
Abstract
Bacteriophages offer a promising alternative to drug-based treatments due to their effectiveness and host specificity. This is particularly important in agriculture as a biocontrol agent of plant diseases. Phage engineering is facilitated by structural knowledge. However, structural information regarding bacteriophages infecting plant pathogens is limited. Here, we present the cryo-EM structure of bacteriophage φTE that infects plant pathogen Pectobacterium atrosepticum. The structure reveals a distinct neck topology compared with other myophages, where tail terminator proteins compensate for reduced connectivity between sheath subunits. A contact network between tail fibers, the sheath initiator, and baseplate wedge proteins provides insights into triggers that transduce conformational changes from the baseplate to the sheath to orchestrate contraction. We observe two distinct oligomeric states of the tape measure protein (TMP), which is six-fold in regions proximal to the N-terminus and throughout most of the tail, while three-fold at the C-terminus, indicating that the TMP may be proteolytically cleaved. Our results provide a structural atlas of the model bacteriophage φTE, enhancing future interpretation of phage host interactions in pectobacteria. We anticipate that our structure will inform rational design of biocontrol agents against plant pathogens that cause diseases such as soft rot and blackleg disease in potatoes.
Similar content being viewed by others

Introduction
Bacteriophages of the class Caudoviricetes represent the most prolific group of viruses in the biosphere1,2. These highly abundant tailed bacteriophages are found in nearly every environmental niche. They are essential for the stability of their ecosystems, contributing to genetic diversity, promoting bacterial evolution, and influencing microbial-induced biogeochemical cycling3. In addition, several of their structural and genetic properties have gained much interest in medical, biotechnological, and agricultural settings as therapeutics and biotechnology scaffolds4,5,6. Bacteriophages also serve as vital tools in studying bacterial biology, providing valuable insights into bacterial evolution, genetic exchange, and defense mechanisms.
One of the most promising applications of bacteriophages is their use to control diseases of bacterial origin as an alternative therapy to antibiotics6,7. However, it is often the case that bacteriophages display suboptimal properties for applications of interest, such as modest infectivity, limited stability under the required conditions, or inadequate host range. While phage engineering is a promising approach to overcome these shortcomings and expand the scope of phage-based disease control, its application remains limited due to the lack of knowledge on phage architecture and mechanisms of infection.
Recent studies have determined the full-virion structure for various long-tailed phages, providing valuable information for rational phage engineering projects8,9,10,11,12,13,14,15,16,17. These include both siphophages (DT57C, T5, JBD30, lambda, R4C) and myophages (Pam3, E217, Milano, Pa193, XM1, A-1(L)). Despite these advancements, the detailed molecular architecture of bacteriophages remains a fertile ground for exploration. For instance, there is limited information regarding the role of individual proteins in signal transduction from lateral fibers governing tail contraction or the functional role of the tape measure protein in this process.
The phage φTE, first isolated in 2012, is a lytic flagellotropic phage that infects Pectobacterium atrosepticum. The phage φTE belongs to the Certrevirus genus of the Vequintavirinae subfamily and has a relatively large genome consisting of ~142 kb. The Pectobacterium host of φTE is of significant economic importance due to its role in causing soft rot and blackleg disease in potatoes, leading to substantial annual financial losses18,19. A variety of phages infecting Pectobacterium atrosepticum have been discovered, including siphophages, myophages, and podophages, and have been analyzed from a biochemical and microbiological perspective20,21,22,23. Nevertheless, among these phages, only ϕM1 (a podophage lacking a long tail) has been studied at a structural level24. Furthermore, no other vequintaviruses have been explored structurally. Phage φTE has been well characterized regarding phage-host interactions, especially concerning bacterial defense systems. φTE has demonstrated a remarkable ability to evolve or acquire RNA antitoxins to counteract toxin-antitoxin (TA) system defenses18. In addition, φTE was used to study CRISPR-Cas spacer acquisition and CRISPR escape, wherein large deletions in structural proteins permitted primed CRISPR escape while also inducing morphological changes to virions23. More recently, φTE was used as a model phage in a landmark study that described the presence and function of RNA-based anti-CRISPR (Racr) elements25. Thus, φTE has become an excellent model for the study of phage-bacterial interactions with both innate (TA) and adaptive (CRISPR-Cas) defense systems.
In this study, we used electron cryo-microscopy (cryo-EM) to reveal the atomic structure of the entire φTE virion. Our structural atlas obtained through a comprehensive analysis of the various virion sub-structures reveals the particle in its native state. Since we could obtain near-atomic resolution throughout most parts of the structure, we were able to build and refine atomic models corresponding to 5 composite structures, cumulatively containing 19 genetically distinct protein chains, some of which assume multiple heterogeneous conformations. AlphaFold models were fit to regions of poor local map resolution to speculate further on these structures. We reconstructed the entire phage particle from single particle images at 12.7 Å resolution, enabling the deposition of a merged atomic model of the complete φTE myophage based on our high-resolution sub-maps together with a single cryo-EM envelope in the protein data bank (PDB). Our structures reveal insights into general principles of phage architecture, such as the presence of a proteolytically processed tape measure protein (TMP) in myophages with two distinct oligomerization states. The reconstruction of the native state particle clearly resolves the TMP as six long alpha helices in the central part of the tail.
Results
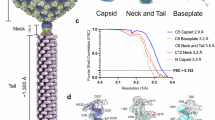
We used cryo-EM to reconstruct the complete φTE particle. In its native state, the particle is composed of a large T = 13 d icosahedral capsid adjoined to a narrow connector region, a helical rigid sheathed tail ~1020 Å in length, and a baseplate module (Fig. 1a, b, Supplementary Movie 1). Each structure was reconstructed separately to take advantage of its local symmetry. Candidate gene products were then identified using a combination of mass spectrometry abundance, AlphaFold2 and AlphaFold3 predictions, and Dali structural homology searches.

a Composite reconstruction (left) and composite deposited model (right) of full φTE virion. Structural dimensions and reconstruction details are annotated. Individual protein names are annotated at the center. This composite model is composed of 3,824,499 atoms, 494,665 residues and 1984 chains. b Schematic of the φTE genome annotated and color coded to match panel (a). Numbers that follow protein names correspond to genome locus tags. NCBI reference sequence: NC_020201.1.
The φTE capsid
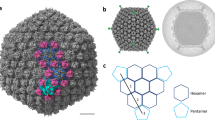
The capsid of the mature φTE virion was reconstructed at 3.6 Å resolution thanks to its icosahedral symmetry. The mature φTE capsid has a diameter of ~1050 Å (vertex to vertex). Each asymmetric unit (ASU) is composed of 13 copies of the major capsid protein (MCP), 13 copies of minor capsid protein (DEC, Fig. 2a, b), and up to 2 copies of the hexamer-centric decoration protein we describe as the Pagoda protein based on its morphology (Fig. 2b, purple). Twelve MCPs in the ASU form two hexameric capsomers referred to as P-Hexamer (pentamer-adjacent hexamer) and H-Hexamer (hexamer-adjacent hexamer). The 13th MCP is dedicated to forming part of the pentamer at the icosahedral five-fold axis. The ASU is then decorated by DEC proteins that form trimers at three-fold and quasi-three-fold axes, establishing reciprocal contacts along the two-fold axis. While the Pagoda protein could not be modeled, a disparity in Pagoda density was observed between H-hexamers and P-hexamers, with the latter showing reduced density. Furthermore, density corresponding to the interaction region between the Pagoda protein and MCPs lacks symmetry and order, indicating that the Pagoda protein may bind in several quasi-equivalent rotated positions. The putative Pagoda protein was assigned as pTE217 based on very strong mass spectrometry abundance; the abundance of pTE217 was the fifth highest after MCP, DEC, Sheath and tube, and was significantly higher than the next unidentified peptide in mass spectrometry data (exponentially modified peptide abundance index: pTE217 = 79,432, next highest pTE203 = 533) (Fig. 1b, Supplementary Table 2). AlphaFold predictions of pTE217 resulted in low confidence predictions with no detectable homology to other well-described hexamer centric decoration proteins.

a The phage φTE T = 13 capsid symmetrized model. Twofold axes are shown in blue ellipses. The three-fold axis is shown as a green triangle. Five-fold axes are shown as cyan pentagons. b Schematic of the capsid ASU. H-Hex = hexamer adjacent hexamer, P-Hex = pentamer adjacent hexamer. MCPs are shown in various shades of blue depending on the environment. DEC proteins are shown in light green. Density corresponding to the Pagoda protein is shown in purple. Map: EMD-45937 (3.6 Å) threshold 0.77 (3.2 σ). c Annotated structure of the φTE MCP. d Structural alignment of all ASU MCPs. Regions of greatest change are indicated with arrows. e Annotated structure of the DEC protein. f Hydrophobic representation of the capsid binding surface of the DEC protein. g The DEC-MCP Binding interface. MCP rendered as white surface, and interactions shown as dark blue cartoons.
The major capsid protein shows a canonical HK97 fold26, a strongly conserved protein fold that forms the main capsid protein of all tailed bacteriophages, herpesviruses, and some archaeal viruses (Fig. 2c); each MCP is found in a range of quasi-equivalent conformations (Fig. 2c, d)26,27. The trimeric decoration protein was found to belong to the β-tulip family through DALI homology analysis (Fig. 2e). Unlike other β-tulip proteins, the DEC features a C-terminal extension that wraps over a conserved intercapsomeric β-sheet, making contacts with all strands of the β-sheet (Fig. 2g). The topological contributions of β-strands in the αβ-domain of DEC are also different from those observed in other known β-tulips28,29,30,31,32. Each DEC protein forms a binding interface through these motifs, contacting eight MCPs with a combined surface area of 3056 Å2,33. Mapping the calculated hydrophobic surface properties of DEC proteins reveals a concentration of non-polar residues at the MCP binding interface indicative of predominant hydrophobic interactions (Fig. 2f).
The phage φTE capsid constitutes the fifth atomic model of a T = 13 bacteriophage capsid with HK97 fold, including the mutated isometric T4 capsid10,34,35,36,37. To investigate variations in capsid geometry among reported similar architectures (bacteriophages Mic1, T5, and T4, these are HK97 fold phage capsids with the same triangulation number T, i.e., the same number of capsid monomers per asymmetric unit), structures with a resolution better than 4 Å were used to generate whole particle protein point arrays. These arrays were constructed by placing markers at the center of mass of all protein chains throughout the capsid, allowing precise, relative measurements of geometric properties such as particle radius, surface area, volume, and sphericity (Fig. 3a). Assuming properly calibrated pixel sizes of those published datasets, analysis of the T = 13 bacteriophage capsids revealed that the φTE capsid has a radius ~8% greater than other T = 13 phages and ~7% greater than the next largest capsid analyzed (Fig. 3b). This expanded radius results in an average surface area ~15% larger and a ~ 27% volume increase (Fig. 3d, e). Thus, relative to structurally categorized phages, the φTE capsid represents an oversized T = 13 architecture. Interestingly, φTE contains no insertion domains. The pairwise alignment of MCP chains demonstrated that the φTE MCP is similar in mass and physical size to other T = 13 phage capsids (Fig. 3c, g). This indicates that similar to the domain packing in thermo phage P74-26, the oversized φTE capsid is likely due to planar A protein domain packing that increases hexamer diameter, not due to insertion of domains (Fig. 3f)38.

a Schematic diagram of geometry analysis methods. b Capsid radius measurements of all T = 13 capsid models used for geometric analysis. Radius measurements are calculated as the distance between centroids of pentamers (vertex-vertex), or between centroids of 3-fold axes MCPs (face-face). c Molecular weights of all T = 13 capsid models used for geometric analysis. d Surface area of all T = 13 capsid models used for geometric analysis. e Internal volume of all T = 13 capsid models used for geometric analysis. f) Diameter measurements of T = 13 hexamers (images are to scale). All genome sizes are listed except for T4 as this model represents an aberrant T = 13 architecture (indicated by *). g Structural alignment and to scale depictions of all MCP’s used in geometric analysis to show relative MCP size. The A-domains of each model were aligned pairwise to the equivalently positioned ASU chain in phage T5 (T5 chain A). PDBs of models analyzed: φTE = 9CUL, Mic1 = 6J3Q, T5 = 6OMC, T4 = 5VF3. Source data are provided as a Source Data file.
The φTE tail
The φTE tail comprises a helical tube encased in a sheath, with structurally distinct ends designed to interface with the phage head and baseplate. It is assumed that the assembly of the φTE head and tail structures occurs independently and are then joined at the interface between the head completion protein and the tail terminator protein—similar to other myophages8,39. In this section, we first describe the helical tail component so that the reader can better understand interactions between connector proteins and tail proteins.
The φTE tail follows the typical architecture of myovirus morphotype bacteriophages, consisting of a rigid inner tube and an external contractile sheath assembly. Each helical assembly is composed of a series of hexameric rings. The inner tube is formed by tube protein subunits consisting of a primary eight-stranded β-sandwich, flanked at one side with a large α-helix (α-1, Supplementary Fig. 2a). The luminal component of each tube proteins β-sandwich also intercalate with adjacent tube proteins in the hexameric disk, augmenting one another to form a continuous 24-stranded β-barrel on the internal surface of each ring (Supplementary Fig. 2a). Inspection of the electrostatic surface potential at the inner tube lumen reveals a strong negative charge distribution, typical of bacteriophage DNA conduits (Supplementary Fig. 2b)13,40,41. An extended β-hairpin motif extends between strands 2 and 5 of the β-sandwich. This motif wraps around the tube in the head distal direction, making contacts with loops between strands of β-sandwiches from several head distal tube subunits, forming a significant part of the tube inter-ring interface. An 18 amino acid N-terminal extension wraps similarly around the β-hairpin in the head proximal direction, wrapping over the β-hairpin of the closest tube subunit in the adjacent head proximal ring, providing stability to the inter-ring interface. The C-terminal arm of the rigid inner tail tube protein features a second α-helix (α-2); a similar helix was observed in the phage 80α gp53. However, the relative position of each helix differs42. The α-2 helix in tube protein wraps laterally around the α-1 helix of an adjacent intra-ring tube subunit, contacting the β-sandwich of the same subunit while also forming strong hydrophobic contacts with helices from the sheath D1 domain (described below).
The outer helix, composed of the tail sheath protein, was observed in both a native and contracted conformation (Fig. 4a, b). Structurally, the sheath assumes a type 2 tail sheath protein fold, featuring a central conserved core composed of three domains: D1, D2, D4, and an external domain referred to as D3 (Fig. 4d)43. Subunits of the sheath protein contact virtually every adjacent subunit in the sheath lattice; however, a primary and significant point of contact occurs by forming a four-stranded β-sheet referred to here as the hinge sheet (Fig. 5i). The hinge sheet is formed by three sheath monomers utilizing strands S1-4, formed by the D1 and D4 domains (Fig. 4d). Strands in the hinge sheet were found to be the most prominent point of conformational change during contraction (Fig. 4e). It appears that through the rotation of strands in the hinge sheet, sheath subunits can undergo rigid body rotation during contraction while maintaining lattice connectivity.

a Depiction of extended tail reconstruction with sheath subunits shown in pale cyan and tube subunits shown in yellow. Map: EMD-45419 (3.5 Å), threshold = 0.72 (5.5 σ). b Depiction of contracted tail reconstruction with sheath subunits shown in blue. Map: EMD-45420 (3.5 Å), threshold = 0.611 (5.2 σ). c Top-down view of native and contracted tail reconstructions showing expansion in sheath radius during contraction. d Domain organization of sheath protein. Strands marked with “S” describe various strands that participate in the formation of β-strands found in the hinge β-sheet. e Overlay of extended and contracted sheath proteins, with conformational shifts annotated.

a N-terminal tape measure protein densities shown in 6 colors for visual separation. Map: EMD-45435 (C6 map component 3.3 Å), threshold = 4.32 (3.9 σ) (b) Portal assembly with alternating portal monomers shown in red and adapters shown in blue. Portal monomers shown in cartoons contact the single adapter protein shown in cartoons. Map density is shown as an iso-surface. Map: EMD-45435 (C12 component map 3.1 Å), cyan adapter region threshold = 7.04 (6.3 σ), red portal region threshold 3.48 (3.1 σ). Adapter internal hydrophobic surface is shown to the right. c Consensus structure of connector region with sheath and tail terminator proteins shown in cartoons. Colored boxes relate to panels below (g–i), depicting various conformations of the hinge-sheet. d Head completion protein positively charged residues shown facing the connecter lumen. e Cross section of head completion proteins shown with electrostatic surface representation. f Side view of HC positively charged residues. g Hinge-sheet formed between TT and first head proximal sheath ring. h Hinge-sheet formed between first and second head proximal sheath rings. i Prototypical hinge-sheet formed by all other sheath rings in the tail.
The D1 domain creates the inner surface of the sheath and most of the hinge sheet. Firstly, the C-terminus of D1 forms the S3 strand (Y443-V451) of the hinge sheet; the strand continues to nestle between the D1 and D4 domains of the adjacent sheath that provides the S4 strand to the same hinge sheet. The D1 component of the sheath then forms two helices (helices α-9, α-10, Fig. 4d) and two strands; the strands establish S1 and S2 of the hinge sheet (S1: D407-P413, S2: I427-V436) and are separated by a short loop that features a small helix. Helices α-9 and α-10 then make extensive contacts with tube proteins, particularly in the outward face of the tube β-sandwich motif and the C-terminal α-helix. The only sheath-tube contacts not mediated by these helices are constituted by a loop extending from D1 (I394-P408) found immediately upstream of S1. As such, the meta-stable adherence of the sheath monomers to tube proteins appears entirely mediated by D1. While strands S1-3 are all part of D1, these strands appear to undergo minimal structural reorganization during contraction, which is primarily facilitated by rotations in the S4 pivot point, situated in D4 (discussed further below). The D2 domain is globular and rich in helices, featuring a six-stranded β-sheet at its center, where D4 contributes three strands. The D3 domain is not present in all described myophages and is composed of a seven-stranded β-sandwich, flanked with an α-helix connecting strands 3 and 4 of the β-sandwich. D3 is positioned laterally relative to the sheath-core domain in both native and contracted states. Since D3 doesn’t contact adjacent subunits in either conformation, it is unlikely D3 plays a role in sheath in φTE contraction. Homologous domains were also observed in phages A-1(L), Milano, XM1, E217 and Pa193 (Supplementary Fig. 3a). Amongst these phages, the D3 domain appears to come in two topologically linked classes (class I: φTE, Milano, A-1(L), class II: Pa193, XM1, E217) (Supplementary Fig. 3b). The relative position of D3 domains is found in a range of orientations, including a capsid proximal direction (XM1), a lateral position (A-1(L)), and a capsid distal position (Milano) (Supplementary Fig. 3a)9,11,14,15,16,44.
Finally, the D4 domain is conserved in many structurally described myophage tails8,9,11,16,44. The N-terminal strand of D4 forms the S4 strand of the hinge sheet (E3-L15), which undergoes significant rotation during contraction (Fig. 4e). It is this rotation that allows the pivoting of whole sheath subunits, making this strand essential for sheath contraction. The remainder of D4 is globular and is nestled against D2 in the native conformation.
Sheath subunits undergo a significant multi-axial rotation during contraction and are radially shifted away from the helical axis by 22 Å. The result of this rotation and translation is a substantial change in helical parameters, resulting in a change in helical twist and rise from 24.67° and 37.77 Å in the native state to 32.67° and 15.93 Å in the contracted state (Fig. 4c). Accompanied by these changes is a substantial increase in contact surface area between sheath units by 36%, a more than doubling of predicted H-bonds, and a substantial net decrease in Gibbs free energy (extended = -66.1 ΔG kcal/mol, contracted = -94 ΔG kcal/mol) indicative of a stronger binding interface33.
The φTE connector
The φTE connector is structurally formed as a quaternary structure consisting of a portal protein ring, an adapter protein ring, a head completion protein (HC) ring, a tail terminator protein (TT), and several disks of conformationally distinct sheath and tube proteins (Fig. 5b, c).
The dodecameric portal assembly is integrated into one vertex on the φTE capsid. The portal protein features the typical crown, wing, stem, and clip domains found in myophage portals45. An interesting interaction occurs with the second C12 ring of adapter proteins. The adapter protein contains an N-terminal domain closely resembling the core adapter domains observed in other phages. The globular N-terminal domain is rich in helices. It features a β-hairpin motif that extends in the head distal direction, forming a 24-stranded β-barrel that inserts into the head completion protein, facilitating the transition from 12-fold to 6-fold symmetry. The N-terminal portion of the adapter protein intercalates between the clip domains of two adjacent portal proteins. This pocket is highly hydrophobic and is predicted to form a strong interaction interface. Unlike other described myophages, the φTE adapter extends through this pocket, forming first a short α-helix that coils in a z-plane around the portal, followed by a series of looping structures. Accordingly, each adapter protein contacts five portal subunits (Figs. 5b, P1–5). This structure twists around the stem loops of the portal, taking on the character of a continuous helical ring, which we call the “bouquet motif” as it forms a sinch around portal stem helices.
The HC comprises an eight-stranded β-sandwich, similar in structure to tube proteins. The HC N-terminus forms a continuous loop around the adapter β-barrel facilitating adaption. The protein features two β-hairpins that appear significant in function; the inner β-hairpin (P57-Y81) seems to be conserved in myophage connectors, facing the inward DNA channel. The φTE HC also features an additional loop extending from this hairpin into the DNA channel (aperture loop), narrowing the constriction point to 24 Å (Supplementary Fig. 2d). Electrostatic inspection of the entire internal HC interface shows a strong positive charge distribution, with three positively charged residues extending off the aperture loop (Fig. 5d–f). The outer β-hairpin extends over a loop in the lower head distal TT disk; this TT loop (E38-G45) appears then to be clamped between both β-hairpins. This constitutes the primary interface between the HC and TT.
The tail is terminated at the distal end by addition of the TT protein. While the overall protein core fold is similar to other myophage TT’s, the φTE TT features an additional C-terminal extension that mimics interactions made by sheath proteins in lower sheath disks. This extension contributes two strands to the first head proximal hinge sheet, replacing S3 and S4, usually provided by two head-proximal sheath proteins (Fig. 5g). The extension then extends further head distal, replacing the S4 strand in the hinge sheet between sheath rings 1 and 2 (Fig. 5h). All subsequent sheath rings then assume a regular hinge sheet topology (Fig. 5i). In the φTE connector, the N-terminal arm of sheath proteins in the first sheath ring that would normally provide the S4 strand to the hinge sheet between sheath rings 1 and 2 is pinned in an alternate conformation, rendering it sterically incapable of augmenting this hinge sheet. This alternate topology significantly disrupts direct sheath lattice connectivity in the φTE connector (Supplementary Fig. 2c).
Finally, inspection of the luminal space of the head proximal tail segments revealed a clearly defined six-fold helical structure (Fig. 5a). The positioning of this structure is indicative of the TMP. While a clear α-helical secondary structure was evident, side chains could not be resolved clearly, leading us to believe that TMP N-terminal strands form a loose helical bundle that is not firmly anchored to adjacent tube proteins. To confirm that the 6-fold symmetry observed here was not a symmetry artifact, we reconstructed a local map of the entire tail with C3 symmetry, using correctly aligned baseplate particles with restricted rotational searches. In this reconstruction, six strands can be observed clearly at the N-terminus and several other locations along the tail, though the helical clarity was reduced in C3 reconstructions (Supplementary Fig. 7). Based on this observation, we propose that the φTE TMP features a six-stranded stoichiometry at its N-terminus. Assuming a predominantly α-helical fold of the TMP along the tail tube, the predicted length of the TMP correlates favorably with the observed length. The six-fold structure of the TMP in the connector region would roughly map to residues 60-180.
The φTE baseplate
The φΤE baseplate was modeled as a composite reconstruction comprising four local reconstructions due to baseplate mobility. The φTE baseplate is a large molecular compound composed of nine structurally distinct proteins, with 81 chains per complex totaling 4 MDa molecular weight (Supplementary Movie 2). The φTE baseplate follows the modular organization observed in phages T4, XM1, Pam3, фCD508, E217, Milano, A-1(L) and Pa193 with three major components: the central hub, which forms the baseplate core; six wedge protomers (sometimes referred to as the nut), which form the base and sidewalls of the baseplate; and the fiber network, which is attached to the outer radius of the wedge (Fig. 6a).

a Summary side view depiction of the φTE baseplate with individual peptides annotated. Structures in white represent unmodelled AlphaFold predictions rigid body-fit to density. Translucent colored densities represent SASA representations of peptide chains. b Top view of the wedge module fitted to composite density used for modeling. White cartoons show AlphaFold predicted unmodelled domains of Wedge 2B rigid body-fit to density. Map: EMD-45953 (C6 map component 3.3 Å), threshold 6.51 (6.0 σ). c Side view of wedge module showing the trifurcation unit and contact between the wedge helical bundle and sheath initiator proteins. d Schematic of STF with modeled residues shown in color and predicted domains shown in white. e Schematic of LTF with modeled residues shown in color and predicted domains shown in white (does not represent extended conformation). f Binding site of STF to outer wedge via Wedge 1 proteins. g Binding site of LTF to outer wedge via Wedge 2 A and sheath initiator proteins. h Putative di-sulfide bond between sheath initiator C143 and Wedge 1 C143.
The wedge complex comprises six lobed protomeric units. Each protomer contains four protein chains: a single copy of wedge 1 protein, two copies of wedge 2 protein in alternate conformations, and a singular IG-like protein we refer to here as the spacer (Fig. 6b). The wedge 1 and wedge 2 proteins form the bulk of the wedge, constituting the canonical trifurcation unit and helical bundle described in previous work (Fig. 6c)46. The trifurcation unit acts as a coordination platform that adheres wedge 1 and 2 proteins into a continuous ring while the helical bundle contacts the sheath initiator.
The φTE inner hub shows C3 symmetry and comprises four proteins (Fig. 7b). The first ring of the hub is a six-fold ring of tube initiator (TI) proteins. Despite a six-fold stoichiometry, TI proteins adopt significant deviations in loops that contact the hub to enable the transition from C6 to C3 symmetry. As such, they are best described using C3 symmetry. The core of TI shares the fold of the tube protein described above, with additional large insertions at the N and C termini that pervade much of the baseplate, forming contacts with adjacent TI, sheath, sheath initiator (SI), spacer, wedge 2, and hub proteins (Supplementary Fig. 4). The trimeric hub ring is located head-distal from the TI protein, composed of a large β-sandwich and a head-distal α-β domain that forms the interface for 1:1 binding with the puncture apparatus (PA) trimer below. The PA itself shows close structural homology to the PAM3 homolog gp20, composed of an N-terminal tri-helical bundle (formed by three monomers), a putative oligonucleotide/oligosaccharide binding site, and a C-terminal β-helical structure terminated by an α-helical domain8. While modest local resolution at the C-terminal tip prevented modeling, the PA AlphaFold-predicted structure could be well accommodated in the density, indicating that its tip structure is similar to PAM3 gp20. The spacer protein sits between hub proteins and the outer wedge, acting as an adapter between the outer C6 and inner C3 environments (Fig. 7a)

a Top-down view of the hub module showing spacer mediated adaption of wedge protomers to the hub. b Consensus image of the hub module. White translucent density represents the composite map used for modeling. Map: EMD-45953, threshold 6.51 (6.0 σ). c TMP shown fitted to its respective density. d TMP C-terminal residues interacting with two PA monomers (blue mesh shows composite density map). Map for (c, d): EMD-45953, threshold 2.69 (2.5 σ). e TMP interactions with PA tri-helical bundle and hub side walls.
Also, within the hub module, we identified three helical densities we assigned as the C-terminus of the TMP, observed to intercalate with the PA helical bundle (Fig. 7c). Due to the high map quality of our C3 baseplate reconstructions, we were able to model this TMP fragment and inspect its interactions with hub and PA rings. Contact analysis showed that each TMP fragment is deeply buried within the hub, contacting all three PA subunits and two of three hub subunits in each baseplate, with a total buried surface area of 1,440 Å2 per monomer. The helical portion of the TMP fragment forms a strong hydrophobic core with the reciprocal PA helical bundle via a network of phenylalanine, tyrosine, and leucine residues, many likely forming π-stacking interactions while also forming strong hydrophobic and stacking interactions with isoleucines and tyrosines on the hub protein inner surface (Fig. 7e). The very C-terminal tip of the TMP is then buried deeper between the hub and PA subunits (Fig. 7d).
Finally, φTE features an intricate fiber network composed of a short-tail fiber (STF), a long-tail fiber (LTF), and a third putative fiber candidate previously annotated in the φTE genome (Fig. 5d, e, Supplementary Fig. 5c). Both STF and LTF had sufficient density for modeling most N-terminal residues (STF: residues 3-92, LTF: residues 14-171), except for some N-terminal loops in the LTF (Supplementary Fig. 5b). AlphaFold3 multimer predictions produced extended models with structural features that fit map regions with low local resolution (Supplementary Fig. 5d). Predicted structures are indicated in all figures.
The short tail fiber shows a trimeric configuration predicted to extend ~300 Å. Its N-terminus contains two loops (L1, L2) that wrap in an anticlockwise direction around one another, forming a globular joint that attaches to wedge 1 monomers through electrostatic and hydrophobic interactions (Fig. 6f). The AlphaFold model of STF predicts a long α-helical coiled-coil terminated with a large globular region rich in Ig-like domains; these features could not be modeled but are supported by low-pass filtered experimental reconstructions (Supplementary Fig. 5d).
The LTF has a predicted total length of 512 Å and forms trimers (Fig. 6e). Each trimer features an N-terminal α-helical bundle, a globular domain, and a short β-helix followed by a short α-helical bundle. The AlphaFold model of LTF then predicts a highly extended β-helix with several flexible regions (Supplementary Fig. 5d). It is unclear exactly how the C-terminal half of the LTF is organized. Still, based on predictions and low-pass filtered baseplate maps, we hypothesize it first contacts the STF before bending toward the capsid and contacting the 3rd putative fiber (Supplementary Fig. 5d).
The modeled N-terminal region of LTF shows some interesting features; most notably, the N-terminal helical bundle is augmented with a 4th helix derived from the SI C-terminus (Fig. 6g). We refer to this feature as the rip-chord bundle. Finally, we identified a putative disulfide bond between the wedge monomer wedge 1 and the SI, immediately N-terminal relative to the rip-chord bundle (GP7:C143, SI:C143) (Fig. 6h). The putative covalent bond is proximal to the attachment site of STF on wedge 1.
Discussion
In the present work, we have applied cryo-EM analysis with a set of focused data-processing strategies to obtain a structural atlas of the entire φTE virion. Surprisingly, the φTE capsid has a larger volume than other T = 13 phages despite no increase in triangulation and minimal changes to MCP size and topology. The larger volume is likely required to package the phage genome, which is larger than the T = 13 phages compared here (genome sizes in Fig. 3f). A similar phenomenon has been observed in phage P74-26, where it was proposed that increased capsid volume was caused by increased hexamer size to avoid the transition to a larger triangulation number associated with greater capsid weakness28. However, changes in triangulation may sometimes provide fitness advantages in headful packaging phages through increasing redundant DNA, permitting the insertion of novel genes without disrupting the current functioning genome47. Our results support a combination of these mechanisms by which capsid volume can be evolutionarily fine-tuned across a range of sizes and volumes, highlighting the plasticity of the HK97 fold.
The φTE extended sheath displays an intricate mesh structure primarily held together by a four-stranded hinging β-sheet. Examining the contracted sheath subunits shows that rotations in the S4 sheet strand predominantly mediate sheath contraction and that the function of the sheath proteins is primarily to maintain the shape and connectivity between strands of the hinge sheet. We found that the four-stranded hinge sheath is maintained in every sheath ring of the tail, with strands donated by the tail terminator in the two head proximal sheath rings and by the sheath initiator in the baseplate proximal sheath ring. Though a similar phenomenon has been observed in other phages, the tail terminator generally contributes a single strand to hinge sheets, while φTE contributes three8,9,16,44. This may be necessary in the case of φTE, as the steric blocking of the head proximal sheath ring S4 strand results in reduced connectivity between the first and second sheath rings (Fig. 5g–i). This seems likely, as stated above; the S4 strand allows sheath subunit rotation while maintaining connectivity during contraction. Thus, the extended tail terminator C-terminus may cement the top two sheath rings to facilitate complete contraction. The fact that this four-stranded β-sheet is maintained along the whole length of the tail suggests that the formation of strong hinge β-sheets is important for stability and proper function.
The presence of the non-conserved D3 domain represents another point of interest in the sheath. These domains have now been observed in bacteriophages infecting Vibrio parahaemolyticus (XM1), Agrobacterium tumefaciens (Milano), Pseudomonas aeruginosa (E217, Pa193), Anabaena sp. PCC 7120 (A-1(L)), Pectobacterium atrosepticum (φTE)9,14,15,16,44. Phages amongst this group utilize a range of receptors (LPS, flagella) and infect hosts that occupy a range of dynamic environments (plant tissue, mammal gut, freshwater). Despite the apparent common presence of the D3 domain in bacteriophage sheaths, cyanophage Pam3 and most described contractile ejection systems (AFP, R2 pyocin, Diffocin, PVC) lack this domain48,49,50,51. While in the φTE sheath, D3 domains do not appear to contact adjacent sheath monomers in the extended or contracted conformations, the equivalent domain in phage Milano encodes disulfide bonds that bind the sheath into a locked state that must undergo reduction prior to contraction14. Thus, it appears that in some circumstances, the D3 domains play a significant role in contraction, while in other cases, they do not. In several described instances, D3 domains are also found to facilitate interactions with tail fibers, such as in φTE, A-1(L), and Bas63 (Bas63 unpublished data: Hodgkinson-Bean, J. Ayala, R. McJarrow-Keller, K. Cassin, L. Rutter, G.L. Crowe, A. Wolf, M. and Bostina, M). It is unclear if these domains provide some broadly conserved function, however it does appear that D3 domains can be co-opted for non-conserved functions in individual species.
The connector plays an essential role in acting as a conduit during DNA entry and exit; however, this process is complicated by the independent assembly of the capsid and tail, requiring that DNA is constrained in the capsid prior to tail attachment. In the φTE connector, we observed strong positive charge distribution in the head completion protein and a narrow aperture, approximately ~ 24 Å diameter, created by inward-facing loops. Similar loops were described in phage XM1, forming a larger aperture (Supplementary Fig. 2d)16. The combined effect of channel constriction and positive charge distribution may aid in arresting DNA in the connector before the connection of the head and tail complexes and subsequent plugging by the TMP. Due to the disorder of the TMP N-terminus and DNA disorder, it is hard to identify the point at which the DNA is extended in the native connector complex; this constriction point may confine the DNA until genome ejection occurs.
An important element required for the assembly and function of the phage tail is the TMP. The TMP determines the tail morphology and likely participates in the injection of the genome across the host membrane52,53,54; however, in most myophage reconstructions to date, the TMP remains disordered, preventing analysis of TMP stoichiometry and structure. Interestingly, we observed the TMP in two distinct oligomerization states along the tail. For the region of the tail extending from the neck region to just above the baseplate, six copies of the TMP are present, adopting in parts a clear helical bundle arrangement consistent with prior hypotheses55 (Fig. 5a). On the other hand, only three copies of the TMP were present in the inner cavity enclosed by the baseplate. These fragments, showing no clear density connecting them to the six copies present along the rest of the tail tube, were identified as a C-terminal fragment of the TMP based on a predicted tri-helical structure from AlphaFold and quality of map fit. The different number of copies of the bulk of the TMP and its C-terminal fragment suggests that proteolytic cleavage takes place on the full-length TMP peptide, leading to the formation of large and small fragments that can be integrated differently in the virion. Proteolytic cleavage of the TMP has indeed been confirmed for siphophages56,57 and was recently theorized to occur in myophages15,42,44,58,59. Our findings suggest that TMP cleavage might be a conserved feature between major groups of bacteriophages that utilize highly variable mechanisms of infection. Whether or not TMP cleavage serves the same purpose in relation to cellular infection requires further investigation.
The myophage baseplate is thought to recognize host receptors via its fiber network. Upon binding of the fibers to cellular receptors, a conformational change is thought to transduce signals to the tail sheath and baseplate structures to initiate contraction and re-arrangements required for injection of the phage genome. Our study revealed an intricate network of contacts between fiber proteins and core baseplate components that would partly explain how these signals are transduced. Firstly, the direct contact observed between the LTF and the STF ensures that these two fibers move concertedly, possibly linking the receptor binding process and the conformational changes triggered by it. Secondly, the rip-cord motif formed by the N-terminal helical bundle of the LTF and the C-terminal helix of the SI directly links the fiber network to the φTE sheath (Fig. 6g). As a result, any rotation of the rip-cord motif (triggered, for example, by receptor binding or baseplate re-arrangement) would be directly transmitted to the sheath, possibly initiating tail contraction. Thirdly, the putative disulfide bond between the SI and wedge 1, located near the rip-chord bundle, could provide a pivoting point that transduces signals associated with receptor binding between the LTF and STF, enabling cooperativity of the effects triggered by the binding of each of them to their receptor (Fig. 6h). Finally, both the STF and LTF are directly bound to the periphery of baseplate wedge lobes and are thus capable of transducing binding signals directly to the baseplate, which must also undergo rearrangement during contraction to release the baseplate hub. The additional unmodelled fiber may also play a role in these processes.
Our findings regarding the φTE virion, in conjunction with previous studies focused on contractile systems, allow us to create a hypothesis regarding the initiation of tail contraction in φTE (summarized in Supplementary Fig. 8). Firstly, STFs and LTFs interact with host receptors. The binding order is unclear, but must involve adhesion to flagella as φTE is flagellotropic and lipopolysaccharide independent60. Following receptor binding, the movement of fibers is transmitted to the baseplate wedge through the site of fiber attachment, inducing baseplate reorganization as observed in other systems54,61 releasing the internal baseplate hub from the external baseplate wedge. Hub release uncouples the tip of the tube from the baseplate. The mechanical motion of fibers directly destabilizes the rip-cord bundle. This destabilization exerts a force on the C-terminal helix of the SI protein, which is transmitted to the first sheath ring via the hinge sheet, resulting in the contraction of the first baseplate proximal sheath ring. Destabilization of this sheath ring is propagated along the sheath, leading to sheath collapse. The SI protein destabilization is likely affected by baseplate reorganization, and multiple fibers must be activated in concert. At the capsid end, the conformation change in the sheath is applied directly to the strands of the TT protein that form part of the two head proximal hinge sheets. This would allow a conformational signal to be transmitted from the TT to the HC protein, facilitating dsDNA release, possibly through conformational change in aperture loops widening the central channel as was observed in the transition from extended to contracted T4 phage connectors62. It is unclear whether ejection of the TMP represents a secondary gating mechanism.
As a final point, our φTE baseplate reconstruction constitutes the second flagellotropic myophage baseplate described (the first being phage Milano14). Despite recent surges in bacteriophage research, little is known about the process of host surface recruitment of flagellotropic myophages. These must somehow migrate from their initial site of flagella binding (in most cases in the flagellin filament), to the host surface, followed by re-alignment of the virion perpendicular to the host surface, primed for contraction63. These constraints are not imposed on primarily LPS-dependent myophages like T4, which bind directly to the surface through LTF mediated random receptor searches that result in LTF binding, inducing STF deployment and tight binding to secondary surface receptors, resulting in perpendicular alignment to the host surface54.
The core baseplate components of our φTE structure show strong structural similarity to both phages Milano and T4, and essentially all known contractile systems, suggesting that the core mechanism of baseplate reorganization and sheath contraction is conserved in contractile ejection systems8,9,14,15,16,44,48,49,50,51. However, a point of divergence between the ΦΤE and Milano baseplates relative to T4 is found in the structure and orientation of tail fibers. Both φTE and Milano have short tail fibers that face in a head distal direction (referred to from here as downward-facing), unlike the retracted fibers observed in phage T4, and several other LPS dependent phages44,46. Similar observations have been made for flagellotropic phages F341 and 7-7-1, which were both observed to adhere to their respective host flagella via short downward-facing tail fibers that bind perpendicular to the flagellar filament64,65. This would suggest that perhaps flagellotropic phages often initiate host binding not through random receptor searches, but through direct binding of pre-deployed short tail fibers to flagella. In such a scenario, it is not clear what initiates the deployment of additional tail fibers or the triggering of base plate rearrangement. Thus, it seems very plausible that flagellotropic myophages may deviate in the sequential nature of tail contraction relative to LPS dependent phages. This paradigm makes sense in the context of phage φTE for several reasons. Firstly, φTE STFs are downward facing, primed and ready for perpendicular attachment to the flagellar filament. Secondly, a STF first binding scenario better suits our rip-cord bundle hypothesis, as transient disassociation of φTE LTFs for receptor binding would be expected to exert force on the SI C-terminal helix leading to sheath contraction and would thus be expected to occur as a secondary event. For these reasons, we deem it likely that initial φTE receptor binding is mediated by STFs. This theory can be tested further through EM analysis between phage φTE and flagella filaments.
To conclude, our comprehensive model provides a solid structural basis for performing studies of the phage infection mechanism.
Methods
Sample purification
For lysate preparation, 6 ml of Pectobacterium atrosepticum overnight culture was added to 200 mL low-NaCl LB (10 g/ml tryptone, 5 g/mL yeast extract, 5 g/mL NaCl). It was incubated for 1.5 hours at 25 °C. 400 ml of high titer φTE sample was added to the culture, followed by incubation at 25 °C for 17 hours for phage growth and cell lysis. Crude lysates were centrifuged at 10,000 × g for 20 minutes (Thermo Fisher Scientific (TFS) LYNX 6000, Fiberlite F12-6 x 500 LEX rotor) at 20 °C; then the supernatant was decanted into an autoclaved Schott bottle. The supernatant containing phage was filtered using a 0.45 µm filter. The sample was then loaded into polypropylene thin-walled 38.5 mL ultracentrifuge tubes (ref 326823) undelayed with 3 mL of 20% w/v sucrose. Tubes were filled to the top and topped with φTE buffer (10 mM Tris – HCl pH 7.4, 10 mM MgSO4, and 0.01% w v21 gelatin). Tubes were loaded into SW32 swing buckets. The sucrose cushion was spun at 50,000 × g for 1 hour 30 minutes at 20 °C (TFS Sorvall™ WX+, Beckman SW32 TI rotor). The sample supernatant was decanted, and the residual buffer was tapped out of tubes. All six pellets were resuspended in 500 ml φTE buffer overnight at 4 °C. Pellets were pooled into a fresh 50 mL falcon (without any up and down pipetting) and were diluted with φTE buffer to a total volume of 22.5 mL.
Two CsCl gradients were prepared; each gradient contained 1.5 ml of CsCl at densities of 1.33, 1.45, 1.6 and 1.7 g/cm3. Gradients were prepared in thin wall 17 mL polypropylene ultracentrifuge tubes (ref 337986). To both gradients, 11 mL of the sample was decanted. Gradients were transferred into Beckman SW32.1 swing buckets and were centrifuged at 100,000 × g for 4 hours at 20 °C. Thick bands were visible post-centrifugation at the interface between 1.45 and 1.6 g/cm3. Bands were carefully harvested using a filtered p1000 pipette. Samples were initially diluted 1:2 with φTE buffer and were then concentrated and fully buffer exchanged into φTE buffer using Amicon 100 kDa cutoff 500 mL concentrators centrifuged at 14,000 × g at room temperature. Samples were stored at 4 °C for later use.
Cryo-EM data collection
Samples for cryo-EM were prepared by depositing 3.5 μL of purified phage solution onto Quantifoil R2/1 gold grids previously plasma cleaned with a 20% hydrogen, 80% oxygen plasma using a Gatan Solarus (Gatan Inc, USA) for 30 s. Samples were then blotted at 4 °C and 100% humidity with a FEI Vitrobot Mark IV (waiting time 30 s, blotting time 6 s, blotting force 0) before flash-freezing in liquid ethane-propane (50:50).
A set of 52,662 movies were collected on a Titan Krios cryo electron microscope (TFS) operating at an acceleration voltage of 300 kV. Images were recorded semi-automatically with EPU software (TFS) with a Falcon 3EC direct electron detector (TFS) in linear mode at a nominal magnification of 78,000, resulting in a pixel size of 1.387 Å/pixel. A defocus range from -0.5 μm to -3.5 μm was applied. The total dose of 50 e−/Å2 was fractionated evenly over 42 frames.
Image processing
All φTE reconstructions were performed in cryoSPARC V4.4.166. All 52,662 micrographs were subjected to patch-based motion correction and patch CTF correction using default parameters (Supplementary Fig. 9a). Downstream workflows were performed as follows.
For the capsid reconstruction, a subset of 5000 micrographs were picked using a Gaussian blob as a reference. The resulting picks were extracted using a box size of 1000 pixels, binned to 100 pixels and subjected to 2D classification. This resulted in 2D class averages of both full and empty capsids, which were then used as templates for two additional, separate picking rounds. Particles were then extracted in both cases using a box size of 1000 pixels and subjected to 2D classification. Particles corresponding to class averages showing high-resolution features were selected for 3D reconstruction (Supplementary Fig. 9b).
For the full capsids, an initial homogeneous refinement using I2 symmetry was performed. The resulting reconstruction was used to perform local CTF refinement for its particles, after which they were cropped to a box size of 900 pixels and downsampled to 808 pixels. The resulting particles were used to perform a final non-uniform refinement with I2 symmetry.
The WALC (Walking ALignment and Classification) procedure was applied to extract particles of the connector and baseplate regions. The WALC workflow consists of iterative cycles of helical reconstruction, shifting of the resulting map along the Z-axis and re-extraction of particles with the applied shift in 3D coordinates, together with 2D classification after each re-extraction. This workflow was applied to a starting stack of tube helical segments. In each round, the helical reconstruction is performed against a reconstruction of the tail tube, using the appropriate helical parameters. A total of seven iterations with both positive and negative Z-axis shifts were performed, each iteration resulting in a shift of 230 Å along the tail. Particles showing features corresponding to either the baseplate or the connector were selected from each 2D classification round. After pooling together the baseplate and connector particles obtained from each WALC round (without combining particles corresponding to the two different parts), duplicates were removed with a proximity threshold of 300 Å. This concept is briefly outlined in Supplementary Fig. 10a.
We reconstructed the baseplate using four separate reconstructions to improve the local resolution of various components since alternating symmetries were present in the baseplate and baseplate mobility was observed through 3D variability analysis:
-C6 upper reconstruction - The C6 upper base plate reconstruction was generated using homogenous refinement, followed by local CTF refinement and non-uniform refinement with C6 point group symmetry. This structure was used for modeling the tube, sheath segments, and the SI and Spacer proteins.
-C6 lower reconstruction - The C6 lower reconstruction utilized a focused local alignment applied to particles subjected to density subtraction of all regions outside of the baseplate wedge; a focus mask covering the wedge module was utilized in this local alignment. The local reconstruction was then submitted to cryoSPARC deep EM enhancer. This map was used to model the Wedge 1 and Wedge 2 proteins.
-C3 reconstruction - The C3 reconstruction was performed using the same methods and focused on the same region as the C6 upper reconstruction but with C3 point group symmetry. This map was used to model the TI, TMP, hub, and PA structures.
-C6 fiber reconstruction - The C6 fiber reconstruction was performed using the same methods as the C6 upper reconstruction, but with a box shift implemented that centered the fiber region more central in the box.
A summary of baseplate maps used for modeling is shown in Supplementary Fig. 6. We then used the Phenix combine_focused_maps tool to create the composite map used for the deposition of the baseplate composite model (Supplementary Fig. 10b).
For the connector region, additional particles were picked through the following separate procedure. Firstly, previously aligned capsid reconstructions were symmetry expanded by I2 symmetry, followed by box re-centering and re-extraction on an arbitrary vertex using the volume alignment tools job in cryoSPARC. New Box center coordinates were set ~20 Å outward from vertices as we found this improved downstream results. The 3D Vertex coordinates were established using the capsid_vertex_finder.py script in ChimeraX, available on GitHub (https://github.com/JimHBean/capsid_vertex_finder)67. Particles were re-extracted with a box size of 336 and were submitted to a delete duplicate particles job. Particles were then submitted to 3D classification, resulting in a class of pooled connector particles with a stack size approximately equal to the initial capsid particle stack size. These particles were combined with those obtained through the WALC procedure, and an additional job to remove duplicate particles was performed. We found the combination of these methods yielded the largest number of usable particles. The connector assembly was then reconstructed as two separate maps depending on the local symmetry and environment. The lower portion of the connector was reconstructed first with C6 point group symmetry imposed via a homogenous refinement job, followed by local CTF correction and non-uniform refinement, yielding a 3.3 Å reconstruction. These aligned particles were then submitted to a volume alignment tools job to shift particle images in Z such that the portal and adapter proteins were centered. We then performed homogenous reconstruction without alignment to identify the approximate portal/adapter complex position, followed by local CTF estimation and masked local refinement with C12 point group symmetry, yielding a map with a resolution of 3.1 Å. We then used the Phenix combine_focused_maps tool to create the composite map used for the refinement68 and deposition of connector models (Supplementary Fig. 10a).
Both the native and contracted conformations of the φTE tail were reconstructed using the cryoSPARC helical reconstruction workflow. Particles were picked using the template-free filament tracer tool with particle dimensions measured from raw micrographs. For both structures, initial helical parameters were estimated from low-resolution C6 connector/baseplate reconstructions that included several rings of sheath proteins. Helical parameters were then automatically refined and implemented using cryoSPARC helical reconstruction with C6 point group symmetry imposed and otherwise default parameters (Supplementary Fig. 9c).
In order to obtain a reconstruction covering the entire length of the tails in the extended state, a total shift of 510 Å was applied to the particles used for the reconstruction of the baseplate along the Z axis and towards the capsid, which placed the resulting particle coordinates in the center of the extended tail. Particles were then extracted with a box size of 860 pixels, subjected to local CTF refinement and used to perform an homogeneous refinement with C3 symmetry only.
To obtain the entire phage reconstruction, an additional shift of 580 Å along the Z axis towards the capsid was applied to the particles used for the entire tail reconstruction. Particles were then extracted using a box size of 2000 pixels and binned to a box size of 500 pixels. An initial reconstruction was then obtained by backprojecting the particles with the alignment parameters determined during the entire tail reconstruction, without applying any symmetry. This first reconstruction was then used to perform an homogeneous refinement without symmetry. The particles were then subjected to 2D classification, and 12,638 particles corresponding to class averages containing density for both the capsid and the entire tail were selected for further processing.
A non-uniform refinement was then performed against the previously obtained first reconstruction of the entire phage, without imposing any symmetry. The resulting reconstruction and its corresponding particles were then used to train a 3D-flex model using a training box size of 200 pixels. Finally, a reconstruction taking into account the flexible motions learnt by the model was performed with particles downsampled to a box size of 440 pixels.
Local resolutions, FSC curves and angular distribution plots for all deposited maps are shown in Supplementary Figs. 11, 12 and 13 respectively.
Mass spectrometry
For mass spectrometry, the gel lanes from three independent gels of separated φTE proteins were fractionated each into five molecular weight fractions and in-gel trypsin digested using a robotic workstation for automated protein digestion (DigestPro Msi, Intavis AG). Tryptic peptides were extracted and concentrated using a centrifugal vacuum concentrator. The peptides of each fraction were reconstituted in water containing 5% acetonitrile and 0.2% formic acid and injected into an Ultimate 3000 nano-flow UHPLC-System that was inline-coupled to a LTQ Orbitrap XL mass spectrometer (Thermo Scientific, San Jose, CA). The obtained raw data were processed and searched against a combined phage and bacterial amino acid sequence database containing 484 φTE and 4739 Pectobacterium atrosepticum sequence entries respectively18, using Proteome Discoverer 2.1 (Thermo Scientific, San Jose, CA). Peptide hits were filtered for a strict FDR (false discovery rate) of q < 0.01 using the Percolator algorithm69 and only proteins that were identified by two or more peptides at a FDR of q < 0.01 were considered as significant identifications. The abundance of the proteins and peptides was quantified using two methods: Exponentially modified protein abundance index (emPAI)70 and Top371. The major difference between these two abundance values is that emPAI is based on the number of observed peptide spectrum matches as a fraction of the theoretical number of observable peptides in the sample while Top3 is based on the measured chromatographic peak area of the three strongest peptides hits from a given protein.
A table summarizing highly abundant peptides determined by mass spectrometry can be found in Supplementary Table 2.
Model building, refinement and validation
Initial models were generated using AlphaFold-2 via the Google Colab platform72,73 and more recently AlphaFold-3 via the AlphaFold-3 server74 (Supplementary Data 1). Plots of pLDDT and PAE for each initial model can be found in Supplementary Figs. 14 and 15 respectively. The initial model generated for the TMP was truncated to keep only the region for which clear, unequivocal density was observed in the baseplate reconstructions (Supplementary Fig. 16). Models were rigid body fit into density using ChimeraX Fit-To-Map. Manual model refinement was performed in Coot and ISOLDE for all protein chains, followed by refinement in Phenix_real_space_refine68,75,76. All individual chains were refined in their respective maps zoned around each model. Map zoning was implemented with a padding factor of ~8 Å around the density associated with the model being refined. Models were also padded at their periphery with additional symmetry-related models to constrain the side chains at the periphery of models being refined. No specific routine for atomic B-factor was performed beyond the automatic refinement performed by Phenix real space refinement (AlphaFold pLDDT values were used as initial B-factor values). B-factors were not interpreted or analyzed in any manner. Individual refined models were then combined into their respective composite ASU models which were subsequently submitted to comprehensive validation by Phenix to test for accurate model geometry, peptide orientation, alpha carbon placement and model-map fit. Validation results are summarized in Supplementary Table 1.
Capsid volume calculations
For each T = 13 capsid analyzed, a protein center of mass coordinate array was constructed using Chimera X. These arrays represent the outer surface of the capsid as defined by the center of mass of each MCP. Delaunay triangulation was performed by generating a Python script that acts on input coordinate arrays, grouping coordinates into a continuous array of simplices that describe particle geometry. The script utilized the SciPy Delaunay module to calculate Delaunay triangulation77.
Internal volume measurements were calculated as the sum of internal volumes of all irregular tetrahedra in each model. The surface area was calculated as the sum of surface area measurements for only externally facing simplex facets.
Rv and Rf were calculated using ChimeraX. Rv was calculated as double the distance from the particle center to the center of mass of pentamers. Rf was calculated as double the distance from the particle center to the center of mass of true 3-fold axes.
Sphericity was calculated as a ratio of surface area to volume using Eq. (1):
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The atomic models used in this study have been deposited in the Protein Data Bank (PDB) under accession codes 9CUL (capsid), 9CC7 (connector), 9CB9 (extended tail), 9CBA (contracted tail), 9CUY (baseplate), 9MJN (full virion), 6J3Q (Mic1 siphophage capsid), 6OMC (T5 siphophage capsid) and 5VF3 (T4 myophage capsid). Electron density maps have been deposited in the Electron Microscopy Data Bank (EMDB) under accession codes EMD-45937 (capsid map), EMD-45435 (connector composite map), EMD-45436 (connector C6 map), EMD-45439 (connector C12 map), EMD-45419 (extended tail helical map), EMD-45420 (contracted tail helical map), EMD-45953 (baseplate composite map), EMD-45486 (baseplate C6 upper map), EMD-45488 (baseplate C6 lower map), EMD-45487 (baseplate C3 map), EMD-45491 (baseplate C6 fiber map), EMD-45613 (entire tail C3 map), and EMD-48317 (entire virion composite map). Mass spectrometry data have been deposited in the MassiVE repository under accession code MSV000097283 [https://doi.org/10.25345/C5VM43895]. Source data are provided as a Source Data file. Source data are provided with this paper.
Code availability
Two Python scripts used in this study have been uploaded and are available via GitHub. Script 1: A Python script executable in ChimeraX. Generates a marker set that corresponds to the center of mass coordinates of icosahedral symmetrized capsid proteins. This marker set can be saved as a.cmm file which contains marker coordinates. Script 1 and instructions for use can be found at: https://doi.org/10.5281/zenodo.15062753. Script 2: A Python script that takes.cmm files generated from Script 1 and performs Delaunay triangulation for capsid volume and surface area measurement. Script 2 and instructions for use can be found at: https://doi.org/10.5281/zenodo.15062882.
References
Cobián Güemes, A. G. et al. Viruses as winners in the game of life. Annu Rev. Virol. 3, 197–214 (2016).
Dion, M. B., Oechslin, F. & Moineau, S. Phage diversity, genomics and phylogeny. Nat. Rev. Microbiol. 18, 125–138 (2020).
Wilhelm, S. W. & Suttle, C. A. Viruses and nutrient cycles in the sea: viruses play critical roles in the structure and function of aquatic food webs. BioScience 49, 781–788 (1999).
Salmond, G. P. & Fineran, P. C. A century of the phage: past, present and future. Nat. Rev. Microbiol 13, 777–786 (2015).
Oromi-Bosch, A., Antani, J. D. & Turner, P. E. Developing phage therapy that overcomes the evolution of bacterial resistance. Annu Rev. Virol. 10, 503–524 (2023).
Dedrick, R. M. et al. Phage therapy of mycobacterium infections: compassionate use of phages in 20 patients with drug-resistant mycobacterial disease. Clin. Infect. Dis. 76, 103–112 (2022).
Sulakvelidze, A., Alavidze, Z. & Morris, J. G. Bacteriophage Therapy. Antimicrobial Agents Chemother. 45, 649–659 (2001).
Yang, F. et al. Fine structure and assembly pattern of a minimal myophage Pam3. Proc. Natl Acad. Sci. USA 120, e2213727120 (2023).
Li, F. et al. High-resolution cryo-EM structure of the Pseudomonas bacteriophage E217. Nat. Commun. 14, 4052 (2023).
Ayala, R. et al. Nearly complete structure of bacteriophage DT57C reveals architecture of head-to-tail interface and lateral tail fibers. Nat. Commun. 14, 8205 (2023).
Sonani, R. R. et al. Neck and capsid architecture of the robust Agrobacterium phage Milano. Commun. Biol. 6, 921 (2023).
Xiao, H. et al. Structure of the siphophage neck-Tail complex suggests that conserved tail tip proteins facilitate receptor binding and tail assembly. PLoS Biol. 21, e3002441 (2023).
Huang, Y. et al. Structure and proposed DNA delivery mechanism of a marine roseophage. Nat. Commun. 14, 3609 (2023).
Sonani, R. R. et al. An extensive disulfide bond network prevents tail contraction in Agrobacterium tumefaciens phage Milano. Nat. Commun. 15, 756 (2024).
Iglesias, S. M. et al. Cryo-EM analysis of pseudomonas phage Pa193 structural components. Commun. Biol. 7, 1275 (2024).
Wang, Z. et al. Structure of vibrio phage XM1, a simple contractile DNA injection machine. Viruses 15, 1673 (2023).
Peng, Y. et al. Structures of mature and urea-treated empty bacteriophage t5: insights into siphophage infection and DNA ejection. Int J. Mol. Sci. 25, 8479 (2024).
Blower, T. R., Evans, T. J., Przybilski, R., Fineran, P. C. & Salmond, G. P. Viral evasion of a bacterial suicide system by RNA-based molecular mimicry enables infectious altruism. PLoS Genet 8, e1003023 (2012).
Perombelon, M. C. M. & Kelman, A. Ecology of the soft rot Erwinias. Annu. Rev. Phytopathol. 18, 361–387 (1980).
Buttimer, C. et al. Novel N4-like bacteriophages of pectobacterium atrosepticum. Pharm. (Basel) 11, 45 (2018).
Wu, J. et al. Isolation and characterization of lytic bacteriophages infecting Pectobacterium atrosepticum. Eur. J. Plant Pathol. 169, 121–130 (2024).
Buttimer, C. et al. Pectobacterium atrosepticum Phage vB_PatP_CB5: a member of the proposed genus ‘phimunavirus. Viruses 10, 394 (2018).
Watson, B. N. J. et al. Different genetic and morphological outcomes for phages targeted by single or multiple CRISPR-Cas spacers. Philos. Trans. R. Soc. Lond. B Biol. Sci. 374, 20180090 (2019).
Eruera, A.-R. et al. Ejectosome of Pectobacterium bacteriophage ΦM1. PNAS Nexus 3, 416 (2024).
Camara-Wilpert, S. et al. Bacteriophages suppress CRISPR-Cas immunity using RNA-based anti-CRISPRs. Nature 623, 601–607 (2023).
Duda, R. L. & Teschke, C. M. The amazing HK97 fold: versatile results of modest differences. Curr. Opin. Virol. 36, 9–16 (2019).
Helgstrand, C. et al. The refined structure of a protein catenane: the HK97 bacteriophage capsid at 3.44 A resolution. J. Mol. Biol. 334, 885–899 (2003).
Stone, N. P. et al. A hyperthermophilic phage decoration protein suggests common evolutionary origin with herpesvirus triplex proteins and an anti-CRISPR Protein. Structure 26, 936–947 e3 (2018).
Zheng, J. et al. Asymmetric structure of podophage gp4 reveals a novel architecture of three types of tail fibers. J. Mol. Biol. 435, 168258 (2023).
Yang, F. et al. Novel fold and capsid-binding properties of the λ-phage display platform protein gpD. Nat. Struct. Biol. 7, 230–237 (2000).
Xu, J., Wang, D., Gui, M. & Xiang, Y. Structural assembly of the tailed bacteriophage varphi29. Nat. Commun. 10, 2366 (2019).
Dedeo, C. L., Teschke, C. M. & Alexandrescu, A. T. Keeping it together: structures, functions, and applications of viral decoration proteins. Viruses 12, 1163 (2020).
Krissinel, E. & Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 (2007).
Jin, H. et al. Capsid structure of a freshwater cyanophage Siphoviridae mic1. Structure 27, 1508–1516.e3 (2019).
Effantin, G., Boulanger, P., Neumann, E., Letellier, L. & Conway, J. F. Bacteriophage T5 structure reveals similarities with HK97 and T4 suggesting evolutionary relationships. J. Mol. Biol. 361, 993–1002 (2006).
Huet, A., Duda, R. L., Boulanger, P. & Conway, J. F. Capsid expansion of bacteriophage T5 revealed by high resolution cryoelectron microscopy. Proc. Natl. Acad. Sci.116, 21037–21046 (2019).
Chen, Z. et al. Cryo-EM structure of the bacteriophage T4 isometric head at 3.3-A resolution and its relevance to the assembly of icosahedral viruses. Proc. Natl Acad. Sci. USA 114, E8184–E8193 (2017).
Stone, N. P., Demo, G., Agnello, E. & Kelch, B. A. Principles for enhancing virus capsid capacity and stability from a thermophilic virus capsid structure. Nat. Commun. 10, 4471 (2019).
Arisaka, F. Assembly and infection process of bacteriophage T4. Chaos 15, 047502 (2005).
Hardy, J. M., Dunstan, R. A., Lithgow, T. & Coulibaly, F. Tall tails: cryo-electron microscopy of phage tail DNA ejection conduits. Biochem Soc. Trans. 50, 459–22W (2022).
Zinke, M. et al. Architecture of the flexible tail tube of bacteriophage SPP1. Nat. Commun. 11, 5759 (2020).
Kizziah, J. L., Manning, K. A., Dearborn, A. D. & Dokland, T. Structure of the host cell recognition and penetration machinery of a Staphylococcus aureus bacteriophage. PLoS Pathog. 16, e1008314 (2020).
Evseev, P., Shneider, M. & Miroshnikov, K. Evolution of phage tail sheath protein. Viruses 14, 1148 (2022).
Yu, R. C. et al. Structure of the intact tail machine of Anabaena myophage A-1(L). Nat. Commun. 15, 2654 (2024).
Dedeo, C. L., Cingolani, G. & Teschke, C. M. Portal protein: the orchestrator of capsid assembly for the dsDNA tailed bacteriophages and herpesviruses. Annu Rev. Virol. 6, 141–160 (2019).
Taylor, N. M. I. et al. Structure of the T4 baseplate and its function in triggering sheath contraction. Nature 533, 346–352 (2016).
Hua, J. et al. Capsids and genomes of jumbo-sized bacteriophages reveal the evolutionary reach of the HK97 Fold. mBio 8, e01579–17 (2017).
Cai, X. et al. Atomic structures of a bacteriocin targeting gram-positive bacteria. Nat. Commun. 15, 7057 (2024).
Ge, P. et al. Action of a minimal contractile bactericidal nanomachine. Nature 580, 658–662 (2020).
Desfosses, A. et al. Atomic structures of an entire contractile injection system in both the extended and contracted states. Nat. Microbiol. 4, 1885–1894 (2019).
Jiang, F. et al. Cryo-EM structure and assembly of an extracellular contractile injection system. Cell 177, 370–383.e15 (2019).
Plisson, C. et al. Structure of bacteriophage SPP1 tail reveals trigger for DNA ejection. EMBO J. 26, 3720–3728 (2007).
Boulanger, P. et al. Phage T5 straight tail fiber is a multifunctional protein acting as a tape measure and carrying fusogenic and muralytic activities. J. Biol. Chem. 283, 13556–13564 (2008).
Hu, B., Margolin, W., Molineux, I. J. & Liu, J. Structural remodeling of bacteriophage T4 and host membranes during infection initiation. Proc. Natl Acad. Sci. USA 112, E4919–E4928 (2015).
Hendrix, R. Tail length determination in double-stranded DNA bacteriophages. Mol. Biol. Bact. Virus Syst. 136, 21–29 (1988).
Tsui, L. C. & Hendrix, R. W. Proteolytic processing of phage lambda tail protein gpH: timing of the cleavage. Virology 125, 257–264 (1983).
Zivanovic, Y. et al. Insights into bacteriophage T5 structure from analysis of its morphogenesis genes and protein components. J. Virol. 88, 1162–1174 (2014).
Linares, R. et al. Structural basis of bacteriophage T5 infection trigger and E. coli cell wall perforation. Sci. Adv. 9, eade9674 (2023).
Wang, C. et al. Architecture of the bacteriophage lambda tail. Structure 32, 35–46(2023).
Blower, T. R., Evans, T. J., Przybilski, R., Fineran, P. C. & Salmond, G. P. C. Viral evasion of a bacterial suicide system by RNA–based molecular mimicry enables infectious altruism. PLoS Genet 8, e1003023 (2012).
Fraser, A. et al. Quantitative description of a contractile macromolecular machine. Sci. Adv. 7, eabf9601 (2021).
Kostyuchenko, V. A. et al. The tail structure of bacteriophage T4 and its mechanism of contraction. Nat. Struct. Mol. Biol. 12, 810–813 (2005).
Gambino, M. & Sorensen, M. C. H. Flagellotropic phages: common yet diverse host interaction strategies. Curr. Opin. Microbiol 78, 102451 (2024).
Ostenfeld, L. J. et al. A hybrid receptor binding protein enables phage F341 infection of Campylobacter by binding to flagella and lipooligosaccharides. Front Microbiol 15, 1358909 (2024).
Gonzalez, F., Helm Richard, F., Broadway Katherine, M. & Scharf Birgit, E. More than rotating flagella: lipopolysaccharide as a secondary receptor for flagellotropic phage 7-7-1. J. Bacteriol. 200, https://doi.org/10.1128/jb.00363-18 (2018).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. methods 14, 290–296 (2017).
Meng, E. C. et al. UCSF ChimeraX: Tools for structure building and analysis. Protein Sci. 32, e4792 (2023).
Afonine, P. V. et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr D. Biol. Crystallogr 68, 352–367 (2012).
Kall, L. et al. Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat. methods 4, 923–925 (2007).
Ishihama, Y. et al. Exponentially modified protein abundance index (emPAI) for estimation of absolute protein amount in proteomics by the number of sequenced peptides per protein. Mol. Cell Proteom. 4, 1265–1272 (2005).
Silva, J. C. et al. Absolute quantification of proteins by LCMSE: a virtue of parallel MS acquisition. Mol. Cell Proteom. 5, 144–156 (2006).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Bisong, E. Building Machine Learning and Deep Learning Models on Google Cloud Platform: A Comprehensive Guide for Beginners, 59-64 (Apress, Berkeley, 2019).
Abramson, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493–500 (2024).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr D. Biol. Crystallogr 66, 486–501 (2010).
Croll, T. I. ISOLDE: a physically realistic environment for model building into low-resolution electron-density maps. Acta Crystallogr D. Struct. Biol. 74, 519–530 (2018).
Jones, E., Oliphant, T. & Peterson, P. SciPy: Open Source Scientific Tools for Python. (2001).
Acknowledgements
We thank Torsten Kleffman for help with Mass Spec analysis and also Oraya Zinder, Brian Tong, Richard Easingwood and Fátima Jorge for technical help during the project. PCF was supported by Bioprotection Aotearoa (Tertiary Education Comission, NZ) and a James Cook Research Fellowship from the Royal Society Te Apārangi of New Zealand. JHB was supported by a University of Otago Doctoral scholarship. We are grateful to the OIST Imaging Section for using the cryo-EM facility, and to the Scientific Computing Section for using the HPC cluster. This work was supported by the Japan Society for the Promotion of Science (JSPS) (grant 21K20645 to RA and 24K01675 to MW and RA), and by direct funding from OIST. BNJW acknowledges support from the UKRI Biotechnology and Biological Sciences Research Council (grant BB/X010600/1).
Author information
Authors and Affiliations
Contributions
J.H.-B.: co-first author, Viral purification, Cryo-EM screening, 3D reconstruction, model building and refinement, figure production, paper writing, paper revision, bioinformatic analysis. R.A.: co-first author, Viral purification, Cryo-EM data collection, 3D reconstruction, model building and refinement, figure production, paper writing, paper revision, bioinformatic analysis. Nadishka Jayawardena: 3D reconstruction, paper revision, G.L.R.: model building and refinement, paper revision, B.W.: viral purification, paper revision, D.M.M.: viral purification, paper revision, J.K.: Python scripting and geometric analysis of capsids. P.F.: advisory, paper revision, M.W. advisory, data collection, paper writing, paper revision, supervision, funding, supervision M.B.: advisory, paper writing, paper revision, supervision.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hodgkinson-Bean, J., Ayala, R., Jayawardena, N. et al. Global structural survey of the flagellotropic myophage φTE infecting agricultural pathogen Pectobacterium atrosepticum. Nat Commun 16, 3257 (2025). https://doi.org/10.1038/s41467-025-58514-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-58514-x
