
Abstract
The development of photoelectrochemical cells for reduction of nitrate to ammonia under solar light is of significant interest for the production of clean chemicals and fuels but has remained a daunting challenge. Here, we investigate various metal catalysts supported on GaN nanowires grown on n+-p Si wafer – an emerging functional platform for scalable artificial photosynthesis – and demonstrate highly stable and efficient photoelectrochemical nitrate reduction reaction. We find that Co and Ni catalysts on GaN/Si exhibit the best performance, with an onset potential >0.3 VRHE and a faradaic efficiency of NH3 of 99% at 0.2 VRHE. These results highlight the advantage of photoelectrochemical system in achieving efficient nitrate reduction under more positive potentials. In-situ measurements and theoretical calculations reveal that the binding modes of the \({{{\rm{NO}}}}_{2}^{{-}}\) intermediate play a key role in the NH3 synthetic process. These results demonstrate that the rational design of catalysts on photoelectrodes can construct synergistic metal-semiconductor interactions for efficient and stable photoelectrochemical NH3 synthesis.
Similar content being viewed by others

Introduction
Nitrate (\({{{\rm{NO}}}}_{3}^{{-}}\)) is a common pollutant found in wastewater, industrial runoffs, and groundwater1. It represents a significant threat to human health since it induces diseases upon conversion to nitrite (\({{{\rm{NO}}}}_{2}^{{-}}\)) in the body2,3. The electrochemical \({{{\rm{NO}}}}_{3}^{{-}}\) reduction reaction (\({{{\rm{NO}}}}_{3}^{{-}}\)RR) is a promising method removing \({{{\rm{NO}}}}_{3}^{{-}}\) by producing ammonia (NH3), a valuable fertilizer and industrial chemical. The \({{{\rm{NO}}}}_{3}^{{-}}\) RR is also an environment-friendly method to close the \({{{\rm{NO}}}}_{3}^{{-}}\)N cycle, reducing the need for the Haber-Bosch process which is energy-intensive and produces greenhouse gases4,5,6. From the energy perspective, it is advantageous when compared to NH3 synthesis by N2 reduction due to the lower dissociation energy of N=O bond (204 kJ/mol) of \({{{\rm{NO}}}}_{3}^{{-}}\) when compared to the N ≡ N bond (941 kJ/mol)1,7. \({{{\rm{NO}}}}_{3}^{{-}}\)-to-NH3 conversion in aqueous electrolyte is a complex 9-proton and 8-electron reduction reaction, where protons are provided by water dissociation. Therefore, the catalysts should have both activity for the water dissociation and the \({{{\rm{NO}}}}_{3}^{{-}}\) conversion to promote the \({{{\rm{NO}}}}_{3}^{{-}}\) RR8. In addition, the \({{{\rm{NO}}}}_{3}^{{-}}\)-to-NH3 reduction is a complicated reaction with different stages of intermediates having various binding modes. Electrocatalysts have been investigated for the \({{{\rm{NO}}}}_{3}^{{-}}\) RR, including metal nanoparticles (NPs)9,10, alloys11,12,13, and compounds14,15,16,17,18,19,20,21,22,23, 2D materials24, and single atom catalysts25,26. However, due to their limited functionality of catalyzing the water dissociation, \({{{\rm{NO}}}}_{3}^{{-}}\) activation, or reduction of intermediates to the NH3, the overpotential still needs to be significantly improved for any practical consideration. In order to mitigate this issue, multiple functional sites for different catalytic reactions were integrated by decorating one catalyst onto another18,27. For instance, an electrocatalyst consisting of two phases of Cu/CuOx and Co/CoO showed promising NH3 synthetic performance due to efficient \({{{\rm{NO}}}}_{3}^{{-}}\)-to-\({{{\rm{NO}}}}_{2}^{{-}}\) conversion on Cu/CuOx and the subsequent \({{{\rm{NO}}}}_{2}^{{-}}\)-to-NH3 conversion on Co/CoO18. This synergistic relay catalysis or intermediate spillover improved the performance compared to their single phases.
Photoelectrodes, fabricated by coupling the catalysts with photo-responsive semiconductor, can convert solar energy directly to the chemical energy and further reduce the overpotential of the \({{{\rm{NO}}}}_{3}^{{-}}\) RR. Previously, Au-decorated Si nanowires (NWs) photoelectrode demonstrated photoelectrochemical (PEC) \({{{\rm{NO}}}}_{3}^{{-}}\) RR28. However, the maximum production rate of NH3 (YNH3) was only ~0.4 μmol/h/cm2 and the stability has remained very limited. Recent advancements in PEC \({{{\rm{NO}}}}_{3}^{{-}}\) RR using chalcogenide light absorbers demonstrated the potential of binary metal catalysts to enhance catalytic activity and efficiency. For instance, CuSn29 and CoCu30 catalysts integrated with Sb2S3 and Sb2Se3 photocathodes have shown exceptional faradaic efficiency of NH3 (~97% at 0.4 VRHE and 88% at −0.2 VRHE, respectively) and low onset potentials (0.62 VRHE and 0.43 VRHE), highlighting their ability to optimize reaction kinetics and improve charge transfer efficiency. These studies illustrate the importance of catalyst design in achieving high performance in PEC \({{{\rm{NO}}}}_{3}^{{-}}\) RR systems.
When catalysts are decorated on the surface of semiconductors, the photoelectrodes have two or more different functional sites at the surface, leading to distinct electronic structures and unique catalytic activity. This determines the reaction thermodynamics, intermediate binding modes, and final products in various catalytic reactions, such as water splitting, carbon dioxide reduction, N2 reduction, and \({{{\rm{NO}}}}_{3}^{{-}}\) reduction31,32,33,34,35,36. Recent studies have shown that GaN nanostructures, e.g., nanowires (NWs), inherently possess the excellent water dissociation activity due to characteristic Ga-N bond at the m-plane crystal facet37,38,39. This suggests that GaN NWs can be an ideal supporting materials to promote the proton-coupled electron transfer reaction including \({{{\rm{NO}}}}_{3}^{{-}}\) reduction to NH3. If the integration of metal catalyst on the semiconductor nanostructures can demonstrate synergetic effects by reducing the thermodynamic energy barriers, controlling the binding modes of adsorbates, and providing a sufficient number of protons with effective transfer of photogenerated electrons, the photoelectrodes will further reduce the overpotential and enhance the productivity of NH3 compared to electrocatalysts. Therefore, rational design of photoelectrodes by selecting a suitable catalyst on the semiconductor surface and the understanding of reaction pathways are urgently required to achieve more efficient and productive solar-driven \({{{\rm{NO}}}}_{3}^{{-}}\) conversion to NH3.
In this work, we investigated the metal catalysts supported on GaN NWs vertically grown on n+-p Si photoelectrodes for PEC \({{{\rm{NO}}}}_{3}^{{-}}\) RR to NH3 to demonstrate the synergistic effects of metal catalysts and GaN NWs. We first screened various noble metals (Pt, Ir, Rh, Pd, Au, Ru, Ag) and transition metals (Cu, Ni, Co) on GaN NWs for NH3 synthesis. Co-loaded GaN/Si (Co/GaN/Si) showed the highest faradaic efficiency of NH3 (FENH3 > 90%) at the widest potential window (0.2 to −0.7 VRHE). Ni/GaN/Si achieved the NH3 production rate of 83.3 μmol/h/cm2 and the maximum value of 201.6 μmol/h/cm2 at potentials of 0 and −0.4 VRHE, respectively. We characterized the performance of photoelectrodes based on the presence of GaN NWs and found that the catalysts loaded on GaN NWs showed much lower onset potential and higher productivity of NH3 than those on planar n+-p Si photoelectrodes. This is due to the synergetic metal-GaN interaction which enhances water dissociation and catalytic activity of nitrogen-containing intermediates. GaN NWs rapidly dissociate water, providing a sufficient number of protons near the surface, which in turn activates \({{{\rm{NO}}}}_{3}^{{-}}\) and facilitates its efficient conversion to \({{{\rm{NO}}}}_{2}^{{-}}\). Then, following \({{{\rm{NO}}}}_{2}^{{-}}\) to NH3 conversion is further promoted by catalysts. A series of in-situ experiments and theoretical calculations were performed to interpret reaction mechanisms and revealed that the binding modes of the NO2− intermediate (*ONO−* or *NO2−) on the metal catalysts were the key to the further deoxygenation process. This work provides insights into the design of efficient photoelectrode for green NH3 production.
Results
Metal catalysts loaded on GaN/Si photoelectrodes
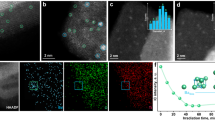
Photoelectrodes were fabricated by plasma-assisted molecular-beam epitaxy growth of GaN NWs on n+–p Si wafers, followed by photodeposition of metal catalysts on GaN NWs. A 45-degree tilt-view scanning electron microscopy (SEM) image of Co/GaN/Si showed that GaN NWs were vertically aligned on the n+-p Si wafer, with a length of ~400 nm and diameter of ~50 nm (Fig. 1a). Since the particle size of metal catalysts was too small to be observed in the SEM image, we performed high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) and energy-dispersive spectroscopy (EDS) elemental mapping to image the Co/GaN/Si (Fig. 1b and Fig. S1) and Ni/GaN/Si photoelectrodes (Fig. 1c and Fig. S2). The HAADF-STEM images showed the segregated Co and Ni nanoparticles (NPs) on both the top and sidewalls of GaN NWs. The atomic number difference between Co (27), Ni (28), and Ga (31) results in a Z-contrast that allows the NPs to be distinguished from the GaN NWs. The EDS elemental maps confirmed the presence of the metal catalysts, with locally aggregated Co (shown in yellow in Fig. 1b) and Ni (shown in orange in Fig. 1c). Ga (blue) and N (cyan) were uniformly distributed over the GaN NWs. The presence of oxygen indicates that the surface metal catalysts were oxidized prior to the analysis.

a 45 degree-tilt-view SEM image of Co/GaN/Si. STEM-HADDF images and EDS elemental maps of b Co/GaN and c Ni/GaN. Ga (blue) and N (cyan) were uniformly detected on GaN NWs while Co (yellow), Ni (orange), O (green) elements were segregated on the surface of GaN NWs. XPS spectra of d Ga 2p2/3, e N 1 s, f Co 2p, g Ni 2p, and h O 1s for GaN/Si, Co/GaN/Si, and Ni/GaN/Si.
To investigate the chemical bonding states of photoelectrodes with different metal catalysts, X-ray photoelectron spectroscopy (XPS) was performed. The Ga 2p3/2 XPS spectra were deconvoluted into a major peak of the Ga-N bond (1116.8 eV) and a minor peak of the Ga-O bond (1118.0 eV) (Fig. 1d)40. Similarly, the N 1s XPS spectra could be separated into a major N-Ga bond (396.9 eV) and a minor N-H bond (398.5 eV), likely due to native oxide or hydroxide on the surface (Fig. 1e)41. It should be noted that although Co, Ni, or other catalysts were loaded on the GaN NWs, the intensities of the Ga 2p3/2 and N 1s spectra were almost preserved. This means that the metal catalysts were partially covered over the GaN and the GaN surface was still exposed. The Co catalysts exhibited Co2+ (782.7 and 798.3 eV) and Co3+ (781.2 and 796.8 eV) oxidation states in the Co 2p spectrum (Fig. 1f). The Ni catalysts revealed Ni-O (853.5 and 871.1 eV), Ni-OH (854.7 and 872.3 eV), and their satellite bonds in the Ni 2p spectrum (Fig. 1g)42,43. In the O 1 s XPS spectra, O-Co and O-Ni bonds from the oxidized phases of the catalysts were observed (Fig. 1h). This is in good agreement with the STEM-EDS results. The metal oxides were also formed on other metal catalysts, such as Pt, Ir, Rh, Pd, Cu, Ag, Ru, and Au (Figs. S3–S10), since the catalysts were deposited under aqueous conditions and exposed to ambient air prior to the analysis.
Photoelectrochemical \({{{\rm{NO}}}}_{3}^{{-}}\) reduction reaction
Photoelectrochemical (PEC) nitrate reduction to ammonia (\({{{\rm{NO}}}}_{3}^{{-}}\)RR) was performed in an H-type cell under AM1.5G 1-sun solar light. When light was illuminated on the n+-p Si wafer through optically transparent GaN NWs, electrons and holes were generated in the conduction and valance bands of Si, respectively (Fig. 2a). The conduction bands of n-Si and n-GaN are approximately aligned44, so the photogenerated electrons in n-Si spontaneously migrate to the conduction band of n-GaN by the built-in potential of the Si p-n junction. These photoelectrons then move to the metal catalysts and participate in reduction reaction. After the reaction was completed, the liquid products of NH3, \({{{\rm{NO}}}}_{2}^{{-}}\), and N2H4 in the cathodic compartment of the reactor were quantified through calibrated spectrocolorimetric measurements (Figs. S11–S13). The gas products were analyzed by gas chromatography.

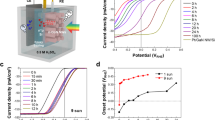
a Energy band diagram of metal/GaN/Si photoelectrode under light illumination. b Faradaic efficiency of NH3 for metal catalysts loaded on GaN/Si. The measurement was conducted in 0.1 M K2SO4 with 0.5 M KNO3 at various cathodic potentials (0.3 to −0.8 VRHE) under 1 sun light illumination. Faradaic efficiency of H2, NH3, \({{{\rm{NO}}}}_{2}^{{-}}\), and N2H4 for c GaN/Si, d Cu/GaN/Si, e Co/GaN/Si, and f Ni/GaN/Si. Co and Ni catalysts showed the highest FENH3 (>98%) among the tested samples.
In the electrolyte, a substantial number of water molecules coexist with \({{{\rm{NO}}}}_{3}^{{-}}\) ions. Therefore, when applying a cathodic potential to the photoelectrode, reduction reactions of \({{{\rm{NO}}}}_{3}^{{-}}\) conversion and hydrogen evolution may occur. To evaluate the photoelectrodes with respect to these two competing reactions, we performed linear sweep voltammetry (LSV) measurement of GaN/Si in 0.1 M K2SO4 with and without 0.5 M KNO3 (Fig. S14). The onset potential (Vonset) at −1 mA/cm2 for GaN/Si shifted positively from −0.63 to −0.21 VRHE and the photocurrent density was as high as ~40 mA/cm2 at −0.8 VRHE when 0.5 M KNO3 was added to 0.1 M K2SO4. Moreover, GaN/Si exhibited fairly high FENH3 = 61% at −0.7 VRHE, suggesting that GaN surface is more catalytically active for \({{{\rm{NO}}}}_{3}^{{-}}\) reduction to NH3 (\({{{\rm{NO}}}}_{3}^{{-}}\)RR) than for the hydrogen evolution reaction. However, Pt, Ir, and Rh catalysts (Figs. S15–S17) loaded on GaN/Si showed little difference in Vonset and photocurrent density regardless of \({{{\rm{NO}}}}_{3}^{{-}}\) in the electrolyte since these metals possess a high affinity for hydrogen adsorption rather than \({{{\rm{NO}}}}_{3}^{{-}}\) and good catalytic activity for hydrogen evolution reaction. As a result, these three metal catalysts showed very low faradaic efficiency of NH3 (FENH3) <13% with high FE of H2 (FEH2) >80% at all potentials (0.3 to −0.8 VRHE) (Fig. 2b). The photoelectrodes with Pd, Ag, Ru, and Cu catalyst showed FENH3 of 37%, 41%, 60%, and 13%, respectively at −0.7 VRHE (Figs. S18–S21). The predominant side product was \({{{\rm{NO}}}}_{2}^{{-}}\), indicating the good catalytic activity in converting \({{{\rm{NO}}}}_{3}^{{-}}\) to \({{{\rm{NO}}}}_{2}^{{-}}\) compared to hydrogen evolution, but the further reduction to NH3 was limited. Interestingly, when Au, Co, and Ni catalysts were coated on GaN/Si, the maximum FENH3 can approach ~100% (Figs. S22–S24).
In more detail, GaN/Si selectively converted \({{{\rm{NO}}}}_{3}^{{-}}\) to \({{{\rm{NO}}}}_{2}^{{-}}\) at low potential (−0.1 VRHE), with FENO2- = 74% (Fig. 2c). As the potential was increased to −0.7 VRHE, FENO2- monotonically decreased to 35% and FENH3 gradually increased to 61%. This demonstrated that GaN has excellent catalytic activity for the conversion of \({{{\rm{NO}}}}_{3}^{{-}}\) to \({{{\rm{NO}}}}_{2}^{{-}}\) and can further reduce \({{{\rm{NO}}}}_{2}^{{-}}\) to NH3 under large cathodic potential. In the case of Cu catalysts supported on GaN/Si, \({{{\rm{NO}}}}_{2}^{{-}}\) was selectively produced (Fig. 2d) with high FENO2- = 94% at 0.3 VRHE. However, unlike GaN/Si, \({{{\rm{NO}}}}_{2}^{{-}}\) was still the major product (FENO2- = 75%) with FENH3 = 13% and FEN2H4 = 14% at −0.7 VRHE. This suggests that the Cu catalyst coated on GaN has a negative effect on NH3 synthesis by desorbing the \({{{\rm{NO}}}}_{2}^{{-}}\) intermediate from the surface and preventing further reduction of \({{{\rm{NO}}}}_{2}^{{-}}\). This is discussed further in our theoretical analysis. In stark contrast, Co/GaN/Si dramatically improved FENH3 to 72% and 99% at 0.3 and 0.2 VRHE, respectively (Fig. 2e). These potentials are more positive than the standard reduction potential of \({{{\rm{NO}}}}_{3}^{{-}}\), meaning that the photovoltage generated by GaN/n+-p Si photoelectrode (~0.5 V)37,38,39 biases the reduction reaction and shifts the operating potential window positively. At the large cathodic potential of −0.8 VRHE, there was a decline of FENH3 to 68% due to increase in the FEH2. In contrast, Ni/GaN/Si selectively produced NH3 over other competing reactions although Ni/GaN/Si required a more negative potential (0 VRHE) to achieve FENH3 > 90% (Fig. 2f). The total faradaic efficiency was less than 100% especially at anodic potentials (+0.2–0 VRHE) because some electrons could be used for the reduction of \({{{\rm{NO}}}}_{3}^{{-}}\) to other nitrogen-containing species, such as N2 and NO, which were not fully analyzed. Additionally, the non-faradaic capacitive current density from ionic adsorption and desorption at the electrical double layer could affect the derivation of faradaic efficiency. We further investigated the effects of the loading amount of metal catalysts coated on GaN NWs on catalytic properties (Fig. S25). Within the precursor concentration range in this work, there was no significant difference in catalytic performance, indicating that the determining factor for catalyst characteristics is the type of metal catalyst rather than the loading amount of metal catalysts.
To further study the metal-support interactions, we compared the LSV curves of n+-p Si, GaN/Si, and metal catalysts loaded n+-p Si and GaN/Si in 0.1 M K2SO4 with 0.5 M KNO3 (Fig. 3a). In the order of Si, Co/Si, and Ni/Si, Vonset and photocurrent density were improved. GaN/Si showed a 1.1 V positive shift of Vonset compared to Si. Remarkably, GaN/Si loaded with Co and Ni catalysts showed much better Vonset = 0.22 and 0.27 VRHE, respectively. Charge transfer kinetics from the photoelectrodes to reactants were investigated by the PEC impedance spectroscopy at 0 VRHE under solar irradiation (Fig. 3b). The radius of the semicircle in the Nyquist plot correlates to the charge-transfer resistance during the PEC reaction45. For Si and Co/Si, the imaginary part of impedance diverged as the real part of impedance increased, indicating no faradaic charge transfer between the photoelectrode and the electrolyte. In contrast, for Ni/Si, GaN/Si, Co/GaN/Si, and Ni/GaN/Si, the radius of the semicircle decreased sequentially, implying a gradual reduction in charge transfer resistance. For GaN/Si, Co/GaN/Si, and Ni/GaN/Si photoelectrodes, the Nyquist plots were further analyzed at a more negative potential of −0.2 VRHE (Fig. S26). Under this condition, the radius of the semicircle significantly reduced, indicating accelerated charge transfer under high bias. From the impedance results, the synergetic metal-GaN interaction facilitated the \({{{\rm{NO}}}}_{3}^{{-}}\) RR likely due to catalytic enhancement and increased electrochemical surface area (Fig. S27).

a LSV curves and b Nyquist impedance plots of Si, Co/Si, Ni/Si, GaN/Si, Co/GaN/Si, and Ni/GaN/Si measured in 0.1 M K2SO4 with 0.5 M KNO3 under 1 sun light illumination. c Faradaic efficiency and d production rate of NH3 plotted as a function of cathodic potentials. Co/GaN/Si exhibited the widest potential window (−0.2 to −0.7 VRHE) for high FENH3 > 90% and Ni/GaN/Si revealed the best YNH3 = 201.6 μmol/cm2/h at −0.4 VRHE.
It should be noted that Co/GaN/Si showed the widest potential window (0.2 to −0.7 VRHE) for the selective NH3 production (FENH3 > 90%) and Ni/GaN/Si also exhibited selective conversion of \({{{\rm{NO}}}}_{3}^{{-}}\) to NH3 (FENH3 > 90%) at potential ≤ 0 VRHE (Fig. 3c). However, n+-p Si (Fig. S28) and those with Co (Fig. S29) and Ni (Fig. S30) catalysts showed low FENH3 < 54% with noticeably increased FEH2 > 42% or FENO2- >43% at −0.8 VRHE. This clearly demonstrated that GaN NWs play an important role in reducing the overpotential and increasing the NH3 selectivity of \({{{\rm{NO}}}}_{3}^{{-}}\) RR. Since Co/GaN/Si and Ni/GaN/Si exhibited the promising Vonset (>0.3 VRHE), high photocurrent density (>38 mA/cm2), and FENH3 (~100%) over the wide potential window, high production rates of 17.5 and 83.3 μmol/h/cm2 were achieved, respectively at 0 VRHE. In addition, the maximum production rate of NH3 = 166.7 μmol/h/cm2 at −0.7 VRHE and 201.6 μmol/h/cm2 at −0.4 VRHE were demonstrated, respectively (Fig. 3d). Applied bias photon-to-current efficiency (ABPE) was calculated for the tested photoelectrodes (Fig. S31). In the order of Si, Co/Si, Ni/Si, GaN/Si, Co/GaN/Si, and Ni/GaN/Si, the maximum ABPE gradually increased up to 2.41%, indicating that GaN NWs are the promising platforms for loading the metal catalysts and facilitating the \({{{\rm{NO}}}}_{3}^{{-}}\)RR. The higher ABPE and NH3 production rate for Ni/GaN/Si over Co/GaN/Si originated from more positive flat band potential likely due to more efficient electron injection from GaN NWs to Ni catalysts and better alignment for electron transfer from the Ni catalyst to the reactants (Fig. S32). Transient photocurrent measurements were conducted to further investigate charge carrier dynamics of photoelectrode with and without catalyst (Fig. S33). The results revealed that Co/GaN/Si exhibited superior charge retention and reduced recombination compared to GaN/Si, suggesting the critical role of Co catalyst in improving charge transfer efficiency and sustaining PEC activity during \({{{\rm{NO}}}}_{3}^{{-}}\)RR.
To investigate the influence of water dissociation in \({{{\rm{NO}}}}_{3}^{{-}}\)RR, we conducted kinetic isotopic effect (KIE) experiments by changing H2O and D2O in the electrolyte (Fig. S34). The KIE value was then calculated as the ratio of NH3 production rates in electrolytes with H2O and D2O. For Co/Si, the KIE value was calculated at 1.63, indicating that the reaction was constrained by water dissociation46. Upon the introduction of GaN NWs, the KIE value for Co/GaN/Si significantly dropped to 1.15. Once protons became readily available at the surface through water dissociation by GaN NWs, it appears that Co catalysts efficiently reduce \({{{\rm{NO}}}}_{3}^{{-}}\) to NH3 through deoxygenation and hydrogenation processes due to the intrinsic selectivity for producing NH3 rather than hydrogen evolution. Consequently, using GaN nanowires as a support for metal catalysts presents a promising approach to demonstrating the synergy between proton supply and the catalysis of nitrogen-containing intermediates.
Control experiments in 0.1 M K2SO4 were carried out using Co/GaN/Si to confirm whether NH3 is produced from \({{{\rm{NO}}}}_{3}^{{-}}\) reactant, and there was no NH3 produced without \({{{\rm{NO}}}}_{3}^{{-}}\) in the electrolyte (Fig. S35). Moreover, 15N isotope labeling clearly evidenced that 15NH3 was produced from 15\({{{\rm{NO}}}}_{3}^{{-}}\) (Fig. S36). The measured Vonset, FENH3, and YNH3 of Co or Ni catalysts on GaN NWs were comparable to those of recent photoelectrodes and superior to state-of-the-art electrocatalysts for \({{{\rm{NO}}}}_{3}^{{-}}\)RR (Table S1).
To study the effects of metal catalysts on catalytic \({{{\rm{NO}}}}_{3}^{{-}}\)RR activity, in-situ Fourier transform infrared (FTIR) measurements were performed for two representative photoelectrodes of GaN/Si (Fig. S37) and Co/GaN/Si (Fig. 38). The potential-dependent spectra (−0.2 to −0.7 VRHE, Figs. S37a, S38a) revealed distinct behaviors between the two photoelectrodes. As \({{{\rm{NO}}}}_{3}^{{-}}\)RR proceeded, both samples exhibited a gradual decrease in the peak intensity at 1348 cm−1 47, which attributes to the consumption of \({{{\rm{NO}}}}_{3}^{{-}}\) ions in the solution. For GaN/Si (Fig. S37a), a characteristic band at 1230 cm−1 corresponding to \({{{\rm{NO}}}}_{2}^{{-}}\) intermediates, was clearly observed. This band became more pronounced at more negative potentials, indicating the accumulation of \({{{\rm{NO}}}}_{2}^{{-}}\) as a primary product. Time-resolved measurements at −0.5 VRHE (Fig. S37b) further confirmed the buildup of \({{{\rm{NO}}}}_{2}^{{-}}\) near the surface as the intensity of \({{{\rm{NO}}}}_{2}^{{-}}\) band gradually increased over time. In contrast, Co/GaN/Si (Fig. S38a) demonstrated no signal of the \({{{\rm{NO}}}}_{2}^{{-}}\) band, even at highly cathodic potentials, suggesting efficient conversion of NO2 intermediates. This trend was further supported by the time-resolved measurements (Fig. S38b), where the absence of the \({{{\rm{NO}}}}_{2}^{{-}}\) band highlighted the catalytic role of Co catalyst in facilitating the PEC \({{{\rm{NO}}}}_{3}^{{-}}\)RR to NH3.
Stability of Co/GaN/Si and Ni/GaN/Si
We recorded the photocurrent density of Co/GaN/Si (Fig. 4a) and Ni/GaN/Si (Fig. 4b) at 0 and −0.6 VRHE under solar irradiation. At 0 VRHE, the reaction was conducted for 5 cycles in 0.1 M K2SO4 with 0.5 M KNO3 and each cycle was performed for 1 h with repeated replacement of fresh electrolyte. At −0.6 VRHE, 10 cycles of reactions were performed for 5 h. The average photocurrent densities of Co/GaN/Si were 3.5 mA/cm2 at 0 VRHE and 35.8 mA/cm2 at −0.6 VRHE. Ni/GaN/Si showed higher average photocurrent densities of 16.1 mA/cm2 at 0 VRHE and 39.2 mA/cm2 at −0.6 VRHE. Both Co/GaN/Si (Fig. 4c) and Ni/GaN/Si (Fig. 4d) showed very high FENH3 > 91% for 10 h. Co/GaN/Si continuously produced NH3 with YNH3 = 18.0 and 162.6 μmol/cm2/h at 0 and −0.6 VRHE, respectively. Ni/GaN/Si also consistently converted \({{{\rm{NO}}}}_{3}^{{-}}\) to NH3 with YNH3 = 73.5 μmol/cm2/h at 0 VRHE. At −0.6 VRHE, YNH3 of Ni/GaN/Si was ~170 μmol/cm2/h. There was no noticeable degradation of catalytic activity or change of morphology during the reaction, demonstrating that metal catalysts supported on GaN/Si photoelectrodes are promising platform for efficient and stable \({{{\rm{NO}}}}_{3}^{{-}}\)RR to NH3. Evaluating the stability at high potential of −0.6 VRHE is crucial from a material perspective since the accelerated degradation due to the detachment of metal catalysts from the photoelectrodes under harsh reaction conditions has been a significant issue48. The excellent stability of both Co/GaN/Si and Ni/GaN/Si highlights the potential advantage for designing a practical device.

Photocurrent density of a Co/GaN/Si and b Ni/GaN/Si in 0.1 M K2SO4 with 0.5 M KNO3 under 1 sun light illumination. Measurements were performed 5 times at 0 VRHE for 1 h each, and then 10 times at −0.6 VRHE for 30 min each. Insets show SEM images of photoelectrodes after 10 h reaction. FENH3 and YNH3 for c Co/GaN/Si and d Ni/GaN/Si.
Theoretical calculation
DFT investigations of the catalytic mechanism were performed to elucidate the origin of the different catalytic activities of Co and Cu catalysts for PEC \({{{\rm{NO}}}}_{3}^{{-}}\)RR. While the Cu catalyst maintains its metal character (see the XPS data in Fig. S7c that shows predominantly Cu0), the Co derived cluster is oxidized to Co3O4 as seen in the XPS spectrum (Fig. 1f) which shows Co2+ and Co3+ peaks. The same holds true for Ni, where the XPS spectrum shows peaks for both Ni-O and Ni-OH (Fig. 1g). In addition to the mechanism on metallic Cu (Fig. S39 and Table S2) and Co (Fig. S40 and Table S3), we therefore performed a mechanistic study of \({{{\rm{NO}}}}_{3}^{{-}}\) reduction on Co3O4 (Figs. S41–S44 and Table S4) as well as NiOOH with Ni3+ and Ni(OH)2 with Ni2+ (Figs. S45–S47 and Table S5).
The intermediates involved in the PEC process for Cu are shown in Fig. 5a, with the energetics of the preferred reaction path shown in Fig. 5b. The initial reduction of \({{{\rm{NO}}}}_{3}^{{-}}\) proceeds through oxidation of the surface and subsequent reductive removal of oxygen to regenerate the active material. The formal reactions in the PEC reduction of \({{{\rm{NO}}}}_{3}^{{-}}\) to NH3 on a metal surface such as Cu and Co are given in Eqs. (1) to (11).
While the reduction of \({{{\rm{NO}}}}_{3}^{{-}}\) to \({{{\rm{NO}}}}_{2}^{{-}}\) is thermodynamically favorable on both metallic Cu and Co surfaces (Fig. 5b and S40, respectively), further reduction to the NO intermediate and subsequent NH3 production depends on the thermal accessibility of the *NO2− adsorption mode at this crucial intermediate. This thermal equilibrium between nonactive *ONO−* and reactive *NO2− adsorption modes, which does not involve an electron transfer and therefore cannot be supported by an applied potential, has different thermodynamic properties on Co and Cu catalysts. For the Cu catalyst, the NO2− intermediate prefers to bind to the surface in the *ONO−* adsorption mode. However, for the second N = O bond dissociation and subsequent deoxygenation, the *ONO−* mode must be switched to the *NO2− adsorption mode. The switching to the *NO2− mode on the Cu catalyst is highly endergonic, with a free energy change of 0.14 eV (=13.15 kJ/mol) (Fig. 5c). This means that the *NO2− mode is not accessible on the Cu catalyst at room temperature and can only be generated as promoted by the energy released from the dissociation reaction. Furthermore, switching from the *ONO−* mode to the *NO2− adsorption mode is in competition with desorption of \({{{\rm{NO}}}}_{2}^{{-}}\) from the surface. This process of expulsion and replacement of the \({{{\rm{NO}}}}_{2}^{{-}}\) by an additional \({{{\rm{NO}}}}_{3}^{{-}}\) is slightly exergonic on Cu catalyst by −0.04 eV (=−3.36 kJ/mol). This much lower energy for the replacement of \({{{\rm{NO}}}}_{2}^{{-}}\) with \({{{\rm{NO}}}}_{3}^{{-}}\) in comparison to switching to the reactive *NO2− intermediate mode explains why the main product of \({{{\rm{NO}}}}_{3}^{{-}}\)RR on Cu is \({{{\rm{NO}}}}_{2}^{{-}}\). Once the \({{{\rm{NO}}}}_{2}^{{-}}\) is formed, it is much more likely to be desorbed rather than further reduction to NO through dissociation of the N = O bond.

a Full mechanism of \({{{\rm{NO}}}}_{3}^{{-}}\) reduction on the catalyst surfaces including reduction to NH3 and incomplete reduction to \({{{\rm{NO}}}}_{2}^{{-}}\) and subsequent desorption. The key intermediate and its two binding modes determining the outcome are encircled. b Reduction from \({{{\rm{NO}}}}_{3}^{{-}}\) to \({{{\rm{NO}}}}_{2}^{{-}}\) and subsequent desorption on Cu catalyst. c Thermodynamics of desorption and further reduction through change of binding mode on Cu catalyst.
For the oxidized species of Co3O4 and Ni(OH)2, we found different reaction mechanism: reductive formation of defects which leads to the subsequent interaction with oxygen containing species, resulting in dissociation of N-O bond. This is different from the metal surfaces where dissociation of the N-O bond led to oxidation of the surface that needed to be reactivated by reduction. Therefore, the order of reaction, with first reducing the surface and then oxidizing it by reaction with the oxygen containing species is reversed on the oxidized species in comparison to the metallic surfaces. On the Co3O4, defect formation is favorable with −0.06 eV for first hydrogenation and −0.55 eV for water release under additional reduction (Fig. S42). Nitrate, nitrite, and NO can directly react with formed oxygen defects (Fig. S43). Notably, the *ONO−* adsorption mode can dissociate since the defect can directly interact with the adsorbed oxygen. The reaction scheme and energetics of nitrate reduction on Co3O4 can be seen in Fig. 6, and the formal reactions in the PEC reduction of \({{{\rm{NO}}}}_{3}^{{-}}\) to NH3 on Co3O4 are given in Fig. S44. Upon adsorption of nitrate on the surface, a defect is reductively formed, that reacts with the adsorbed \({{{\rm{NO}}}}_{3}^{{-}}\) to extract one oxygen, dissociating the first N-O bond and refilling the defect. This leads to *NO2 on the surface, that gets reduced to \({{{\rm{NO}}}}_{2}^{{-}}\). Further defect formation leads to dissociation of the second N-O bond, resulting in *NO. Further reduction of NO to NH3 is thermodynamically downhill. Therefore, full reduction to NH3 is the energetically favored pathway on Co3O4 catalyst, making NH3 the main product.

a Full mechanism of \({{{\rm{NO}}}}_{3}^{{-}}\) reduction on Co3O4. Defect formation leads to N-O bond dissociation. b Free energy of the reduction from \({{{\rm{NO}}}}_{3}^{{-}}\) to NH3 on Co3O4.
As shown in Fig. S45, NiOOH readily reduces towards Ni(OH)2 at low applied potentials, meaning the active species is most likely Ni(OH)2 (Fig. S46). As Ni(OH)2 has no active Ni surface sites available, oxygen defect formation is necessary for its reactivity. Ni(OH)2 is a base that can be attacked by acids. The reaction mechanism determined by DFT is shown in Fig. 7a and the corresponding free energy profiles of the reaction steps are shown in Fig. 7b. HNO3 can react with Ni(OH)2, releasing water and leaving a positively charged surface with an oxygen defect. This defect directly interacts with the remaining nitrate to form the Ni(OH)O*NO2 intermediate with one of the oxygen atoms from nitrate being incorporated into the surface. Further reduction breaks the N-O bond, restoring the Ni(OH)2 and releasing NO2. Two NO2 molecules can readily react with water and form HNO3 and HNO2. These can then interact with the surface again, releasing water and forming Ni(OH)O*NO species. Upon reduction, NO is released, followed by protonation and water release. The NO then reacts, forming Ni(OH)O*N intermediate that is subsequently reduced to Ni(OH)O*NH and Ni(OH)O*NH2. Continued reduction leads to release of NH3, leaving behind a hydrogen defect that is filled in the next reduction step, completing the cycle. All steps are thermodynamically downhill with the exception of the release of NO that is 0.02 eV uphill- an energy easily provided by the photoexcited electrons. Therefore, the main product of nitrate reduction on Ni(OH)2 is NH3. The full reaction paths and energetics of reduction to NH3 as well as reduction to \({{{\rm{NO}}}}_{2}^{{-}}\) and subsequent desorption for all catalytic systems are given in Figs. S39–S47 and summarized in Tables S2–S6.

a Mechanism of \({{{\rm{NO}}}}_{3}^{{-}}\) reduction to NH3 on Ni(OH)2. Defect formation allows for attachment of nitrate, further reduction regenerates the Ni(OH)2 surface and dissociates the N-O bond. The mechanism proceeds similarly with HNO2 and NO, with full reduction towards NH3. b Free energy of the \({{{\rm{NO}}}}_{3}^{{-}}\) reduction on Ni(OH)2.
Discussion
In summary, we have investigated various metal catalysts for the PEC \({{{\rm{NO}}}}_{3}^{{-}}\)RR to NH3. Remarkably, we found that first-row earth-abundant transition metals Co and Ni catalysts outperformed noble metals (Pt, Ir, Rh, Pd, Ru, Au, and Ag) and the widely used Cu catalyst. In-situ IR measurement showed that a large number of \({{{\rm{NO}}}}_{2}^{{-}}\) intermediates were desorbed from Cu catalysts but further reduced to NH3 on Co catalysts. Theoretical calculations confirmed that the deoxygenation process of the *NO2− intermediate is feasible on Co3O4 and Ni(OH)2 catalysts via reaction with oxygen defects. In contrast, \({{{\rm{NO}}}}_{2}^{{-}}\) molecules desorbed from the Cu surface due to the *ONO−* binding mode with a lower barrier for desorption than for switching to the *NO2− mode that is needed for further deoxygenation processes. This leads to incomplete reduction of nitrate to nitrite on Cu, while the Co and Ni containing catalysts are capable of full reduction of \({{{\rm{NO}}}}_{3}^{{-}}\) to NH3. Owing to the strong catalytic activities of Co and Ni and the synergistic effects of metal-GaN interactions, high performance was achieved, including a low onset potential Vonset = 0.3 VRHE, high faradaic efficiency of NH3 (~99% at 0.2 VRHE), and high production rate of NH3 (201.6 μmol/h/cm2 at −0.4 VRHE). Moreover, the photoelectrodes were stable and performed for 10 h of reaction without any noticeable degradation. The photoelectrodes, which consist of the two most popular semiconductors (Si and GaN) and inexpensive transition metals (Co and Ni), have the potential to be used in industry-scale conversion of \({{{\rm{NO}}}}_{3}^{{-}}\) pollutant to value-added NH3 under solar light.
Methods
Preparation of GaN nanowires on n+-p Si wafer
Starting p-type Si (100) wafer was purchased from Purewafer and has a resistivity of 1–10 Ω·cm. The n+-p Si was demonstrated by spin-coating of phosphorus dopant on the front side of the polished p-type Si wafer and then thermal annealing at 950 °C under nitrogen atmosphere for 4 h. N-type GaN NWs were grown on n+-p Si wafer by plasma-assisted molecular-beam epitaxy under nitrogen-rich condition to promote the formation of an N-terminated surface. The growth temperature was 790 °C and the duration was ~2 h. The forward plasma power was 350 W with Ga flux beam equivalent pressure of 5 × 10−8 Torr.
Loading of metal catalysts on GaN/Si
Metal catalysts were loaded on GaN/Si by a photodeposition method. GaN/Si wafer on a Teflon holder was placed in the bottom of a quartz reactor containing 66 mL of 20 vol% methanol aqueous solution. Then, 10 μl of 0.2 M H2PtCl6 (Sigma-Aldrich, 99.9%), 0.2 M IrCl3 (Sigma-Aldrich, 99.8%), 0.2 M Na3RhCl6 (Sigma-Aldrich), 0.1 M K2PdCl4 (Sigma-Aldrich, 99.99%), 0.2 M CuCl2 (Sigma-Aldrich, 99.999%), 0.2 M AgNO3 (Sigma-Aldrich, ≥99.0%), 0.4 M RuCl3 (Sigma-Aldrich, 99.99%), 0.2 M HAuCl4 (Sigma-Aldrich, 99.99%), 0.2 M Ni(NO3)2 (Sigma-Aldrich, 99.999%), or 0.2 M CoCl2 (Sigma-Aldrich, 97%) solutions were added into the chamber to deposit Pt, Ir, Rh, Pd, Cu, Ag, Ru, Au, Ni, or Co catalysts, respectively. The reactor was evacuated for 5 min using a rotary pump. A 300 W Xe lamp (Cermax, PE300BUV) was used as a light source which irradiated the chamber for 30 min. When light illuminated the GaN/Si wafer in the precursor solution, electrons are photoexcited to the conduction band, creating holes in the valence band of the GaN NWs. The photoexcited electrons then migrated to the GaN surface, participating in the reduction of metal ions. Simultaneously, photogenerated holes in the valence band migrated to the surface, oxidizing the methanol sacrificial agent in the solution. Since the bandgap of GaN (~3.4 eV) was suitable to absorb ultraviolet light and drive the redox reactions, there was no need for an external bias in the photodeposition process. After the photodeposition, the samples were rinsed with deionized water and dried by air blowing.
Characterization
The scanning electron microscopy (SEM) was performed using a field-emission scanning electron microscope (MIRA3 TESCAN) with 10 kV acceleration voltage. The high-angle annular dark-field scanning-transmission electron microscopy (HAADF-STEM) and energy dispersive X-ray spectroscopy (EDS) images were collected at 200 kV using JEOL 3100R05 Double Cs Corrected TEM/STEM with a 200 kV accelerating voltage. The X-ray photoelectron spectroscopy (XPS) was measured using a Kratos Axis Ultra XPS with a monochromatic Al Kα source. The ultraviolet-visible (UV-vis) spectroscopy (Varian 50-Bio) was used to quantify the products in the electrolytes. The In-situ Fourier transform infrared spectroscopy was performed on an INVENIO-R Fourier transform infrared spectrometer (FTIR) equipped with a mercury cadmium telluride (MCT) detector. Isotope lableing test was performed by 1H nuclear magnetic resonance (NMR) measurment using Varian Vnmrs 500. 525 μL of solution was mixed with 50 μl of 0.5 M H2SO4 (Sigma-Aldrich, 95.0–98.0%), 50 μl of DMSO-d6 (Sigma-Aldrich, 99.9%), 25 μl of 12.5 mM maleic acid (Sigma-Aldrich, ≥99%), and 25 μl of 27 mM Gd3+ solution (Sigma-Aldrich, 99.9%) for NMR analysis49.
Photoelectrochemical \({{{\rm{NO}}}}_{3}^{{-}}\) reduction
All photoelectrochemical measurements were performed in an H-type cell separated by a Nafion membrane with a three-electrode system using a potentiostat (Bio Logic SAS SP-200). Ag/AgCl filled with 3 M KCl was used for the reference electrode and Pt wire was used for the counter electrode. GaN/Si as well as other metal-loaded photoelectrodes were used as the working electrodes. The measured potentials (VAg/AgCl) (V) were converted to the reversible hydrogen electrode (VRHE) (V) by using the Nernst function: VRHE = VAg/AgCl + 0.197 + 0.0591 × pH. The electrolytes were aqueous solutions of 0.1 M K2SO4 (Sigma-Aldrich, ≥99%) or 0.1 K2SO4 with 0.5 M KNO3 (Sigma-Aldrich, ≥99%) prepared by dissolving the solid salts in deionized water. pH values of electrolytes were measured using a pH meter (Mettler Toledo). 8 ml of electrolyte was filled in the cathodic compartment of H-type cell and purged with Ar for 30 min before the measurement. The light source for the photoelectrochemical reaction was LCS-100 (ORIEL) and the intensity with AM 1.5G filter was calibrated by adjusting the distance from the sample to the light source. Linear sweep voltamnetry curves were obtained at a rate of 10 mV/s. Chronoamerometic curves were recorded every 1 s. All measurements were conducted at ambient pressure and room temperature. After the reaction, gas products were analyzed using gas chromatography (Shimadzu GC-8A) equipped with a thermal conductivity detector. Then, the electrolytes were collected to quantify the NH3, \({{{\rm{NO}}}}_{2}^{{-}}\), and N2H4 products using UV-Vis spectrophotometry.
Determination of NH3
The concentration of NH3 product was spectrocolorimetrically determined by the indophenol blue method25. 1 ml of electrolyte was taken out from the cathodic compartment in H-typce cell (and diluted if needed). Then, 1 ml of a 1 M NaOH (Sigma-Aldrich, ≥97%) solution containing 5 wt% salicylic acid (Sigma-Aldrich, ≥99%) and 5 wt% sodium citrate (Sigma-Aldrich, ≥99%) was added to the 1 ml of electrolyte, followed by the addition of 0.5 ml of 0.05 M NaClO (Sigma-Aldrich, available chlorine 4.00–4.99%) and 0.1 ml of 1 wt% C5FeN6Na2O (Sigma-Aldrich, ≥99%) solution. The solution was stored in dark for ~2 h at room temperature before the UV-vis measurement. The concentration of NH3 was determined by absorbance at a wavelength of 655 nm. NH4Cl (Sigma-Aldrich, ≥99.5%) standard solutions with known concentration was used for the calibration.
Determination of \({{{\rm{NO}}}}_{2}^{{-}}\)
0.2 g of N-(1-naphthyl) ethylenediamine dihydrochloride (Sigma-Aldrich, ≥98%), 4 g of p-aminobenzenesulfonamide (Sigma-Aldrich, ≥98%), and 10 ml of phosphoric acid (Sigma-Aldrich, ≥85 wt% in H2O) were dissolved into 50 ml of deionized water as the color reagent25. Then, 2.5 ml of the diluted electrolyte were mixed with 0.05 ml of color reagent and stored ~30 min prior to the measurement. The absorbance at a wavelength of 540 nm was collected by UV-vis spectrometer to quantify the concentration of \({{{\rm{NO}}}}_{2}^{{-}}\). KNO2 (Sigma-Aldrich, ≥96%) standard solutions with known concentration was used for the calibration.
Determination of N2H4
1.5 g of C9H11NO (Sigma-Aldrich, 97%) and 7.5 ml of HCl (Sigma-Aldrich, 37%) were added into 75 ml of ethanol (Sigma-Aldrich, ≥99.5%) as the color agent50. Then, 1 ml of the electrolyte was mixed with 1 mL of color reagent and stored for ~1 h before the measurment. The absorbance at a wavelength of 455 nm was collected by UV-vis spectrometer to determinte the concentration of N2H4. Hydrazine hydrate (Sigma-Aldrich, 80% in water) standard solutions with known concentration was used for the calibration.
Data availability
Data supporting the findings of this study are available within the article and supplementary information. A source data file for data presented in the main text is provided with the manuscript. All other data supporting the findings are available from the corresponding authors upon request. Source data are provided with this paper.
References
van Langevelde, P. H., Katsounaros, I. & Koper, M. T. Electrocatalytic nitrate reduction for sustainable ammonia production. Joule 5, 290–294 (2021).
Rogan, W. J. & Brady, M. T. Committee on Environmental Health, and the Committee on Infectious Diseases. Drinking water from private wells and risks to children. Pediatrics 123, e1123–e1137 (2009).
Martínez, J., Ortiz, A. & Ortiz, I. State-of-the-art and perspectives of the catalytic and electrocatalytic reduction of aqueous nitrates. Appl Catal. B 207, 42–59 (2017).
MacFarlane, D. R. et al. A roadmap to the ammonia economy. Joule 4, 1186–1205 (2020).
Soloveichik, G. Electrochemical synthesis of ammonia as a potential alternative to the Haber–Bosch process. Nat. Catal. 2, 377–380 (2019).
Wang, P. et al. Breaking scaling relations to achieve low-temperature ammonia synthesis through LiH-mediated nitrogen transfer and hydrogenation. Nat. Chem. 9, 64–70 (2017).
Chen, G. F. et al. Advances in electrocatalytic N2 reduction—strategies to tackle the selectivity challenge. Small Methods 3, 1800337 (2019).
Zhang, G. et al. Tandem electrocatalytic nitrate reduction to ammonia on MBenes. Angew. Chem. Int Ed. 62, e202300054 (2023).
Li, J. et al. Efficient ammonia electrosynthesis from nitrate on strained ruthenium nanoclusters. J. Am. Chem. Soc. 142, 7036–7046 (2020).
Song, Z. et al. Efficient electroreduction of nitrate into ammonia at ultra‐low concentrations via enrichment effect. Adv. Mater. 34, 2204306 (2022).
Wang, Y. et al. Enhanced nitrate-to-ammonia activity on copper–nickel alloys via tuning of intermediate adsorption. J. Am. Chem. Soc. 142, 5702–5708 (2020).
Liu, H. et al. Efficient electrochemical nitrate reduction to ammonia with copper‐supported rhodium cluster and single‐atom catalysts. Angew. Chem. 7, e202202556 (2022).
Ge, Z. X. et al. Interfacial engineering enhances the electroactivity of frame‐like concave RhCu bimetallic nanocubes for nitrate reduction. Adv. Energy Mater. 12, 2103916 (2022).
Chen, G.-F. et al. Electrochemical reduction of nitrate to ammonia via direct eight-electron transfer using a copper–molecular solid catalyst. Nat. Energy 5, 605–613 (2020).
Wang, Y., Zhou, W., Jia, R., Yu, Y. & Zhang, B. Unveiling the activity origin of a copper‐based electrocatalyst for selective nitrate reduction to ammonia. Angew. Chem. Int Ed. 59, 5350–5354 (2020).
Li, J. et al. 3.4% solar‐to‐ammonia efficiency from nitrate using fe single atomic catalyst supported on MoS2 nanosheets. Adv. Funct. Mater. 32, 2108316 (2022).
Kani, N. C. et al. Solar-driven electrochemical synthesis of ammonia using nitrate with 11% solar-to-fuel efficiency at ambient conditions. Energy Environ. Sci. 14, 6349–6359 (2021).
He, W. et al. Splicing the active phases of copper/cobalt-based catalysts achieves high-rate tandem electroreduction of nitrate to ammonia. Nat. Commun. 13, 1–13 (2022).
Zhang, N. et al. Governing interlayer strain in bismuth nanocrystals for efficient ammonia electrosynthesis from nitrate reduction. ACS Nano 16, 4795–4804 (2022).
Ye, S. et al. Elucidating the activity, mechanism and application of selective electrosynthesis of ammonia from nitrate on cobalt phosphide. Energy Environ. Sci. 15, 760–770 (2022).
Jiang, M. et al. Interfacial reduction nucleation of noble metal nanodots on redox-active metal–organic frameworks for high-efficiency electrocatalytic conversion of nitrate to ammonia. Nano Lett. 22, 2529–2537 (2022).
Liu, Q. et al. Ambient ammonia synthesis via electrochemical reduction of nitrate enabled by NiCo2O4 nanowire array. Small 18, 2106961 (2022).
Cheng, X. F. et al. Coordination symmetry breaking of single atom catalysts for robust and efficient nitrate electroreduction to ammonia. Adv. Mater. 34, 2205767 (2022).
Sun, W. J. et al. Built‐in electric field triggered interfacial accumulation effect for efficient nitrate removal at ultra‐low concentration and electroreduction to ammonia. Angew. Chem. Int Ed. 60, 22933–22939 (2021).
Wu, Z.-Y. et al. Electrochemical ammonia synthesis via nitrate reduction on Fe single atom catalyst. Nat. Commun. 12, 1–10 (2021).
Li, P., Jin, Z., Fang, Z. & Yu, G. A single-site iron catalyst with preoccupied active centers that achieves selective ammonia electrosynthesis from nitrate. Energy Environ. Sci. 14, 3522–3531 (2021).
Li, X., Shen, P., Li, X., Ma, D. & Chu, K. Sub-nm RuO x clusters on Pd metallene for synergistically enhanced nitrate electroreduction to ammonia. ACS Nano 17, 1081–1090 (2023).
Kim, H. E. et al. Photoelectrochemical nitrate reduction to ammonia on ordered silicon nanowire array photocathodes. Angew. Chem. Int Ed. 134, e202204117 (2022).
Ren, S. et al. Enhanced charge‐carrier dynamics and efficient photoelectrochemical nitrate‐to‐ammonia conversion on antimony sulfide‐based photocathodes. Angew. Chem. Int. Ed. 63, e202409693 (2024).
Ren, S., Gao, R. T., Nguyen, N. T. & Wang, L. Enhanced charge carrier dynamics on Sb2Se3 photocathodes for efficient photoelectrochemical nitrate reduction to ammonia. Angew. Chem. 136, e202317414 (2024).
van Deelen, T. W., Hernández Mejía, C. & de Jong, K. P. Control of metal-support interactions in heterogeneous catalysts to enhance activity and selectivity. Nat. Catal. 2, 955–970 (2019).
Dong, W. J. et al. Tailoring electronic structure of bifunctional Cu/Ag layered electrocatalysts for selective CO2 reduction to CO and CH4. Nano Energy 78, 105168 (2020).
Shi, L., Yin, Y., Wang, S. & Sun, H. Rational catalyst design for N2 reduction under ambient conditions: strategies toward enhanced conversion efficiency. ACS Catal. 10, 6870–6899 (2020).
Wang, S. et al. High-throughput identification of highly active and selective single-atom catalysts for electrochemical ammonia synthesis through nitrate reduction. Nano Energy 100, 107517 (2022).
Parastaev, A. et al. Boosting CO2 hydrogenation via size-dependent metal–support interactions in cobalt/ceria-based catalysts. Nat. Catal. 3, 526–533 (2020).
Zhang, J., Zhang, Q. & Feng, X. Support and interface effects in water‐splitting electrocatalysts. Adv. Mater. 31, 1808167 (2019).
Dong, W. J. et al. Pt nanoclusters on GaN nanowires for solar-asssisted seawater hydrogen evolution. Nat. Commun. 14, 179 (2023).
Xiao, Y. et al. Oxynitrides enabled photoelectrochemical water splitting with over 3000 hrs stable operation in practical two-electrode configuration. Nat. Commun. 14, 2047 (2023).
Xiao, Y. et al. Crystallographic effects of GaN nanostructures in photoelectrochemical reaction. Nano Lett. 22, 2236–2243 (2022).
Wolter, S. et al. X-ray photoelectron spectroscopy and x-ray diffraction study of the thermal oxide on gallium nitride. Appl Phys. Lett. 70, 2156–2158 (1997).
Duan, T., Pan, J. & Ang, D. S. Interfacial chemistry and valence band offset between GaN and Al2O3 studied by X-ray photoelectron spectroscopy. Appl Phys. Lett. 102, 201604 (2013).
Chastain, J. & King, R. C. Jr Handbook of X-ray photoelectron spectroscopy. Perkin Elmer Corp. 40, 221 (1992).
Weidler, N. et al. X-ray photoelectron spectroscopic investigation of plasma-enhanced chemical vapor deposited NiO x, NiO x (OH) y, and CoNiO x (OH) y: influence of the chemical composition on the catalytic activity for the oxygen evolution reaction. J. Phys. Chem. C. 121, 6455–6463 (2017).
Vanka, S. et al. High efficiency Si photocathode protected by multifunctional GaN nanostructures. Nano Lett. 18, 6530–6537 (2018).
Dong, W. J. et al. Silver halide catalysts on GaN nanowires/Si heterojunction photocathodes for CO2 reduction to syngas at high current density. ACS Catal. 12, 2671–2680 (2022).
Deng, W., Zhang, P., Seger, B. & Gong, J. Unraveling the rate-limiting step of two-electron transfer electrochemical reduction of carbon dioxide. Nat. Commun. 13, 803 (2022).
Pérez-Gallent, E., Figueiredo, M. C., Katsounaros, I. & Koper, M. T. Electrocatalytic reduction of Nitrate on Copper single crystals in acidic and alkaline solutions. Electrochim. Acta 227, 77–84 (2017).
Tan, J. et al. Hydrogel protection strategy to stabilize water-splitting photoelectrodes. Nat. Energy 7, 537–547 (2022).
Kolen, M., Smith, W. A. & Mulder, F. M. Accelerating 1H NMR detection of aqueous ammonia. ACS Omega 6, 5698–5704 (2021).
Watt, G. W. & Chrisp, J. D. Spectrophotometric method for determination of hydrazine. Anal. Chem. 24, 2006–2008 (1952).
Acknowledgements
This work was supported by United States Army Research Office Award W911NF2110337, awarded to V.S.B. and Z.M. The authors wish to thank Dr. Peng Zhou for discussions on the catalytic reactions.
Author information
Authors and Affiliations
Contributions
W.J.D. conducted the photoelectrochemical tests. J.P.M. and Z.L. performed the DFT calculations. V.S.B. and Z.M. supervised the project. K.L. conducted photoelectrochemical analysis. Z.Y. performed XPS and TEM measurement. I.A.N. performed MBE growth. Y.X. discussed the catalytic reactions. K.R.Y. performed initial calculations and discussed possible catalytic mechanisms. W.J.D., J.P.M., V.S.B., and Z.M. wrote the manuscript. All authors discussed the results and commented on the manuscript.
Corresponding authors
Ethics declarations
Competing interests
Some IP related to this work was licensed to NS Nanotech, Inc. and NX Fuels, Inc., which were co-founded by Z.M. The University of Michigan and MI have a financial interest in the company. The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Wooseok Yang, Lei Wang and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Dong, W.J., Menzel, J.P., Li, K. et al. Nitrate reduction to ammonia catalyzed by GaN/Si photoelectrodes with metal clusters. Nat Commun 16, 3383 (2025). https://doi.org/10.1038/s41467-025-58372-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-025-58372-7
