
Abstract
The solute carrier protein SLC19A1 is crucial for transporting folate nutrients, antifolate chemotherapeutics, and more recently cyclic dinucleotides (CDNs) immune transmitters, influencing various physiological and pathological processes. While the inward-open state of human SLC19A1 (hSLC19A1) has been previously described, key aspects regarding its conformational dynamics, substrate selectivity, and precise mechanisms underlying CDNs transport remain elusive. Using an antibody-facilitated conformation screening strategy, we present cryo-electron microscopy structures of hSLC19A1 in its outward-open state with and without bound substrates, revealing detailed mechanisms of substrate recognition and conformational changes during transport. We identify both general and specific features for folate/antifolate recognition, including an SLC19A1-specific pocket for accommodating γ-carboxylate-modified antifolates. Intriguingly, CDNs bind as monomers within the canonical pocket of outward-open hSLC19A1, contrasting with dimeric binding in inward-open structures. Together with functional assays, these findings provide a framework for developing antifolate drugs and CDN-targeted therapies, advancing our understanding of SLC19A1’s physiological and therapeutic functions.
Similar content being viewed by others


Introduction
Folate, an essential nutrient for mammalian growth and development, acts as a crucial one-carbon unit donor in fundamental metabolic processes and plays pivotal roles in synthesizing key biomolecules such as thymidine, purine nucleotides, serine, and methionine1,2. The solute carrier protein SLC19A1, also known as the reduced folate carrier, has been extensively studied for its role in folate transport over several decades3,4,5. Additionally, SLC19A1 facilitates the transport of antifolate drugs, such as methotrexate and pemetrexed, which are widely used in the treatment of cancer, rheumatoid arthritis, and psoriasis6. Interestingly, recent studies have unveiled a novel function of SLC19A1 in the transport of cyclic dinucleotides (CDNs)7,8. CDNs serve as pivotal immune signaling molecules capable of eliciting broad downstream immune responses by binding to the stimulator of interferon genes (STING)9,10,11,12,13. SLC19A1 transports not only mammalian endogenous CDN: 2’3’-cGAMP synthesized by cyclic GMP–AMP synthase (cGAS)14,15,16, but also bacterial CDN and synthetic CDN-type drugs11,17,18,19. The dual role of SLC19A1 in folate/antifolate and CDN transport underscores its significance in fundamental metabolism, infection, immunopathology, cancer chemotherapy, and immunotherapy4,5,20,21,22.
In line with the major facilitator superfamily (MFS) fold, SLC19A1 comprises 12 transmembrane (TM) helixes, with TM1-6 forming the N-terminal domain (NTD) and TM7-12 constituting the C-terminal domain (CTD)23. The transmembrane transport of substrates by MFS protein normally occurs through a rocker-switch rotation model, wherein the two domains undergo conformational changes from an outward-open to an inward-open state24. Previous cryo-electron microscopy (cryo-EM) studies have provided insights into the substrate binding details of human SLC19A1 (hSLC19A1) in its inward-open state, with folate and antifolate binding as monomers within a canonical pocket25,26,27 and CDNs forming a compact dimeric unit within a non-canonical cavity25. However, all the reported structures exhibit the same inward-open conformation, leaving unresolved questions about how hSLC19A1 changes conformation and recognizes substrates during the transport cycle. Moreover, it is still unclear whether the captured CDN dimer in the inward-open state represents the actual form in which CDNs are transported by hSLC19A1 or just a potential substrate binding mode, since CDNs are not located in the typical pocket of SLC proteins25. Therefore, whether CDNs are transported in the dimeric form remains an open question25. Further structural studies of hSLC19A1 in complex with different substrates at the outward-open conformation are warranted to fully elucidate the transport cycle and diverse substrate recognition mechanisms of hSLC19A1, thereby advancing understanding of its physiological roles and potential therapeutic implications.
Results
Protein engineering for structure determination
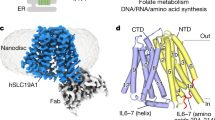
Previously reported hSLC19A1 structures all exhibit the inward-open conformation, suggesting that purified hSLC19A1 may have a preference for this state in vitro. To capture hSLC19A1 in other conformations, we sought to systematically introduce mutations at the NTD-CTD interface aimed at destabilizing the inward-open state without affecting the substrate recognition tunnel. By taking advantage of our previously isolated inward-open specific antibody to hSLC19A1 (mAb70#)25, we were able to efficiently distinguish the conformation of each mutant in cells using flow cytometry, rather than going through tedious purification and structure determination procedures for each sample (Fig. 1a). In addition, we also generated two conformation-unspecific antibodies recognizing linear epitopes as control antibodies, mAb5# and mAb6#, targeting the disordered N-terminal tail and intracellular loop between TM6 and TM7 (IL6-7), respectively (Supplementary Fig. 1a). Subsequent flow cytometry results show that, among multiple mutants tested, the G307F mutation completely disrupts the binding between hSLC19A1 and mAb70# while leaving interactions with mAb5# and mAb6# unaffected (Supplementary Fig. 1b, c). Size-exclusion chromatography experiments with purified hSLC19A1WT and hSLC19A1G307F further confirm these results (Supplementary Fig. 1d, e). Consistently, the 3H-labelled antifolate uptake of hSLC19A1G307F is abolished in the cellular uptake assay (Fig. 1b and Supplementary Fig. 2), highlighting that the G307F mutation may have trapped hSLC19A1 in a conformation other than the inward-open state.

a Schematic diagram of the antibody-facilitated strategy for identifying conformationally altered hSLC19A1 mutants. b [3H]-MTX uptake assays using SLC19A1−/− HeLa cells stably expressing either WT hSLC19A1 or G307F mutant. Data were normalized to WT and are mean ± s.e.m. of n = 4 biologically independent experiments. Statistical analysis was performed using two-tailed unpaired Student’s t-tests. EV, empty vector; WT, wild type. c Cryo-EM density map of outward-open hSLC19A1. d The overall structure of outward-open hSLC19A1. The N- and C-terminal domains (NTD and CTD) are coloured in blue and yellow, respectively. e Cut-open side view of the electrostatic potential surface of the hSLC19A1 outward-open structure. f Domain-wise alignments between outward- (blue and yellow) and inward-open (grey) hSLC19A1. g Rotation of representative TM segments between outward- (blue and yellow) and inward-open (grey) hSLC19A1 relative to the membrane norm. h Cytosolic bottom view of the intracellular gate in the outward-open conformation. Key residues are shown as sticks. i Conformational changes of Phe141 (left), Gln150 (middle) and Arg157 (right) on the basis of NTD-centered structural superimposition of outward- (blue and yellow) and inward-open hSLC19A1 (grey). Key residues are shown as sticks and hydrogen bonds are represented as dashed lines. j [3H]-MTX uptake assays using SLC19A1−/− HeLa cells stably expressing either WT hSLC19A1 or intracellular-gate residue mutants. Data were normalized to WT and are mean ± s.e.m. of n = 4 biologically independent experiments. Statistical analysis was performed using two-tailed unpaired Student’s t-tests. ***P ≤ 0.001, ****P ≤ 0.0001. Source data are provided as a Source Data file.
hSLC19A1 is a relatively small membrane protein and lacks rigid soluble domains, making it challenging for high-resolution structure determination. To facilitate cryo-EM reconstructions, we designed a hSLC19A1G307F construct carrying an N-terminal BRIL domain and a C-terminal specific nanobody (termed hSLC19A1EM-G307F; Supplementary Fig. 3a), both of which can specifically bind to a widely used anti-BRIL Fab fragment. In this way, the subsequent addition of anti-BRIL Fab may bridge the N- and C-terminus of hSLC19A1 together, thereby enhancing sample stability and facilitating cryo-EM particle alignment. It should be noted that the N- and C-terminal fusions to the wild-type protein still largely preserve the substrate cellular uptake activity (Supplementary Figs. 2 and 3b), suggesting that the fusion strategy per se does not significantly affect hSLC19A1 function.
Structure of hSLC19A1 in outward-open state
To verify the conformational details of the engineered protein, we reconstituted the complex of hSLC19A1EM-G307F and anti-BRIL Fab (Supplementary Fig. 3c, d) and successfully determined its cryo-EM structure at a resolution of 2.94 Å (Fig. 1c-e, Supplementary Fig. 3e–i and Supplementary Table 1). The density is of sufficient quality to trace the transmembrane regions of hSLC19A1 from residues 24 to 457 (Supplementary Fig. 4a). Some parts of the intracellular loop between TM6 and TM7 (IL6-7, residues 213-248) and the extracellular loop between TM7 and TM8 (EL7-8, residues 295-302) were not observed in the cryo-EM map, possibly due to the dynamic nature of these regions (Fig. 1d). Interestingly, in contrast to previously reported structures, the engineered hSLC19A1 adopts an outward-open conformation (Fig. 1d). In this conformation, TM1 and TM2 tilt away from TM7 and TM8, exposing the entrance to a large solvent-accessible and highly conserved cavity that extends more than halfway across the cell membrane (Fig. 1d and Supplementary Fig. 4b). Characterized by a narrow extracellular entrance, the upper portion of the cavity extends toward the NTD and presents an overall negative electrostatic surface (Fig. 1e). Conversely, the lower half of the cavity contracts and shows an overall positive charge, consistent with the anionic properties of CDN and folate (Fig. 1e). On the intracellular side, TM4 and TM5, along with TM10 and TM11, snugly interlock to seal the cavity from the cytoplasm (Supplementary Fig. 4c). Additionally, IL2-3, IL4-5, IL6-7 and IL10-11 closely associate to further stabilize the outward-open conformation (Supplementary Fig. 4c).
Domain-wise alignments between outward- and inward-open structures of hSLC19A1 show that the alternate access is achieved through a rocker-switch motion of the NTD and CTD domains. Individual NTD and CTD of outward-open hSLC19A1 align well with those in inward-open state, with root-mean-square deviation (RMSD) values of 0.69 Å over 168 Cα atoms and 0.71 Å over 155 Cα atoms, respectively (Fig. 1f). Both domains of hSLC19A1 rotate relative to an axis perpendicular to the membrane plane by approximately 18°, allowing the transmembrane regions to remain embedded within the membrane (Fig. 1g). The highly conserved and functionally important N-terminal region of IL6-728 (aa203-aa212), which is tightly embedded in a surface groove formed by the intracellular ends of TM2, TM3, TM4 and TM11, also undergoes relative motion to accommodate the movement of CTD in the outward-open state (Supplementary Fig. 4d). In the current outward-open conformation, three pairs of hydrogen bonds (Gln150-Ala324/Val327 and Arg157-Gln377) and a set of hydrophobic interactions (Phe141-Ala388-Phe400), presumably contribute to the closure of the intracellular gate (Fig. 1h, i). Interestingly, structural superimposition with the inward-open structure reveals dramatic conformational changes in Phe141, Gln150 and Arg157, which would disrupt the intracellular gate and trigger the closure of the extracellular gate as well as the release of substrates (Fig. 1i and Supplementary Fig. 4e). To validate these structural observations, we substituted these gate residues and assessed the cellular uptake of 3H-labelled antifolate in SLC19A1−/− HeLa cells stably expressing either wild-type or mutant hSLC19A1. The F141S and R157A substitutions nearly abolish antifolate uptake, Q150A, A388S and F400S reduce antifolate uptake by about 70–80%, and Q377A decreases the activity by around 25% (Fig. 1j, Supplementary Figs. 2 and 4f).
Our structure also provides a detailed explanation of why the G307F single point mutation drives conformational changes (Supplementary Fig. 1b, c). In the inward-open hSLC19A1 structure, Gly307 on TM8 forms a hydrogen bond with the carbonyl group of Gln172 on TM5 (Supplementary Fig. 4g). However, by wedging into the lateral interface between NTD and CTD, Phe307 disrupts the hydrogen bond and stacks with Gln172 to lock the outward-open conformation (Supplementary Fig. 4g). To check whether substitutions of Gly307 with residues of different side chain sizes can enable hSLC19A1 in alternative intermediate states of the transport cycle, we further generated G307W, G307S, and G307A mutations. Cryo-EM analysis shows that G307W and G307S both adopt the same outward-open structures as observed with G307F, while a smaller side chain mutation G307A maintains the inward-open conformation similar to the wild-type protein (Supplementary Fig. 4h). These results suggest that the outward- and inward-open conformations likely represent two relatively stable states of the SLC19A1 transport cycle.
Recognition of 5-MTHF and MTX in outward-open state
While previous studies have provided insights into the interaction pattern of inward-open hSLC19A1 with folate and antifolate, a comprehensive understanding of the transport cycle of these substrates necessitates further structural elucidation. Therefore, using the aforementioned engineering strategy, we determined the cryo-EM structures of outward-open hSLC19A1 in complex with the reduced folate 5-methyltetrahydrofolate (5-MTHF) and the antifolate methotrexate (MTX) at resolutions of 3.43 Å and 3.44 Å, respectively (Fig. 2a–f, Supplementary Fig. 5 and Supplementary Table 1).

a, d Cryo-EM density maps of outward-open hSLC19A1 bound to 5-MTHF (a) and MTX (d). b, e The overall structures of outward-open hSLC19A1 bound to 5-MTHF (b) and MTX (e). The NTD and CTD are coloured in blue and yellow, respectively. 5-MTHF and MTX are shown as a space-filling representation and are coloured in cyan and wheat, respectively. c, f A cut-open side view of the electrostatic potential surface of outward-open hSLC19A1 bound to 5-MTHF (c) and MTX (f). g Different conformations of the bound 5-MTHF in the outward- (cyan) and inward-open state (grey) of hSLC19A1 on the basis of NTD-centered structural superimposition. h Hydrogen bonding interactions between 5-MTHF (sticks; cyan) and outward-open hSLC19A1 (grey). Key residues are shown as sticks and hydrogen bonds are represented as dashed lines. i Hydrophobic interactions between 5-MTHF (sticks; cyan) and outward-open hSLC19A1 (grey). Key residues are shown as sticks and dots. j Different conformations of the MTX in the outward-open (wheat) and inward-open state (grey) on the basis of NTD-centered structural superimposition. k [3H]-MTX uptake assays using SLC19A1−/− HeLa cells stably expressing either WT hSLC19A1 or folate-binding pocket mutants. Data were normalized to WT and are mean ± s.e.m. of n = 4 biologically independent experiments. Statistical analysis was performed using two-tailed unpaired Student’s t-tests. ***P ≤ 0.001, ****P ≤ 0.0001. Source data are provided as a Source Data file.
5-MTHF is a predominant natural folate in diet and blood and plays a crucial role in normal metabolism and development. The well-resolved density map reveals that 5-MTHF is situated at the polar cavity in an approximately upright conformation, with its long axis aligned parallel to the transmembrane helices (Fig. 2a–c, g and Supplementary Fig. 6a, b). Anchored by hydrogen bonds with Thr49 of TM1 and Glu123 of TM4 (Fig. 2h), the pterin moiety of 5-MTHF inserts diagonally into the upper negatively charged pocket within the NTD (Fig. 2c). The pterin ring and methyl group establish extensive hydrophobic contacts with Glu45/Ile48 of TM1, Leu72 of TM2 and Tyr126 of TM4 (Fig. 2i). Meanwhile, the benzoyl moiety extends downward perpendicular to the plane of the pterin ring, forming a hydrogen bond with Arg133 of TM4, along with hydrophobic interactions with Tyr126/Met130 of TM4 and Tyr286 of TM7 (Fig. 2h, i). In addition, the glutamate moiety faces the intracellular side and reaches the positively charged region (Fig. 2c, h). The β-carboxylate group faces the CTD and interacts with Tyr281 of TM7, Arg373 and Gln377 of TM10, while the γ-carboxylate group turns back towards the NTD and interacts with Arg133 of TM4 (Fig. 2c, h).
It is noteworthy that SLC19A1 shows a pronounced preference for reduced folate over folic acid4. Compared to folic acid, 5-MTHF contains an additional methyl group at position N5 (Supplementary Fig. 6a). Our previous inward-open structure reveals that this extra methyl group is embedded in a hydrophobic pocket formed by Glu45, Ile48 and Tyr126, thereby enhancing the affinity of 5-MTHF for hSLC19A125. Although the overall binding site and interaction mode between 5-MTHF and the current outward-open hSLC19A1 are similar to those of the inward-open structure, the detailed conformations of the pterin ring, benzoyl and glutamate moieties of 5-MTHF exhibit notable differences (Fig. 2g). Specifically, the pterin ring of 5-MTHF rotates ~180° with the N5 methyl group serving as the pivot, maintaining the methyl group within the hydrophobic pocket (Fig. 2g, i). The benzoyl and glutamate moieties also undergo appropriate adjustments accordingly (Fig. 2g). These findings highlight the contribution of the N5 methyl group to the reduced folate selectivity preference of SLC19A1 and indicate that 5-MTHF likely possesses some degree of flexibility to accommodate the transport cycle.
MTX is a widely used antifolate drug for the treatment of cancer and autoimmune diseases. Consistent with the 5-MTHF-bound structure, MTX also adopts an upright conformation and binds within the central cavity, with the pteridine ring occupying the electronegative pocket at NTD and the glutamate moiety extending toward the electropositive region (Fig. 2d–f and Supplementary Fig. 6c). The interactions between MTX and the outward-open hSLC19A1 are similar to those of 5-MTHF (Supplementary Fig. 6d, e). Interestingly, in the previously reported inward-open hSLC19A1 structure bound to an N-hydroxysuccinimide (NHS)-conjugated MTX26, the glutamate moiety of MTX was covalently linked to Lys411 of TM11 via NHS-mediated crosslinking, resulting in a 180° rotation of the glutamate and benzoyl groups when aligning the NTD of SLC19A1 of the two structures (Fig. 2j). These results indicate that the glutamate moiety of antifolate exhibits high dynamics within the electropositive cavity to accommodate the binding and dissociation with hSLC19A1.
The residues lining the binding sites for 5-MTHF and MTX are highly conserved among vertebrates (Supplementary Fig. 7). To investigate the contribution of the observed SLC19A1-substrate interactions, we introduced mutations into the corresponding residues of hSLC19A1 (Supplementary Fig. 2) and examined the effects on cellular 3H-labelled antifolate uptake. The representative substitutions lead to a substantial reduction in antifolate uptake, with E123A, R133A and R373A mutations almost abolishing the activity (Fig. 2k). Furthermore, the initial-rate analysis confirms the critical roles of representative residues for folate/antifolate recognition (Supplementary Fig. 4f).
Selective recognition of SLC19A1-specific antifolate PT523
Tumor cells tend to use different types of folate transporters to sustain the elevated folate levels necessary for their rapid growth. Studies have shown that SLC19A1 primarily functions in hematologic malignancies and solid tumors, including acute lymphoblastic leukemia and osteosarcoma3,29,30. In contrast, SLC46A1 (also known as proton-coupled folate transporter, PCFT), another crucial pathway for folate and antifolate drug delivery, is prominently expressed in solid tumors, particularly in breast, lung and ovarian tumor cell lines4,31,32,33. The widely used antifolate drugs, including MTX and pemetrexed (PMX), can be absorbed by healthy cells through both SLC19A1 and SLC46A1, resulting in significant cytotoxicity34. Therefore, selective targeting of SLC19A1 or SLC46A1 through designer drugs would facilitate more effective treatments while mitigating cytotoxic effects35.
The γ-carboxylate groups of folates and antifolates have been demonstrated to be important for their uptake by SLC19A1 and SLC46A125,33. However, PT523, an atypical antifolate featuring a substituted γ-carboxylate (Supplementary Fig. 8a), emerges as an even more favorable substrate for SLC19A1 while exhibiting minimal transport by SLC46A14,36,37. To elucidate the SLC19A1-specific recognition mechanism of PT523, we determined the cryo-EM structure of outward-open hSLC19A1 in complex with PT523 at a resolution of 3.25 Å (Fig. 3a–c, Supplementary Fig. 8b–g and Supplementary Table 1). The cryo-EM density is of high quality and corresponds well to the molecular shape of PT523 (Supplementary Fig. 8a, g). An additional smeared density was also observed near PT523 in the pocket, likely due to non-specific attachment, which diminishes with decreasing PT523 concentration during cryo-EM sample preparation (Supplementary Fig. 9a, b). Similar to 5-MTHF, PT523 is tightly embedded in the central polar cavity with its long axis aligning parallel to the transmembrane helices (Fig. 3b, c and Supplementary Fig. 9c). The pteridine ring of PT523 occupies the electronegative pocket of the NTD, forming interactions with Glu45/Ile48/Thr49 of TM1, Leu72 of TM2 and Glu123 of TM4 (Fig. 3d, e). The benzoyl moiety forms hydrophobic interactions with Tyr126/Met130 of TM4 and the carbonyl is hydrogen bonded by Arg133 of TM4 (Fig. 3d, e). The β-carboxylate group faces the CTD and engages in hydrogen bonds with Tyr281 of TM7 and Arg373/Gln377 of TM10 (Fig. 3c, d). Remarkably, the γ-substituted hemiphthaloyl group delves into a distinct pocket within the NTD at the bottom of the central cavity (Fig. 3c, f, g and Supplementary Fig. 9d). The aromatic ring is supported from below by Tyr136 of TM4 and is flanked on both sides by hydrophobic interactions with the side chains of Arg133 of TM4 and Val160 of TM5 (Fig. 3f, g). Furthermore, the carbonyl and carboxyl are further stabilized by Arg133/Ser137 of TM4 and Arg157 of TM5 through hydrogen bonds (Fig. 3f, g). Thus, these additional contacts appear to contribute to the high transport activity of PT523. In addition, the existence of the specific and conserved pocket (Supplementary Fig. 7), lined by Arg133, Tyr136, Ser137, Arg157 and Val160, provides structural insights into the preference of SLC19A1 for antifolates with γ-carboxylate modifications over those with β-carboxylate modifications38.

a Cryo-EM density map of outward-open hSLC19A1 bound to PT523. b The overall structure of outward-open hSLC19A1 bound to PT523. The NTD and CTD are coloured in blue and yellow, respectively. PT523 is shown as a space-filling representation and is coloured in salmon. c A cut-open side view of the electrostatic potential surface of outward-open hSLC19A1 bound to PT523. d, f Hydrogen bonding interactions between PT523 (sticks; salmon) and outward-open hSLC19A1 (grey). Key residues are shown as sticks and hydrogen bonds are represented as dashed lines. e, g Hydrophobic interactions between PT523 (sticks; salmon) and outward-open hSLC19A1 (grey). Key residues are shown as sticks and dots.
In contrast, in the recently solved SLC46A1-PMX outward-open structure, PMX binds at the base of the polar cavity in an orientation nearly parallel to the membrane plane33. The pyrimidine moiety of SLC46A1-bound PMX resides in a negatively charged cavity within the CTD, while the glutamate group extends toward the NTD with the γ-carboxylate group nestled in a positively charged pocket (Supplementary Fig. 9e). Consequently, SLC46A1 ligands must establish an appropriate distance between the ring and the γ-carboxylate group to adapt to the minimal distance (~16 Å) of the functional groups within the binding site33,35. The length of the long axis of PT523 (~23 Å) exceeds the capacity of the substrate pocket of SLC46A1, thus the γ-substituted hemiphthaloyl group will apparently create significant steric hindrance with the positively charged pocket within the NTD (Supplementary Fig. 9c, e). Taken together, the additional interactions of PT523’s hemiphthaloyl group with SLC19A1, coupled with its steric hindrance with the positively charged pocket of SLC46A1, may contribute to the high affinity and substrate selectivity preference of PT523 for SLC19A1. These results provide new clues for the design of next-generation antifolate drugs with enhanced transporter specificity and reduced side effects. Moreover, besides serving as delivery systems for antifolate drugs, SLC19A1 and SLC46A1 also represent attractive targets for modulating folate uptake in tumor cells. A comparative analysis of substrate binding modes between SLC19A1 and SLC46A1, both in their outward-open conformation, will likely provide a framework for the rational design of specific exofacial inhibitors aimed at blocking folate uptake in different tumor types, as the substrate binding pocket of the outward-open conformation is readily accessible on the extracellular surface39,40.
Recognition of CDN monomer in outward-open state
In our previously reported hSLC19A1-CDN inward-open structures, CDNs were observed to bind as a compact dual-molecule unit near the intracellular side of hSLC19A1, even at low CDN:transporter molar ratios25. However, the compact CDN dimer does not bind within the canonical substrate cavity of SLC proteins and is likely to hinder closure of the intracellular gate of hSLC19A1 due to its numerous interactions with gate residues (Supplementary Fig. 11a). Therefore, the CDN dimer may only represent a potential substrate binding mode, and it remains unclear whether CDNs are transported by SLC19A1 in such a dimeric form. To further understand the precise transport mechanism of CDNs by SLC19A1, we determined the structure of outward-open hSLC19A1 bound to a representative CDN-type drug, 2′3′-CDAS, at a resolution of 3.34 Å (Fig. 4a–c, Supplementary Fig. 10 and Supplementary Table 1).

a Cryo-EM density map of outward-open hSLC19A1 bound to 2′3′-CDAS. b, The overall structure of outward-open hSLC19A1 bound to 2′3′-CDAS. The NTD and CTD are coloured in blue and yellow, respectively. 2′3′-CDAS is shown as a space-filling representation and is coloured in green. c, A cut-open side view of the electrostatic potential surface of outward-open hSLC19A1 bound to 2′3′-CDAS. d Hydrogen bonding and hydrophobic interactions between 2′3′-CDAS (sticks; green) and outward-open hSLC19A1 (grey). Key residues are shown as sticks and dots, and hydrogen bonds are represented as dashed lines. e SLC19A1−/− THP-1 cells stably expressing either WT hSLC19A1 or CDN-binding-pocket mutants were exposed to 2′3′-cGAMP. The induction of IFNB1 mRNA was measured by quantitative PCR with reverse transcription (RT-qPCR). Data are mean ± s.e.m. of n = 4 biological replicates. Statistical analysis was performed using two-tailed unpaired Student’s t-tests. f, h 2′3′-cGAMP (f) and [3H]-MTX (h) uptake assays using SLC19A1−/− HeLa cells stably expressing either WT hSLC19A1 or mutants of folate-binding and CDN-binding pocket. Data were normalized to WT and are mean ± s.e.m. of n = 4 biologically independent experiments. Statistical analysis was performed using two-tailed unpaired Student’s t-tests. **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001. Source data are provided as a Source Data file. g Structural superimposition of 5-MTHF (cyan) and 2′3′-CDAS (green) bound in the canonical substrate pocket of the outward-open hSLC19A1.
2′3′-CDAS represents one of the most extensively evaluated synthetic CDN-type drugs which consists of two AMP moieties connected by 2′-5′/3′-5′ mixed linkages with bisphosphothioate substitutions (Supplementary Fig. 10f). The cryo-EM density is of good quality to distinguish 2′-5′ and 3′-5′ phosphodiester bonds and different chemical groups (Supplementary Fig. 10g). Intriguingly, in contrast to inward-open structures, only one 2′3′-CDAS molecule binds within the canonical substrate cavity of hSLC19A1 (Fig. 4a–d). Positioned within the central canonical cavity of hSLC19A1, 2′3′-CDAS monomer adopts a U-type conformation (Fig. 4a–d). The sugar-phosphate ring faces the intracellular side and wedges diagonally into the positively charged region of the cavity, with the 3′-5′ thiophosphate interacting with Lys411 of TM11 and the 2′-5′ thiophosphate interacting with Tyr281 of TM7 (Fig. 4d and Supplementary Fig. 11b). Moreover, the free 3′-OH is held in place by a hydrogen bond with Arg133 of TM4 (Fig. 4d and Supplementary Fig. 11b). Consistent with the orientation of the sugar-phosphate ring, the 2′-5′ side adenine base occupies a lower position and is tightly packed to the NTD by hydrophobic interactions with Leu72 of TM2 as well as Tyr126/Met130 of TM4 (Fig. 4d). On the other hand, the 3′-5′ side adenine base is located at a higher position by interacting with His289 of TM7 in the CTD and is almost perpendicular to the 2′-5′ side adenine base (Fig. 4d). The backbones of 2′3′-CDAS-bound hSLC19A1 align well with those in substrate-free and folate-bound structures, with the side chains within the cavity exhibiting reasonable adjustments to adapt different type of substrates (Supplementary Fig. 11c, d).
To verify the importance of residues involved in CDN recognition, we assessed the effect of hSLC19A1 mutants on extracellular CDN-mediated STING pathway activation by cellular assays (Fig. 4e and Supplementary Fig. 2). In SLC19A1−/− THP-1 cells stably expressing WT or mutant hSLC19A1, IFNB1 mRNA production levels induced by extracellular 2′3′-cGAMP are reduced to varying extents by representative mutations (Fig. 4e). In particular, substitutions of Arg133, His289 and Lys411 to alanine nearly abolish STING pathway activation (Fig. 4e). Direct measurement of intracellular 2′3′-cGAMP accumulation41 using the cGAMP ELISA Kit further confirms the importance of Arg133, His289 and Lys411 in CDN recognition and cellular uptake (Fig. 4f).
Distinctive interaction strategies for folate and CDN
Although both folate/antifolate and CDN occupy the canonical substrate cavity of the outward-open hSLC19A1, their interaction details differ significantly (Fig. 4g). The pterin moiety of 5-MTHF occupies the upper negatively charged pocket within the NTD, and the glutamate moiety extends to the intracellular side and reaches the positively charged region (Fig. 4g). Despite sharing partial structural similarities with the pterin group of 5-MTHF (Supplementary Figs. 6a and 10f), the adenine bases of 2′3′-CDAS occupy lower positions and establish contacts with both domains instead of fitting into the negatively charged pocket of the NTD as folate does (Fig. 4g). Structural superimposition reveals certain overlap between the 2′-5′ side adenine base of 2′3′-CDAS and the benzoyl moiety of 5-MTHF (Fig. 4g), and therefore, not surprisingly, the two types of substrates may compete with each other in cellular uptake processes. Mutations of residues involved in the interaction of hSLC19A1 with both CDN and folate (e.g., Arg133) markedly reduce the uptake of both types of substrates (Fig. 4f, h, Supplementary Figs. 2 and 4f). However, mutations targeting CDN pocket-specific residues (e.g., His289 and Lys411) almost abolish CDN uptake while only mildly affecting folate uptake, highlighting the specific role of these residues in CDN recognition (Fig. 4f, h, Supplementary Figs. 2 and 4f). These findings lay the groundwork for the development of SLC19A1-specific inhibitors that selectively block CDN uptake without significantly affecting folate, which will be constructive in the treatment of autoimmune diseases.
Recognition of exported organic anion in outward-open state
As an antiporter, SLC19A1 mediates the import of folates or CDNs by coupling the export of organic anions such as ATP, ADP, AMP, 5-aminoimidazole-4-carboxamide ribotide monophosphate (ZMP), and glucose 6-phosphate (G6P)4,5. Among these, thiamine pyrophosphate (TPP) stands out as one of the preferred coupled substrates of SLC19A1, binding to inward-open SLC19A1 in a manner similar to folates (Supplementary Fig. 11e)27. To understand how SLC19A1 recognizes the exported organic anion in its outward-open conformation, we further determined the cryo-EM structure of outward-open hSLC19A1 in complex with TPP at 3.74 Å (Supplementary Fig. 11f, g and Supplementary Table 1). The pyrimidine ring of TPP aligns well with the density at the folate pocket and its interactions with SLC19A1 are similar to those of pterin ring in 5-MTHF (Supplementary Fig. 11h, i and Fig. 2h, i). However, the thiazolium ring and the pyrophosphate moiety remain unresolved due to the weak densities (Supplementary Fig. 11h). After the intracellular release of substrates in the inward-open conformation, organic anions bind to the inward-open SLC19A1, triggering the transporter to switch to its outward-open conformation, which drives the transport of folates and CDNs (Fig. 5). It is possible that the affinity of TPP for the outward-open cavity becomes weaker, facilitating the extracellular release of TPP. Interestingly, our cellular results indicate that the mutation in the folate-specific pocket (e.g., E123A) significantly reduces the uptake of both folates and CDNs (Fig. 4f, h and Supplementary Fig. 2), probably because the mutation affects the recognition of not only folates but also the coupled organic anions.

Schematic diagram of the alternating-access model for SLC19A1-mediated folate/antifolate and CDN transport. Alternating access of SLC19A1 is achieved through conformational changes alternately expose central substrate binding sites to each side of the membrane. Key residues involved in substrate recognition are highlighted.
Discussion
Originally recognized as a transporter for folate and antifolate, and recently identified as an immune transmitter transporter, SLC19A1 plays a pivotal role in basal metabolism, antitumor therapy, infection and immunopathology. Although previous works have established a structural framework for understanding the interaction modes of folate/antifolate and CDN in the inward-open conformation, further elucidation of the conformational change, substrate selectivity, and precise transport mechanism of CDN remain necessary. In the current work, using an antibody-facilitated screening strategy, we successfully identified a mutation capable of fixing SLC19A1 in the outward-open state and determined the structures of SLC19A1 in the substrate-free state and in complex with different types of substrates. This work also demonstrates the potential of an antibody-facilitated mutant screening strategy in elucidating the transport cycle of other SLC family proteins.
In the outward-open conformation of SLC19A1, TM1 and TM2, as well as TM7 and TM8, diverge to reveal the extracellular entrance to the hydrophilic cavity, facilitating substrate access and binding. Hydrogen bonds of Gln150-Ala324/Val327 and Arg157-Gln377, along with hydrophobic interactions of Phe141-Ala388-Phe400, form the intracellular gates, sealing off the cavity from the intracellular side. Notably, structural superimposition reveals that Phe141, Gln150 and Arg157 undergo dramatic conformational changes during the transport cycle, which would open the intracellular gate and trigger the intracellular release of substrates. In addition to the conformational changes during the transport cycle, the 5-MTHF-, MTX- and PT523-bound structures also shed light on the molecular mechanisms underlying substrate preference for SLC19A1 (Fig. 5). Remarkably, SLC19A1 NTD forms a specific pocket at the bottom of the cavity, lined by Arg133, Tyr136, Ser137, Arg157 and Val160, to accommodate the γ-substituted hemiphthaloyl group of PT523. However, comparison with the recently determined outward-open structure of SLC46A133, another folate transporter, reveals a potentially strong steric hindrance between the cavity of SLC46A1 and the γ-carboxylate modifications of PT523. Given the different expression levels of SLC19A1 and SLC46A1 in different tumor types, the integration of current molecular insights may accelerate the development of novel antifolates targeting specific transporters with minimized side effects.
Different from inward-open structures, the 2′3′-CDAS-bound outward-open structure shows that only one CDN molecule binds within the canonical cavity of SLC19A1 in a U-shaped conformation. The detailed binding pattern of CDN differs significantly from that of folate/antifolate, despite of sharing some similarities in chemical structure (Fig. 5). Specifically, CDN is sandwiched between the two domains of SLC19A1 at the positively charged position, whereas folate and antifolate interact more prominently with the NTD, with the pterin moiety extending into the upper negatively charged pocket. Through systematic mutant analysis, we identified that mutations targeting CDN-specific residues (e.g., H289A and K411A) nearly abolish SLC19A1-mediated CDN uptake while only slightly affecting folate/antifolate transport. These results highlight the distinctive recognition strategies of SLC19A1 for different substrate types, and provide critical insights for the development of potential small molecule drugs with selective regulatory activity for CDN and folate uptake processes. Inspired by the CDN-bound outward-open cryo-EM structure, we performed molecular docking and found that CDN may also be able to bind as a monomer to the canonical cavity of inward-open SLC19A1 (Supplementary Fig. 11j). The calculated binding free energy suggests that CDN binds to this pocket with low affinity (Supplementary Fig. 11k), which probably facilitates the release of substrates into cytoplasm and also makes it challenging to capture the inward-open structure with CDN monomer bound within the canonical pocket. Taken together, the structural, cellular and computational results indicate that CDN can be transported in its monomeric form by SLC19A1 via a canonical mechanism of SLC family proteins (Fig. 5).
Considering that in our previously reported SLC19A1-CDN inward-open structures25, the compact CDN dimer binds in a non-canonical pocket near the intracellular side and probably hinders the efficient intracellular release of CDN due to stronger interactions, the CDN dimer captured in these structures may not represent an effective transport unit of SLC19A1. On the other hand, given that CDN dimer or oligomer exhibits dynamic behavior in solution and plays important roles in other biological processes42,43,44, and that CDN can bind as a dimer even at low CDN:SLC19A1 molar ratios, it is reasonable to speculate that the dimer-bound conformation may represent a substrate binding mode of SLC19A1 with potential regulatory functions. Further studies will be required to determine whether this dimeric binding state functions in different immune activation or suppression scenarios. In addition, the non-canonical dimeric binding mode may also provide an unexpected avenue and strategy for the design of new classes of SLC19A1-specific drugs (Fig. 5).
In summary, the data elucidated in this study reveal the molecular basis for the transport mechanism and substrate preference of SLC19A1, establishing the framework for the development of antineoplastic and anti-autoimmune drugs.
Methods
Epitope mapping of SLC19A1 specific antibodies
Monoclonal antibodies targeting human SLC19A1 (hSLC19A1) were screened using single-cell B cell receptor (BCR) sequencing of antigen-specific cells, following a previously established protocol25. Approximately 10,000 antigen specific B cells (hSLC19A1-bound CD3-B220+CD19+CD38−GL7+ B cells) were isolated from the spleens and lymph nodes of immunized mice using the BD FACSAria III cell sorter. These cells were barcoded through the 10x Chromium Single Cell platform (10x Genomics). The libraries were sequenced on the Illumina Novaseq X Plus platform in a 150 bp pair-ended manner (Berry Genomics Corporation). A total of 54 VH and VL sequences were selected, synthesized, and cloned into mammalian expression vectors with constant regions of mouse IgG2a heavy chain and κ light chain respectively. The recombinant antibodies were then transiently expressed in HEK-293T cells using lipo2000 (Thermo Fisher Scientific) according to the manufacturer’s instruction. Culture supernatants were collected 24 hours post transfection. HEK-293T cells overexpressing hSLC19A1, mouse SLC19A1 (mSLC19A1) and chimeras of hSLC19A1 with specific regions replaced by mSLC19A1 were stained with the culture supernatants as the primary antibodies and PE goat anti-mouse IgG (BioLegend) as the secondary antibody. Epitope mapping of the recombinant antibodies was performed using western blot and flow cytometry on an Attune NxT Flow Cytometer (Thermo Fisher Scientific). The monoclonal antibodies (mAb70#, mAb5#, and mAb6# targeting different binding epitopes) were produced in HEK-293F Human Embryonic Kidney cells and purified using a protein A agarose prepacked column.
Antibody-facilitated screening of conformationally altered hSLC19A1 mutations
The DNA sequences encoding engineered mutants of hSLC19A1, designed to be locked in different conformations, were cloned into a modified pTT3 vector containing a GFP reporter gene linked with an IRES sequence. HEK-293T cells were transfected with these constructs using lipo2000 (Thermo Fisher Scientific). Following transfection, the medium was replaced with fresh DMEM supplemented with 10% (v/v) fetal bovine serum (FBS) and antibiotics (100 U/ml penicillin and 100 μg/ml streptomycin) after 6 hours. After 24 hours, the transfected cells were collected, labeled with hSLC19A1-specific antibodies targeting different epitopes (mAb70#, mAb5#, and mAb6#) as the primary antibodies and PE goat anti-mouse IgG (BioLegend) as the secondary antibody. Flow cytometry screening for outward-open proteins was conducted using an Attune NxT Flow Cytometer (Thermo Fisher Scientific).
Cloning, expression and purification of hSLC19A1
The cDNA of hSLC19A1 (Uniport ID: P41440) was cloned into a modified PTT5 vector with a C-terminal 10×His tag using the ClonExpress II One Step Cloning Kit (Vazyme) for overexpressing in HEK-293F. To facilitate cryo-EM structures determination, the gene encoding BRIL fragment was fused to the N-terminus of TM1 of hSLC19A1 and the gene encoding anti-Fab nanobody was fused into the C-terminus of TM12 by a helical linker (AEEEKRKAEEEKRK). All mutants were generated with a standard PCR-based strategy. To produce hSLC19A1 protein, 0.5 mg hSLC19A1 plasmids were pre-incubated with 1.5 mg PEI in 50 mL Opti-MEM medium at 37 °C. After 20 mins, the mixture was added drop by drop into 500 mL HEK-293F cells at a density of 2 ~ 2.5 × 106 cells/mL. The transfected cells were cultured for 72 hours before harvesting by centrifugation. Harvested HEK-293F cells were resuspended in lysis buffer containing 50 mM Tris-HCl pH 7.5, 300 mM NaCl, 10 mM imidazole, 5% glycerin and 1 mM PMSF. The cells were disrupted by sonication, and 1% (w/v) n-dodecyl-b-D-maltoside (DDM, Anatrace) and 0.1% (w/v) cholesteryl hemisuccinate (Anatrace) were added for protein solubilizing at 4 °C for 2 hours. After centrifugation (30,000 × g, 30 mins, 4 °C), the supernatant was incubated with nickel affinity resin at 4 °C for 1 hour, and then the resin was washed with the washing buffer containing 50 mM Tris-HCl pH 7.5, 300 mM NaCl, 60 mM imidazole, 0.05% (w/v) n-dodecyl-b-D-maltoside (DDM, Anatrace) and 0.005% (w/v) cholesteryl hemisuccinate (Anatrace) and 5% glycerin. The protein was eluted with 50 mM HEPES pH 7.5, 300 mM NaCl, 400 mM imidazole, 0.05% (w/v) n-dodecyl-b-D-maltoside (DDM, Anatrace) and 0.005% (w/v) cholesteryl hemisuccinate (Anatrace) and 5% glycerin. The eluted protein was further purified through a Superdex 200 Increase 10/300 GL column (GE Healthcare) in the SEC buffer containing 20 mM HEPES pH 7.5, 150 mM NaCl, 0.01% (w/v) GDN. Peak fractions were collected and concentrated to ~2 mg/mL.
The hSLC19A1EM-Fab complex assembly
For complex assembly, the purified hSLC19A1EM and anti-BRIL Fab were mixed on ice at a molar ratio of 1:1.1. After incubation for 20 mins, the mixture was subjected to a Superdex 200 Increase 10/300 GL column (GE Healthcare) equilibrated with SEC buffer (20 mM HEPES pH 7.5, 150 mM NaCl and 0.01% GDN) to remove the excess anti-Bril Fab. For nanodisc reconstitution, hSLC19A1EM, MSP1D1, POPG and anti-BRIL Fab were mixed at a molar ratio of 1:2.1:84:1.1. Detergents were removed by 100 mg/mL Bio-beads with gentle agitation overnight. Subsequently, the Bio-beads were removed and the nanodisc reconstitution mixture was loaded onto a Superdex 200 Increase 10/300 GL column equilibrated with 20 mM HEPES pH 7.5 and 150 mM NaCl. Peak fractions were analyzed by SDS-PAGE and then concentrated for cryo-EM sample preparation.
Cryo-EM sample preparation and data collection
For apo hSLC19A1 samples, the peak fractions of hSLC19A1EM-Fab in GDN (20 mM HEPES pH 7.5, 150 mM NaCl and 0.01% GDN) were concentrated to 8.4 mg/mL. For substrate-bound samples, the peak fractions of the SLC19A1EM-Fab in nanodisc (20 mM HEPES pH 7.5 and 150 mM NaCl) were concentrated to 10.7 mg/mL and separately incubated with 5 mM 5-MTHF, 8 mM MTX, 5 mM PT523, 3 mM 2’3’-CDAS and 5 mM TPP for 3 h. 3 μL of the protein sample was deposited onto the glow-discharged holey carbon, 300 mesh R1.2/1.3 Au grids (Quantifoil). Subsequently, the grids were blotted for 2.5 s and plunged into liquid ethane for quick freezing using the Vitrobot Mark IV (Thermo Fisher Scientific). All cryo-EM datasets were collected under the dose rate of 9.2 e− Å−2 s−1 and dose-fractioned into 32 frames, yielding a total dose of 60 e− Å−2 by SerialEM45 on the Talos Arctica 200 kV FEG (Thermo Fisher Scientific) with a K2 summit direct electron elector (Gatan) and a GIF quantum energy filter (Gatan). The pixel size was calibrated at 0.5 Å (×130,000) under super-resolution mode, with the defocus ranging from −0.8 to −1.5 μm. Images were recorded using beam–image shift data collection methods46.
Cryo-EM data processing
3301 micrographs were collected for outward-open apo-hSLC19A1 structure dataset. Beam-induced motion correction and dose weighting were performed with MotionCor247. Corrected micrographs were then imported into cryoSPARC48 for contrast transfer function (CTF) estimation with CTFFIND449. A total of 2,001,272 particles were picked and extracted. After two rounds of 2D classification, 854,517 particles with clear features were picked and subjected to the Ab initio reconstruction. The following two rounds of heterogeneous refinement yielded 345,903 good particles which resulted in a reconstruction with an overall resolution of 3.26 Å. The particles were then imported into RELION50 for Bayesian polishing, followed by non-uniform refinement and local refinement in cryoSPARC, which yielded a final reconstruction to 2.94 Å resolution. 5-MTHF-bound (6,961 micrographs; 220,166 particles), MTX-bound (2,333 micrographs; 97,106 particles), PT523-bound (2,082 micrographs; 94,583 particles), 2′3′-CDAS-bound (5,257 micrographs; 301,880 particles) and TPP-bound (1,389 micrographs; 50,812 particles) datasets were processed using a similar procedure and yielded reconstructions at 3.43 Å, 3.44 Å, 3.25 Å, 3.34 Å and 3.74 Å respectively. The local resolution estimation was performed to determine the local resolution of the final map in cryoSPARC.
Model building and refinement
The inward-open hSLC19A1 (PDB: 7XPZ)25 was used as initial template for model building. The NTD and CTD were roughly fitted into the hSLC19A1 outward-open maps by UCSF Chimera51. After manually adjustment by Coot52, the resulting model was further refined in PHENIX53. The atomic model of ligand-bound hSLC19A1 was generated by several rounds of real space refinement in COOT and PHENIX with the apo outward-open hSLC19A1 as the initial model. 5-MTHF, MTX, PT523, 2′3′-CDAS or TPP was fitted into the density using COOT, then the resulting model was manually rebuilt in COOT and further refined in PHENIX. The models were validated through evaluation of the Clash scores, MolProbity scores and statistics of the Ramachandran plots by PHENIX. All the structural figures were prepared by PyMOL54 and UCSF ChimeraX55.
hSLC19A1 overexpression cell line generation
The experiments were carried out as previously described in ref. 25. In brief, the DNA sequence encoding human SLC19A1 or its mutants was cloned into a lentiviral plasmid containing a GFP reporter gene connected by a 2 A sequence. The mutant-carrying lentiviruses were produced in HEK-293T cells and used to infect SLC19A1−/− Hela or SLC19A1−/− THP-1 cells in the presence of 8 μg/ml polybrene (Sigma-Aldrich). After 72 hours of culture, lentiviral-infected cells showing similar levels of GFP expression were isolated using a BD FACSAria III cell sorter (BD Biosciences).
[3H]-labelled antifolate uptake assay
The cellular [3H]-labelled antifolate uptake assay was performed under published protocols25,56. The cell lines were generated as mentioned above. For uptake assays, 2 × 105 WT or SLC19A1 mutant cells per well were seeded into 24-well plates. After 12 h growth, the cells were washed twice with 500 µL HBSS buffer at pH 7.4 (Pricella). The buffer contains 140 mg/L CaCl2, 100 mg/L MgCl2•6H2O, 100 mg/L MgSO4•7H2O, 350 mg/L NaHCO3 and 1000 mg/L D-glucose. The cells were then incubated with 200 µL HBSS buffer supplemented with 25 nM [3H]-labelled methotrexate (American Radiolabeled Chemicals) for 10 mins. The uptake was terminated and washed twice by 500 µL ice-cold HBSS buffer. Lysed by 200 µL 0.2 M NaOH, the amount of accumulated [3H]-labelled methotrexate was calculated by scintillation counting in Ultima Gold (Perkin Elmer). For measurement of the initial uptake rates, the amount of accumulated [3H]-labelled methotrexate was calculated at 0.5 min, 1 min, 1.5 min and 2 min respectively.
CDN stimulation assay
The experiments were according to established protocols25. Briefly, SLC19A1−/− THP-1 cells stably expressing either wild-type or mutant hSLC19A1 were seeded in a 24-well plate at a density of 3 × 105 cells per well in 400 µL medium. The cells were then stimulated with 30 μM 2′3′-cGAMP for 3 hours. Cells were collected and total RNAs were extracted using TRIzol reagent (Thermo Fisher Scientific). Genomic DNA was removed by DNase I treatment (RQ1, Promega). Reverse transcription was carried out using M-MLV Reverse Transcriptase (Promega). Real-time PCR was performed with 1x PowerUp™ SYBR™ Green Master Mix (Applied Biosystems) according to the manufacturer’s instructions. The primers used were as follows: β-actin, 5’-ACCGAGCGCGGCTACAG-3’ and 5’-CTTAATGTCACGCACGATTTCC-3’; IFNB1: 5’-GTCTCCTCCAAATTGCTCTC-3’ and 5’-ACAGGAGCTTCTGACACTGA-3’. Samples were analyzed on a QuantStudio Q7 (Applied Biosystems).
Cell based CDN uptake assay
SLC19A1−/− Hela cells stably expressing either wild-type or mutant hSLC19A1 were seeded into 24-well plates at a density of 3 × 105 cells per well and cultured for 12 hours. After being washed twice with PBS, the cells were exposed to fresh DMEM complete medium containing 50 μM cGAMP for 4 hours. Following this, the cells were washed five times with PBS, lysed in 100 μL of RIPA buffer (150 mM NaCl, 1% Triton-X-100, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris pH 8.0) on ice for 15 mins, and then centrifuged to remove insoluble material before cGAMP quantification. The intracellular cGAMP levels were measured using the Direct 2’3’-Cyclic GAMP ELISA Kit (Arbor Assays).
Molecular docking
The binding modes between hSLC19A1 and 2’3’-cGAMP were determined by using molecular docking method. Structural models of inward-open (PDB: 7XPZ) and outward-open (PDB: 9JOZ) hSLC19A1 were set to be rigid in the docking process. The ligand 2’3’-cGAMP was set to be flexible. The docking process was accomplished by using the AutoDock Vina software and performed within 25 Å × 25 Å × 25 Å cubes centered on the center points of the chosen canonical substrate binding pocket with exhaustiveness set to 32. The results were ranked based on the lowest binding energy. Structural drawings were generated using PyMol.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data that support this study are available from the corresponding authors upon request. The cryo-EM maps have been deposited into the Electron Microscopy Data Bank under accession numbers EMD-61690 (outward-open hSLC19A1), EMD-61756 (outward-open hSLC19A1 + 5-MTHF), EMD-61758 (outward-open hSLC19A1+MTX), EMD-61759 (outward-open hSLC19A1 + PT523), EMD-61760 (outward-open hSLC19A1 + 2'3’-CDAS) and EMD-61761 (outward-open hSLC19A1+TPP). The coordinates have been deposited into the Protein Data Bank under accession numbers PDB: 9JOZ (outward-open hSLC19A1), PDB: 9JRI (outward-open hSLC19A1 + 5-MTHF), PDB: 9JRK (outward-open hSLC19A1+MTX), PDB: 9JRL (outward-open hSLC19A1 + PT523), PDD: 9JRM (outward-open hSLC19A1 + 2'3’-CDAS) and PDB: 9JRN (outward-open hSLC19A1+TPP). A source Data file is included with this manuscript. Source data are provided with this paper.
References
Zheng, Y. & Cantley, L. C. Toward a better understanding of folate metabolism in health and disease. J. Exp. Med. 216, 253–266 (2019).
Clare, C. E., Brassington, A. H., Kwong, W. Y. & Sinclair, K. D. One-carbon metabolism: linking nutritional biochemistry to epigenetic programming of long-term development. Annu. Rev. Anim. Biosci. 7, 263–287 (2019).
Matherly, L. H., Hou, Z. & Deng, Y. Human reduced folate carrier: translation of basic biology to cancer etiology and therapy. Cancer metastasis Rev. 26, 111–128 (2007).
Hou, Z. & Matherly, L. H. Biology of the major facilitative folate transporters SLC19A1 and SLC46A1. Curr. Top. Membr. 73, 175–204 (2014).
Matherly, L. H. & Hou, Z. Structure and function of the reduced folate carrier: a paradigm of a major facilitator superfamily mammalian nutrient transporter. Vitam. hormones 79, 145–184 (2008).
Goldman, I. D., Chattopadhyay, S., Zhao, R. & Moran, R. The antifolates: evolution, new agents in the clinic, and how targeting delivery via specific membrane transporters is driving the development of a next generation of folate analogs. Current opinion in investigational drugs (London, England: 2000) 11, 1409–1423 (2010).
Luteijn, R. D. et al. SLC19A1 transports immunoreactive cyclic dinucleotides. Nature 573, 434–438 (2019).
Ritchie, C., Cordova, A. F., Hess, G. T., Bassik, M. C. & Li, L. SLC19A1 is an importer of the immunotransmitter cGAMP. Mol. cell 75, 372–381. e375 (2019).
Danilchanka, O. & Mekalanos, J. J. Cyclic dinucleotides and the innate immune response. Cell 154, 962–970 (2013).
Zaver, S. A. & Woodward, J. J. Cyclic dinucleotides at the forefront of innate immunity. Curr. Opin. cell Biol. 63, 49–56 (2020).
Zhang, X. et al. Cyclic GMP-AMP containing mixed phosphodiester linkages is an endogenous high-affinity ligand for STING. Mol. cell 51, 226–235 (2013).
Gao, P. et al. Structure-function analysis of STING activation by c [G (2′, 5′) pA (3′, 5′) p] and targeting by antiviral DMXAA. Cell 154, 748–762 (2013).
Shang, G., Zhang, C., Chen, Z. J., Bai, X.-C. & Zhang, X. Cryo-EM structures of STING reveal its mechanism of activation by cyclic GMP–AMP. Nature 567, 389–393 (2019).
Wu, J. et al. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 339, 826–830 (2013).
Sun, L., Wu, J., Du, F., Chen, X. & Chen, Z. J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786–791 (2013).
Gao, P. et al. Cyclic [G (2′, 5′) pA (3′, 5′) p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell 153, 1094–1107 (2013).
Burdette, D. L. et al. STING is a direct innate immune sensor of cyclic di-GMP. Nature 478, 515–518 (2011).
Diner, E. J. et al. The innate immune DNA sensor cGAS produces a noncanonical cyclic dinucleotide that activates human STING. Cell Rep. 3, 1355–1361 (2013).
Fu, J. et al. STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade. Sci. Transl. Med. 7, 283ra252–283ra252 (2015).
Mekers, V. E., Kho, V. M., Ansems, M. & Adema, G. J. cGAS/cGAMP/STING signal propagation in the tumor microenvironment: key role for myeloid cells in antitumor immunity. Radiother. Oncol. 174, 158–167 (2022).
Xie, W. & Patel, D. J. Structure-based mechanisms of 2’ 3’-cGAMP intercellular transport in the cGAS–STING immune pathway. Trends Immunol. 44, 50–467 (2023).
Matherly, L. H. & Hou, Z. Folate transporter offers clues for anticancer drugs. Nature 612, 39–41 (2022).
Hou, Z., Ye, J., Haska, C. L. & Matherly, L. H. Transmembrane domains 4, 5, 7, 8, and 10 of the human reduced folate carrier are important structural or functional components of the transmembrane channel for folate substrates. J. Biol. Chem. 281, 33588–33596 (2006).
Quistgaard, E. M., Löw, C., Guettou, F. & Nordlund, P. Understanding transport by the major facilitator superfamily (MFS): structures pave the way. Nat. Rev. Mol. Cell Biol. 17, 123–132 (2016).
Zhang, Q. et al. Recognition of cyclic dinucleotides and folates by human SLC19A1. Nature 612, 170–176 (2022).
Wright, N. J. et al. Methotrexate recognition by the human reduced folate carrier SLC19A1. Nature 609, 1056–1062 (2022).
Dang, Y. et al. Molecular mechanism of substrate recognition by folate transporter SLC19A1. Cell Discov. 8, 141 (2022).
Liu, X. Y., Witt, T. L. & Matherly, L. H. Restoration of high-level transport activity by human reduced folate carrier/ThTr1 thiamine transporter chimaeras: role of the transmembrane domain 6/7 linker region in reduced folate carrier function. Biochem. J. 369, 31–37 (2003).
Liu, M. et al. Transcriptional regulation of the human reduced folate carrier in childhood acute lymphoblastic leukemia cells. Clin. cancer Res. 12, 608–616 (2006).
Ifergan, I., Meller, I., Issakov, J. & Assaraf, Y. G. Reduced folate carrier protein expression in osteosarcoma: implications for the prediction of tumor chemosensitivity. Cancer 98, 1958–1966 (2003).
Qiu, A. et al. Identification of an intestinal folate transporter and the molecular basis for hereditary folate malabsorption. Cell 127, 917–928 (2006).
Zhao, R., Aluri, S. & Goldman, I. D. The proton-coupled folate transporter (PCFT-SLC46A1) and the syndrome of systemic and cerebral folate deficiency of infancy: hereditary folate malabsorption. Mol. Asp. Med. 53, 57–72 (2017).
Parker, J. L. et al. Structural basis of antifolate recognition and transport by PCFT. Nature 595, 130–134 (2021).
Zhao, R. et al. The proton-coupled folate transporter: impact on pemetrexed transport and on antifolates activities compared with the reduced folate carrier. Mol. Pharmacol. 74, 854–862 (2008).
Newstead, S. Structural basis for recognition and transport of folic acid in mammalian cells. Curr. Opin. Struct. Biol. 74, 102353 (2022).
Rhee, M. S., Galivan, J., Wright, J. E. & Rosowsky, A. Biochemical studies on PT523, a potent nonpolyglutamatable antifolate, in cultured cells. Mol. Pharmacol. 45, 783–791 (1994).
Rosowsky, A., Wright, J. E., Vaidya, C. M. & Forsch, R. A. The effect of side-chain, para-aminobenzoyl region, and B-ring modifications on dihydrofolate reductase binding, influx via the reduced folate carrier, and cytotoxicity of the potent nonpolyglutamatable antifolate Nα-(4-amino-4-deoxypteroyl)-Nδ-hemiphthaloyl-l-ornithine. Pharmacol. Therapeutics 85, 191–205 (2000).
Deng, Y. et al. Role of lysine 411 in substrate carboxyl group binding to the human reduced folate carrier, as determined by site-directed mutagenesis and affinity inhibition. Mol. Pharmacol. 73, 1274–1281 (2008).
Wang, J. et al. Glucose transporter inhibitor-conjugated insulin mitigates hypoglycemia. Proc. Natl Acad. Sci. 116, 10744–10748 (2019).
Wang, N. et al. Molecular basis for inhibiting human glucose transporters by exofacial inhibitors. Nat. Commun. 13, 2632 (2022).
Maltbaek, J. H., Cambier, S., Snyder, J. M. & Stetson, D. B. ABCC1 transporter exports the immunostimulatory cyclic dinucleotide cGAMP. Immunity 55, 1799–1812.e1794 (2022).
Zhang, Z., Kim, S., Gaffney, B. L. & Jones, R. A. Polymorphism of the signaling molecule c-di-GMP. J. Am. Chem. Soc. 128, 7015–7024 (2006).
Chan, C. et al. Structural basis of activity and allosteric control of diguanylate cyclase. Proc. Natl Acad. Sci. 101, 17084–17089 (2004).
Tschowri, N. et al. Tetrameric c-di-GMP mediates effective transcription factor dimerization to control Streptomyces development. Cell 158, 1136–1147 (2014).
Mastronarde, D. N. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36–51 (2005).
Wu, C., Huang, X., Cheng, J., Zhu, D. & Zhang, X. High-quality, high-throughput cryo-electron microscopy data collection via beam tilt and astigmatism-free beam-image shift. J. Struct. Biol. 208, 107396 (2019).
Zheng, S. Q. et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. methods 14, 331–332 (2017).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. methods 14, 290–296 (2017).
Rohou, A. & Grigorieff, N. CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 (2015).
Zivanov, J. et al. New tools for automated high-resolution cryo-EM structure determination in RELION-3. elife 7, e42166 (2018).
Pettersen, E. F. et al. UCSF Chimera—a visualization system for exploratory research and analysis. J. Computational Chem. 25, 1605–1612 (2004).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D: Biol. Crystallogr. 66, 486–501 (2010).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D: Biol. Crystallogr. 66, 213–221 (2010).
Schrödinger, L. The PyMOL Molecular Graphics System, Version 1.8. (No Title) (2015).
Goddard, T. D. et al. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 27, 14–25 (2018).
Visentin, M., Zhao, R. & Goldman, I. D. Augmentation of reduced folate carrier-mediated folate/antifolate transport through an antiport mechanism with 5-aminoimidazole-4-carboxamide riboside monophosphate. Mol. Pharmacol. 82, 209–216 (2012).
Acknowledgements
Cryo-EM data collection was carried out at the Center for Biological Imaging, Core Facilities for Protein Science at the Institute of Biophysics, Chinese Academy of Sciences. The computation work was performed using High-performance computing resources, the Center for Biological Imaging, Institute of Biophysics, Chinese Academy of Science. All radioactivity experiments were performed at the radioactive isotope laboratory (Institute of Biophysics, CAS), with guidance from H.J. Zhang in handling radioactive materials. The plasmid expressing membrane scaffold protein 1D1 (MSP1D1) was a gift from Dr. Z. Liu’s group. We also thank Cryo-Electron Microscopy Platform of Medical Science and Technology Innovation Center of Shandong First Medical University for the support of Cryo-EM data collection. This work was supported by grants from National Natural Science Foundation of China (32325028 & 32130057 to P.G., 32171219 to A.G., 32401005 to Q.Z., 82371853 to X.Z.), National Key R&D Program of China (2024YFA1307400 to P.G.), Beijing Natural Science Foundation (Z220018 to P.G.), Basic Research Program Based on Major Scientific Infrastructures-CAS (JZHKYPT-2021-05 to P.G.), CAS Project for Young Scientists in Basic Research (YSBR-074 to P.G.), and China Postdoctoral Science Foundation (2023M740257 & BX20230468 to Q.Z.).
Author information
Authors and Affiliations
Contributions
Q.Z. and K.L. purified proteins, prepared cryo-EM samples, collected and processed cryo-EM data, and reconstructed density maps. Y.G. assisted with cryo-EM data processing. Q.Z. and P.G. built and refined models. X.Z. and Q.Z. performed antibody screening and validation. X.Z., Q.Z. and K.L. performed cellular assays. Y.Z., X.N., and J.M. assisted with antibody screening. P.S. and S.L. assisted with cell culture and protein expression. Z.L. performed molecular docking assay. P.G., L.Z., and A.G. initiated the project and directed the research. P.G. and Q.Z. wrote the manuscript with help of all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Larry Matherly and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, Q., Zhang, X., Liu, K. et al. Molecular basis of SLC19A1-mediated folate and cyclic dinucleotide transport. Nat Commun 16, 3146 (2025). https://doi.org/10.1038/s41467-025-58378-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-58378-1
This article is cited by
-
Opportunities and challenges of targeting cGAS–STING in cancer
Nature Reviews Cancer (2026)
