from each cell type for the identification of biomarkers and transcriptional signatures of cure and TF.

PCA for all samples were performed using log2 transformed, quantile and CPM-normalized, and filtered counts of annotated protein-coding genes (A–C). The first two principal components are shown on the X and Y-axes, with the proportion of total variance attributable to that PC indicated. D–F show correlation plots of the log2FC values for DEGs identified using DESeq2 with the default model including cure/TF as factors with SVA-adjustment (x-axis), and SVA-adjusted data where cure/TF and visit (Pre-Tx, Mid-Tx, and End-Tx) were considered as factors. Pearson correlation coefficient (r) is shown for DEGs and Spearman correlation coefficient (ρ) for associated P-values. (G–I) Correlation plots of the log2FC values for DEGs using Dream (x-axis) and DESeq2 using SVA-adjusted data. Pearson correlation coefficient (r) is shown for DEGs and R2 of the linear regression.
Because we sampled the same patients before treatment and twice during the course of several weeks post-treatment, we wanted to rule out that the multiple data points were not amplifying the effects observed. To that end, we included “patient” as a random effect variable in a series of linear mixed models (LMM) using the dream19 functionality from variancePartition20. Our LMM included visit, final outcome, and cell type as fixed effects; and donor as a random effect. We then compared the DE outputs of dream (log2FC and P-values in the contrast of Cure vs. TF; Supplementary Data 4) against the DESeq2 data. High correlations of log2FC values (≥ 0.84) were observed for all cell types (Fig. 4G–I), indicating a strong similarity among DEG lists obtained with the two analytical approaches. Up to 40% of genes deemed significant by DESeq2 in any of the three cell types were also significant using dream. Network analyses using as input the DEGs resulting from dreams also yielded consistent results (genes and pathways) to those derived from the DESeq2 data (Supplementary Fig. 6).
Monocyte, neutrophil and eosinophil transcriptomes from CL patients who cure differ from those with TF
Examination of Nφ, Mo, and Eφ transcriptomes showed a clear clustering of samples by treatment outcome, in each of the three cell types, revealing a distinct separation of samples along the first principal component (PC1) (Fig. 5A). On average, PC1 explained 18% of the variance across the three cell types. A DE analysis in each of the three cell type transcriptomes revealed 3 to 4 times more significant DEGs between cures and TF in Mo, when compared to Eφ and Nφ (Supplementary Data 5), consistent with higher transcriptional activity of Mo21,22. Those DE profiles are represented in the form of volcano plots (Fig. 5B). When a |log2FC | ≥ 1 threshold was established, a comparable number of DEGs was observed between cures and TF among the three cell types: 191 in Eφ, 160 in Nφ and 112 in Mo (Supplementary Data 5 and Fig. 5C).

PCA plots, after batch correction estimated by SVA, of transcriptomes collected at all-time points from monocytes (Mo), eosinophils (Eφ), and neutrophils (Nφ) (A). Volcano plots of DEGs using DEseq2 (Wald test and its Benjamini-Hochberg adjusted P-value < 0.05 and |log2FC | ≥ 1) for each cell type, with genes up-regulated in TF vs. cures colored pink, and those down-regulated in TF vs. cures labeled purple. The top 10 up- and down-regulated genes are labeled (B). Venn diagram showing the intersection of common DEGs (P < 0.05 and |log2FC | ≥ 1) between cell types (C). The inner and outer colored ellipses represent the 90% and 95% confidence intervals, respectively. These are only shown for classes containing more than 3 samples.
In Eφ, a robust induction of IFNα/β signaling was revealed by gene up-regulation of innate immune receptors, signaling molecules, transcription factors, and regulators of signaling pathways (Supplementary Data 5). The gene encoding IRF7 was up-regulated in TF Eφ. Together with IRF3, IRF7 is the canonical transcriptional regulator of Type I IFNs23. IFIH1 (gene encoding MDA5), a RIG-I-like receptor and cytoplasmic sensor of dsRNAs, was also up-regulated in TF. MDA5 has been demonstrated to be an amplifier of innate immune responses and associated with autoinflammation24. Genes encoding downstream effectors (including OAS1, OAS2, OAS3, OASL, BST2, MX1, MX2, IFI6, XAF1, GBP2, and IFI27), and pathway regulators (IFIT5, ISG15, RSAD2, USP18, HLA-G, DHX58, and DDX60) were found in enriched gene categories in Eφ (Fig. 6A, B), substantiating elements of a Type I IFN signature in TF. Type I IFN response pathway genes were also enriched in Nφ transcriptomes from TF (Supplementary Data 6 and Supplementary Data 7). Notably, genes encoding HERC6, IFI44L, ISG15, USP18, IFI27 and DDX60 (Fig. 6C, D) were also expressed at higher levels in TF Nφ. Consistently, Mo transcriptomes from TF patients were also enriched in mRNAs encoding Type I IFN-related genes (Fig. 6E, F), suggesting synergistic innate cell functions towards hightened type I IFN inflammation in TF. Downregulation of IL1R1 and IL1R2 (decoy) receptors was observed in Mo from TF patients, and downregulation of molecules related to MHCII antigen presentation was found in all cell types from TF patients (Supplementary Data 5, Supplementary Data 6 and Fig. 6A, C, E). Genes involved in wound healing and cell proliferation (HBGEF, EGR1, and EGR3) were down-regulated in Eφ of TF patients. In Mo from TF, down-regulated antimicrobial peptide genes (including CTSG, LTF, CAMP, DEFA3, and LCN2) and immune receptor activity genes (including IL2RB, IL1R2, HLA-DQA1, HLA-DQB1, IL1R1, and CXCR4) were significantly enriched. Altogether, these results suggest that mechanisms underlying TF are associated with an enhancement of Type I IFN signaling, a dampening of antimicrobial effectors (antimicrobial peptides), and functions linking the innate and adaptive immune systems (antigen presentation).

Panels A, C, E, and G. Significantly DEGs (Padj < 0.05 and |log2FC | ≥ 1) were used as input for network analyses. STRING V12.0 was used to construct networks based on co-expression, databases, and experiment terms, with line thickness representing the confidence of the interaction. Enriched categories shown overlaying the networks are those selected from KEGG, GO, or Reactome (one-sided Fisher test with g:SCS-corrected P-values) terms with FDR < 0.05 and strength > 1. The blue border in nodes represents up-regulated genes in TF compared to cures; red borders depict downregulated genes. The intensity of the border color reflects the magnitude of the DE in our dataset. Genes belonging to the Type I IFN cluster are grouped under a blue shade, MHCII in red, and others in gray. Panels B, D, F, H. The same significant DEGs were used for over-representation analyses by gProfiler2. Terms with adjusted P-values ≤ 0.05 (g:SCS method in gProfiler) from the GO biological processes were clustered by term similarity, and the resulting trees were plotted. Groups of nodes are colored by their parent GO term, the size of terminal nodes denotes the number of genes observed in the category, and the color of each node represents the adjusted significance of the observed over-representation.
Twelve DEGs were common among all cell types: IFI44L, IFI27, PRR5, PRR5-ARHGAP8, RHCE, FBXO39, RSAD2, SMTNL1, USP18, AFAP1, SIRPG and OTOF (Fig. 5C). Although more than 70% of significant DEGs with |log2FC | > 1 were unique to each cell type (underscoring a cell-specific response) gene enrichment analyses showed common significantly enriched features between the different innate cells (Supplementary Data 6, Fig. 6B, D, F). Among up-regulated genes, enrichment of type I IFN responses was common to Mo, Eφ, and Nφ from TF patients, and this was also supported by GSVA (Supplementary Data 7). No similarities in gene enrichment analysis were found for down-regulated genes among cell types, with the exception of MHCII-related genes (Supplementary Data 6).
Considering the overlap of DEGs among cell types, we evaluated whether an IFN gene signature could be detected in a combined analysis of transcriptomes derived from all innate cell types. As observed in the PCA before SVA (Supplementary Fig. 7A), most of the variance in the data (81% for PC1 and PC2 combined) could be attributed to the cell type. When SVA was used (Supplementary Fig. 7B), a partial separation of transcriptomes was observed between cells isolated from CL patients who cured versus those with TF. Differential expression analyses revealed 210 genes ( | log2FC | ≥ 1; 137 upregulated in TF compared to cures and 73 downregulated (Supplementary Data 5). Consistently, the IFN and MHC signatures identified in the analyses of each cell population were also observed in the combined analysis (Fig. 6G, H).
In a complementary approach to DE analyses, we employed weighted gene co-expression network analysis (WGCNA) to identify co-expressed gene clusters associated with therapeutic response. The Mo data resulted in four gene modules with a significant association. The four modules comprised 478 genes and were enriched in genes encoding ribosomal, mitochondrial and DNA nuclear activity and IFNα/β-inducible proteins (Supplementary Data 8and Supplementary Fig. 8). In Nφ, three modules comprising 581 genes were associated with therapeutic response and were enriched in cellular pathways similar to those found in our DE analyses: Type I IFN, MHC-II, DNA nuclear activity, glycolytic metabolism, cell migration, vesicular transport, cell death and the immunoproteasome. For Eφ, both associated modules comprised 200 genes, with an enrichment of genes related to Type I IFN responses, consistent with DE analysis, and Nφ and Mo WGCNA. Of the total genes contained within the significant modules from the three cell types, 36 were shared, and all were up-regulated in TF. Among those, 24 were related to the IFN responses (Supplementary Fig. 8C, D), and this was consistent with the DE analyses. Prominent in WGCNA were STAT1 and STAT2, two known transcription factors involved in IFN signaling and expression of interferon-stimulated genes (ISGs). Both transcription factors were up-regulated in TF, suggesting coordinated and sustained IFN effector functions elicited downstream of IFN receptor ligation. Together, WGCNA and DE analyses support the participation of a systemic pro-inflammatory environment sustained in TF, mediated in part by Type I IFNs.
To explore whether these IFN signatures observed in innate cells were also detected at the lesion site, we compared CPM values of any differentially expressed ISGs identified in any of the innate cell populations analyzed, including those common to all cell types. Significantly higher expression of OAS1, OASL, GBP2, MX1, DDX60, IFIT5 and XAF1 was observed in lesion biopsies from TF patients (Supplementary Fig. 9A). Higher expression of MX1 and OASL was corroborated in an independent transcriptomic data set from lesion biopsies of other 11 patients previously published by our group25 (Supplementary Fig. 9B). In a combined analysis of all lesion biopsy transcriptomes (Supplementary Fig. 9C; cures n = 14, TF n = 11), OAS1, OAS3, OASL, RSAD2 and MX1 were significantly higher in TF biopsies, suggesting involvement of local and systemic IFN responses in treatment failure.
IFNα/β stimulated genes constitute a hallmark signature of TF in CL
Based on the functional commonalities between the cell-specific transcriptional profiles, we recognized a common innate gene signature, from which we were able to identify biomarkers that predict TF. From the gene lists derived from our WGCNA and DE analyses, we selected the 12 common DE genes in Mo, Nφ and Eφ: IFI44L, IFI27, PRR5, PRR5-ARHGAP8, RHCE, FBXO39, RSAD2, SMTNL1, USP18, AFAP1, OTOF and SIRPG. Each of those common genes was up-regulated in TF, with the exception of AFAP1, which was down-regulated. We, therefore, constructed a composite score for each patient based on this innate gene signature of up-regulated genes (which excluded AFAP1). RPKM values were extracted from all Pre-Tx samples for each cell type. Those were selected for their predictive value in guiding early therapeutic interventions. RPKM values were used to construct raw and normalized scores (Z-score), the latter to account for possible outliers within groups. Both raw and Z-scores were higher in TF patients, and this difference was statistically significant for the scores derived from Nφ, and for the Mo + Nφ composite scores (Fig. 7A–D and Supplementary Fig. 10). Consistently, raw and Z-scores from Pre-Tx Nφ samples and the Mo + Nφ scores, were significantly predictive of the therapeutic response (Fig. 7E, F).

RPKM data of the 11 up-regulated innate signature genes from Pre-Tx samples was used to construct neutrophil (A, C) and multi-cell (monocyte + neutrophil panels B, D) composite scores. Raw (A, B) and normalized scores (C, D) are shown. Statistical significance was evaluated by analysis of variance. P-values from two-tailed analyses are shown. Representation of receiver operating characteristic (ROC) curves for raw (filled line and black circles) and normalized (dashed line and open circles) scores from neutrophils (E) and a combined score of monocytes plus neutrophils (F). ROC curves represent the area under the curve (AUC) of the false positive rate versus the true positive rate. P-values, AUC, and cutoff values based on Youden’s J statistic are shown in the graph plots. Mono+Neutro: combined scores of monocytes and neutrophils.
We next applied a machine learning approach to carry out a comprehensive analysis of all data on hand and complement our focused DE analyses. The expression matrices of all samples obtained from Tumaco or both clinics (Tumaco and Cali) were split into 10 rounds of training (40%) and testing sets (60%), through random partitioning and cross-validation. The training sets were used to generate K-nearest neighbors (KNN), logistic regression (GLM), gradient boost (XGBoosted GLM), and random forest models26. Following an evaluation of the resulting models by comparing the predictions of the training data to the known clinical outcomes (Supplementary Data 9), we used those models to predict clinical outcomes in the test partition and evaluated their performance with an emphasis on a dataset that only included Pre-Tx samples as these would be the most translatable for clinical and therapeutic decision making. Overall, the GLM models showed better performance. Using data from all blood cell types collected from all visits in the Tumaco clinic only, specificity, sensitivity, and accuracy, were 0.83, 0.84, and 0.82, respectively (Supplementary Data 9). When the models were restricted to include only Pre-Tx samples, the metrics were impacted (as an example, GLM metrics changed to specificity = 0.78, sensitivity = 0.70, accuracy = 0.69). A second round of the dual analysis described above included samples from both clinics, substantially improving the performance of all models with accuracy up to 0.89, sensitivity 0.87, specificity 0.95, and AUC 0.85 (for GLM) (Supplementary Data 9). As expected, the addition of samples from both study sites, and from all subsequent visits enhanced the performance of all models (Supplementary Data 9), possibly overcoming the clinic-based batch effect observed for DE analyses.
To explore the relationship between the predictive features identified by ML and DE, we conducted a “pseudo-bulk” DE analysis of all innate cell samples obtained at the three time points from patients from Cali and Tumaco (Fig. 8A, B). A total of 282 genes from the combined DE analysis in the Cure/TF contrast were observed, among which were the 12 innate biosignature genes (Supplementary Data 10, Fig. 8C). The top 300 most variable importance genes for each of the four ML algorithms used was contrasted against the DE gene set (Supplementary Data 10). Interestingly, the most significant overlap between ML and DE analyses resulted from the KNN model, sharing 18 genes, five of which also feature in the innate biosignature (IFI27, USP18, OTOF, SIRPG, and RSAD2). Although the three other algorithms performed better than KNN in terms of specificity, sensitivity, and accuracy metrics, they fared worse in their overlap with the DE analyses. This likely results from the requirement of significantly larger datasets for ML compared to DE analyses.

PCA for all samples collected from patients in Cali and Tumaco (Mo, Nϕ, and Eϕ, collected Pre-Tx, Mid-Tx, and at End-Tx) were performed using log2 transformed, quantile and CPM-normalized, and filtered counts of annotated protein-coding genes (A), SVA-adjusted data considering cell-type for variance adjustment (B). The first two PCs are shown on the X and Y-axes, with the proportion of total variance attributable to that PC indicated. The inner and outer colored ellipses represent the 90% and 95% confidence intervals, respectively. C Volcano plot of DEGs, using DEseq (Wald test and its Benjamini-Hochberg adjusted P-value < 0.05 and |log2FC | ≥ 1), highlighting in labels the top 10 up- or down-regulated genes in TF compared to cures.
Discussion
The response to antileishmanial chemotherapy has been primarily attributed to the drug susceptibility of the etiological agent, patient adherence to the therapeutic scheme, and intrinsic differences in drug exposure (pharmacokinetics). However, the extent of the contribution of in vitro drug susceptibility of Leishmania to the outcome of in vivo treatment remains controversial27, largely due to lack of harmonized methods for susceptibility testing and large-scale clinical evaluations of these associations. It is known that immunosuppression negatively impacts the efficacy of antileishmanials, both in murine models of infection28,29,30,31,32 and in humans33,34, indicating a substantial effect of the immune response on the outcome of treatment. However, approximately 25% of immunocompetent individuals present with TF in controlled clinical trials35,36,37. In this study, we present the comprehensive transcriptomic profiling of the most abundant innate cells from peripheral blood (Mo, Nφ, and Eφ) in immunocompetent CL patients undergoing antileishmanial chemotherapy. Our results reveal transcriptomic differences in all innate cell populations when comparing patients who cured and those with TF, and these were constant throughout the course of treatment. Notably, evidence of the participation of Type I IFN innate inflammatory responses in TF during systemic treatment with Glucantime® was common to Mo, Nφ, and Eφ transcriptomes. This instigated the search for an innate biosignature of TF, resulting in a score derived from the expression of 11 common DEGs, including a subset of Type I ISGs. This score was predictive of TF.
Type I IFNs (IFNα and IFNβ) are rapidly induced during viral infections and are central to the antiviral response23. However, their role in infections with intracellular bacteria or protozoan parasites remains elusive. A low dose of IFNβ protected mice from progressive CL caused by L. major38, and this was related to the induction of iNOS, NK cytotoxicity, and early production of IFNγ39. In visceral leishmaniasis (VL) caused by L. donovani, IFNα/β acts as an upstream suppressor of anti-parasitic TH1 cells, and IFNAR1 -/- mice better control infection compared to wild type40. These results provide evidence of both protective and pathogenic roles of Type I IFNs in leishmaniasis, which are likely dependent on the IFNα/β concentration and downstream regulation of the response. In human PBMCs, pharmacological blocking of IFNα/β resulted in an antigen-specific increase of IFNγ production, and this was reverted by inhibition of MHCII (HLA-DR) antigen presentation40. Interestingly, heightened expression of IFNα/β-stimulated genes in Mo, Nφ, and Eφ of TF patients was consistently accompanied by significant dampening of MHCII gene expression. This suggests that similar to what observed in VL, an impaired protective immunity mediated by Type I IFNs via antigen-presenting cells, could be occurring in CL. The transcriptomic signatures of Type I and Type II IFN responses have substantial overlaps41,42. Whether the observed responses in TF patients are uniquely attributable to Type I IFNs remains to be determined. However, mediators of canonical Type I IFN signaling pathways such as IFIH1 (gene encoding MDA5), a RIG-I-like receptor and cytoplasmic sensor of dsRNAs, MX1, DDX60, and USP18, strongly support a substantial contribution of Type I IFNs driving the response.
Recent evidence shows that Type I IFNs promote the pathogenesis and severity of M. tuberculosis infection in both mice and humans43,44. This was associated with the dampening of protective IL-1 signaling via eicosanoid imbalance. In monocytes from TF, high Type I IFN-inducible gene expression was accompanied by repression of IL1R1 and IL1R2 (decoy receptor), and increased IL1β. Downregulation of IL1R1, even in the context of up-regulated IL1β, suggests that the cross-balance of Type I IFNs and IL1 is not only relevant to the pathogenesis and severity of TB but also to the outcome of CL treatment.
Severity and tissue damage during infectious diseases is often mediated by immunopathology caused by exacerbated and uncontrolled inflammatory responses. During viral infections, IFNα/β promotes CD8+ T cell longevity and clonal expansion45, as well as NK cell functions23. Notably, Pre-Tx lesion biopsies from TF patients exhibited a transcriptional profile compatible with enhanced cytolytic activity mediated by CD8+T and NK cells, similar to what observed in TF patients infected with L.V. braziliensis15. It is plausible that the heightened Type-I IFN responses, mediated by systemic innate inflammatory cells (and to a lesser extent found directly in the affected tissue), could contribute to skin immunopathology driven by CD8+T and NK cells in lesions of CL patients who do not respond to treatment.
The mechanisms by which IFNα/β-inducible genes are modulated during Leishmania infection remain unknown and constitute part of our ongoing investigations. In L. Viannia infections, expression of IFNα/β and its contribution to disease severity has been proposed to be mediated by the Leishmania RNA virus (LRV). A systematic evaluation of the presence of LRV in more than 100 L.V. panamensis clinical strains did not show evidence of the presence of LRV in this species46. This was consistent with the absence of LRV in all L.V. panamensis, as well as in L.V. braziliensis clinical isolates from our study participants, ruling out any contribution of LRV to the IFN signature observed in TF. We have previously shown that L.V. panamensis induces TLR4 gene expression as early as 8 h after interaction with human monocyte-derived macrophages47. Intracellular parasite survival and TNFα production were found to be dependent on TLR447. Furthermore, using RNA-seq, we have shown induction of a Type-I IFN signature in human PBMCs, occurring as early as 24 h after L.V. panamensis infection with strains associated with chronic CL, and not with those causing self-healing disease48. These previous findings lead us to hypothesize that rapid (8 h) and, likely, strain-specific TLR engagement by Leishmania could induce IFNα/β gene expression, leading to a second wave of IFNα/β-inducible genes as early as 24 h post-infection.
The slow discovery pipeline for novel antimicrobials, and especially for those causing neglected tropical diseases, has driven the development of innovative approaches such as host-directed therapies (HDTs). The basis of HDTs relies on a detailed understanding of the role of host responses in the pathogenesis and clinical outcome of infections. However, HDTs for leishmaniasis, including immunomodulation, have often been based on knowledge of the contribution of immune responses to disease in animal models, resulting in failed clinical trials49,50,51,52. Understanding the innate factors driving therapeutic healing of CL offers a unique opportunity for rational identification of HDTs that optimize available therapeutic regimens, and can capitalize on past and current pharmaceutical developments in modifiers of innate immune functions. Interestingly, antibody-mediated blocking of IFNαR in mice, or the use of FDA-approved ruxolitinib (a small molecule inhibitor of JAK1 and JAK2), synergized with amphotericin B to control L. donovani infection40. Our data suggest that modulation of Type I-IFN responses is a likely target for host-directed therapy in CL caused by L. Viannia.
The high frequency and severity of adverse events and of TF during antileishmanial treatment demands stratification of therapeutic interventions to populations where they will be most effective. Results from this study revealed a transcriptional innate signature of TF. This signature allowed the construction of a composite score, with significant specificity and sensitivity to predict TF before initiation of treatment. Although our sample size presents an evident limitation for the successful implementation of machine learning techniques, GLM models were good predictors of outcome, albeit using the top 3000 most variable genes. The AUC achieved using our composite scores, which incorporate 11 of the 12 signature genes, closely mirrored the AUCs obtained with machine learning models that included all 3000 genes. This similarity not only underscores the robustness of our selected genes but also validates the effectiveness of our transcriptional hallmark in capturing the essential genomic signatures.
The score derived from Nφ, and the composite score from Mo + Nφ were both predictive of TF. That Nφ are the most abundant white blood cells in the blood, and monocytes one of the most transcriptionally active, supports the likelihood of developing whole blood tests for future validation, which also facilitates access in remote rural populations. Consistent with this hypothesis was the finding that a “pseudo-bulk” analysis of all transcriptomes (generated from samples from all cell types, all time points, and even both recruitment clinics) derived DEGs and enrichment categories with IFN signatures overrepresented in TF. Scoring systems have assisted screening in other immunological systems. IFN-scores based on gene expression profiles of Type-I ISGs have been used as screening tools for monogenic interpheronopathies, and to stratify patients with systemic lupus erythematosus53,54. Genes reported in these scoring systems often differ between diseases. However, four of the twelve genes that composed our innate biosignature (IFI27, IFI44L, USP18 and RSAD2), have been consistently reported as signature members in other autoinflammatory diseases, and used for clinical assessments53,55.
Personalized medicine is anticipated to be restricted to developed countries widening the disparity between “the rich and the poor”56. Technology-driven research, systems medicine, and genetic knowledge should reduce healthcare disparities rather than exacerbating them. Our study provides a solid first step towards validation and implementation of personalized medicine for CL, one of the most neglected tropical infectious diseases of global importance.
Methods
Ethics statement
This study (IRB code #1273) was approved and monitored by the Institutional Review Board for ethical conduct of research involving human subjects of the Centro Internacional de Entrenamiento e Investigaciones Médicas (CIDEIM) in accordance with national (Resolution 008430, República de Colombia, Ministry of Health, 1993) and international (Declaration of Helsinki and amendments, World Medical Association, Fortaleza, Brazil, October 2013) guidelines. All individuals voluntarily participated in the study, and written informed consent was obtained for each participant.
Study design and subjects
This study was designed to identify host biomarkers and innate immune functions that participate in the response to antileishmanial treatment in patients with localized CL. Transcriptional profiling of innate immune cells and lesion biopsies was conducted. Adult patients (18 to 60 years of age) with the parasitological diagnosis of active CL with a time of evolution < 6 months, and without apparent immune deficiencies (negative HIV test, no evidence of immunological disorder or treatment with medication having immunomodulating effects), who received standard-of-care treatment with Glucantime (GLUC, 20 mg/kg/day for 20 days) or miltefosine (MLF, 1.8–2.5 mg/kg daily dose for 28 days) were included in this study. Treatment outcome was evaluated at week 13 following initiation of treatment for GLUC and at week 26 for MLF. The cure was defined as complete re-epithelization and the absence of inflammatory signs for all lesions. TF was defined as incomplete re-epithelization and/or the presence of induration, raised borders, or redness in any lesion, reactivation of the original lesion(s), or the appearance of new lesions.
Skin lesions biopsies samples
Skin lesion punch biopsies were obtained before initiation of treatment. Biopsy punches of 3 mm were obtained under local anesthetic, taking into account the following ratio: 1/3 of healthy skin and 2/3 of the edge of the lesion (the indurated edge, which does not include necrotic tissue). Skin biopsies were immediately stored in 1 mL Allprotect® (Qiagen, cat. 76405). Samples were equilibrated overnight at 4 °C and then stored at − 20 °C until processing57.
Isolation of monocytes, neutrophils and eosinophils from peripheral blood samples
Ninety mL of whole blood anticoagulated with EDTA were obtained from each patient. PBMCs and polymorphonuclear leukocytes (PMNs) were isolated by centrifugation over a PolymorphprepTM (Axis-Shield, cat. 1114683) gradient according to the manufacturer’s instructions. CD14 + Mo were purified from PBMCs using the CD14 microbeads ultrapure kit (Milteny Biotec, cat. 130118906) coupled to magnetic cell sorting (MACS). CD16 + Nφ were purified from PMNs using CD16 microbeads human kit (Milteny Biotec, cat. 130045701), and Eφ were obtained by negative selection using the eosinophil isolation kit human (Milteny Biotec, cat. 130092010). Cells were washed with cold PBS, precipitated by centrifugation, and the cell pellet was resuspended in 100–200 µL of RNAlaterTM (Invitrogen, cat. AM7020) and stored at − 80 °C for later use. The purity of isolated cell populations was evaluated by flow cytometry and light microscopy. One million cells of each subpopulation were used for staining. Mo, Nφ, and Eφ were stained with CD14-FITC/CD3-APC, CD66-FITC/CD16-PE and CD16-PE, respectively. Flow cytometry acquisition was performed on a BD Accuri C6 (BD Biosciences) cytometer; 50,000 events were collected for each processed sample. Data analysis was done using Flow-Jo (Treestar) version 10.0. The gating strategy included verification by FSC/SSC (forward and side scatter features) and expression of cell-specific markers: CD14 + for Mo; CD16 + /CD66 + Nφ; and CD16- for Eφ. Samples were used only when purity was > 95%.
Leishmania strains, typing and drug susceptibility testing
Leishmania isolates were obtained from all patients and propagated in Senekjie´s biphasic blood agar and immediately stored in liquid nitrogen until use. Strains were typed by immunoreactivity to monoclonal antibodies as previously described58. Drug susceptibility of intracellular amastigotes was estimated by evaluation of % parasite survival in PMA-differentiated U-937 cells after exposure to pentavalent antimony (SbV) at a final concentration of 32 µg/mL, compared to control without drug exposure. Leishmania strains were defined as Sb-resistant when the percent reduction of the parasite burden after drug exposure was < 78%. Susceptibility cutoff was defined based on a panel of well-characterized clinical isolates presenting with intrinsic resistance or susceptibility to SbV 59,60.
RNA isolation and cDNA synthesis
RNA isolation from purified cell populations (Mo, Nφ, and Eφ) stored in RNAlater was performed using TRIzolTM (Invitrogen, cat. 15596026), followed by RNA cleanup with RNeasy Mini Kit columns (Qiagen, cat. 74104). Isopropanol/water (1:1) was used for RNA precipitation. RNA isolation from lesion biopsies was performed by tissue disruption, homogenization, and extraction using TRIzolTM reagent as previously described57. RNA integrity was assessed using an Agilent 2100 bioanalyzer (RNA 6000 Nano LabChip, Agilent Technologies, Cat. 5067-1511). For RNA-seq, poly(A)-enriched cDNA libraries were generated using the Illumina TruSeq v2 sample preparation kit (San Diego, CA. Cat. 20020594) and checked for quality and quantity using bioanalyzer and quantitative PCR.
RNA-seq data generation, preprocessing, and quality trimming
Single or paired-end reads were obtained on an Illumina NovaSeq 6000 at the Genetic Resources Core Facility, Johns Hopkins Department of Genetic Medicine, Baltimore, MD; or on an Illumina HiSeq1000 at the Brain & Behavior Institute - Advanced Genomic Technologies Core (BBI-AGTC) at the University of Maryland, College Park, MD. Trimmomatic61 was used to remove Illumina adapter sequences, discard reads shorter than 40 nucleotides, and trim any 4 nucleotide rolling window with a mean Phred quality score less than or equal to 20. Sequence quality metrics were assessed using FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Raw data and project information is available via NCBI-dbGaP study ID # 38338; phs003545.v1. All code and analytical pipelines are available at https://doi.org/10.5281/zenodo.15002596.
Mapping cDNA fragments and abundance estimation
Reads were aligned against the human (hg38 revision 100), L.V. panamensis (TriTrypDb release 36), and L.V. braziliensis (release 26) genomes with HISAT2 (2.1.0)62 using the default parameters. The resulting accepted hits and mapped reads were sorted and indexed via SAMtools63 and passed to HTSeq64 for generating count tables.
Global data assessment, visualization and differential expression analysis
Biological replicates and batch effects were assessed and visualized using an R package, hpgltools (https://github.com/elsayed-lab/hpgltools), developed and maintained in the El-Sayed lab. Normalized data were visualized using log2 transformed counts per million (CPM) reads following filtering to remove low counts (defined as any gene with a sum less than twice the number of samples or when all samples had fewer than 2 counts). Samples in which fewer than 11,000 genes were observed in a non-zero genes plot, were removed. Following data filtration, visualizations were performed to observe the sample relationships. These included density plots, boxplots of depth, coefficient of variance plots, hierarchical clustering analyses based on Pearson’s correlation coefficient and Euclidean distance, variance partition analyses, and principal component analyses (PCA) before and after normalization. Several combinations of normalization and batch adjustment strategies were evaluated along with surrogate variable estimation via SVA65. Samples were queried via cure/TF status, visit number, clinic, and cell type in order to calculate the surrogate variable (SV) loadings, and the F-statistic was calculated for each variable with respect to each SV.
Differential expression analyses were performed using a single pipeline, which performed all pairwise comparisons using the Bioconductor packages: limma66, edgeR67, DESeq268, EBSeq69, NOISeq70, and a basic analysis using only log2CPM values. In each case (except EBSeq and the basic analysis), the surrogate variable estimates provided by SVA were used to adjust the statistical model in an attempt to address the batch/surrogate effects. Dream19 was also used to evaluate a linear mixed effects model, with the donor as the random effect (with fixed slope and variable intercept) to account for the variability introduced by multiple samples per person. This iteration of the analyses sought to relate the expression data as a function of the final outcome, visit, cell type, and donor. Each contrast was evaluated in the context of its agreement with other methods, but the interpretations were primarily informed by the DESeq2 results. Genes with significant changes in abundance ( | log2 fold change | ≥ 1 and false discovery rate adjusted P-values ≤ 0.05) were passed to gProfileR71 and clusterProfiler72 (one-sided Fisher test with g:SCS-corrected P-values). Gene ontology analyses were supplemented with manual data curation. Network analyses were performed with STRING 12.073. Simultaneously, gene set variation analysis (GSVA)74 was performed to produce an enrichment score (using the Wald test) against the mSigDB75 datasets (C2, C7, and H) on a per-sample basis. These scores were passed to limma to evaluate the difference in GSVA score distributions for each gene set in the samples (limma and Benjamini-Hochberg adjusted P-values used to compare sample categories). Results from limma were then filtered according to log2 fold change, adjusted P-value, and maximum GSVA score mean.
Detection of virus sequences
Kraken 2, with a supplemented version of its viral database76, was used to check each sample specifically for the Leishmania RNA virus (LRV), as well as any other putative viral reads. Confirmation of LRV absence was performed by qRT-PCR as described46.
Weighted gene co-expression network analysis (WGCNA)
WGCNA co-expression networks were generated and examined using low-count filtered, SVA-adjusted, RPKM (Reads per kilobase per million) by average CDS length, log2-transformed counts as input. Pairwise Pearson correlations between each gene pair were calculated and transformed into a signed adjacency matrix using the minimum power that resulted in a scale-free R2 fit of 0.8. The resulting modules and associated eigengenes were produced via the default correlation matrix blockwise module detection methods from WGCNA. Module scores by sample were queried against the metadata factors (cure and TF) via Pearson’s correlations and scored using the P-value metrics provided by WGCNA. The eigengenes were extracted from modules with scores deemed significant, manually examined, and passed to gene set enrichment methods. Modules significantly associated with the outcome and containing more than 1000 genes were excluded from the analyses.
Composite scores
Pre-Tx RPKM values were used to construct raw and normalized (Z-score) composite scores. The selection of genes that constitute each individual composite was based on the DE genes between cures and TF that were common among Nφ, Eφ, and Mo (signature genes). Raw scores were calculated per patient, as the sum of RPKM values of the signature genes in each cell type (Eq. 1). A normalized score was also computed based on the sum of normalized RPKM values (Eq. 2). For raw and normalized scores, “I” indexes genes and “k” patients.
For each cell type:
where
and
Receiver operating characteristic (ROC) curves were used to explore the predictive potential of raw scores and Z-scores to discriminate therapeutic cure and failure. Sensitivity and specificity parameters were calculated for scores. Youden’s J statistic was used to define cutoff values.
Machine learning models
A series of machine learning models were generated and examined via the caret R package26 to create transcriptome-informed classifiers of patients likely to cure or fail treatment. Initial expression sets were selected to include all data from innate cells from patients recruited in Cali and Tumaco, or data only from Tumaco patients. The starting data were log2-transformed CPM values, normalized, filtered to exclude genes with CV < 0.1, centered, and filtered to exclude genes with correlations ≥ 0.95. The most variable 3000 genes were then selected. The remaining data was split into training (0.4) and testing (0.6) sets 10 times. The training datasets were used to create k-nearest neighbor, random forest, GLM, and gradient boost models with an arbitrarily chosen mix of bootstrap and CV sampling. The test partitions were evaluated for accuracy and sensitivity/specificity with respect to the known outcome of each patient.
Statistical analyses
For the exploration and description of the sociodemographic and clinical variables, univariate analyses were performed. Categorical variables were described with frequencies and percentages. Quantitative variables were described as means ( ± SD) or medians (IQR) according to the distribution of the data. For the comparison of qualitative variables, Fisher’s exact test or the chi2 test was used according to the distribution of the data. Fisher´s test was also used for comparison of tables larger than 2 × 277. Quantitative variables were compared using t-test or U-Mann-Whitney tests. Normality was determined with qq plots and the Shapiro-Wilk test. In all analyses, P-values < 0.05 were considered significant. Statistical analysis was performed using GraphPad Prism version 9 and R version 4.1.3.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All sequence data is available at NCBI-dbGaP study ID # 38338; phs003545.v1 (https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs003545.v1.p1).
Code availability
All code and analytical pipelines are available at https://doi.org/10.5281/zenodo.15002596.
References
WHO. Vector-borne diseases. https://www.who.int/news-room/fact-sheets/detail/vector-borne-diseases (2020).
WHO/TDR. Global Report for Research on Infectious Diseases of Poverty. (2012).
WHO. Leishmaniasis: Status of endemicity of cutaneous leishmaniasis 2020. https://apps.who.int/neglected_diseases/ntddata/leishmaniasis/leishmaniasis.html (2020).
Oliveira, L. F. et al. Systematic review of the adverse effects of cutaneous leishmaniasis treatment in the New World. Acta Trop. 118, 87–96 (2011).
PAHO. Guideline for the Treatment of Leishmaniasis in the Americas. (2022).
Uribe-Restrepo, A. F., Prieto, M. D., Cossio, A., Desai, M. M. & Del Mar Castro, M. Eligibility for local therapies in adolescents and adults with cutaneous leishmaniasis from southwestern Colombia: A cross-sectional study. Am. J. Trop. Med. Hyg. 100, 306–310 (2019).
Rosales-Chilama, M. et al. Parasitological confirmation and analysis of Leishmania diversity in asymptomatic and subclinical infection following resolution of cutaneous Leishmaniasis. PLoS Negl. Trop. Dis. 9, 1–20 (2015).
Vergel, C. et al. Evidence for Leishmania (Viannia) parasites in the skin and blood of patients before and after treatment. J. Infect. Dis. 194, 503–511 (2006).
Camera, P. et al. Haematogenous dissemination of Leishmania (Viannia) braziliensis in human American tegumentary leishmaniasis. Trans. R. Soc. Trop. Med. Hyg. 100, 1112–1117 (2006).
Scott, P. & Novais, F. O. Cutaneous leishmaniasis: Immune responses in protection and pathogenesis. Nat. Rev. Immunol. 16, 581–592 (2016).
Gómez, M. A. et al. Immuno-pharmacokinetics of meglumine antimoniate in patients with cutaneous Leishmaniasis caused by Leishmania (Viannia). Clin. Infect. Dis. 72, E484–E492 (2021).
Del Mar Castro, M. et al. Pharmacokinetics of miltefosine in children and adults with Cutaneous leishmaniasis. Antimicrob. Agents Chemother. 61, 1–11 (2017).
Navas, A. et al. Profiles of local and systemic inflammation in the outcome of treatment of human cutaneous leishmaniasis caused by L. Viannia. Infect. Immun. 88, 1–12 (2020).
Kip, A. E. et al. Systematic review of biomarkers to monitor therapeutic response in leishmaniasis. Antimicrob. Agents Chemother. 59, 1–14 (2015).
Amorim, C. F. et al. Variable gene expression and parasite load predict treatment outcome in cutaneous leishmaniasis. Sci. Transl. Med. 11, 1–9 (2019).
Adaui, V. et al. Association of the endobiont double-stranded RNA virus LRV1 with treatment failure for human Leishmaniasis caused by Leishmania braziliensis in Peru and Bolivia. J. Infect. Dis. 213, 112–121 (2016).
Valencia, B. M. et al. Leishmania RNA virus-1 is similarly detected among metastatic and non-metastatic phenotypes in a prospective cohort of American Tegumentary Leishmaniasis. PLoS Negl. Trop. Dis. 16, e0010162 (2022).
Campos, T. M. et al. Granzyme B produced by natural killer cells enhances inflammatory response and contributes to the immunopathology of cutaneous Leishmaniasis. J. Infect. Dis. 221, 973–982 (2020).
Hoffman, G. E. & Roussos, P. Dream: powerful differential expression analysis for repeated measures designs. Bioinformatics 37, 192–201 (2021).
Hoffman, G. E. & Schadt, E. E. variancePartition: interpreting drivers of variation in complex gene expression studies. BMC Bioinforma. 17, 483 (2016).
Garratt, L. W. Current understanding of the neutrophil transcriptome in health and disease. Cells 10, 2406 (2021).
Monaco, G. et al. RNA-Seq signatures normalized by mRNA abundance allow absolute deconvolution of human immune cell types. Cell Rep. 26, 1627–1640 (2019).
Mcnab, F., Mayer-barber, K., Sher, A., Wack, A. & Garra, A. O. Type I interferons in infectious disease. Nat. Rev. Immunol. 15, 87–103 (2020).
Gregorio, A., Junior, D., Sampaio, N. G. & Rehwinkel, J. A balancing act: MDA5 in antiviral immunity and autoinflammation. Trends Microbiol. 27, 75–85 (2019).
Giraldo-Parra, L. et al. Consolidation of a molecular signature of healing in cutaneous Leishmaniasis Is achieved during the first 10 days of treatment. J. Immunol. 212, 894–903 (2024).
Kuhn, M. Building predictive models in R using the caret package. J. Stat. Softw. 28, 1–26 (2008).
Domagalska, M. A., Barrett, M. P. & Dujardin, J.-C. Drug resistance in Leishmania: does it really matter? Trends Parasitol. 39, 251–259 (2023).
Murray, H. W. & Delph-Etienne, S. Roles of endogenous gamma interferon and macrophage microbicidal mechanisms in host response to chemotherapy in experimental visceral leishmaniasis. Infect. Immun. 68, 288–293 (2000).
Murray, H. W., Jungbluth, A., Ritter, E., Montelibano, C. & Marino, M. W. Visceral leishmaniasis in mice devoid of tumor necrosis factor and response to treatment. Infect. Immun. 68, 6289–6293 (2000).
Murray, H. W., Montelibano, C., Peterson, R. & Sypek, J. P. Interleukin-12 regulates the response to chemotherapy in experimental visceral Leishmaniasis. J. Infect. Dis. 182, 1497–1502 (2000).
Murray, H. W. & Nathan, C. F. Macrophage microbicidal mechanisms in vivo: reactive nitrogen versus oxygen intermediates in the killing of intracellular visceral Leishmania donovani. J. Exp. Med. 189, 741–746 (1999).
Murray, H. W., Oca, M. J., Granger, A. M. & Schreiber, R. D. Requirement for T cells and effect of lymphokines in successful chemotherapy for an intracellular infection. Experimental visceral leishmaniasis. J. Clin. Invest. 83, 1253–1257 (1989).
Laguna, F. Treatment of leishmaniasis in HIV-positive patients. Ann. Trop. Med. Parasitol. 97, 135–142 (2003).
Laguna, F. et al. Amphotericin B lipid complex versus meglumine antimoniate in the treatment of visceral leishmaniasis in patients infected with HIV: a randomized pilot study. J. Antimicrob. Chemother. 52, 464–468 (2003).
Palacios, R., Osorio, L. E., Grajales, L. F. & Ochoa, M. T. Treatment failure in children in a randomized clinical trial with 10 and 20 days of meglumine antimonate for cutaneous leishmaniasis due to Leishmania viannia species. Am. J. Trop. Med. Hyg. 64, 187–193 (2001).
Soto, J., Toledo, J., Vega, J. & Berman, J. Short report: efficacy of pentavalent antimony for treatment of colombian cutaneous leishmaniasis. Am. J. Trop. Med. Hyg. 72, 421–422 (2005).
Velez, I. et al. Efficacy of miltefosine for the treatment of American cutaneous leishmaniasis. Am. J. Trop. Med. Hyg. 83, 351–356 (2010).
Mattner, J. et al. Protection against progressive leishmaniasis by IFN-beta. J. Immunol. 172, 7574–7582 (2004).
Diefenbach, A. et al. Type 1 interferon (IFNalpha/beta) and type 2 nitric oxide synthase regulate the innate immune response to a protozoan parasite. Immunity 8, 77–87 (1998).
Kumar, R. et al. Type I interferons suppress anti-parasitic immunity and can be targeted to improve treatment of visceral Leishmaniasis. Cell Rep. 30, 2512–2525.e9 (2020).
Schneider, W. M., Chevillotte, M. D. & Rice, C. M. Interferon-stimulated genes: a complex web of host defenses. Annu. Rev. Immunol. 32, 513–545 (2014).
Barrat, F. J., Crow, M. K. & Ivashkiv, L. B. Interferon target-gene expression and epigenomic signatures in health and disease. Nat. Immunol. 20, 1574–1583 (2019).
Mayer-Barber, K. D. et al. Host-directed therapy of tuberculosis based on interleukin-1 and type I interferon crosstalk. Nature 511, 99–103 (2014).
Berry, M. P. R. et al. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature 466, 973–977 (2010).
Kolumam, G. A., Thomas, S., Thompson, L. J., Sprent, J. & Murali-Krishna, K. Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection. J. Exp. Med. 202, 637–650 (2005).
Rosales-Chilama, M., Oviedo, M. Y., Quintero, Y. K., Fernandez, O. L. & Gomez, M. A. Leishmania RNA Virus Is Not Detected in All Species of the Leishmania Viannia Subgenus: The Case of L. (V.) panamensis in Colombia. Am. J. Trop. Med. Hyg. 108, 555–560 (2023).
Gallego, C., Golenbock, D., Gomez, M. A. & Saravia, N. G. Toll-like receptors participate in macrophage activation and intracellular control of Leishmania (Viannia) panamensis. Infect. Immun. 79, 2871–2879 (2011).
Gomez, M. A. et al. Early leukocyte responses in ex-vivo models of healing and non-healing human Leishmania (Viannia) panamensis infections. Front. Cell Infect. Microbiol. 11, 687607 (2021).
Castro, M. D. M. et al. Pentoxifylline in the treatment of cutaneous Leishmaniasis: A randomized clinical trial in Colombia. Pathogens 11, 378 (2022).
Pinart, M. et al. Interventions for American cutaneous and mucocutaneous leishmaniasis. Cochrane Database Syst. Rev. 8, CD004834 (2020).
Firooz, A. et al. Imiquimod in combination with meglumine antimoniate for cutaneous leishmaniasis: a randomized assessor-blind controlled trial. Arch. Dermatol. 142, 1575–1579 (2006).
Sundar, S. & Murray, H. W. Effect of treatment with interferon-γ alone in visceral Leishmaniasis. J. Infect. Dis. 172, 1627–1629 (1995).
Pin, A. et al. An easy and reliable strategy for making type I interferon signature analysis comparable among research centers. Diagnostics 9, 113 (2019).
Baechler, E. C. et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc. Natl. Acad. Sci. USA 100, 2610–2615 (2003).
Kim, H. et al. Development of a validated interferon score using NanoString Technology. J. Interf. Cytokine Res. 38, 171–185 (2018).
Alyass, A., Turcotte, M. & Meyre, D. From big data analysis to personalized medicine for all: challenges and opportunities. BMC Med. Genomics 8, 33 (2015).
Giraldo-Parra, L., Ramirez, L. G., Navas, A. & Gómez, M. A. Quality parameters for RNA preparations from biopsies of ulcerated human skin. Wellcome Open Res. 7, 1–13 (2023).
Grimaldi, G. & McMahon-Pratt, D. Monoclonal antibodies for the identification of New World Leishmania species. Mem. Inst. Oswaldo Cruz 91, 37–42 (1996).
Fernandez, O. L. et al. Miltefosine and antimonial drug susceptibility of Leishmania Viannia species and populations in regions of high transmission in Colombia. PLoS Negl. Trop. Dis. 8, e2871 (2014).
Fernández, O. et al. Novel approach to in vitro drug susceptibility assessment of clinical strains of Leishmania spp. J. Clin. Microbiol. 50, 2207–2211 (2012).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915 (2019).
Li, H., Handsaker, B., Wysoker, A. & Fennell, T. The sequence alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Anders, S., Theodor Pyl, P. & Huber, W. HTSeq-a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015).
Leek, J. T., Johnson, W. E., Parker, H. S., Jaffe, A. E. & Storey, J. D. The SVA package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 28, 882–883 (2012).
Ritchie, M. E. et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, 1–13 (2015).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 1–21 (2014).
Leng, N. et al. EBSeq: An empirical Bayes hierarchical model for inference in RNA-seq experiments. Bioinformatics 29, 1035–1043 (2013).
Tarazona, S., García, F., Ferrer, A., Dopazo, J. & Conesa, A. NOIseq: a RNA-seq differential expression method robust for sequencing depth biases. EMBnet. J. 17, 18 (2012).
Kolberg, L. et al. G:Profiler-interoperable web service for functional enrichment analysis and gene identifier mapping (2023 update). Nucleic Acids Res. 51, W207–W212 (2023).
Yu, G., Wang, L.-G., Han, Y. & He, Q.-Y. clusterProfiler: an R Package for comparing biological themes among gene clusters. J. Integr. Biol. 16, 284–287 (2012).
Szklarczyk, D. et al. The STRING database in 2021: customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 49, D605–D612 (2021).
Hänzelmann, S., Castelo, R. & Guinney, J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinforma. 14, 7 (2013).
Liberzon, A. et al. Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740 (2011).
Wood, D. E., Lu, J. & Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 20, 257 (2019).
Mehta, C. R. & Patel, N. R. A network algorithm for performing fisher’s exact test in r × c contingency tables. J. Am. Stat. Assoc. 78, 427–434 (1983).
Acknowledgements
We gratefully acknowledge all patients who participated in this study and members of the CIDEIM Clinical Unit. We also acknowledge the support of the personnel of the CIDEIM BioBank for their technical assistance in the phenotyping of strains processed in this study, and Mariana Rosales for support with the identification of LRV in Leishmania strains. NIAID/NIH awards U19AI129910 and R01AI172887, M.A.G., and N.E.S. Fogarty International Center U.S. National Institutes of Health D43TW006589, M.A.G. and D.E.R. Wellcome Trust 107595/Z/15/Z, M.A.G. Ministerio de Ciencia, Tecnología e Innovación - Minciencias (contract no. 848−2019, code no. 222984368586), M.A.G. and D.A.V. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author information
Authors and Affiliations
Contributions
Conceptualization: M.A.G. and N.E.S. Methodology: A.T.B., D.A.V., L.G.P., T.A., and N.A. Investigation: M.A.G., A.T.B., D.A.V., L.G.P., D.E.R., T.A., N.E.S., and N.A. Visualization: M.A.G., A.T.B., and D.A.V. Funding acquisition: M.A.G. and N.E.S. Project administration: M.A.G. and N.E.S. Supervision: M.A.G. and N.E.S. Writing – original draft: M.A.G., A.T.B., D.A.V., D.E.R., and N.E.S. Writing – review & editing: M.A.G., A.T.B., D.A.V., L.G.P., D.E.R., T.A., N.E.S., and N.A.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Wim Adriaensen, and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Gómez, M.A., Belew, A.T., Vargas, D.A. et al. Innate biosignature of treatment failure in human cutaneous leishmaniasis. Nat Commun 16, 3235 (2025). https://doi.org/10.1038/s41467-025-58330-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-58330-3
