
Abstract
The electrochemical reduction of carbon dioxide (CO2) to carbon monoxide (CO) is challenged by a selectivity decline at high current densities. Here we report a class of indigo-based molecular promoters with redox-active CO2 binding sites to enhance the high-rate conversion of CO2 to CO on silver (Ag) catalysts. Theoretical calculations and in situ spectroscopy analyses demonstrate that the synergistic effect at the interface of indigo-derived compounds and Ag nanoparticles could activate CO2 molecules and accelerate the formation of key intermediates (*CO2– and *COOH) in the CO pathway. Indigo derivatives with electron-withdrawing groups further reduce the overpotential for CO production upon optimizing the interfacial CO2 binding affinity. By integrating the molecular design of redox-active centres with the defect engineering of Ag structures, we achieve a Faradaic efficiency for CO exceeding 90% across a current density range of 0.10 − 1.20 A cm–2. The Ag mass activity toward CO increases to 174 A mg–1Ag. This work showcases that employing redox-active CO2 sorbents as surface modification agents is a highly effective strategy to intensify the reactivity of electrochemical CO2 reduction.
Similar content being viewed by others


Introduction
The electrocatalytic carbon dioxide reduction reaction (CO2RR) powered by renewable electricity offers an attractive carbon-negative route for producing industrial chemicals and fuels1. Among the various products of CO2RR, carbon monoxide (CO) stands out as the most competitive target when considering the market value and cost efficiency2,3,4. To render CO2 electrolysis profitable compared to conventional petrochemical processes, techno-economic analyses indicate that the Faradaic efficiency (FE) for CO should exceed 90% at current densities above 500 mA cm–2 (refs 3,4). However, achieving high-rate, high-selectivity CO production in CO2RR remains challenging, as near-unity selectivity is only observed at limited partial current densities2. This limitation arises from the large overpotential required to initiate CO2 conversion, coupled with the competing hydrogen evolution reaction (HER)2.
The linear CO2 molecule features a robust C=O bond of 1.163 Å in length and 750 kJ mol–1 in dissociation energy, making it thermodynamically stable and difficult to activate5. The two-electron, two-proton conversion of CO2 to CO typically involves the adsorption and activation of CO2 to form the *CO2– intermediate, which is subsequently hydrogenated to produce the carbon-bound *COOH species, followed by further reduction and desorption steps to yield CO (refs. 1,2). The initial activation of CO2 is demonstrated to be the possible rate-determining step, especially for silver (Ag) catalysts, due to their weak interaction with CO2 and the sluggish kinetics toward *CO2– and *COOH formation1,6,7. Therefore, ample efforts have been devoted to promoting CO2 stabilization and activation on Ag-based catalysts through approaches such as nanostructuring, doping, and alloying2,8.
Beyond solely tuning metal catalysts, surface modification with organic compounds has emerged as a promising alternative strategy9,10. This strategy leverages synergistic effects at the interface between organic molecules and heterogeneous active sites, which regulates the interfacial reactant concentrations, intermediates adsorption, and reaction energy barriers10. In this context, compounds exhibiting reactive interactions with CO2 have been explored for modifying Ag surfaces in CO2-to-CO conversion11,12. For instance, amine-confined Ag has been developed to enrich local CO2 concentration during oxygen-containing flue gas electro-reduction13, and grafting pyridine groups onto Ag electrodes has been shown to induce a ten-fold activity enhancement toward CO by stabilizing the *COOH intermediate14. Similar improvement has also been observed with Ag catalysts upon the addition of CO2-binding imidazolium ionic liquids in nonaqueous electrolytes15. These studies demonstrate the potential of using organic molecules to modulate the reactivity profiles of heterogeneous CO2RR. However, a comprehensive understanding of the structure-function relationships in molecularly enhanced catalysts remains elusive, as precise synthetic controls over their interactions with CO2 and/or relevant intermediates are still lacking16.
In our recent study, we discovered indigo (Id), a redox-active Lewis base, as an effective CO2 sorbent for electrochemically mediated carbon capture (Fig. 1a)17. Specifically, the carbonyl groups in Id can undergo electro-reduction to form a nucleophilic dianion (Id2–), which then reacts with electrophilic CO2 to afford a carbonate adduct (Id-2CO22–)17,18,19,20. Inspired by the ability of redox-active Id to capture CO2 via carbonate formation, we seek to explore the possibility of employing indigo-based compounds as molecular promoters to accelerate CO2-to-CO conversion in conjunction with Ag catalysts. We envisage that the unique binding interaction between electrochemically reduced Id and CO2 could facilitate the activation of linear CO2 molecule into a bent conformation, consequently stabilizing key reaction intermediates to lower the energy barriers for protonation steps6. Importantly, molecular engineering allows us to finetune the chemical properties of the CO2-binding sites, resulting in more favorable intermediate adsorption behaviors18.

a Reaction mechanism of redox-active Id for CO2 capture. b CV of Id under N2 and CO2 atmospheres. The experiments were conducted by dissolving 2.5 mM Id in dimethyl sulfoxide (DMSO) with 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6) at a scan rate of −20 mV s–1. Ferrocene/ferrocenium (Fc0/+) was used as an internal reference. Potentials are non-iR corrected. c DFT-optimized structure of Id-2CO22– adduct showing the bent CO2 configuration at the redox-active oxygen center. The corresponding bond angles and lengths are listed next to the structure. The black, red, blue, and white spheres represent C, O, N, and H atoms, respectively. d, e Comparison of CO FE as a function of jtotal (d), and jCO as a function of potential (e) for AgNP and AgNP+Id. Potentials have been 100% iR corrected. The flow cell was operated with 1 M KOH (pH = 14.0 ± 0.2). Data are presented as mean ± s.d. Error bars represent s.d. from measurements of three independent electrodes. f Adsorption configuration of the *CO2– intermediate at the Ag/Id interface and comparison of *CO2– adsorption energies for AgNP and AgNP+Id. The black, red, blue, white, and purple spheres represent C, O, N, H, and Ag atoms, respectively. g Potential-dependent ATR-SEIRAS contour map for AgNP and AgNP/Id. ΔR/R0 = (R − R0)/R0, where R and R0 are spectra collected at the sample potential and the open circuit potential, respectively. Source data are provided as a Source Data file.
Herein, through a combination of precise molecular design, in situ spectroscopy characterizations, and theoretical calculations, we systematically investigate a series of indigo derivatives with varying CO2 affinities to unravel the interplay between electro-activated CO2 sorbents and the catalytic performance at the Ag/organic interface. Moreover, by further immobilizing CO2-binding moieties into a macromolecular structure and interfacing them with defect-rich Ag particles on a carbon support, the resulting optimized hybrid catalyst markedly improves CO2RR reactivity and Ag mass activity toward CO. In particular, steady CO FEs of over 90% can be reached at ampere-level current densities up to nearly 1.2 A cm–2, accompanied by a notable Ag mass activity of 174 A mg–1Ag toward CO production.
Results
Probing the promotional effect of indigo on Ag catalyst for CO2RR toward CO
To confirm the reactivity of reduced Id toward CO2, we conducted cyclic voltammetry (CV) studies of Id in an aprotic electrolyte under nitrogen (N2) and CO2 atmospheres. Analogous to other redox-active CO2 sorbents, the positive shift in the reduction onset potential and the merging of the two redox waves in the presence of CO2 strongly imply a chemical interaction between reduced Id and CO2 (Fig. 1b)17,18,19,20. The formation of the Id-2CO22– adduct was also validated in our precious study17. Similarly, an anodic shift in redox potential under CO2 was observed in an aqueous electrolyte (Supplementary Fig. 1), suggesting that Id2– is also capable of binding with CO2 under typical CO2RR conditions21,22. Density functional theory (DFT) calculation further supports that the complexation with Id2– induces considerable restructuring of CO2 molecules (Fig. 1c and Supplementary data 1). The CO2 adduct adopts a bent configuration with a bond angle of 132.4° (O–C–O), and the two C–O bonds (bonds a(′)and b(′)) are elongated to 1.236–1.261 Å. The bent CO2 and the redox-active oxygen center form a C–O bond (bonds c(′)) with a length of ~ 1.486 Å. Given that the onset potential for CO2 capture by Id (0.25 V vs. the reversible hydrogen electrode (RHE), Supplementary Fig. 1) is more positive than the equilibrium potential for CO2RR-to-CO conversion (–0.10 V vs. RHE)7, we hypothesize that the introduction of CO2-binding Id to Ag catalysts could effectively active CO2 by weakening its C=O bond strength during CO2RR, thereby facilitating the subsequent reductive transformation.
To test our hypothesis, we ultrasonically mixed Id molecules with commercial Ag nanoparticles (AgNP, 20–40 nm in diameter) in isopropanol and spray-coated the resulting ink onto a carbon gas diffusion layer (GDL) to obtain the Id-modified Ag electrode (AgNP+Id). Scanning electron microscopy (SEM) images reveal that Id crystals are interspersed among agglomerated AgNP, and energy-dispersive X-ray spectroscopy (EDS) elemental mapping confirms the uniform distribution of both components across the electrode (Supplementary Fig. 2). The successful modification of the AgNP electrode with Id is also verified by X-ray photoelectron spectroscopy (XPS), which shows strong N 1s signals (Supplementary Fig. 3a). The Ag 3d XPS spectra indicate the chemical state of metallic Ag0 for both bare AgNP and AgNP+Id (Supplementary Fig. 3b). These results suggest that Id modification by physical mixing has minimal impact on the morphology or electronic structure of AgNP.
The CO2RR performance was accessed in a flow cell using 1 M potassium hydroxide (KOH) aqueous electrolyte. Compared to bare AgNP, the AgNP+Id electrode promotes CO selectivity by suppressing the competing HER to below 5%, especially at higher current densities (Supplementary Figs. 4 and 5). Specifically, AgNP+Id achieves a CO FE of 92% at a total current density (jtotal) of 283 mA cm–2, whereas CO FEs for AgNP rapidly drop to below 87% when jtotal exceeds 173 mA cm–2 (Fig. 1d). AgNP+Id also exhibits enhanced CO activity, as manifested by a ~ 70 mV decrease in overpotential at a jtotal of 30 mA cm–2 and a ~ 2.7-fold increase in CO partial current density ( jCO) at –0.57 V vs. RHE, compared to AgNP under the same conditions (Fig. 1e). Control experiments without AgNP indicate that improved reactivity upon the addition of Id shall be attributed to a synergistic effect between Id and AgNP since the Id-only sample predominantly generates hydrogen (H2) with negligible CO production (Supplementary Fig. 6). The incorporation of Id molecules slightly decreases the electrochemically active surface area (ECSA), likely due to the blocking of the Ag surface by organic moieties (Supplementary Fig. 7 and Supplementary Table 1)23. Therefore, AgNP+Id delivers an even higher ECSA-normalized jCO compared to bare AgNP (Supplementary Fig. 7d), excluding surface roughness as the contributor to the improved CO2RR performance.
We conducted DFT calculations to gain a deeper understanding of the cooperativity between Id and Ag in CO2 activation (Supplementary data 1). The results demonstrate that the configuration of adsorbed CO2 at the Ag/Id interface resembles that of the Id-2CO22– adduct (Fig. 1f). The bent CO2 molecule is stabilized through the redox-active oxygen center of reduced Id, with a *CO2– adsorption energy of –0.635 eV. By contrast, *CO2– exhibits much weaker adsorption energy of merely –0.211 eV on a pristine Ag surface, which is insufficient to initiate the CO2 reduction pathway24. These preliminary findings indicate that the electro-activated Id moiety is a promising promoter to collectively stabilize CO2 near the Ag surface, which is pivotal for boosting CO2-to-CO conversion24.
Building on the insights from DFT calculations, we then employed in situ attenuated total reflection surface-enhanced infrared absorbance spectroscopy (ATR-SEIRAS) to identify intermediate adsorption features during CO2RR. Potential-dependent spectra were collected in CO2-saturated D2O with 0.1 M potassium phosphate as the electrolyte to minimize interference from adsorbed H2O and (bi)carbonate ions (Fig. 1g and Supplementary Fig. 8). Both the AgNP and AgNP+Id samples show strong bands at ~ 1550 cm–1, ascribed to the adsorbed CO32– species25,26. The stronger intensity of this band for AgNP+Id could originate from the faster local proton consumption during CO2RR. Bare AgNP displays a very weak band at ~ 1640 cm–1 (Fig. 1g), which corresponds to the C=O stretching of the *COOD intermediate (ν(*COOD))27. The redshift of this band relative to the reported wavenumber for *COOH might be due to the deuteration effect25,28. By contrast, we observe intensified ν(*COOD) bands emerging at lower overpotentials for AgNP+Id, implying accelerated kinetics of *COOD formation.
Importantly, additional bands arise exclusively for the Id-modified sample (Fig. 1g). We assign the band at ~1698 cm–1 to the carbonate C=O stretching of the Id-2CO22– adduct (ν(C=O)), which closely matches the DFT-simulated vibration of 1675 cm–1 at the catalytic interface (Supplementary Fig. 9). The band intensity peaks at around –0.5 V vs. RHE and gradually decreases as the potential becomes more negative. Such a phenomenon reflects the progressive accumulation and conversion of activated *CO2– species. In addition, the band at ~ 1590 cm–1 can likely be attributed to the asymmetric O−C−O stretching of *CO2– evoked by the resonance structure of Id-2CO22– (νas(O−C−O)), which merges with the CO32– band at more cathodic potentials. It is noteworthy that the νas(O−C−O) band has been reported as weak or even undetectable on bare Ag, especially at low overpotentials27. Our observation of a strong νas(O−C−O) band implies the existence of an unconventional adsorption configuration of *CO2– at the Ag/Id interface, such as partial Ag−O coordination28,29, as also suggested by our DFT calculations (Fig. 1f). We also observe broader bands in the range of 1460–1530 cm–1, though their precise assignments remain debatable. In some reports, these bands are associated with adsorbed CO2 and/or protonated carbonate species25,26. Collectively, the in situ ATR-SEIRAS results support that the redox-active Id molecule can significantly facilitate CO2 activation to form key intermediates at the Ag/Id interface, namely *CO2– and *COOH, ultimately leading to promoted CO generation.
Optimizing CO2 binding affinity on indigo derivatives
Our combined experimental and theoretical results confirmed the hypothesis that modifying Ag catalysts with CO2-binding Id moieties can create reactive interfaces conducive to CO2 activation and subsequent conversion to CO. Notably, the advantage of employing redox-active CO2-binding organic molecules as promoters lies in the ability to leverage precise synthetic control to finetune their CO2 affinities18,20. This enables the insightful interrogation of the correlations between molecular properties and catalytic performance and thus directs the optimization of intermediate adsorptions in the CO2RR-to-CO pathway. Our previous studies demonstrated that introducing electron-withdrawing groups (EWGs, such as −Br and −COOH) to the structure of redox-active compounds weakens their interaction with CO2 by reducing the electron density at the binding sites while introducing electron-donating groups (EDGs, such as −OCH3 and −OC3H7) enhances CO2 affinity18. Therefore, we synthesized a series of indigo derivatives with different substituent groups (see Supplementary Note 1 for synthesis details), including 5,5′,6,6′-tetramethoxylindigo (TMId), 5,5′-dipropoxyindigo (DPId), 6,6′-dibromoindigo (DBId), and indigo-6,6′-dicarboxylic acid (DCId), to investigate their impact on Ag-catalyzed CO2RR.
CV curves of the synthesized indigo derivatives were examined in both aprotic and aqueous electrolytes to assess their CO2 complexation behaviors (Supplementary Figs. 10 and 11). Similar to Id, all of these indigo derivatives exhibit chemical reactions with CO2 upon electro-reduction in both environments, as evidenced by the positive shifts in redox potential under CO2 compared to N2 atmospheres. It is well-established that, in aprotic solvents, a linear scaling relationship exists between the reduction potential and the CO2 affinity of redox-active CO2-binding molecules30. Specifically, species with more anodic reduction potentials generally show weaker CO2 affinities. Here, we find that the onset potentials for CO2 capture in aprotic and aqueous electrolytes also display a strong linear correlation (Fig. 2a). This finding implies that molecular engineering remains effective in tuning the interaction strength between CO2 and indigo derivatives in aqueous electrolytes, which is also supported by our DFT calculation (Supplementary Table 2). Thus, the CO2 affinity of indigo derivatives gradually decreases across the series of TMId, DPId, Id, DBId, and DCId. Furthermore, it is reasonable to infer that this trend in CO2 affinity can extend to the corresponding hybrid interface with Ag catalysts for CO2RR, as corroborated by the AgNP+Id case discussed above.

a Linear relationship between the onset potentials for CO2 capture in aprotic and aqueous electrolytes for indigos functionalized with EDGs or EWGs. b, c Comparison of CO FE at different jtotal (b), and jCO at different potentials (c) for AgNP modified with various indigos. d Correlation between the CO2RR potential at a jtotal of ~ 30 mA cm–2 for modified AgNP catalysts and the onset potential for CO2 capture by various indigos. Potentials have been 100% iR corrected. The flow cell was operated with 1 M KOH (pH = 14.0 ± 0.2). Data are presented as mean ± s.d. Error bars represent s.d. from measurements of three independent electrodes. Source data are provided as a Source Data file.
The indigo derivatives were then used to modify AgNP catalysts following the same procedure as that used for AgNP+Id. Again, there are no apparent changes in the structure or chemical state of AgNP after modification (Supplementary Figs. 12–17). The as-prepared hybrids are denoted as AgNP+TMId, AgNP+DPId, AgNP+DBId, and AgNP+DCId, respectively. Introducing these indigo derivatives to AgNP drastically promotes CO2RR compared to bare AgNP (Supplementary Figs. 18 and 19). All modified samples display similar enhancement in CO selectivity, achieving FEs of ~ 93% across the investigated current densities (Fig. 2b). Nevertheless, pronounced variations in jCO with respect to overpotential can be observed. Using the CO activity of AgNP+Id as a benchmark, we found that indigos functionalized with EWGs (DBId and DCId) can further accelerate CO generation on Ag, while those functionalized with EDGs (TMId and DPId) demand higher overpotentials for CO2-to-CO conversion (Fig. 2c). The best CO2RR performance is achieved with DCId, whose CO2 affinity is the weakest among the indigo derivatives. Specifically, AgNP+DCId attains a jCO of 273 mA cm–2 at –0.54 V vs. RHE, which is ~ 2.6 times higher than that of AgNP+TMId (the derivative with the strongest CO2 affinity) at a similar potential.
Intriguingly, we identified a linear relationship between the CO2RR potential of the modified catalysts and the onset potential for CO2 capture of various indigos (Fig. 2d). This indicates that the CO2RR activity is strongly correlated with the varying *CO2– adsorption energy at the catalytic interface. DFT calculations reveal that the *CO2– adsorption energy at the Ag/DCId interface (–0.583 eV) falls between those of the Ag/Id interface (–0.635 eV) and the bare Ag surface (–0.211 eV, Supplementary Fig. 20). We postulate that, for an optimal CO2RR performance, the CO2-binding capabilities of molecular modifiers should be sufficiently strong to effectively stabilize the *CO2– intermediate at the molecule/catalyst interface, yet not too strong to potentially slow down the subsequent hydrogenation and/or *CO desorption steps in the CO pathway31. To validate this hypothesis, we evaluated the CO2RR performance of thioindigo-modified AgNP. Thioindigo has a much weaker interaction with CO2 due to its electron-deficient nature32, as evidenced by its nearly identical redox behavior under CO2 and N2 conditions in an aqueous electrolyte (Supplementary Fig. 21b and Supplementary Table 2). As a result, thioindigo shows no promotional effect on AgNP for CO2-to-CO conversion (Supplementary Fig. 21). Plotting the CO2RR potential against the onset potential for CO2 capture over AgNP electrodes modified with various indigos affords a volcano-like trend (Supplementary Fig. 22), with AgNP+DCId exhibiting the most favorable *CO2– adsorption energy toward CO formation in our case.
As a final note, no trends in CO2RR activity were observed in relation to surface hydrophobicity or roughness across the different modified electrodes (Supplementary Figs. 23–25). The comparable ECSAs of these samples also suggest that the availability of Ag active sites is not substantially affected by the modification (Supplementary Table 1). These results rule out other possible factors as main contributors to the enhanced CO2RR performance of AgNP modified with indigo derivatives.
Immobilizing CO2-binding moieties via polymerization
Albeit being the most effective promoter, molecular DCId is susceptible to dissolution into the electrolyte during CO2 electrolysis at relatively high current densities (Supplementary Fig. 26). To achieve a more stable molecular modification, we immobilized the redox-active CO2-binding moieties into a macromolecular structure. As shown in Fig. 3a, we synthesized an indigo-based polymer (P-Id) by amidation between DCId and 2,2′-(ethylenedioxy)diethylamine (DODA, see Methods for synthesis details). Fourier-transform infrared (FTIR) spectroscopy of P-Id displays a vibration band associated with the amide C−N stretching at ~ 1540 cm–1, accompanied by the disappearance of the C=O stretching band of carboxylic acid from the DCId precursor at ~ 1680 cm–1 (Fig. 3b). These findings indicate the successful amidation polymerization between the two precursors. The formation of the proposed P-Id structure is further verified by solid-state 13C nuclear magnetic resonance (NMR) spectroscopy (Fig. 3c), where the observed peaks can be assigned to the different types of carbon marked in Fig. 3a.

a Synthesis of P-Id. HBTU: 2-(1H-benzotriazol-1-yl)−1,1,3,3-tetramethyluronium hexafluorophosphate; DIPEA: N,N-diisopropylethylamine; DMF: N,N-dimethylformamide. b FTIR spectra of DCId, DODA, and P-Id. c Solid-state 13C NMR spectrum of P-Id. The peaks are assigned to the corresponding carbons labeled in (a). Asteroids denote spinning sidebands. d, e Comparison of CO FE as a function of jtotal (d), and jCO as a function of potential (e) for AgNP, AgNP+DCId, and AgNP+P-Id. Potentials have been 100% iR corrected. The flow cell was operated with 1 M KOH (pH = 14.0 ± 0.2). Data are presented as mean ± s.d. Error bars represent s.d. from measurements of three independent electrodes. Source data are provided as a Source Data file.
It is important to note that the onset potential for CO2 capture by P-Id closely matches that of DCId, confirming that the polymer structure retains the redox-active oxygen center without compromising its favorable CO2 binding affinity (Supplementary Figs. 27 and 28). As anticipated, modifying AgNP with P-Id (AgNP+P-Id) by ultrasonic mixing results in substantially enhanced CO2RR reactivity, with the CO FEs exceeding 90% across a wide range of jtotal from 30 to 850 mA cm–2 (Fig. 3d and Supplementary Fig. 29). Particularly, AgNP+P-Id achieves a CO FE of 99% at a jtotal of 100 mA cm–2, slightly outperforming AgNP+DCId (95%) at a similar jtotal. This additional improvement is likely due to further HER inhibition in the more hydrophobic microenvironment induced by P-Id (Supplementary Fig. 30). Given the comparable CO2 affinity of the redox-active centers, AgNP+P-Id exhibits almost identical overpotentials to AgNP+DCId at relatively low current densities (Fig. 3e). However, AgNP+P-Id enables CO2 electrolysis at much higher current densities (jCO up to nearly 700 mA cm–2), thanks to the suppressed solubility and enhanced robustness of P-Id, as substantiated by post-electrolytic characterizations (Supplementary Fig. 31).
Engineering Ag catalyst for enhanced utilization of Ag/indigo interfaces
To enhance the availability of catalytic interfaces, we sought to improve the dispersion and utilization of Ag active sites by incorporating highly porous carbon black as the catalyst support (Supplementary Fig. 32). Carbon supports typically offer a better conductive network, mitigate nanoparticle agglomeration, and increase mass activity by reducing catalyst loading33,34. Therefore, we prepared a carbon-supported Ag catalyst with abundant surface defects (D-Ag/C) via in situ electrodeposition (see Methods for synthesis details).
The Ag loading on the GDL was 6 μg cm–2 (equivalent to 2 wt% on carbon black) measured by the inductively coupled plasma optical emission spectrometry (ICP-OES). High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) and corresponding STEM-EDS mapping show that the Ag particles obtained via in situ electrodeposition have an average diameter of ~28 nm and are primarily isolated and well dispersed on the carbon matrix (Fig. 4a and Supplementary Fig. 33). High-resolution transmission electron microscopy (HRTEM) images indicate the presence of planar defects on the Ag surface (Fig. 4b and Supplementary Fig. 34), which can provide uncoordinated sites to potentially enhance CO2 conversion35,36. X-ray absorption spectroscopy (XAS) was applied to probe the electronic state and coordination structure of the D-Ag/C catalyst. The normalized X-ray absorption near-edge structure (XANES) spectra at the Ag K-edge confirm the metallic Ag nature of D-Ag/C (Fig. 4c), consistent with the XPS result (Supplementary Fig. 35). The Ag K-edge extended X-ray absorption fine structure (EXAFS) spectra (R-space) show the lowest intensity of peaks between 1.5 and 3.3 Å, corresponding to the first nearest neighboring Ag−Ag bond for D-Ag/C, in comparison to Ag foil and commercial AgNP (Fig. 4d). Corresponding fitting analysis indicates that the coordination numbers (CNs) of Ag decrease in the order: Ag foil (12) > commercial AgNP (10.8 ± 0.4) > D-Ag/C (8.0 ± 0.6) (Fig. 4e, Supplementary Fig. 36 and Supplementary Table 3). The slight CN diminution in AgNP relative to Ag foil is ascribed to the small particle size, while the further diminution in CN for D-Ag/C shall be mainly driven by the existence of abundant planar defects37.

a HAADF-STEM and STEM-EDS mapping of the D-Ag/C catalyst showing isolated Ag particles on carbon support. b HRTEM image of the D-Ag/C catalyst showing abundant planar defects (denoted by arrows). c, d XANES data (c) and Fourier transform magnitudes of the k2-weighted EXAFS spectra (d) at the Ag K-edge of AgNP and D-Ag/C. The spectra of Ag foil were shown as a reference. e The Ag−Ag coordination numbers obtained by theoretical fits to the EXAFS data. Source data are provided as a Source Data file.
We subsequently benchmarked the CO2RR performance of D-Ag/C in a flow cell. D-Ag/C possesses superior CO2RR reactivity compared to carbon-supported AgNP (AgNP/C) with the same Ag loading of 6 μg cm–2 (Supplementary Fig. 37). Furthermore, D-Ag/C achieves a maximum CO FE of 94% and a jCO of 279 mA cm–2 at –0.73 V vs. RHE (Fig. 5a and b). These values are ~ 1.1- and ~ 1.5-fold higher, respectively than those of commercial AgNP with a much higher Ag loading of 0.4 mg cm–2 at the same potential (Supplementary Fig. 37). The combination of the carbon support and the in situ electrodeposition method effectively prevents the aggregation of defective Ag particles and increases the density of highly active sites participating in CO2RR, leading to a significant enhancement in CO production for D-Ag/C.

a, b Comparison of CO FE as a function of jtotal (a), and jCO as a function of potential (b) for the D-Ag/C and D-Ag/C + P-Id catalysts. c Ag mass activity toward CO product at different potentials for the D-Ag/C + P-Id catalyst. Potentials have been 100% iR corrected. The flow cell was operated with 1 M KOH (pH =14.0 ± 0.2). Data are presented as mean ± s.d. Error bars represent s.d. from measurements of three independent electrodes. d Performance comparison of D-Ag/C+P-Id with reported electrocatalysts for CO2RR to CO in the gas diffusion electrode (GDE)-based electrolyzer. Details are given in Supplementary Table 4. Source data are provided as a Source Data file.
The highly dispersed D-Ag/C catalyst provides exciting opportunities for creating rich catalytic interfaces when modified with P-Id. Indeed, introducing P-Id to D-Ag/C (D-Ag/C+P-Id) increases the jCO to 818 mA cm–2 at –0.73 V vs. RHE, nearly tripling the performance of bare D-Ag/C at the same potential, meanwhile maintaining a high CO FE of 93% (Fig. 5a, b and Supplementary Fig. 38). Analogous to the AgNP+P-Id case, P-Id modification drastically extends the CO2-to-CO conversion across a wider current density range (Fig. 5a). The FEs for CO exceed 95% at a jtotal ranging from 188 to 780 mA cm–2. Remarkably, the FE for CO remains at 91% when the jtotal is ramped up to 1.17 A cm–2 at –0.80 V vs. RHE, ranking among the highest performances reported to date (Fig. 5c). The low Ag loading gives rise to a high mass activity of 174 A mg–1Ag for CO production at –0.80 V vs. RHE (Fig. 5c), which is nearly two orders of magnitude higher than previously reported AgNP catalysts and even outperforms state-of-the-art single-atom catalysts (SACs) for the CO2RR to CO (Fig. 5d and Supplementary Table 4). Note that H2 is the only product from electrolysis under an argon atmosphere, which confirms that the carbon source of CO evolution originates from the CO2 feedstock (Supplementary Fig. 39). Such a collective strategy could potentially pave the way for large-scale, cost-effective, and sustainable CO2-to-CO conversion (see Supplementary Note 2 for more discussions).
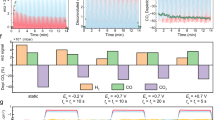
To improve the full-cell energy efficiency (EE), the D-Ag/C+P-Id catalyst was integrated into a zero-gap membrane electrode assembly (MEA) cell. The catalyst-coated membrane (CCM) method was utilized on the cathode and paired with a highly efficient Ni foam-supported Ni-Fe oxide as the anode to minimize voltage loss (Supplementary Fig. 40)38,39,40. The MEA cell, operated at 50 °C with 0.2 M cesium hydroxide (CsOH) as the anolyte, yields steady CO selectivity across the investigated current density range of 0.10−1.20 A cm–2 at cell voltages between 2.34 and 3.89 V (Fig. 6a). A peak CO FE of 94% with a jCO of 375 mA cm–2 is achieved at a cell voltage of 2.89 V, corresponding to an EE of 43% (Fig. 6b). The MEA cell also exhibits marginal decay in CO production performance over at least 20 hours at a jtotal of 400 mA cm–2 (Fig. 6c). Post-reaction characterizations demonstrate the promising durability of both the D-Ag/C and P-Id components (Supplementary Figs. 41 and 42).

a FE and cell voltage at different jtotal. b jCO and EE for CO at different cell voltages without iR compensation. Data are presented as mean ± s.d. Error bars represent s.d. from three independent measurements. c CO2RR stability at a jtotal of 400 mA cm–2 when operating the MEA cell at 50 °C using 0.2 M CsOH (pH = 13.2 ± 0.1) as the anolyte.
Discussion
In summary, using redox-active indigo molecules as a model system, we demonstrate that decorating Ag-based catalysts with electro-activated CO2-binding organics creates synergistic interfaces that significantly enhance both the selectivity and activity of CO2-to-CO conversion. The dynamic complexation interactions between these organic promoters and CO2 readily activate CO2 molecules and effectively enrich the *CO2– and *COOH intermediates at the nearby Ag catalytic sites. Importantly, by precisely tuning the CO2 binding affinities of indigo derivatives via molecular engineering, we unravel a critical volcano-like relationship between the *CO2– adsorption energy induced by organic modifiers and their promotional effect on CO2RR. This mechanistic insight culminates in the development of a hybrid catalyst that couples polymerized indigo moieties with the optimal CO2 affinity and highly dispersed, defect-rich Ag particles, achieving impressive CO2RR performance at ampere-level current densities. Furthermore, we anticipate that this redox-active molecular platform can be extended to benefit CO2 electrolysis to high-value multi-carbon products when incorporated into copper-based catalysts. Our work opens new avenues for the rational design of highly efficient, molecularly tailored catalysts. Moreover, the integration of CO2-binding species with CO2RR catalysts could potentially enable a desirable reactive carbon capture scheme where CO2 from dilute sources is directly converted into chemicals and fuels without prior concentration, offering advantages in process intensification and energy efficiency.
Methods
Chemicals and materials
Silver nanopowder (20−40 nm, 99.9%) and silver nitrate (AgNO3, 99.9%) were obtained from Thermo Scientific. Indigo powder (97.0%) was purchased from TCI America. Chemicals for polymerization, including HBTU (99.0%), DIPEA (99.5%), DODA (98.0%), and DMF (99.9%), were purchased from Sigma Aldrich. All chemicals are used as received without purification.
Organic synthesis
Synthesis procedures for indigo derivatives are provided in Supplementary Note 1. P-Id was prepared via amidation polymerization. Specifically, to a mixture of DCId (0.7 g, 2 mmol) and HBTU (1.64 g, 2.16 mmol) in anhydrous DMF (30 ml) was added DIPEA (2.09 ml, 12 mmol) dropwise. The mixture was stirred for 30 mins before DODA (2 mmol, 292 μl) was added. The reaction was stirred for another 24 hours at room temperature (20 °C) and poured into a saturated NaHCO3 aqueous solution (200 ml). The precipitate was collected by filtration, washed sequentially with saturated NaHCO3 (aq), water, 0.2 M HCl (aq), cold methanol, and dichloromethane, and dried in vacuo to give a dark blue solid (784 mg, 85% yield).
D-Ag/C catalyst synthesis
The D-Ag/C catalyst was synthesized using the in situ electrodeposition method. Vulcan XC-72 carbon black suspension (10 mg ml–1) was added to AgNO3 aqueous solution (0.33 mg ml–1) and sonicated in an ice bath for 2 h to allow the uniform dispersion of Ag+ on the carbon black support. The resulting slurry was added to isopropanol with 5 wt% Nafion solution to obtain the catalyst ink, which was then spray-coated on GDL (Sigracet 39BB) until reaching a total mass loading of 0.3 mg cm–2. The D-Ag/C catalyst was formed in situ by reducing the as-prepared GDE at 30 mA cm–2 for 15 min in the flow cell supplied with CO2 gas and 1 M KOH. The final Ag loading on the GDE was ~ 6 μg cm–2 measured by ICP-OES.
Material characterizations
FTIR spectra were collected on a Thermo Fischer Nicolet iS5 spectrometer. Solid-state 13C NMR spectrum was obtained from a Bruker Ascend 500 MHz Solids at a spin rate of 10000 Hz. Liquid 1H NMR spectra were recorded on a Bruker Avance 400 MHz with solvent residue peak as the internal reference. SEM images were collected on a Helios G5-focused ion beam SEM. XPS measurements were carried out using a PHI Quantera XPS equipped with the Al Kα radiation source. HAADF-STEM, STEM-EDS elemental mapping, and HRTEM were collected from a FEI Titan Themis3 S/TEM. XAS measurements were performed on the Beamline 7-BM of the National Synchrotron Light Source II (NSLS II) at Brookhaven National Laboratory. The Athena and Artemis software in the Demeter package was applied for data processing and analysis41. The theoretical EXAFS signal was fitted to the experimental EXAFS data in R-space by Fourier transforming both the theoretical and experimental data.
Electrochemical measurements
CV measurements were carried out on a BioLogic VSP potentiostat (BioLogic Science Instruments). Glassy carbon (3 mm in diameter) was used as the working electrode, and Pt wire was used as the counter electrode. For measurements conducted in aprotic electrolytes, Ag wire was used as a pseudo-reference electrode, and ferrocene was used as an internal reference. The organic compound (2.5 mM) was dissolved in DMSO with 100 mM TBAPF6 as the supporting salt. For measurements conducted in aqueous electrolytes, Ag/AgCl (3 M KCl) was used as the reference electrode. The reference electrode was calibrated using a standard hydrogen electrode before measurements. The compound was mixed with Vulcan XC-72 (mass ratio of 1:1) in isopropanol and drop-casted onto the glassy carbon electrode. The pH of the electrolyte (1 M KHCO3 saturated with either N2 or CO2) was measured by a pH meter (SevenCompact). The potential was converted to the RHE scale using ERHE = EAg/AgCl + 0.209 V + 0.0591 × pH. CV curves were collected at the scan rate of –20 and –10 mV s–1 for the aprotic and aqueous conditions, respectively.
Electrocatalytic CO2RR measurements
CO2RR performances were evaluated in a flow cell at 20 °C with 1 M KOH (pH = 14.0 ± 0.2) as the electrolyte. The fresh electrolyte was prepared using a volumetric flask before the testing. For the GDE fabrication, AgNP or AgNP mixed with organic species of interest were dispersed in isopropanol with 5 wt% Nafion. The concentration of AgNP was 5 mg ml–1. The molar ratio of small molecules to AgNP was kept at 10%. The P-Id content was 50 wt% relative to AgNP. After sonication for 2 h, the ink was spray-coated onto the GDL. The Ag loading was controlled to be ~ 0.4 mg cm–2 for all samples. The GDE cathode and Ni foam anode were separated by an anion exchange membrane (AEM; Sustainion X37-50, 50 μm, 3 × 3 cm2). AEM was submerged into 1 M KOH for 12 h and rinsed with deionized water before assembling into the cell. A multichannel peristaltic pump (Ismatec) was applied to feed the electrolyte (30 ml) at 1 ml min–1 and 2 ml min–1 to the cathodic and anodic compartments, respectively. A mass flow controller (Alicat Scientific) was employed to control the CO2 gas flow rate at 50 standard cubic centimeters per minute (sccm) to the cathode. The CO2RR experiments were operated in the chronoamperometry mode under ambient conditions on a BioLogic VSP potentiostat. When the current density exceeded 1 A cm–2, a d.c. power supply (B&K Precision) was used instead of the potentiostat. The cathodic potential was determined relative to the Ag/AgCl (3 M) reference electrode and converted to the RHE scale accordingly. Because of the rapid dynamic evolutions of local environments, the resistance (R) between the cathode and reference electrode was immediately measured using potentiostatic electrochemical impedance spectroscopy (PEIS) after each electrolysis, and then the ohmic drop (iR) was 100% manually compensated to each potential. The R was around 3.3 ± 0.3 Ω for an electrode area of 1 cm2. Non-iR corrected data were provided in Supplementary Table 5.
In the MEA setup, a more robust AEM (PiperION, 40 μm, 3 × 3 cm2)42 was used for the CCM fabrication. In brief, the catalyst was airbrushed onto the AEM directly, and combined with a highly hydrophobic GDL (Tory 090) to form the cathode38. The anode, Ni-Fe oxide, was prepared using a reported electrodeposition method on Ni foam39. The MEA cell was operated at 50 °C with 0.2 M CsOH (pH = 13.2 ± 0.1) as the anolyte to mitigate salt precipitation and the consequent electrode flooding43. The catalyst, membrane, and cell setup were initially activated at 30 mA cm–2 for 2 h before starting the performance measurements. The cell voltage was recorded without the iR correction.
During CO2 electrolysis, an online gas chromatograph (GC, Shimadzu GC-2014) equipped with a thermal conductivity detector was employed to monitor the gas products. The FEs of gas products were calculated as follows:
where z is the number of electrons transferred to form a target product; F is the Faraday constant; x is the molar fraction of a target product determined by GC; V is the molar flow rate of effluent gas measured using a digital flow meter (Omega); and jtotal is the total current density.
The EEs of CO were calculated as follows:
where \({E}_{{{{\rm{CO}}}}}^{0}\) is the equilibrium potential for CO2RR to CO (– 0.10 V vs. RHE)7; \({E}_{{{{\rm{cell}}}}}\) is the applied cell voltage; and \({{FE}}_{{{{\rm{CO}}}}}\) is the FE for CO.
In situ ATR-SEIRAS measurements
The ATR-SEIRAS setup was assembled according to prior works44,45. The catalyst ink was drop-casted onto a silicon ATR wafer with a thermal-evaporated Ag film (30 nm). The catalyst loading was controlled to be 0.4 mg cm–2. Graphite rod and Ag/AgCl (3 M KCl) were used as the counter and reference electrodes, respectively. The SEIRAS spectra were recorded using a Thermo Fischer Nicolet iS50 spectrometer equipped with an N2 cooled HgCdTe (MCT) detector and a Veemax III IR attachment from PIKE. The spectrometer was operated at a scan rate of 30 kHz. Spectra were acquired with a spectral resolution of 4 cm–1, and 16 interferograms were coadded for each spectrum. During in situ measurements, the electrolyte (0.1 M potassium phosphate in D2O) was continuously sparged with CO2. CO2 electrolysis was carried out at potentials ranging from –0.1 to –1.0 V vs. RHE. The spectrum collected at open circuit potential was used as the reference.
DFT calculations
DFT calculations were performed using the Vienna Ab initio Simulation Package (VASP)46. The Perdew-Burke-Ernzerhof (PBE) functional47 was applied, with a plane wave energy cutoff of 400 eV. Van der Waals interactions were accounted for using the DFT-D3 dispersion correction48. A constant potential of –0.3 V vs. RHE was used in the simulations, and the pH was set to 14, reflecting the alkaline conditions of the electrolyte. Silver relaxation was modeled using a (6 × 6 × 6) supercell with a 15 Å vacuum gap, and the Gamma point was employed for k-point sampling. The adsorption of CO2 and indigo on the Ag(111) surface was studied using the constant-potential model (CPM)49 with a VASPsol patch to consider the implicit solvation50,51. The adsorption of two CO2 molecules on the indigo and its derivatives was calculated with two extra electrons. Considering the increased complexity of the derivative molecules and the need for a much larger Ag surface, we calculated the adsorption on isolated molecules for qualitative analysis (Supplementary Table 2).
Data availability
All the data that support the findings of this study are available in the main text and the Supplementary Information. Data are also available from the corresponding author upon request. Source data are provided in this paper.
References
Birdja, Y. Y. et al. Advances and challenges in understanding the electrocatalytic conversion of carbon dioxide to fuels. Nat. Energy 4, 732–745 (2019).
Jin, S., Hao, Z., Zhang, K., Yan, Z. & Chen, J. Advances and challenges for the electrochemical reduction of CO2 to CO: from fundamentals to industrialization. Angew. Chem. Int. Ed. 60, 20627–20648 (2021).
De Luna, P. et al. What would it take for renewably powered electrosynthesis to displace petrochemical processes? Science 364, eaav3506 (2019).
Shin, H., Hansen, K. U. & Jiao, F. Techno-economic assessment of low-temperature carbon dioxide electrolysis. Nat. Sustain. 4, 911–919 (2021).
Yu, S. & Jain, P. K. Plasmonic photosynthesis of C1–C3 hydrocarbons from carbon dioxide assisted by an ionic liquid. Nat. Commun. 10, 2022 (2019).
Singh, M. R., Goodpaster, J. D., Weber, A. Z., Head-Gordon, M. & Bell, A. T. Mechanistic insights into electrochemical reduction of CO2 over Ag using density functional theory and transport models. Proc. Natl. Acad. Sci. USA 114, E8812–E8821 (2017).
Kuhl, K. P. et al. Electrocatalytic conversion of carbon dioxide to methane and methanol on transition metal surfaces. J. Am. Chem. Soc. 136, 14107–14113 (2014).
Zhao, S., Jin, R. & Jin, R. Opportunities and challenges in CO2 reduction by gold- and silver-based electrocatalysts: from bulk metals to nanoparticles and atomically precise nanoclusters. ACS Energy Lett. 3, 452–462 (2018).
Nam, D.-H. et al. Molecular enhancement of heterogeneous CO2 reduction. Nat. Mater. 19, 266–276 (2020).
Zhang, J. et al. Molecular tuning for electrochemical CO2 reduction. Joule 7, 1700–1744 (2023).
Sanz-Pérez, E. S., Murdock, C. R., Didas, S. A. & Jones, C. W. Direct capture of CO2 from ambient air. Chem. Rev. 116, 11840–11876 (2016).
Zeng, S. et al. Ionic-liquid-based CO2 capture systems: structure, interaction and process. Chem. Rev. 117, 9625–9673 (2017).
Sun, J. W. et al. Direct oxygen-containing simulated flue gas electrolysis over amine-confined Ag catalyst in a flow cell. Chem. Catal. 4, 100923 (2024).
Abdinejad, M. et al. CO2 electrolysis via surface-engineering electrografted pyridines on silver catalysts. ACS Catal. 12, 7862–7876 (2022).
Dongare, S. et al. A bifunctional ionic liquid for capture and electrochemical conversion of CO2 to CO over silver. ACS Catal. 13, 7812–7821 (2023).
Campos, K. R. Direct sp3 C–H bond activation adjacent to nitrogen in heterocycles. Chem. Soc. Rev. 36, 1069–1084 (2007).
Jayarapu, K. N. et al. Indigo as a low-cost redox-active sorbent for electrochemically mediated carbon capture. Adv. Funct. Mater. 34, 2402355 (2024).
Li, X., Zhao, X., Liu, Y., Hatton, T. A. & Liu, Y. Redox-tunable Lewis bases for electrochemical carbon dioxide capture. Nat. Energy 7, 1065–1075 (2022).
Li, X. et al. Redox-tunable isoindigos for electrochemically mediated carbon capture. Nat. Commun. 15, 1175 (2024).
Li, X., Mathur, A., Liu, A. & Liu, Y. Electrifying carbon capture by developing nanomaterials at the interface of molecular and process engineering. Acc. Chem. Res. 56, 2763–2775 (2023).
Liu, Y., Ye, H.-Z., Diederichsen, K. M., Van Voorhis, T. & Hatton, T. A. Electrochemically mediated carbon dioxide separation with quinone chemistry in salt-concentrated aqueous media. Nat. Commun. 11, 2278 (2020).
Jing, Y. et al. Electrochemically induced CO2 capture enabled by aqueous quinone flow chemistry. ACS Energy Lett. 9, 3526–3535 (2024).
Chen, B. et al. Molecular enhancement of direct electrolysis of dilute CO2. ACS Energy Lett. 9, 911–918 (2024).
Monteiro, M. C. O. et al. Absence of CO2 electroreduction on copper, gold and silver electrodes without metal cations in solution. Nat. Catal. 4, 654–662 (2021).
Zhang, Z.-M. et al. Probing electrolyte effects on cation-enhanced CO2 reduction on copper in acidic media. Nat. Catal. 7, 807–817 (2024).
Moradzaman, M. & Mul, G. Infrared analysis of interfacial phenomena during electrochemical reduction of CO2 over polycrystalline copper electrodes. ACS Catal. 10, 8049–8057 (2020).
Firet, N. J. & Smith, W. A. Probing the reaction mechanism of CO2 electroreduction over Ag films via operando infrared spectroscopy. ACS Catal. 7, 606–612 (2017).
Delmo, E. P. et al. In situ infrared spectroscopic evidence of enhanced electrochemical CO2 reduction and C–C coupling on oxide-derived copper. J. Am. Chem. Soc. 146, 1935–1945 (2024).
Chernyshova, I. V., Somasundaran, P. & Ponnurangam, S. On the origin of the elusive first intermediate of CO2 electroreduction. Proc. Natl. Acad. Sci. USA 115, E9261–E9270 (2018).
Barlow, J. M. & Yang, J. Y. Oxygen-stable electrochemical CO2 capture and concentration with quinones using alcohol additives. J. Am. Chem. Soc. 144, 14161–14169 (2022).
Hansen, H. A., Varley, J. B., Peterson, A. A. & Nørskov, J. K. Understanding trends in the electrocatalytic activity of metals and enzymes for CO2 reduction to CO. J. Phys. Chem. Lett. 4, 388–392 (2013).
Bauer, H., Kowski, K., Kuhn, H., Lüttke, W. & Rademacher, P. Photoelectron spectra and electronic structures of some indigo dyes. J. Mol. Struct. 445, 277–286 (1998).
Ma, S. et al. Carbon nanotube containing Ag catalyst layers for efficient and selective reduction of carbon dioxide. J. Mater. Chem. A 4, 8573–8578 (2016).
Sun, H. et al. Promoting ethylene production over a wide potential window on Cu crystallites induced and stabilized via current shock and charge delocalization. Nat. Commun. 12, 6823 (2021).
Wu, X. et al. Fast operando spectroscopy tracking in situ generation of rich defects in silver nanocrystals for highly selective electrochemical CO2 reduction. Nat. Commun. 12, 660 (2021).
Mariano, R. G., McKelvey, K., White, H. S. & Kanan, M. W. Selective increase in CO2 electroreduction activity at grain-boundary surface terminations. Science 358, 1187–1192 (2017).
She, X. et al. Pure-water-fed, electrocatalytic CO2 reduction to ethylene beyond 1,000 h stability at 10 A. Nat. Energy 9, 81–91 (2024).
Rios Amador, I. et al. Protocol for assembling and operating bipolar membrane water electrolyzers. STAR Protoc. 4, 102606 (2023).
Lu, X. & Zhao, C. Electrodeposition of hierarchically structured three-dimensional nickel–iron electrodes for efficient oxygen evolution at high current densities. Nat. Commun. 6, 6616 (2015).
Hansen, K. U., Cherniack, L. H. & Jiao, F. Voltage loss diagnosis in CO2 electrolyzers using five-electrode technique. ACS Energy Lett. 7, 4504–4511 (2022).
Ravel, B. & Newville, M. ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Radiat. 12, 537–541 (2005).
Endrődi, B. et al. High carbonate ion conductance of a robust PiperION membrane allows industrial current density and conversion in a zero-gap carbon dioxide electrolyzer cell. Energy Environ. Sci. 13, 4098–4105 (2020).
Cofell, E. R., Nwabara, U. O., Bhargava, S. S., Henckel, D. E. & Kenis, P. J. A. Investigation of electrolyte-dependent carbonate formation on gas diffusion electrodes for CO2 electrolysis. ACS Appl. Mater. Interfaces 13, 15132–15142 (2021).
Gerke, C. S. et al. Electrochemical C–N bond formation within boron imidazolate cages featuring single copper sites. J. Am. Chem. Soc. 145, 26144–26151 (2023).
Gerke, C. S., Klenk, M., Zapol, P. & Thoi, V. S. Pulsed-potential electrolysis enhances electrochemical C–N coupling by reorienting interfacial ions. ACS Catal. 13, 14540–14547 (2023).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Zhao, X. & Liu, Y. Origin of selective production of hydrogen peroxide by electrochemical oxygen reduction. J. Am. Chem. Soc. 143, 9423–9428 (2021).
Mathew, K., Sundararaman, R., Letchworth-Weaver, K., Arias, T. A. & Hennig, R. G. Implicit solvation model for density-functional study of nanocrystal surfaces and reaction pathways. J. Chem. Phys. 140, 084106 (2014).
Mathew, K., Kolluru, V. S. C., Mula, S., Steinmann, S. N. & Hennig, R. G. Implicit self-consistent electrolyte model in plane-wave density-functional theory. J. Chem. Phys. 151, 234101 (2019).
Acknowledgements
We acknowledge financial support from the Johns Hopkins University, the David and Lucile Packard Foundation, the Arnold and Mabel Beckman Foundation, and the National Science Foundation (NSF grant number 2237096). This work was partially performed at the Materials Characterization and Processing Center in the Whiting School of Engineering at Johns Hopkins University. Yayuan Liu and V.S.T. are grateful for the support of the Ralph S. O’Connor Sustainable Energy Institute (ROSEI). R.W. and Yuanyue Liu acknowledge the support by Welch Foundation (F-1959), and the computational resources provided by ACCESS and NREL. C.S.G and V.S.T. acknowledge the U.S. Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences, Catalysis Science program, under grant DE-SC0021955. S.R. acknowledges support from IIT Kanpur (Project number 2024098) and the Chandrakanta Kesavan Center for Energy Policy and Climate Solutions, IITK (Project number 2021136H). A.C.M. would like to acknowledge CONAHCyT for the doctoral scholarship provided under the program (CVU1051087). A.I.F. and S.X. acknowledge support by the NSF grant CHE 2102299. The work carried out at Brookhaven National Laboratory was supported by the DOE under contract DE-SC0012704. XAS measurements used resource 7-BM of the National Synchrotron Light Source II, a DOE Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under contract DE-SC0012704. The 7-BM beamline operations were supported in part by the Synchrotron Catalysis Consortium (DOE Office of Basic Energy Sciences grant DE-SC0012335). We appreciate beamline support by L. Ma, D. Yang and A. Tayal. Z.L. would like to thank T. Zhang and X. She for their valuable suggestions.
Author information
Authors and Affiliations
Contributions
Z.L. and X.L. conceptualized the project under the supervision of Yayuan Liu; Z.L. synthesized catalysts, performed electrochemical tests, and analyzed experimental data. X.L. synthesized and characterized organic compounds. R.W. performed DFT calculations under the supervision of Yuanyue Liu; C.S.G and Z.L. conducted in situ ATR-SEIRAS measurements under the supervision of V.S.T.; S.R., A.C.M., and P.M.A. performed XPS and TEM characterizations. S.X. and A.I.F. carried out XAS measurements and analyses. L.Z. and K.N.J. conducted SEM characterizations. A.M., D.L., A.L., and L.G. helped with data processing. T.L. carried out ICP-OES measurements. Z.L., X.L., and Yayuan Liu wrote the paper. All authors discussed the results and commented on the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Peer review
Peer review information
Nature Communications thanks Yafei Li, and the other anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, Z., Li, X., Wang, R. et al. Electro-activated indigos intensify ampere-level CO2 reduction to CO on silver catalysts. Nat Commun 16, 3206 (2025). https://doi.org/10.1038/s41467-025-58593-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41467-025-58593-w
